
Exploration of the two-step crystallization of organic micro/nano crystalline materials by fluorescence spectroscopy†
 b
and
Qing-Zheng
Yang
*a
b
and
Qing-Zheng
Yang
*a
Abstract
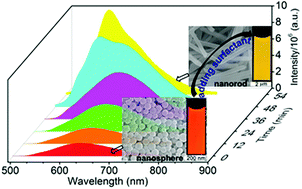
Clearly understanding the crystallization process of organic micro/nano crystalline (OMC) materials in solution is a long-standing challenge because of the difficulty in the separation of intermediates and monitoring the process in situ and in real-time. Herein, we report the exploration of the crystallization process of OMC materials from the amorphous intermediates by taking advantage of the spectral change of an environment-sensitive emission dye BF2bcz. The intermediate for the formation of the OMC materials by the solvent-exchange method was separated as amorphous nanospheres which were transformed into crystalline nanorods by adding the surfactant to their aqueous dispersion. The distinct emission properties of the amorphous molecular aggregates and nanorods, used as a fingerprint for each species, allow for in situ and real-time monitoring of the crystallization process by using fluorescence spectroscopy. Such a facile method readily identified that increasing the concentration of surfactant and temperature both accelerated the crystallization process of BF2bcz in aqueous solution, while the size of the nanorods increased with a decrease in the concentration of surfactant. Our work provided direct experimental evidence to support the two-step nucleation mechanism in the preparation of OMC by the solvent-exchange method.
 Please wait while we load your content...
Please wait while we load your content...

