DOI:
10.1039/C8QM00112J
(Research Article)
Mater. Chem. Front., 2018,
2, 1165-1174
Charge-transfer complexes based on C2v-symmetric benzo[ghi]perylene: comparison of their dynamic and electronic properties with those of D6h-symmetric coronene†
Received
16th March 2018
, Accepted 13th April 2018
First published on 16th April 2018
Abstract
Single crystals of three neutral charge-transfer complexes and a cation radical salt based on a C2v-symmetric polycyclic aromatic hydrocarbon, benzo[ghi]perylene (bper), were obtained. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with 7,7,8,8-tetracyanoquinodimethane (TCNQ) involves DA-type alternating π-columns, whereas the alternating π-columns in the 2:1 TCNQ complex are flanked by another bper molecule. The in-plane rotation of bper in the 1:1 complex was significantly suppressed compared with that of coronene in (coronene) (TCNQ), which is associated with the lower molecular symmetry of bper. Whereas the 3:1 TCNQ complex involves DDA-type alternating π-columns flanked by another bper molecule, bper molecules in the 3:1 cation radical salt with Mo6O192− have a columnar structure with a [101]-like charge-ordered pattern associated with intermolecular interactions in the bay region of bper. The dimerisation of charge-rich bper molecules results in an increased energy gap at the Fermi level, and consequently, semiconducting behaviour of the salt has a larger activation energy than that of the isostructural coronene salt in the partially charged state. The lower molecular symmetry of bper also affects the degeneracy of the frontier-orbitals; the energy difference between the HOMO and the HOMO−1 of bper is significantly larger than that of coronene, which is comparable to the intermolecular transfer integrals.
:1 complex with 7,7,8,8-tetracyanoquinodimethane (TCNQ) involves DA-type alternating π-columns, whereas the alternating π-columns in the 2:1 TCNQ complex are flanked by another bper molecule. The in-plane rotation of bper in the 1:1 complex was significantly suppressed compared with that of coronene in (coronene) (TCNQ), which is associated with the lower molecular symmetry of bper. Whereas the 3:1 TCNQ complex involves DDA-type alternating π-columns flanked by another bper molecule, bper molecules in the 3:1 cation radical salt with Mo6O192− have a columnar structure with a [101]-like charge-ordered pattern associated with intermolecular interactions in the bay region of bper. The dimerisation of charge-rich bper molecules results in an increased energy gap at the Fermi level, and consequently, semiconducting behaviour of the salt has a larger activation energy than that of the isostructural coronene salt in the partially charged state. The lower molecular symmetry of bper also affects the degeneracy of the frontier-orbitals; the energy difference between the HOMO and the HOMO−1 of bper is significantly larger than that of coronene, which is comparable to the intermolecular transfer integrals.
Introduction
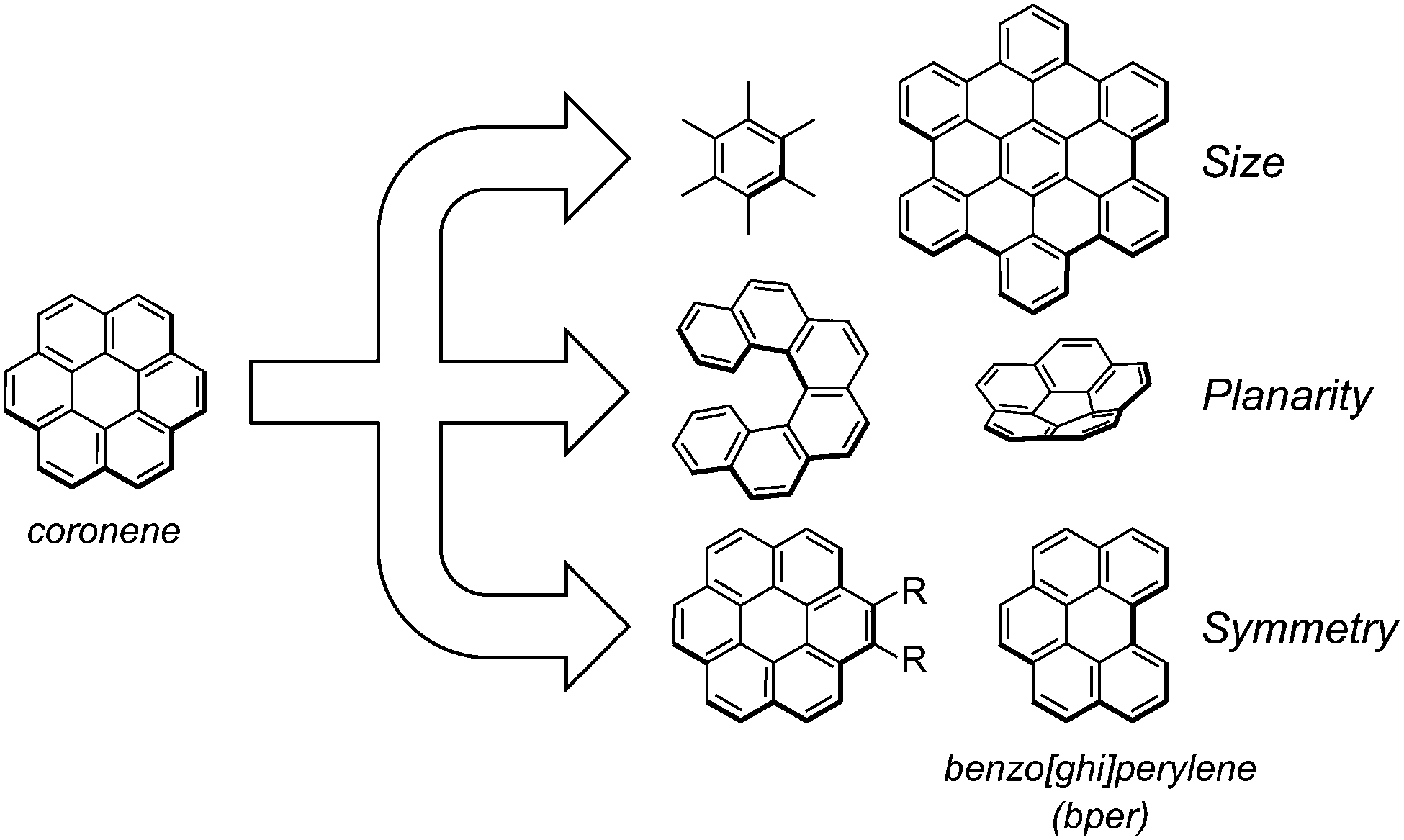
Highly symmetric polycyclic aromatic hydrocarbons (PAHs) are attractive as potential components of high-Tc superconductors owing to their degenerate frontier orbitals1–3 and are a promising platform for artificial molecular rotors with electronic functionalities in the solid state.3–6 Because the dynamic and electronic properties of solids largely depend on the molecular and supramolecular structures of the components, a better understanding of the structure–property relationship with a focus on the molecular symmetry of PAHs would allow the rational design of solids with desired functionalities. Over the past few years, our group has prepared a variety of charge-transfer (CT) complexes composed of the smallest D6h-symmetric PAH molecule, coronene (Scheme 1) as an electron donor (D). Notably, both dicationic7 and dianionic8 coronene are in a thermally accessible triplet state at room temperature associated with the doubly degenerate HOMO (e2u) and LUMO (e1g) levels. The CT complexes range from insulating neutral complexes9–11 to conducting cation radical salts,12–14 and especially, some of the latter with π-stacking columns of cationic coronene show semiconducting behaviour with a high room-temperature conductivity (>1 S cm−1). In addition, we found that the in-plane rotational rate of coronene molecules in CT solids can be varied by more than six orders of magnitude (1 kHz to 7 GHz at room temperature) by modifying intermolecular interactions, such as van der Waals, π–π, and hydrogen bonding interactions.
 |
| | Scheme 1 Molecular structures of coronene and its analogues. | |
Investigation of complexes composed of coronene analogues and comparison with the corresponding coronene-based CT complexes will provide a deeper understanding of how the high symmetry of coronene affects the dynamic and electronic properties in the solid state. There are three apparent categories of coronene analogues (Scheme 1): (1) D6h-symmetric polyaromatic hydrocarbons with different π-conjugated sizes (e.g., hexamethylbenzene and hexa-peri-hexabenzocoronene),15,16 (2) nonplanar PAHs with curved or twisted π-conjugated planes (e.g., [n]circulene (n ≠ 6) and [n]helicene),17–21 and (3) planar PAHs with reduced symmetries (e.g., 1,2-disubstituted coronene and benzo[ghi]perylene (bper)).22,23 Within the second group, we have recently realised peculiar π-electron networks in neutral CT complexes by combining corannulene (i.e., [5]circulene)24 or [6]helicene25 as D with planar 7,7,8,8-tetracyanoquinodimethane (TCNQ) analogues as electron acceptors (A's). In search of CT complexes using PAHs in the third group, we focus on C2v-symmetric bper, which is regarded as a coronene analogue that has lost a benzene ring; unlike 1,2-disubstituted coronene, bper has no bulky substituents and has the higher electron donating ability (E11/2(D) = 1.01 V vs. saturated calomel electrode (SCE)) than that of coronene (1.23 V).26 In the solid state, bulky substituents inevitably inhibit intermolecular π–π interactions, which are indispensable for the formation of a π-conduction network, and the appropriate electron-donating ability allows the formation of stable CT complexes under aerobic conditions. Herein, we report the synthesis and the structural, optical, and transport properties of neutral CT complexes with TCNQ having different compositions (1–3) and a cation radical salt with a hexamolybdate (Mo6O192−) cluster unit (4), with the support of theoretical calculations. Crystallographic data were also used to assess the thermal fluctuations, which are indicative of in-plane molecular rotations, of bper molecules in the solid state.
Results
(bper)(TCNQ) (1) and (bper)3(TCNQ) (3) were prepared by the solvent evaporation method, whereas (bper)2(TCNQ) (2) was obtained preferentially by the co-sublimation method. (bper)3Mo6O19 (4) was obtained by the electrochemical oxidation of bper in the presence of an electrolyte, (Bu4N)2Mo6O19 (see Experimental and computational for details). The crystal structures of 1–4 were determined at 100 K (Table 1).
Table 1 Crystallographic data of 1–4
|
|
(bper)(TCNQ) (1) |
(bper)2(TCNQ) (2) |
(bper)3(TCNQ) (3) |
(bper)3Mo6O19 (4) |
|
R
1 = ∑(|F0| − |Fc|)/∑|F0|.
wR2 = [∑w(F02 − Fc2)2/∑w(F02)2]0.5.
|
| Formula |
C34H16N4 |
C56H28N4 |
C78H40N4 |
C66H36Mo6O19 |
| Formula weight |
480.53 |
756.87 |
1033.20 |
1708.64 |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
| Crystal size (mm3) |
0.49 × 0.09 × 0.06 |
0.20 × 0.08 × 0.07 |
0.50 × 0.07 × 0.04 |
0.56 × 0.08 × 0.04 |
|
a (Å) |
7.1719(4) |
7.1246(8) |
10.0725(14) |
9.8455(9) |
|
b (Å) |
10.8284(5) |
12.3495(14) |
10.8786(15) |
11.0998(10) |
|
c (Å) |
29.7864(15) |
21.257(3) |
12.9332(18) |
12.8243(11) |
|
α (deg) |
90 |
78.598(2) |
113.110(2) |
86.101(1) |
|
β (deg) |
90 |
80.637(2) |
105.578(2) |
67.763(1) |
|
γ (deg) |
90 |
80.207(2) |
90.301(2) |
89.093(1) |
|
V (Å3) |
2313.2(2) |
1790.4(4) |
1245.7(3) |
1294.2(2) |
|
Z
|
4 |
2 |
1 |
1 |
| Temperature (K) |
100 |
100 |
100 |
100 |
|
d
calc (g cm−3) |
1.380 |
1.404 |
1.377 |
2.192 |
|
μ(Mo Kα) (mm−1) |
0.083 |
0.083 |
0.080 |
1.499 |
| Reflns used |
4716 |
6295 |
5057 |
5122 |
| Refined params |
343 |
541 |
469 |
511 |
|
R
1 (for I > 2σ(I))a |
0.0386 |
0.0783 |
0.0490 |
0.0204 |
| wR2 (for all data)b |
0.0799 |
0.1755 |
0.1189 |
0.0515 |
| GOF on F2 |
1.030 |
1.003 |
1.003 |
1.026 |
| CCDC |
1537813
|
1537814
|
1537815
|
1537816
|
Charge transfer
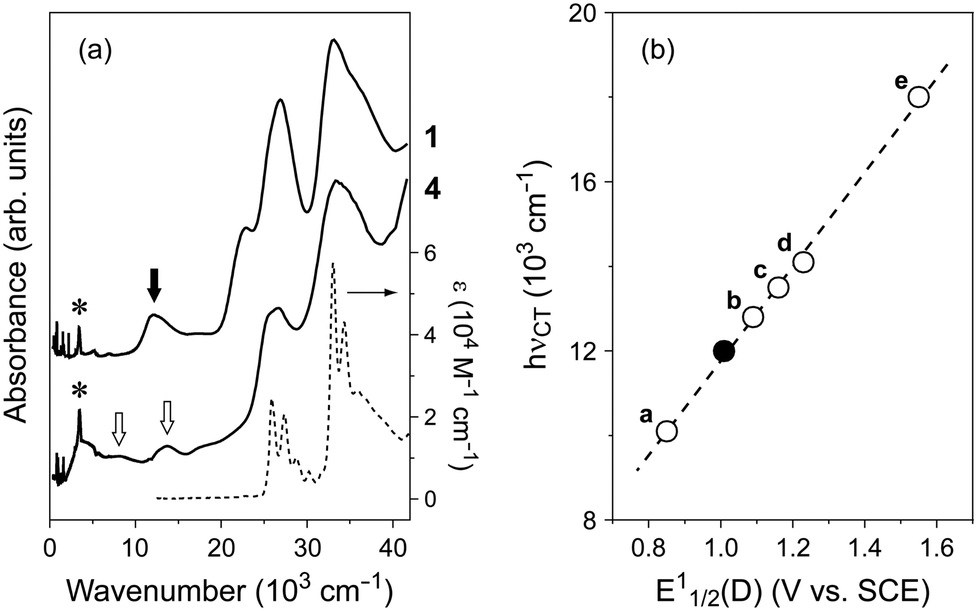
Regardless of composition, complexes 1–3 exhibit a CT absorption band at around hνCT = 12 × 103 cm−1 in addition to the intramolecular transition bands of the component molecules above 20 × 103 cm−1 (Fig. 1a). These hνCT values are lower than those of coronene–TCNQ complexes (14 × 103 cm−1)9 because the electron-donating ability of bper is stronger than that of coronene.27,28 As shown in Fig. 1b, complexes 1–3 lie on the straight line obtained by applying a least-squares method to the plots of hνCT against E11/2(D) for the TCNQ complexes of typical PAH molecules (perylene,29 anthracene,30 pyrene,31 coronene,9,32 and triphenylene33). Bond length analysis based on crystallographic data34 estimates the charge of the TCNQ molecules to be −0.14(3) for 1, −0.23(9) for 2, and −0.17(4) for 3, which are the same within experimental error. Infrared spectra support the neutral nature (Fig. S1 in ESI†).
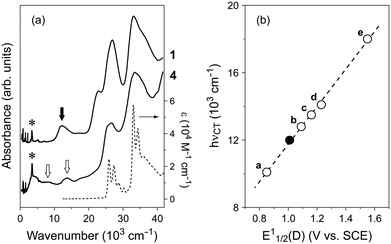 |
| | Fig. 1 (a) Absorption spectra of 1 and 4 measured on KBr pellets (solid lines) together with pristine bper in 10−5 M chloroform solution (dotted line). Closed and open arrows indicate the bands originating from an intermolecular charge transfer transition between neutral bper and TCNQ and an intramolecular transition of monocationic bper, respectively. Asterisks indicate the band originating from the O–H stretching mode of water molecules absorbed in the KBr powder. (b) Plot of CT energy (hνCT) against the first redox potential of PAH molecules (E11/2(D)) in TCNQ complexes (closed circle: 1–3, a: (perylene)(TCNQ),29 b: (anthracene)(TCNQ),30 c: (pyrene)(TCNQ),31 d: (coronene)(TCNQ),9,32 e: (triphenylene)2(TCNQ)(toluene)33). | |
Salt 4 exhibits the distinct bands at 8.1 and 13.7 × 103 cm−1, which are reminiscent of the absorption spectra of the bper monocation isolated in matrices.35–37 This spectral feature is firm evidence that this salt is the first example of a solid composed of cationic bper. The salt also exhibits a low-lying band with a maximum at approximately 4 × 103 cm−1, which arises from the intermolecular transition between neutral and monocationic bper molecules,38 as expected from the X-ray diffraction measurements (vide infra).
(bper)(TCNQ) (1).
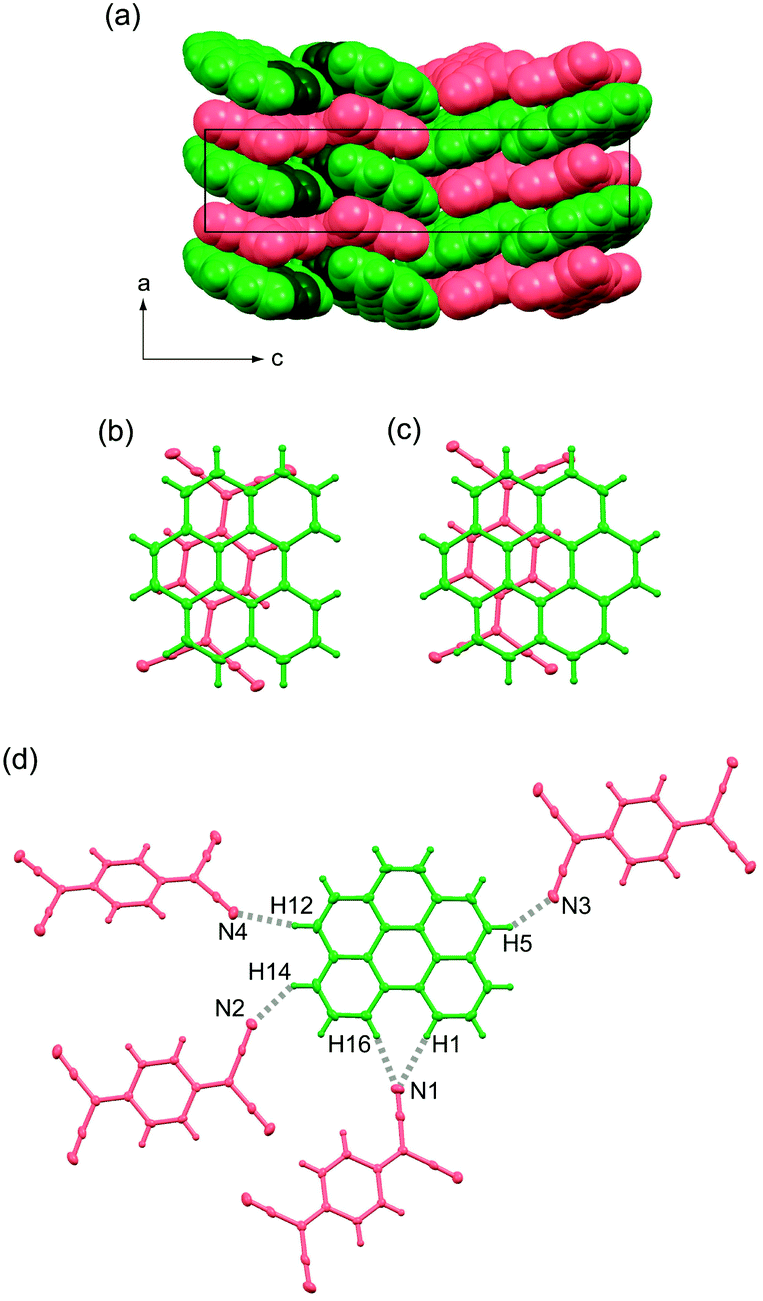
Complex 1 crystallises in a non-centrosymmetric orthorhombic lattice with space group P212121, and each bper and TCNQ molecule is crystallographically independent. The complex involves DA-type alternating π-stacking columns (Fig. 2a) with a ring-over-bond pattern (Fig. 2b) along the a axis, as in (coronene)(TCNQ) (Fig. 2c).9,32 Within the column, bper molecules are aligned in the same direction because each unit cell along the a direction contains only one bper molecule. Adjacent bper and TCNQ molecules are arranged in a nonparallel fashion, and the interplanar distance, which is defined as half the distance between bper molecules across a TCNQ molecule, was estimated to be 3.27 Å. It should be noted that the dihedral angle (θ) between bper and TCNQ molecules within the column (3.36°) is apparently larger than that in (coronene)(TCNQ) (2.40°), but the interplanar distance is comparable to that in (coronene)(TCNQ) (3.26 Å).9 Along the side-by-side direction, each bper molecule is connected to four adjacent TCNQ molecules (Fig. 2d) through C–H⋯N hydrogen bonds (H1⋯N1: 2.72 Å, H16⋯N1: 2.63 Å, H14⋯N2: 2.75 Å, H12⋯N4: 2.61 Å, H5⋯N3: 2.69 Å vs. sum of van der Waals radii: 2.75 Å39).
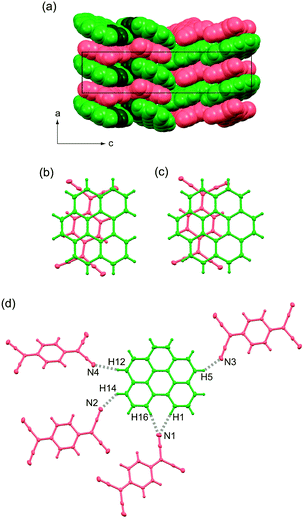 |
| | Fig. 2 (a) Molecular packing of 1 viewed along the b axis in a space-fill representation. Bper and TCNQ molecules appear in green and red, respectively, where carbon and hydrogen atoms in the bay region are darkened. Overlap patterns of adjacent PAH and TCNQ molecules within a column viewed perpendicularly to the molecular plane of PAH in (b) 1 and (c) (coronene)(TCNQ).9,32 (d) C–H⋯N hydrogen bonds around bper molecule in 1 along the side-by-side direction. | |
(bper)2(TCNQ) (2).
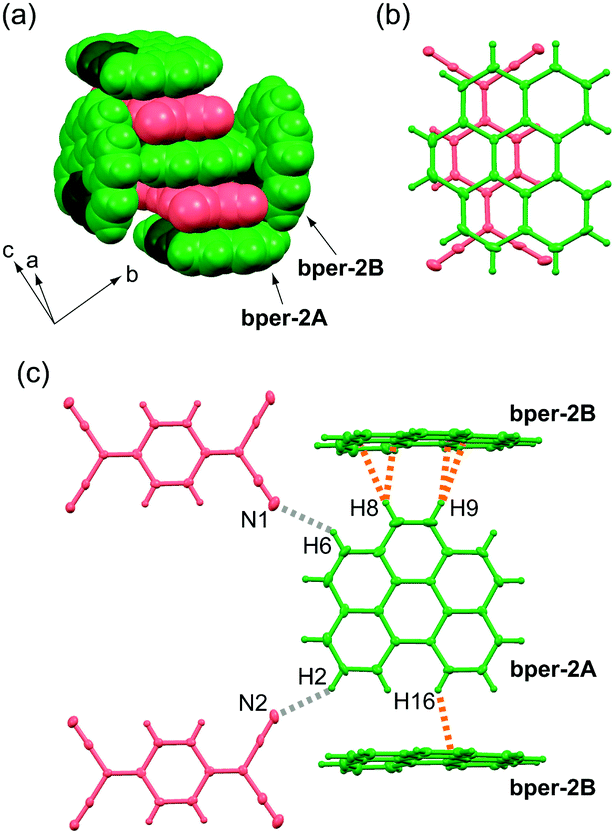
Complex 2 has lower crystal symmetry (centrosymmetric triclinic P) than 1, and the asymmetric unit contains two bper molecules (denoted as bper-2A and bper-2B) and one TCNQ molecule. In the crystal, the bper-2A molecules construct a DA-type alternating π-stacking column with TCNQ along the a axis, whereas bper-2B, which has a tilt angle with respect to bper-2A of 84.3°, separates the neighbouring columns approximately along the [1 −1 0] direction (Fig. 3a). A coronene–TCNQ CT complex with a 2:1 ratio has not been obtained at present, but (perylene)2TCNQ with the same packing motif was recently prepared by Vermeulen et al.40 Within the column, adjacent bper-2A and TCNQ molecules, whose molecular planes are slightly inclined to each other (θ = 0.79°), display a ring-over-bond overlap pattern (Fig. 3b). Along the side-by-side direction, each bper-2A molecule forms short interatomic contacts with two bper-2B molecules through C–H⋯π hydrogen bonds (≈2.9 Å;39,41,42 the observed C–H⋯C distances are 2.65–2.89 Å) and two TCNQ molecules through C–H⋯N hydrogen bonds (H2⋯N2: 2.66 Å, H6⋯N1: 2.73 Å), as shown in Fig. 3c.
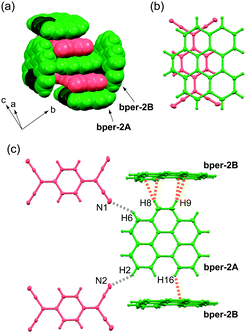 |
| | Fig. 3 (a) Molecular arrangement of 2 in a space-fill representation. Bper and TCNQ molecules appear in green and red, respectively, where carbon and hydrogen atoms in the bay region are darkened. (b) Overlap pattern of adjacent bper-2A and TCNQ molecules within a column viewed perpendicularly to the molecular plane of bper-2A in 2. (c) Short interatomic contacts around one bper-2A molecule with surrounding bper-2B and TCNQ molecules in 2. The gray and orange dotted lines show C–H⋯N and C–H⋯C (or C–H⋯π) hydrogen bonds, respectively. | |
(bper)3(TCNQ) (3).
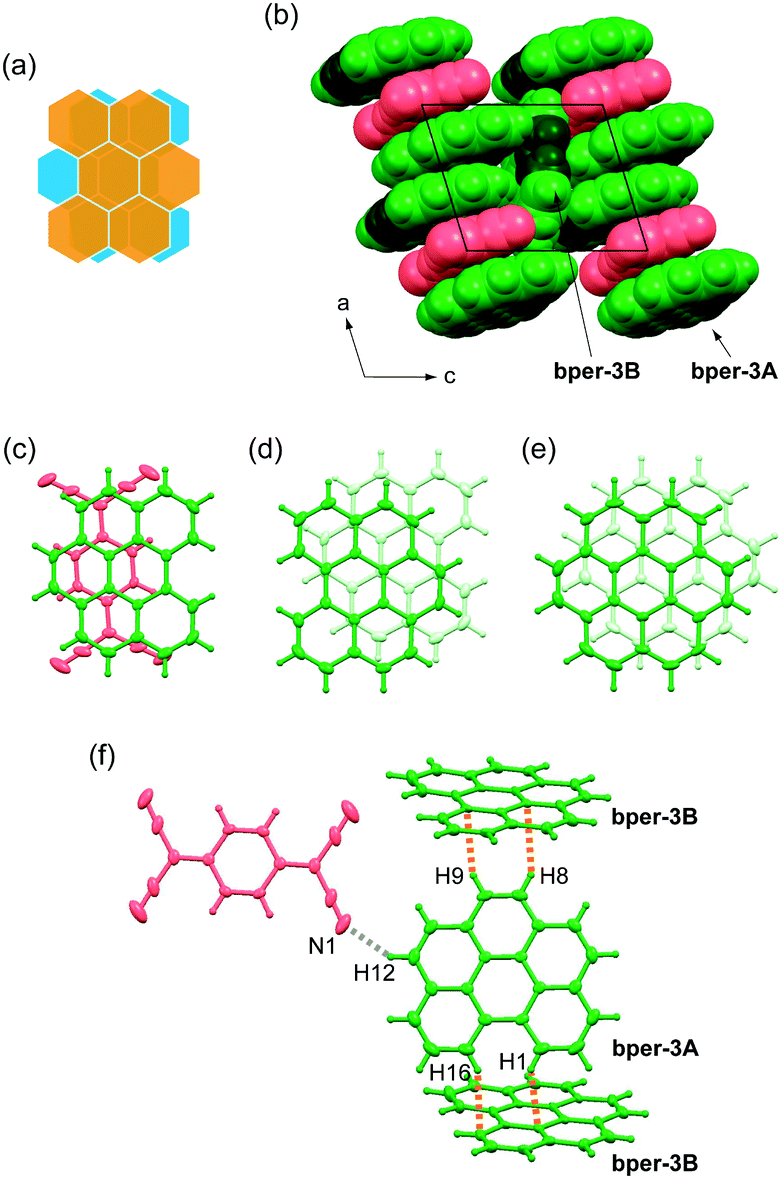
Complex 3 also belongs to the centrosymmetric triclinic system with space group P and is isostructural with (coronene)3(TCNQ).9 The asymmetric unit includes one and a half bper molecules (denoted as bper-3A and bper-3B, respectively) and half a TCNQ molecule; bper-3B is disordered over two orientations, which are related by an inversion center, with equivalent occupancy factors of 0.5 (Fig. 4a). As shown in Fig. 4b, the complex involves DDA-type alternating π-stacking columns with a [bper-3A⋯bper-3A⋯TCNQ] repeating unit along the a axis. As in the case of 2, the column is flanked by another bper molecule (bper-3B) with a tilt angle of 69.8° with respect to bper-3A. The ring-over-bond-type overlap pattern between bper-3A and TCNQ (Fig. 4c) is reminiscent of those in 1 and 2, and the molecular planes are slightly inclined to each other (θ = 0.97°). Adjacent bper-3A molecules within the column are oriented in the opposite direction with a ring-over-atom overlap pattern on an inversion center (Fig. 4d), in contrast to the ring-over-bond overlap pattern in pristine bper crystal43,44 with a “sandwich herringbone structure”45 and the same interplanar distance (3.37 Å). A shift along the in-plane direction (2.67 Å) is significantly larger than that in (coronene)3(TCNQ) (1.50 Å; Fig. 4e) with a longer interplanar distance (3.43 Å).9 As shown in Fig. 4f, each bper-3A molecule is connected to two bper-3B molecules through C–H⋯π hydrogen bonds (the observed C–H⋯C distances are 2.70–2.89 Å) and one TCNQ molecule through a C–H⋯N hydrogen bond (H12⋯N1: 2.70 Å).
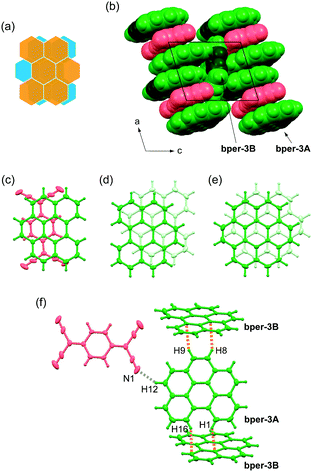 |
| | Fig. 4 (a) Schematic representation of the disorder of bper-3B molecule in 3. (b) Molecular packing of 3 viewed along the b axis in a space-fill representation. Bper and TCNQ molecules appear in green and red, respectively, where carbon and hydrogen atoms in the bay region are darkened. (c) Overlap pattern of adjacent bper-3A and TCNQ molecules within a column viewed perpendicularly to the molecular plane of bper-3A in 3. Overlap patterns of adjacent PAH molecules within a column viewed perpendicularly to the molecular plane of PAH in (d) 3 and (e) (coronene)3(TCNQ).9,32 (f) Short interatomic contacts around one bper-3A molecule with surrounding bper-3B and TCNQ molecules in 3. The gray and orange dotted lines show C–H⋯N and C–H⋯C (or C–H⋯π) hydrogen bonds, respectively. | |
(bper)3Mo6O19 (4).
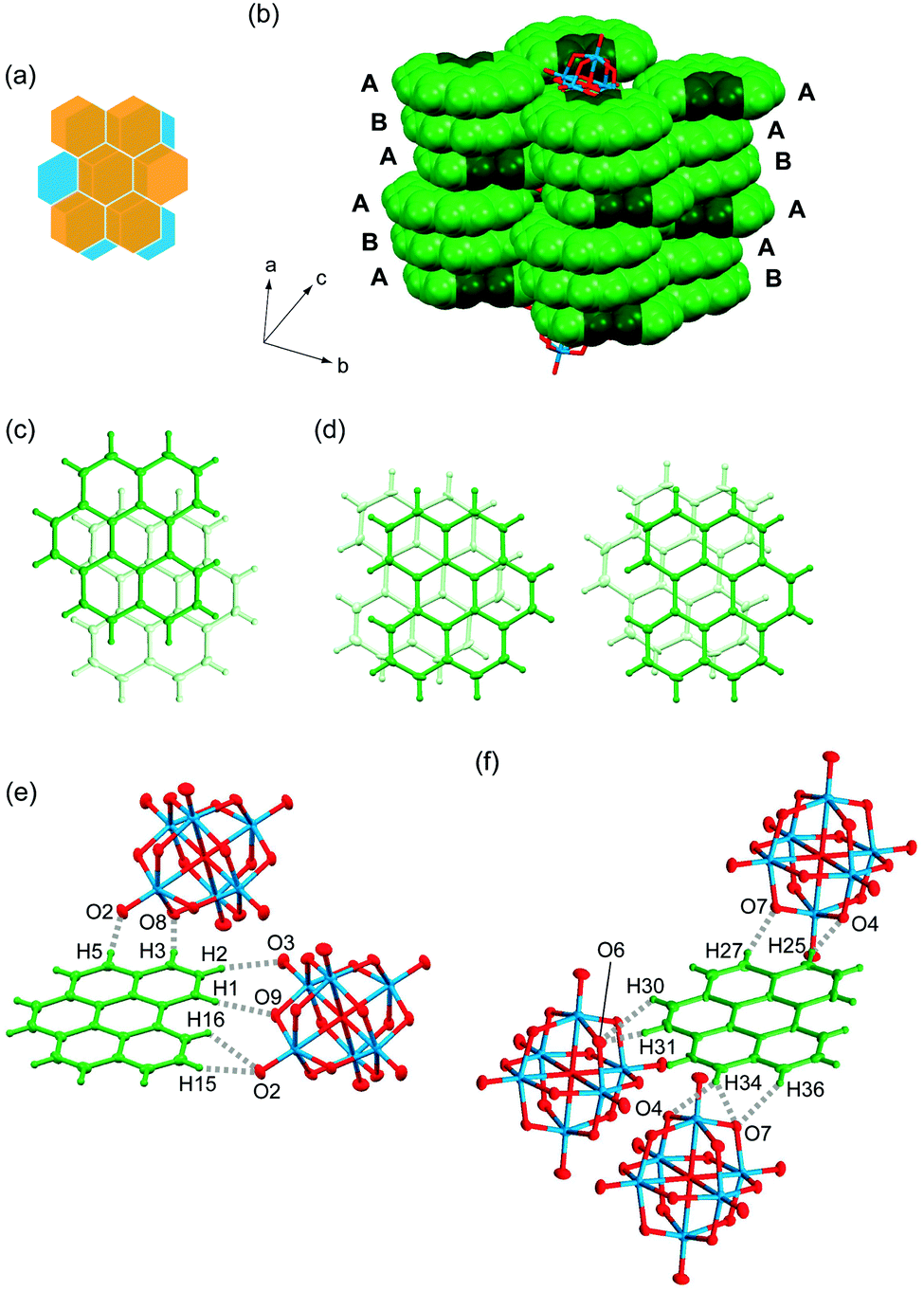
Salt 4 is the first cation radical solid based on bper, and the triclinic lattice (space group P) is isostructural with (coronene)3Mo6O19.13 One and a half bper molecules (denoted as bper-4A and bper-4B, respectively) and half a Mo6O192− cluster unit are crystallographically independent; namely, bper-4B and Mo6O192− are located on an inversion center. Similar to bper-3B in 3, bper-4B is disordered over two orientations with equivalent occupancy factors of 0.5 (Fig. 5a). Notably, the salt forms a segregated π-stacking column with a [bper-4A⋯bper-4A⋯bper-4B] repeating unit along the a axis (Fig. 5b). Such a cationic column has been reported for several PAH molecules.46–50 Within the column, adjacent bper-4A molecules oriented in the opposite direction display a ring-over-bond overlap pattern (Fig. 5c) with an interplanar distance of 3.14 Å, which is similar to that in isostructural (coronene)3Mo6O19 (3.16 Å; ring-over-atom).13 This distance is significantly shorter than those in natural graphite (3.35 Å; ring-over-atom),51 pristine bper crystal (3.36 Å; ring-over-bond),43,44 and 3 (3.37 Å; ring-over-atom), possibly due to stabilisation of the partially occupied HOMO of each bper-4A. In contrast, adjacent bper-4A and bper-4B molecules are nonparallel with a dihedral angle of 3.9°; one of the disordered bper-4B molecules is oriented in the same direction as bper-4A, whereas the other is oriented in the opposite direction (Fig. 5d). Notably, the interplanar distance, which is defined as half the distance between two bper-4A molecules separated by a bper-4B molecule, was estimated to be 3.28 Å, which is significantly longer than that in (coronene)3Mo6O19 (3.19 Å).13
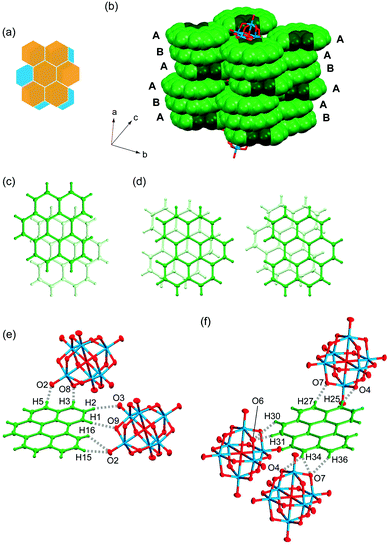 |
| | Fig. 5 (a) Schematic representation of the disorder of bper-4B molecule in 4. (b) Columnar structure of bper molecules in 4 in a space-fill representation, where carbon and hydrogen atoms in the bay region are darkened. A and B indicate the bper-4A and bper-4B molecules, respectively. Overlap patterns of (c) adjacent bper-4A molecules and (d) adjacent bper-4A and bper-4B molecules with two possible orientations within a column viewed perpendicularly to the molecular plane of bper-4A in 4. C–H⋯O hydrogen bonds around (e) bper-4A and (f) one orientation of disordered bper-4B in 4 along the side-by-side direction. | |
Each bper-4A molecule interacts with two adjacent Mo6O192− cluster units along the side-by-side direction through C–H⋯O hydrogen bonds (H15⋯O2: 2.67 Å, H16⋯O2: 2.58 Å, H1⋯O9: 2.49 Å, H2⋯O3: 2.50 Å, H3⋯O8: 2.43 Å, H5⋯O2: 2.41 Å vs. sum of van der Waals radii: 2.72 Å;39Fig. 5e). Notably, hydrogen atoms in the bay region of bper-4A are essential for the formation of quadruple hydrogen bonds with an adjacent Mo6O192− cluster unit. Fig. 5f shows the short interatomic contacts between one orientation of disordered bper-4B and Mo6O192− along the side-by-side direction. Each bper-4B molecule forms C–H⋯O hydrogen bonds with three adjacent Mo6O192− cluster units through double or triple C–H⋯O hydrogen bonds (H36⋯O7: 2.68 Å, H34⋯O7: 2.52 Å, H34⋯O4: 2.50 Å, H31⋯O6: 2.63 Å, H30⋯O6: 2.64 Å, H27⋯O7: 2.67 Å, H25⋯O4: 2.37 Å). Unlike the case of bper-4A, hydrogen atoms in the bay region of bper-4B make no contribution to hydrogen bond formation.
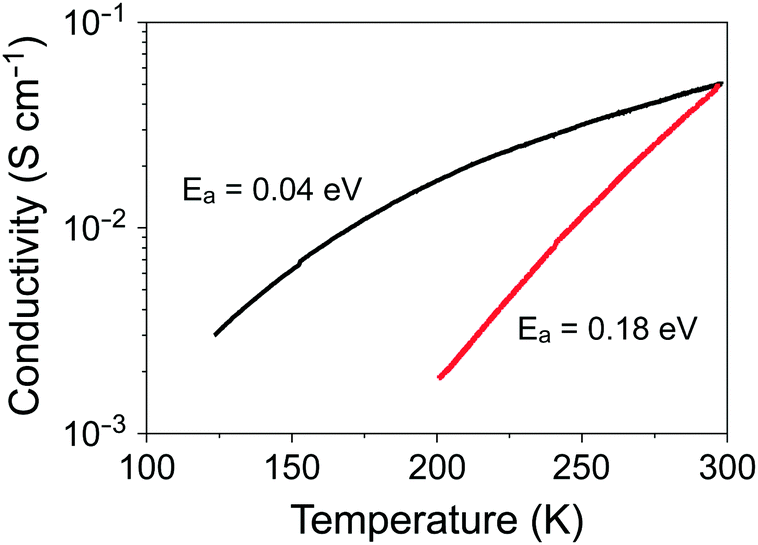
Resistivity measurements were performed to investigate the effect of molecular symmetry on the electronic properties. Fig. 6 shows the temperature dependence of the electrical conductivity (σ) of a single crystal of 4, which has a room-temperature value of 5 × 10−2 S cm−1 similar to that of (coronene)3Mo6O19.13 However, a marked difference between the two salts was observed upon cooling; namely, the σ value of 4 decreases steeply with decreasing temperature. The activation energy of 4 was estimated to be 0.18 eV, which is more than four times that of (coronene)3Mo6O19 (0.04 eV).
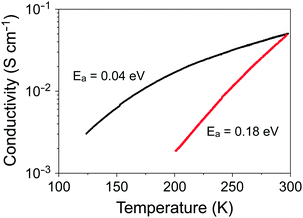 |
| | Fig. 6 Temperature dependence of the electrical conductivity of a single crystal of 4 (red line) and (coronene)3Mo6O19 (black line).13 | |
Discussion
Effect of molecular structure on dynamic properties
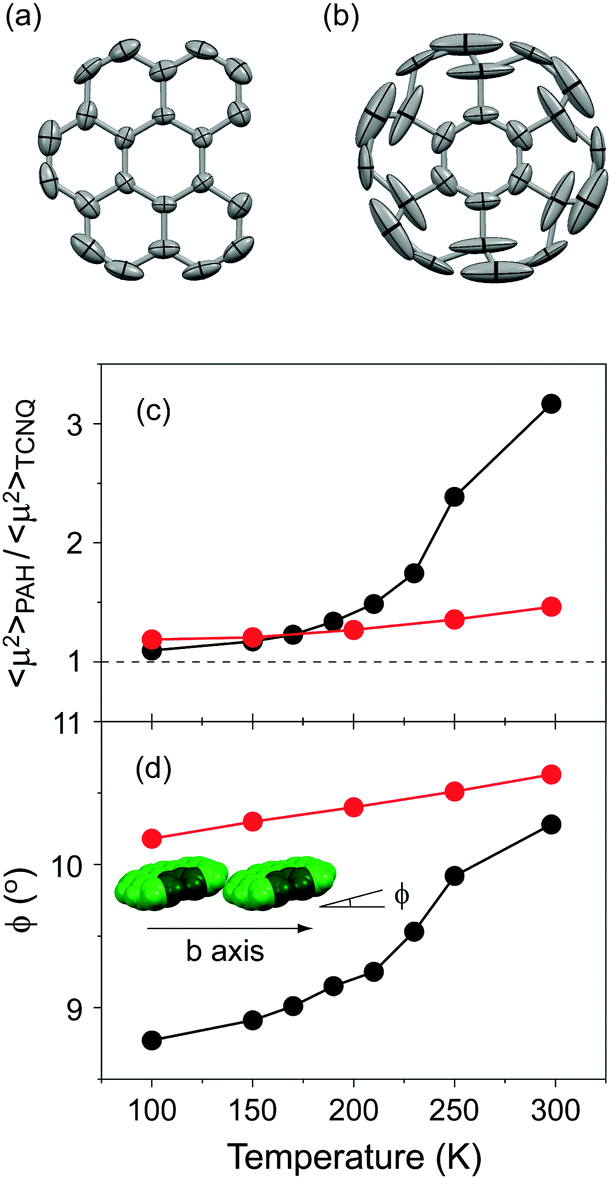
Solid-state NMR spectroscopy has become a definitive experimental technique for investigating the dynamic properties of molecular rotators in the solid state.3,5,6 Despite this demonstrated utility, the deuteration of bper is unavailable at the present stage. Another way to assess the dynamic behaviour of molecules is to examine the magnitude and anisotropy of thermal fluctuations based on crystallographic data. Indeed, coronene molecules, whose fast in-plane 6-fold flipping motion was confirmed by the 2H NMR spectra of the deuterated complexes, exhibit large thermal fluctuations along the in-plane direction.9,11 As shown in Fig. 7a, the in-plane thermal fluctuations of bper molecules in 1 at room temperature are less pronounced than those of (coronene)(TCNQ) (Fig. 7b);9 the average mean-square displacement of component carbon atoms, 〈μ2〉, was estimated to be 0.062 Å2 for bper in 1 and 0.145 Å2 for coronene in (coronene)(TCNQ) at 298 K. Thus, the in-plane rotation of bper is significantly suppressed by the loss of a benzene ring, even though 1 has a larger θ value than (coronene)(TCNQ).
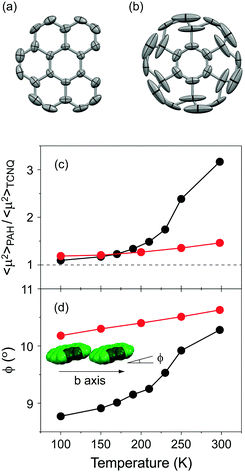 |
| | Fig. 7 Thermal fluctuations of (a) bper in 1 and (b) coronene in (coronene)(TCNQ)9 at 298 K viewed perpendicular to the molecular plane. Hydrogen atoms are omitted for clarity, and the 50% probability thermal ellipsoids are shown. Temperature dependence of (c) relative 〈μ2〉 values of PAH (bper or coronene) with respect of that of TCNQ (〈μ2〉PAH/〈μ2〉TCNQ) and (d) angles between the PAH plane and the crystallographic axis along the side-by-side direction (ϕ) in each CT solid (red circles: 1, black circles: (coronene)(TCNQ)).9 The inset shows the arrangement of bper molecules in 1 along the b axis. | |
To shed light on this phenomenon, we investigate the temperature dependence of the relative 〈μ2〉 value with respect of that of TCNQ in 1, as previously performed for (coronene)(TCNQ).9 As shown in Fig. 7c, the coronene complex shows a rapid increase in 〈μ2〉PAH/〈μ2〉TCNQ at around 200 K, above which the angle between the coronene plane and the crystallographic a axis (ϕ; Fig. 7d) also steeply increases to accelerate molecular rotation. In contrast, for 1, neither parameter shows such an anomaly in the measured temperature region (100–298 K). The origin of the effect on the dynamic properties of these complexes is not determined from the crystallographic data. However, the difference in the relationship between 〈μ2〉PAH/〈μ2〉TCNQ and ϕ provides clear evidence for the importance of intermolecular interactions in determining the dynamic properties. Considering that the decreased axial symmetry of a rotator leads to an increase in the rotational barrier as well as a decrease in the number of energy maxima and minima,6 it is apparent that the lower molecular symmetry of bper is a dominant factor in suppressing the displacement that accompanies molecular rotation.
Effect of molecular structure on charge state
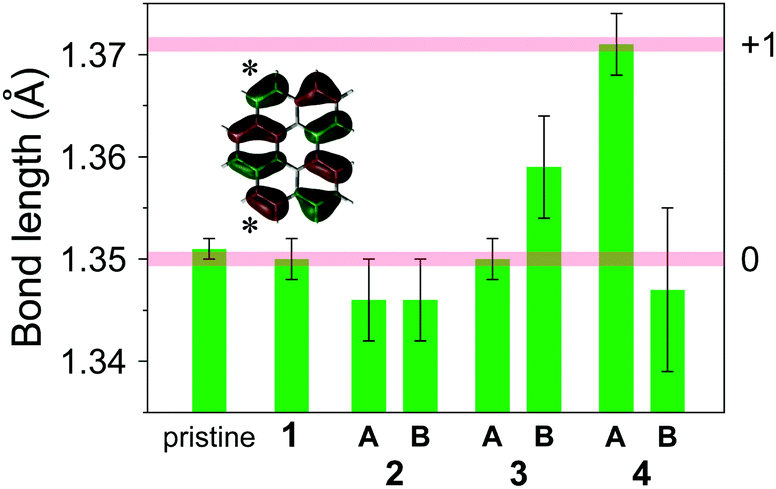
The bonds marked by an asterisk in the inset of Fig. 8, which have bonding character in the HOMO level, were found to have similar lengths in pristine bper crystal and neutral CT complexes 1–3 (except for bper-3B), whereas those in bper-4A are longer than them by ca. 0.02 Å (Fig. 8). Considering that the bond lengths in bper-4B are comparable to those observed in the neutral species, bper-4B is in a nearly neutral state and bper-4A must be in a nearly monocationic state to construct a [101]-like charge-ordered column.52,53 Thus, the low-lying absorption band at around 4 × 103 cm−1 shown in Fig. 1a can be assigned to electron transfer between cationic bper-4A and neutral bper-4B. Although the reason that the bonds in bper-3B are longer than expected is unclear, density functional theory (DFT) calculations at the B3LYP/6-31+G(d,p) level of theory (Fig. S3 in ESI†) show that the bonds in the bper monocation are elongated by 0.017 Å relative to those in neutral bper, which quantitatively supports the experimental results. First principles calculations within DFT based on the crystallographic data estimate the charges of bper-4A and bper-4B to be +0.82 and +0.37, respectively.54
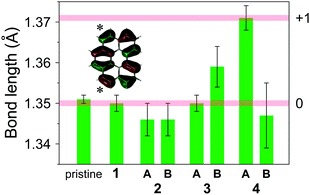 |
| | Fig. 8 Lengths of the bonds marked by an asterisk in the inset for pristine bper crystal and 1–4. The pale red horizontal lines mark the estimated values for monocationic (upper) and neutral (lower) bper molecules. The inset shows the HOMO distribution of a bper molecule calculated by DFT methods at the RB3LYP/6-31+G(d,p) level of theory. | |
Apparently, compared with (coronene)3Mo6O19, the removal of a benzene ring from a coronene molecule results in looser molecular packing in 4, as manifested by the orientational disorder of bper-4B. This disorder is an indication that the Coulombic interactions of bper-4B with Mo6O192− through hydrogen bonds are weaker than those for bper-4A, which forms quadruple hydrogen bonds with Mo6O192− in the bay region. It is possible that the difference in intermolecular interactions plays a key role in the emergence of charge disproportionation, namely, charge-rich bper-4A and charge-poor bper-4B.
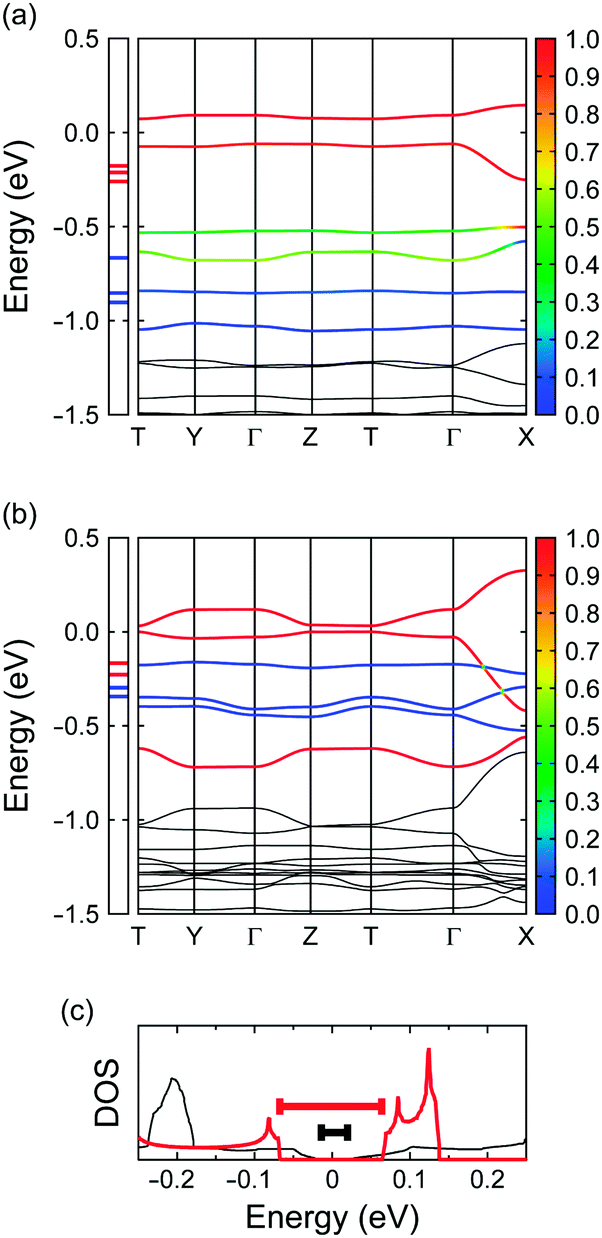
To elucidate the relationship between molecular symmetry and the electronic properties, we focus on the interplanar distances within the column as described above. It appears that those in 4 and (coronene)3Mo6O1913 bear some similarities together with striking differences; namely, the distance between adjacent monocationic bper-4A molecules is similar to that in (coronene)3Mo6O19 to stabilise the partially occupied HOMO of bper-4A (3.14 Å in 4vs. 3.16 Å in (coronene)3Mo6O19), whereas the distance between bper-4A and bper-4B (3.28 Å) is significantly longer than that in (coronene)3Mo6O19 (3.19 Å) possibly due to the neutral character of bper-4B. The intermolecular transfer integrals for 4 at 100 K calculated by the first principles method within DFT (277 meV between bper-4A and bper-4A and 100–159 meV between bper-4A and bper-4B) have a remarkable alternation relative to the case in (coronene)3Mo6O19 (257 and 208–219 meV, respectively) owing to the increased distance between bper-4A and bper-4B. As shown in Fig. 9a and b, this alternation inevitably gives rise to decreased band dispersion accompanied by an increased energy gap at the Fermi level (124 meV) compared with that in (coronene)3Mo6O19 (32 meV). It appears that the increased energy gap is responsible for the semiconducting behaviour with a larger activation energy, as mentioned above; namely, these salts can be regarded as band insulators. The exceptionally small activation energy of (coronene)3Mo6O19 largely depends on the highly symmetric molecular structure of coronene.
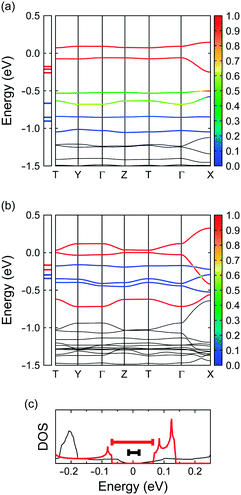 |
| | Fig. 9 DFT energy band structures of (a) 4 and (b) (coronene)3Mo6O19,13 calculated by first principles methods using the crystallographic data at 100 K, where T is (0, 1/2, 1/2) and the energy is given relative to the Fermi energy. The coloration of the six highest branches represents the fraction of HOMO (red) and HOMO−1 (blue) components. Red and blue horizontal ticks in the left box indicate the energy levels of the HOMO and the HOMO−1, respectively. (c) Density of states (DOS) of 4 (red) and (coronene)3Mo6O19 (black)13 near the Fermi energy, where the thick red and black bars indicate the energy gaps. | |
Effect of molecular structure on orbital degeneracy
One of the most striking effects of molecular symmetry on π-electronic systems, which has an impact on electronic properties, is orbital degeneracy. As reported previously, the energy difference between the HOMO and the HOMO−1 of coronene (Jahn–Teller splitting) in (coronene)3Mo6O19 is very small (ca. 0.12 eV; Fig. 9a and c) and comparable to the intermolecular transfer integrals.13 It is noteworthy that this value is similar to that in C60 anions (0.06–0.12 eV).55 In contrast, the energy difference in 4 (ca. 0.59 eV; Fig. 9b and c) is about five times greater than that in (coronene)3Mo6O19 due to the decreased molecular symmetry. This marked difference distinctly indicates that the HOMO and HOMO−1 energy levels in (coronene)3Mo6O19 are almost degenerate despite the reduced symmetry of the crystal field (space group P). If cationic coronene molecules could be stacked uniformly in solids, the density of states (DOS) of the HOMO/HOMO−1 band would likely be enhanced because of the orbital degeneracy. Subsequent adjustment of the Fermi level to the high DOS region may produce a new class of organic superconductors within the Bardeen–Cooper–Schrieffer (BCS) framework through electron–phonon coupling.1–3 Studies along this line are now in progress.
Conclusions
In the present study, the influence of the molecular symmetry of planar PAH molecules on dynamic and electronic properties was investigated by comparing compounds of C2v-symmetric bper with corresponding compounds of D6h-symmetric coronene. It was found that the removal of a benzene ring from the coronene molecule significantly suppresses molecular rotation along the in-plane direction in the solid state. In addition, the lower molecular symmetry of bper leads to charge ordering associated with the formation of significant hydrogen bonds with counter anions in the bay region, which is responsible for enhanced alternation of interplanar distances and transfer integrals within the column. These features inevitably result in an increased energy gap at the Fermi level, which corresponds to semiconducting behaviour with a large activation energy. Isostructural cation radical salts composed of PAH molecules with different symmetries enable investigations of the effect of molecular symmetry on the degeneracy of frontier orbitals in the solid state; namely, a comparison with the present bper-based cation radical salt revealed that the doubly degenerate HOMO of coronene molecules is maintained, even though the cation radical salt has a low-symmetry crystal field.
Experimental and computational
Materials and methods
Dichloromethane and pentane were used as received (Wako Chemicals), whereas methanol, chloroform, and acetone were distilled prior to use. Bper was purchased from Aldrich and used without any further purification. TCNQ was purified by sublimation prior to use, and (Bu4N)2Mo6O19 was synthesized and recrystallised from acetone according to the literature procedure.56
Synthesis of (bper)(TCNQ) (1).
Equimolar amounts of bper and TCNQ (0.017 mmol) was dissolved in 50 mL of dichloromethane/pentane (1:1). Natural evaporation of the solution at room temperature yielded ca. 3 mg (0.006 mmol) of black needle-shaped crystals of 1 (yield: 37%).
Synthesis of (bper)2(TCNQ) (2).
Equimolar amounts of bper and TCNQ (0.020 mmol) were ground together in an agate mortar and transferred into a 12 mm-diameter borosilicate glass tube. After sealing under vacuum (5.8 × 10−5 mbar), the 13 cm-long tube was placed in a two-zone furnace with a temperature gradient from 220 °C (sample side) to 150 °C (opposite side). After heating for 2 days, black thick plate crystals of 2 were grown on the center area in the tube (yield: ca. 50%).
Synthesis of (bper)3(TCNQ) (3).
Equimolar amounts of bper and TCNQ (0.050 mmol) were added to 20 mL of dichloromethane/pentane (1:1). After stirring at room temperature, the suspension was filtered to remove any traces of undissolved TCNQ. The filtrate was removed by natural evaporation at room temperature to yield ca. 10 mg (0.010 mmol) of black needle-shaped crystals of 3 (yield: 58%).
Synthesis of (bper)3Mo6O19 (4).
Bper (0.016 mmol) and (Bu4N)2Mo6O19 (0.022 mmol) were added to the anodic and cathodic compartments, respectively, in an H-shaped cell. After the cell was vacuum-dried, 18 mL of dichloromethane and a few drops of methanol were added to the cell. A constant current (1.0 μA) was passed between the two platinum electrodes. Black needle-shaped crystals of 4 were grown on the anodic electrode over a period of 2 weeks at 15 °C (yield: ca. 50%).
Spectroscopy.
UV-Vis-NIR absorption spectra were measured on KBr pellets (3.8–42 × 103 cm−1) or in a chloroform solution (12–50 × 103 cm−1) using a Shimadzu UV-3100 spectrophotometer.
X-Ray structural analysis.
Single-crystal X-ray diffraction experiments were performed on a CCD-type diffractometer (Bruker SMART APEX II) with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 100 K. A single crystal was mounted on a glass capillary and cooled by a stream of cooled nitrogen gas. The crystal structures were solved by a direct method using the SIR2004 program57 and were refined by a full-matrix least-squares method on F2 using the SHELXL program.58 All non-hydrogen atoms were anisotropically refined. The positional parameters of the hydrogen atoms were calculated under the assumption of fixed C–H bond lengths of 0.93 Å with sp2 configuration of the parent atoms.
Resistivity measurements.
Electrical resistivity of 4 was measured using a conventional four-probe method using a Keithley 2000 multimeter. Gold wires (ϕ = 10 μm) were attached to a single crystal with carbon paint (DOTITE XC-12). A DC current of less than 10 μA was applied along the stacking direction (crystal long axis).
Theoretical calculations (single molecule).
DFT calculations with the B3LYP functional59 were carried out with the 6-31+G(d,p) basis set.60,61 To ensure the reliability of the frequencies, both “Opt = Tight” and “Int = Ultrafine” were specified. The stabilities of the wave functions were confirmed by specifying the “Stable = Opt” keyword in the present DFT calculations. All the computations were performed using the Gaussian 09 program package,62 in which the bper molecule lies on the yz plane.
Theoretical calculations (band).
Using the experimental structure, we performed the electronic structure calculations within the generalised gradient approximation63 in the framework of the density functional theory as implemented in the quantum-ESPRESSO code.64 Ultrasoft pseudopotentials65 and the plane-wave basis set with cutoff energies of 30 Ry for wave functions and 150 Ry for charge densities were used. To obtain the transfer integrals, we constructed six maximally-localized Wannier orbitals66,67 from the six bands at the top of the valence band, which correspond to HOMO and HOMO−1 of each bper molecule. Charge disproportionation was discussed by diagonalising this six-band model.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Numbers JP25288041 and JP16H04139 (YY), JP16H00924 (TK), JP15K17901 and JP17H05153 (Y. Nakano), JP26288035 (HY), JP26110512 and JP16H00964 (HK), and JP23225005 (GS) and by JST PRESTO (TK). Theoretical calculations were partly performed at Research Center for Computational Science, Okazaki, Japan, and the SuperComputer System, Institute for Chemical Research, Kyoto University, Japan.
Notes and references
- O. Gunnarsson, Rev. Mod. Phys., 1997, 69, 575 CrossRef CAS.
- M. J. Rosseinsky, Chem. Mater., 1998, 10, 2665 CrossRef CAS.
- Y. Yoshida, K. Isomura, Y. Kumagai, M. Maesato, H. Kishida, M. Mizuno and G. Saito, J. Phys.: Condens. Matter, 2016, 28, 304001 CrossRef PubMed.
- R. K. Boyd, C. A. Fyfe and D. A. Wright, J. Phys. Chem. Solids, 1974, 35, 1355 CrossRef.
- T. Akutagawa, Mater. Chem. Front., 2018 10.1039/C7QM00603A.
- C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892 RSC.
- P. J. Krusic and E. Wasserman, J. Am. Chem. Soc., 1991, 113, 2322 CrossRef CAS.
- M. Glasbeek, J. D. W. van Voorst and G. J. Holjtink, J. Chem. Phys., 1966, 45, 1852 CrossRef CAS.
- Y. Yoshida, Y. Shimizu, T. Yajima, G. Maruta, S. Takeda, Y. Nakano, T. Hiramatsu, H. Kageyama, H. Yamochi and G. Saito, Chem. – Eur. J., 2013, 19, 12313 CrossRef CAS PubMed.
- Y. Yoshida, Y. Kumagai, M. Mizuno and G. Saito, Cryst. Growth Des., 2015, 15, 1389 CAS.
- Y. Yoshida, Y. Kumagai, M. Mizuno, K. Isomura, Y. Nakamura, H. Kishida and G. Saito, Cryst. Growth Des., 2015, 15, 5513 CAS.
- Y. Yoshida, M. Maesato, Y. Kumagai, M. Mizuno, K. Isomura, H. Kishida, M. Izumi, Y. Kubozono, A. Otsuka, H. Yamochi, G. Saito, K. Kirakci, S. Cordier and C. Perrin, Eur. J. Inorg. Chem., 2014, 3871 CrossRef CAS.
- Y. Yoshida, K. Isomura, H. Kishida, Y. Kumagai, M. Mizuno, M. Sakata, T. Koretsune, Y. Nakano, H. Yamochi, M. Maesato and G. Saito, Chem. – Eur. J., 2016, 22, 6023 CrossRef CAS PubMed.
- Y. Yoshida, K. Isomura, M. Maesato, T. Koretsune, Y. Nakano, H. Yamochi, H. Kishida and G. Saito, Cryst. Growth Des., 2016, 16, 5994 CAS.
-
R. Foster, Organic Charge-Transfer Complexes, Academic Press, London, 1969 Search PubMed.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS PubMed.
-
Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry, ed. M. A. Petrukhina and L. T. Scott, Wiley, Hoboken, 2012 Search PubMed.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS PubMed.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS PubMed.
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763 CrossRef CAS PubMed.
- R. Rieger, M. Kastler, V. Enkelmann and K. Müllen, Chem. – Eur. J., 2008, 14, 6322 CrossRef CAS PubMed.
- S. Hirayama, H. Sakai, Y. Araki, M. Tanaka, M. Imakawa, T. Wada, T. Takenobu and T. Hasobe, Chem. – Eur. J., 2014, 20, 9081 CrossRef CAS PubMed.
- Y. Yoshida, K. Isomura, Y. Nakamura, H. Kishida and G. Saito, Chem. Lett., 2015, 44, 709 CrossRef CAS.
- Y. Yoshida, Y. Nakamura, H. Kishida, H. Hayama, Y. Nakano, H. Yamochi and G. Saito, CrystEngComm, 2017, 19, 3626 RSC.
- E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124 CrossRef CAS.
- J. B. Torrance, J. E. Vazquez, J. J. Mayerle and V. Y. Lee, Phys. Rev. Lett., 1981, 46, 253 CrossRef CAS.
- G. Saito and Y. Yoshida, Bull. Chem. Soc. Jpn., 2007, 80, 1 CrossRef CAS.
- I. J. Tickle and C. K. Prout, J. Chem. Soc., Perkin Trans. 2, 1973, 720 RSC.
- R. M. Williams and S. C. Wallwork, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 168 CrossRef CAS.
- C. K. Prout, I. J. Tickle and J. D. Wright, J. Chem. Soc., Perkin Trans. 2, 1973, 528 RSC.
- X. Chi, C. Besnard, V. K. Thorsmlle, V. Y. Butko, A. J. Taylor, T. Siegrist and A. P. Ramirez, Chem. Mater., 2004, 16, 5751 CrossRef CAS.
-
Y. Yoshida
et al.
, unpublished data.
- T. J. Kistenmacher, T. J. Emge, A. N. Bloch and O. O. Cowan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 1193 CrossRef.
- D. M. Hudgins and L. J. Allamandola, J. Phys. Chem., 1995, 99, 3033 CrossRef CAS PubMed.
- F. Salama, G. A. Galazutdinov, J. Krełowski, L. J. Allamandola and F. A. Musaev, Astrophys. J., 1999, 526, 265 CrossRef CAS.
- A. L. Mattioda, D. M. Hudgins and L. J. Allamandola, Astrophys. J., 2005, 629, 1188 CrossRef CAS.
- T. Sugano, Y. Yakushi and H. Kuroda, Bull. Chem. Soc. Jpn., 1978, 51, 1041 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- D. Vermeulen, L. Y. Zhu, K. P. Goetz, P. Hu, H. Jiang, C. S. Day, O. D. Jurchescu, V. Coropceanu, C. Kloc and L. E. McNeil, J. Phys. Chem. C, 2014, 118, 24688 CAS.
- D. Braga, F. Grepioni and E. Tedesco, Organometallics, 1998, 17, 2669 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- J. G. White, J. Chem. Soc., 1948, 1398 RSC.
- Crystal data for pristine bper: C22H12, M = 276.34, monoclinic space group P21/c, a = 9.7377(8) Å, b = 11.9539(10) Å, c = 11.3813(10) Å, β = 97.968(1)°, V = 1312.03(19) Å3, Z = 4, T = 100 K, dcalc = 1.399 g cm−3, μ(Mo Kα) = 0.079 mm−1, 2672 reflections used, 199 refined parameters, R1 = 0.0401 [for I > 2σ(I)], wR2 = 0.1267 (all data), GOF = 1.048; CCDC 1537817†.
- G. R. Desiraju and A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 473 CrossRef.
- C. Kröhnke, V. Enkelmann and G. Wegner, Angew. Chem., Int. Ed. Engl., 1980, 19, 912 CrossRef.
- V. Enkelmann, K. Göckelmann, G. Wieners and M. Monkenbusch, Mol. Cryst. Liq. Cryst., 1985, 120, 195 CrossRef CAS.
- T. Sugano and M. Kinoshita, Bull. Chem. Soc. Jpn., 1989, 62, 2273 CrossRef CAS.
- J. B. Torrance, P. S. Bagus, I. Johannsen, A. I. Nazzal, S. S. P. Parkin and P. Batail, J. Appl. Phys., 1988, 63, 2962 CrossRef CAS.
-
M. Almeida and R. T. Henriques, in Handbook of Organic Conductive Molecules and Polymers, ed. H. S. Nalwa, Wiley, 1997, vol. 1, pp. 87–149 Search PubMed.
- G. E. Bacon, Acta Crystallogr., 1951, 4, 558 CrossRef CAS.
- (bper)(GaCl4), which was recently obtained by the electrochemical oxidation of bper in the presence of (Et4N)(GaCl4) (Fig. S2 in ESI†), involves monocationic bper molecules according to the composition. The length of the bond, which is indicated by an asterisk in the inset of Fig. 8, in (bper)(GaCl4) was estimated to be 1.37 Å. Notably, the value is equal to that of charge-rich bper-4A in 4.
- For several CT complexes, the charge states of electron donor molecules have been estimated on the basis of the bond lengths. For example, see: P. Guionneau, C. J. Kepert, G. Bravic, D. Chasseau, M. R. Truter, M. Kurmoo and P. Day, Synth. Met., 1997, 86, 1973 CrossRef CAS; G. Fischer and E. Dormann, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7792 CrossRef.
- Because the charge estimation was performed under the assumption of one orientation of the disordered bper-4B, two neighbouring bper-4A molecules within the column become nonequivalent. Thus, charges of the two bper-4A were estimated to be +0.85 and +0.78 (average charge: +0.82).
- M. Capone, M. Fabrizio, C. Castellani and E. Tosatti, Rev. Mod. Phys., 2009, 81, 943 CrossRef and references therein.
- M. Che, M. Fournier and J. P. Launay, J. Chem. Phys., 1979, 71, 1954 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381 CrossRef CAS.
-
G. M. Sheldrick, SHELXL-2013, University of Göttingen, Germany, 2013 Search PubMed.
- K. Raghavachari, Theor. Chem. Acc., 2000, 103, 361 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. von Ragué Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
-
M. J. Frisch, et al.Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. Giannozzi,
et al.
, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- N. Marzari and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12847 CrossRef CAS.
- A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari, Comput. Phys. Commun., 2008, 178, 685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Infrared spectra of TCNQ complexes (Fig. S1), crystal structure of (bper)(GaCl4) (Fig. S2), Kohn–Sham orbitals obtained from DFT calculations (Fig. S3). CCDC 1537813–1537817 (1–4 and pristine bper). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00112j |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Shunsuke
Tango
c,
Kazuhide
Isomura
c,
Yuto
Nakamura
c,
Hideo
Kishida
c,
Takashi
Koretsune
de,
Masafumi
Sakata
f,
Yoshiaki
Nakano
*ab,
Shunsuke
Tango
c,
Kazuhide
Isomura
c,
Yuto
Nakamura
c,
Hideo
Kishida
c,
Takashi
Koretsune
de,
Masafumi
Sakata
f,
Yoshiaki
Nakano