
Exploration of the two-step crystallization of organic micro/nano crystalline materials by fluorescence spectroscopy†
Peng-Zhong
Chen
a,
Li-Ya
Niu
a,
Han
Zhang
a,
Yu-Zhe
Chen
 b and
Qing-Zheng
Yang
*a
b and
Qing-Zheng
Yang
*a
aKey Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: qzyang@bnu.edu.cn
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 11th April 2018
Abstract
Clearly understanding the crystallization process of organic micro/nano crystalline (OMC) materials in solution is a long-standing challenge because of the difficulty in the separation of intermediates and monitoring the process in situ and in real-time. Herein, we report the exploration of the crystallization process of OMC materials from the amorphous intermediates by taking advantage of the spectral change of an environment-sensitive emission dye BF2bcz. The intermediate for the formation of the OMC materials by the solvent-exchange method was separated as amorphous nanospheres which were transformed into crystalline nanorods by adding the surfactant to their aqueous dispersion. The distinct emission properties of the amorphous molecular aggregates and nanorods, used as a fingerprint for each species, allow for in situ and real-time monitoring of the crystallization process by using fluorescence spectroscopy. Such a facile method readily identified that increasing the concentration of surfactant and temperature both accelerated the crystallization process of BF2bcz in aqueous solution, while the size of the nanorods increased with a decrease in the concentration of surfactant. Our work provided direct experimental evidence to support the two-step nucleation mechanism in the preparation of OMC by the solvent-exchange method.
Introduction
Organic micro/nano crystalline (OMC) materials have attracted much attention during the past decades because of their wide applications in electronics, optics, catalysis, and the pharmaceutical industry.1 The OMC materials are mainly prepared from organic molecules through a self-assembly process driven by the weak interactions among molecules, such as hydrogen bonds, van der Waals forces, π–π interactions, etc. The self-assembly methods significantly affect the mode of the weak interactions in the OMC materials.2 Solvent-exchange is an important and commonly used method in preparing OMC materials because of its merits of being simple, facile and low cost.1a,3 Various architectures of OMC materials have been obtained through the solvent-exchange method.1a,f The morphology of OMC materials can significantly influence their functional properties; for example, the morphology-dependent emission and charge transport behavior in opto-electronic materials.4,5 Accordingly, understanding the formation mechanism of OMC materials in solution is of great importance to obtain the materials with desired morphology and properties in a reproducible way. Although much effort has been devoted to the final crystal structures based on different building blocks, mechanistic insights into the crystallization process and the control of the morphology of OMC materials are still elusive.In crystal engineering, the study of the solution crystallization process mainly focuses on its early stages of molecular self-assembly associated with nucleation.6 Two kinds of mechanisms, i.e. the classical nucleation theory and the two-step mechanism have been proposed to describe the dynamics of aggregates during the nucleation process in solution.7 The classical nucleation theory considers that the initially formed molecular aggregates, namely the crystal's embryo, have the same molecular arrangements as the mature crystal.8 Lately, the two-step nucleation mechanism describes the initial formation of a dense disordered cluster of solute molecules in the supersaturated solution which further reorganized into an ordered structure.9 Although the two-step nucleation mechanism is well accepted in the formation of OMC materials, there is still a lack of direct experimental evidence to strongly support this mechanism.10 First, it is a challenge to separate the initial amorphous molecular intermediates because of their dynamic and metastable natures. Another reason is the difficulty in in situ and real-time monitoring of the transformation from the amorphous intermediate to the crystalline state.
Herein, we report the separation and characterization of the intermediate for the formation of the OMC materials. Such intermediates were the metastable amorphous molecular aggregates which were able to transform into crystalline nanorods by adding the surfactant to their aqueous dispersions. Fluorescence spectroscopy was utilized to in situ and real-time monitor this conversion process. The results indicate that increasing the concentration of the surfactant (Csurfactant) and temperature both accelerated the crystallization process. Nevertheless, the size of the nanorods was independent of temperature, which increased with the decrease of Csurfactant. Our work provided direct experimental evidence to support the two-step nucleation mechanism.
Results and discussion
The difluoroboron β-diketonate compound (BF2bcz) (Fig. 1 and Scheme S1, ESI†) containing two carbazole groups was selected to shed light on the crystallization process in solution because of its environment-sensitive emission properties and its tendency to self-assemble into ordered structures in tetrahydrofuran (THF) aqueous solutions (Fig. S1, ESI†).11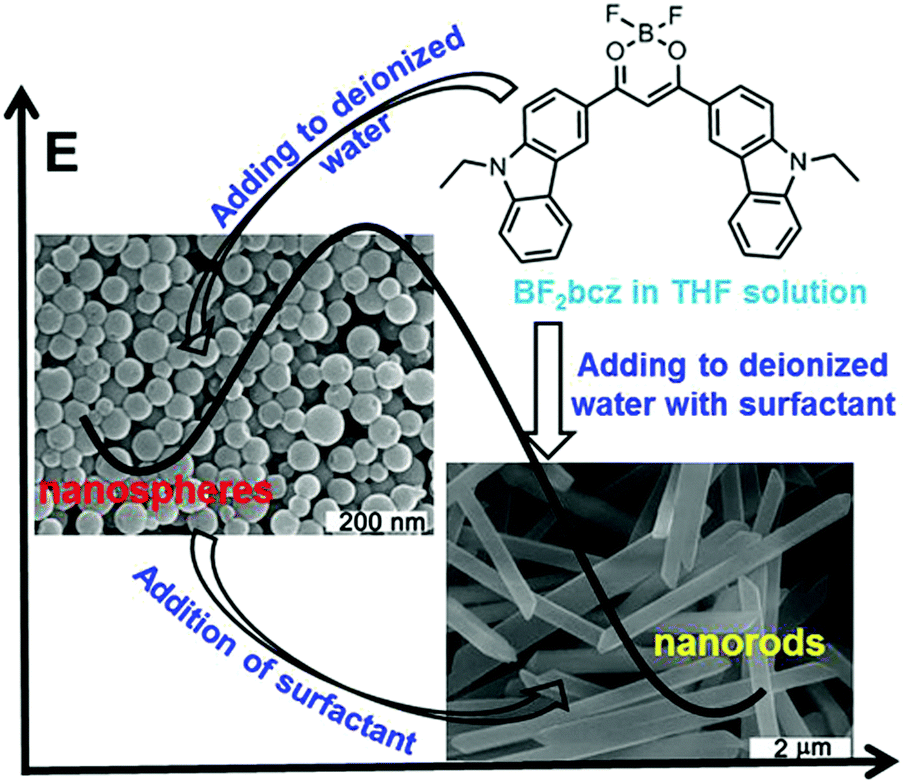 | ||
| Fig. 1 The image representation of the preparation of the BF2bcz nanospheres and nanorods from the BF2bcz monomer in THF solution, the morphological transition from nanospheres to nanorods, and the schematic energy diagram depicting the energy barrier between the nanospheres and nanorods. | ||
Previously, we described the preparation of nanorods by the fast addition of BF2bcz THF solution into deionized water in the presence of a surfactant such as sodium dodecyl sulfate (SDS) at room temperature.11d The surfactant served as the solubilizer and the template directing the formation of the BF2bcz nanorods.12 We envisaged that the metastable intermediate could be trapped in the absence of the surfactant and we may be able to obtain such an intermediate. The metastable intermediate was obtained by the injection of the BF2bcz solution in THF into deionized water without surfactant under the identical conditions as used for the preparation of the nanorods. The scanning electron microscopy (SEM) images indicated that the isolated intermediates were nanospheres with a regular shape and uniform size (Fig. 1). The average diameter estimated from SEM was approximately 100 nm, similar to that determined by dynamic light scattering (DLS) (Fig. S2, ESI†). The nanospheres displayed no noticeable X-ray diffraction peaks, indicating the amorphous feature of the nanospheres (Fig. 2a). The above results indicate that the rapid aggregation of BF2bcz driven by the strong hydrophobic interaction upon addition of its THF solution to water kinetically trapped the metastable amorphous nanospheres. The BF2bcz molecules in the nanospheres were not able to reorganize into thermodynamically stable crystalline nanorods without the help of the surfactant to overcome the strong hydrophobic interactions among the BF2bcz molecules.
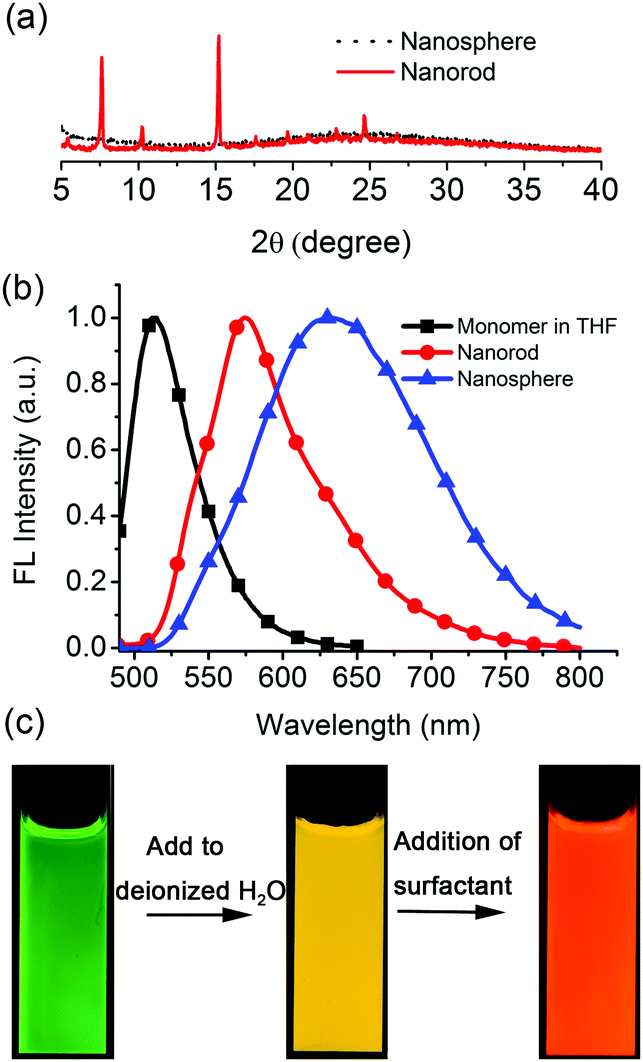 | ||
| Fig. 2 (a) The XRD patterns of the nanospheres and nanorods. (b) The emission spectra of the BF2bcz monomer in THF, aqueous dispersions of the nanospheres and the nanorods. (c) The fluorescence images of the BF2bcz monomer in THF (left), aqueous dispersions of nanospheres (middle) and nanorods (right) under illumination with a UV lamp. | ||
The fact that the nanospheres were the precursors of the crystalline nanorods was confirmed by the phase transition from the amorphous nanospheres to the crystalline nanorods by the addition of the surfactant to the aqueous dispersion of the nanospheres. Both anionic (e.g. SDS) and cationic surfactants (e.g. hexadecyltrimethylammonium bromide, CTAB) yielded such phase transition with high efficiency (Fig. S3, ESI†). The surfactant helped the nanospheres to overcome the hydrophobic interactions among the BF2bcz molecules and guided the BF2bcz molecules to reorganize into nanorods.
The amorphous nanospheres showed drastically different emission properties compared to the crystalline nanorods because of their distinct molecular arrangements (Fig. 2b and Fig. S4, ESI†). The nanospheres displayed a symmetrical and broad emission band centered at approximately λ = 640 nm, accompanied by a fluorescence quantum yield (Φf) of 0.08 and a fluorescence lifetime (τ) of 30.3 ns. It was worthy to note that the emission spectrum and fluorescence lifetime were quite similar to that of the dilute BF2bcz@SDS solution, in which the fluorescence from the excimers was observed (Fig. S5, ESI†). Given the random orientation of the molecules in the amorphous nanospheres, we speculated that the “excimer” species might possess the lowest energy levels and serve as energy acceptors to efficiently harvest the energies from the other higher energetic species. Therefore, the nanospheres always exhibited excimeric emission. With respect to the crystalline nanorods, the emission spectrum blue shifted to λ = 580 nm compared to that of the nanospheres, with Φf = 0.30 and τ = 5.6 ns (Fig. S6, ESI†). Both nanospheres and nanorods showed red-shifted emission compared with the BF2bcz monomer in dilute THF solution (λ = 489 nm). The conversion processes from the BF2bcz monomer and nanospheres to nanorods were apparent by their distinct emission colors under illumination with a UV lamp (Fig. 2c).
A comprehensive control of phase conversion and the unique emission properties of the nanospheres and nanorods allowed monitoring of the evolution process from nanospheres to nanorods by using fluorescence spectroscopy.13 After the addition of the surfactant, the initial emission maxima of the nanospheres at λ = 640 nm gradually enhanced and slightly red-shifted with the increase of time (Fig. 3a and Fig. S7, ESI†). Subsequently, a shoulder band at approximately λ = 580 nm emerged, similar to that of the mature nanorods, and then grew rapidly, and ultimately became constant. By plotting the emission intensities at λ = 580 nm against the evolution time, the resulting profile was non-linear (Fig. 3b). It displayed an initial slow growth, representing the nucleation stage, which was also the rate-determining step for the formation of the crystalline nanorods. After such a period of time, a rapid increase of the profile was observed and ascribed to the growth step.
 | ||
| Fig. 3 (a) The emission changes during the morphological transition from nanospheres to nanorods by adding CTAB (1.5 mg mL−1) to the aqueous dispersion of nanospheres. (b) The time-dependent emission intensities monitored at λ = 580 nm during the process of nanosphere–nanorod transition. | ||
The utilization of fluorescence spectroscopy to study the mechanism for the formation of the crystalline nanorods in our systems showed great advantages over electron microscopy, since it allowed for in situ and real-time monitoring of this process very conveniently. The effect of the external conditions such as temperatures and Csurfactant has been studied by fluorescence spectroscopy. Firstly, keeping all the conditions constant except the temperature, the crystallization process of BF2bcz was monitored by fluorescence spectroscopy at a temperature gradient from 15 to 30 °C with a 5 °C interval. After injecting the BF2bcz THF solution into the CTAB aqueous solution (CCTAB = 1.0 mg mL−1), the resulting molecular aggregates showed identical size and emission spectra to the nanospheres, further confirming that the nanospheres were the precursors of the crystalline nanorods (Fig. S8 and S9, ESI†). With the increase of the evolution time, a new peak at λ = 580 nm emerged and increased in intensity, ultimately becoming constant, implying the complete formation of the nanorods. At lower temperature, it took a longer time to reach the plateau of the emission spectra, e.g. from approximately 170 min at 15 °C decreasing to 26 min at 30 °C (Fig. 4 and Fig. S10, ESI†). This phenomenon indicated that the nanorods formed quickly at high temperature. The SEM images of the nanorods formed at temperatures from 15–30 °C revealed similar morphology and size, with a width in the range of 200–300 nm and a length of approximately 6 μm (Fig. S11, ESI†).
 | ||
| Fig. 4 The emission changes at different temperatures during the formation of the nanorods. (a) 15 °C, (b) 20 °C, (c) 25 °C, and (d) 30 °C. CCTAB = 1.0 mg mL−1. The spectra were recorded every 3 min. | ||
The CCTAB influenced the rate of the formation of the nanorods, as well as the sizes of the resulting nanorods. Decreasing the CCTAB from 2.0 mg mL−1 to 0.5 mg mL−1 induced the evolution time for the formation of the nanorods to increase from 20 min to 170 min (Fig. S12 and S13, ESI†). Meanwhile, the nanorods increased their length from 5 μm at the CCTAB of 2.0 mg mL−1 to about 30 μm with the CCTAB reduced to 0.5 mg mL−1 (Fig. 5). The final nanorods formed at different conditions exhibited the same emission spectra, indicating the identical molecular packing inside the nanorods (Fig. S14, ESI†). Similar phenomena were also observed when the surfactant (SDS) was exploited in the investigation (Fig. S15–S20, ESI†).
 | ||
| Fig. 5 The SEM images of the BF2bcz nanorods under different CCTAB. (a) 2.0 mg mL−1; (b) 1.0 mg mL−1; (c) 0.5 mg mL−1; (d) 0.25 mg mL−1. Scale bar: 5 μm. | ||
BF2bcz was also able to fabricate into nanorods when CCTAB and CSDS were below their respective critical aggregation concentrations (CAC) (Fig. S21 and S22, ESI†). This implied that the micelles were not prerequisite for inducing crystallization in our system. Combining this with the results obtained from the emission spectra during the crystallization process, we envisaged the possible mechanism for the formation of the nanorods. The emission spectra exhibited almost the same change tendency regardless of the addition order of the surfactant. This indicated that the BF2bcz molecules firstly spontaneously aggregated in the diffusion-limited aggregation regime to form amorphous nanospheres in an extremely short time. Subsequently, the nanospheres were covered by the surfactants. The protective layer of surfactants stabilized the nanospheres and lowered their surface energy, which further induced the commencement of the nucleation and growth of nanorods. The Csurfactant-dependent size of the nanorods was probably related to the surfactant coverage area on the surface of the assemblies. At high Csurfactant, a wide coverage area inhibited the sustained growth of the nanorods, while the narrow coverage at low Csurfactant caused the exposed parts of the assemblies to continuously elongate. This was the possible reason for the inverse relationship between the size of the nanorods and Csurfactant.
Then, we further investigated the dynamic system of the aqueous dispersion of the microrods by the addition of a probe molecule BF2cna, at a BF2cna/BF2bcz molar ratio of 0.1% (Fig. S23, ESI†).11d Without BF2cna, the pristine microrods exhibited a yellow emission color. After adding BF2cna, the emission color gradually red shifted with increasing aging time (Fig. 6). For example, the microrods showed an orange color after standing for 30 min, probably because the BF2cna molecules were adsorbed on the surface of the BF2bcz nanorods, and an inefficient energy transfer from BF2bcz to BF2cna has taken place. However, after increasing the standing time to approximately 100 hours, the emission color of the microrods became red, implying an efficient energy transfer. This result indicated that the BF2cna molecules penetrated into the interior part of the microrods. We concluded that there may involve a dynamic equilibrium between the assembly units in the formed microrods and the units in the surrounding media.
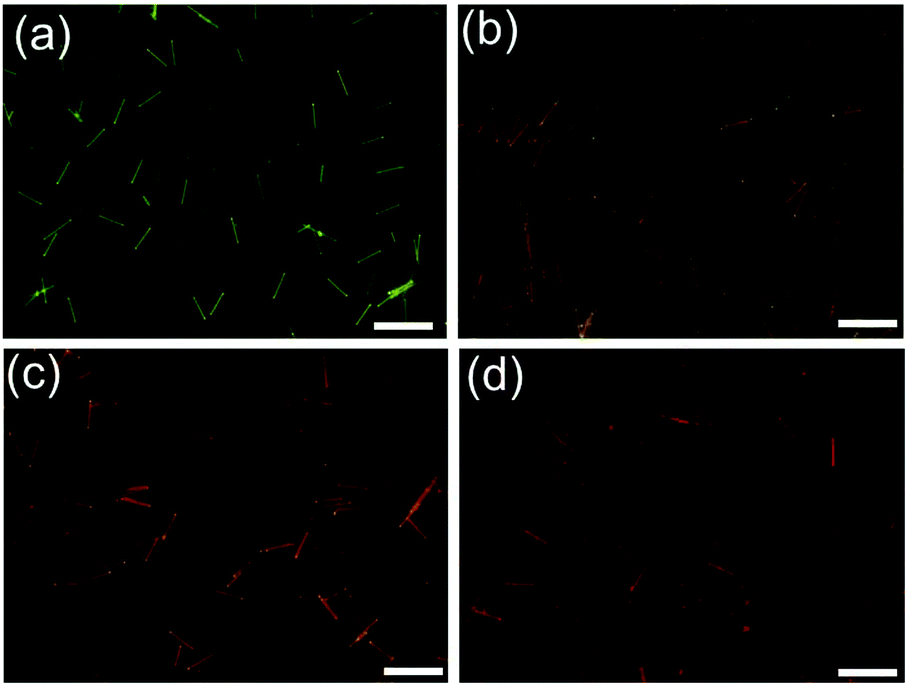 | ||
| Fig. 6 The fluorescence microscopy images of the BF2bcz nanorods after adding 0.1 mol% of BF2cna to the aqueous dispersion of the BF2bcz nanorods and viewed at different times. (a) 30 min; (b) 2 h; (c) 8 h; (d) 100 h. Scale bar: 10 μm. | ||
Conclusions
In conclusion, our work provides solid evidence for a two-step mechanism during the solution crystallization, in which the first formation of amorphous molecular aggregates precedes the nucleation. We have successfully separated the initial molecular aggregates in the absence of the surfactant and characterized their properties. The amorphous molecular aggregates were able to transform into crystalline nanorods by adding a surfactant to their aqueous dispersions. Fluorescence spectroscopy was utilized to in situ and real-time monitor the morphological transition process because of the distinct emission properties of the molecular aggregates and nanorods. In addition, the fluorescence technique was facile to identify the influence of external conditions, e.g. temperature and concentration of the surfactant, on the process of solution crystallization. It clarified that increasing the concentration of the surfactant and temperature both accelerated the crystallization process. This may inspire the study of other crystallization processes by using fluorescence techniques. We anticipate that our studies will help to understand the crystallization process of OMC materials in-depth, and may provide a guideline for the preparation of OMC materials with desired properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21525206, 21472202).Notes and references
- (a) C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC; (b) Z. Yu, Y. Wu, L. Xiao, J. Chen, Q. Liao, J. Yao and H. Fu, J. Am. Chem. Soc., 2017, 139, 6376–6381 CrossRef CAS PubMed; (c) W. Zhang, J. Yao and Y. S. Zhao, Acc. Chem. Res., 2016, 49, 1691–1700 CrossRef CAS PubMed; (d) M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed; (e) N. K. Duggirala, M. L. Perry, O. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC; (f) Y. Li, T. Liu, H. Liu, M.-Z. Tian and Y. Li, Acc. Chem. Res., 2014, 47, 1186–1198 CrossRef CAS PubMed.
- (a) R. J. Li, X. T. Zhang, H. L. Dong, Q. K. Li, Z. G. Shuai and W. P. Hu, Adv. Mater., 2016, 28, 1697–1702 CrossRef CAS PubMed; (b) Y. Wang, J. Liu, H. D. Tran, M. Mecklenburg, X. N. Guan, A. Z. Stieg, B. C. Regan, D. C. Martin and R. B. Kaner, J. Am. Chem. Soc., 2012, 134, 9251–9262 CrossRef CAS PubMed.
- Y. Guo, L. Xu, H. Liu, Y. Li, C.-M. Che and Y. Li, Adv. Mater., 2015, 27, 985–1013 CrossRef CAS PubMed.
- (a) X. Yang, X. Lin, Y. Zhao, Y. S. Zhao and D. Yan, Angew. Chem., Int. Ed., 2017, 56, 7853–7857 CrossRef CAS PubMed; (b) J. H. Kim, S. K. Park, J. H. Kim, D. R. Whang, W. S. Yoon and S. Y. Park, Adv. Mater., 2016, 28, 6011–6015 CrossRef CAS PubMed; (c) Y. Yan and Y. S. Zhao, Chem. Soc. Rev., 2014, 43, 4325–4340 RSC.
- (a) Z. Chen, Y. Liu, W. Wagner, V. Stepanenko, X. Ren, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 5729–5733 CrossRef CAS PubMed; (b) T. Brixner, R. Hildner, J. Kohler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef; (c) Q. Song, Y. Jiao, Z. Wang and X. Zhang, Small, 2016, 12, 24–31 CrossRef CAS PubMed.
- (a) T. Yamazaki, Y. Kimura, P. G. Vekilov, E. Furukawa, M. Shirai, H. Matsumoto, A. E. S. Van Driessche and K. Tsukamoto, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2154–2159 CrossRef CAS PubMed; (b) G. C. Sosso, J. Chen, S. J. Cox, M. Fitzner, P. Pedevilla, A. Zen and A. Michaelides, Chem. Rev., 2016, 116, 7078–7116 CrossRef CAS PubMed; (c) R. E. Schreiber, L. Houben, S. G. Wolf, G. Leitus, Z.-L. Lang, J. J. Carbó, J. M. Poblet and R. Neumann, Nat. Chem., 2016, 9, 369 CrossRef PubMed; (d) J. Ihli, W. C. Wong, E. H. Noel, Y.-Y. Kim, A. N. Kulak, H. K. Christenson, M. J. Duer and F. C. Meldrum, Nat. Commun., 2014, 5, 3169 Search PubMed; (e) P. Tan, N. Xu and L. Xu, Nat. Phys., 2013, 10, 73 CrossRef.
- (a) D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergstrom and H. Colfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC; (b) R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166–2179 CrossRef CAS PubMed; (c) P. G. Vekilov, Nanoscale, 2010, 2, 2346–2357 RSC; (d) D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed.
- M. Sleutel, J. Lutsko, A. E. S. Van Driessche, M. A. Durán-Olivencia and D. Maes, Nat. Commun., 2014, 5, 5598 CrossRef CAS PubMed.
- (a) M. Warzecha, R. Guo, R. M. Bhardwaj, S. M. Reutzel-Edens, S. L. Price, D. A. Lamprou and A. J. Florence, Cryst. Growth Des., 2017, 17, 6382–6393 CrossRef CAS; (b) H. Li, A. D. Chavez, H. Li, H. Li, W. R. Dichtel and J.-L. Bredas, J. Am. Chem. Soc., 2017, 139, 16310–16318 CrossRef CAS PubMed; (c) M. K. Bera and M. R. Antonio, J. Am. Chem. Soc., 2016, 138, 7282–7288 CrossRef CAS PubMed; (d) M. Agthe, T. S. Plivelic, A. Labrador, L. Bergström and G. Salazar-Alvarez, Nano Lett., 2016, 16, 6838–6843 CrossRef CAS PubMed; (e) X. Ye, Y. Liu, Y. Lv, G. Liu, X. Zheng, Q. Han, K. A. Jackson and X. Tao, Angew. Chem., Int. Ed., 2015, 54, 7976–7980 CrossRef CAS PubMed.
- (a) S. M. A. Fateminia, Z. Wang, C. C. Goh, P. N. Manghnani, W. Wu, D. Mao, L. G. Ng, Z. Zhao, B. Z. Tang and B. Liu, Adv. Mater., 2017, 29, 1604100 CrossRef PubMed; (b) C. Shahar, S. Dutta, H. Weissman, L. J. W. Shimon, H. Ott and B. Rybtchinski, Angew. Chem., Int. Ed., 2016, 55, 179–182 CrossRef CAS PubMed.
- (a) P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 1700332 CrossRef; (b) P.-Z. Chen, J.-X. Wang, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, J. Mater. Chem. C, 2017, 5, 12538–12546 RSC; (c) P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS; (d) P.-Z. Chen, Y.-X. Weng, L.-Y. Niu, Y.-Z. Chen, L.-Z. Wu, C.-H. Tung and Q.-Z. Yang, Angew. Chem., Int. Ed., 2016, 55, 2759–2763 CrossRef CAS PubMed.
- (a) Z.-Q. Lin, P.-J. Sun, Y.-Y. Tay, J. Liang, Y. Liu, N.-E. Shi, L.-H. Xie, M.-D. Yi, Y. Qian, Q.-L. Fan, H. Zhang, H. H. Hng, J. Ma, Q. Zhang and W. Huang, ACS Nano, 2012, 6, 5309–5319 CrossRef CAS PubMed; (b) X. Gu, J. Yao, G. Zhang and D. Zhang, Small, 2012, 8, 3406–3411 CrossRef CAS PubMed; (c) H. Fu, D. Xiao, J. Yao and G. Yang, Angew. Chem., Int. Ed., 2003, 42, 2883–2886 CrossRef CAS PubMed.
- A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2015, 8, 10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00118a |
| This journal is © the Partner Organisations 2018 |