
Surface engineering of nickel selenide for an enhanced intrinsic overall water splitting ability†
Peng Fei
Liu
 a,
Le
Zhang
a,
Li Rong
Zheng
b and
Hua Gui
Yang
*a
a,
Le
Zhang
a,
Li Rong
Zheng
b and
Hua Gui
Yang
*a
aKey Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: hgyang@ecust.edu.cn
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China
First published on 18th July 2018
Abstract
The development of efficient catalytic electrodes towards the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) is at the heart of renewable-energy technologies. Despite the tremendous efforts towards engineering electrode schemes for increasing exposed surface areas and active sites, improving intrinsic catalytic activity still remains a great challenge. Here, we develop a surface-polyaniline (PANI) functionalized nickel selenide (NiSe–PANI) electrode with great performance enhancement for both the HER and OER. The decorated PANI layer subtly modulates the surface electronic structures of NiSe, with a surface-optimized selenium-enriched configuration for the HER and enhanced generation of NiIII/IV active species when oxidized for the OER. When used as a bifunctional electrocatalyst for overall water splitting, the NiSe–PANI electrode displays excellent performance, with a current density of ∼10 mA cm−2 at an applied voltage of 1.53 V during a long-term electrolysis test, and outperforms the Pt and IrO2 combination as the benchmark and most of the earth-abundant material-based bifunctional catalysts. Similar PANI–functionalization on other bifunctional nickel chalcogenide electrodes also exhibits obviously enhanced performance for overall water splitting, demonstrating the wider applicability of intrinsic activity enhancement via a surface electronic modulation strategy.
1. Introduction
Electrochemical water splitting has attracted worldwide attention, owing to its renewable-electricity-powered path to hydrogen production and chemical feedstock.1–4 Unfortunately, the large overpotentials (η) of the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) greatly hinder the promotion of water-electrolysis application.5–11 Therefore, tremendous efforts have been made towards developing efficient HER and OER catalysts with earth-abundant materials.1,2 Single bifunctional electrocatalysts in the same pH range, which can simplify the electrolyzer system and lower the total cost,12–14 are extremely essential and have recently been developed, including 3d transition metal-based oxides,15,16 layered double hydroxides (LDHs),17 sulfides,18 selenides,19 nitrides20,21 and phosphides.22,23 Furthermore, design strategies like increasing catalyst loading, optimizing catalyst morphologies and incorporating with carbon-based materials have been demonstrated to expose more surface areas, leading to more population of active sites.24–27 However, such approaches can ultimately become limited by the mass transfer of reagents and/or the conductivity of catalysts.1,15,28 Thus, it is urgently imperative to explore novel and simple modulation methods to tune the properties of these bifunctional catalysts, further improving their intrinsic electrocatalytic activity and surface charge transfer ability.Polyaniline (PANI) has attracted great attention in electrocatalysis owing to its unique π-conjugated structures, which lead to good electrochemical stability and favourable hydrophilic properties.29–33 Meanwhile, PANI can also interact with electrocatalysts and thus modulate the electronic structures of the catalysts.31,34 Motivated by that, we reason that PANI modification might be able to tune the surface electronic structures of bifunctional electrocatalysts with an enhanced charge transfer ability, and subsequently facilitate the overall water splitting ability. Since nickel (Ni)-based chalcogenides have been proved as classical bifunctional electrocatalysts with excellent activity for both the HER and OER,19,35–37 we examine whether the intrinsic activities of these Ni-based bifunctional catalysts could be improved systematically by modulating their electronic structures. To the best of our knowledge, such a surface modulation strategy with enhancements for both HER and OER activity has never been reported.
Herein, we prepare porous nickel selenide (NiSe) microsphere arrays on nickel foam, followed by rational electropolymerization of PANI (NiSe–PANI), with apparent enhancement of intrinsic electrocatalytic activity (based on electrochemical surface areas) and the charge transfer ability. Induced by electron delocalization between Ni d-orbitals and PANI π-conjugated ligands, and reasonable electron transfer from NiSe to PANI, the modified PANI layers significantly promote the surface-formation of selenium-enriched structures for improved HER activity, and enhance the in situ generation of NiIII/IV high-valence centers for NiSe precatalysts when oxidized for improved OER activity. Thus, the bifunctional NiSe–PANI electrocatalyst exhibits remarkable performance for both the HER and OER in an alkaline electrolyte (1 M KOH), affording a current density (j) of 10 mA cm−2 at η of −120 mV for the HER and 180 mV for the OER. Furthermore, an alkaline water electrolyzer based on NiSe–PANI catalysts as both an anode and a cathode gives j of ∼10 mA cm−2 at a cell voltage of 1.53 V, with excellent stability for long-term electrolysis for more than 100 hours (h), which is superior to most reported systems employing nonprecious bifunctional electrocatalysts. More importantly, the surface-PANI-engineering strategy could also be extended to bifunctional Ni3S2 electrocatalysts. The resulting Ni3S2–PANI electrodes require 1.64 V to achieve j of ∼10 mA cm−2, which is 100 mV lower than that of the initial Ni3S2 electrodes to promote overall water splitting.
2. Results and discussion
2.1 Structural characterization
To fabricate the NiSe–PANI electrocatalyst, porous metallic Ni superstructures with open spaces were first electrodeposited on commercial 3D porous Ni foam using concomitantly evolved hydrogen as a template (Fig. 1a). Subsequently, the resulting porous Ni microspheres were subjected to hydrothermal selenylation in NaHSe solution (Caution: the toxic gas H2Se would be released during the formation of NaHSe), followed by electropolymerization of PANI as a functionalization layer. Scanning electron microscopy (SEM) images are shown in Fig. S1 (ESI†), and clearly show the 3D hierarchically porous morphology of the pristine Ni microspheres. After selenylation and subsequent electrodeposition with PANI, the NiSe–PANI microspheres nearly maintained the original morphology, with uniformly stacked microspheres of 4–6 μm particle size on the Ni foam (Fig. 1b). High-magnification SEM images (Fig. 1c and d) reveal the presence of numerous mesopores on the surfaces of the NiSe–PANI microspheres. The hierarchically porous structure with a 3D configuration might offer large interfacial area, reduced ionic diffusion distance, convenient charge separation and effective accessibility of active sites, which would accelerate the electrocatalytic process.24 After coating with PANI, chiffon-like layers were observed on the NiSe–PANI microspheres. In order to identify the uniformity of the PANI coating, energy dispersive spectroscopy (EDS) elemental mapping was carried out. In Fig. S2 (ESI†), the N, Ni and Se elements are uniformly distributed on the NiSe–PANI microspheres, verifying the successful selenylation and electropolymerization process. A typical transmission electron microscopy (TEM) image of NiSe–PANI is shown in Fig. 1e, which displays that NiSe–PANI spheres consist of small nanoparticles surrounded with PANI layers. A high-resolution TEM (HRTEM) image (Fig. 1f) further confirms that the crystalline NiSe component is significantly embedded in the PANI layer. In the magnified HRTEM images of NiSe–PANI (Fig. 1g and h), two sets of lattice fringes were observed, giving interplanar distances of 0.273 and 0.302 nm corresponding to the (101) and (100) crystallographic planes of the NiSe phase. | ||
| Fig. 1 (a) Schematic synthesis outline of a hierarchically porous NiSe–PANI structure. (b–d) Top-view SEM images of the porous NiSe–PANI microspheres arrays on Ni foam at different magnifications. (e) TEM image and (f–h) HRTEM images of NiSe–PANI, illustrating that the NiSe microspheres consist of small particles surrounded by PANI layers. | ||
The crystal structures of the as-prepared samples were determined by X-ray diffraction (XRD). The XRD pattern of the pre-electrodeposited Ni superstructures on the Ni foam demonstrates the formation of metallic Ni, which is consistent with previous reports (Fig. 2a).38 After selenylation in the NaHSe solution, the metallic Ni microspheres were transferred into NiSe (PDF #020892). Aside from the peaks which are due to the Ni foam substrate, the characteristic peaks at 32.7° and 49.9° originate from NiSe (101) and (110) facets. After coating with PANI layers, no extra diffraction peaks were seen in the NiSe–PANI sample, indicating that the PANI layers are amorphous (Fig. 2a and Fig. S3, ESI†). The above XRD results coincide well with the HRTEM results for NiSe–PANI. Furthermore, micro-Raman spectra (Fig. 2b) show four peaks at 150.6, 170.8, 214.4 and 241.3 cm−1 for the NiSe sample, which are consistent with previous reports.39,40 On the other hand, for the NiSe–PANI sample, an obvious peak centered at 253.8 cm−1 was observed, which is the resonance peak of amorphous elemental selenium, further revealing the newly formed Se–Se bond in NiSe–PANI.39,41 The apparent peak at 482.4 cm−1 in both samples confirms the possible existence of Ni–O vibrational modes,19,40 which might result from surface oxidation.19
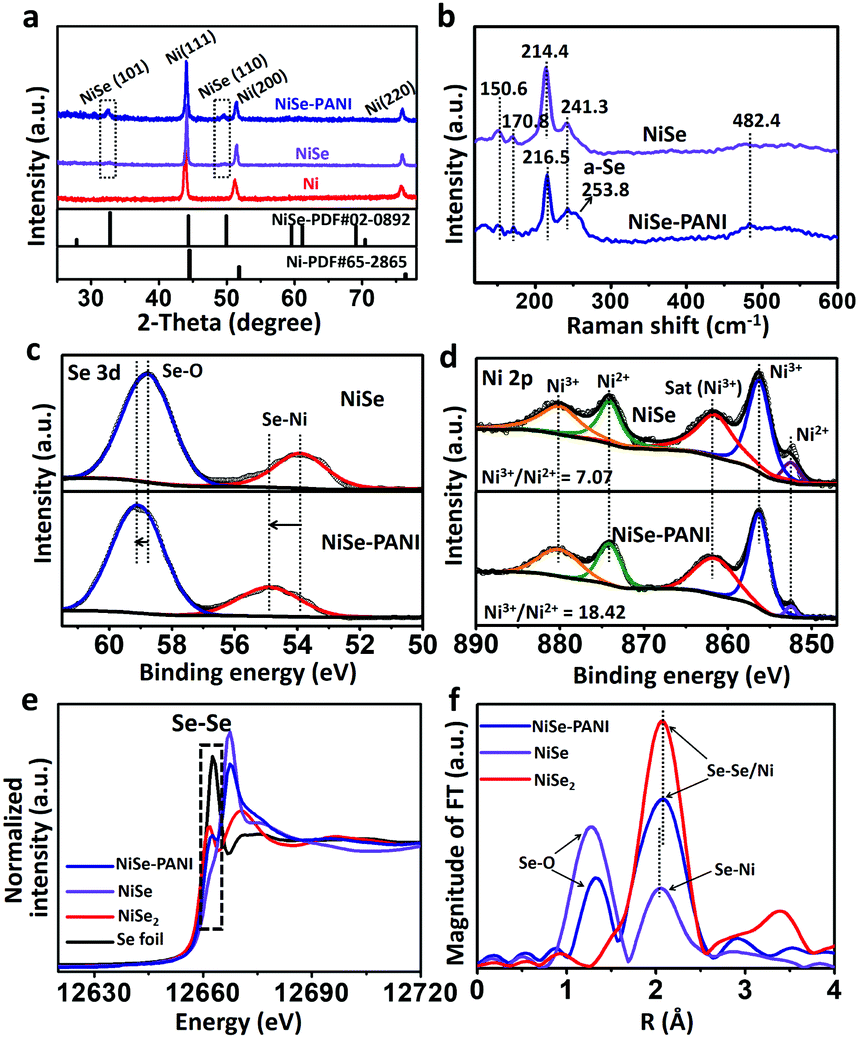 | ||
| Fig. 2 (a) XRD patterns of the pristine electrodeposited Ni precursor, NiSe and NiSe–PANI samples. (b) Raman spectra of the NiSe and NiSe–PANI catalysts, and the obvious Raman shift at around 253.8 cm−1 indicates the presence of amorphous selenium for Se–Se configurations (named a-Se). XPS spectra of (c) Se 3d and (d) Ni 2p regions for the NiSe and NiSe–PANI samples. (e) XANES spectra of the Se K-edge for the NiSe–PANI, NiSe, NiSe2 and Se foil samples, revealing the existence of Se–Se bonds in NiSe–PANI. (f) The k3-weighted Fourier transform spectra of extended X-ray absorption fine structures. | ||
To gain further insight into the changes in the elemental bonding configuration after electropolymerization of PANI, we characterized the samples by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 2c, the peaks at 53.9 and 58.8 eV in the NiSe sample are ascribed to the Se–Ni bond and the surface oxidation composition of Se.19,39,40,42 After electropolymerization with PANI on the surface, the binding energies of NiSe–PANI in the Se 3d region underwent a positive shift. Considering the possible electron delocalization between Ni d-orbitals and PANI π-conjugated ligands and electron transfer from NiSe to PANI,29,30 the peak assigned to the Se–Ni bond shifted from 53.9 to 54.9 eV, suggesting the formation of amorphous selenium for Se–Se configurations.19,39,40,42 Fig. S4 (ESI†) shows the O 1s spectra of both NiSe and NiSe–PANI samples. The peak fitting analysis of Ni 2p (Fig. 2d) shows that the chemical species of Ni can be identified as Ni2+ (852.5 eV) and Ni3+ (856.3 eV), with binding energies that are close to those of other reported nickel chalcogenides (NiSe, NiSe2 and (Ni, Co)0.85Se).19,40,43 The relative Ni3+/Ni2+ atomic ratio on the surfaces of both samples could be obtained by comparing the areas that the fitted curves covered. It could be clearly seen that the Ni3+/Ni2+ atomic ratio (18.42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) on the NiSe–PANI surface is notably higher than that (7.07:1) on NiSe, indicating that more Ni3+ species than Ni2+ are present on NiSe–PANI – that is, strong electronic interactions between PANI and NiSe (electron transfer from Ni to PANI) occurred after electropolymerization modification (Fig. S5, ESI†). Simultaneously, due to the electron acceptability, the introduction of PANI could give rise to a small electron transfer from NiSe to PANI, making NiSe more easily oxidized and facilitating the surface-formation of active NiIII/IV species for the OER process.19,35,42,44–46
:1) on the NiSe–PANI surface is notably higher than that (7.07:1) on NiSe, indicating that more Ni3+ species than Ni2+ are present on NiSe–PANI – that is, strong electronic interactions between PANI and NiSe (electron transfer from Ni to PANI) occurred after electropolymerization modification (Fig. S5, ESI†). Simultaneously, due to the electron acceptability, the introduction of PANI could give rise to a small electron transfer from NiSe to PANI, making NiSe more easily oxidized and facilitating the surface-formation of active NiIII/IV species for the OER process.19,35,42,44–46
To further probe the electronic structures of these catalysts, NiSe, NiSe–PANI and controlled samples were characterized using X-ray absorption near edge structure (XANES) spectra. As shown in Fig. 2e, partial oxidation of Se2− to Se0 was detected in the NiSe–PANI sample as indicated by a reduction in the intensity of the XANES peak of the Se K-edge around 12667 eV and an increase in the white line feature (12662 eV). This feature unambiguously demonstrates the formation of Se–Se bonds in the NiSe–PANI sample,47 which benefited from the surface electron transfer from NiSe to PANI. The extended X-ray absorption fine structure (EXAFS) spectra were recorded to detect changes in coordination structures (Fig. 2f). In the local structure of Se sites, the pristine Se–Ni bonds shifted to a longer distance after PANI modification. We ascribe this change to the surface formation of Se–Se bonds, mainly because of the similar local coordination environments like NiSe2 controlled samples.
Based on the above results of Raman characterization, XPS spectra, XANES spectra and EXAFS spectra, we conclude that the surface-PANI-engineering strategy promotes the surface-formation of Se–Se components, leading to a selenium-enriched structure in the NiSe structure, which may potentially benefit an efficient HER process. Meanwhile, as the electron acceptor, PANI might enhance the generation of high valence NiIII/IV centers of NiSe when oxidized for OER activity. In general, the electropolymerization of PANI notably optimized the NiSe electronic structure, improving the overall electrocatalytic water splitting activity.
2.2 HER electrocatalytic performance
The electrocatalytic performances were evaluated via a typical three-electrode system connected to an electrochemical workstation in an H-type cell with 1 M KOH electrolyte. The catalysts were electrochemically pre-conditioned before the test (see more details in the Experimental section) to reach a stable state. The linear sweep voltammetry (LSV) curves in Fig. 3a show the current density (j) values normalized to the geometric area of the electrode versus potential (vs. RHE) for blank Ni foam, PANI electrodeposited on Ni foam (named PANI), NiSe, commercial Pt/C deposited on Ni foam (5 wt% Pt on Vulcan carbon black, ∼1 mg cm−2) and NiSe–PANI, respectively. The NiSe–PANI sample produced j of −10, −20 and −100 mA cm−2 at η of −120, −152 and −217 mV (η10 = −120 mV, η20 = −152 mV, η100 = −217 mV), respectively. These values indicate its more efficient HER activity than NiSe before PANI functionalization, which simultaneously surpasses the HER activities of other controlled samples. Meanwhile, LSV curves of NiSe decorated with PANI with different times of electropolymerization are shown in Fig. S6 (ESI†).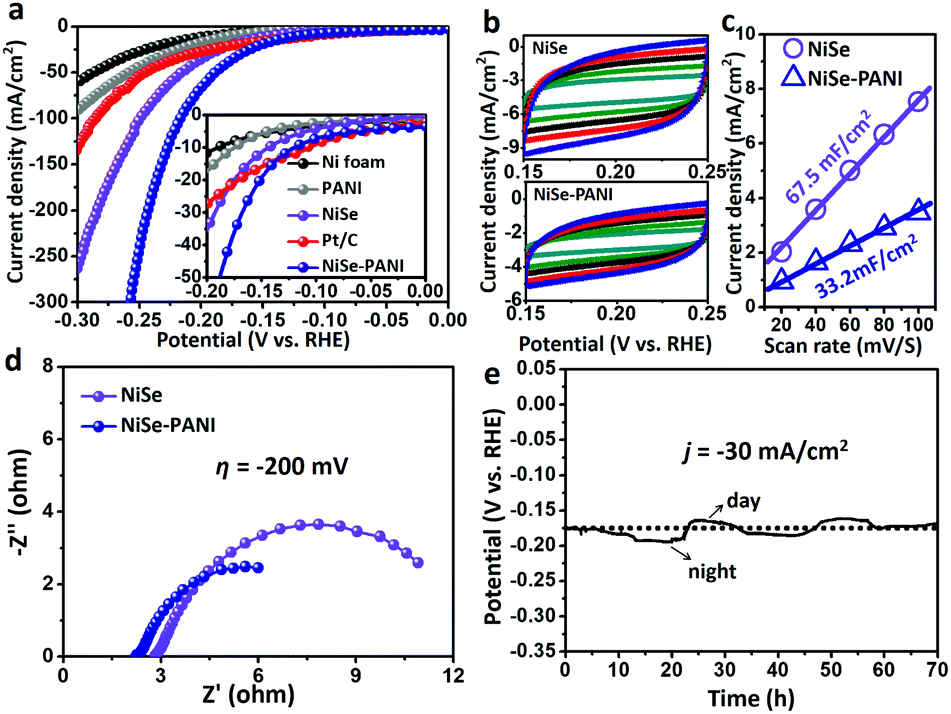 | ||
| Fig. 3 HER electrocatalytic performance. (a) LSV curves for the HER of blank Ni foam, PANI electrodeposited on Ni foam (named PANI), NiSe, commercial Pt/C deposited on Ni foam (5 wt% Pt on Vulcan carbon black, ∼1 mg cm−2) and NiSe–PANI. Inset of (a): enlarged LSV curves over a small HER potential range. (b) Cyclic voltammograms of NiSe and NiSe–PANI at different scan rates (from 20 to 100 mV s−1 with an increment of 20 mV s−1). (c) Scan rate dependence of the current densities for NiSe and NiSe–PANI at 0.20 V vs. RHE. (d) Nyquist plots of NiSe and NiSe–PANI at η of −200 mV. (e) Chronopotentiometric curves of NiSe–PANI at j of −30 mA cm−2 for a continuous HER process. | ||
In order to compare the electrochemical surface areas (ECSAs) of the NiSe catalysts, the double-layer capacitances (Cdl) of the samples were estimated using a simple cyclic voltammetry (CV) method. As shown in Fig. 3b and c, the plots of Δj = (ja − jc) at 0.20 V (vs. RHE) against the scan rates were recorded. Apparently, after PANI modification, the slope (equivalent to twice the Cdl) decreased (from the initial 67.5 mF cm−2 to 33.2 mF cm−2), indicating that the PANI layer would somewhat reduce the active sites exposed to the electrolyte. However, considering the better HER performance and lower ECSAs of NiSe–PANI than those of NiSe, we conclude that the activity of each HER active site was significantly enhanced after PANI functionalization (Table 1). Thus, the intrinsic catalytic activity for the HER was strongly improved.
| Sample | HER | OER | ||
|---|---|---|---|---|
| ECSA [cm2] | ECSA-corrected j200 [mA cm−2] | ECSA [cm2] | ECSA-corrected j350 [mA cm−2] | |
| NiSe | 210.9 | −0.043 | 485.6 | 0.020 |
| NiSe–PANI | 103.8 | −0.162 | 454.4 | 0.075 |
Electrical impedance spectroscopy (EIS; Fig. 3d) was used to evaluate the electron transfer ability of NiSe–PANI. When operated at η of −200 mV, the NiSe–PANI catalyst exhibited a much smaller radius of the semicircle in the Nyquist plots than NiSe, which can accelerate the exchange of charged species in NiSe–PANI. The reason can be attributed to the presence of PANI, which could subtly modify the surface electronic structures of NiSe.
To avoid the background current interfering the evaluation of HER performance, a multi-step chronoamperometric test was conducted (Fig. S7, ESI†). The potentials started at −0.04 V to −0.20 V with an increment of −0.02 V every 600 s. In the low overpotential range (below −200 mV, with no iR-correction), the η and the corresponding j matched well with those tested in the polarization curves, suggesting that low capacitance current existed in the HER potential range.
To evaluate the stability of NiSe–PANI as a HER catalyst, chronopotentiometric curves in 1 M KOH were recorded (Fig. 3e). To sustainably achieve j of −30 mA cm−2, the η should be retained around −175 mV, without any obvious increment after a long-term stability test of 70 h, suggesting NiSe–PANI's potential as a sustainable HER catalyst. Notably, we ascribe the time-dependent potential variation to the periodic diurnal temperature variation and the rapid potential fluctuation to the release of hydrogen bubbles off the electrode.48 Meanwhile, Raman spectra after the HER test confirm the existence of amorphous selenium species (Fig. S8, ESI†). Moreover, SEM, XRD and XPS spectra exhibited no apparent changes compared with pristine samples (Fig. S9–S12, ESI†).
The synthesized NiSe possesses a hexagonal structure, in which Se atoms are only surrounded by adjacent Ni atoms. After surface functionalization with PANI, the coordination environments of Se changed, with the formation of local Se–Se configurations. Previously, numerous reports have identified that local S–S or Se–Se ligands of metal chalcogenides significantly contribute to the HER activity.39,49,50 Moreover, previous density functional theory (DFT) calculations have demonstrated that the Se sites and the Se–Se bonds are of critical importance for striking HER activity.39 Thus, we also attribute the enhanced HER performance to the surface enriched Se–Se bonds, and we emphasize the importance of this surface electronic modulation strategy to subtly tune the HER activity.
2.3 OER electrocatalytic performance
Likewise, the OER electrocatalytic performances of the NiSe–PANI and controlled samples were also evaluated in a typical three-electrode system in 1 M KOH electrolyte. Previous reports have proved that NiSe as a pre-catalyst can be activated to form high-valence Ni species on the surface to promote the OER process.19,42 Therefore, the NiSe–PANI and controlled samples were all pre-treated to achieve a stable state for the OER evaluation (see more details in the Experimental section). Compared with blank Ni foam, PANI, NiSe and commercial IrO2 deposited on Ni foam (∼1 mg cm−2), NiSe–PANI showed the best OER activity, requiring η50 = 303 mV, η100 = 337 mV and η300 = 385 mV, respectively (Fig. 4a). | ||
| Fig. 4 OER electrocatalytic performance. (a) LSV curves for the OER of blank Ni foam, PANI, NiSe, commercial IrO2 deposited on Ni foam (∼1 mg cm−2) and NiSe–PANI. (b) Cyclic voltammograms of NiSe and NiSe–PANI at different scan rates (from 20 to 100 mV s−1 with an increment of 20 mV s−1). (c) Scan rate dependence of the current densities for NiSe and NiSe–PANI at 1.25 V vs. RHE. (d) Nyquist plots of NiSe and NiSe–PANI at η of 350 mV. (e) Chronopotentiometric curves of NiSe–PANI at j of 30 mA cm−2 for a continuous OER process. (f) Schematic mechanism of NiSe–PANI for an efficient OER. The enhanced generation of NiIII/IV active species when oxidized promotes the OER process. (g) Normalized transformation of NiII to NiIII/IV on the basis of NiSe, revealing the enhanced generation of NiIII/IV due to PANI functionalization. | ||
In Fig. 4b and c, the plots of Δj = (ja − jc) at 1.25 V (vs. RHE) against the scan rates were also recorded to determine the ECSAs of the NiSe catalysts. Obviously, after electropolymerization of PANI, the slope slightly decreased (from the initial 155.4 mF cm−2 to 145.4 mF cm−2) as that was tested for the HER, due to the PANI functionalization. Nonetheless, the better OER performance and lower ECSAs of the NiSe–PANI catalyst further prove that the activity of each OER active site was significantly improved after PANI modification (Table 1).
The Nyquist plots (Fig. 4d) exhibited that the NiSe–PANI catalyst when operated at η of 350 mV possessed smaller ionic and ohmic resistances, indicating the role of PANI in improving the charged species (electrons and OH−) communication between the electrolyte and the electrode.32
In addition, a multi-step chronoamperometric test for the OER was implemented to evaluate OER current densities at different overpotentials (Fig. S13, ESI†). The potentials started from 1.39 V to 1.61 V with an increment of 0.02 V. The results reveal that the current remained very stable at each potential in the entire range, and the current could switch quite rapidly. In the low overpotential range (below 300 mV), the η and the corresponding j also matched well with those tested in the polarization curves. Moreover, the small j and the corresponding η, which could not be directly obtained in the LSV test because of the oxidation peak of NiII to NiIII/IV, were identified with approximately η10 = 180 mV, η17 = 220 mV and η36 = 260 mV.
The stability of NiSe–PANI as an OER catalyst was investigated using a chronopotentiometric test at j of 30 mA cm−2 (Fig. 4e). To sustain the OER process, the η should be retained around 250 mV, without any obvious increment after a 70 hours test, revealing the durability of NiSe–PANI as the OER catalyst. The morphology of NiSe–PANI nearly remained unchanged after the OER test (Fig. S9, ESI†). The HRTEM image of NiSe–PANI revealed the in situ generation of NiOOH during the OER process, which might contribute to the OER activity (Fig. S14, ESI†). The XRD patterns of NiSe–PANI after the OER test shown in Fig. S10 (ESI†) demonstrate the existence of the NiSe structure. In addition, the XPS spectra of NiSe–PANI for the Se 3d and Ni 2p regions indicate that both the Se and Ni species underwent the oxidization process on the surface (Fig. S11 and S12, ESI†). It is notable that the oxidized surface species (Ni-based oxides and oxyhydroxides) are real active OER sites for 3d transition metal-based compound OER precatalysts.
In conclusion, we ascribe the improved intrinsic OER activity of NiSe–PANI to the enhanced generation of NiIII/IV, because PANI as an electron acceptor can activate the NiSe catalyst to obtain high-valence metal centers (Fig. 4f). Notably, the remarkable enhancement of NiIII/IV generation could be reasonably speculated as shown in Fig. 4g, as a result of PANI functionalization.
2.4 Electrocatalytic overall water splitting performance
Based on the results mentioned above, we anticipate that NiSe–PANI could act as a bifunctional electrocatalyst for overall water splitting. Hence, a two-electrode configuration was employed. When NiSe–PANI was used as both an anode and a cathode catalyst for an alkaline water electrolyzer, j of 10 mA cm−2 could be afforded at an applied voltage of 1.53 V, which is better than blank Ni foam, PANI, NiSe and the Pt/C–IrO2 combination (Fig. 5a). Remarkably, the bifunctional electrocatalytic activity of NiSe–PANI is comparable to those of chalcogenide-based bifunctional catalysts and other state-of-the-art overall water splitting catalysts (Table S1, ESI†). The Nyquist plots of the two-electrode configuration for NiSe and NiSe–PANI catalysts also demonstrate a more efficient charge transfer ability of the hybrid NiSe–PANI as both an anode and a cathode (Fig. S15, ESI†). In addition, NiSe–PANI bifunctional electrocatalysts can maintain the high activity to achieve j of 10 mA cm−2 at an applied voltage of around 1.53 V for more than 100 h (Fig. 5b). Overall, these performances and cost-effectiveness of NiSe–PANI make it a promising material for large-scale alkaline water splitting. | ||
| Fig. 5 Bifunctional electrocatalytic water splitting performance. (a) Overall water splitting characteristics of NiSe, NiSe–PANI and controlled samples with a two-electrode configuration. (b) Chronopotentiometric curves of NiSe–PANI in a two-electrode configuration with constant j of 10 mA cm−2. | ||
To extend the effect of PANI functionalization to other Ni-based chalcogenides, Ni3S2 nanorods were prepared (Fig. S16, ESI†) according to a previous report51 and then electropolymerized with a PANI layer (Ni3S2–PANI). The obtained Ni3S2–PANI exhibited enhanced HER and OER activities in 1 M KOH solution (Fig. S17 and S18, ESI†). Remarkably, a two-electrode configuration electrode with Ni3S2–PANI as a bifunctional electrocatalyst gave j of 10 mA cm−2 at an applied voltage of 1.64 V (Fig. S19, ESI†), which is 100 mV higher than those of pristine Ni3S2 bifunctional catalysts, further confirming that the PANI–functionalization effect can be extended to other Ni-based chalcogenides.
3. Conclusions
In summary, hierarchically porous NiSe microspheres functionalized with PANI layers have been synthesized. Attributed to the strong chemical interactions between NiSe and PANI, the surface formation of selenium-enriched structures improves the HER activity, and the enhanced generation of high-valence NiIII/IV centers contributes greatly to improve the OER activity. Equally, the PANI layers simultaneously provide efficient charge transfer toward the embedded NiSe nanoparticles. Benefitting from the enhanced intrinsic activity and surface charge transfer ability, the bifunctional NiSe–PANI catalyst outperforms most of the overall water splitting electrocatalysts, with a high current density and excellent stability. Furthermore, the PANI–functionalization strategy can be extended to other Ni-based chalcogenides to enhance the overall water splitting ability. Therefore, we believe that the surface electronic modulation strategy provides a novel design strategy to fabricate bifunctional electrocatalysts for large-scale alkaline water splitting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21573068), the National Natural Science Fund for Distinguished Young Scholars (51725201) and the Fundamental Research Funds for the Central Universities (222201718002). The authors also thank the crew of the 1W1B beamline of Beijing Synchrotron Radiation Facility for the constructive assistance with the XAFS measurements and data analyses.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef.
- Y. Hou, X. Chen, S. Yang, Y. L. Zhong, C. Li, H. Zhao and H. G. Yang, Nano Energy, 2017, 36, 102–109 CrossRef.
- S.-H. Hsu, J. Miao, L. Zhang, J. Gao, H. Wang, H. Tao, S.-F. Hung, A. Vasileff, S. Z. Qiao and B. Liu, Adv. Mater., 2018, 30, 1707261 CrossRef PubMed.
- J. Qi, X. Lai, J. Wang, H. Tang, H. Ren, Y. Yang, Q. Jin, L. Zhang, R. Yu, G. Ma, Z. Su, H. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- Y. Zheng, Y. Jiao, S. Qiao and A. Vasileff, Angew. Chem., Int. Ed., 2017, 57, 7568–7579 CrossRef PubMed.
- H. Zhang, H. Jiang, Y. Hu, P. Saha and C. Li, Mater. Chem. Front., 2018, 2, 1462–1466 RSC.
- L. Xie, X. Ren, Q. Liu, G. Cui, R. Ge, A. M. Asiri, X. Sun, Q. Zhang and L. Chen, J. Mater. Chem. A, 2018, 6, 1967–1970 RSC.
- J. Zhao, X. Li, G. Cui and X. Sun, Chem. Commun., 2018, 54, 5462–5465 RSC.
- X. Xiong, C. You, Z. Liu, A. M. Asiri and X. Sun, ACS Sustainable Chem. Eng., 2018, 6, 2883–2887 CrossRef.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef PubMed.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef PubMed.
- L. Wang, C. Gu, X. Ge, J. Zhang, H. Zhu and J. Tu, Adv. Mater. Interfaces, 2017, 4, 1700481 CrossRef.
- J. Li, Y. Wang, T. Zhou, H. Zhang, X. Sun, J. Tang, L. Zhang, A. M. Al-Enizi, Z. Yang and G. Zheng, J. Am. Chem. Soc., 2015, 137, 14305–14312 CrossRef PubMed.
- L. Wang, C. Gu, X. Ge, J. Zhang, H. Zhu and J. Tu, ChemNanoMat, 2018, 4, 124–131 CrossRef.
- P. F. Liu, S. Yang, B. Zhang and H. G. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 34474–34481 CrossRef PubMed.
- Q. Xiong, Y. Wang, P.-F. Liu, L.-R. Zheng, G. Wang, Y. H.-G. Yang., P.-K. Wong, H. Zhang and H. Zhao, Adv. Mater., 2018, 30, 1801450 CrossRef PubMed.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef PubMed.
- Y. Zhang, B. Ouyang, J. Xu, G. Jia, S. Chen, R. S. Rawat and H. J. Fan, Angew. Chem., Int. Ed., 2016, 55, 8670–8674 CrossRef PubMed.
- Q. Liu, L. Xie, F. Qu, Z. Liu, G. Du, A. M. Asiri and X. Sun, Inorg. Chem. Front., 2017, 4, 1120–1124 RSC.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef PubMed.
- T. Liu, L. Xie, J. Yang, R. Kong, G. Du, A. M. Asiri, X. Sun and L. Chen, ChemElectroChem, 2017, 4, 1840–1845 CrossRef.
- B. You and Y. J. Sun, Adv. Energy Mater., 2016, 6, 1502333 CrossRef.
- X. Zhang, X. Zhang, H. M. Xu, Z. S. Wu, H. L. Wang and Y. Y. Liang, Adv. Funct. Mater., 2017, 27, 1606635 CrossRef.
- E. Hu, Y. Feng, J. Nai, D. Zhao, Y. Hu and X. W. Lou, Energy Environ. Sci., 2018, 11, 872–880 RSC.
- X. Ji, R. Zhang, X. Shi, A. M. Asiri, B. Zheng and X. Sun, Nanoscale, 2018, 10, 7941–7945 RSC.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Nørskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef PubMed.
- S. Chen, Z. Wei, X. Qi, L. Dong, Y. G. Guo, L. Wan, Z. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef PubMed.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef PubMed.
- J. X. Feng, L. X. Ding, S. H. Ye, X. J. He, H. Xu, Y. X. Tong and G. R. Li, Adv. Mater., 2015, 27, 7051–7057 CrossRef PubMed.
- Y. Liu, J. Li, F. Li, W. Z. Li, H. D. Yang, X. Y. Zhang, Y. S. Liu and J. T. Ma, J. Mater. Chem. A, 2016, 4, 4472–4478 RSC.
- J. He, M. Wang, W. Wang, R. Miao, W. Zhong, S.-Y. Chen, S. Poges, T. Jafari, W. Song, J. Liu and S. L. Suib, ACS Appl. Mater. Interfaces, 2017, 9, 42676–42687 CrossRef PubMed.
- J.-X. Feng, S.-Y. Tong, Y.-X. Tong and G.-R. Li, J. Am. Chem. Soc., 2018, 140, 5118–5126 CrossRef PubMed.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef PubMed.
- C. Hu, L. Zhang, Z. J. Zhao, A. Li, X. Chang and J. Gong, Adv. Mater., 2018, 30, 1705538 CrossRef PubMed.
- Z. Li, W. Niu, L. Zhou and Y. Yang, ACS Energy Lett., 2018, 3, 892–898 CrossRef.
- B. You, N. Jiang, M. L. Sheng, M. W. Bhushan and Y. J. Sun, ACS Catal., 2016, 6, 714–721 CrossRef.
- F. Wang, Y. Li, T. A. Shifa, K. Liu, F. Wang, Z. Wang, P. Xu, Q. Wang and J. He, Angew. Chem., Int. Ed., 2016, 55, 6919–6924 CrossRef PubMed.
- I. H. Kwak, H. S. Im, D. M. Jang, Y. W. Kim, K. Park, Y. R. Lim, E. H. Cha and J. Park, ACS Appl. Mater. Interfaces, 2016, 8, 5327–5334 CrossRef PubMed.
- X. M. Zhou, P. Gao, S. C. Sun, D. Bao, Y. Wang, X. B. Li, T. T. Wu, Y. J. Chen and P. P. Yang, Chem. Mater., 2015, 27, 6730–6736 CrossRef.
- R. Xu, R. Wu, Y. Shi, J. Zhang and B. Zhang, Nano Energy, 2016, 24, 103–110 CrossRef.
- C. Xia, Q. Jiang, C. Zhao, M. N. Hedhili and H. N. Alshareef, Adv. Mater., 2016, 28, 77–85 CrossRef PubMed.
- Y. Y. Wu, G. D. Li, Y. P. Liu, L. Yang, X. R. Lian, T. Asefa and X. X. Zou, Adv. Funct. Mater., 2016, 26, 4839–4847 CrossRef.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. Waterhouse, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, J. Am. Chem. Soc., 2016, 138, 6517–6524 CrossRef PubMed.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef PubMed.
- J. T. Olegario, N. Yee, M. Miller, J. Sczepaniak and B. Manning, J. Nanopart. Res., 2010, 12, 2057–2068 CrossRef.
- J. Huang, Y. Sun, Y. Zhang, G. Zou, C. Yan, S. Cong, T. Lei, X. Dai, J. Guo, R. Lu, Y. Li and J. Xiong, Adv. Mater., 2018, 30, 1705045 CrossRef PubMed.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef PubMed.
- J. Hu, B. L. Huang, C. X. Zhang, Z. L. Wang, Y. M. An, D. Zhou, H. Lin, M. K. H. Leung and S. H. Yang, Energy Environ. Sci., 2017, 10, 593–603 RSC.
- W. J. Zhou, X. J. Wu, X. H. Cao, X. Huang, C. L. Tan, J. Tian, H. Liu, J. Y. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and EDS elemental mapping images; XRD patterns; XPS analysis; Raman spectra; EIS spectra; electrochemical data of bifunctional Ni3S2 catalysts; and table for comparison of NiSe–PANI and other bifunctional water splitting electrocatalysts. See DOI: 10.1039/c8qm00292d |
| This journal is © the Partner Organisations 2018 |