
Negatively charged 2D black phosphorus for highly efficient covalent functionalization†
Lei
Zhang
a,
Lin-Feng
Gao
a,
Liuxiao
Li
a,
Chen-Xia
Hu
a,
Qi-Qi
Yang
a,
Zhi-Yuan
Zhu
a,
Rui
Peng
b,
Qiang
Wang
 *a,
Yong
Peng
c,
Jun
Jin
*b and
Hao-Li
Zhang
*ad
*a,
Yong
Peng
c,
Jun
Jin
*b and
Hao-Li
Zhang
*ad
aState Key Laboratory of Applied Organic Chemistry (SKLAOC), Key Laboratory of Special Function Materials and Structure Design (MOE), College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: haoli.zhang@lzu.edu.cn; qiangwang@lzu.edu.cn
bCollege of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: jinjun@lzu.edu.cn
cKey Laboratory of Magnetism and Magnetic Materials of Ministry of Education, Lanzhou University, Lanzhou, 730000, P. R. China
dTianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin 300072, P. R. China
First published on 11th July 2018
Abstract
Two dimensional (2D) black phosphorus (BP) has attracted great attention in recent years, but its applications have been hindered by poor stability and difficulty in functionalization. In this work, we demonstrate that negatively charged BP fabricated via lithium ion intercalation exhibits much higher reactivity to diazonium modification than commonly studied neutral BP. The diazonium functionalized BP nanosheets show high surface coverage and remain stable under ambient conditions for more than 200 days. Such covalently modified BP nanosheets also provide a versatile platform for further chemical decoration toward a wide range of functionalities. As a proof of concept, we demonstrate that the highly stable and covalently functionalized 2D BP materials can be applied in nonlinear optics and catalysis. Together with the high versatility and scale-up potential, the surface functionalization method based on negatively charged 2D BP could significantly expand the applications of BP in a wide range of fields.
Introduction
Black phosphorus (BP), known as an allotropy of phosphorus, has recently been explored extensively as a new candidate for nanoelectronics1–3 and optoelectronics.4 BP exhibits similar structural properties to graphite, i.e., strong covalent bonds in plane and weak van der Waals forces between layers, enabling it to be a new monoelemental 2D material after graphene. Moreover, the electronic bandgap of BP varies from 0.3 eV in the bulk to 2.0 eV in a monolayer, which connects the zero band gap of graphene and the large band gap of transition metal dichalcogenides (TMDs).5 With a tunable direct band gap dependent on layer thickness, strong anisotropy,6 high on/off current ratios,4 and impressive charge carrier mobility,7 BP has quickly risen as a new star in the 2D material field.Hitherto, 2D BP nanosheets can be fabricated using several methods, including mechanical cleavage,4,8 liquid exfoliation,2 electrochemical approach,9 solvothermal reaction,10 and pulsed laser deposition.11 However, the poor stability of 2D BP is still the major obstacle limiting its applications.12 A few strategies have been proposed in order to depress BP degradation, including using various capping layers (aluminum oxide,13 fluoro-polymer14 and hexagonal boron nitride7) or surface coordination protection.15 Covalent functionalization has recently emerged as a new and effective method to produce stable 2D BP sheets.16–19
Developing an efficient covalent functionalization strategy of BP nanosheets is attractive as the surface functional groups may alter the chemical stability and physical properties of pristine BP to meet the requirements for different applications. Ryder et al. reported that BP nanosheets can be functionalized with benzene diazonium derivatives.18 Tan et al. used an organic aluminum compound to modify the surface of BP nanosheets on silicon substrates for field-effect transistors.20 However, these strategies based on mechanically cleaved BP nanosheets are generally time-consuming and not scalable. Zhao et al. reported the surface coordination between BP quantum dots and titanium sulfonate ligand in solution.15 Most recently, Sofer et al. studied the chemical modification of liquid exfoliated BP nanoparticles and reported the low reactivities between BP nanoparticles and diazonium salts or organolithium species.16 To date, direct formation of a C–P bond on large size BP sheets in a scalable manner remains a big challenge.
Herein, we report a new finding that negatively charged ultrathin 2D BP nanosheets exhibit significantly higher reactivity to diazonium addition than neutral BP nanosheets. The functionalized BP shows significantly improved air stability. Importantly, this method also provides a versatile platform for further surface functionalization to fabricate more complicated BP-based nanomaterials. We have demonstrated that the covalent functionalized 2D BP nanosheets can exhibit tunable nonlinear optical behaviors and excellent catalytic properties.
Results and discussion
Diazonium functionalization has been widely utilized in the modification of various nanomaterials, such as carbon nanotubes21–23 and graphene,24–26 owning to its easy operation and potential for further modification. The reaction between diazonium and nanomaterials is generally believed to follow a radical addition mechanism (Scheme 1a). However, the diazonium reaction showed little success in functionalizing liquid exfoliated BP nanosheets.16 Herein, we propose that the reaction between BP and aryl diazonium salts can be prompted by using negatively charged BP sheets based on two considerations. First, negatively charged BP sheets can attract diazonium cations by electrostatic interaction to obtain a higher local concentration than that of neutral substrates. Secondly, after the release of nitrogen, the aryl cation produced from the diazonium can readily react with the negative BP substrate under cation addition mechanisms.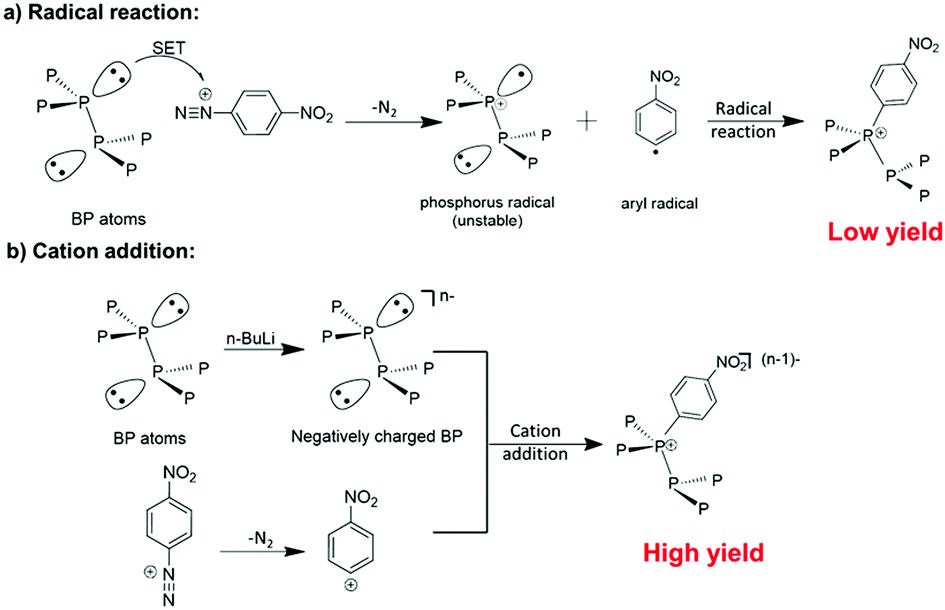 | ||
| Scheme 1 Two different mechanisms may be involved in the covalent functionalization of BP by diazonium. (a) Radical addition mechanism: nitrogen release and a single electron transfer (SET) from neutral BP leading to the formation of an aryl radical and a phosphorus radical. Then the phosphorus radical can bond to the aryl radical via a radical addition reaction. (b) Cation addition mechanism: an aryl cation is formed after the release of nitrogen, and then bonds to the negatively charged BP surface via cation addition. | ||
Scheme 1 illustrates the two possible reaction mechanisms between the aryl diazonium and BP. Under the radical addition mechanism, nitrogen release from the diazonium is accompanied by a single electron transfer from neutral BP leading to the formation of an aryl radical and a phosphorus radical. Then the phosphorus radical can bond to the aryl radical via a radical addition reaction. However, it is known by organic chemists that to stabilize phosphorous radicals large ligands providing spatial hindrance and p–π conjugation are essential. Therefore, low reactivity of the BP to radical addition is expected as phosphorus radical formation will be difficult in the basal plane of BP due to low stability. Consequently, a radical addition reaction on BP is very likely to be hindered by the poor stability of the phosphorus radical. Meanwhile, organic chemistry tells us that the reaction between diazonium and BP could also follow the cation addition mechanism, in which the aryl cation is formed after the release of nitrogen and then react with BP. It is expected that the cation addition route may give a higher yield as the aryl cation can easily react with the negatively charged BP to form a neutral additive product. In fact, previous research on graphene and TMDs has suggested that negatively charged nanomaterials may show high reactivity towards electrophiles like aryl cations.27–29 However, the fabrication of negatively charged BP and subsequent reaction with aryl cations has not been experimentally investigated.30
To verify the above hypothesis, we have prepared negatively charged 2D BP nanosheets using ion intercalation, and then tested their reaction with 4-nitrobenzene-diazonium (4-NBD) tetrafluoroborate salt. Scheme 2 illustrates the process from preparation of negatively charged BP sheets to the subsequent covalent functionalization. Preparation of negatively charged graphene27,31,32 and TMDs28,29,33 has been applied for covalent modification, but has not yet been reported in the preparation of negatively charged BP. Herein, we choose n-BuLi to exfoliate bulk BP into negatively charged 2D nanosheets (Scheme 2a–c). Following the successful exfoliation, the negatively charged BP nanosheets were then reacted with 4-NBD to give nitrophenyl modified BP nanosheets (BP-NO2) (Scheme 2d). The surface nitro groups can be transformed into amino groups via nonmetallic facile reduction (Scheme 2e). The amino functionalized BP (BP-NH2) can be further modified by other nanomaterials, like gold nanoparticles (Au NPs), to give a BP-NH2–Au nanohybrid catalyst (Scheme 2f).
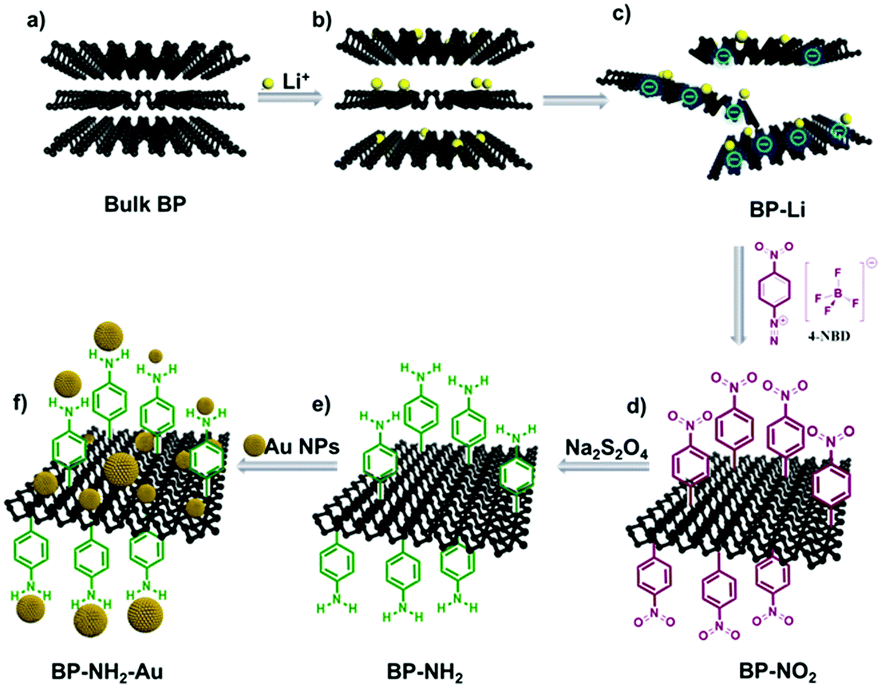 | ||
| Scheme 2 Schematic representation of functionalization of BP. (a–c) Bulk BP was intercalated by lithium ions to produce negatively charged BP-Li nanosheets, which can react with 4-NBD to form BP-NO2. (d and e) The BP-NO2 can be transferred into BP-NH2via reduction. (f) Finally, BP-NH2 was decorated with Au NPs abbreviated as BP-NH2–Au. | ||
The inset of Fig. 1a shows the corresponding color evolution of the BP n-hexane suspension before (colorless in the top), and after (brown) the reaction with n-BuLi. We found that n-BuLi can successfully exfoliate the bulk BP into a brown homogeneous suspension in hexane (referred to as BP-Li). The obtained BP-Li was carefully washed with n-hexane and deionized water prior to characterization. The representative transmission electron microscope (TEM) images of the BP-Li are shown in Fig. 1a, which indicates a large flat structure with a lateral size of around 2 μm. The selected area electron diffraction (SAED) pattern confirms the orthorhombic crystal structure of the BP-Li (Fig. 1b). Moreover, the lattice fringes of 0.140 nm in the high-resolution TEM (HR-TEM) images are assigned to the (204) planes of the BP crystal (Fig. 1c). Overall the TEM analysis proves that the exfoliated BP nanosheets maintain their original crystalline nature.
 | ||
| Fig. 1 (a) Representative TEM image of BP-Li nanosheets. The digital photograph of the dispersion before (left) and after the reaction (right) is shown in the inset; (b) SAED pattern image of lithium ion intercalated BP nanosheets; (c) HR-TEM image showing the lattice spacings; (d) AFM image and height profiles (inset) of BP-Li nanosheets. | ||
The obtained BP-Li were characterized by UV-vis-NIR and Raman spectroscopy. In the UV-vis-NIR absorption spectra, the samples exhibit broad absorption spanning from the high-energy UV-vis to low-energy near-infrared (NIR) range (Fig. S1, ESI†). The Tauc plot derives a band gap of 1.75 eV for the high-energy transition and implied a direct transition.34 Raman spectra show three major peaks located at 361.9, 437.5 and 464.8 cm−1, corresponding to the A1g, B2g and A2g phonon modes of BP, respectively (Fig. S2, ESI†). In comparison with bulk BP, the three peaks of BP-Li slightly shifted to higher wavenumbers for ∼5–10 cm−1, indicating that the bulk material was successfully exfoliated into ultrathin 2D nanosheets.35 In addition, the size morphology and height information were studied using atomic force microscopy (AFM). The profile shown in Fig. 1d displays a typical height of ∼4–8 nm. Considering that one monolayer is 0.53 nm thick, the observed thickness implies 8–15 layers, confirming the ultrathin feature of the BP-Li nanosheets.36
Fig. 2a shows the FT-IR spectra of the BP-Li and BP-NO2 samples. The samples were washed with n-hexane, iso-propanol, acetone and deionized water to remove excess diazonium salt and physisorbed materials prior to the measurements. Several intense new peaks appeared in the FT-IR spectra of the BP-NO2 samples compared with that of BP-Li. The peaks at 1378–1344 cm−1 and 1537–1519 cm−1 correspond to the symmetric and asymmetric vibrations of the NO2 group, respectively.37 Additionally, the weak peaks at ∼864 cm−1 and ∼1440 cm−1 can be assigned to the P–C bond.1,16 There is no peak in the region of 2250–2350 cm−1, indicating that unreacted diazonium compounds have been removed completely. These characteristic peaks associated with the attached 4-NBD moieties show increased intensity with the increase of the added 4-NBD ratios. The FT-IR data strongly corroborated the successful modification of 2D BP with nitro functional groups.
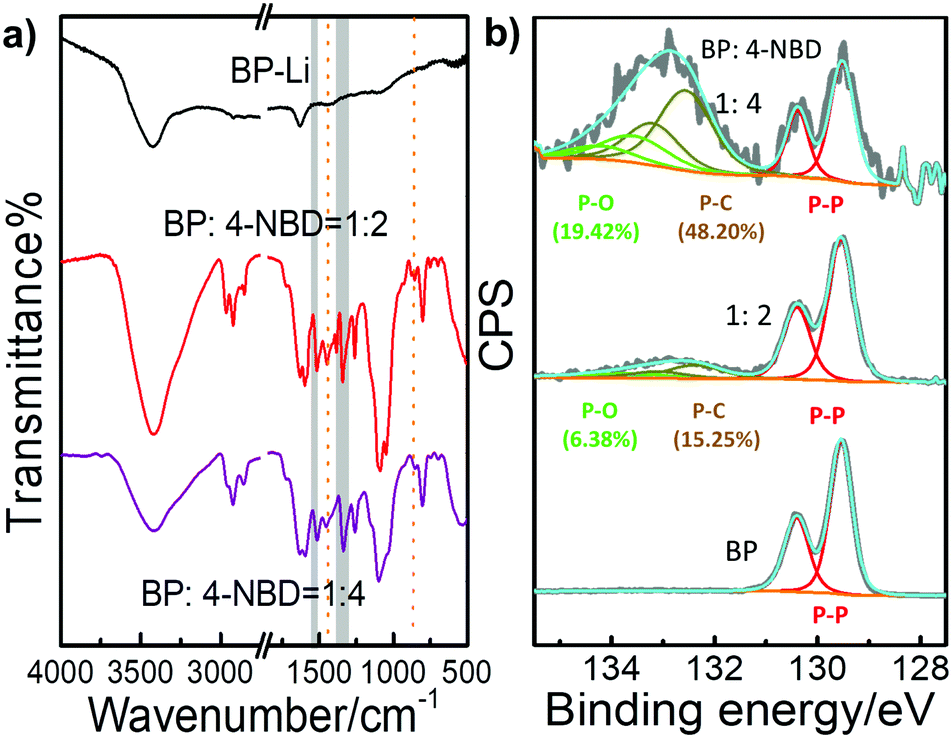 | ||
Fig. 2 (a) FT-IR spectra and (b) high resolution P 2p XPS spectra of BP-Li and BP-NO2 in different ratios (the mole ratios of BP to 4-NBD are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:4, respectively). :2 and 1:4, respectively). | ||
Raman spectra are very sensitive to the thickness of 2D materials. The corresponding Raman spectra of the BP-Li and BP-NO2 are displayed in Fig. S3b (ESI†). All spectra exhibit similar characteristic peaks of BP nanosheets. Compared with BP-Li, all of the three peaks displayed a shift to lower frequencies in BP-NO2. The red shift of BP-NO2via functional attachments is consistent with literature reports, which is due to the energy increase of the system.15,33 The above FT-IR and Raman spectra confirm the existence of NO2 groups and the structural integrity of the 2D BP structure after chemical modification.
Fig. 2b and Fig. S4, S5 (ESI†) demonstrate the XPS spectra of the modified BP. The XPS survey spectra show signals from the elements O, N, C and P in BP-NO2 (Fig. S4a, ESI†). To obtain more detailed information on the chemical state, high resolution XPS spectra of C 1s and P 2p were recorded (Fig. S5, ESI†). In BP-Li, the doublet at ∼130 eV is assigned as the P 1/2p and P 3/2p level. After reacting with 4-NBD, a broad peak at ∼133 eV appears, which agrees well with the presence of phosphorus–aryl compounds, evident for covalent bonding with nitrophenyl groups (Fig. 2b).10,16,18 The increase in the intensity of the P–C bond parallels the degree of functionalization (Table S1, ESI†). In conclusion, XPS data confirm that the negatively charged BP nanosheets exhibit enhanced reactivity to 4-NBD, and the C–P covalent bond can be successfully formed.
To verify whether negative charges are essential for the reaction between BP and 4-NBD, a series of control experiments using neutral BP nanosheets have been conducted (Fig. S6 and S7, ESI†). These experiments include the reaction between 4-NBD and (1) BP-Li with negative charges quenched by water; (2) N-methyl-2-pyrrolidone (NMP) exfoliated neutral BP nanosheets; (3) NMP exfoliated neutral BP nanosheets with copper iodide (CuI) (Table S2, ESI†). All these control experiments unambiguously indicate that neutral BP nanosheets have low reactivity to the radical addition reaction, which is consistent with previous literature results.16,17
Covalent functionalization has been proved to be one of the most promising ways to prevent air oxidation of BP.18 Previous research has shown that BP nanosheets can react with oxygen.12 The ambient stability of BP-NO2 nanosheets was monitored by the absorption spectra (Fig. 3a and b). In the beginning, both unmodified BP and BP-NO2 demonstrated a typical broad absorption band. After being exposed to ambient conditions for 200 days, the absorption peak of the unmodified BP completely vanished as a result of oxidation. No Tyndall effect can be observed (inset of Fig. 3a), indicating that the 2D nanostructure of BP has degraded completely in 200 days. In contrast, the BP-NO2 still maintains broad absorption, and the Tyndall effect can be clearly observed after exposure to air for 200 days (Fig. 3b). Moreover, TEM images showed that the BP-NO2 maintains the layered structures after 200 days (Fig. S10, ESI†). The BP-NO2 exhibits an extraordinary stability for over 200 days compared to unmodified BP nanosheets.
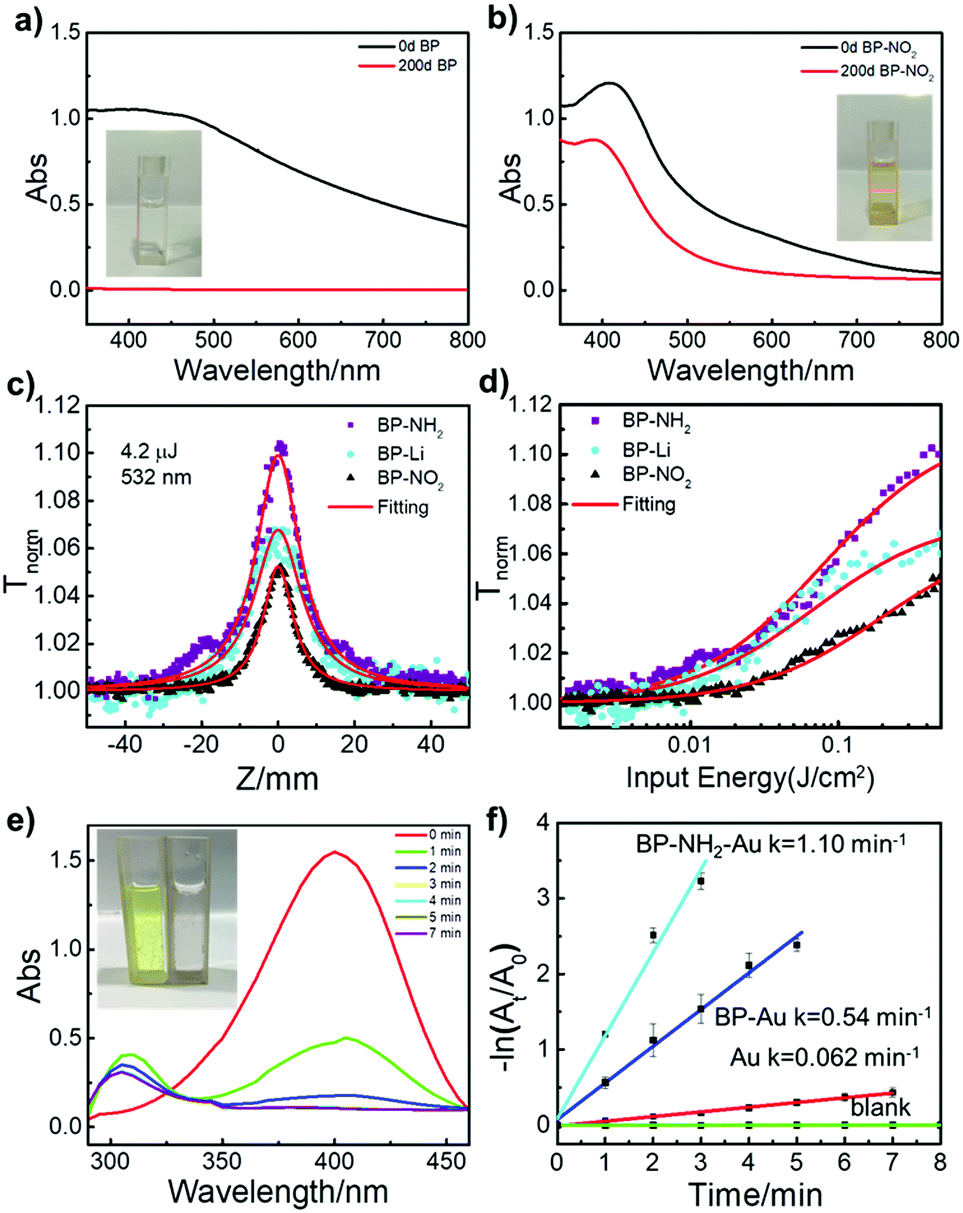 | ||
| Fig. 3 (a and b) Stability test of BP nanosheets and BP-NO2 under ambient conditions for 0 days and 200 days (solvent: NMP). The inset photographs are the BP nanosheets and BP-NO2 dispersion taken after 200 days. (c) Open aperture Z-scan data (symbols) and fitted curves (solid curves) for the samples with similar linear transmittance of ∼0.77 in NMP at the excitation of 532 nm, and 4.2 μJ, and (d) corresponding curves of transmittance versus input fluence. (e) UV-vis absorption spectra of 4-nitrophenol reduced by NaBH4 in the presence of BP-NH2–Au. (f) The corresponding plots of −ln(At/A0) as a function of time obtained from different catalysts (BP-NH2–Au: cyan, BP-Au: blue, Au: red) and without a catalyst (blank: green), respectively. | ||
The covalently functionalized BP not only shows enhanced stability but also provides a useful platform for versatile chemical reactions. For instance, the BP-NO2 could easily be reduced to BP-NH2 by sodium hyposulfite. Raman and FT-IR spectra of BP-NH2 are presented in Fig. S9 (ESI†). As a proof of concept, we demonstrate the applications of the modified BP in nonlinear optics and catalysis.
Nonlinear optical (NLO) properties of the 2D BP sheets with superficial functional groups on the 2D BP surface were tested. All of the three samples, BP-Li, BP-NO2 and BP-NH2 demonstrated excellent third-order NLO responses using an open-aperture Z-scan technique. The results are shown in Fig. 3c, where the transmission of all of the samples increases as the input light intensity increases (input intensity is maximum at Z = 0), signifying strong saturable absorption (SA) behavior. A control experiment of solvent NMP showed no SA, confirming that the NLO behavior originates from the suspended 2D materials. The peak of BP-NH2 is significantly higher than that of BP-Li under the same linear absorption. By contrast, the peak of BP-NO2 exhibits lower intensity than that of BP-Li, implying that the superficial functional groups may impose strong effects on the optical properties of BP.
Fig. 3d displays the corresponding data of normalized transmittance versus input fluence. The saturation intensity of BP-NH2 is the lowest among the three samples, suggesting that the saturation condition was easily met for BP-NH2. As the superficial functional groups on 2D BP may have significant effects on their electron delocalization state, the universal strategy for chemical modification of 2D BP opens new avenues for tuning their opto-electronic properties. Indeed, in this work, we found that the covalently functionalized 2D BP by both electron-donating (–NO2) and accepting functional groups (–NH2) demonstrate stable but distinguished NLO responses.
Laser photolysis measurement and the photoluminescence (PL) lifetimes of the three samples were further measured to disclose the mechanism of the SA behaviors, as shown in Fig. S11 and S12 (ESI†). It suggested that the PL lifetime has a positive correlation with SA performance of the modified BP nanosheets, and by changing the functional electron-donating (–NH2) or accepting (–NO2) groups on the BP surface, the NLO response can be readily tuned.
The functionalized BP also provides an efficient platform for catalyst design. For instance, BP-NH2 can easily complex with Au NPs to produce BP-NH2–Au, which may be used as a new catalyst. To demonstrate its catalytic capability, we chose the reduction of 4-nitrophenol as a model reaction,38 which is an important reaction in addressing environmental pollution caused by nitro compounds.39Fig. 3e shows a typical time-dependent UV-vis absorption for the catalysed reduction of 4-nitrophenol in the presence of BP-NH2–Au. The corresponding apparent reaction rate constant (k) was calculated via linear fitting (Fig. 3f). The reaction is evident via the sample color change from yellow to colorless (Fig. 3e, inset). As control experiments, a series of time-dependent UV-vis absorption spectra were also collected for comparison, including reaction without a catalyst, with BP-Au (unmodified BP-Li nanosheets with Au NPs) and with Au NPs as catalysts, (Fig. S13, ESI†). BP-NH2–Au took the shortest period of time and displayed the highest k value compared to that of BP-Au or Au NPs. The k value of BP-NH2–Au is approximately 1.10 min−1. Meanwhile, the TEM studies indicate that the morphology of the BP-NH2–Au remains nearly unchanged after the catalytic reaction, suggesting good structural stability (Fig. S14, ESI†). These results suggest that functionalized BP nanosheets could be used as an excellent platform for the development of novel catalysts.
Moreover, the introduction of Au NPs provides a visual tool to evaluate the functionalization degree of the BP-NH2 nanosheets. The TEM images of BP-NH2–Au and BP-Au show that Au NPs are distributed homogeneously over the BP-NH2 surface while Au NPs were mainly deposited at the sheet edges on the unfunctionalized BP-Li nanosheets (Fig. S14, ESI†). Au NPs are known to bind with amino groups.40 Therefore, the high coverage of Au NPs on the amino-functionalized BP surface reveals the very high yield of the addition reaction of the 4-NBD on the negatively charged BP nanosheets.
Conclusions
In conclusion, we have developed an efficient surface functionalization strategy for BP taking advantage of the high reactivity of diazonium salts on the negatively charged ultrathin BP nanosheets. This new method combines the exfoliation and efficient covalent functionalization of BP nanosheets in one continuous process. It was found that unmodified 2D BP, and BP with electron-donating groups of –NH2 (BP-NH2) and accepting groups of –NO2 (BP-NO2) attached demonstrate distinct nonlinear optical responses, indicating that the surface modification is an efficient method to tune the electronic properties of BP nanosheets. In addition, the Au NP decorated BP-NH2 nanosheets (BP-NH2–Au) also showed efficient catalytic reduction of 4-nitrophenol. The successful modification of BP provides a new toolbox for tailoring their chemical and optoelectronic properties. This new and efficient chemical functionalization method is expected to facilitate the applications of BP in various fields, such as optoelectronics and catalysis.Experimental section
Materials
The BP crystals were purchased from Smart-Elements. N-Hexane was from Tianjin Rionlon Bohua Pharmaceutical Chemical Company Limited and was distilled and refluxed over a sodium wire under argon atmosphere before use. N-BuLi (2.5 M) in hexane was purchased from Beijing Ino-chem Technology Company Limited. N-Methyl-2-pyrrolidone (NMP) was from Chengdu Kelong Chemical Reagent Factory and was dried with CaH2 and distilled under reduced pressure before use. 4-Nitrobenzene-diazonium (4-NBD) tetrafluoroborate salt was purchased from Tianjin Heowns Biochemical Technology Company Limited. Isopropanol and acetone were purchased from Tianjin Fuyu Fine Chemical Company Limited. Sodium hyposulfite (Na2S2O4) was purchased from Shanghai Sulphuric Acid Factory. Gold(III) chloride (AuCl3·HCl·4H2O) was obtained from Chengdu Kelong Chemical Reagent Factory. Trisodium citrate dihydrate (Na3C6H5O7·2H2O) was purchased from Tianjin Shengao Chemical reagent Company Limited. Sodium hydroxide (NaOH) was purchased from Tianjin Kaitong Chemical Reagent Company Limited. Deionized water (resistivity: 18.3 MΩ cm) was used throughout the experiment.Synthetic procedures
000 rpm for 20 min. The precipitate (BP-NH2) was collected and redispersed into 1 mL Au NP solution, and the solution was centrifuged at 15000 rpm for 20 min for purity and the precipitate was redispersed in 1 mL deionized water to obtain BP-NH2–Au (0.1 mg P, determined by ICP-MS). As a control, a catalyst consisting of 1 mL Au NPs mixed with 0.1 mg BP-Li was prepared (referred to as BP-Au).
Characterization
UV-vis-NIR absorption spectra were collected on a TU-1810 Spectrophotometer (Beijing Purkinje General Instrument, China). FT-IR spectra were recorded on a Bruker Vertex 70v vacuum spectrometer. The Raman spectra were obtained on a Renishaw Raman Microscope System equipped with the excitation wavelength of 633 nm. XPS measurements were obtained on a PHI-5702 apparatus (Physical Electronics, USA), which has a monochromated AlKα source. The TEM micrographs were acquired using Talos F200C and Tecnai G2 F30 Field Emission Transmission Electron Microscopes (FEI, USA) at operating voltages of 200 kV and 300 kV, respectively. The height and amplitude features of the products were invetigated on an Atomic Force Microscope (AFM) of Agilent 5500. P element content was determined via inductively coupled plasma atomic emission spectroscopy (ICP-AES) on an Agilent 725-ES. The laser photolysis experiments were performed on a LP920 (Edinburgh Instrument) transient absorption spectrometer. Before the measurements, the samples were purged with nitrogen to remove oxygen (∼10 min) in the solution. The Z-scan setup which was equipped with a Q-switched Nd:YAG laser (Continuum, Model Surelite SL-I-10) was performed to measure the nonlinear optical responses of the as-prepared samples. For a description in detail, please read our previous publication.43Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Ministry of Science and Technology of China (2017YFA0204903), the National Natural Science Foundation of China (NSFC. 51733004, 51525303, 21673106, 21702085, 21602093, and 21572086), the 111 Project and the Fundamental Research Funds for the Central Universities (lzujbky-2017-109). The authors thank beam line BL14B1 (Shanghai Synchrotron Radiation Facility) for providing the beam time. We wish to thank the Electron Microscopy Center of Lanzhou University for the microscopy and microanalysis of our specimens.Notes and references
- H. Liu, Y. Zou, L. Tao, Z. Ma, D. Liu, P. Zhou, H. Liu and S. Wang, Small, 2017, 13, 1700758 CrossRef PubMed.
- J. Kang, J. D. Wood, S. A. Wells, J. H. Lee, X. Liu, K. S. Chen and M. C. Hersam, ACS Nano, 2015, 9, 3596–3604 CrossRef PubMed.
- C. Wang, Q. He, U. Halim, Y. Liu, E. Zhu, Z. Lin, H. Xiao, X. Duan, Z. Feng, R. Cheng, N. O. Weiss, G. Ye, Y. C. Huang, H. Wu, H. C. Cheng, I. Shakir, L. Liao, X. Chen, W. A. Goddard Iii, Y. Huang and X. Duan, Nature, 2018, 555, 231–236 CrossRef PubMed.
- J. Wu, G. K. Koon, D. Xiang, C. Han, C. T. Toh, E. S. Kulkarni, I. Verzhbitskiy, A. Carvalho, A. S. Rodin, S. P. Koenig, G. Eda, W. Chen, A. H. Neto and B. Ozyilmaz, ACS Nano, 2015, 9, 8070–8077 CrossRef PubMed.
- X. Ling, H. Wang, S. Huang, F. Xia and M. S. Dresselhaus, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4523–4530 CrossRef PubMed.
- X. Wang, A. M. Jones, K. L. Seyler, V. Tran, Y. Jia, H. Zhao, H. Wang, L. Yang, X. Xu and F. Xia, Nat. Nanotechnol., 2015, 10, 517–521 CrossRef PubMed.
- A. Avsar, I. J. Vera-Marun, J. Y. Tan, K. Watanabe, T. Taniguchi, A. H. Castro Neto and B. Özyilmaz, ACS Nano, 2015, 9, 4138–4145 CrossRef PubMed.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef PubMed.
- A. Ambrosi, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 10443–10445 CrossRef PubMed.
- Y. Zhang, X. Rui, Y. Tang, Y. Liu, J. Wei, S. Chen, W. R. Leow, W. Li, Y. Liu, J. Deng, B. Ma, Q. Yan and X. Chen, Adv. Energy Mater., 2016, 6, 1502409 CrossRef.
- Z. Yang, J. Hao, S. Yuan, S. Lin, H. M. Yau, J. Dai and S. P. Lau, Adv. Mater., 2015, 27, 3748–3754 CrossRef PubMed.
- R. A. Doganov, E. C. O'Farrell, S. P. Koenig, Y. Yeo, A. Ziletti, A. Carvalho, D. K. Campbell, D. F. Coker, K. Watanabe, T. Taniguchi, A. H. Castro Neto and B. Ozyilmaz, Nat. Commun., 2015, 6, 6647 CrossRef PubMed.
- J. D. Wood, S. A. Wells, D. Jariwala, K. S. Chen, E. Cho, V. K. Sangwan, X. Liu, L. J. Lauhon, T. J. Marks and M. C. Hersam, Nano Lett., 2014, 14, 6964–6970 CrossRef PubMed.
- J. S. Kim, Y. Liu, W. Zhu, S. Kim, D. Wu, L. Tao, A. Dodabalapur, K. Lai and D. Akinwande, Sci. Rep., 2015, 5, 8989 CrossRef PubMed.
- Y. Zhao, H. Wang, H. Huang, Q. Xiao, Y. Xu, Z. Guo, H. Xie, J. Shao, Z. Sun, W. Han, X. F. Yu, P. Li and P. K. Chu, Angew. Chem., Int. Ed., 2016, 55, 5003–5007 CrossRef PubMed.
- Z. Sofer, J. Luxa, D. Bousa, D. Sedmidubsky, P. Lazar, T. Hartman, H. Hardtdegen and M. Pumera, Angew. Chem., Int. Ed., 2017, 56, 9891–9896 CrossRef PubMed.
- H. Hu, H. Gao, L. Gao, F. Li, N. Xu, X. Long, Y. Hu, J. Jin and J. Ma, Nanoscale, 2018, 10, 5834–5839 RSC.
- C. R. Ryder, J. D. Wood, S. A. Wells, Y. Yang, D. Jariwala, T. J. Marks, G. C. Schatz and M. C. Hersam, Nat. Chem., 2016, 8, 597–602 CrossRef PubMed.
- Y. T. Zhao, L. P. Tong, Z. B. Li, N. Yang, H. D. Fu, L. Wu, H. D. Cui, W. H. Zhou, J. H. Wang, H. Y. Wang, P. K. Chu and X. F. Yu, Chem. Mater., 2017, 29, 7131–7139 CrossRef.
- W. C. Tan, Y. Cai, R. J. Ng, L. Huang, X. Feng, G. Zhang, Y. W. Zhang, C. A. Nijhuis, X. Liu and K. W. Ang, Adv. Mater., 2017, 29, 1700503 CrossRef PubMed.
- P. Salice, E. Fabris, C. Sartorio, D. Fenaroli, V. Figà, M. P. Casaletto, S. Cataldo, B. Pignataro and E. Menna, Carbon, 2014, 74, 73–82 CrossRef.
- S. Banerjee, T. Hemraj-Benny and S. S. Wong, Adv. Mater., 2005, 17, 17–29 CrossRef.
- C. A. Dyke and J. M. Tour, J. Am. Chem. Soc., 2003, 125, 1156–1157 CrossRef PubMed.
- J. Park and M. Yan, Acc. Chem. Res., 2013, 46, 181–189 CrossRef PubMed.
- G. L. Paulus, Q. H. Wang and M. S. Strano, Acc. Chem. Res., 2013, 46, 160–170 CrossRef PubMed.
- D. Bousa, O. Jankovsky, D. Sedmidubsky, J. Luxa, J. Sturala, M. Pumera and Z. Sofer, Chem. – Eur. J., 2015, 21, 17728–17738 CrossRef PubMed.
- K. C. Knirsch, R. A. Schafer, F. Hauke and A. Hirsch, Angew. Chem., Int. Ed., 2016, 55, 5861–5864 CrossRef PubMed.
- K. C. Knirsch, N. C. Berner, H. C. Nerl, C. S. Cucinotta, Z. Gholamvand, N. McEvoy, Z. Wang, I. Abramovic, P. Vecera, M. Halik, S. Sanvito, G. S. Duesberg, V. Nicolosi, F. Hauke, A. Hirsch, J. N. Coleman and C. Backes, ACS Nano, 2015, 9, 6018–6030 CrossRef PubMed.
- D. Voiry, A. Goswami, R. Kappera, C. e Silva Cde, D. Kaplan, T. Fujita, M. Chen, T. Asefa and M. Chhowalla, Nat. Chem., 2015, 7, 45–49 CrossRef PubMed.
- A. Hirsch and F. Hauke, Angew. Chem., Int. Ed., 2018, 57, 4338–4354 CrossRef PubMed.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H. P. Steinruck, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef PubMed.
- P. Vecera, J. Holzwarth, K. F. Edelthalhammer, U. Mundloch, H. Peterlik, F. Hauke and A. Hirsch, Nat. Commun., 2016, 7, 12411 CrossRef PubMed.
- P. Vishnoi, A. Sampath, U. V. Waghmare and C. N. Rao, Chem. – Eur. J., 2017, 23, 886–895 CrossRef PubMed.
- A. H. Woomer, T. W. Farnsworth, J. Hu, R. A. Wells, C. L. Donley and S. C. Warren, ACS Nano, 2015, 9, 8869–8884 CrossRef PubMed.
- Z. Guo, H. Zhang, S. Lu, Z. Wang, S. Tang, J. Shao, Z. Sun, H. Xie, H. Wang, X.-F. Yu and P. K. Chu, Adv. Funct. Mater., 2015, 25, 6996–7002 CrossRef.
- D. Hanlon, C. Backes, E. Doherty, C. S. Cucinotta, N. C. Berner, C. Boland, K. Lee, A. Harvey, P. Lynch, Z. Gholamvand, S. Zhang, K. Wang, G. Moynihan, A. Pokle, Q. M. Ramasse, N. McEvoy, W. J. Blau, J. Wang, G. Abellan, F. Hauke, A. Hirsch, S. Sanvito, D. D. O'Regan, G. S. Duesberg, V. Nicolosi and J. N. Coleman, Nat. Commun., 2015, 6, 8563 CrossRef PubMed.
- E. Bekyarova, M. E. Itkis, P. Ramesh, C. Berger, M. Sprinkle, W. A. de Heer and R. C. Haddon, J. Am. Chem. Soc., 2009, 131, 1336–1337 CrossRef PubMed.
- J. Peng, Y. Lai, Y. Chen, J. Xu, L. Sun and J. Weng, Small, 2017, 13, 1603589 CrossRef PubMed.
- H. Hu, J. H. Xin, H. Hu, X. Wang, D. Miao and Y. Liu, J. Mater. Chem. A, 2015, 3, 11157–11182 RSC.
- M. Quintana, E. Vazquez and M. Prato, Acc. Chem. Res., 2013, 46, 138–148 CrossRef PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef.
- Y. Wang, H. Li, J. Zhang, X. Yan and Z. Chen, Phys. Chem. Chem. Phys., 2016, 18, 615–623 RSC.
- L. F. Gao, J. Y. Xu, Z. Y. Zhu, C. X. Hu, L. Zhang, Q. Wang and H. L. Zhang, Nanoscale, 2016, 8, 15132–15136 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and associated discussions. See DOI: 10.1039/c8qm00237a |
| This journal is © the Partner Organisations 2018 |