
Ratiometric real-time monitoring of hydroxyapatite–doxorubicin nanotheranostic agents for on-demand tumor targeted chemotherapy†
Yao
Kang‡
,
Wen
Sun‡
,
Jiangli
Fan
 *,
Zimu
Wei
,
Suzhen
Wang
,
Mingle
Li
,
Zhen
Zhang
,
Yahui
Xie
,
Jianjun
Du
and
Xiaojun
Peng
*,
Zimu
Wei
,
Suzhen
Wang
,
Mingle
Li
,
Zhen
Zhang
,
Yahui
Xie
,
Jianjun
Du
and
Xiaojun
Peng
State Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Linggong Road, Dalian 116024, China. E-mail: fanjl@dlut.edu.cn
First published on 29th June 2018
Abstract
We reported dual-fluorescent hydroxyapatite–doxorubicin (DOX) (DDHAP) nanocomposites for tumor-targeted therapy. A newly designed fluorescent tumor-targeting group, DFA1, is grafted onto the nanoparticle surface, which can enhance the cellular uptake of DDHAP by binding to γ-glutamyl transpeptidase (GGT), a cell surface-associated enzyme that is overexpressed on cancer cell membranes. This DFA1 moiety could undergo fluorescence quenching after binding to GGT, and the whole nanocomposite collapsed under the cancerous pH condition, thereby releasing the free fluorescent DOX as an effective anticancer drug. Thus, ratiometric fluorescence tracking can be built up by measuring the DOX/DFA1 fluorescence ratio. The dual fluorescence for ratiometric real-time tracking of the nanotherapeutic agents provides a new platform for better understanding the detailed process of their cellular uptake and intracellular dissociation. Moreover, as confirmed by in vivo studies, hydroxyapatite–DOX nanotheranostic agents demonstrate specific tumor-targeting, efficient tumor tissue penetrating and excellent tumor inhibiting effects. Nanotheranostic agents based on DDHAP show high potential for effective cancer treatment in future clinical settings.
Introduction
In recent decades, considerable efforts have been devoted to conventional chemotherapy, which is one of the main approaches for anticancer treatment.1 However, clinical translation is hindered by the poor pharmacokinetic profiles and poor selectivity of chemical drugs for tumors, resulting in low chemotherapeutic efficacy.2,3 Conventional chemotherapy also has serious side effects on healthy tissues.2,4 The rational design of nanotheranostic agents combining nanocarriers and anticancer drugs provides a promising approach to addressing the aforementioned problems.5–8 Nanotheranostic agents functionalized with tumor-targeting groups on their surfaces can accumulate at tumor sites through a positive approach. The dissociation of the nanocomposites to release free drugs can be precisely controlled through stimulus factors including pH, temperature, redox potential, enzymes, and light.7,9–15 Therefore, nanotheranostic agents can greatly improve the chemotherapeutic efficacy and minimize the undesirable side effects of drugs on healthy tissues.Recently, various inorganic nanostructures, including silicon nanoparticles, carbon nanotubes, gold nanoparticles, and quantum dots, have been reported to function as nanocarriers.16–23 Among them, hydroxyapatite [Ca10(PO4)6(OH)2, HAP] has been considered as a potentially suitable material for this purpose due to its excellent biocompatibility and good drug-loading capacity.21,24–28 Moreover, HAP easily degrades to calcium (Ca) and phosphorus (P) under weakly acidic conditions.21,25,26 Thus, the dissociation of the nanocomposites can be achieved through pH mediation.
To achieve optimal therapeutic efficacy, real-time spatial and temporal tracking of cellular uptake and intracellular dissociation of nanocarriers within living cells is important.28–30 Thus, it is necessary to produce traceable nanocarriers through rigorous chemical modifications. Non-destructive fluorescence imaging with high selectivity and high sensitivity has been widely used for the localization and monitoring of drug release.31–35 However, the fluorescence imaging response is severely affected by the local concentrations of fluorescent sensors as well as the microenvironments, which can cause misunderstanding of the cellular uptake process and misevaluating the drug release quality, and finally affect the treatment efficiency.36,37 A ratiometric approach has been developed to eliminate the effects of the above-mentioned factors by measuring the intensity ratio at two different wavelengths, which provides a built-in self-correction regardless of the environmental effects.38–42 However, few studies have been reported on the ratiometric-monitoring of cellular uptake and intracellular dissociation of nanotheranostic agents.43,44 Besides, most nanotheranostic agents have a complex structural design, in which tumor targeting and imaging are separately implemented.45–48 These issues complicate the fabrication of these systems, and thereby hinder their application in real use.
Herein, we prepare a simple structured and multifunctional HAP system for anticancer treatment, in which ratiometric real-time tracking of cellular uptake and intracellular dissociation of the nanotheranostic agent was achieved (Scheme 1). In this design, the anticancer drug (doxorubicin, DOX) is loaded into the HAP nanostructure rather than employing conventional surface adsorption. Furthermore, a newly synthesized tumor-targeting group, DFA1, is grafted onto the nanoparticle surface. DFA1 can enhance the cellular uptake by binding to γ-glutamyl transpeptidase (GGT), a cell surface-associated enzyme specifically expressed on cancer cell membranes. Interestingly, the green fluorescence of DFA1 is quenched after interacting with GGT. Therefore, DFA1 is employed as both a tumor-targeting group and a fluorescent sensor. More importantly, ratiometric fluorescence can be built up when the nanocomposites are dissociated through measuring the DOX/DFA1 fluorescence ratio. Therefore, the proposed system realized ratiometric real-time tracking of the nanotheranostic agents in living cells. Moreover, as confirmed by in vivo studies, the HAP nanoparticles demonstrate specific tumor targeting, efficient tumor tissue penetrating and excellent tumor inhibiting effects. In addition, the nanotheranostic agents did not cause any toxic side effects on mice during the treatment. Hence, the design of the HAP nanoparticles provides a simple platform for efficient anticancer treatment and a detailed understanding of their cellular uptake and intracellular dissociation through ratiometric real-time monitoring.
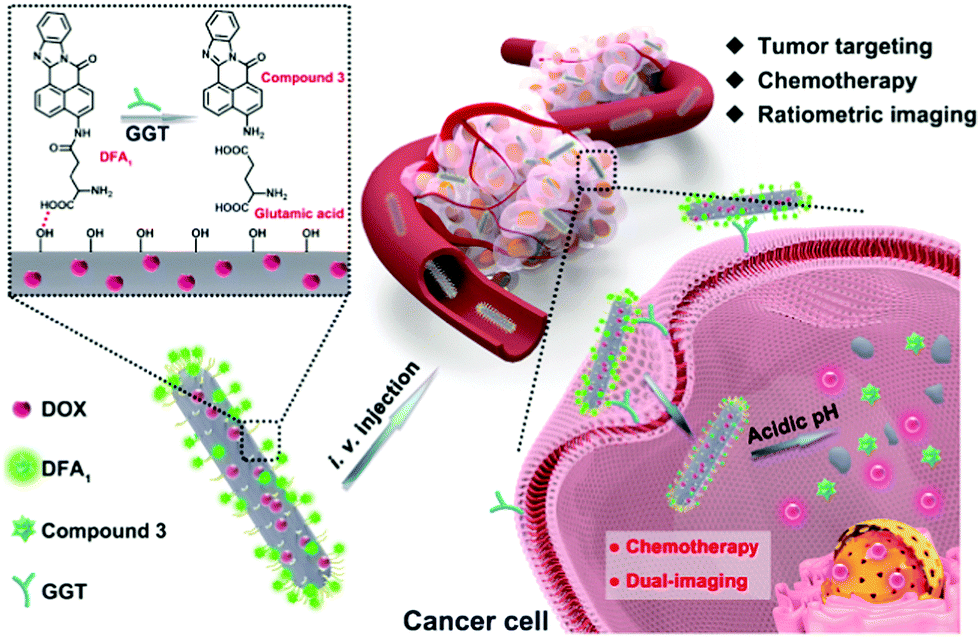 | ||
| Scheme 1 Schematic illustration of cancer treatment using DDHAP. | ||
Results and discussion
Preparation and characterization of DDHAP
The preparation of dual-fluorescent HAP–DOX (DDHAP) is shown in Schemes S1 and S2 (ESI†) and described in the Experimental section. DOX was applied as a template for the nucleation of HAP due to its positively charged amino group and was thus loaded into HAP materials (denoted DOX@HAP). A new sensor (DFA1) with a glutamic acid linkage was synthesized (Scheme S1, ESI†), which was believed to undergo GGT digestion, leading to a fluorescence response. The chemical structure of DFA1 was well characterized by proton nuclear magnetic resonance (1H NMR and 13C NMR) and electrospray ionization mass spectrometry (ESI-MS) (Fig. S1–S3, ESI†). DFA1 was grafted onto the nanoparticle surface through hydrogen bonding, which could promote the accumulation of nanoparticles at tumor sites through a positive tumor-targeting approach.A series of experiments was performed to characterize their structural properties. First, HAP, DOX@HAP, and DDHAP were investigated using a transmission electron microscope (TEM). Pure HAP nanoparticles were rod-shaped with a uniform size distribution (Fig. 1a). The morphologies and sizes of DOX@HAP and DDHAP were similar to those of pure HAP, suggesting that drug loading and surface modification did not influence the HAP nanostructure (Fig. 1a–c). The TEM images also show an excellent dispersion of DDHAP nanoparticles, and a distinct aggregation of HAP nanorods. The aggregation of HAP nanorods is due to strong hydrogen binding since a large number of OH groups exist on the nanoparticle surface. However, after surface modification with DFA1, hydrogen bonding between nanorods is significantly reduced. Thus the dispersity of DDHAP is much better than that of bare HAP. Scanning electron microscopy (SEM) also revealed a similar nanoparticle structure (Fig. 1d). According to the SEM image, the average length, width and height of DDHAP were 142, 37, and 28 nm, respectively (Fig. 1d). Energy dispersive X-ray (EDX) analysis of HAP revealed the presence of oxygen (O), phosphorus (P), and calcium (Ca) in the nanoparticles (Fig. 1e). In addition, the Ca/P ratio of DDHAP was 1.66, which is close to the ideal ratio of 1.67 (Fig. S4, ESI†). These results confirmed the successful preparation of the nanotherapeutic agents.
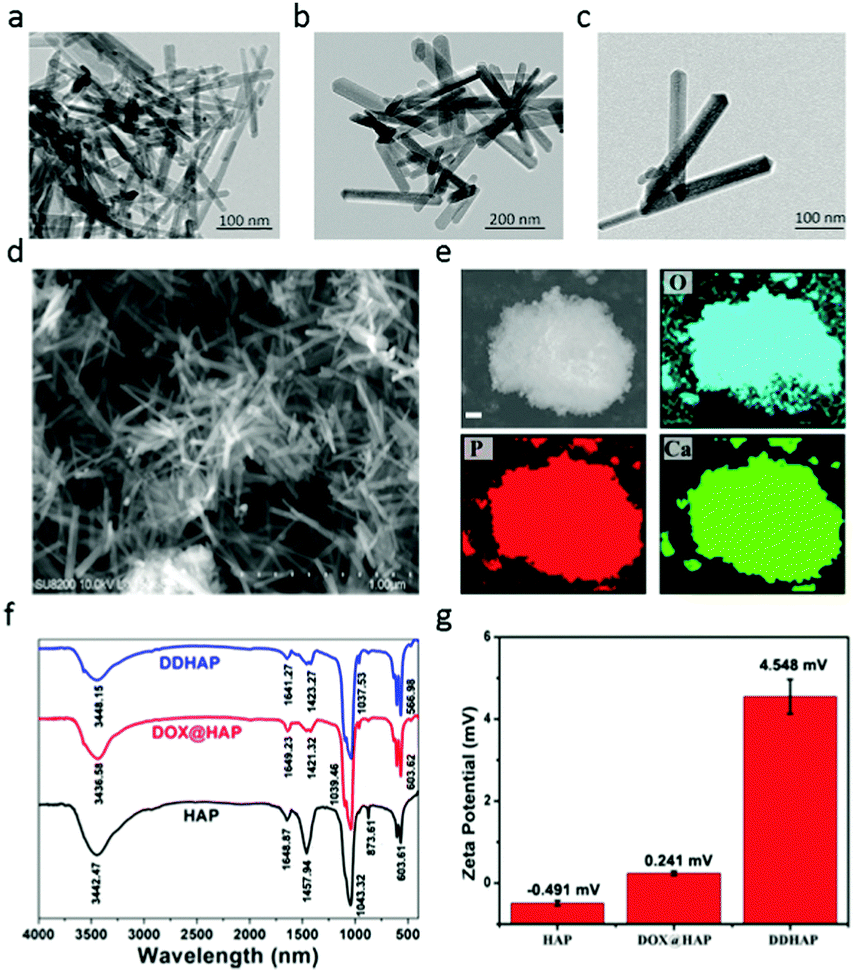 | ||
| Fig. 1 (a–c) TEM images of HAP (a), DOX@HAP (b) and DDHAP (c). (d) SEM image of DDHAP. (e) EDX image data analysis. The nanoparticles contain oxygen (O), phosphorus (P), and calcium (Ca). (f) FT-IR spectra measured in each step of the preparation process. (g) Zeta-potentials of HAP, DOX@HAP and DDHAP (error bars are based on five duplicable tests). | ||
Successful drug-loading was confirmed by Fourier transform infrared spectroscopy (FT-IR). Stretching vibrations of P–O and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1458 and 873 cm−1 in DOX@HAP and DDHAP showed a significant decrease compared with that in pure HAP. The reduction in transmittance after DOX loading is due to the existence of DOX in HAP, which occupied some original areas of Ca2+ and PO43− units, and finally resulted in low P–O and PO contents in the structure. Furthermore, X-ray powder diffraction (XRD) of the nanoparticles revealed distinct differences between pure HAP, DOX@HAP and DDHAP (Fig. S5, ESI†). The diffraction peaks of the (211), (002), (222), and (213) planes in DOX-loaded nanoparticles decreased, further demonstrating that the anticancer drug DOX was successfully loaded into the HAP structure. The loading content of DOX was 107.12 mg g−1, calculated from the absorption analysis of DDHAP, which is much higher than that used for the conventional loading method through surface absorption (approximately 40 mg g−1) (Fig. S6 and S7, ESI†).49–52 The high DOX content in HAP nanoparticles is superior for enhanced therapeutic efficiency. The N2 adsorption–desorption isotherm experiment indicates that the nanoparticles have a high specific surface area (108.45 m2 g−1) (Fig. S8, ESI†). To investigate whether the DFA1 sensor was decorated on the nanoparticle surface, zeta-potential tests were conducted (Fig. 1g). HAP and DOX@HAP exhibited low potentials (−0.419 and 0.241), whereas the potential of DDHAP significantly increased to 4.548, due to the reduction in the number of hydroxyl groups on the nanoparticle surface. The result demonstrated that DFA1 was successfully grafted onto the nanoparticle surface. The DFA1 content on the DDHAP surface was 2.12 mg g−1 calculated through fluorescence analysis (Fig. S9 and S10, ESI†).
O at 1458 and 873 cm−1 in DOX@HAP and DDHAP showed a significant decrease compared with that in pure HAP. The reduction in transmittance after DOX loading is due to the existence of DOX in HAP, which occupied some original areas of Ca2+ and PO43− units, and finally resulted in low P–O and PO contents in the structure. Furthermore, X-ray powder diffraction (XRD) of the nanoparticles revealed distinct differences between pure HAP, DOX@HAP and DDHAP (Fig. S5, ESI†). The diffraction peaks of the (211), (002), (222), and (213) planes in DOX-loaded nanoparticles decreased, further demonstrating that the anticancer drug DOX was successfully loaded into the HAP structure. The loading content of DOX was 107.12 mg g−1, calculated from the absorption analysis of DDHAP, which is much higher than that used for the conventional loading method through surface absorption (approximately 40 mg g−1) (Fig. S6 and S7, ESI†).49–52 The high DOX content in HAP nanoparticles is superior for enhanced therapeutic efficiency. The N2 adsorption–desorption isotherm experiment indicates that the nanoparticles have a high specific surface area (108.45 m2 g−1) (Fig. S8, ESI†). To investigate whether the DFA1 sensor was decorated on the nanoparticle surface, zeta-potential tests were conducted (Fig. 1g). HAP and DOX@HAP exhibited low potentials (−0.419 and 0.241), whereas the potential of DDHAP significantly increased to 4.548, due to the reduction in the number of hydroxyl groups on the nanoparticle surface. The result demonstrated that DFA1 was successfully grafted onto the nanoparticle surface. The DFA1 content on the DDHAP surface was 2.12 mg g−1 calculated through fluorescence analysis (Fig. S9 and S10, ESI†).
Optical response of DFA1 and DDHAP to GGT
The optical response of the DFA1 sensor to GGT was initially investigated. As shown in Fig. S11a (ESI†), DFA1 displayed one absorption band centered at 440 nm. Upon addition of GGT (50 U L−1), the original absorption peak vanished, while a new absorption peak appeared at 490 nm. More importantly, the sensor also showed fluorescence changes in the presence of GGT (Fig. S11b, ESI†). When it is excited at 440 nm, the emission intensity at 531 nm gradually decreased as the GGT concentration increased (Fig. 2a). The fluorescence quenching was due to the enzyme reaction, which cleaved the amino acid moiety from DFA1 and yielded compound 3 with limited solubility in water. The time course of the sensor (DFA1) response to GGT was also recorded (Fig. 2b). The fluorescence intensity at 531 nm decreased with reaction time and then remained constant after 45 min. These results demonstrate that DFA1 can serve as a rapid fluorescence sensor for GGT. The selectivity of DFA1 toward GGT was investigated by employing a number of putative interferents including essential metal ions and different amino acids (Fig. 2c and Fig. S12, ESI†). Obviously, no change in the emission intensity was observed in the presence of the other analytes. Thus, the newly synthesized DFA1 was considered to be potentially suitable for detection of GGT with negligible interference from other biologically-relevant substances. Moreover, the photostability of DFA1 was measured under continuous light irradiation (Fig. S13, ESI†). A high intensity UV lamp (365 nm, 15 W) was used for this measurement. A decrease of <5% of the initial fluorescence intensity was observed after 2 h irradiation, suggesting good photostability of the sensor.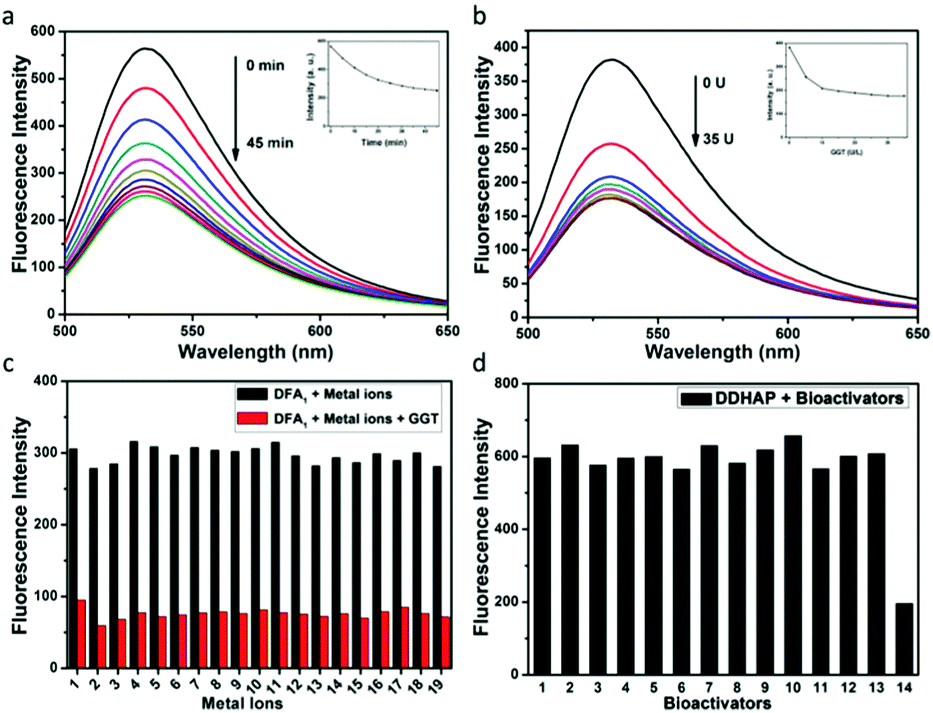 | ||
| Fig. 2 (a) Time courses of the fluorescence spectra of the sensor DFA1 (10 μM) in the presence of GGT (50 U L−1). (b) Fluorescence spectra of the sensor DFA1 (10 μM) after addition of GGT with different concentrations. Data were collected at 45 min after addition of GGT in PBS buffer (10 mM, pH 7.4) at 37 °C. (c) Fluorescence intensities (λem = 540 nm) of DFA1 (10 μM) in the presence of GGT with or without different interferents (1, NaHCO3; 2, PbCl2; 3, CoCl2; 4, KCl; 5, BaCl2; 6, NaOAc; 7, NaSCN; 8, NaBr; 9, NaCl; 10, Na2SO4; 11, CuCl2; 12, NiCl2; 13, NaF; 14, Na2SO3; 15, CrCl3; 16, Na2CO3; 17, NaBaO3; 18, MnCl2; and 19, FeCl3). (d) Fluorescence intensities (λem = 540 nm) of DDHAP (10 μM) in the presence of different bioactivators (1, control; 2, HSA; 3, RNA; 4, DNA; 5, triacylglycerol acylhydrolase; 6, lysozyme; 7, proteinase K; 8, histone; 9, collagen; 10, hemoglobin; 11, BSA; 12, β-amylase; 13, trypsin; and 14, GGT). | ||
Having known that DFA1 is suitable for recognizing and detecting GGT, we further tested the optical response of DDHAP toward GGT after grafting DFA1 onto the HAP nanoparticle surface. As expected, the nanoparticles also exhibited fluorescence quenching in the presence of GGT, suggesting that DDHAP inherited the ability of DFA1 to detect GGT without interference from other substances (Fig. 2d). Thus, DDHAP nanoparticles were expected to achieve positive tumor targeting through a specific interaction between DFA1 and GGT, which can be visualized through the fluorescence response.
Cascaded drug release from DDHAP
A key performance indicator of a nanotherapeutic agent in anticancer treatment is the controlled release of loaded-drugs at the tumor site. We assessed DOX release from DDHAP nanoparticles at different pH values. As shown in Fig. 3a, the release percentage of DOX was less than 20% even after stirring for 24 h under neutral conditions (pH = 7.4), confirming the high stability of DDHAP, which can reduce drug leakage from the nanocarriers. The low drug leakage from DDHAP was due to the drug-loading behavior, i.e. DOX was encapsulated inside the HAP materials rather than being adsorbed on the surface. In contrast, DOX release from DDHAP sharply increased to 79% and 80% at pH 6 and 5.5, respectively, during incubation (Fig. 3a). Thus, acidic conditions with a pH value lower than 6.0 could result in almost complete dissociation of the nanoparticles and efficiently release the drug. This phenomenon is attributed to the degradation of HAP under the acidic conditions, which weakens the interaction between the drug and HAP nanoparticles. Therefore, we speculated that the nanoparticles are highly toxic to cancer cells but minimally toxic to normal cells, as the pH of cancer cells is more acidic (approximately 6.4–6.8) than that of normal cells (approximately 7.2–7.6).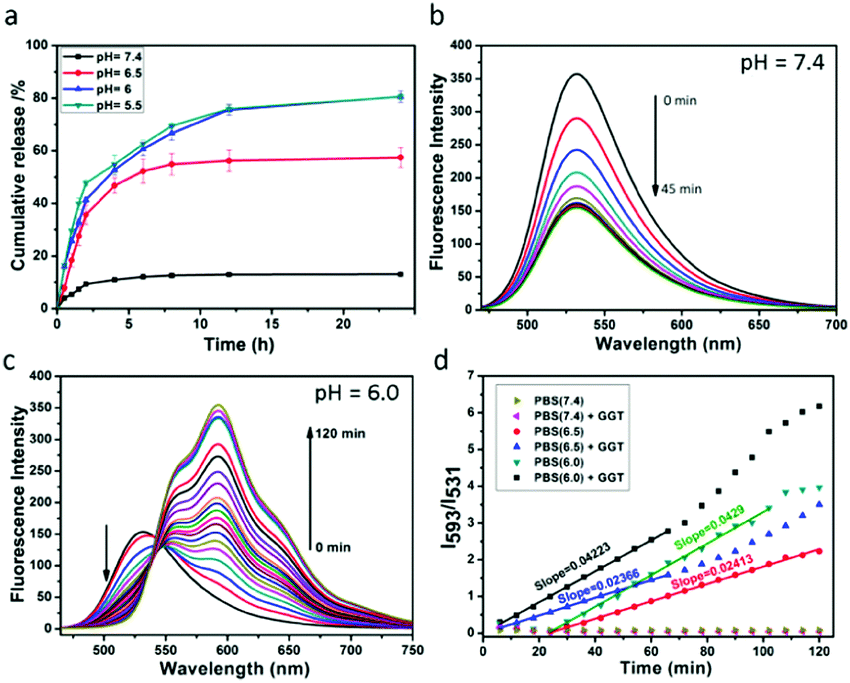 | ||
| Fig. 3 (a) Release profiles of DOX from DDHAP in PBS buffer with different pH values (neutral: 7.4; acidic: 6.5, 6 and 5.5). (b) Changes in the emission spectra of DDHAP upon addition with GGT (50 U L−1) for 45 min (λex = 440 nm). (c) Changes in the emission spectra of DDHAP under acidic conditions (pH = 6.0) for 120 min (λex = 440 nm). (d) The ratios of the fluorescence intensities at 593 and 531 nm (I593/I531) under different conditions. | ||
To better understand the drug release from DDHAP nanoparticles, a further series of experiments were conducted by varying two factors, including GGT and the pH value. First, GGT (50 U L−1) was added to a solution containing DDHAP nanoparticles, and the fluorescence spectra were recorded over time (Fig. 3b). The fluorescence intensity of DFA1 gradually decreased due to the specific interaction with GGT, which was identical to the behavior of free DFA1. However, no fluorescence from DOX was observed during this measurement. The result demonstrated that the interaction between DFA1 and GGT did not result in the release of DOX. We further adjusted the pH of the above mixture from a neutral (pH = 7.4) to acidic condition (pH = 6.0), and the fluorescence changes were monitored every 6 min over a period of 120 min (Fig. 3c). Apparently, a specific fluorescence peak of DOX centered at 593 nm was detected, indicative of successful drug release from the HAP nanoparticles (Fig. 3c and Fig. S14, ESI†). The result also proved that DOX was loaded into the HAP nanoparticles rather than surface adsorbed. Moreover, the ratios of the intensities of DFA1 to DOX (I593/I531) at different pH values were determined over time (Fig. 3d). Under neutral conditions, the intensity remained constant because only a small amount of DOX could be released. However, the intensity ratio versus time displayed a linear relationship in acidic environments and the slope was determined by only the pH value without interference from GGT. In addition, the slope of the linear relationship at pH 6.0 was higher than that at pH 6.5, suggesting that acidic conditions lead to the degradation of DDHAP and the release of drugs and lower pHs allow for more thorough degradation of DDHAP. Overall, the rationally designed nanotherapeutic agents provided a platform for achieving ratiometric real-time monitoring of the drug release.
In vitro anticancer assessment of DDHAP
The cytotoxicity of DDHAP nanoparticles was investigated using two normal cell lines (COS-7 and HL-7702) and four cancer cell lines (A2780, HepG2, MCF-7, and HCT-116). Cells were incubated with DDHAP at different concentrations for 24 h, and cell viability was evaluated by the MTT assay. The viability of normal cells remained at 80% even when the concentration of DDHAP reached 20 mg DOX per mL (Fig. 4a). Notably, HL-7702 normal liver cells express higher GGT levels than COS-7, which may result in a more efficient cellular uptake.53 However, no significant cytotoxicity was observed because the nanoparticles cannot be hydrolyzed in the neutral cellular microenvironment. Conversely, the viability of cancer cells decreased in the presence of DDHAP due to the enhanced cellular uptake and low intracellular pH-induced drug release (Fig. 4b and Tables S1, S2, ESI†). The cell viability further decreased when the concentration of DDHAP was increased. Thus, the toxicity of DDHAP to cancer cells was concentration-dependent, and the surface grafted DFA1 made DDHAP demonstrate distinct cytotoxicity toward GGT-overexpressing cancer cells. Subsequently, the viabilities of cancer (HepG2) and normal (HL-7702) cells treated with free DOX, DOX@HAP and DDHAP were further evaluated and compared. As shown in Fig. 4d, DDHAP had an IC50 of 3.617 mg DOX per L with HepG2, which was similar to that of the free DOX (3.587 mg L−1). However, DOX@HAP showed a much higher IC50 (12.323 mg DOX per L) due to the lack of tumor-targeting groups. Therefore, the presence of DFA1 enhanced the cellular uptake of HAP nanoparticles, leading to efficient drug release into the cancer cells. In contrast, both DOX@HAP and DDHAP were nontoxic to normal cells, although free DOX was highly toxic to normal cells. Therefore, DDHAP nanoparticles are promising for anticancer treatment, while reducing the toxic side effects of the loaded drugs on normal cells.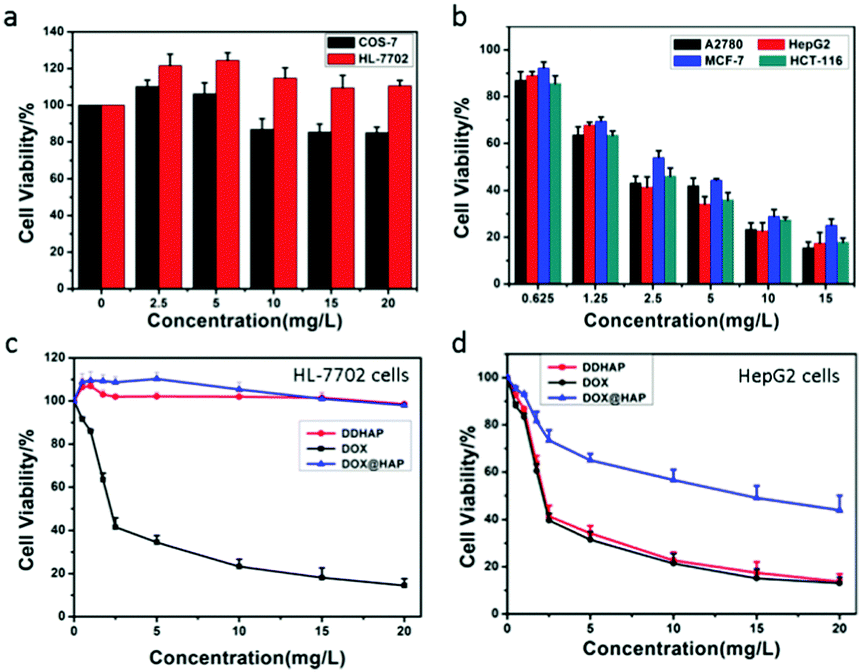 | ||
| Fig. 4 (a) Viabilities of normal cells (COS-7 and HL-7702) treated with DDHAP at various concentrations (n = 5). (b) Viabilities of cancer cells (A2780, HepG2, MCF-7 and HCT-116) treated with DDHAP at various concentrations. Cells without any treatment were used as the control (n = 5). (c and d) Viabilities of HL-7702 (c) and HepG2 (d) treated with DOX, DOX@HAP and DDHAP at various concentrations (n = 5). | ||
The nanoparticles were then incubated with the above-mentioned six cell lines (four cancer and two normal cell lines) for different periods of time before thoroughly washing the cells and observing them using confocal laser scanning microscopy (CLSM) (Fig. 5a and Fig. S15–S19, ESI†). The observation of the green fluorescence (λem = 510–530 nm, λex = 458 nm) of DFA1 after 15 min incubation confirmed that DDHAP could recognize GGT-overexpressing cancer cells and internalize into the cells. Subsequently, the fluorescence gradually decreased over time (120 min) due to the interaction between GGT and DDHAP, which is in accordance with the phenomenon observed in solution (Fig. 5a and b). However, a distinct process of drug release from DDHAP nanoparticles could be observed from the DOX channel (λex = 488 nm, λem = 590–620 nm). The fluorescence intensity of DOX gradually increased and then transferred from the cytoplasm to cell nucleus after 90 min incubation (Fig. 5a and b). More importantly, the process of drug delivery was visualized by ratiometric imaging through monitoring the green and red channels (Ired/Igreen) (Fig. 5c). The ratio was linearly related to the incubation time, providing a platform to monitor the process of drug release into the living cells.
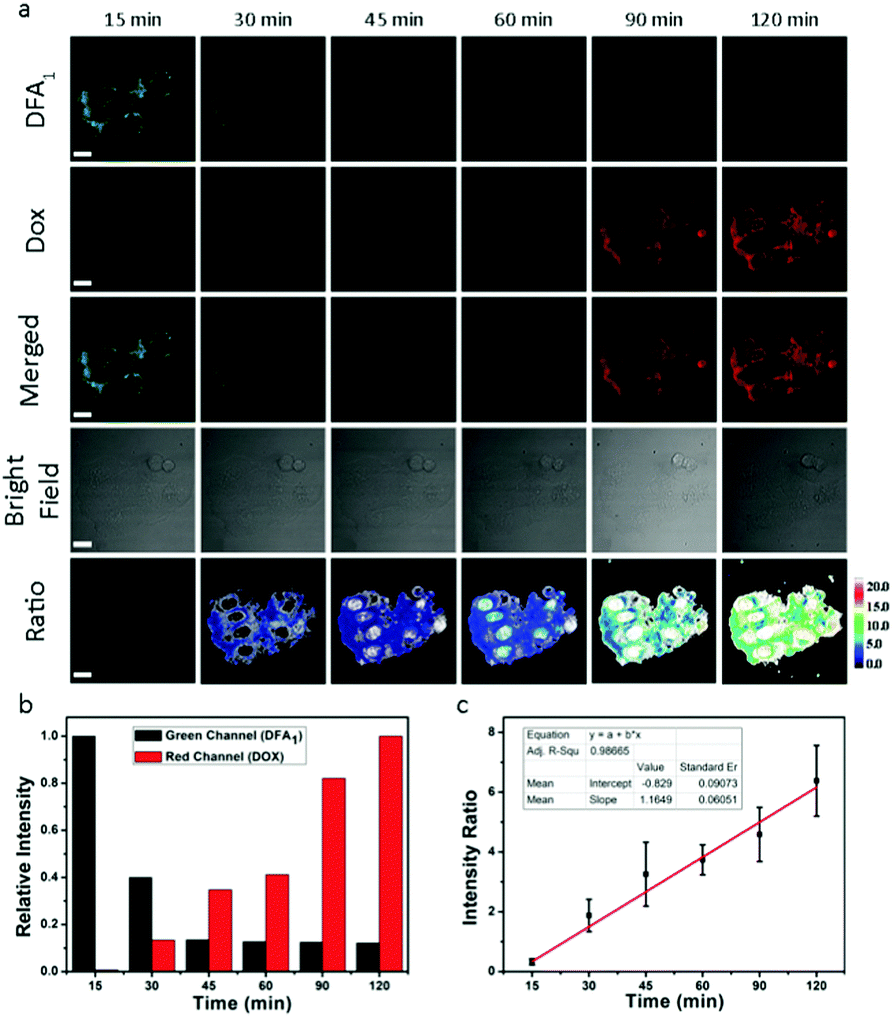 | ||
| Fig. 5 (a) CLSM images of HepG2 cells treated with DDHAP for 15–120 min. Row 1: green channel of DFA1; row 2: red channel of DOX; row 3: merged results of the green and red channels; row 4: bright field of the cells; row 5: ratio of DOX (red channel) to DFA1 (green channel). (b) Relative fluorescence intensities of DOX and DFA1 in HepG2 after incubation with DDHAP for different time intervals. (c) The DOX/DFA1 fluorescence ratio was linearly related to the incubation time (r2 = 0.9867). Green channel: λex = 458 nm, λem = 510–530 nm; red channel: λex = 488 nm, λem = 590–620 nm. | ||
The cytotoxicity of DDHAP nanoparticles was further examined with Hoechst 33342 and propidium iodide (PI), which have been widely used to stain live and dead cells, respectively (Fig. 6a). After incubation of HepG2 with DDHAP nanoparticles, the cells were stained with Hoechst 33342 and PI. Although DDHAP nanoparticles were already accumulated in the nucleus (green fluorescence) after 4 h incubation, apparent blue fluorescence could still be observed from the Hoechst channel, suggesting slight cytotoxicity of the nanoparticles after this short-time treatment. However, the blue fluorescence completely disappeared and a strong red fluorescence was observed after 8 or 12 h of incubation with DDHAP, indicating high toxicity of the nanoparticles after 8 h treatment. A further flow cytometry (FCM) experiment was conducted on HepG2 cells after different treatments (Fig. 6b). The cell content confirmed by FCM showed that 96.0% and 2.12% cells were in the late and early apoptotic stages in the DDHAP group. The apoptotic cell content of the DDHAP group was much higher than those of the free DOX and the DOX@HAP groups due to the enhanced cellular uptake of the nanoparticles and efficient intracellular drug release. Moreover, almost no cell necrosis occurred in the DOX@HAP and DDHAP groups. Thus, HAP materials can avoid abnormal cell necrosis due to their excellent biocompatibility and non-toxicity to biosystems. In contrast, cells treated with free DOX showed a large necrotic cell content (37.1%). These results demonstrated that HAP could realize efficient drug delivery, enhance the anticancer efficacy, and reduce the toxic side effects of anticancer drugs.
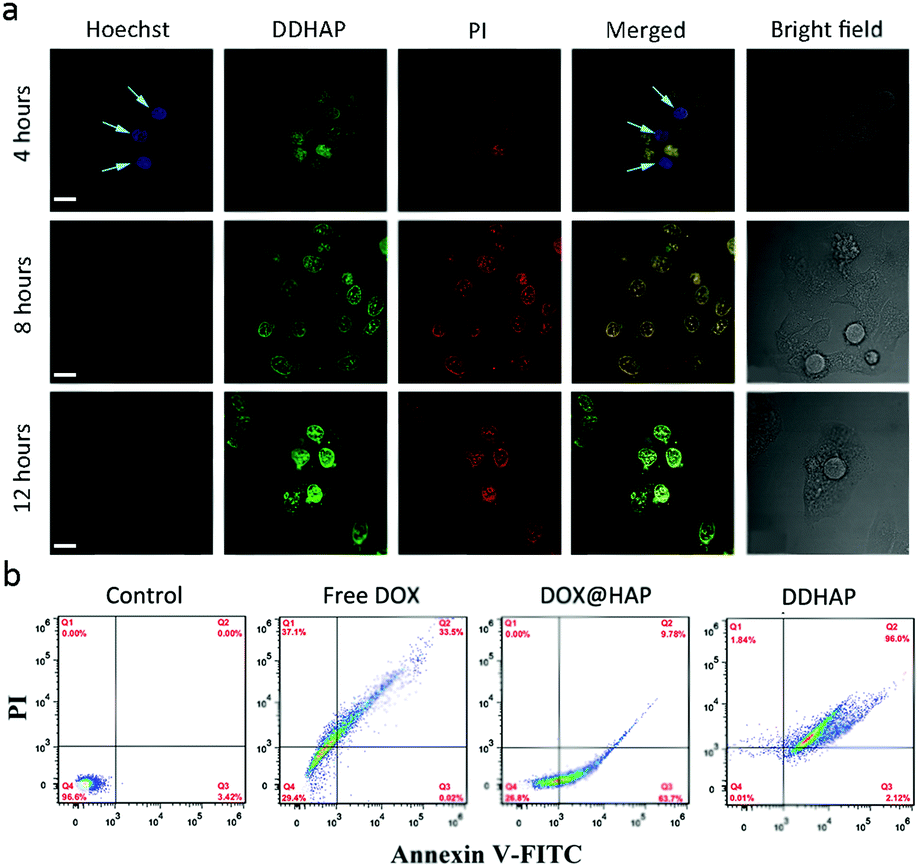 | ||
| Fig. 6 (a) CLSM images of HepG2 cells after being incubated with DDHAP for 4–12 h. The live cells were stained with Hoechst 33342 (blue, rank 1), and dead cells were stained with PI (red, rank 3). (b) Flow cytometric examination of the drug-induced apoptosis of DDHAP-treated HepG2 cells (time: 24 h). | ||
In vitro tissue imaging and dissociation of DDHAP in tumors
We conducted in vitro tissue imaging to investigate the cellular uptake of the nanoparticles in fresh tissues. Slices of the tumor and other organs (kidneys, stomach, heart, and liver) were immersed in solutions of DDHAP (10 mg DOX L−1) for 4 h. Subsequently, these tissues were rinsed with phosphate-buffered saline (PBS) three times, and their fluorescence images were obtained by CLSM (Fig. S20–S24, ESI†). As shown in Fig. S20 (ESI†), DFA1 and DOX fluorescence emissions were detected after incubating the tumor tissue with DDHAP. The fluorescence spectra of DFA1 and DOX can be observed using CLSM under the excitation at 458 nm, and demonstrate that HAP nanoparticles were able to enter tumor cells and efficiently dissociated within cells (Fig. S20a–d, ESI†). However, negligible DOX fluorescence was detected in healthy organ slices, due to the low cellular uptake efficacy and the neutral cellular microenvironment (Fig. S21–S24, ESI†). Furthermore, we performed a depth scanning of the tissue slice to investigate the tumor penetration ability of the nanoparticles (Fig. S20e, ESI†). The penetration depth of DDHAP into the tumor tissue was 470 μm, which is better than those reported for other nanoparticle systems.54,55 Thus, DDHAP demonstrated excellent permeability toward the tumor tissue, indicating its potential for use in in vivo applications. In addition, tissue imaging experiments were also carried out in healthy organs. The penetration depths of DDHAP into these organs were approximately 20–50 μm, which were much less than that of the tumor tissue (Fig. S21–S24, ESI†). The GGT-modified nanoparticles demonstrated better permeability toward the tumor tissue due to the specific cancer cell targeting.Taken together, the above studies indicated that DDHAP nanoparticles displayed a specific tumor targeting ability, excellent tumor penetration, and efficient intracellular dissociation to release drugs into tumor tissues. Encouraged by the successful in vitro experiments, we decided to explore the potential of the nanoparticles in in vivo applications.
In vivo antitumor efficacy of DDHAP
The ability of DDHAP to accumulate at the tumor site was initially investigated. DDHAP nanoparticles and free DOX were intravenously administered into HepG2-tumor-bearing mice via the tail vein. The mice were monitored over 12 h by whole-body optical imaging using a small animal in vivo imaging system. Biodistribution of nanoparticles (5 mg DOX per kg) or DOX (5 mg kg−1) was imaged 1, 2, 4, 8, and 12 h after the intravenous injection (Fig. 7a). In comparison with the whole-body distribution of free DOX, clear tumor delineations with intense fluorescence were observed at 8 h after the injection of DDHAP, indicating efficient accumulation of DDHAP at the tumor site. The fluorescence was retained at the tumor site even 12 h after the injection of DDHAP, in contrast to the much weaker fluorescence signals in the other parts of the mouse body, demonstrating a capacity for efficient and long-term passive tumor-targeted accumulation. To further prove the tumor accumulation of DDHAP nanoparticles, the tumor and other organs including the heart, lungs, liver, intestine, kidneys, and spleen were harvested for ex vivo imaging. Fluorescence from DDHAP can be observed in the tumor and normal organs (Fig. 7b and c). The fluorescence intensities of the nanoparticles were 2.18%, 3.00%, 3.96%, 7.90%, 4.36%, 3.81% and 74.79% in the heart, lungs, liver, intestine, kidneys, spleen and tumor tissue, respectively. In contrast, only a low intensity of nanoparticles was found in the tumor site in the free DOX group due to the rapid clearance of the drug from the blood circulation. Therefore, DDHAP nanoparticles demonstrated excellent tumor accumulation, which greatly improved the bioavailability of the anticancer drug to tumors.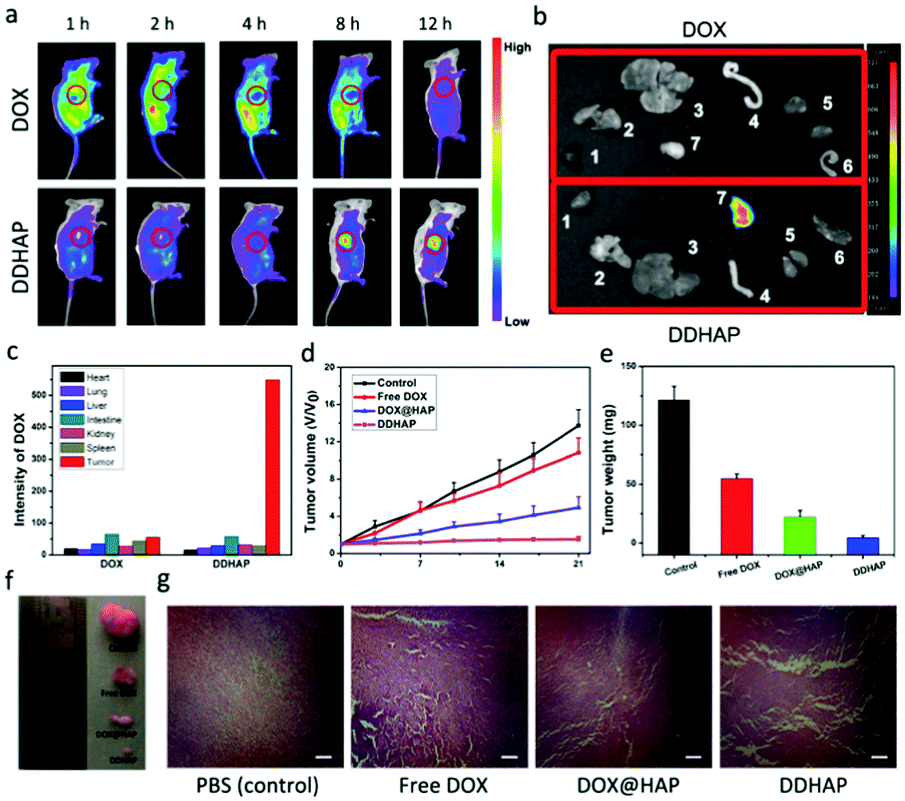 | ||
| Fig. 7 (a) In vivo fluorescence imaging of HepG2 tumor-bearing mice after intravenous injection of DOX (top) and DDHAP nanoparticles (bottom). Images were taken at 1, 2, 4, 8 and 12 h after intravenous injection. (b) Ex vivo fluorescence imaging of important organs and the tumor tissues (1. heart; 2. lungs; 3. liver; 4. intestine; 5. kidneys; 6. spleen; 7. tumor). The organs and tumor tissues were collected 24 h post-injection of DDHAP or DOX. (c) Fluorescence analysis of the different organs and tumor tissues. (d) The relative tumor volumes of tumor-bearing mice during different treatments. Data are given as means ± SD (n = 4). (e) Average weights of tumors after treatment. The mice were sacrificed, and the tumors were isolated for weighing. Data are given as means ± SD (n = 4). (f) Photographic images of excised tumors after treatment. (g) H&E staining images of tumor tissues of each group after treatment. Scale bar: 100 μm. | ||
To assess the in vivo efficacy of DDHAP in anticancer treatment, HepG2 tumor-bearing mice were exposed to different treatments, including saline, free DOX (5 mg kg−1), DOX@HAP (5 mg DOX per kg), and DDHAP (5 mg DOX per kg). The tumor volume of each group was monitored over 21 days. As shown in Fig. 7d, the growth of the tumor was completely inhibited in the group treated with DDHAP nanoparticles, compared with those in the groups treated with saline, free DOX, and DOX@HAP. Mice treated with DOX@HAP showed a moderate tumor inhibition compared with the control group. Notably, the administration of DDHAP resulted in the strongest suppression of tumor growth, which validated the importance of the surface-modified GGT for passive tumor targeting. Free DOX was inefficient in tumor growth inhibition because the drug can be rapidly cleared from the bloodstream, resulting in low bioavailability to tumors. The average tumor weight of each group was also obtained after treatment. The tumor weight in the DDHAP group was much lower than those in the other groups, which is in accordance with the result of relative tumor volumes (Fig. 7e). The above results were also supported by the representative tumor images after treatment (Fig. 7f). Additionally, histological analysis of the tumor tissues was carried out to further investigate antitumor efficacy after different treatments. Hematoxylin and eosin (H&E) staining of tumor tissues showed no obvious destruction in the PBS group, whereas the tissues in the free DOX, DOX@HAP, and DDHAP groups showed different levels of changes, with a certain number of cells in the apoptotic state. Clearly, the highest level of destruction was observed in the DDHAP group, which evidenced the promising tumor ablation activity of the nanoparticles.
The body weights of the mice were also monitored during treatment (Fig. S25a, ESI†). The mice in the free DOX group showed a significant decrease in body weight (20%) due to the toxic side effects caused by the drug. However, no obvious changes in the body weight were observed in the DOX@HAP and DDHAP groups, indicating that the HAP nanocarriers showed negligible systemic toxicity. Moreover, due to the high toxicity of free drug DOX, all mice treated with DOX died within 28 days (Fig. S25b, ESI†). By the end of the study, 100% and 80% of the mice from the control and DOX@HAP groups, respectively, died due to the extremely large tumors. In contrast, 80% of the mice treated with DDHAP survived because one mouse needed to be killed for the above-mentioned in vivo anticancer measurements (Fig. S25b, ESI†). To further access the ex vivo biosafety of these nanoparticles, histological analysis of the major organs (spleen, lungs, kidneys, heart, and liver) was performed after the different treatments (Fig. S25c and S26, ESI†). No obvious damage or physiological or morphological changes were found in the main organ slices from the H&E staining images, confirming excellent biocompatibility of DDHAP in vivo. In addition, DDHAP also showed good compatibility with blood, because no hemolysis occurred after incubating red cells with DDHAP (Fig. S27, ESI†). Collectively, these results confirm that DDHAP nanoparticles demonstrated enhanced therapeutic effects on and excellent biosafety to living mice.
Conclusions
In summary, we reported a DFA1 modified nanotherapeutic agent, DDHAP, for anticancer treatment. An acidic cellular environment can induce the dissociation of the nanotherapeutic agent to release the loaded drugs. The newly developed DFA1 grafted onto the surface of DDHAP selectively interacted with GGT, resulting in fluorescence quenching of the sensor. The specific interaction enabled the nanoparticles to efficiently target the cancer cells and thus realize tumor-targeted therapy. More importantly, ratiometric real-time tracking of drug release was achieved based on the fluorescence ratio between DFA1 and released DOX, which provides a new platform for better understanding the detailed process of intracellular dissociation of the nanotherapeutic agent. The nanoparticles demonstrated distinct cytotoxicity toward GGT-overexpressing cancer cells while remaining safe to normal cells. In vivo antitumor investigation with tumor-bearing mice confirmed the enhanced tumor-targeting and excellent antitumor effects of these nanoparticles. Moreover, DDHAP showed no toxic side effects on the mice during the treatment. Given these encouraging results, this nanotherapeutic agent based on HAP shows high potential as a therapeutic entity in anticancer treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. K. and W. S. contributed equally to this work. This work was financially supported by the National Science Foundation of China (21576037, 21676047, 21421005), the NSFC–Liaoning United Fund (U1608222), and the Fundamental Research Funds for the Central Universities (2018011013).Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2016, 66, 7 CrossRef PubMed.
- P. A. Vasey, S. B. Kaye, R. Morrison, C. Twelves, P. Wilson, R. Duncan, A. H. Thomson, L. S. Murray, T. E. Hilditch, T. Murray, S. Burtles, D. Fraier, E. Frigerio, J. Cassidy and C. R. C. P. I. I. Comm, Clin. Cancer Res., 1999, 5, 83 Search PubMed.
- R. Zhou, R. Mazurchuk and R. M. Straubinger, Cancer Res., 2002, 62, 2561 Search PubMed.
- J. J. Monsuez, J. C. Charniot, N. Vignat and J. Y. Artigou, Int. J. Cardiol., 2010, 144, 3 CrossRef PubMed.
- M. P. Stewart, A. Sharei, X. Y. Ding, G. Sahay, R. Langer and K. F. Jensen, Nature, 2016, 538, 183 CrossRef PubMed.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, J. Am. Chem. Soc., 2016, 138, 704 CrossRef PubMed.
- J. Li, L. T. Mo, C. H. Lu, T. Fu, H. H. Yang and W. H. Tan, Chem. Soc. Rev., 2016, 45, 1410 RSC.
- C. H. Song, Y. J. Zhang, C. Y. Li, G. C. Chen, X. F. Kang and Q. B. Wang, Adv. Funct. Mater., 2016, 26, 4192 CrossRef.
- C. Liang, L. G. Xu, G. S. Song and Z. Liu, Chem. Soc. Rev., 2016, 45, 6250 RSC.
- J. Liu, Y. R. Huang, A. Kumar, A. Tan, S. B. Jin, A. Mozhi and X. J. Liang, Biotechnol. Adv., 2014, 32, 693 CrossRef PubMed.
- P. Huang, D. L. Wang, Y. Su, W. Huang, Y. F. Zhou, D. X. Cui, X. Y. Zhu and D. Y. Yan, J. Am. Chem. Soc., 2014, 136, 11748 CrossRef PubMed.
- M. Z. Ye, Y. X. Han, J. B. Tang, Y. Piao, X. R. Liu, Z. X. Zhou, J. Q. Gao, J. H. Rao and Y. Q. Shen, Adv. Mater., 2017, 29, 1702342 CrossRef PubMed.
- Q. Chen, C. Liang, X. Q. Sun, J. W. Chen, Z. J. Yang, H. Zhao, L. Z. Feng and Z. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5343 CrossRef PubMed.
- K. Han, Q. Lei, S. B. Wang, J. J. Hu, W. X. Qiu, J. Y. Zhu, W. N. Yin, X. Luo and X. Z. Zhang, Adv. Funct. Mater., 2015, 25, 2961 CrossRef.
- R. L. Zhang, S. Gao, Z. L. Wang, D. Han, L. Liu, Q. J. Ma, W. H. Tan, J. Tian and X. Y. Chen, Adv. Funct. Mater., 2017, 27, 1701027 CrossRef PubMed.
- R. K. Singh, K. D. Patel, K. W. Leong and H. W. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 10309 CrossRef PubMed.
- G. X. Lv, W. S. Guo, W. Zhang, T. B. Zhang, S. Y. Li, S. Z. Chen, A. S. Eltahan, D. L. Wang, Y. Q. Wang, J. C. Zhang, P. C. Wang, J. Chang and X. J. Liang, ACS Nano, 2016, 10, 9637 CrossRef PubMed.
- L. P. Qiu, T. Chen, I. Ocsoy, E. Yasun, C. C. Wu, G. Z. Zhu, M. X. You, D. Han, J. H. Jiang, R. Q. Yu and W. H. Tan, Nano Lett., 2015, 15, 457 CrossRef PubMed.
- X. M. Li, T. C. Zhao, Y. Lu, P. Y. Wang, A. M. El-Toni, F. Zhang and D. Y. Zhao, Adv. Mater., 2017, 29, 1701652 CrossRef PubMed.
- R. Wang, L. Zhou, W. X. Wang, X. M. Li and F. Zhang, Nat. Commun., 2017, 8, 14702 CrossRef PubMed.
- X. H. Hao, X. X. Hu, C. M. Zhang, S. Z. Chen, Z. H. Li, X. J. Yang, H. F. Liu, G. Jia, D. D. Liu, K. Ge, X. J. Liang and J. C. Zhang, ACS Nano, 2015, 9, 9614 CrossRef PubMed.
- G. F. Luo, W. H. Chen, Q. Lei, W. X. Qiu, Y. X. Liu, Y. J. Cheng and X. Z. Zhang, Adv. Funct. Mater., 2016, 26, 4339 CrossRef.
- S. D. Huo, S. B. Jin, X. W. Ma, X. D. Xue, K. N. Yang, A. Kumar, P. C. Wang, J. C. Zhang, Z. B. Hu and X. J. Liang, ACS Nano, 2014, 8, 5852 CrossRef PubMed.
- S. S. A. Abidi and Q. Murtaza, J. Mater. Sci. Technol., 2014, 30, 307 CrossRef.
- Y. F. Wang, J. L. Wang, H. Hao, M. L. Cai, S. Y. Wang, J. Ma, Y. Li, C. B. Mao and S. M. Zhang, ACS Nano, 2016, 10, 9927 CrossRef PubMed.
- W. Sun, J. Fan, S. Wang, Y. Kang, J. Du and X. Peng, ACS Appl. Mater. Interfaces, 2018, 10, 7832 CrossRef PubMed.
- P. Venkatesan, N. Puvvada, R. Dash, B. N. P. Kumar, D. Sarkar, B. Azab, A. Pathak, S. C. Kundu, P. B. Fisher and M. Mandal, Biomaterials, 2011, 32, 3794 CrossRef PubMed.
- J. L. Fan, S. Z. Wang, W. Sun, S. G. Guo, Y. Kang, J. J. Du and X. J. Peng, AIChE J., 2018, 64, 835 CrossRef.
- Y. P. Ho and K. W. Leong, Nanoscale, 2010, 2, 60 RSC.
- P. P. Yang, S. L. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679 RSC.
- L. Zhou, R. Wang, C. Yao, X. M. Li, C. L. Wang, X. Y. Zhang, C. J. Xu, A. J. Zeng, D. Y. Zhao and F. Zhang, Nat. Commun., 2015, 6, 6938 CrossRef PubMed.
- H. Zhang, J. L. Fan, J. Y. Wang, S. Z. Zhang, B. R. Dou and X. J. Peng, J. Am. Chem. Soc., 2013, 135, 11663 CrossRef PubMed.
- H. Zhang, J. L. Fan, J. Y. Wang, B. R. Dou, F. Zhou, J. F. Cao, J. L. Qu, Z. Cao, W. J. Zhao and X. J. Peng, J. Am. Chem. Soc., 2013, 135, 17469 CrossRef PubMed.
- H. Zhu, J. L. Fan, J. J. Du and X. J. Peng, Acc. Chem. Res., 2016, 49, 2115 CrossRef PubMed.
- L. Wu and X. G. Qu, Chem. Soc. Rev., 2015, 44, 2963 RSC.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185 RSC.
- Q. Jin, L. Feng, D. D. Wang, Z. R. Dai, P. Wang, L. W. Zou, Z. H. Liu, J. Y. Wang, Y. Yu, G. B. Ge, J. N. Cui and L. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28474 CrossRef PubMed.
- Z. Zhang, J. L. Fan, G. H. Cheng, S. Ghazali, J. J. Du and X. J. Peng, Sens. Actuators, B, 2017, 246, 293 CrossRef.
- J. L. Fan, M. M. Hu, P. Zhan and X. J. Peng, Chem. Soc. Rev., 2013, 42, 29 RSC.
- Y. C. Chen, C. C. Zhu, Z. H. Yang, J. J. Chen, Y. F. He, Y. Jiao, W. J. He, L. Qiu, J. J. Cen and Z. J. Guo, Angew. Chem., Int. Ed., 2013, 52, 1688 CrossRef PubMed.
- N. Jiang, J. L. Fan, F. Xu, X. J. Peng, H. Y. Mu, J. Y. Wang and X. Q. Xiong, Angew. Chem., Int. Ed., 2015, 54, 2510 CrossRef PubMed.
- M. Li, J. Fan, H. Li, J. Du, S. Long and X. Peng, Biomaterials, 2018, 164, 98 CrossRef PubMed.
- K. Han, S. B. Wang, Q. Lei, J. Y. Zhu and X. Z. Zhang, ACS Nano, 2015, 9, 10268 CrossRef PubMed.
- L. Y. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896 CrossRef PubMed.
- J. Y. Li, J. Liu and C. Y. Chen, ACS Nano, 2017, 11, 2403 CrossRef PubMed.
- J. J. Liu, C. Wang, X. J. Wang, X. Wang, L. Cheng, Y. G. Li and Z. Liu, Adv. Funct. Mater., 2015, 25, 384 CrossRef.
- J. T. Xu, P. P. Yang, M. D. Sun, H. T. Bi, B. Liu, D. Yang, S. L. Gai, F. He and J. Lin, ACS Nano, 2017, 11, 4133 CrossRef PubMed.
- T. Y. Jiang, R. Mo, A. Bellotti, J. P. Zhou and Z. Gu, Adv. Funct. Mater., 2014, 24, 2295 CrossRef.
- F. Ye, H. F. Guo, H. J. Zhang and X. L. He, Acta Biomater., 2010, 6, 2212 CrossRef PubMed.
- B. Kundu, D. Ghosh, M. K. Sinha, P. S. Sen, V. K. Balla, N. Das and D. Basu, Ceram. Int., 2013, 39, 9557 CrossRef.
- J. S. Son, M. Appleford, J. L. Ong, J. C. Wenke, J. M. Kim, S. H. Choi and D. S. Oh, J. Controlled Release, 2011, 153, 133 CrossRef PubMed.
- Q. F. Zhao, T. Y. Wang, J. Wang, L. Zheng, T. Y. Jiang, G. Cheng and S. L. Wang, Appl. Surf. Sci., 2011, 257, 10126 CrossRef.
- M. D. Dabeva and D. A. Shafritz, Am. J. Pathol., 1993, 143, 1606 Search PubMed.
- J. Lademann, H. Richter, A. Teichmann, N. Otberg, U. Blume-Peytavi, J. Luengo, B. Weiss, U. F. Schaefer, C. M. Lehr, R. Wepf and W. Sterry, Eur. J. Pharm. Biopharm., 2007, 66, 159 CrossRef PubMed.
- W. Stummer, U. Pichlmeier, T. Meinel, O. D. Wiestler, F. Zanella, R. Hans-Jurgen and A.-G. S. Grp, Lancet Oncol., 2006, 7, 392 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and detailed experimental procedures. See DOI: 10.1039/c8qm00215k |
| ‡ Y. K. and W. S. contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |