
Enhancing light hydrocarbon storage and separation through introducing Lewis basic nitrogen sites within a carboxylate-decorated copper–organic framework†
Xiuping
Liu
,
Weidong
Fan
,
Minghui
Zhang
,
Guixia
Li
,
Haijun
Liu
,
Daofeng
Sun
 ,
Lianming
Zhao
*,
Houyu
Zhu
and
Wenyue
Guo
*
,
Lianming
Zhao
*,
Houyu
Zhu
and
Wenyue
Guo
*
College of Science, China University of Petroleum, Qingdao, Shandong 266580, People's Republic of China. E-mail: lmzhao@upc.edu.cn; wyguo@upc.edu.cn; Fax: +86-532-8698-3363; Tel: +86-532-8698-3363
First published on 9th April 2018
Abstract
A novel nanoporous Cu metal–organic framework (NEM-4) with open CuII sites, Lewis basic nitrogen sites, and uncoordinated –COO− groups exhibits both outstanding uptake capacities (in cm3 (STP) g−1) for C2H2 (204), C2H4 (164.1), C2H6 (172.2), C3H6 (197.4), and C3H8 (196.1) and high selectivities for C2H2/CH4 (63.2), C3H6/CH4 (174.8), and C3H8/CH4 (168.3) under ambient conditions. After eight cycles of adsorption–desorption tests, only 8.2% and 10.3% decrease in the acetylene and propene storage capacities was observed, indicating an excellent repeatability. Compared with 1 (carboxylate decorated NOTT-101), when nitrogen sites are inserted, the C2–C3 hydrocarbon uptakes of NEM-4 can be significantly enhanced. Grand Canonical Monte Carlo and first-principles calculations reveal that not only the open CuII sites but also the uncoordinated –COO− groups and the nitrogen sites play significant roles in its high C2–C3 hydrocarbon uptakes. Moreover, the adsorption and separation of cationic dyes in NEM-4 are highly size and charge state dependent, and the adsorbed methylene blue (MB+) in NEM-4 can be efficiently released in an NaCl-containing CH3OH solution. This study reveals that the combination of open metal sites, carboxylate groups, Lewis basic pyridyl sites, and appropriate pore geometry is responsible for the high adsorption/separation of light hydrocarbons in NEM-4.
1. Introduction
Energy consumption and environmental degradation are dilemmas in industry and society nowadays since fossil energy still accounts for the highest market share.1 Using cleaner energy instead of the conventional fossil energy and capturing carbon dioxide from emission sources are good strategies to solve this issue.2 Natural gas, which consists primarily of methane with varying amounts of C2H6, C3H8, CO2, H2O, etc., is deemed to be a preferable alternative fuel because it is naturally abundant and environmentally friendly compared to petrol and diesel.3 In addition, hydrocarbons are very important raw materials and basic feedstocks for industrial products and fine chemicals. Therefore, the separation of light C2–C3 hydrocarbons and CO2 from methane, with the aim of upgrading the quality of natural gas and providing an alternative chemical source of C2–C3 hydrocarbons for further chemical processing and transformation, has become an important means to increase the utilization efficiency of natural gas and exerts a profound impact on industrial and economic development.4 However, traditional methods (e.g., cryogenic distillation) and materials (e.g., activated carbons and zeolites) are not energy-efficient routes and are generally poor in terms of selectivity.5Metal–organic frameworks (MOFs), which can be straightforwardly self-assembled from organic linkers and metal ions/clusters, possess many excellent characteristics, such as high porosity, tunable pore size/shape, and adjustable surface chemistry.6–10 Therefore, their applications in the gas storage and separation field have been intensively investigated.11 Several strategies including utilizing structural flexibility and surface functionality, tuning pore size and shape, and tailoring the pore surface, have been adopted to enhance the MOFs’ gas adsorption and separation efficiency.12 Among them, the pore surface functionality of MOF sorbents is considered to be foremost for promoting their separation performance.13 In this respect, some researches have shown that adding different functional sites such as accessible metal sites, Lewis basic/acidic sites, and polar functional groups (–COOH, –NH2) and/or forming charged skeletons can be used to fundamentally improve the adsorbate–surface interactions.14–16 In addition, the size/geometry of the pores/cages also plays a significant role in the adsorption/separation of adsorbates. Hasan et al. found that a pore size of about 10 Å is more efficient for rapid gas storage applications.17
The NbO-type Cu MOFs containing basic m-benzenedicarboxylate moieties have gained tremendous attention in the past decade because of their high gas uptake/selectivity, suitable window geometry, and rare framework interpenetration.18 In 2005, Schröder et al. reported that a series of MOF-505 analogues own high H2 adsorption capacity due to the pocket effect.19,20 By a ligand extension strategy, a NbO-type copper–tetracarboxylate framework (NOTT-101) was prepared and found to have a high C2H2 uptake of 184 cm3 g−1 at 1 bar and 298 K.21 To further enhance the interactions with gas molecules, functional sites have been immobilized on the pore surfaces of NbO-type Cu MOFs. Rao et al. synthesized a MOF by using a tetracarboxylate with one Lewis basic pyridyl site to enhance the interaction with acetylene molecules through the H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H⋯N (pyridyl) hydrogen bond, showing a high C2H2 storage capacity (193 cm3 g−1 at 1 bar and 298 K).22 The two Lewis basic pyridyl site decorated MOF-505 (ZJU-40a) synthesized by Wen et al. exhibited a higher gravimetric C2H2 uptake of 216 cm3 g−1 at 1 bar and 298 K, due to the higher concentration of Lewis basic nitrogen sites.21 Li et al. synthesized the pyrimidine-decorated MOF-505 (UTSA-76) and found that the central pyrimidine ring in the linker of UTSA-76a has a much shallower rotational barrier (∼8.2 vs. ∼20.2 kJ mol−1 for the central benzene ring in NOTT-101a), suggesting that decoration with Lewis basic nitrogen sites can enhance the mobility of the central ring favoring CH4 storage (260 cm3 cm−3 at 298 K and 65 bar).23 In our previous work, we synthesized a new analogue MOF-505 decorated with mono-COO− in the central benzene ring of the linker (1), which shows not only high gas uptake but also selective MB+ adsorption.24 This encouraged us to further improve the performance for light hydrocarbon uptake and separation by replacing the central benzene ring in the linker of 1 with a pyridyl ring, in consideration that the modification can not only add Lewis basic pyridyl sites in the pores but also improve the mobility and orientation of the central ring which enhances the accessible pore volume of the material. With this goal in mind, in this paper, we designed and synthesized a new organic linker, 2,5-di(3,5-dicarboxylphenyl)-nicotinic acid (H5N), to build an anionic microporous MOF, {[Cu2(N)·(H2O)2]·[2H2O·3DMA·(CH3)2NH2]}n (NEM-4). Complex NEM-4 possesses a NbO-type framework just like 1 with exposed –COO− groups, Lewis basic pyridyl sites, and open metal sites (OMSs) on the pore surfaces. The new material exhibits both high uptakes and good adsorption selectivities (over CH4) for C2–C3 hydrocarbons, e.g., uptakes at 1 bar and 295 K: C2H2 (204 cm3 g−1), C2H4 (164.1 cm3 g−1), C2H6 (172.2 cm3 g−1), C3H6 (197.4 cm3 g−1), C3H8 (196.1 cm3 g−1); selectivities at 295 K: C2H2/CH4 (63.2), C2H4/CH4 (44.3), C2H6/CH4 (20.1), C3H6/CH4 (174.8), C3H8/CH4 (168.3). Furthermore, the repeatability of NEM-4 was confirmed by only slight losses of the C2H2 (8.2%) and C3H6 (10.3%) storage capacities after eight adsorption–desorption cycles.
C–H⋯N (pyridyl) hydrogen bond, showing a high C2H2 storage capacity (193 cm3 g−1 at 1 bar and 298 K).22 The two Lewis basic pyridyl site decorated MOF-505 (ZJU-40a) synthesized by Wen et al. exhibited a higher gravimetric C2H2 uptake of 216 cm3 g−1 at 1 bar and 298 K, due to the higher concentration of Lewis basic nitrogen sites.21 Li et al. synthesized the pyrimidine-decorated MOF-505 (UTSA-76) and found that the central pyrimidine ring in the linker of UTSA-76a has a much shallower rotational barrier (∼8.2 vs. ∼20.2 kJ mol−1 for the central benzene ring in NOTT-101a), suggesting that decoration with Lewis basic nitrogen sites can enhance the mobility of the central ring favoring CH4 storage (260 cm3 cm−3 at 298 K and 65 bar).23 In our previous work, we synthesized a new analogue MOF-505 decorated with mono-COO− in the central benzene ring of the linker (1), which shows not only high gas uptake but also selective MB+ adsorption.24 This encouraged us to further improve the performance for light hydrocarbon uptake and separation by replacing the central benzene ring in the linker of 1 with a pyridyl ring, in consideration that the modification can not only add Lewis basic pyridyl sites in the pores but also improve the mobility and orientation of the central ring which enhances the accessible pore volume of the material. With this goal in mind, in this paper, we designed and synthesized a new organic linker, 2,5-di(3,5-dicarboxylphenyl)-nicotinic acid (H5N), to build an anionic microporous MOF, {[Cu2(N)·(H2O)2]·[2H2O·3DMA·(CH3)2NH2]}n (NEM-4). Complex NEM-4 possesses a NbO-type framework just like 1 with exposed –COO− groups, Lewis basic pyridyl sites, and open metal sites (OMSs) on the pore surfaces. The new material exhibits both high uptakes and good adsorption selectivities (over CH4) for C2–C3 hydrocarbons, e.g., uptakes at 1 bar and 295 K: C2H2 (204 cm3 g−1), C2H4 (164.1 cm3 g−1), C2H6 (172.2 cm3 g−1), C3H6 (197.4 cm3 g−1), C3H8 (196.1 cm3 g−1); selectivities at 295 K: C2H2/CH4 (63.2), C2H4/CH4 (44.3), C2H6/CH4 (20.1), C3H6/CH4 (174.8), C3H8/CH4 (168.3). Furthermore, the repeatability of NEM-4 was confirmed by only slight losses of the C2H2 (8.2%) and C3H6 (10.3%) storage capacities after eight adsorption–desorption cycles.
2. Experimental and computational section
2.1 Materials and measurements
All chemical reagents were used as commercially obtained without further purification. The ligand H5N was prepared according to the ESI,† Section 1.1 (Scheme S1) and characterized by 1H NMR spectroscopy on a Bruker AVANCE-300 NMR spectrometer (Fig. S1, ESI†). X-ray powder diffraction (XRD) was measured on a Panalytical X-Pert pro diffractometer with Cu-Kα radiation. Infrared spectra were recorded on a Bruker VERTEX-70 spectrometer using KBr pellets in the frequency range 4000–400 cm−1. Elemental (C, H, and N) analyses (EA) were determined on a CE instruments EA 1110 analyzer. Low-pressure (<800 Torr) gas (N2, CO2, CH4, C2H2, C2H4, C2H6, C3H6, and C3H8) sorption isotherms were measured using a Micrometrics ASAP 2020 surface area and pore size analyzer. Thermogravimetric (TG) curves were measured from 40 to 900 °C on a Mettler Toledo TGA instrument at a heating rate of 10 °C min−1 under a N2 atmosphere (100 ml min−1).2.2 Synthesis of complex NEM-4
A mixture of Cu(NO3)2 and H5N was dissolved in DMA/C2H5OH/H2O mixed solvents. Then, all reagents were sealed in a pressure-resistant glass bottle and slowly heated to 85 °C from room temperature. After keeping at 85 °C for 50 hours, the mixture was slowly cooled to 30 °C at a rate of 7 °C h−1. The pale-blue block crystals of NEM-4 were separated in 72.6% yield based on copper. The sample of NEM-4 was insoluble in common solvents such as H2O, dichloromethane or methanol. EA (%) measured (calculated) for [Cu2(N)·(H2O)2]·[2H2O·3DMA·(CH3)2NH2] was C, 45.71 (45.37); H, 5.56 (5.4); N, 7.49 (7.35). Details of the synthesis procedure are given in the ESI,† Section 1.1.2.3 Computational methods
In this work, the low-pressure adsorption isotherm of pure C2H2 in NEM-4 was simulated using the grand canonical Monte Carlo (GCMC) method implemented in the MUSIC code.25 DFT calculations were performed to provide the atomic partial charges on the NEM-4 framework for the GCMC calculations as well as to give the optimized structures and energies of C2H2, C2H4, C2H6, C3H6 and C3H8 interaction with the NEM-4 fragmented cluster. The calculations were carried out by using the Perdew Burke Ernzerhof (PBE) functional under the generalized gradient approximation (GGA) functional with the double-ξ numerical polarization (DNP) basis set implemented in the DMol3 program package in the Materials Studio of Accelrys Inc.26 The details of all the calculations are given in the ESI.†3. Results and discussion
3.1 Crystal structure descriptions
It is revealed that complex NEM-4 possesses a NbO-type topology formulated as {[Cu2(N)·(H2O)2]·[2H2O·3DMA·(CH3)2NH2]}n based on the EA, TGA, and single-crystal XRD studies. NEM-4 crystallizes in a trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and has the same basic structure as NOTT-10119 with all the linkers replaced by H5N (see Fig. 1). Each CuII ion is coordinated by five O atoms in a square pyramidal geometry, and the secondary building units (SBUs) of the dinuclear Cu2(COO)4 paddlewheels connect with organic linkers to obtain a three dimensional NbO-type structure (Fig. 1b). There are two types of nanocages in NEM-4 (Fig. 1c): the smaller spherical-like cage is defined by six inorganic SBUs with a pore diameter of about 11 Å, while the larger shuttle-shaped cage is defined by six organic SBUs at the dimensions of 9.4 × 21 Å. The 3D framework is formed via the alternate stacking of the two types of cages with the ratio of 1
m and has the same basic structure as NOTT-10119 with all the linkers replaced by H5N (see Fig. 1). Each CuII ion is coordinated by five O atoms in a square pyramidal geometry, and the secondary building units (SBUs) of the dinuclear Cu2(COO)4 paddlewheels connect with organic linkers to obtain a three dimensional NbO-type structure (Fig. 1b). There are two types of nanocages in NEM-4 (Fig. 1c): the smaller spherical-like cage is defined by six inorganic SBUs with a pore diameter of about 11 Å, while the larger shuttle-shaped cage is defined by six organic SBUs at the dimensions of 9.4 × 21 Å. The 3D framework is formed via the alternate stacking of the two types of cages with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The accessible pore volume of NEM-4 is 0.98 cm3 g−1 (after the removal of the coordinated water) as calculated based on the single crystal structure using Poreblazer V.3.0.2,27 slightly larger than the experimental value (0.92 cm3 g−1) based on the N2-sorption test (Fig. 2a) due to imperfection of the synthesized crystal structures. As expected, NEM-4 with orientation-adjustable pyridyl rings has a larger accessible pore volume than 1 (0.78 cm3 g−1),24 which is expected to enhance the adsorption capacity for adsorbates. The enlargement of the pore volume in NEM-4 might also be due to the variation of the dihedral angle between the two coplanar end benzene rings and the central ring in the linker, i.e., 35° in NEM-4 and 44° in 1, indicated by single-crystal XRD (Fig. S3, ESI†). More details of the crystal data of complex NEM-4 are given in Table S1 (ESI†).
:1. The accessible pore volume of NEM-4 is 0.98 cm3 g−1 (after the removal of the coordinated water) as calculated based on the single crystal structure using Poreblazer V.3.0.2,27 slightly larger than the experimental value (0.92 cm3 g−1) based on the N2-sorption test (Fig. 2a) due to imperfection of the synthesized crystal structures. As expected, NEM-4 with orientation-adjustable pyridyl rings has a larger accessible pore volume than 1 (0.78 cm3 g−1),24 which is expected to enhance the adsorption capacity for adsorbates. The enlargement of the pore volume in NEM-4 might also be due to the variation of the dihedral angle between the two coplanar end benzene rings and the central ring in the linker, i.e., 35° in NEM-4 and 44° in 1, indicated by single-crystal XRD (Fig. S3, ESI†). More details of the crystal data of complex NEM-4 are given in Table S1 (ESI†).
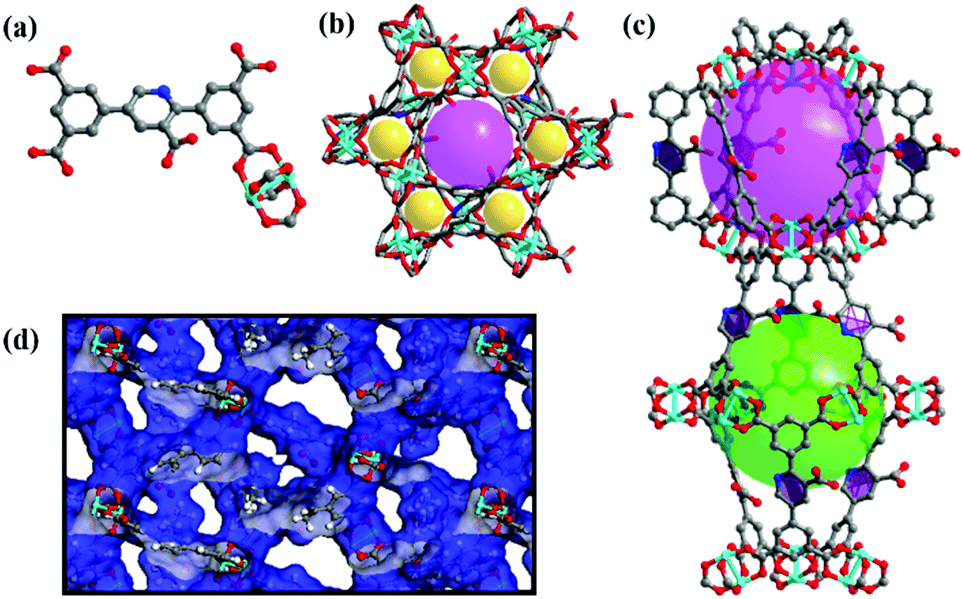 | ||
| Fig. 1 (a) Tetracarboxylate linker and the [Cu2(COO)4] paddlewheel cluster. (b) 3D framework viewed from the c axis. (c) The shuttle-shaped cage and spherical-like cage of NEM-4. (d) Cross-section view of pores of NEM-4 viewed from the b axis, Cu: blue-green; C: grey; O: red; N: blue. | ||
 | ||
| Fig. 2 (a) The N2 adsorption isotherm in NEM-4 at 77 K. The C2H2, C2H4, C2H6, and CH4 adsorption isotherms in NEM-4 at (b) 273 and (c) 295 K. (d) The C3H6 adsorption isotherms in NEM-4 at 273 and 295 K. (e) The C3H8 adsorption isotherms in NEM-4 at 273 and 295 K. (f) The isosteric heats of adsorption of C2H2, C2H4, C2H6, C3H6, C3H8, and CH4. | ||
3.2 Powder X-ray diffraction, thermal analysis, and IR spectra
The powder XRD patterns of the samples were used for checking the phase purity and stability of NEM-4 at room temperature. The experimental XRD patterns are almost identical to the calculated XRD pattern based on the crystal structure using Mercury 3.3 (Fig. S4, ESI†), suggesting that the samples contain pure single phase and the crystal structure remains after adsorption of MB+ or removal of solvent guest molecules. TG analyses of NEM-4 were carried out from 40 to 800 °C under a dry nitrogen atmosphere with a heating rate of 10 °C min−1. A rather good thermal stability of NEM-4 is observed because it remains stable without decomposition up to 245 °C (Fig. S5, ESI†).The IR spectra of NEM-4 in the as-synthesized and activated states are shown in Fig. S6 (ESI†). The deprotonation of –COOH giving –COO− is characterized by the equivalent out-of-phase and in-phase vibration modes of the two C–O bonds, i.e., νas(COO) and νs(COO).24,28 The band at about 1690 cm−1 might be the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration of the pyridyl rings.29 The broad band at about 3450 cm−1 might be due to the characteristic NH stretching vibration of free (CH3)2NH2+ inside the pores of NEM-4,30 since it is preserved in the waterless state. (CH3)2NH2+ should be generated from the decomposition of DMA during the solvothermal reaction to balance the anionic framework.31
N stretching vibration of the pyridyl rings.29 The broad band at about 3450 cm−1 might be due to the characteristic NH stretching vibration of free (CH3)2NH2+ inside the pores of NEM-4,30 since it is preserved in the waterless state. (CH3)2NH2+ should be generated from the decomposition of DMA during the solvothermal reaction to balance the anionic framework.31
3.3 Gas adsorption properties
To confirm the permanent porosity of the material, the methanol solvent-exchanged sample was degassed under a dynamic vacuum at 90 °C for 12 h; the color of the sample in the process changes from pale blue to dark blue, similar to the situation of other frameworks in which open CuII sites can be generated.32 As shown in Fig. 2a, the activated NEM-4 displays a typical type-I adsorption behavior of N2; the steep rise of the N2 uptake below P/P0 = 0.05 confirms the retention of microporous structures after removing solvents from the crystalline sample. The maximum N2 uptake is 597 cm3 g−1 at 77 K, surpassing the value of 502 cm3 g−1 in its structural analogue 1.24 The BET surface area in the activated NEM-4 was estimated as 2278.35 m2 g−1, which also shows remarkable expansion compared to 1 (1919.2 m2 g−1),24 but is smaller than those of the isostructural frameworks NOTT-101 (2805 m2 g−1)19 and ZJU-5a (2823 m2 g−1),22 because some space has been occpuied by the –COO− and [(CH3)2NH2]+. From the N2-sorption isotherm, the pore-size distribution in NEM-4 was calculated on the basis of nonlocal density functional theory (Fig. S8, ESI†). Two peaks are focused at around 11 and 21 Å, in accordance with the internal structural feature of the cages (Fig. 1).In Fig. S9 (ESI†), the CO2 sorption isotherms indicate that NEM-4 possesses high CO2 uptake capacity (173 and 97 cm3 g−1 under 1 bar at 273 and 295 K, respectively), which surpasses some of the promising MOFs reported, such as 1 (140/86 cm3 g−1 at 1 bar and 273/295 K)24 and HNUST-1 (156.4/93.0 cm3 g−1 at 1 bar and 273/298 K).33 To better understand these observations, the CO2 adsorption enthalpies (Qst) were calculated based on the isotherms at 273 and 295 K using the virial method (Fig. S10, ESI†). NEM-4 is found to have a high heat of CO2 adsorption (27.4 kJ mol−1) at zero loading, which does not change obviously at higher coverages. Note that the Qst values of CO2 in the isostructural frameworks 1,24 NOTT-101,19 and NJU-Bai34 are 21, 26, and 24.5 kJ mol−1, respectively.
Considering the specific characters of NEM-4, e.g., –COO− and nitrogen sites on the pore surface and charged skeleton, we further investigated its potential application in the adsorption and separation of light hydrocarbons. Single-component gas adsorption isotherms at 273 and 295 K were measured for various light hydrocarbons (CH4, C2H2, C2H4, C2H6, C3H6, and C3H8). Although all the hydrocarbons show reversible type-I adsorption, the adsorption isotherm has the trend to become steeper when turning from CH4 through C2 to C3 hydrocarbons (see Fig. 2b–e). More specifically, CH4 shows a near-linear isotherm with the lowest slope, the adsorption isotherm of C2 hydrocarbons is more or less a curve with the flex point at aroud 0.4 bar so that it is not saturated at the highest pressure measured, while the adsorption capacity for the C3 hydrocarbons shows a sudden variation below 0.25 bar and then tends to saturation at higher pressures. The different slopes of the adsorption isotherms of hydrocarbons at low pressures might be attributed to the difference in the affinities to the framework, which is further confirmed by the Qst values at zero coverage, i.e., 18.3 kJ mol−1 for CH4, 23.8–38.8 kJ mol−1 for C2 hydrocarbons, and 40.5–44.4 kJ mol−1 for C3 hydrocarbons (Fig. 2f). The more favorable adsorption of the C2–C3 light hydrocarbons is presumably owing to their much higher polarizabilities compared to CH4.35
In practical applications, C2H2 gas is stored at ambient temperatures. Excitingly, NEM-4 exhibits extraordinarily high C2H2 uptake of 204 cm3 g−1 at 1 bar and 295 K, even higher than the values of the isoreticular NOTT-101 (184 cm3 g−1),211 (190 cm3 g−1),36 and ZJU-5a (193 cm3 g−1)22 under the same conditions. Considering the same basic structures with open metal sites of these three isoreticular MOFs, the enhanced C2H2 uptake in NEM-4 might be attributed to the synergistic effect of the –COO− groups and pyridyl sites on the pore surfaces, which induces the specific affinity with C2H2 molecules to further improve the C2H2 uptake, i.e., the H–CC–H⋯O (–COO−) and H–CC–H⋯N (pyridyl sites) hydrogen bonding. Furthermore, the C2H2Qst (38.8 kJ mol−1) for NEM-4 at low loading is higher than those isoreticular MOFs such as NOTT-101 (32.6 kJ mol−1)21 and ZJU-5a (35.8 kJ mol−1),22 indicating the stronger interactions between C2H2 and the framework NEM-4. Importantly, in Table 1, the gravimetric C2H2 uptake of NEM-4 is among the high values reported and is comparable with the best performace of MFM-188a (232 cm3 g−1).37 As we can see in Table 1, these excellent MOFs present a large surface area and high OMS and functional group (FG) densities (except FJI-H8). In other words, the incorporation of OMS and FG has great potential to improve the C2H2 storage capacity. The C2H2 uptake of NEM-4 is 222 cm3 g−1 at 1 bar and 273 K. Meanwhile, the uptake capacities of NEM-4 for C2H4, C2H6, C3H6, and C3H8 are also fairly high with values of 164.1, 172.2, 197.4, and 196.1 cm3 g−1 at 1 bar and 295 K and 207.8, 213.6, 209.1, and 198.2 cm3 g−1 at 1 bar and 273 K, respectively. As shown in Table S2 (ESI†), the uptakes of NEM-4 for the C2–C3 hydrocarbons at 295 K are much higher than some of the best performing MOFs, such as MFM-202a,38 Cu–TDPAT,39 and MFM-300a.38 The CH4 uptake in NEM-4 is 19.3 cm3 g−1 at 295 K and 1 bar and 32.7 cm3 g−1 at 273 K and 1 bar (Fig. 2b and c), which is higher than that of NOTT-101 (19 cm3 g−1 at 295 K and 1 bar) but lower than that of the QI-Cu (23 cm3 g−1 at 295 K and 1 bar).40 Considing the practical application, we also evaluated the repeatability of NEM-4 for C2H2 and C3H6 storage. About 100 mg of desolvated sample was loaded onto an ASAP2020-M analyser, and eight cycles of C2H2 and C3H6 adsorption at 295 K were recorded without the reactivation process between each cycle. There is only 8.2% and 10.3% loss in adsorption capacity of C2H2 and C3H6 after eight cycles, which indicates that NEM-4 is promising in refillable C2 and C3 hydrocarbon storage (Fig. 3). The enormous difference of uptakes for C2–C3 hydrocarbons versus CH4 prompted us to investigate the corresponding C2H2/CH4, C2H4/CH4, C2H6/CH4, C3H6/CH4, and C3H8/CH4 separation performance of NEM-4.
| Material | BET SA (m2 g−1) | OMS density (mmol g−1) | FG density (mmol g−1) | Uptake (cm3 g−1) | Q st (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|
| a FG(s): MFM-188, amide; ZJU-40, pyridyl; NEM-4, pyridyl and carboxylate; ZJU-5a, pyridyl; 1, carboxylate; ZJU-10, hydroxyl. b Value measured at 298 K and 1 bar. | ||||||
| MFM-188 | 2568 | 3.3 | 3.26 | 232 | 32.5 | 39 |
| FJI-H8 | 2025 | 3.6 | 0 | 224 | 32 | 39 |
| ZJU-40a | 2858 | 3.8 | 3.8 | 216b | 34.5 | 23 |
| NEM-4 | 2278 | 3.3 | 3.3 | 204 | 38.8 | This work |
| HKUST-1 | 1781 | 5.0 | 0 | 201 | 30.4 | 11 |
| ZJU-5a | 2823 | 3.8 | 1.9 | 193 | 35.8 | 25 |
| 1 | 1919 | 3.3 | 1.7 | 190 | 35.6 | 38 |
| NOTT-101 | 2880 | 3.8 | 0 | 184 | 32.4 | 23 |
| ZJU-10 | 2392 | 3.7 | 1.9 | 174b | 39 | Fig. S11 (ESI) |
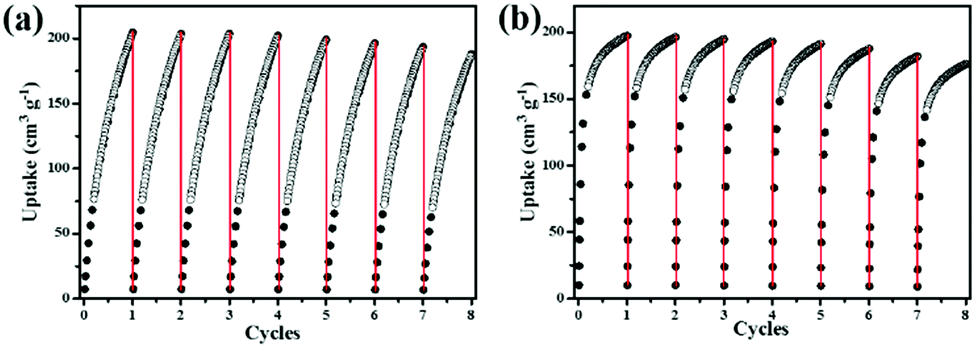 | ||
| Fig. 3 Cycles of C2H2 (a) and C3H6 (b) adsorption for NEM-4 at 295 K. | ||
To predict the selectivities, we used the ideal adsorbed solution theory (IAST) to calculate the multi-component adsorption behavior from the experimental pure-gas isotherms.41 The separation ratios of C2H2, C2H4, C2H6, C3H6, C3H8, and CO2 over CH4 are calculated at the molar ratios of 10:90 and 50:50, respectively. As shown in Fig. 4, as the pressure increases, the selectivity of C2H2/CH4, C2H4/CH4, and CO2/CH4 decreases initially and then tends to be a constant, C2H6/CH4 and C3H8/CH4 at 10:90 show an increasing trend of selectivity, while the selectivity of C3H6/CH4 and C3H8/CH4 at 50:50 increases initially and then slightly decreases. For the molar ratio at 10:90, the selectivity of C2H2/CH4, C2H4/CH4, C2H6/CH4, C3H6/CH4, and C3H8/CH4 at 295 K is calculated as 85.5, 55.1, 16.7, 174.2, and 167.2, respectively. When the C2–C3 hydrocarbons and CH4 are equivalently mixed, the selectivities of C2H2, C2H4, C2H6, C3H6, and C3H8 over CH4 at 295 K are found to be 63.2, 44.3, 20.1, 174.8, and 168.3, respectively. Notably, the selectivity of C3H6/CH4 in NEM-4 (174.8) is much higher than the values of some reported promising MOFs, e.g., UTSA-35a (140)42 and AFM-202a (75).38 In addition, the C2H2/CH4 selectivity of NEM-4 (63.2) is also much higher than those of MOFs with OMSs, such as Zn4(OH)2(1,2,4-BTC)2 (14.7)43 and ZJNU-57 (35).44 For equimolar CO2/CH4 gas mixtures, the predicted selectivity at 1 bar and 273 K is 11.1, higher than the highly selective porous materials reported for CO2 over CH4 such as rht-type MOF (8.5)45 and PCN-88 (7.1).46 The high adsorption selectivities of C2–C3 hydrocarbons over CH4 might be attributed to four factors, that is, (i) the high density of OMSs plays an important role; (ii) the C–H⋯N (pyridyl) and C–H⋯O (–COO−) hydrogen bonding favors the uptake of C2–C3 hydrocarbons; (iii) the charged skeleton47 and cationic counterions (CH3)2NH2+ enhance adsorbent–adsorbate interaction through charge-induced force. In particular, the high polarizability makes C2–C3 hydrocarbons generate stronger interactions with the adsorbent compared with the nonpolarizable CH4;48 and (iv) the suitable pore size of NEM-4 matches with the kinetic diameters of the C2–C3 light hydrocarbons, resulting in the high C2–C3/CH4 sieving effects.49 All these results indicate that NEM-4 is a prospective adsorbent for effective adsorptive separation of CH4 from other gas, and thus is suitable for natural gas upgrading.
 | ||
| Fig. 4 Adsorption selectivities of NEM-4 calculated by the IAST method for mixtures of (a) C2H2/CH4, (b) C2H4/CH4, (c) C2H6/CH4, (d) C3H6/CH4, and (e) C3H8/CH4 at 295 K. (f) The selectivity of CO2/CH4 at 273 K. | ||
Theoretically, confirming C2–C3 hydrocarbon adsorption sites within the MOF skeletons is very important for us to design new MOF-based materials for gas storage and separation. In this respect, theoretical simulation as a powerful tool can provide a lot of useful information.50 First, the C2H2 adsorption property of NEM-4 at 295 K was studied by GCMC simulations. As shown in Fig. S12 (ESI†), the simulated adsorption isotherm is generally in accordance with the experimental one, even though the theoretical uptake at higher pressures is slightly overestimated (by <10.6%). To our knowledge, the open CuII metal sites within MOF materials usually play a favorable role in the high gas uptake.51 In Fig. 5, the GCMC snapshot of the structure of NEM-4 adsorbed with C2H2 molecules at 295 K and 0.2 bar indicates that the open CuII site is preferred for C2H2 adsorption; also, Lewis basic pyridyl and carboxylate groups are very important sites. For open CuII sites, the adsorbate tends to interact with the metal center in a parallel fashion forming Cu⋯C bonds.54 At carboxylate and Lewis basic pyridyl sites, C2H2 is preferentially located with the hydrogen atoms pointing to the oxygen atoms (–COO−) and nitrogen atoms (pyridyl). The other preferential adsorption regions are the entrance windows across the framework pores, which might be due to the gas molecules at the copper sites and the surrounding benzene rings could lead to an additional attractive contribution at the pore windows.52
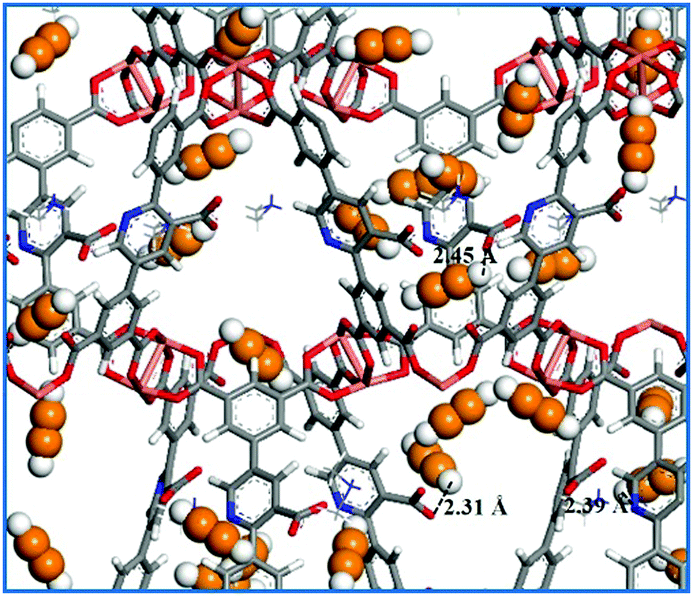 | ||
| Fig. 5 Snapshot of pure C2H2 adsorption in NEM-4 at 0.2 bar and 295 K. The orange and white represent C and H atoms of C2H2 molecules. | ||
To further probe the advantages of the open CuII sites, Lewis basic pyridyl sites, and –COO− groups for C2–C3 hydrocarbons adsorption, DFT calculations were performed on NEM-4, as shown in Fig. 6. The calculated adsorption energies (ΔEad) of hydrocarbons C2H2, C2H4, C2H6, C3H6, and C3H8 at the CuII site are −27.25, −28.19, −23.46, −40.25, and −22.68 kJ mol−1 with the closest CC2/C3⋯Cu distances of 2.47, 2.76, 2.79, 2.78, and 2.74 Å, respectively. At the Lewis basic pyridyl site, the ΔEad of C2H2, C2H4, C2H6, C3H6, and C3H8 is up to −26.59, −17.55, −20.66, −19.47, and −18.92 kJ mol−1 with the HC2/C3⋯N distances of 2.22, 2.61, 2.63, 2.60, and 2.58 Å, respectively, while the affinities of the –COO− site (ΔEad = −24.35, −14.74, −20.37, −19.30, and −18.87 kJ mol−1; O⋯HC2/C3 = 2.41, 2.56, 2.66, 2.49, and 2.45 Å, respectively) are slightly weaker than those of the Lewis basic pyridyl site. In addition, we further studied the CH4 at the open metal site, Lewis basic pyridyl site, and carboxylate group is only −7.65, −14.92, and −11.41 kJ mol−1, respectively (Fig. S13, ESI†). Our calculations confirm that pyridyl and –COO− are powerful C2–C3 recognition sites, especially for C2H2. Such a phenomenon might be due to the enhancement of the polarization on the C2H2 molecule induced by the functional groups via the electrical field–quadrupole interactions.53 The open metal is the strongest adsorption site, especially for C3H6, which might be attributed to the stronger interaction of the C3H6 π-bonding orbital with the open metal as well as the larger dipole moment than that of other C1–C3 light hydrocarbons.54 These results reveal the contributions of all three adsorption sites in NEM-4 to the high uptake of C2–C3 hydrocarbons.
 | ||
| Fig. 6 Preferential C2H2, C2H4, C2H6, C3H6, and C3H8 adsorption sites and corresponding adsorption energies in NEM-4 obtained from first-principles calculations. Orange-red, red, gray, blue, and white represent Cu, O, C, N, and H atoms, respectively. | ||
3.4 Adsorption of dye molecules
Ionic MOFs may have more unique advantages such as selective adsorption of cationic/anionic dyes by host–guest electrostatic interactions and/or guest–guest exchange interactions.55 In this section, we first investigated the charge of the NEM-4 and determined the uptakes of dye molecules with different charged states (Fig. S14, ESI†), i.e., MB+, Sudan II (SD0), and methyl orange (MO−) in their methanol solutions (5 ml, 4 × 10−5 M). After the NEM-4 samples were put into the corresponding solutions, the concentration of the cationic MB+ is drastically decreased (Fig. 7a), whereas both the neutral SD0 and anionic MO− dyes show only a slight decline in concentration after 12 h (Fig. S15c and d, ESI†). Then, the separation performance of NEM-4 for the 1:1 MB+/MO− and MB+/SD0 mixtures in methanol solutions was further explored. As shown in Fig. 7c and d, NEM-4 could only selectively absorb MB+, and the solutions almost show the colors of pure SD0 or MO− at last (Fig. S17c and d, ESI†). Moreover, the MB+ molecules loaded in NEM-4 (Fig. 7b) can be released quickly and efficiently in the NaCl-containing methanol solutions; the release efficiency is up to 62.1% after 150 min. However, in the pure methanol solution only a trace amount of MB+ can be released. These facts indicate that the release of dyes is triggered by the ion-exchange process that occurs between MB+ in NEM-4 and Na+ in the NaCl-containing methanol solution. We also study the influence of dye molecular size on its ability to be absorbed by NEM-4 and all details of dye adsorption are given in the ESI.†
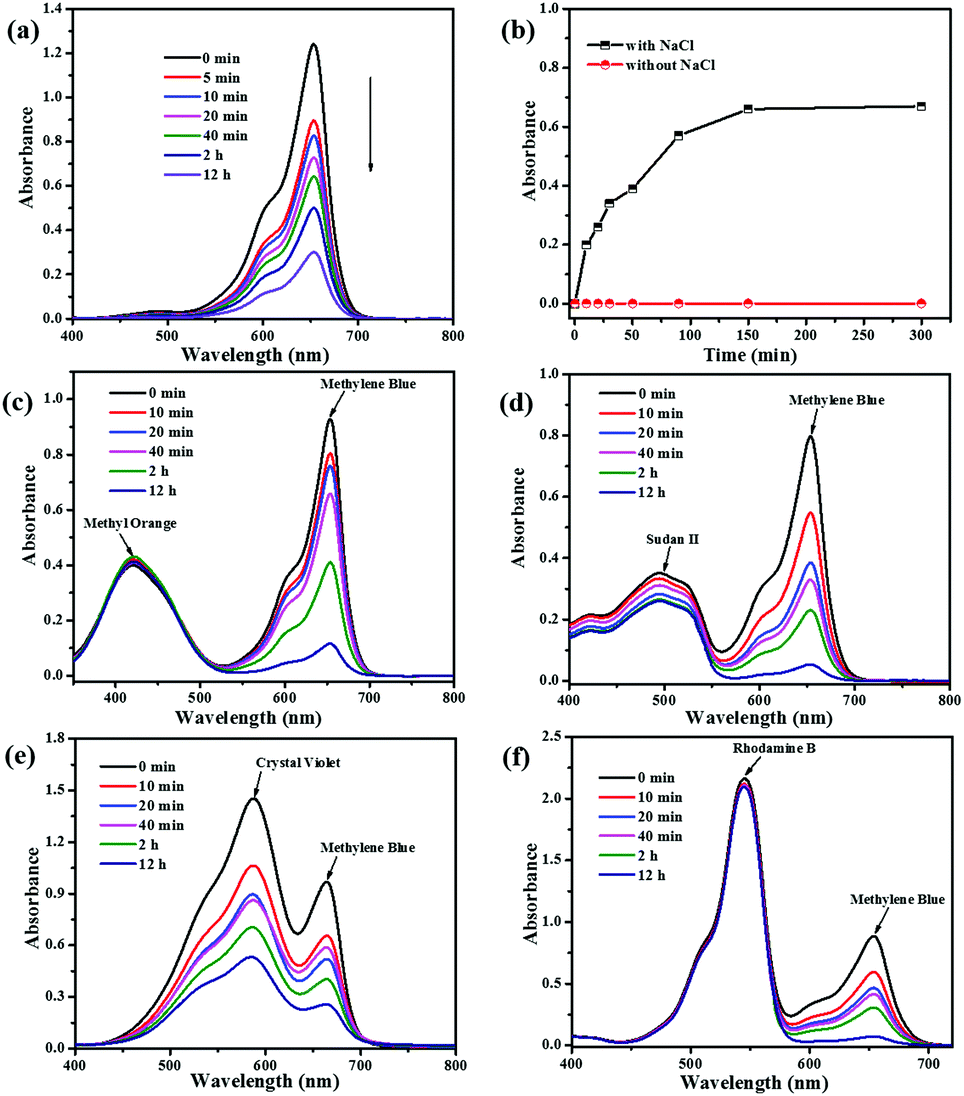 | ||
| Fig. 7 (a) UV-vis spectra of MB+ (5 ml, 4 × 10−5 M) in CH3OH solution during an adsorption test with NEM-4 over 5 min, 10 min, 20 min, 40 min, 2 h, and 12 h. (b) UV-vis absorbance (at 665 nm) of MB+ in CH3OH solutions with and without NaCl after being soaked with the fully loaded MB+@NEM-4. UV-vis spectra of the (c) MB+/MO−, (d) MB+/SD0, (e) MB+/CV+, and (f) MB+/RB+ dye mixtures in CH3OH solution during an adsorption test with NEM-4 over 0 min, 10 min, 20 min, 40 min, 2 h, and 12 h. All the dye mixtures are at the ratio of 1:1. | ||
4. Conclusions
In summary, aiming to improve the light hydrocarbon adsorption/separation, we have developed a new organic linker of the aromatic pentacarboxylic acid used in a three-dimensional Cu porous metal–organic framework (NEM-4). The NEM-4 with open CuII sites, exposed –COO− groups, and Lewis basic pyridyl sites enabled this MOF to take up a large quantity of C2H2 (204 cm3 g−1), C2H4 (164.1 cm3 g−1), C2H6 (172.2 cm3 g−1), C3H6 (197.4 cm3 g−1), and C3H8 (196.1 cm3 g−1) under ambient conditions and it exhibited excellent acetylene and propene repeatability. High adsorption selectivities of NEM-4 were also observed for C2H2/CH4 (33.2), C2H4/CH4 (26), C2H6/CH4 (10), C3H6/CH4 (152.2), and C3H8/CH4 (90.8) based on the gas uptake data as well as the ideal adsorbed solution theory (IAST). The observed enhanced light hydrocarbon uptakes in NEM-4 compared to those of its structural analogues NOTT-101 and 1 indicate that the exposed –COO− groups and Lewis basic pyridyl site functionality can significantly enhance the gas-framework interactions, which is further supported by the GCMC and first-principles calculations. Furthermore, the anionic NEM-4 shows highly selective adsorption of MB+ (over CV+, RB+, MO−, and SD0) in CH3OH solution and efficient release of the adsorbed MB+ in NaCl-containing methanol solution. The incorporation of Lewis basic nitrogen sites within the carboxylate-decorated MOF, therefore, improves the light hydrocarbon uptakes as well as provides some design ideas and technical basis for the synthesis of highly efficient hydrocarbon storage materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21776315), the Program for Natural Science Foundation of Shandong Province (ZR2017MB053, ZR2016BL12), the Fundamental Research Funds for the Central Universities (17CX02031A, 15CX05068A, and 15CX08010A) and Qingdao independent innovation program (16-5-1-88-jch).References
- L. Li, X. S. Wang, J. Liang, Y. B. Huang, H. F. Li, Z. J. Lin and R. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 9777–9781 CAS.
- R. J. Li, M. Li, X. P. Zhou, D. Li and M. O’Keeffe, Chem. Commun., 2014, 50, 4047–4049 RSC.
- X. X. Lv, L. J. Li, S. Tang, C. Wang and X. B. Zhao, Chem. Commun., 2014, 50, 6886–6889 RSC.
- F. Chen, D. Bai, Y. Wang, D. Jiang and Y. He, Mater. Chem. Front., 2017, 1, 2283–2291 RSC.
- Y. Yu, Y. Y. Zhuang, Z. H. Wang and M. Q. Qiu, Ind. Eng. Chem. Res., 2003, 42, 6898–6903 CrossRef CAS.
- Z. J. Lin, J. Lv, M. C. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- Q. X. Luo, B. W. An, M. Ji and J. Zhang, Mater. Chem. Front., 2018, 2, 219–234 RSC.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. A. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- K. Liu, X. Li, D. Ma, Y. Han, B. Li, Z. Shi, Z. Li and L. Wang, Mater. Chem. Front., 2017, 1, 1982–1988 RSC.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- I. Spanopoulos, C. Tsangarakis, E. Klontzas, E. Tylianakis, G. Froudakis, K. Adil, Y. Belmabkhout, M. Eddaoudi and P. N. Trikalitis, J. Am. Chem. Soc., 2016, 138, 1568–1574 CrossRef CAS PubMed.
- P. Yu, Q. Li, Y. Hu, N. Liu, L. Zhang, K. Su, J. Qian, S. Huang and M. Hong, Chem. Commun., 2016, 52, 7978–7981 RSC.
- F. S. Tang, R. B. Lin, R. G. Lin, J. C. G. Zhao and B. Chen, J. Solid State Chem., 2018, 258, 346–350 CrossRef CAS.
- T. A. Makal, J. R. Li, W. Lu and H. C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- P. Q. Liao, X. W. Chen, S. Y. Liu, X. Y. Li, Y. T. Xu, M. Tang, Z. Rui, H. Ji, J. P. Zhang and X. M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- H. Babaei, A. J. H. McGaughey and C. E. Wilmer, Chem. Sci., 2017, 8, 583–589 RSC.
- Y. He, B. Li, M. O′Keeffe and B. Chen, Chem. Soc. Rev., 2014, 43, 5618–5656 RSC.
- X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
- S. Yang, X. Lin, A. Dailly, A. J. Blake, P. Hubberstey, N. R. Champness and M. Schröder, Chem. – Eur. J., 2009, 15, 4829–4835 CrossRef CAS PubMed.
- H. M. Wen, H. Wang, B. Li, Y. Cui, H. Wang, G. Qian and B. Chen, Inorg. Chem., 2016, 55, 7214–7218 CrossRef CAS PubMed.
- X. T. Rao, J. C. Cai, J. C. Yu, Y. B. He, C. D. Wu, W. Zhou, T. Yildirim, B. L. Chen and G. Qian, Chem. Commun., 2013, 49, 6719–6721 RSC.
- B. Li, H. M. Wen, H. Wang, H. Wu, M. Tyagi, T. Yildirim, W. Zhou and B. Chen, J. Am. Chem. Soc., 2014, 136(17), 6207–6210 CrossRef CAS PubMed.
- X. P. Liu, Z. Y. Xiao, J. Xu, W. B. Xu, P. P. Sang, L. M. Zhao, H. Y. Zhu, D. F. Sun and W. Y. Guo, J. Mater. Chem. A, 2016, 4, 13844–13851 CAS.
- A. Gupta, S. Chempath, M. J. Sanborn, A. L. Clark and Q. R. Snurr, Mol. Simul., 2003, 29, 29–46 CrossRef CAS.
- B. J. Delley, Chem. Phys., 2000, 113, 7756–7764 CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- Q. Zhang, J. Yu, J. Cai, R. Song, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2014, 50, 14455–14458 RSC.
- X. Luo, Y. Guo, F. Ding, H. Zhao, G. Cui, H. Li and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 7053–7057 CrossRef CAS PubMed.
- C. Y. Sun, X. L. Wang, C. Qin, J. L. Jin, Z. M. Su, P. Huang and K. Z. Shao, Chem. – Eur. J., 2013, 19, 3639–3645 CrossRef CAS PubMed.
- M. L. Ma, J. H. Qin, C. Ji, H. Xu, R. Wang, B. J. Li, S. Q. Zang, H. W. Hou and S. R. Batten, J. Mater. Chem. C, 2014, 2, 1085–1093 RSC.
- Y. Yan, X. Lin, S. Yang, A. J. Blake, A. Dailly, N. R. Champness, P. Hubbersteya and M. Schröder, Chem. Commun., 2009, 1025–1027 RSC.
- B. Zheng, H. Liu, Z. Wang, X. Yu, P. Yi and J. Bai, CrystEngComm, 2013, 15, 3517–3520 RSC.
- M. Zhang, Q. Wang, Z. Lu, H. Liu, W. Liu and J. Bai, CrystEngComm, 2014, 16, 6287–6290 RSC.
- Z. Bao, G. Chang, H. Xing, R. Krishna, Q. Ren and B. Chen, Energy Environ. Sci., 2016, 9, 3612–3641 CAS.
- X. P. Liu, X. Li, J. Li, G. X. Li, S. Guo, H. Y. Zhu, L. M. Zhao, C. L. Hao and W. Y. Guo, J. Mater. Sci., 2018, 53, 8866–8877 CrossRef CAS.
- F. Moreau, I. da Silva, N. H. Al Smail, T. L. Easun, M. Savage, H. G. Godfrey, S. F. Parker, P. Manuel, S. Yang and M. Schröder, Nat. Commun., 2017, 8, 14085–14094 CrossRef CAS PubMed.
- S. Gao, C. G. Morris, Z. Lu, Y. Yan, H. G. W. Godfrey, C. Murray, C. C. Tang, K. M. Thomas, S. Yang and M. Schröder, Chem. Mater., 2016, 28, 2331–2340 CrossRef CAS.
- K. Liu, D. X. Ma, B. Y. Li, Y. Li, K. X. Yao, Z. J. Zhang, Y. Han and Z. Shi, J. Mater. Chem. A, 2014, 2, 15823–15828 CAS.
- Y. Huang, W. Zhang, J. Wang and Z. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 9307–9313 CAS.
- Y. Nie, L. Li and Z. D. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- Y. He, Z. Zhang, S. Xiang, F. R. Fronczek, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 6493–6495 RSC.
- Z. J. Zhang, S. C. Xiang, X. T. Rao, Q. Zheng, F. R. Fronczek, G. D. Qian and B. L. Chen, Chem. Commun., 2010, 46, 7205–7207 RSC.
- Y. Wang, M. He, Z. Tian, H. Zhong, L. Zhu, Y. Zhang, X. Zhang, D. L. Chen and Y. He, Dalton Trans., 2018, 47, 2444–2452 RSC.
- B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC.
- J. R. Li, J. Yu, W. Lu, L. B. Sun, J. Sculley, P. B. Balbuena and H. C. Zhou, Nat. Commun., 2013, 4, 1538–1546 CrossRef PubMed.
- H. Xu, J. F. Cai, S. C. Xiang, Z. J. Zhang, C. D. Wu, X. T. Rao, Y. G. Cui, Y. Yang, R. Krishna, B. L. Chen and G. D. Qian, J. Mater. Chem. A, 2013, 1, 9916–9921 CAS.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Q. Yang, A. D. Wiersum, P. L. Llewellyn, V. Guillerm, C. Serre and G. Maurin, Chem. Commun., 2011, 47, 9603–9605 RSC.
- H. C. Guo, F. Shi, Z. F. Ma and X. Q. Liu, Mol. Simul., 2013, 40, 349–360 CrossRef.
- Y. G. Lee, H. R. Moon, Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2008, 47, 7741–7745 CrossRef CAS PubMed.
- M. Fischer, F. Hoffmann and M. Froba, Chem. Phys. Chem., 2010, 11, 2220–2229 CrossRef CAS PubMed.
- M. Zhang, B. Li, Y. Li, Q. Wang, W. Zhang, B. Chen, S. Li, Y. Pan, X. You and J. Bai, Chem. Commun., 2016, 52, 7241–7244 RSC.
- Y. S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- X. L. Wang, C. Qin, E. B. Wang and Z. M. Su, Chem. – Eur. J., 2006, 12, 2680–2691 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1552519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00105g |
| This journal is © the Partner Organisations 2018 |