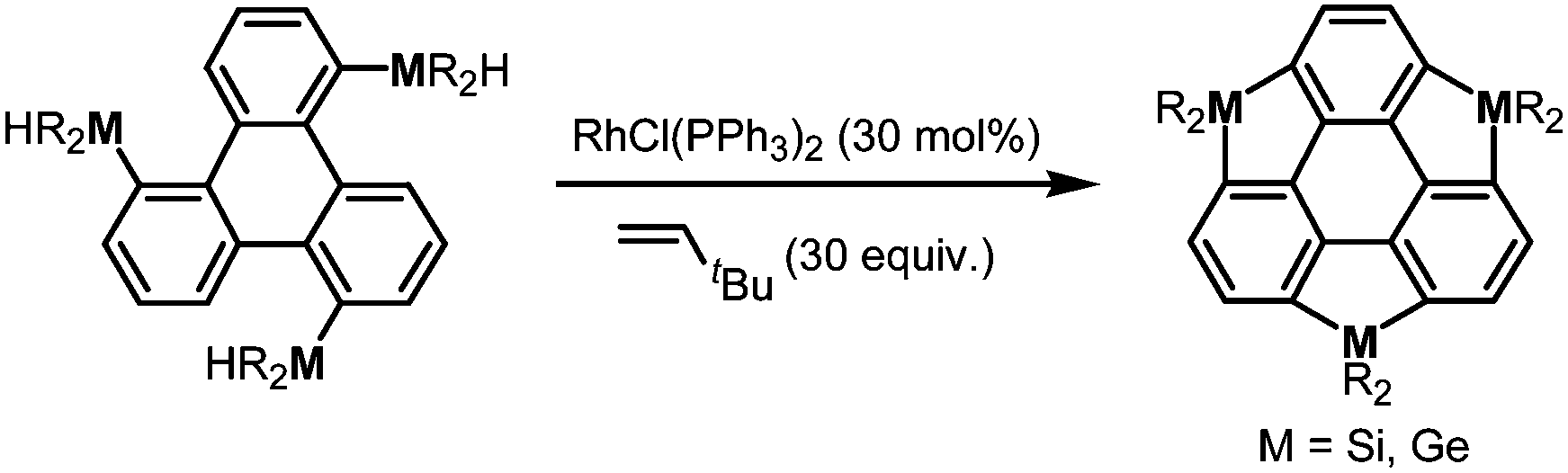DOI:
10.1039/C7QM00593H
(Review Article)
Mater. Chem. Front., 2018,
2, 635-661
Figuration of bowl-shaped π-conjugated molecules: properties and functions
Received
19th December 2017
, Accepted 29th January 2018
First published on 1st February 2018
Abstract
Synthesis, properties, and functions of bowl-shaped pi-conjugated molecules including a hetero atom-doped system are reviewed.

Masaichi Saito
| Masaichi Saito was born in Tokyo, Japan, in 1967. He received his BS and PhD degree from the University of Tokyo in 1991 and 1996, respectively, under the supervision of Professor Renji Okazaki. He started his academic career at Saitama University in 1996 and has been a professor since 2009. He was awarded the CSJ Award for Creative Work (2015) from the Chemical Society of Japan, and the Commendation for Science and Technology Prizes (2017) from the MEXT, Japan. He serves on the editorial advisory board of Applied Organometallic Chemistry, Main Group Metal Chemistry, and European Journal of Inorganic Chemistry. |

Hiroshi Shinokubo
| Hiroshi Shinokubo was born in Kyoto in 1969. He received his BS and PhD degree from Kyoto University in 1992 and 1998, respectively. He became an Assistant Professor with Prof. Oshima in 1995 at Kyoto University. He collaborated with Prof. Atsuhiro Osuka at Kyoto University as an Associate Professor from 2003 to 2008. In 2008, he became a Professor at Nagoya University. He received the CSJ Award for Young Chemists in 2004, the Minister Award for Distinguished Young Scientists from MEXT in 2008, the JSPS Prize in 2012, the SSOCJ Award for Functional Material Chemistry 2012, and Inoue Prize for Science 2018. |

Hidehiro Sakurai
| Hidehiro Sakurai received his PhD degree from The University of Tokyo in 1994 under the direction of Professor Koichi Narasaka. After studying with Professor Narasaka in Tokyo and with Professor C. P. Casey at the University of Wisconsin-Madison, he joined Osaka University in 2000. In 2004, he moved to the Institute for Molecular Science (IMS). In 2014, he returned to Osaka University as a Professor at the Division of Applied Chemistry. Professor Sakurai has studied various topics in organic synthesis and organometallic chemistry. His current research interests include the science of buckybowls, nonplanar π-aromatic compounds, and colloidal nanometal catalysts. |
Introduction
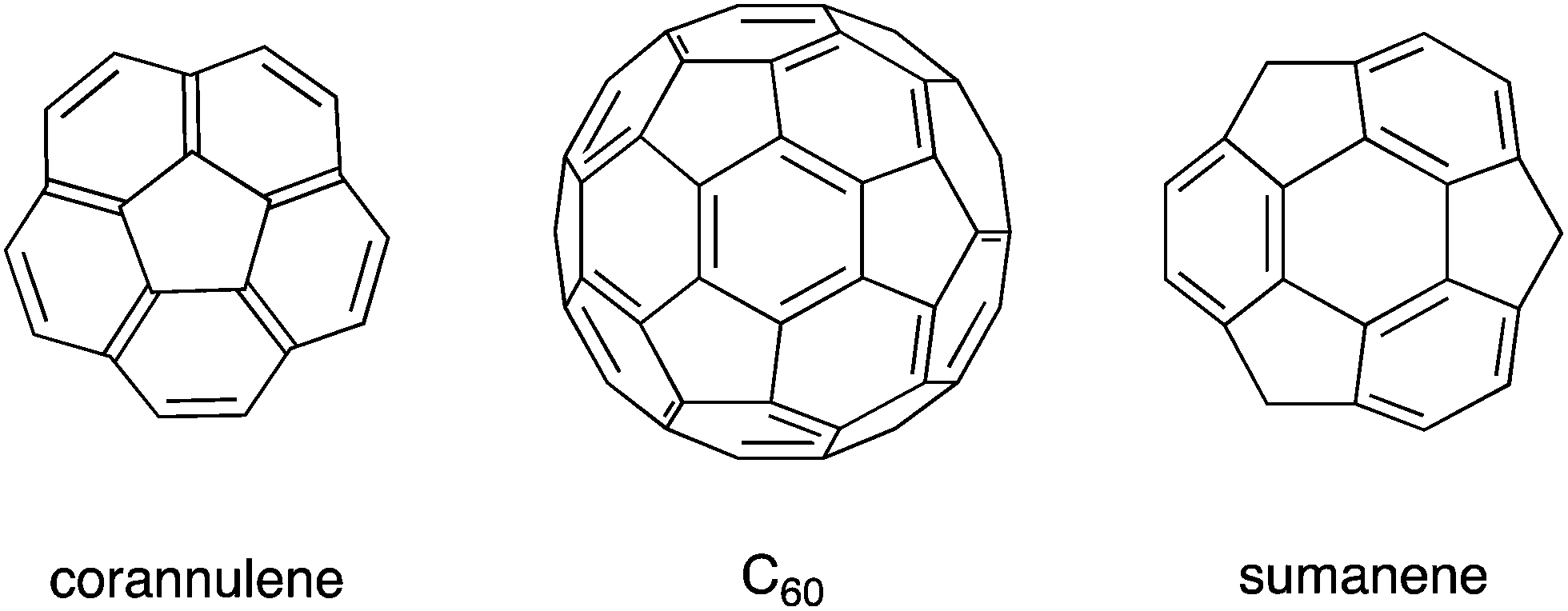
In recent decades, remarkable progress has been made in the field of organic synthesis, and, as a result, researchers have been able to synthesize compounds with complicated structures that could not be synthesized in the past, such as curved π-conjugated molecules. Following the discovery of fullerenes1 and carbon nanotubes,2 bowl-shaped aromatic compounds (so-called buckybowls), which are partial structures of fullerenes and carbon nanotubes, attracted much attention, both as model compounds and precursors for the creation of new materials. For example, corannulene, a pristine buckybowl with C5v symmetry, required multiple steps for synthesis in 1966.3 However, the establishment of the flash vacuum pyrolysis (FVP) method in the 1990s4,5 and the development of kilogram-scale synthesis6 allowed corannulene to become readily and commercially available. Sumanene, another pristine buckybowl with C3v symmetry, took a long time to be synthesized for the first time in 2003,7 but, in 2017, it is also commercially available (Chart 1).
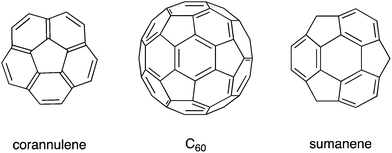 |
| | Chart 1 Structures of C60 and two pristine buckybowls: corannulene and sumanene. | |
Currently, various functionalized derivatives can be synthesized using these pristine structures as starting materials, or through independent routes, due to the recent progress in the field of organometallic chemistry, specifically in aromatic compound synthesis (typically by cross-coupling reactions and aromatic C–H bond activation reactions).8 It should be mentioned, although this fact tends to be ignored, that the development and observation of chemical reactions on metal surfaces through the development of microscope technology also greatly contributed to this field.9
Buckybowl chemistry, at first, mainly focused on the development of synthetic methods; however, as the derivatization of buckybowls has been well established, buckybowl chemists have begun to study the potential applications of buckybowls in the material chemistry field. Herein, we present a focused review of new perspectives on buckybowl chemistry directed toward material chemistry. This review is not comprehensive; particularly, synthetic studies of all-carbon buckybowls that were discussed in previous reviews10–19 are not included in this review. However, synthetic studies are also discussed in the sections of heteroatom-containing molecules because synthetic improvement is still necessary in these fields. Among the various types of non-planar π-conjugated systems, we mainly cover buckybowls, the partial structure of fullerene consisting of a combination of six-membered and five-membered rings, and their heteroatom analogues. Belt-shaped compounds, such as cycloparaphenylenes (CPPs)20 and carbon nanobelts,21 and twist-shaped compounds22 were also excluded from this review.
1. Brief overview of the “bowl” effect
Even if one were able to construct bowl-shaped π-conjugated molecules using new methods, doing so would be meaningless if their properties remained unchanged from those of ordinary, flat π-conjugated molecules. The beginning of this review is a brief overview of the properties affected by the bowl shape and the curvature in π-conjugated systems.
1.1 Electronic and geometric perturbation
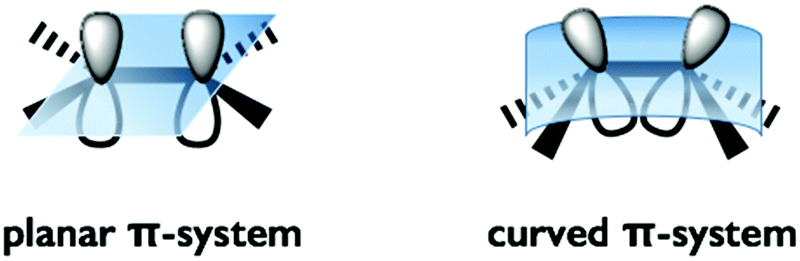
The distortion of the π-conjugation of buckybowls induces an inefficient overlap of each p-orbital and an increase of the s-orbital character in sp2 carbon compared to that of flat π-conjugation. As a result, several phenomena are expected, including a decrease of the LUMO level, enhancement of the electron accepting property and high reactivity toward cycloaddition, most of which are common properties of fullerenes (Fig. 1).
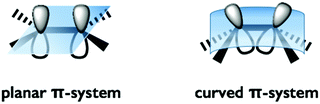 |
| | Fig. 1 Planar-π vs. curved-π systems. | |
1.2 Concave and convex faces
The curvature resulting from π-conjugation causes two distinct faces: a concave face and a convex face. The orbital structures and electron densities of these two faces are very different; therefore, buckybowls intrinsically possess a Janus character. For example, difference of the stereoelectronic effect between two faces was investigated.23
The bowl shape also induces a dipole moment in a perpendicular axis despite the absence of functional groups. Corannulene has a dipole moment of 2.07 D and sumanene has a dipole moment of 2.70 D, which means that ferroelectric materials can be developed if these molecules are assembled in a one-dimensional fashion. In addition, generally speaking, buckybowls are soluble in various solvents, probably due to their dipole moments and the avoidance of insoluble materials induced by strong π–π interactions.
1.3 Curved-π/curved-π interactions
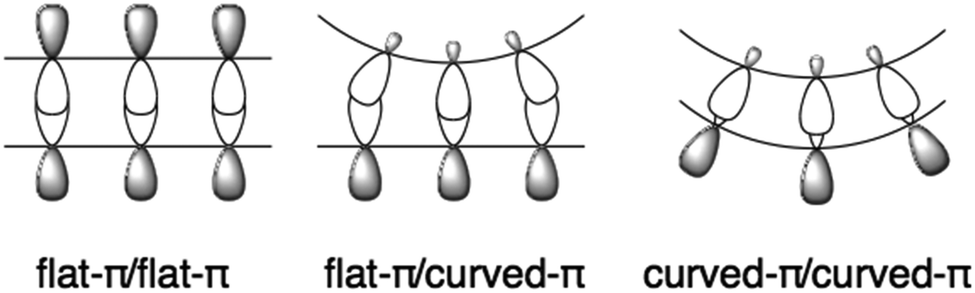
Flat π-conjugated compounds favour stacking through π–π interactions. To prevent electrostatic repulsion, each layer maintains a staggered position, resulting in herringbone packing in a crystal. The π–π interaction in flat systems is sometimes too strong to form an aggregate with poor solubility. In contrast, for geometric reasons, π–π interactions between the flat-π and curved-π may be inefficient; instead, curved surfaces favour interactions with similarly curved surfaces. Curved-π/curved-π interactions compensate for the overlap, and, in the case of bowl/bowl stacking, favour interactions between electrostatically different concave and convex surfaces (Fig. 2).
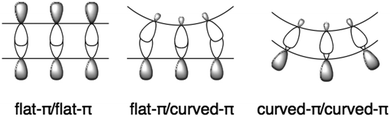 |
| | Fig. 2 Complementary curved-π/curved-π interactions. | |
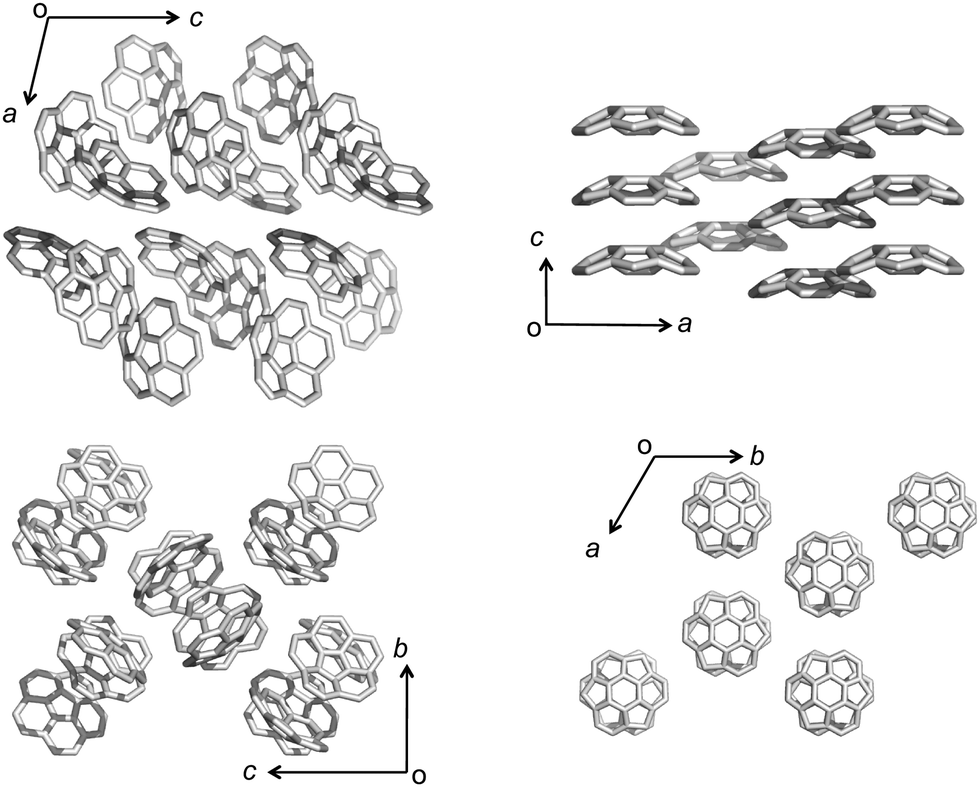
The most representative example of concave/convex interactions is the crystal packing of buckybowls. Unlike five-fold symmetric corannulene,24 three-fold symmetric sumanene forms a single crystal with perpendicularly ordered columnar packing.25,26 Each column faces the same direction, which means that the crystal has a high dipole moment. However, not every sumanene derivative has unidirectional columnar packing; many compounds have bidirectional columnar packing to restrain bias (Fig. 3).
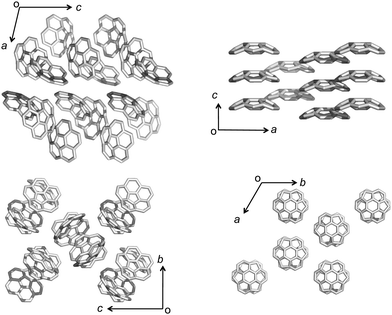 |
| | Fig. 3 Single crystal packing structures of corannulene (left) and sumanene (right). | |
1.4 Dynamic behaviour
Bowl-shaped π-conjugated molecules exhibit unique dynamic behaviour. An example of this is the interconversion of concave and convex faces, the bowl-to-bowl inversion, which occurs when the bowl size is small enough to overcome the high-energy barrier of the transition state. The bowl-to-bowl inversion of corannulene27 and sumanene28 has been well studied both experimentally and theoretically. In most cases, bowl-to-bowl inversion follows the double-well potential model, and the flat state is the transition state. Due to their substituent effect on the peripheral aromatic ring, corannulene derivatives decrease the inversion barrier,27 while sumanene derivatives increase the inversion barrier.28 However, it is also known that some buckybowls do not follow the double-well potential model but instead follow multistep interconversion through a sigmoidal-structured transition state.29
Indeed, interesting outcomes of bowl-shaped aromatics have been induced by the above properties mostly multiple. For example, the dynamic behaviour of bowl-to-bowl inversion results in structural changes, the interchange of the two Janus faces, and changes in the direction of the dipole moment, all of which might allow for the development of a stimuli-responsive system. Furthermore, unidirectional columnar packing in the solid state would introduce isotropic characters to the system (Fig. 4).
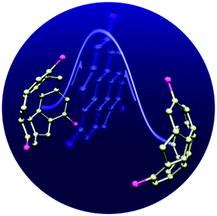 |
| | Fig. 4 Bowl-to-bowl inversion. | |
1.5 Role of heteroatoms in π-conjugation
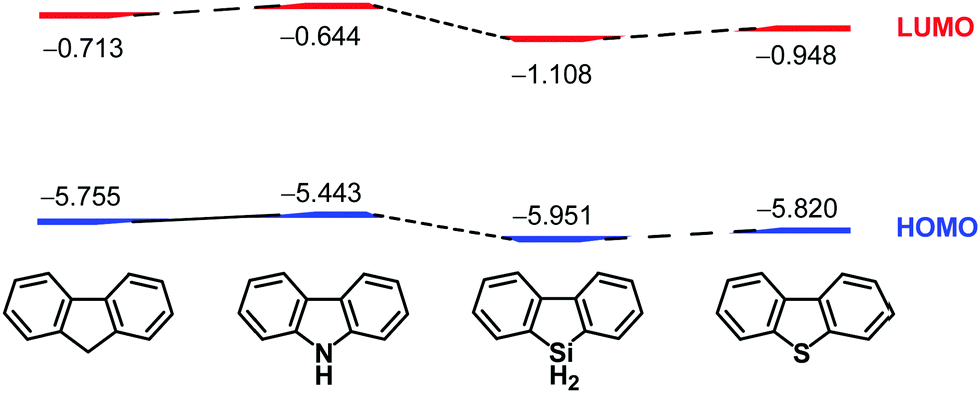
After the chemistry of carbon-based π-conjugated molecules had been established, attention next focused on the substitution of skeletal carbon atoms of the π-conjugated molecules by heteroatoms. This is because heteroatoms can perturb electronic structures of the original carbon-based compounds. For example, carbazole has a high-lying HOMO compared to that of fluorene, while the LUMO of 9-silafluorene is lower than that of fluorene30 because of σ*–π* conjugation.31 That is, the electronic properties of 9-heterafluorene are dependent on the central heteroatoms on the 9-position, and they are utilized as building blocks for intelligent materials such as fluorescent polymers32–34 and chemical sensors.35,36 Accordingly, introduction of heteroatoms into carbon-based π-conjugated molecules is of fundamental importance to create potential functional materials that would possess electronic characteristics different from those of the carbon-based materials. During the course of the studies on heteroatom-bearing π-conjugated compounds, synthesis and investigation of the effects of heteroatoms on the electronic perturbation of curved-π systems are receiving considerable interest (Chart 2).
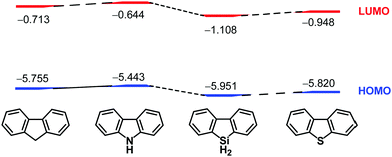 |
| | Chart 2 Changes of HOMO and LUMO levels by heteroatoms of a fluorene family. | |
2. Bowl-shaped polyaromatic hydrocarbons
2.1 Photo-, electric- and energy-storage materials
Similar to fullerenes, buckybowls possess the characteristic of electron acceptors. However, the difference between the two is that buckybowls are more soluble in many of the common organic solvents; buckybowls are also easy to functionalize at the peripheral positions of their frameworks to tune physical and chemical properties. For example, the introduction of F,37,38 or CF3,37,39,40 or the perfluoroalkyl group,41 as well as the fusion of carboximide to the bowl,42 significantly improves electron acceptance. In addition, the introduction of an electron donor group effectively constructs the donor–acceptor system.43,44
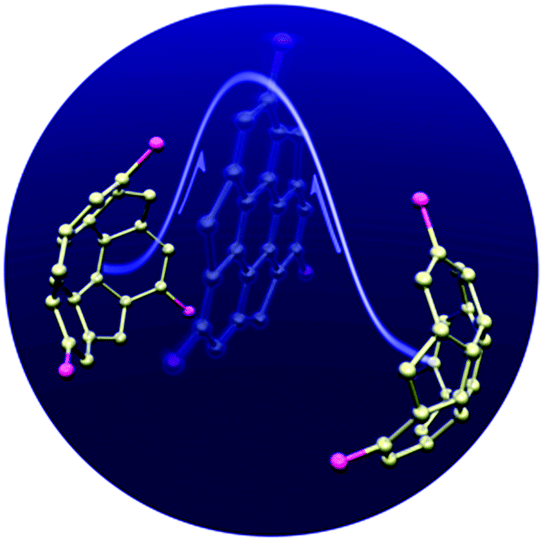
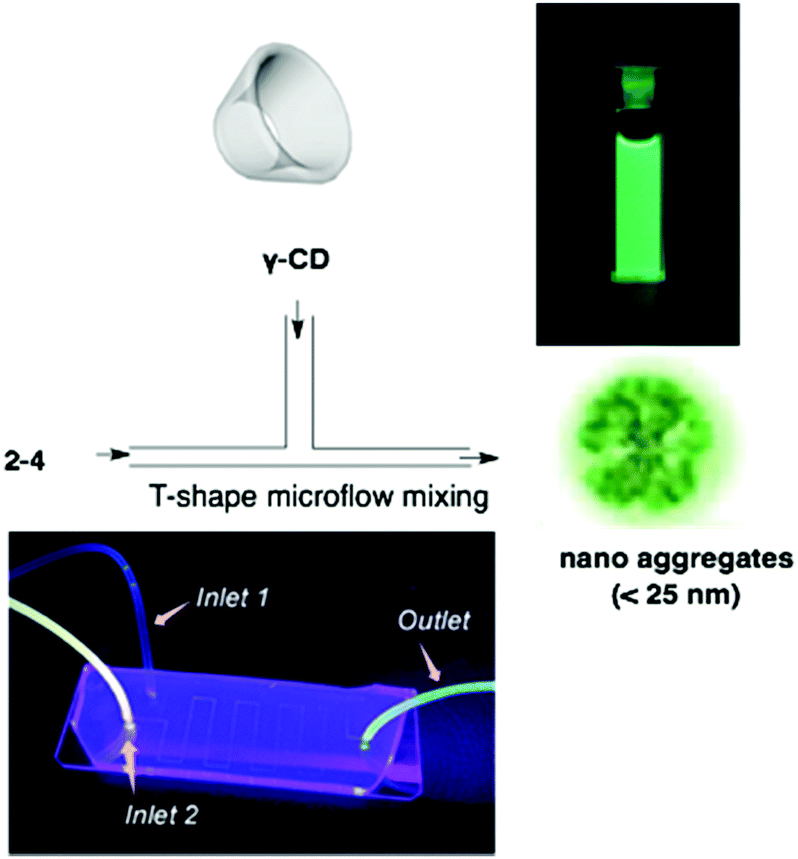
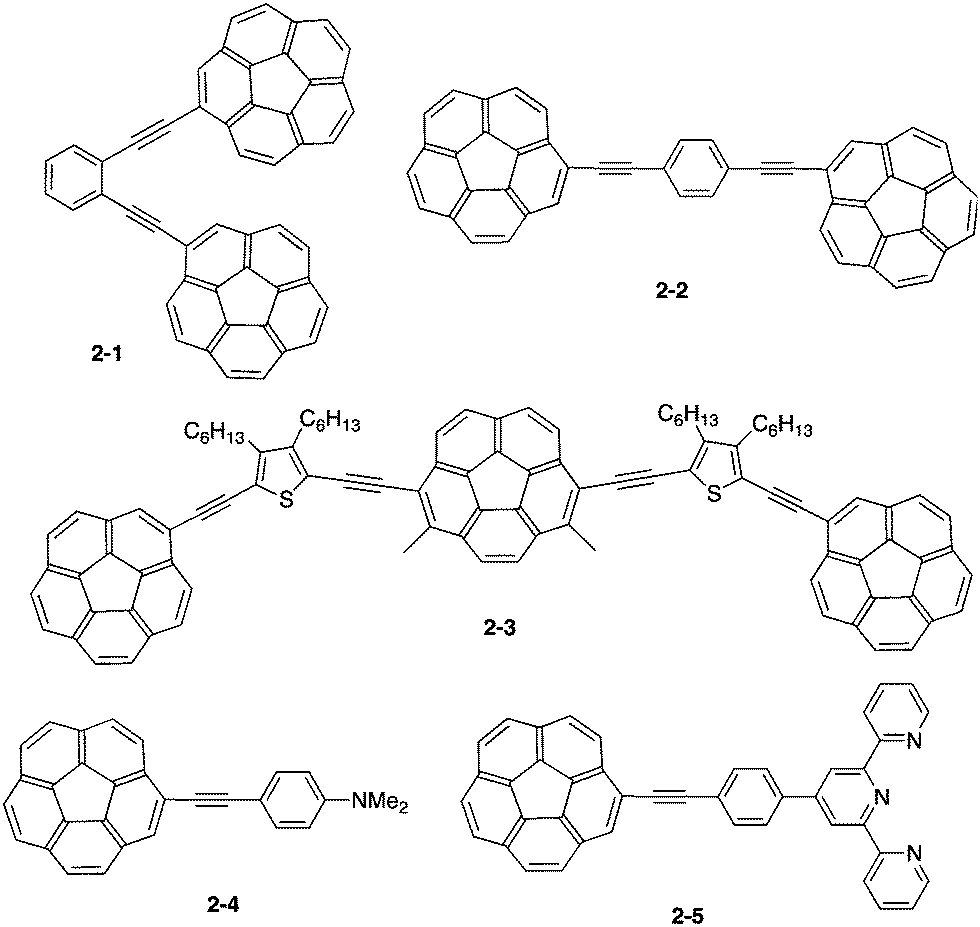
Corannulene was first examined for its application as blue-emitting materials. Although pristine corannulene itself does not show a good fluorescence property (ca. Φ = 0.07),45 an appropriate substitution significantly enhances the fluorescence intensity.46 Mack and co-workers reported that two corannulenes tethered with a diethynylphenylene unit (2-1, 2-2) exhibited high fluorescence (Φ = 0.60) that could be applied as a blue-emitting material.47 Stuparu prepared a series of discrete oligomers containing corannulene as a nonplanar and electron deficient unit and thiophene as a planar and electron rich unit (2-3), which exhibited a two-photon absorption cross section up to 600 GM and two-photon-excited intense green luminescence.48 Based on the mass spectrometric evidence of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex formation of corannulene and γ-CD,49 Stuparu mixed an ethynyl-tethered D–A system of corannulene (2-4) with γ-CD using the microfluidic method to generate nanometer-scale (up to 25 nm) assembly material consisting of only organic materials without any metals, which showed bright green luminescence and multi-photon activity (TPA = 32 GM) in water. This could be applied as a non-toxic bioimaging material.50 A similar ethynyl-tethered system with a terpyridine moiety (2-5) was applied to the responsive materials with metal ions (Fig. 5 and Chart 3).51
:1 inclusion complex formation of corannulene and γ-CD,49 Stuparu mixed an ethynyl-tethered D–A system of corannulene (2-4) with γ-CD using the microfluidic method to generate nanometer-scale (up to 25 nm) assembly material consisting of only organic materials without any metals, which showed bright green luminescence and multi-photon activity (TPA = 32 GM) in water. This could be applied as a non-toxic bioimaging material.50 A similar ethynyl-tethered system with a terpyridine moiety (2-5) was applied to the responsive materials with metal ions (Fig. 5 and Chart 3).51
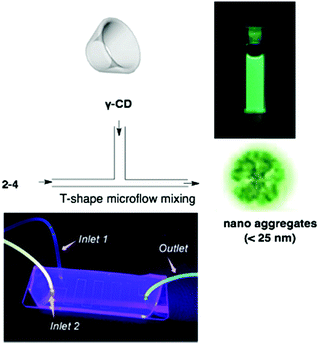 |
| | Fig. 5 Water-dispersible nanoaggregate of 2-4 with γ-CD. | |
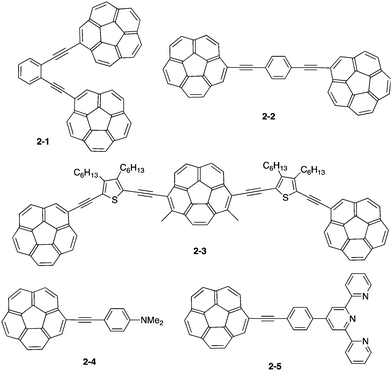 |
| | Chart 3 Corannulene derivatives for photoluminescence materials. | |
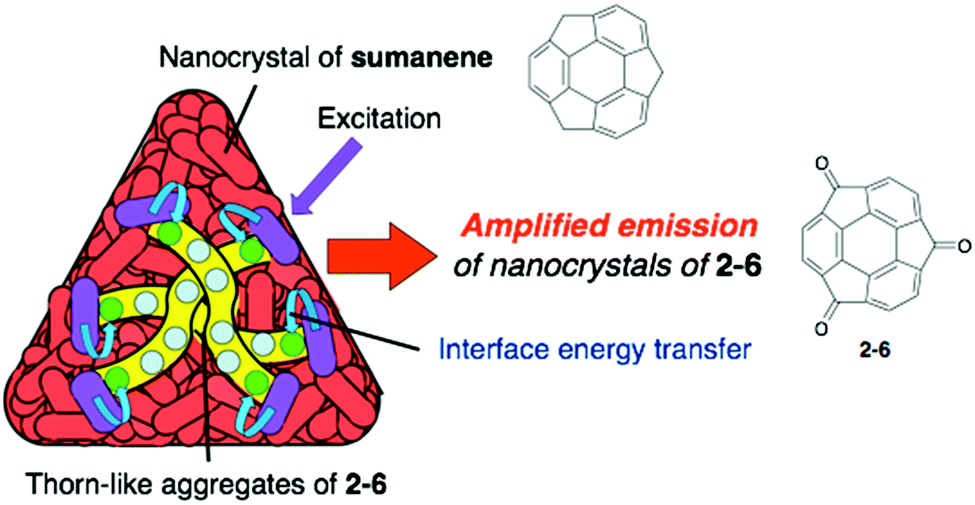
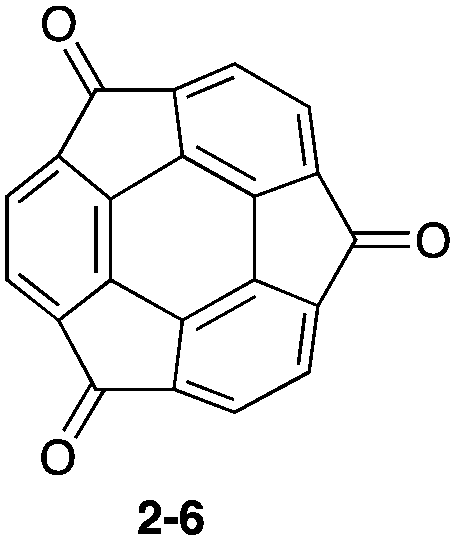
Pristine sumanene also has poor fluorescence properties, but the characteristic method of sumanene functionalization is the utilization of its benzylic position. Sumanenetrione (2-6), which consists of fully conjugated tri-ketone compounds with a multiple reversible redox property,52–54 showed significantly short-lived phosphorescence (53 μs at 293 K) compared with a planer triplet ketone, such as a fluorenone triplet (500 μs). This is due to the fast intersystem crossing process, which is attributed to the curved structure of 2-6, enhancing the spin–orbit coupling.55 Although 2-5 did not emit in the solid state, the core–shell structure of the two nanocrystals of 2-6 (core) and sumanene (shell) showed the emitting ability corresponding to 2-6 (Fig. 6 and Chart 4).56
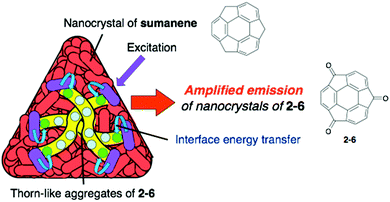 |
| | Fig. 6 Core–shell (Onigiri) nanostructure of 2-6/sumanene. | |
 |
| | Chart 4 Sumanenetrione (2-6). | |
Bowl-shaped π-conjugated molecules are expected to act as electron transporting materials or as related devices. In the pursuit of better materials for conducting electricity, the overlap of intermolecular electronic wave functions and the consequent band formation in the solid states and/or quantum nanostructures are considered for the design of the charge transport mechanism.57 An alternative mechanism of charge transport involving tightly bound π-molecular orbital overlap has been identified—especially in curved aromatic molecules.58–60 A set of diffuse molecular orbitals, super atomic molecular orbitals (SAMOs), which exist universally in curved molecules,58 plays a key role in this electron transporting mechanism. Followed by the investigation with the use of C60,58 the evidence of the enhanced photocurrent response in corannulene films was also reported.61
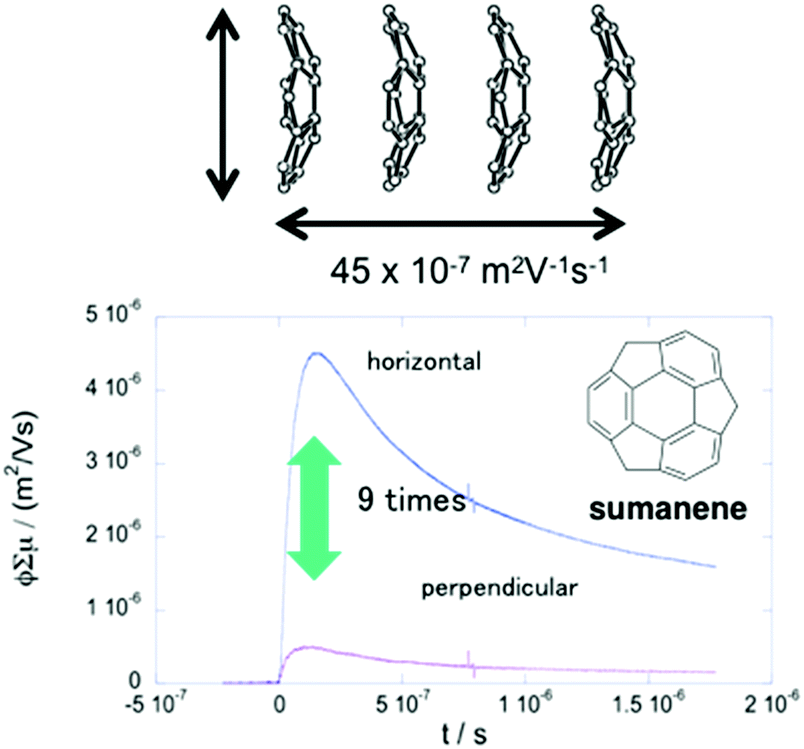
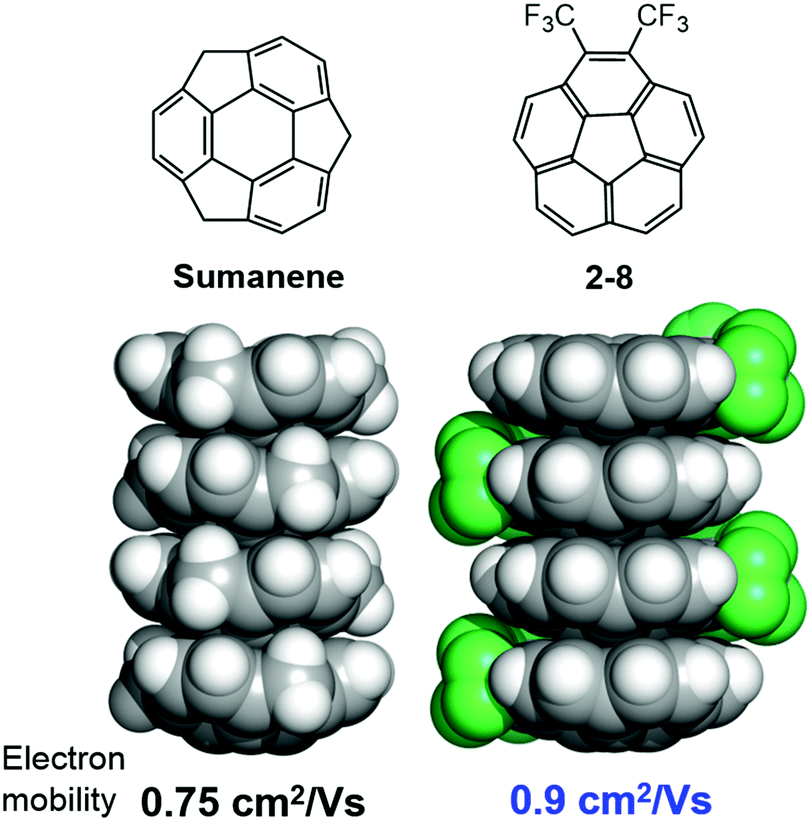
The anisotropic electron transport properties of sumanene crystals were first reported by Seki and Hirao.62 The charge carrier mobility of the needle-like sumanene crystals was measured using the time-resolved microwave conductivity (TRMC) method. A large intracolumnar electron mobility (0.75 cm2 V−1 s−1), which had a 9.2 times larger anisotropy along with the stacking column axis, was observed (Fig. 7).62 The electron conductivity seems to be dependent on the crystal packing structure. Trimethylsumanene (2-7), which formed a typical bi-directional columnar packing structure (cf. sumanene: unidirectional columnar packing), exhibited a mobility of 0.05 cm2 V−1 s−1.63 In contrast, the electron mobility of pristine corannulene, having non-columnar packing, was measured by the TRMC method and was shown to be 1,000 times smaller than that of sumanene (5 × 10−4 cm2 V−1 s−1).63 However, in the case of 1,2-bis(CF3)-corannulene (2-8), which formed a perpendicular columnar packing, the electron mobility improved to 0.9 cm2 V−1 s−1, which was probably due to both the electronic effect of the CF3 group and the geometrical effect.40 Therefore, the rational designs of the buckybowls, size, curvature, crystal packing, and other elements are now being intensively studied both experimentally and theoretically (Fig. 8).64,65
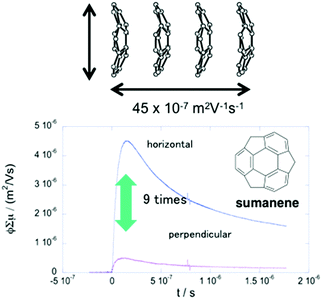 |
| | Fig. 7 Anisotropic electron transport properties of sumanene. | |
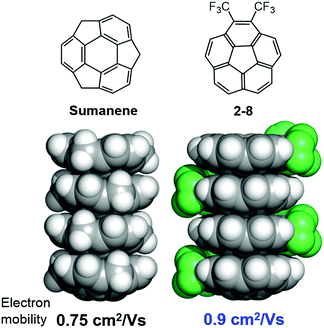 |
| | Fig. 8 Columnar packing is important to induce the electron mobility. | |
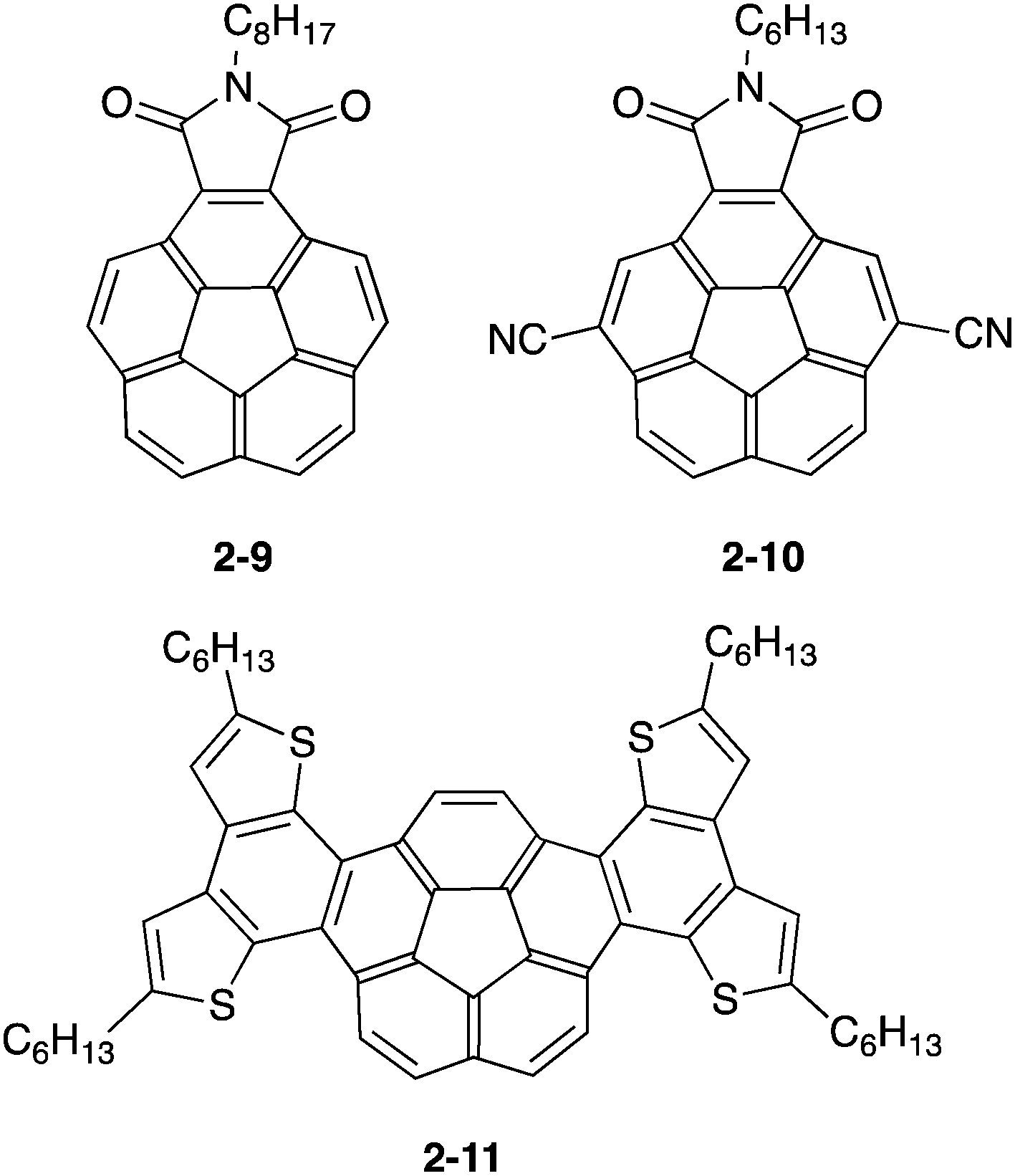
The application of bowl-shaped compounds to the organic electronic devices began with Cao's group and others.66 Tuning of electronic structures and crystal engineering (control of the packing structure) might be crucial to the success of the device. Corannulene derivatives with an electron-accepting imide moiety (2-9,672-1068) or electron-donating fused thiophene (2-11)69 were investigated. Both 2-9 and 2-10 were carefully designed to form a dense columnar packing in crystals due to the consideration of the molecular dipoles. Compound 2-9 displayed an electron mobility of 0.02 cm2 V−1 s−1 in a vacuum, while a hole mobility of up to 0.05 cm2 V−1 s−1 was obtained under air.67 By using 2-10, the n-channel OFET (by lowering the LUMO level and the dense packing in crystals) was achieved.68 In contrast, 2-11 formed the short contact at the thiophene rings as convex/convex interaction in crystals and behaved as hole transporting materials with a mobility of 0.06 cm2 V−1 s−1 (Fig. 9 and Chart 5).69
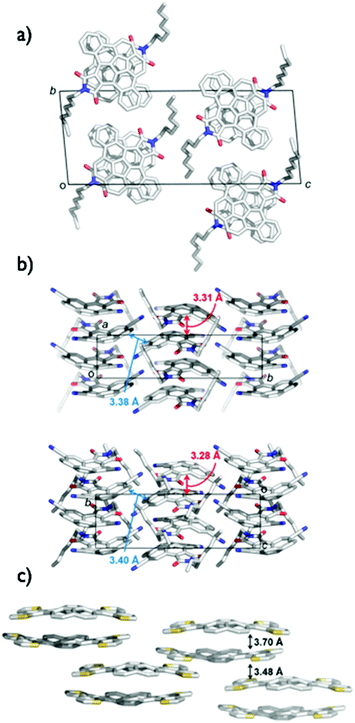 |
| | Fig. 9 Single crystal packing structures: (a) 2-9, (b) 2-10, (c) 2-11. | |
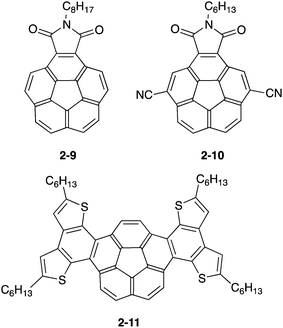 |
| | Chart 5 Corannulene derivatives for OFET. | |
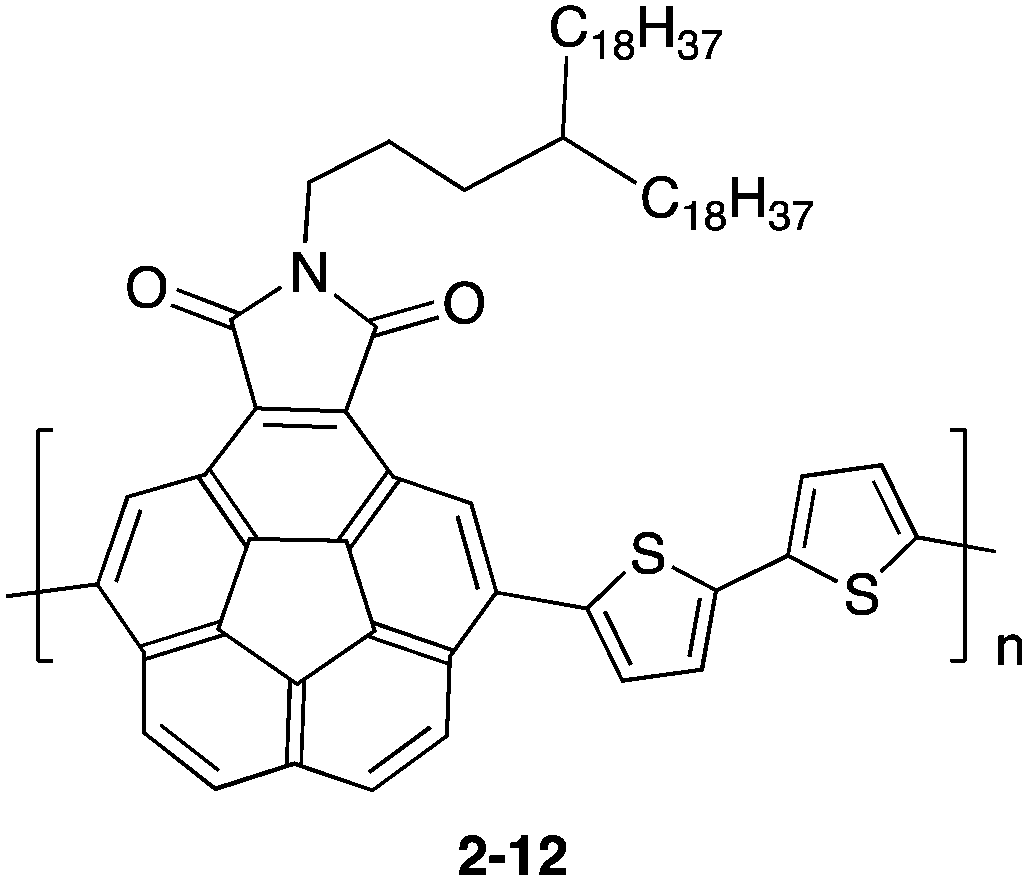
Another approach is using donor–accepter polymers (2-12), in which corannulene imide was used as an electron accepter unit (Chart 6).70
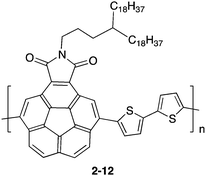 |
| | Chart 6 D–A type polymer containing corannulene (2-12). | |
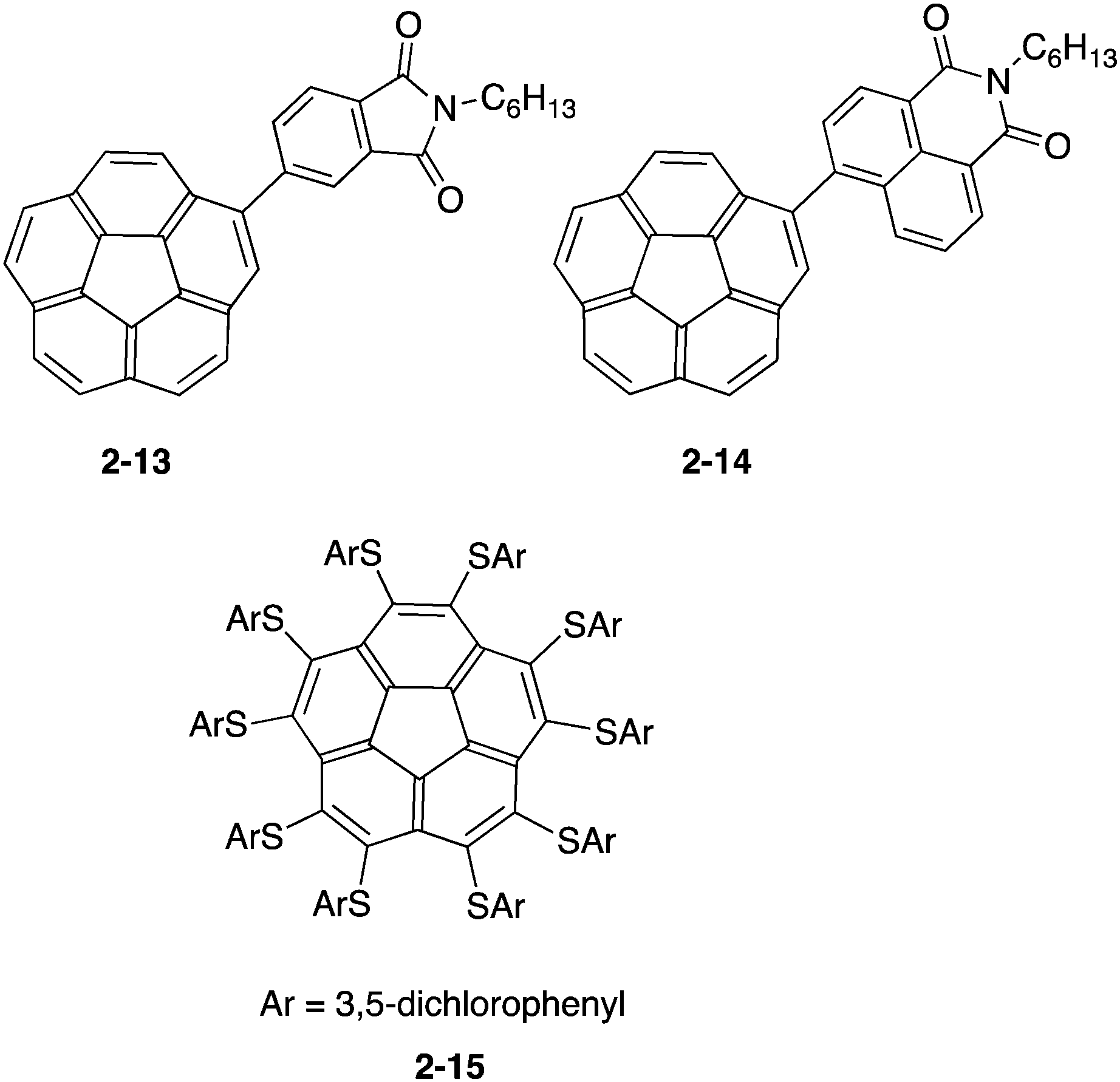
Due to the electron-accepting property being similar to that of fullerenes, buckybowl derivatives were also considered as a substitute of the fullerene derivatives with high tunability. Baldridge and Cao prepared a solution-processed bulk heterojunction solar cell by using two corannulene derivatives (2-13, 2-14) with the imide group. The power conversion efficiencies reached were 0.32% and 1.03%, respectively, indicating the possibility of a non-fullerene device.71 On the other hand, Baldridge and Siegel focused on the property of the organothio-substituent, which raised the HOMO level and lowered the LUMO level, anticipating good electron acceptors with a smaller HOMO–LUMO gap. They prepared a series of organothio-substituted corannulene and investigated the photovoltaic device's performance. Among these derivatives, the power conversion efficiency of 2-15 reached 0.95% (Chart 7).72,73
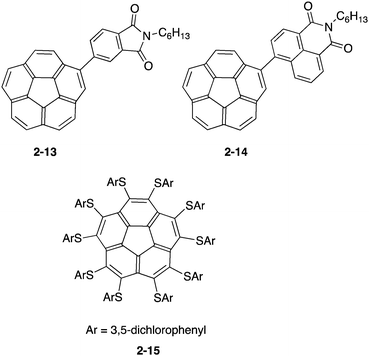 |
| | Chart 7 Corannulene derivatives for photovoltaic devices. | |
Having the unique structure of carbon networks and intrinsically possessing a strain in the framework of bowl-shaped π-conjugation, buckybowls are also utilized as a resource of carbon materials. Madhavi and Stuparu prepared polymeric nanomaterials from the Diels–Alder adduct of corannulyne and furan (2-16) by ring-opening metathesis polymerization, which resulted in porous carbon materials (2-17). They applied it to a supercapacitor device (Scheme 1 and Fig. 10).74
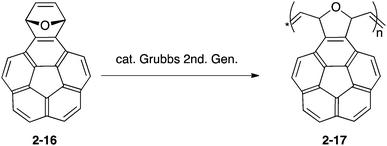 |
| | Scheme 1 | |
 |
| | Fig. 10 SEM image of 2-17. | |
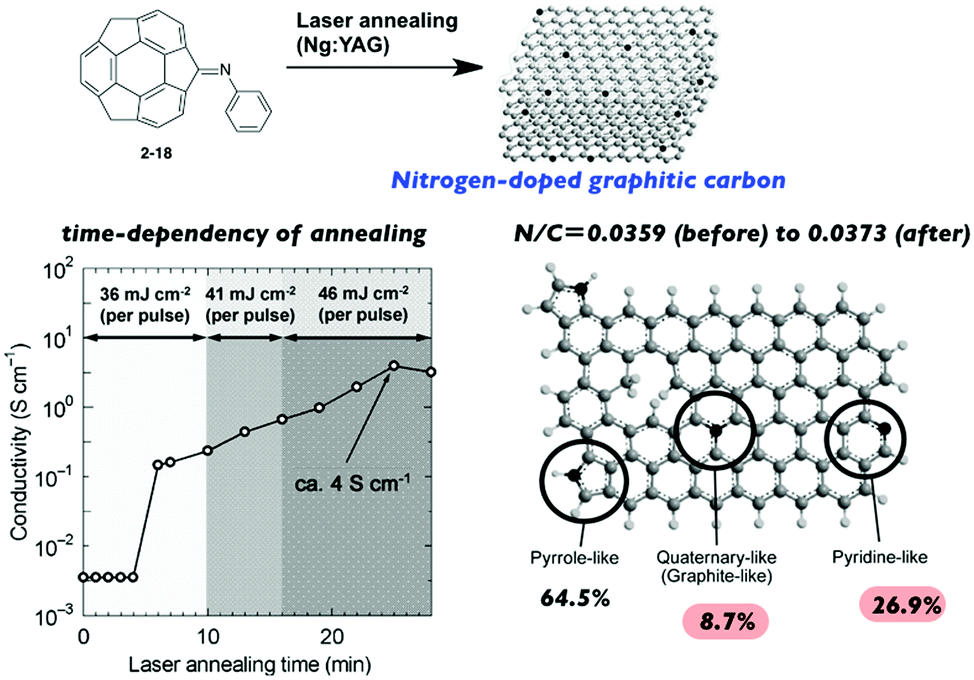
Hirao and co-workers investigated the laser-induced carbonization of sumanene derivatives.75–77 Laser annealing of the sumanenone imine (2-18) resulted in nitrogen-doped graphitic carbon. The properties of the resulting carbon materials (including the electron conductivity) were dependent on the annealing time; it is noteworthy that the N/C ratio remained nearly the same throughout the process to avoid the unnecessary release of nitrogen (Fig. 11).75
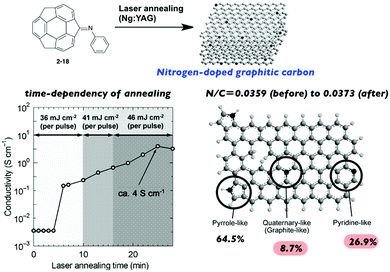 |
| | Fig. 11 Laser-induced carbonization of sumanene derivatives for N-doped nanocarbon. | |
Fujita et al. showed the formation of a graphitic carbon nanocage from sumanene nanocrystals induced by electron-beam irradiation (Fig. 12).78
 |
| | Fig. 12 Electron-beam-irradiated carbon nanocage from sumanene nanocrystals. | |
2.2 Complex formation with fullerenes through a curved-π/curved-π interaction
One of the fundamental problems in fullerene and carbon nanotube chemistry is that these nanocarbons are obtained as a mixture of many isomers, and all of them resemble each other. Therefore, they are extremely difficult to obtain as an identical pure isomer. Since the major difference between isomers is in their curvature on the spherical π-surface, a complementary curved-π/curved-π interaction has long been expected to assist in the molecular recognition and the isolation/purification of fullerenes. In addition, to improve the properties of fullerene derivatives in organic materials, an appropriate dopant to a solid-state fullerene is also desired, and a bowl-shaped compound has been considered as a candidate due to its good affinity to fullerenes. Therefore, in both the solution state and solid state, the host–guest chemistry between fullerenes and bowl-shaped compounds has been intensively studied as described below.
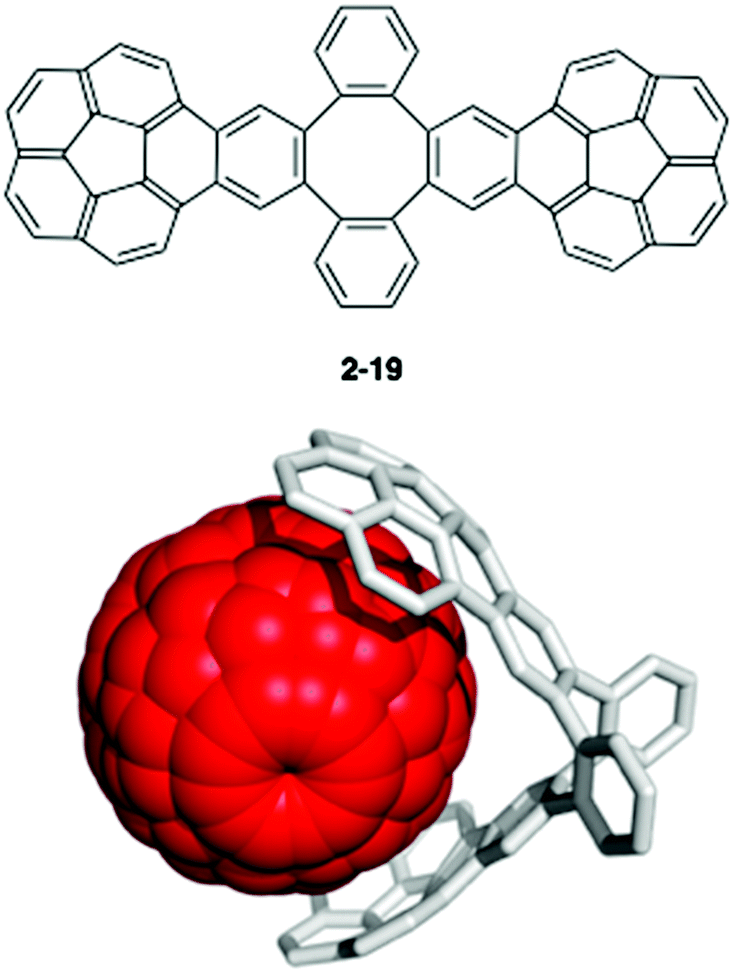
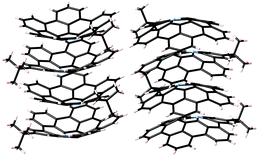
One of the most famous works in this area is Sygula's “buckycatcher” molecules. There is insufficient interaction between one corannulene unit and C60 in the solution state to overcome the solvation and entropy penalty. As a result, the buckycatcher (2-19), having two corannulene pincers on a butterfly-structured cyclooctatetraene framework, was carefully designed; it forms a stable inclusion complex both in organic solvents and in the solid state.79 A recent thermodynamics study by NMR and isothermal titration calorimetry (ITC) revealed that the complexation of C60 or C70 with 2-19 is enthalpically driven and that the association constants (Ka) in toluene at 298 K were 3200 ± 150 M−1 with C60 and 4600 ± 170 M−1 with C70, respectively (Fig. 13).80
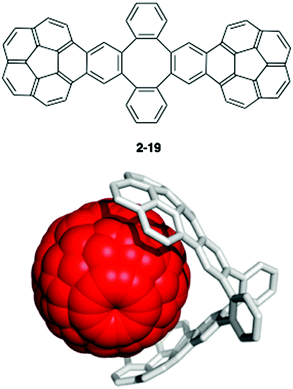 |
| | Fig. 13 Sygula's buckycatcher (2-19) and its complexation with C60. | |
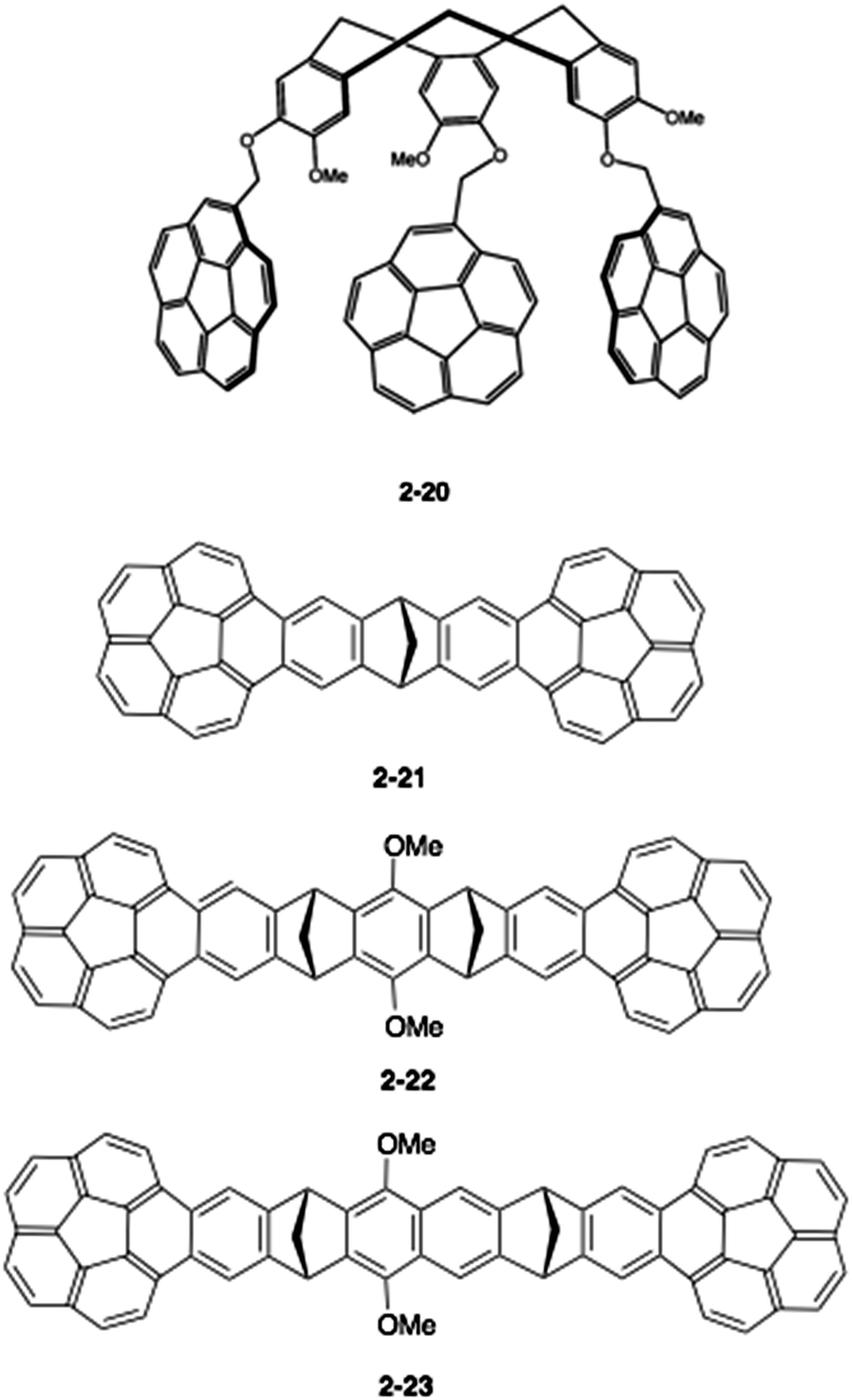
Therefore, a key to the success of capturing fullerenes is the design of the tether units, such as rigidity/flexibility, the number of arms and the type of hand (buckybowls). Sygula's group next synthesized a three-armed flexible host (2-20), but, disappointingly, 2-20 had an inferior interaction to 2-19 for both C60 and C70, (Ka = 1500 ± 50 M−1 and 1180 ± 30 M−1, respectively).81 Then, the group changed the tether to the more rigid dibenzonorbornadiene, and the thus-synthesized “buckycatcher II” (2-21) formed a 2:1 complex with C60 as a nanosized universal joint.82 They also prepared the related host molecules (2-22, 2-23) and carried out comprehensive studies (Chart 8).83,84
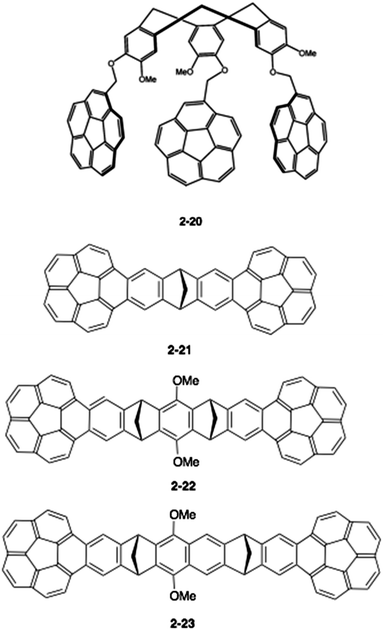 |
| | Chart 8 Second generation buckycatchers. | |
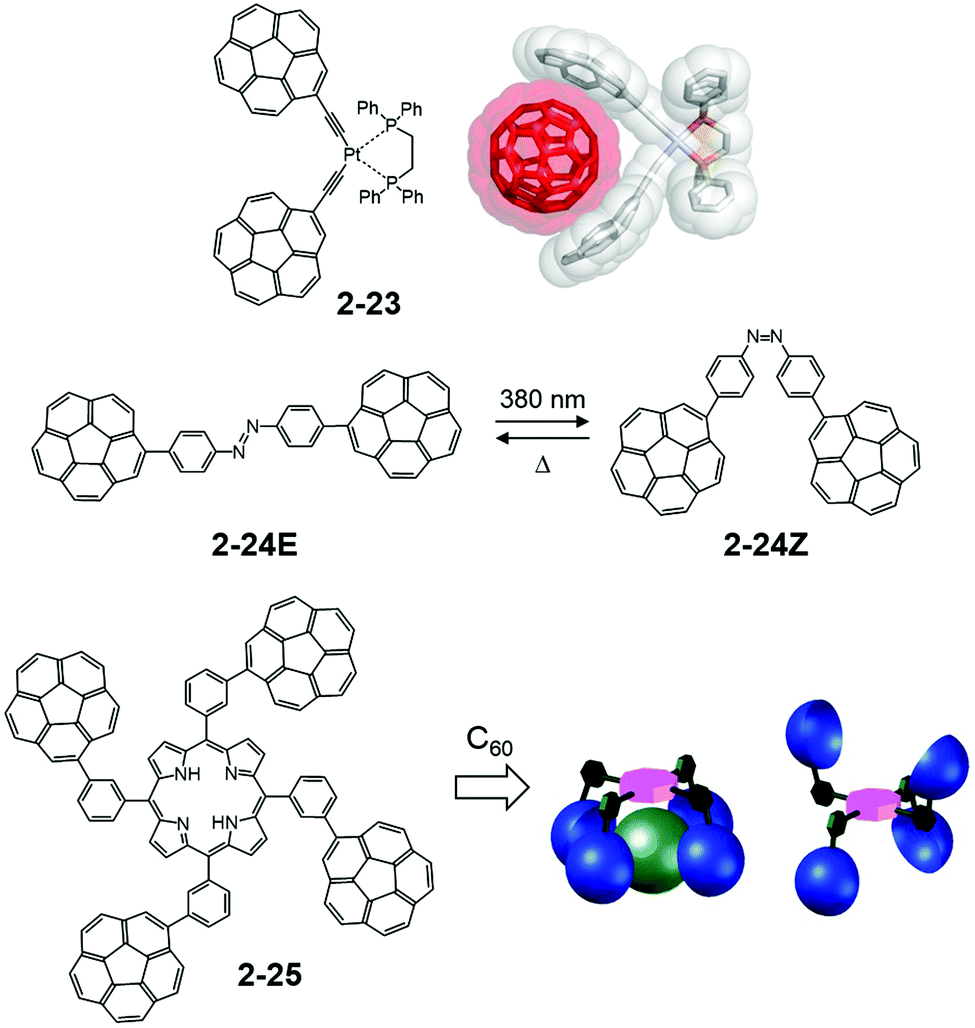
In contrast, Álvarez utilized easy-to-access tethers for the multiple corannulene arms. A square planer platinum complex (2-24) demonstrated an effective ability to capture fullerene both in the solid state and in the solution state—particularly toward C70. The association constant with C70 in toluene-d8 reached up to 20700 ± 600 M−1.85 It is noteworthy that a corannulene “hand” is the most suitable method for capturing C60 compared with other flat or twisted aryl moieties (such as coronene, pyrene, and 6-helicene). The rigid arm structure is also effective. The same group studied the three-armed host using the click chemistry approach,86 diazobenzene-tethered bicorannulene for the light-gated tweezers system (2-25),87 and the tetra-armed system with porphyrins as a tether (2-26).88 Among the four atropisomers distributed statistically, the α4 isomer was found to be an excellent host for C60 (Fig. 14).
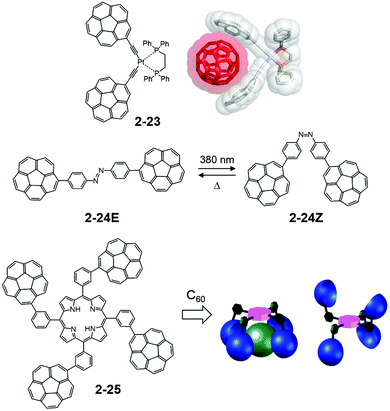 |
| | Fig. 14 Álvarez's C60 capturing systems. | |
A new class of fullerene host is polymer materials. Stuparu reported two different types of polymers for the fullerene receptor. One type is the star polymers with a C5-symmetric corannulene as a core (2-27),89 and another is a block co-polymer of PEG with corannulene-containing methacrylic ester (2-28).90 Both worked as C60 receptors (Chart 9).
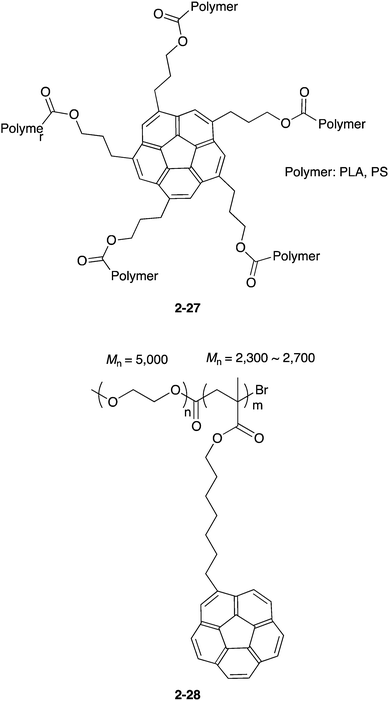 |
| | Chart 9 Stuparu's polymers for C60 capture. | |
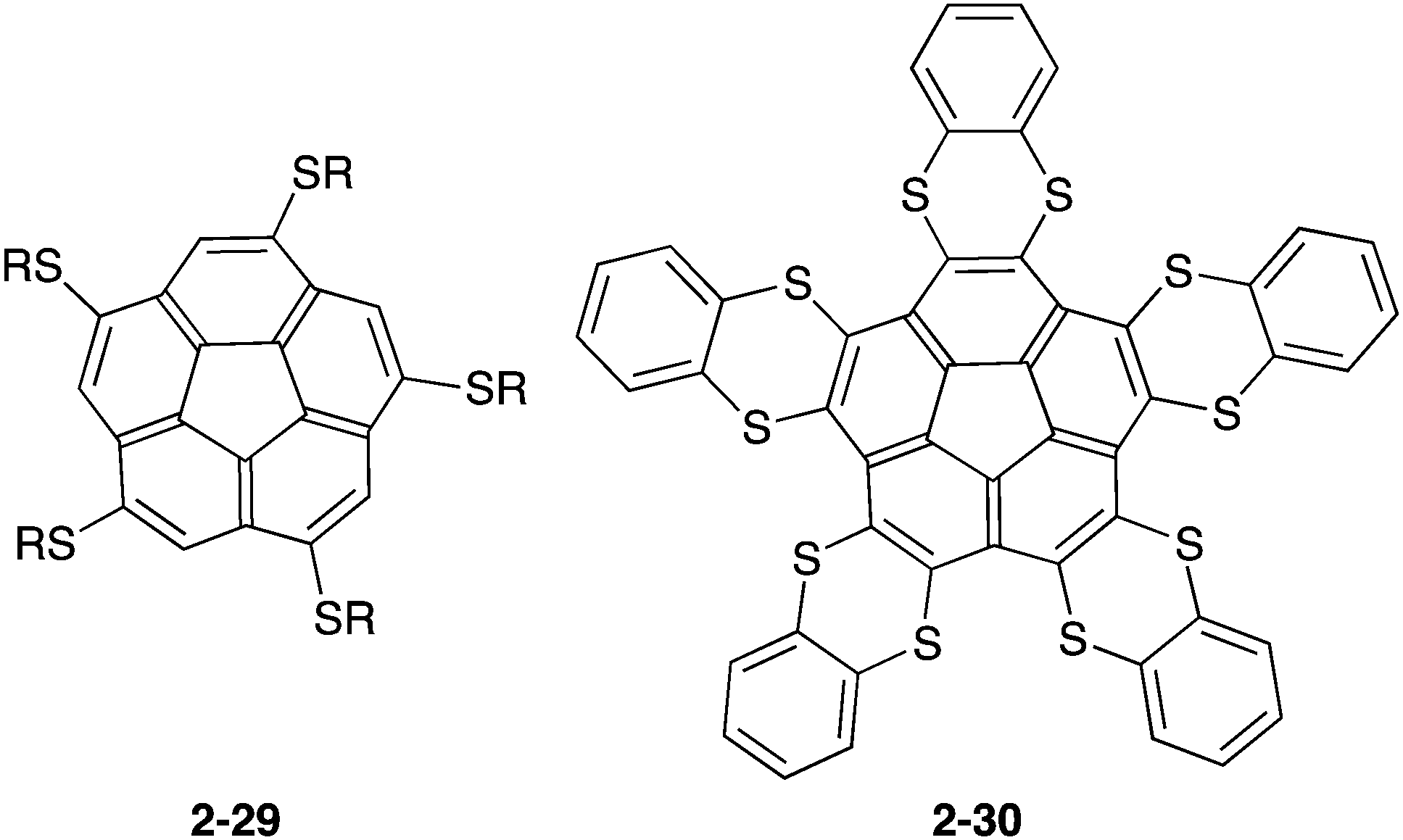
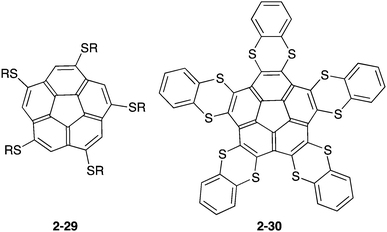
The above-mentioned fullerene hosts possess multiple buckybowl units in order to overcome solvation and the entropy penalty. Only penta- or deca-S-substituted corannulenes (2-29,912-3092), which are more electron-rich corannulenes than the pristine corannulene, were known to form a 1:1 complex in the solution (Chart 10).
 |
| | Chart 10 Electron-rich S-substituted corannulenes. | |
In contrast, in the absence of solvation, the complementary curved-π/curved-π interaction might work more effectively. Indeed, the pristine corannulene formed a complex with C60 in the gas phase,93 and a Cu(110)-surface also supported corannulene with C60.94 In 2012, Dawe and Georghiou demonstrated that 1:1 co-crystallization of pristine corannulene with pristine C60 was possible (Fig. 15).95 After that, Zhang and Hu obtained the one-directional co-crystal of corannulene and fullerene by a solution process, and these nanocrystals displayed high electron transport characteristics of up to 0.11 cm2 V−1 s−1, as well as a good photoresponse of 0.09 A W−1.96 These results strongly indicate that the hybrid system of fullerenes and buckybowls would open the door to wide application in solid-state organic materials.97–99
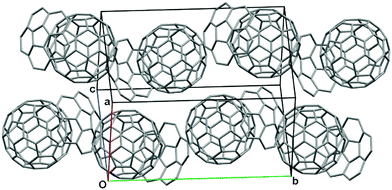 |
| | Fig. 15 Corannulene:C60 = 1:1 co-crystal. | |
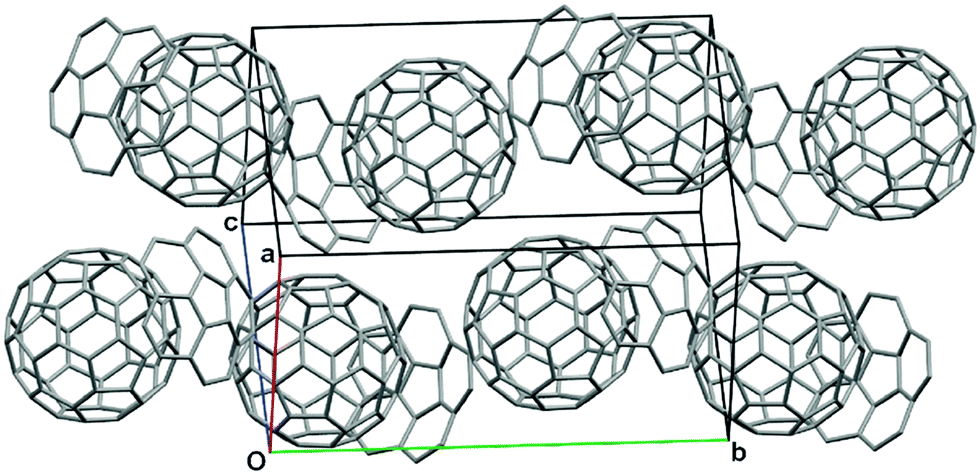
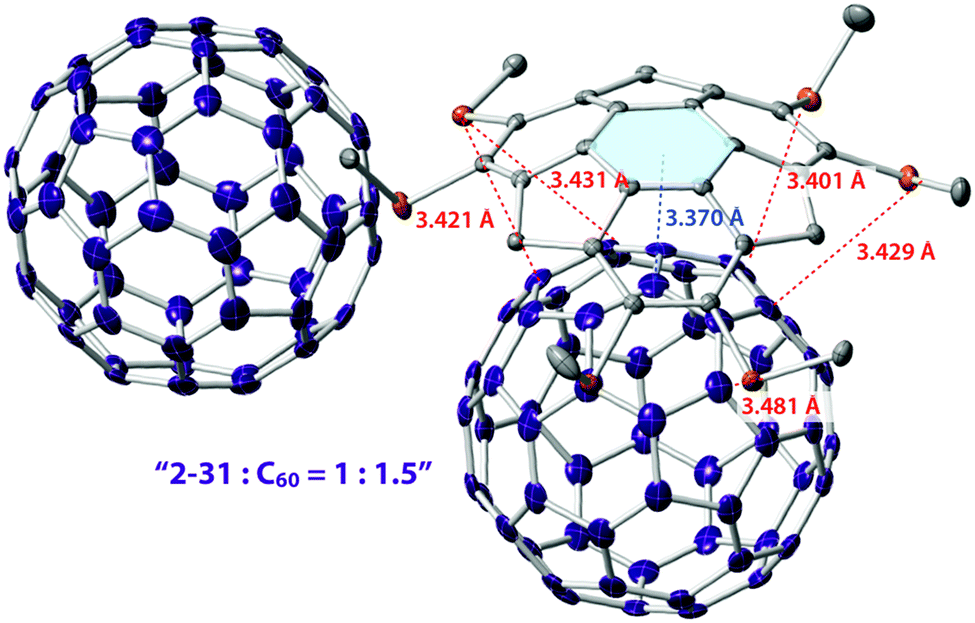
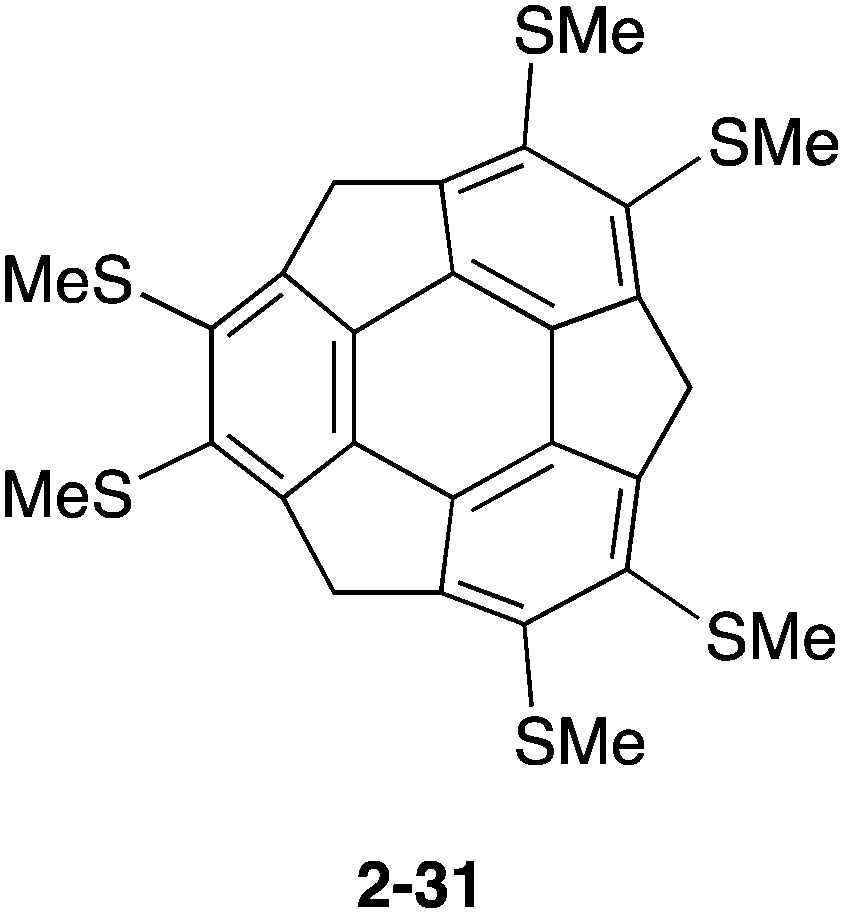
Similar to pristine corannulene, pristine sumanene did not form a complex with fullerenes in the solution state. Fukushima reported the first example of sumanene derivatives that interacted with C60 in a solution state and also formed a co-crystal.100 A hexamethylthiosumanene (2-31), a more electron-rich sumanene than pristine sumanene, formed a 1:1 complex in toluene solution measured by 1H-NMR. In contrast, from a mixture of 2-31 with an equimolar of C60 in toluene, co-crystals were obtained, but at a ratio of 2:3, to constitute a unit cell. In the crystal packing, C60 interacted with a total of 12 methylthio groups of the six neighbouring 2-31 molecules, leading to the formation of a two-dimensional network (Fig. 16 and Chart 11).
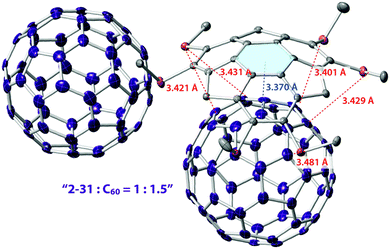 |
| | Fig. 16 Co-crystal structure of 2-31 with C60. | |
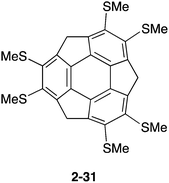 |
| | Chart 11 Hexamethylthiosumanene (2-31) | |
Metal-encapsulated fullerenes enhance electron-accepting properties compared with vacant fullerenes. Li+@C60 is a good substrate to investigate the electron transfer behaviour of the buckybowl and fullerene units. Fukuzumi carried out a comprehensive study on the electron transfer in a charge-transfer complex of corannulenes with Li+@C60.101
2.3 Buckybowls on a metal surface
The two distinct faces of bowl-shaped π-conjugated molecules and their intrinsic electron-accepting property allow us to enjoy the interesting coordination chemistry of metals: the mode of coordination at the concave surface and at the convex surface must be different.
With respect to the property of buckybowls as electron acceptors, multiply-reduced states of buckybowls complexed with metals have been extensively studied (only recent and selected reports are cited).102–111 Also, face-selective π-complexation of transition metals112–116 and transition metal complex-induced self-assembly systems117–120 have also been investigated. However, in this review, we focus specifically on buckybowls on a metal surface. A study of buckybowl behaviour on a metal surface is important for evaluating the geometrical and electronic properties of organic/inorganic interfaces that are relevant to molecular electronics. Furthermore, buckybowl-like fragments are also postulated in the formation of carbon clusters on metal surfaces.121
Fasel and Ernst first reported the adsorption of corannulene on a Cu(110) surface to spontaneously form enantiomorphous 2D lattice structures on the rectangular surface in a bowl-up form.122 It was also shown that the reversible phase transitions easily happened in a monolayer structure.123 The spontaneous bowl-up assembly on the surface motivated investigation into its use as a curved-surface reservoir. Indeed, complexation with C6094 as well as corannulene eclipsed stacking,124 which had been unknown in single crystal form, were observed (Fig. 17). The bowl-down adsorption form was generated when thioether functions were installed on the peripheral position of corannulene.125 In the case of deeper bowls, such as the C70-fragment bowl C38H14 (2-32), the isolated molecule adsorbed in a bowl-up fashion, the same as with corannulenes; however, in the bigger islands, several geometries including the bowl-down and tilted structures were also observed, indicating significant intermolecular interaction through π/π and C-H/π.126
 |
| | Fig. 17 Corannulene on Cu(110) makes a complex with C60. | |
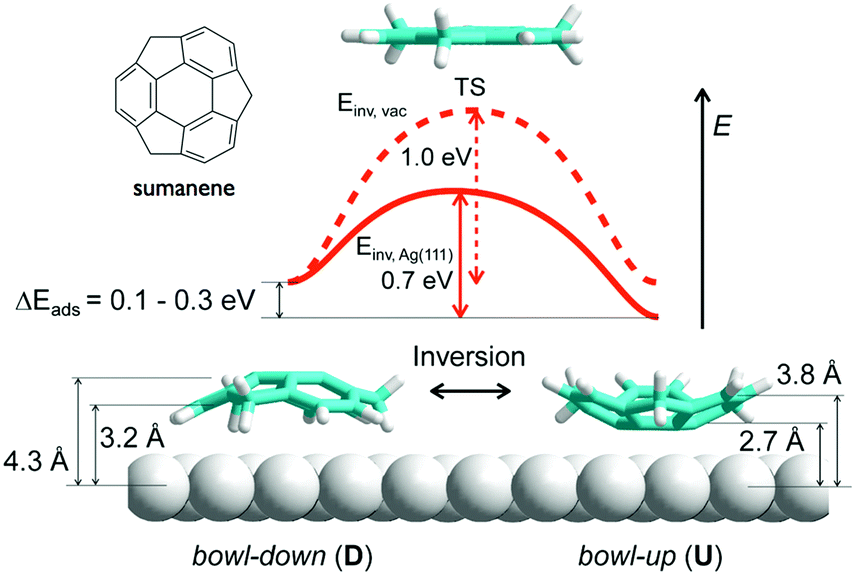
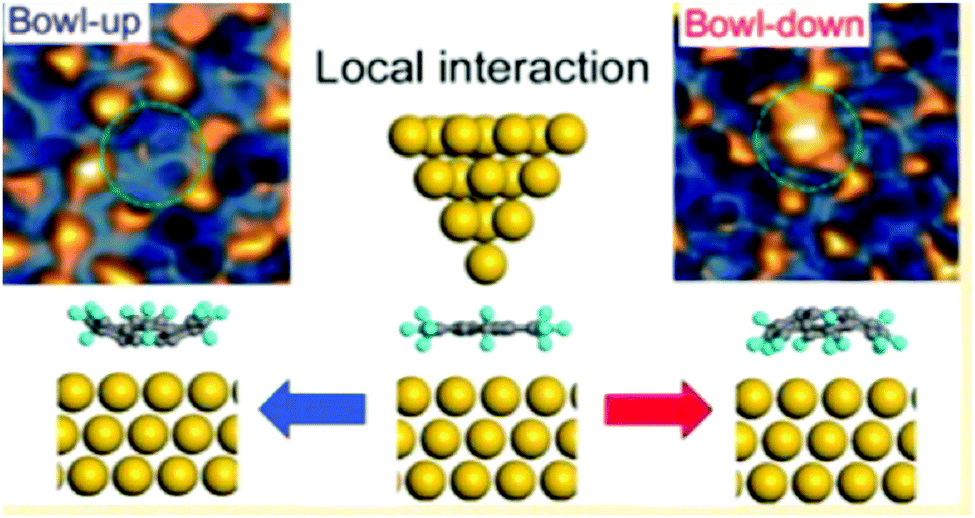
In contrast to corannulene, sumanene not only displayed bowl-up adsorption, but also displayed bowl-down adsorption. In the study of the adsorption on an Ag(111) surface, all sumanene molecules at the smaller islands were aligned as a bowl-up adsorption. However, in the densely packed large islands, significant numbers of sumanene were detected in bowl-down form. The results indicated that one-third of sumanene molecules accumulated strain from the neighbouring molecules, and bowl inversion happened spontaneously to alleviate the strain and stabilize the intermolecular interaction. DFT calculation suggested that the energy barrier of bowl flipping is significantly decreased on metal surfaces (0.7 eV) in comparison with that in a gas phase (1.0 eV) (Fig. 18).127
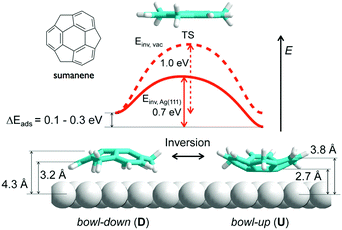 |
| | Fig. 18 Decreasing the bowl-to-bowl energy barrier of sumanene on an Ag(111) surface. | |
In 2016, Fujii and co-workers demonstrated that the bowl inversion of sumanene on a surface can be actively driven by the external force of an STM tip via the sumanene monolayer on Au(111), which was easily prepared by a solution process. By tuning the local metal–organic molecule interaction using the STM tip, the researchers showed sumanene structural bistability with a switching rate that is two orders of magnitude faster than that of the stochastic process. Their results strongly suggest the possibility of molecular-scale memory if the bowl-up or bowl-down state is interpreted as a 0/1 signal (Fig. 19).128
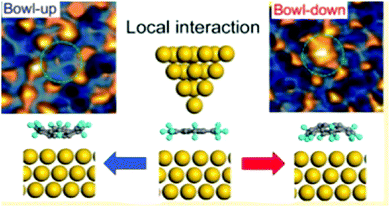 |
| | Fig. 19 Active bowl inversion of sumanene on a surface of Au(111) driven by the external force of an STM tip. | |
The mechanism of the self-assembly of organic molecules is also highly interesting. Indeed, the initial motivation for investigating corannulene adsorption might have been an interest in the mismatch of five-fold molecules, which cannot cover a flat surface. The Ernst group continued to investigate the pentagon-tiling problems and to elucidate several compensation fashions.129–131 With the inherent chirality in the bowl-shape, hemibuckminsterfullerene was displayed for the restructuring of a metal surface in such a way that chirality was imprinted onto the metal. This restructuring is considered as the principal stage of the modification of achiral crystals into a chiral shape.132
2.4 Liquid crystalline and supramolecular chemistry
The unique characteristics of bowl-shaped π-conjugated molecules, such as two distinct π-surfaces, assembly driven by the curved-π/curved-π interaction, and bowl-to-bowl inversion, are fascinating to apply to supramolecular chemistry, especially in anticipation of developing the dynamic and/or stimuli-responsive supramolecule system.
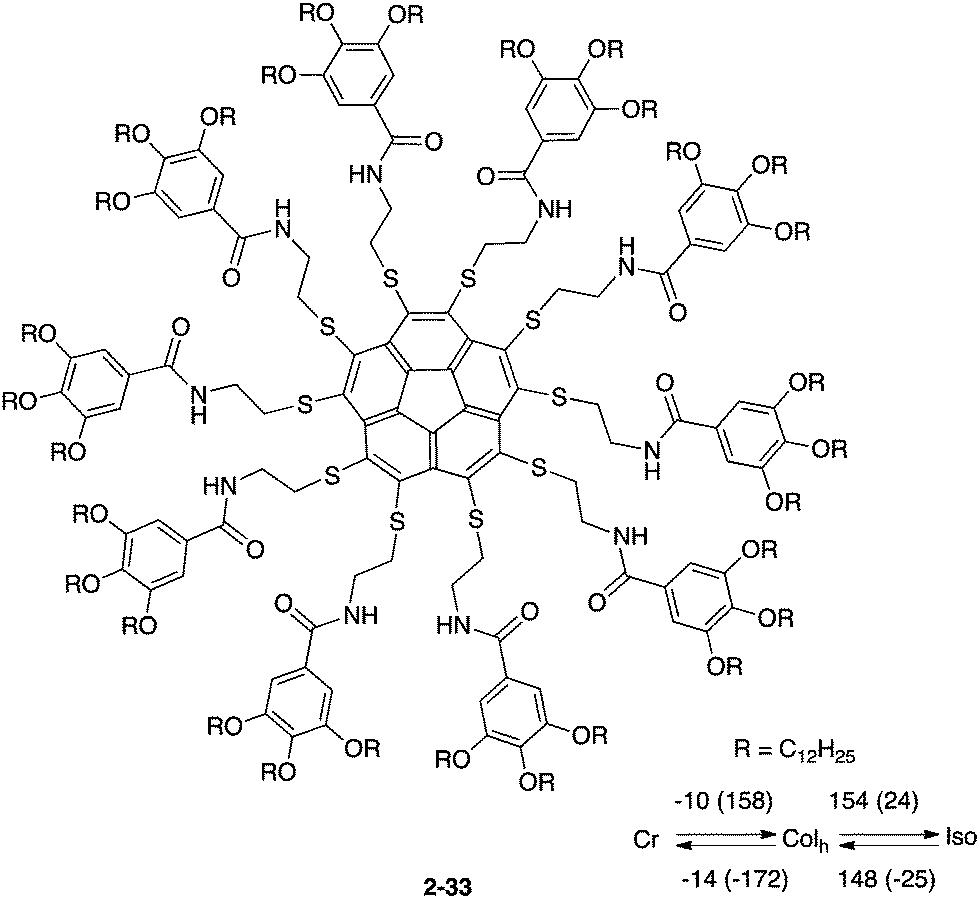
In 2009, Aida and co-workers first reported a liquid crystalline corannulene derivative responsive to an electrical field.133 Because buckybowls possess a dipole moment along the perpendicular axis by their structural demand, ferroelectric properties can be induced if the bowl assembles into a one-dimensional column. Based on that idea, the group prepared a series of deca-substituted corannulene having long alkyl chains, and these showed a liquid crystalline state. Among them, 2-33 formed a hexagonal columnar mesophase over a wide temperature range, including room temperature, and the stacked columns of 2-33 were aligned in response to an applied electrical field (Chart 12).133
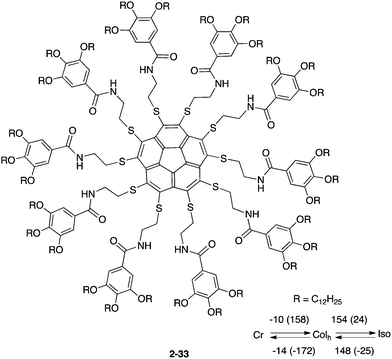 |
| | Chart 12 Corannulene liquid crystals (2-33) responsive to an electrical field. | |
The same group extended the molecular system to realize chain-growth supramolecular polymerization. In this case, C5-symmetric penta-substituted corannulenes (2-34, 2-35) were used as an initiator and a monomer unit, respectively. Both are chiral compounds, and the enantiomers are interconvertible through the bowl-to-bowl inversion process. A key to the success of the chain-growth polymerization is the difference of the hydrogen bonding and the rate of the bowl-to-bowl inversion between 2-34 and 2-35. There is no intramolecular hydrogen bonding in 2-34; therefore, it flips very rapidly even at room temperature, while 2-35 forms strong intramolecular hydrogen bonding that significantly diminishes the rate of bowl inversion and also prevents intermolecular hydrogen bonding. When 2-35 meets 2-34, hydrogen bond reorganization occurs to form the 1:1 complex (initiation step). The resulting 1:1 complex also has five free carbonyl groups, a hydrogen bonding acceptor by the compensation of the original hydrogen bonding with the initiator, and site-selective chain growth through the matched hydrogen bonding network formation (Fig. 20).134
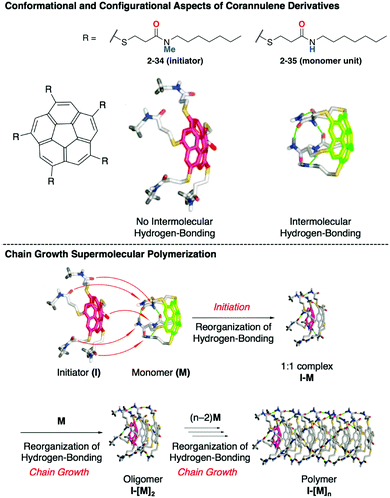 |
| | Fig. 20 Mechanism of the chain-growth supramolecular polymerization of corannulene derivatives. | |
Related to these works, Mahadevegowda and Stuparu prepared corannulene thioether having ethylene glycol units, which showed thermosensitiveness to assemble into larger emissive structures.135,136
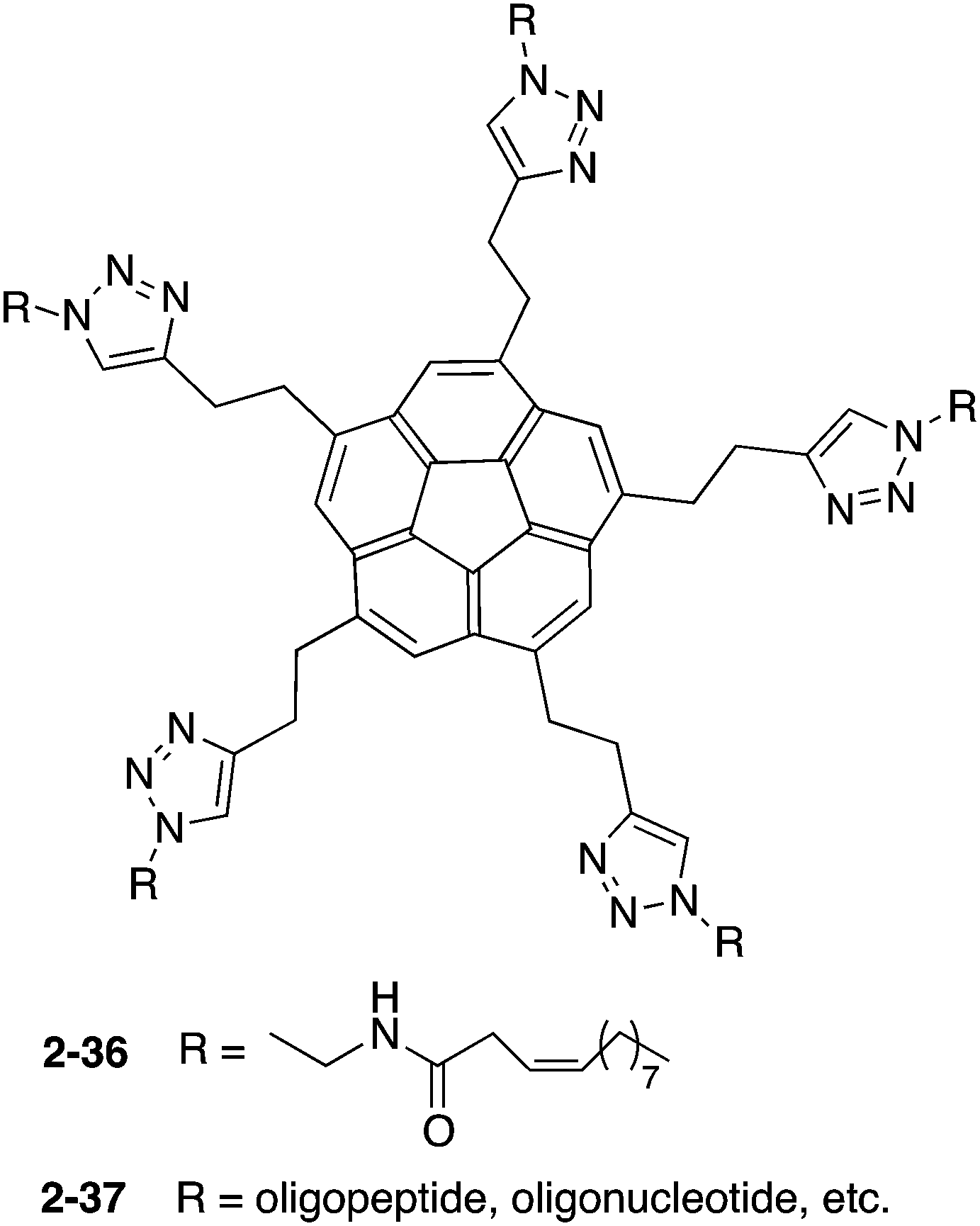
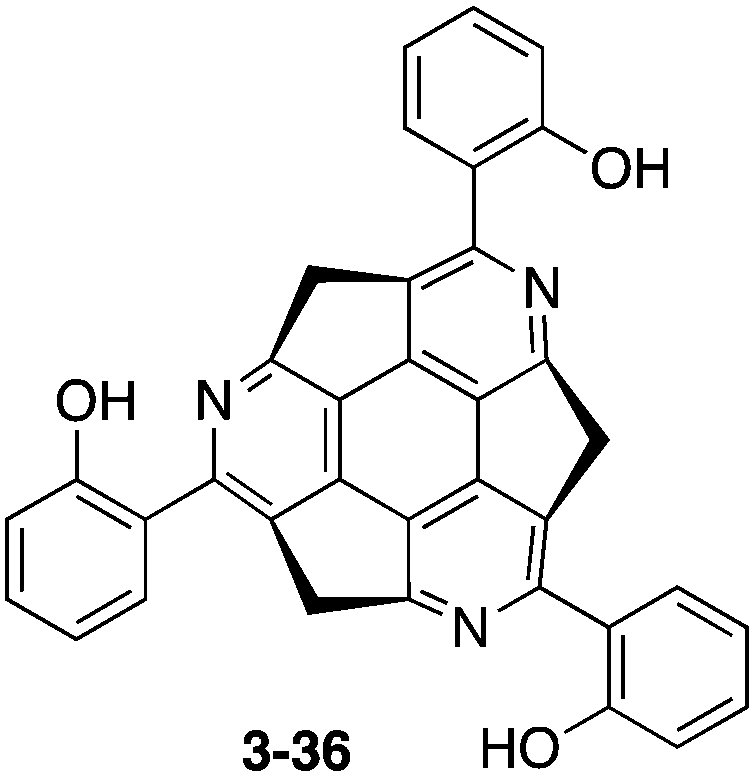
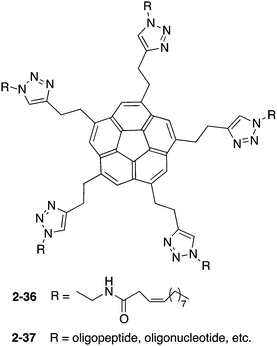
Siegel and co-workers synthesized 2-36, which exhibited gelation properties and a liquid crystalline phase.137 They also prepared 2-37, by changing the chain moieties from the alkyl chain to oligopeptides or oligonucleotides to apply for bioconjugation (Chart 13).138
 |
| | Chart 13 Star-shaped systems for bioconjugation. | |
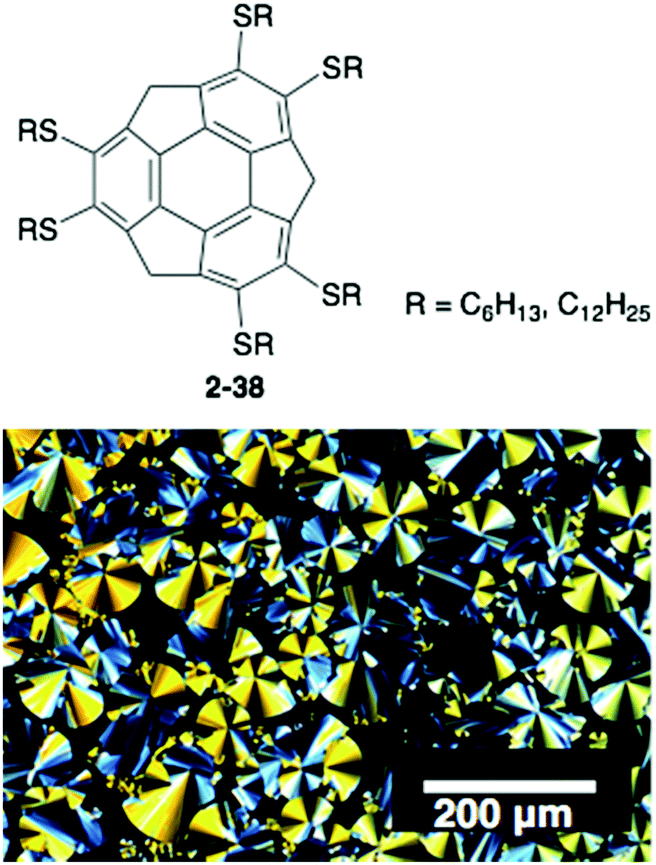
The above examples take advantage of the properties of corannulene; however, the corannulene unit itself does not work as a mesogen, and many side chains are required to generate the LC mesophase. In contrast, very recently sumanene was found to be a potential mesogen with a liquid crystalline state. A series of hexa-alkylthio derivatives of sumanenes (2-38) were synthesized that showed, even 2-38C6, a remarkably high-order columnar LC mesophase over a wide temperature range. The result is encouraging for the development of sumanene-based LC materials (Fig. 21).100
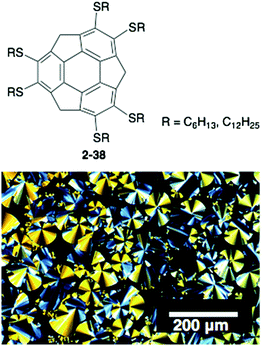 |
| | Fig. 21 Structure and POM image of 2-38C6. | |
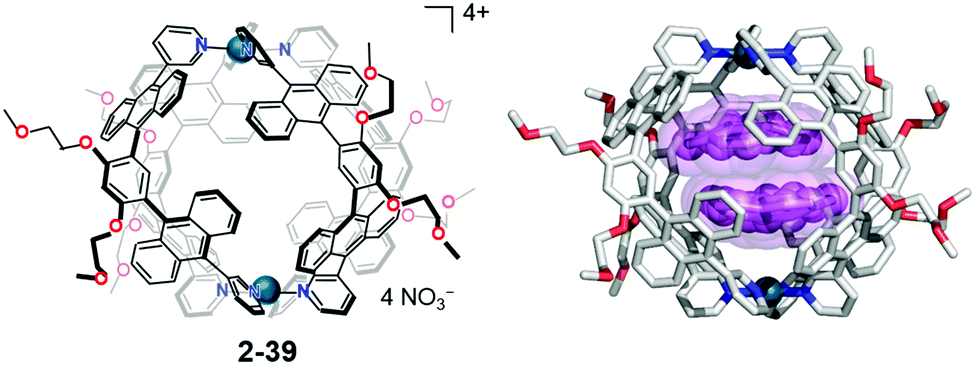
In host–guest chemistry, most of the host molecules as well as the cage compounds consist of flat aromatic molecules; therefore, the resulting void structure should possess a flat boundary. The mismatch of the flat/curve boundary in the restricted cage environments gives rise to a distorted, high-energy state selectively in the cage. Yoshizawa's anthracene-walled PdII-linked M2L4 molecular capsule (2-39) encapsulated two molecules of corannulene in a cage to form an unusual concave–concave dimer structure. Interestingly, 2-38 also formed a selective 2-39:triphenylene:corannulene = 1:1:1 complex (Fig. 22).139
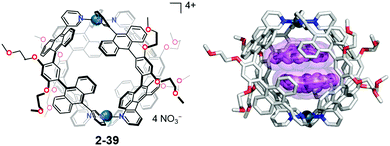 |
| | Fig. 22 Yoshizawa's inclusion complex forms a concave–concave dimer structure of corannulene. | |
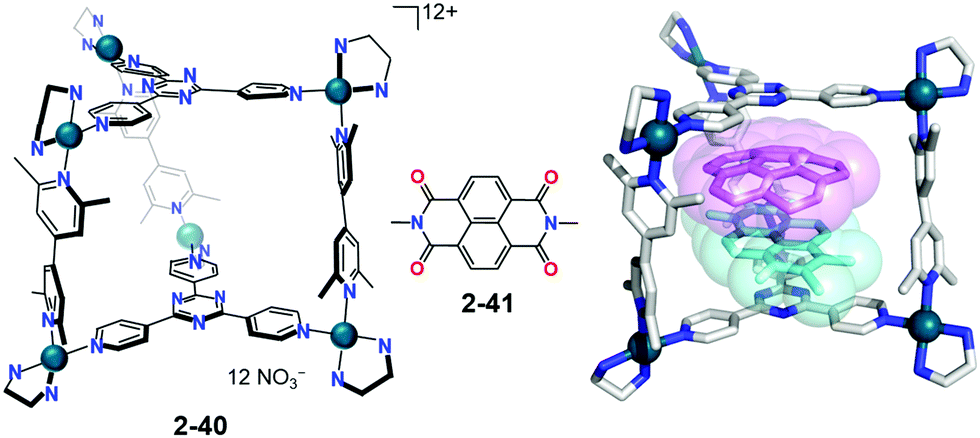
Fujita used his self-assembled coordination cage (2-41) for the co-complexation of corannulene with naphthalene diimide 2-41 based on a prediction that the donor–accepter pairing might assist the co-encapsulation. As expected, the paired inclusion complex showed that corannulene was significantly compressed in a cage of bowl depth from 0.87 Å to 0.56(10) Å. It should be noted that the interaction between corannulene and 2-41 was negligible; instead, corannulene interacted rather strongly with the cage framework, indicating that the metal-coordinated ligand is a better electron accepter (Fig. 23).140
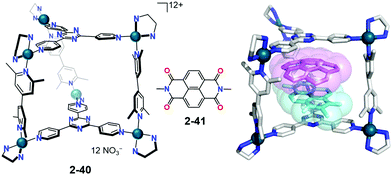 |
| | Fig. 23 Fujita's inclusion complex; corannulene is significantly compressed inside the cage. | |
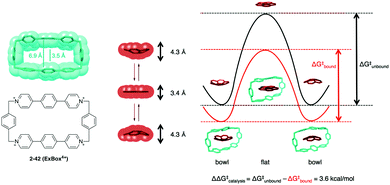
In 2014, Siegel demonstrated a beautiful example of the curved-shape effect in a supramolecular cage, Stoddard's ExBox4+ (2-42) for the induced-fit catalysis of corannulene bowl inversion. The void width of 2-42 is 3.5 Å, which is narrower than the width of the ground state (bowl) of the corannulene (4.3 Å) and fits rather well with that of the transition state (flat) (3.4 Å) for the bowl inversion. Indeed, 2-42 accelerates the bowl inversion of corannulene (3.6 kcal mol−1 smaller) by the effective stabilization of the flat transition state in a cage (Fig. 24).141
 |
| | Fig. 24 Induced-fit catalysis of corannulene bowl-to-bowl inversion. | |
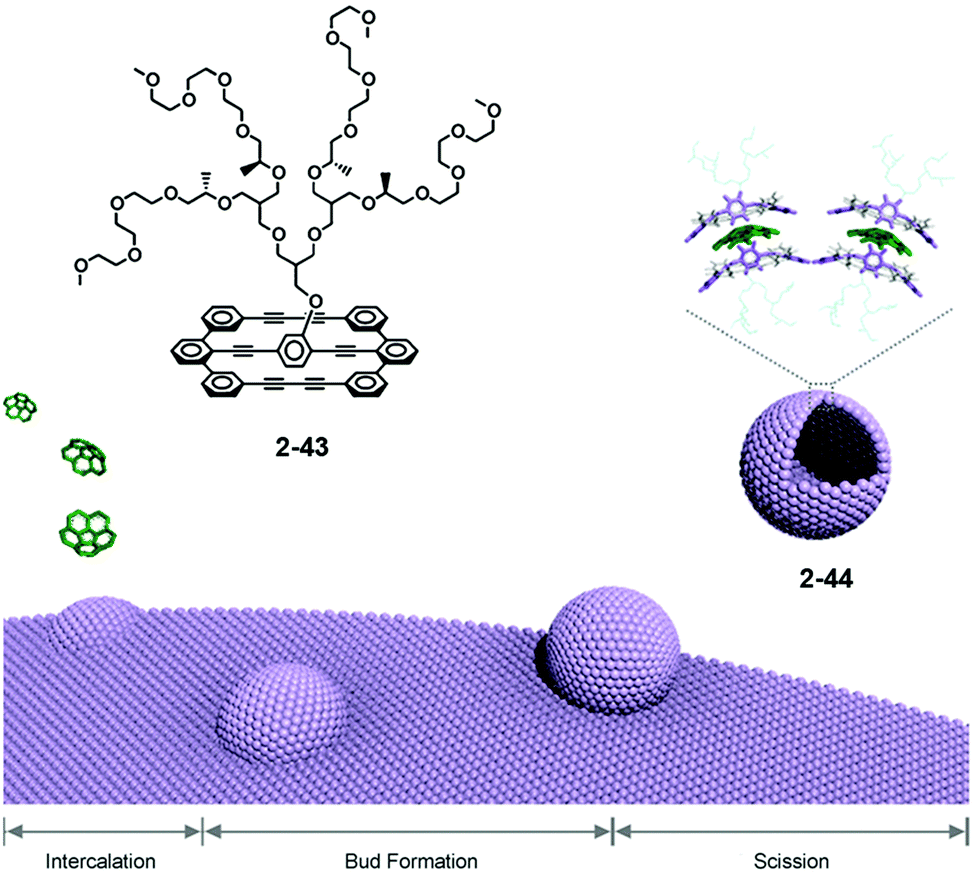
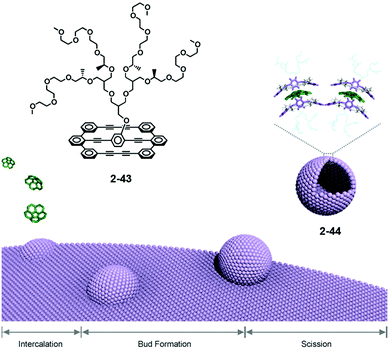
Lee reported the formation of supramolecular capsules from the bilayer membrane driven by the intercalation of corannulene. Effective scission of the bilayered structure of 2-43, followed by the intercalation, occurred to form capsules 2-44 after one month. The curved structure of corannulene might adopt the curved conformation. Consequently, 2-44 could be considered as supramolecular capsules because the framework of the capsule consists of the lateral array of corannulene. 2-44 showed interesting photocatalytic activity to degrade encapsulated fluorescein dye under sunlight irradiation (Fig. 25).142 Recently, Stuparu reported evidence of a 1:1 inclusion complex of corannulene with γ-CD.49
 |
| | Fig. 25 Intercalation-driven capsule formation by corannulene. | |
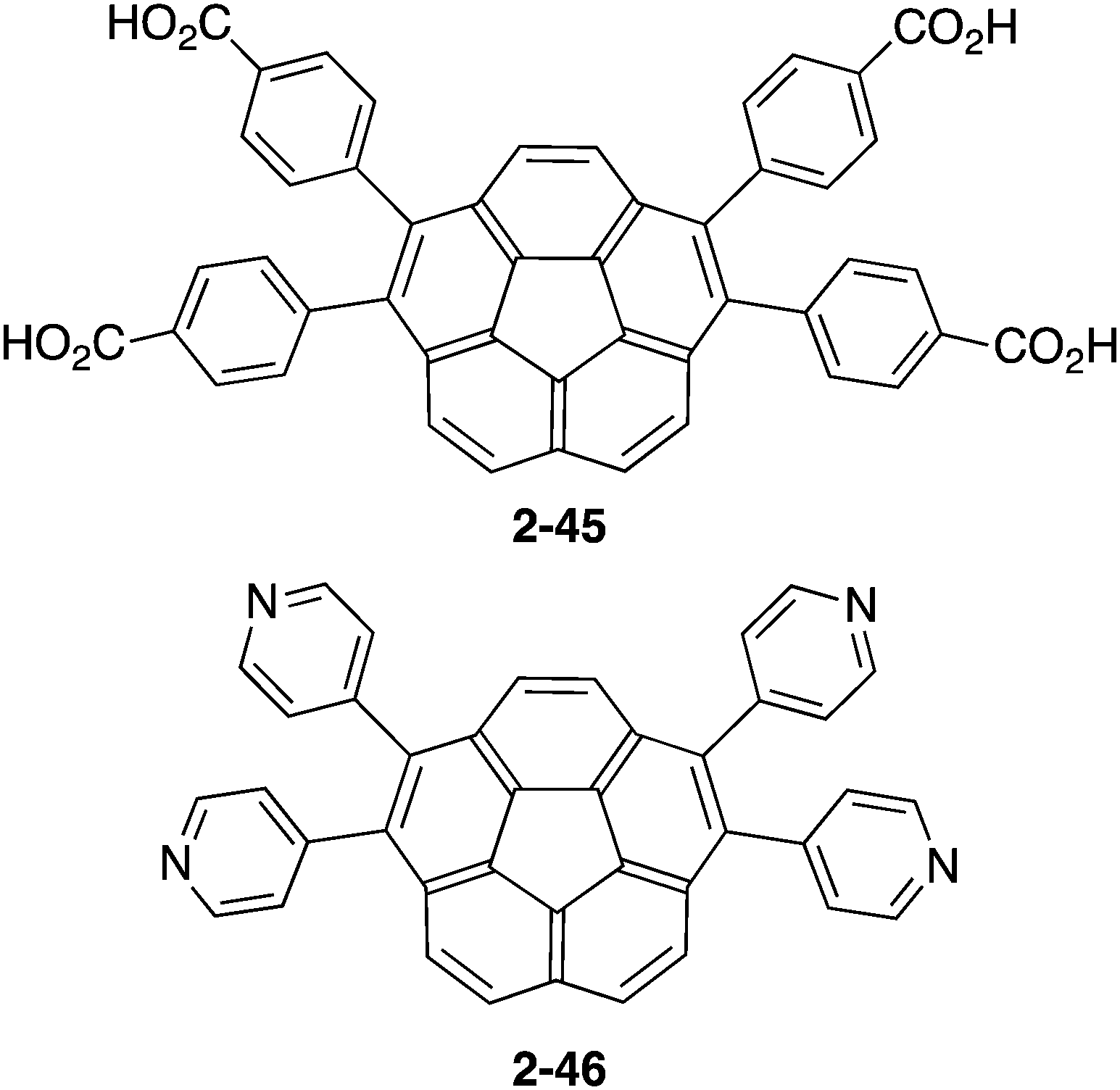
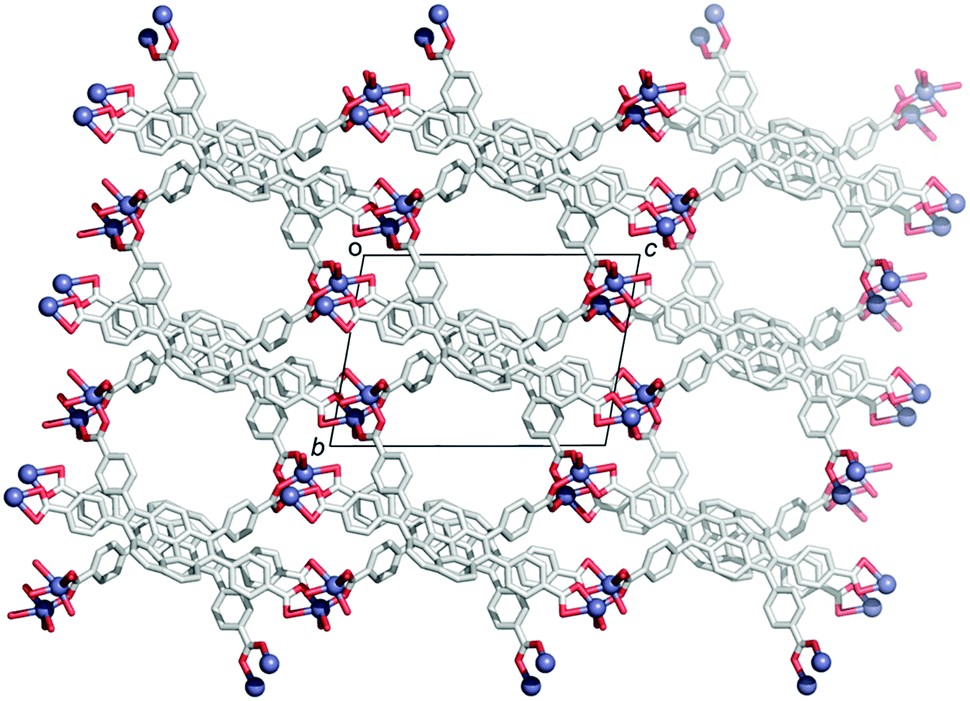
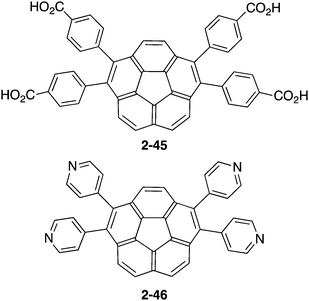
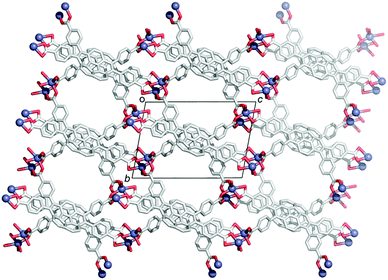
In contrast to the many examples of buckybowls as guest molecules, studies on the buckybowl-containing cage with the curved void are still rare. The first example of the buckybowl-containing metal organic framework (MOF) was reported by Shustova in 2015. Corannulene derivatives having a four-carboxyl group (2-45) formed the MOF structure by the reaction with Zn(NO3)2 in DMF/EtOH mixed solvent. X-ray analysis revealed that the structure of 2-45MOF formed a two-dimensional network, and the distortion of the corannulene unit was negligible. The thus-formed 2-45MOF exhibited superior electron accepting properties and photoluminescence quantum yield in comparison with the unmodified corannulene (Chart 14 and Fig. 26).143
 |
| | Chart 14 Corranulene derivatives (2-45, 2-46) for MOF ligands. | |
 |
| | Fig. 26 Structure of 2-45MOF. | |
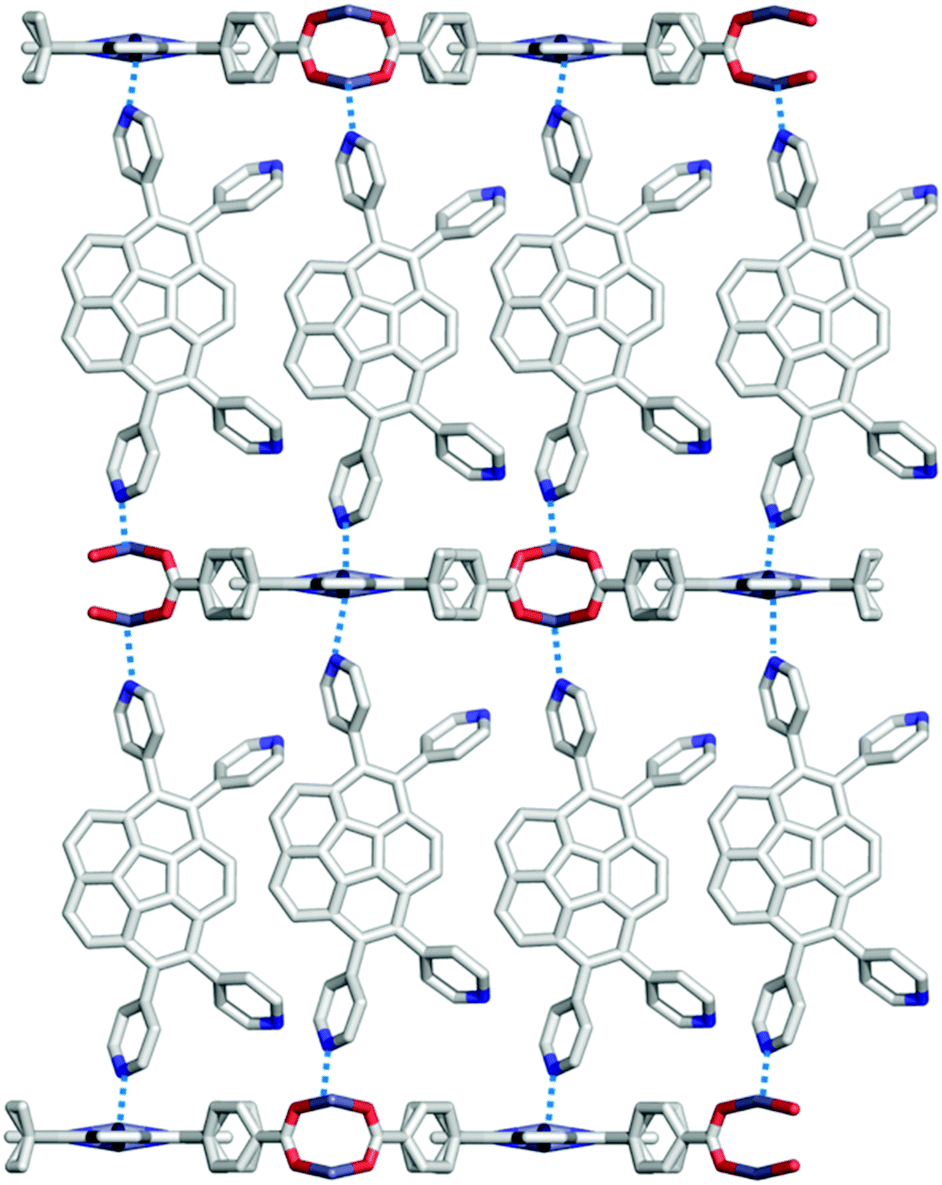
Next, Avdoshenko and Shustova synthesized corannulene with a four γ-pyridyl group (2-46), and made various types of donor–acceptor complexes with metal complexes including the network structures. These complexes exhibited rapid ligand-to-ligand energy transfer as well as solid-state photoluminescence. These research studies clearly demonstrated the potential application of the intrinsic properties of buckybowls in solid-state materials (Fig. 27).144
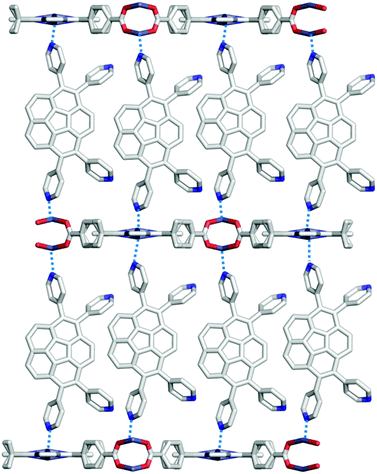 |
| | Fig. 27 Shustova's donor–acceptor complex system. | |
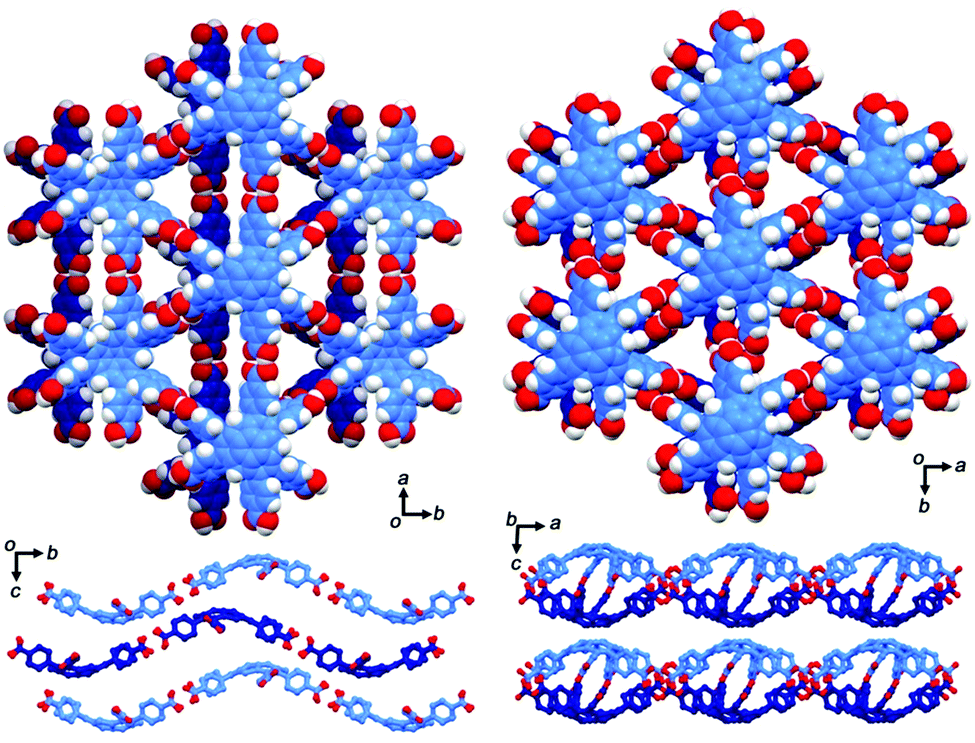
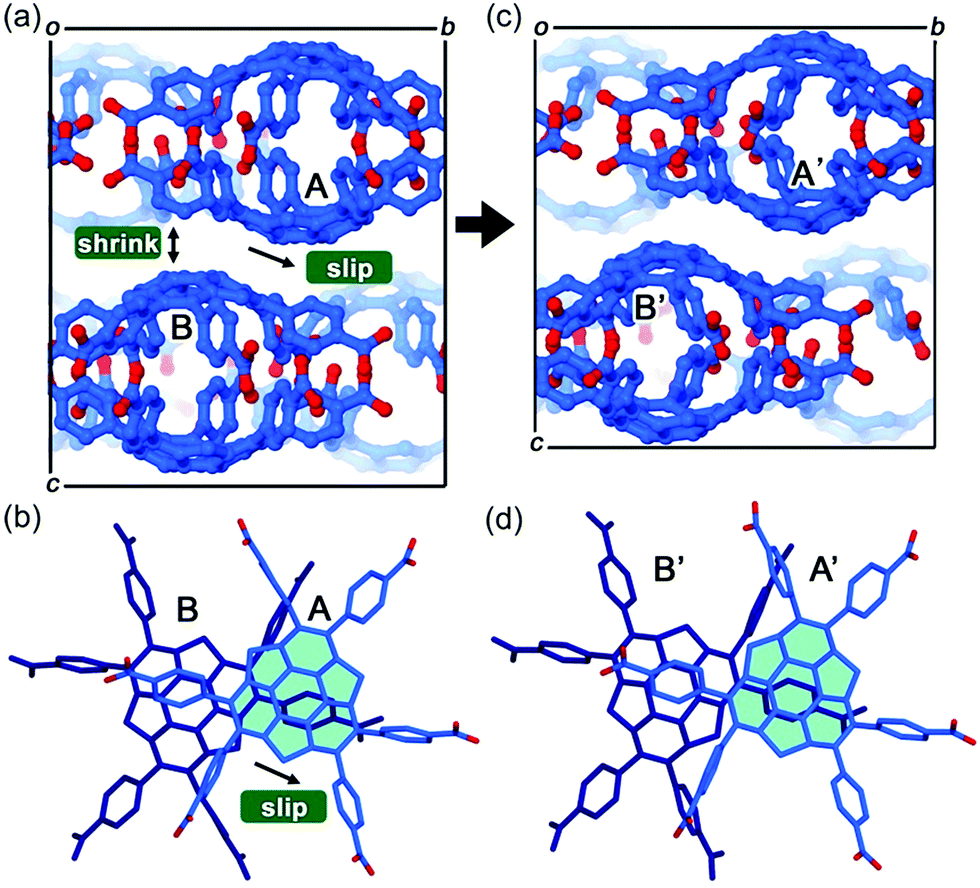
The first example of the sumanene-containing hydrogen-bonded network system was reported by Hisaki and co-workers. Similar to the hexagonal network (HexNet) formation from the triphenylene-based hexacarboxylic acid unit, the core unit was reformed from triphenylene to sumanene (2-47). Two distinct network structures, waved HexNet (2-48) and bilayered HexNet (2-49), were obtained. In 2-48, the sumanene bowl was shallower than the pristine sumanene; however, in 2-49, the bowl shape was nearly the same as the pristine structure. Noteworthy is that 2-49 underwent anisotropic shrinking along the c-axis by 11% under high pressure conditions (970 MPa). The shrinkage was caused by the offset sliding between the spherical π-surfaces of the bilayered sheets (Fig. 28 and 29).145
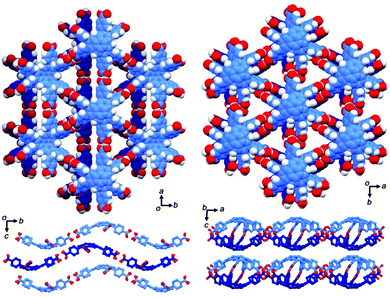 |
| | Fig. 28 Structures of 2-48 (left) and 2-49 (right). | |
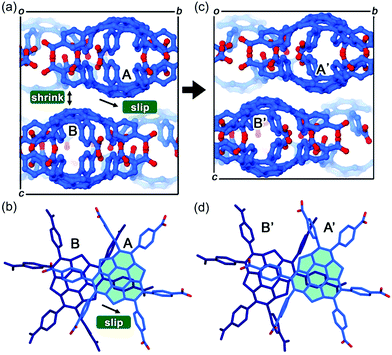 |
| | Fig. 29 Mechanism of the anisotropic shrinking under pressure. | |
3. Nitrogen-containing bowl-shaped π-systems
3.1 Subphthalocyanines
Bowl-shaped π-systems with nitrogen atoms in their conjugation networks have been known for a long time since the discovery of subphthalocyanines (Chart 15).146 Subphthalocyanine 3-1 is a ring-contracted analogue of phthalocyanine consisting of three isoindoline units and one boron atom in its centre. Subphthalocyanines are synthesized efficiently in good yields through cyclotrimerization of phthalonitrile precursors in the presence of boron trihalides. Subphthalocyanine derivatives have found a number of potential applications such as in OLEDs, OFETs, photovoltaic cells, information storage, nonlinear optics and sensors. The syntheses and applications of subphthalocyanines have been summarized in excellent reviews.147–149
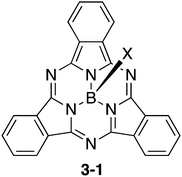 |
| | Chart 15 Subphthalocyanine. | |
3.2 Subporphyrins
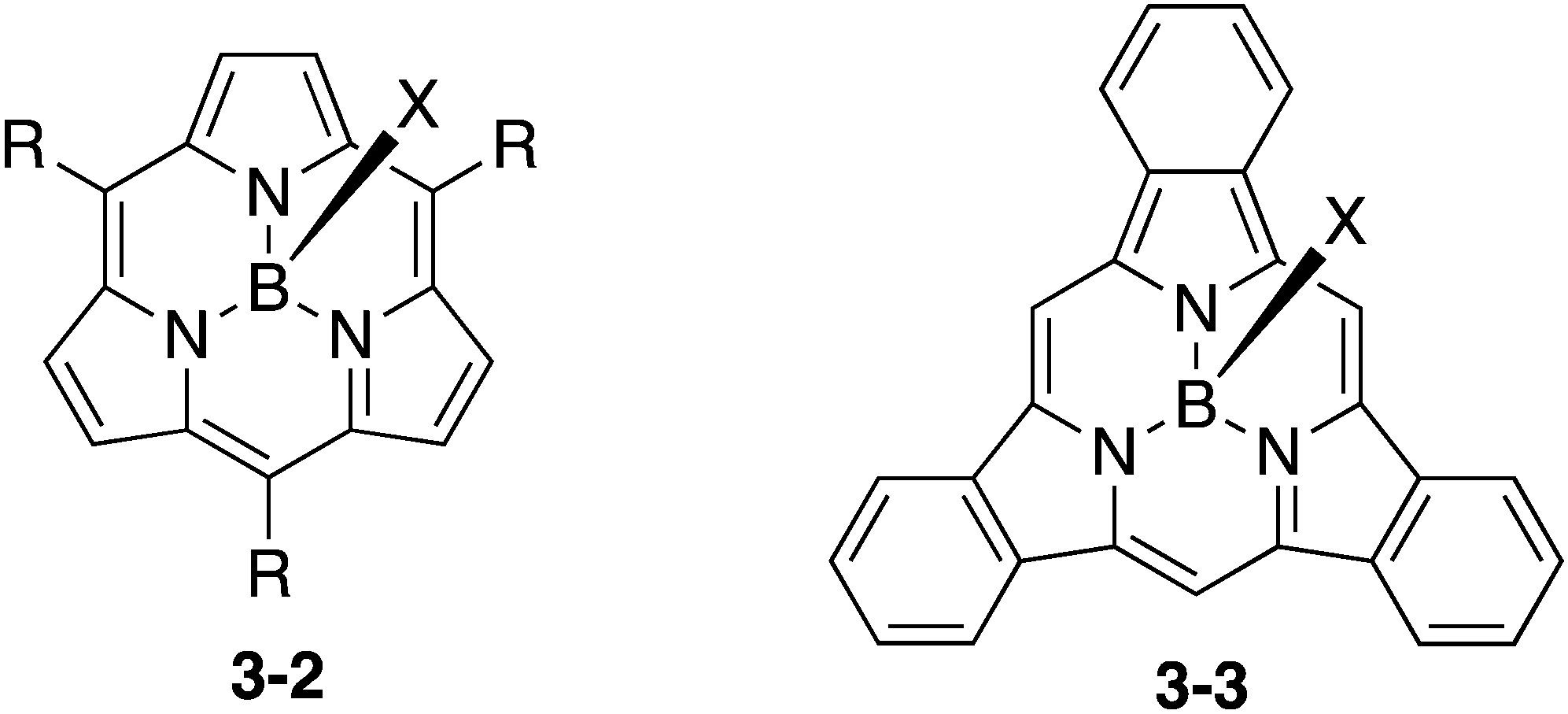
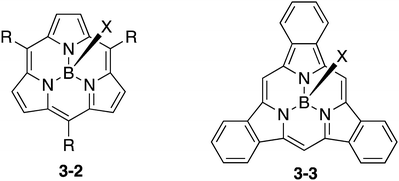
Subporphyrin is a type of ring-contracted porphyrin, which consists of three pyrrole units and three bridging methine carbon atoms (Chart 16). Consequently, subporphyrins exhibit distinct aromaticity due to their 14π electronic conjugation circuits. In sharp contrast to the rather facile preparation of subphthalocyanines, the synthesis of subporphyrin has been challenging and has been achieved only recently by Osuka and Kobayashi, independently.150–153 Similar to subphthalocyanines, subporphyrins 3-2 and 3-3 are also obtained as boron complexes, which adopt bowl-shaped structures. None of the free-base form of subporphyrin has been isolated to date. The use of subporphyrins for fullerene-binding and sensors has been investigated. The syntheses and properties of subporphyrins have been recently reviewed.154–156 In comparison to subphthalocyanines, however, the applications of subporphyrins have been relatively unexplored. Considering the wide applicability of porphyrin analogues, subporphyrins also have huge potential for the future.
 |
| | Chart 16
meso-Substituted subporphyrin and tribenzosubporphyrin. | |
3.3 Norcorrole
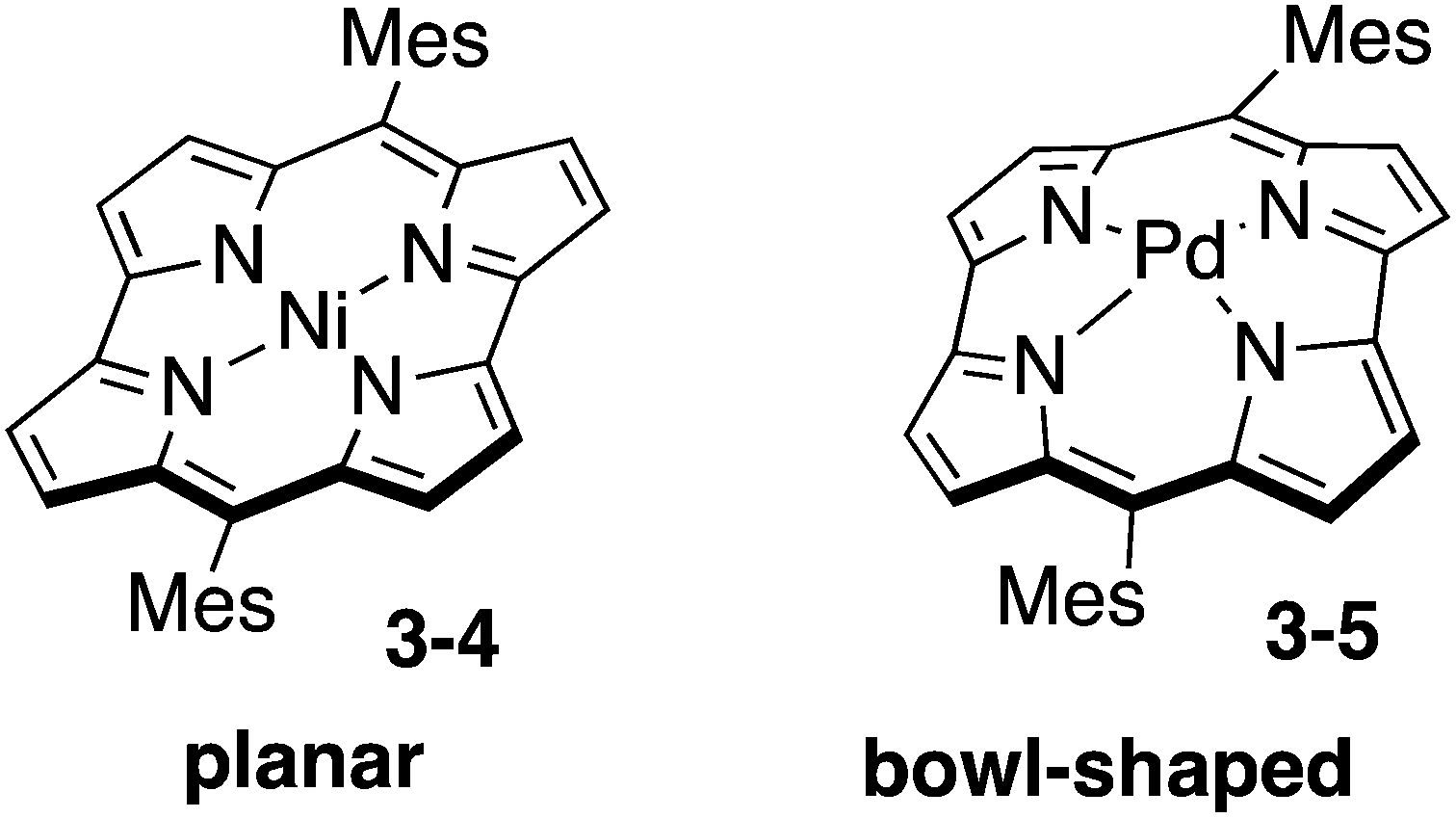
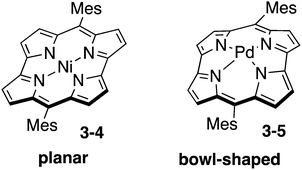
Norcorrole is one of the ring-contracted porphyrins, which lacks two carbon atoms at the meso-positions from regular porphyrin (Chart 17). Because of 16 π-electrons in their cyclic conjugation and highly planar conformation, norcorrole Ni(II) complex 3-4 exhibits distinct antiaromatic character.157 Furthermore, norcorrole Ni(II) complex 3-4 shows intriguing properties and functions originating from their small HOMO–LUMO gap. Recently, norcorrole 3-4 have been investigated as electrode active materials in secondary battery.158 The high single-molecule conductance of a norcorrole Ni(II) complex has been recently revealed.159
 |
| | Chart 17 Ni(II) norcorrole and Pd(II) norcorrole. | |
Insertion of a large Pd(II) cation into the small central cavity of a free-base norcorrole induced severe distortion to result in the formation of bowl-shaped norcorrole Pd(II) complex 3-5.160 Interestingly, norcorrole Pd(II) complex 3-5 maintained strong antiaromaticity in spite of its nonplanar distorted structure. Norcorrole Pd(II) complex 3-5 also exhibited bowl-to-bowl inversion behaviour and face-dependent magnetic properties.
3.4 Azacorannulenes
Nitrogen-embedded bowl-shaped molecules have been receiving much attention as model compounds for azafullerenes and the end-cap structure of nitrogen-doped carbon nanotubes. In addition, drastic changes in electronic properties of buckybowls by nitrogen atoms are expected. However, incorporation of nitrogen atoms into the skeleton of corannulene has been quite challenging due to the reactive nature of heteroatoms and heteroatom-containing functionalities. Recently, Scott summarised their long-term campaign in which the Scott group tackled the synthesis of azacorannulenes.161
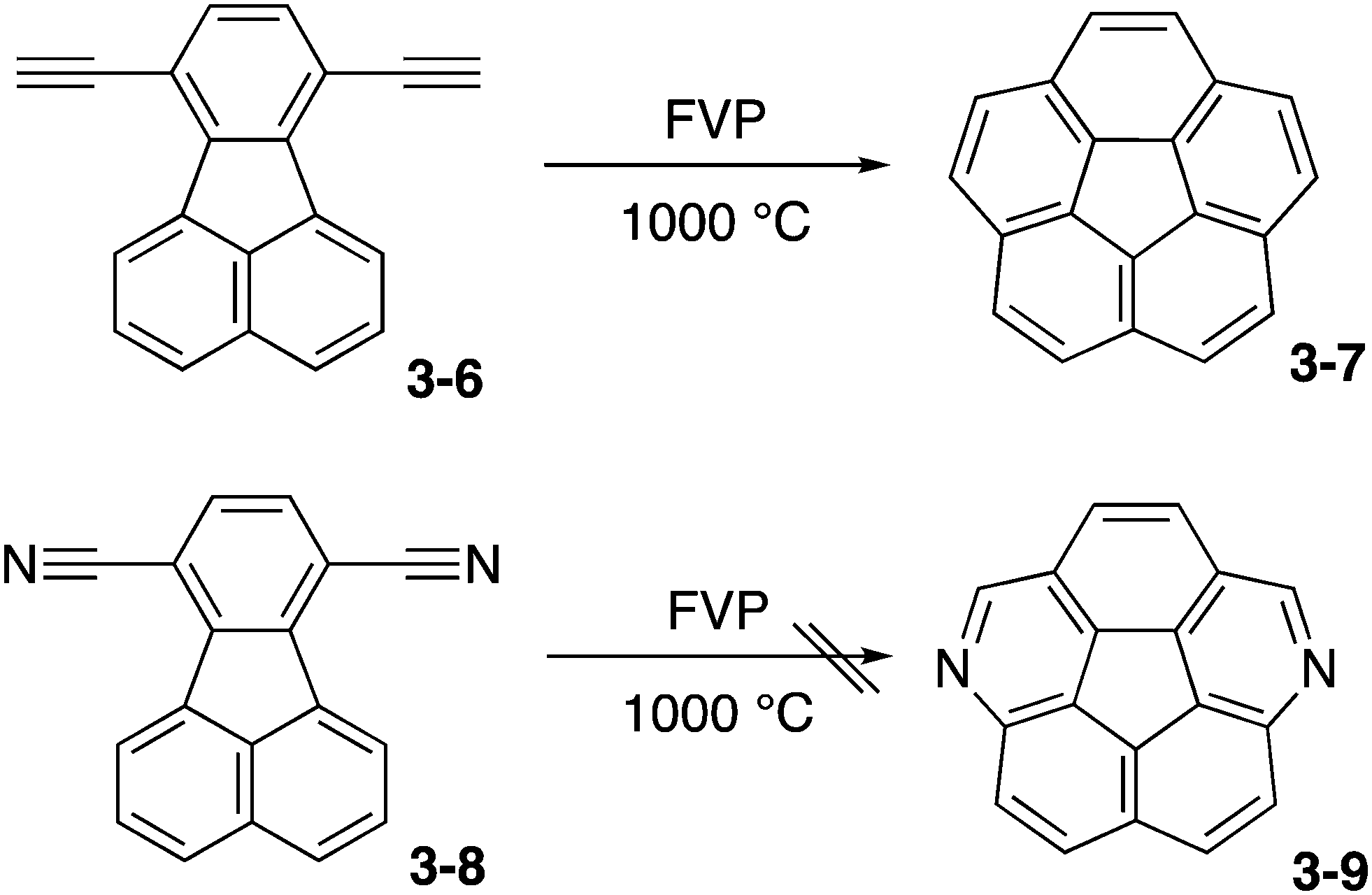
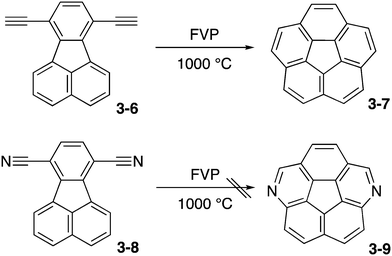
The first challenge was flash vacuum pyrolysis (FVP) of dicyanofluoranthene 3-8 aiming at diazacorannulene 3-9 (Scheme 2). This strategy was based on their successful synthesis of corannulene 3-7 from diethynylfluoranthene 3-6.162 However, the conversion is energetically unfavorable and the reaction did not work.
 |
| | Scheme 2 Challenge toward diazacorannulene 3-9. | |
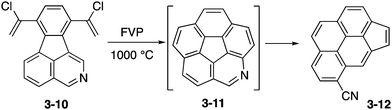
Then Scott and co-workers attempted FVP of a chlorovinylated precursor 3-10 containing an isoquinoline moiety (Scheme 3). In this case, azacorannulene 3-11 was probably formed but the pyridine moiety in the initial product was thermally labile to undergo the ring opening reaction to nitrile 3-12. This example highlights the difficulty in the synthesis of thermally unstable distorted molecules containing heteroatoms.
 |
| | Scheme 3 Challenge toward azacorannulene 3-11. | |
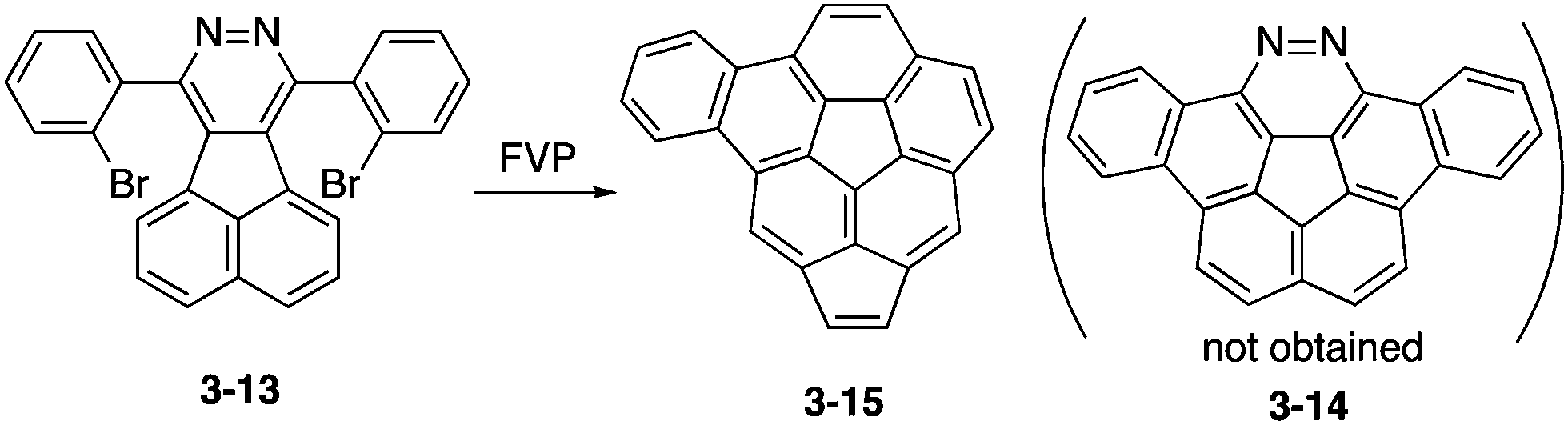
To prepare 1,2-diazacorannulene, Scott, Bodwell and co-workers attempted FVP of acenaphtho[1,2-d]pyridazine 3-13 into diazadibenzocorannulene 3-14 (Scheme 4).163 However, under harsh conditions of the FVP process, nitrogen atoms were extruded and benzoacenocorannulene 3-15 through unexpected rearrangement was obtained instead, which was also an interesting π-expanded corannulene.
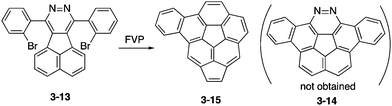 |
| | Scheme 4 Challenge toward diazadibenzocorannulene 3-14. | |
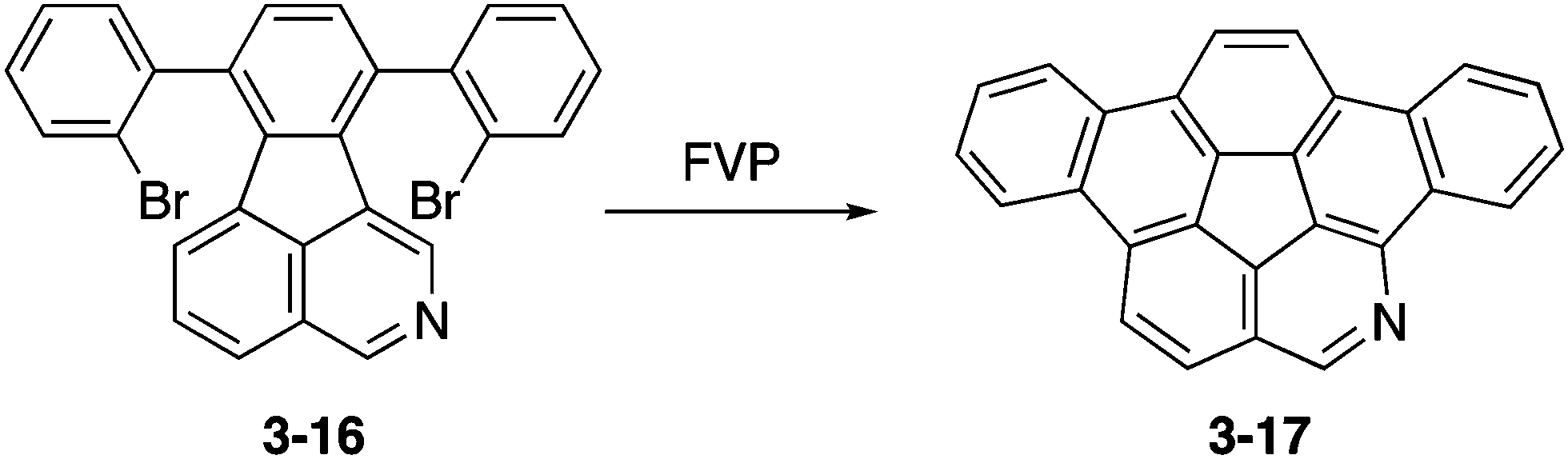
Eventually, peripherally nitrogen-embedded azadibenzocorannulene 3-17 was successfully prepared in 28% yield through FVP of a dibromophenylated precursor 3-16 at 1000 °C (Scheme 5).161 Unfortunately, the obtained azabuckybowl 3-17 was not very stable toward silica gel purification and the ring-opening product amino aldehyde was formed through acid-induced hydrolysis. Consequently, improvement of the synthesis and stability of azacorannulenes with pyridine moieties in the structure are required to explore the properties and applications of these molecules.
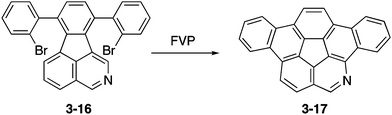 |
| | Scheme 5 Synthesis of azadibenzocorannulene 3-17. | |
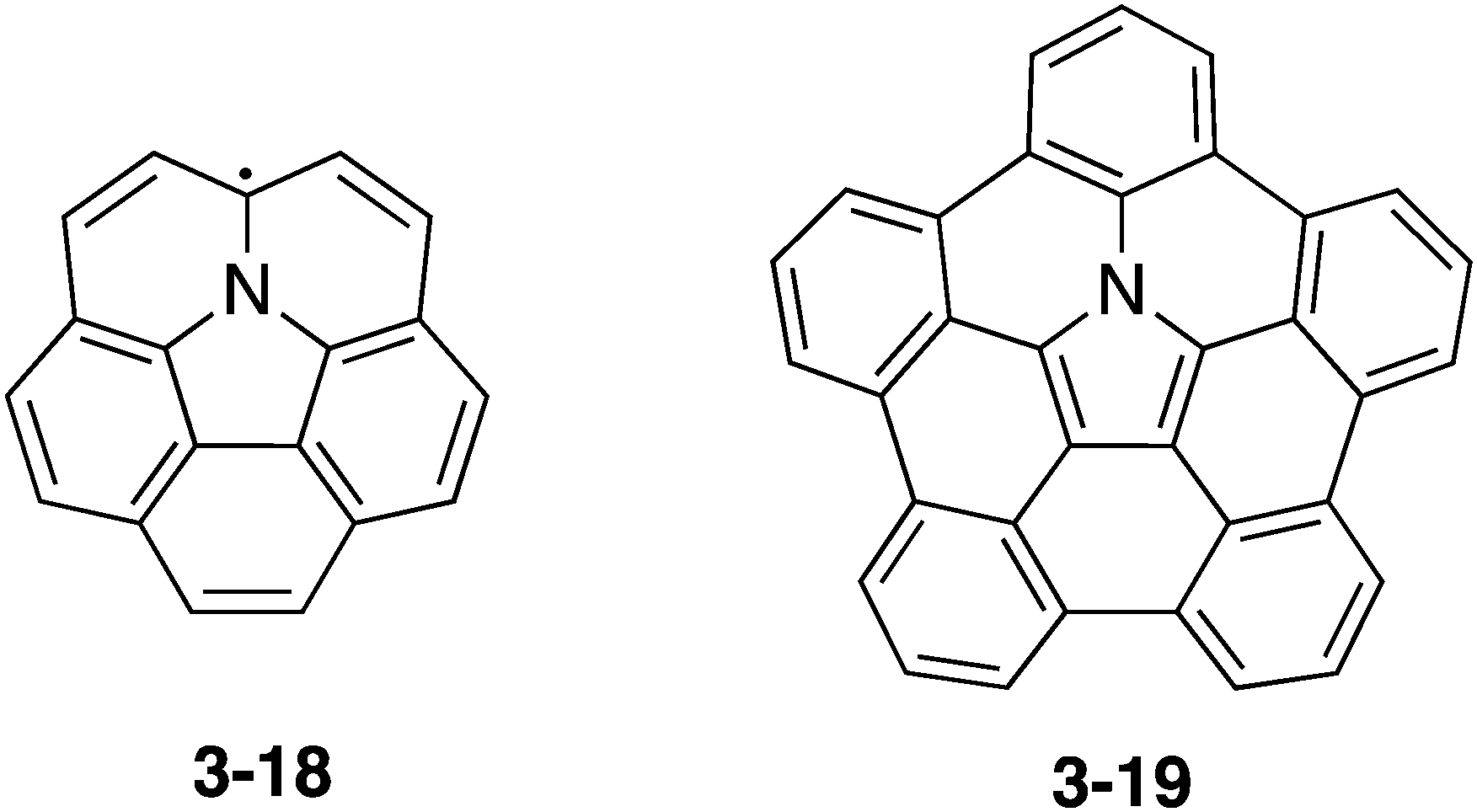
As an azacorannulene bearing a nitrogen atom in the core structure of corannulene, one can consider azacorranulene 3-18 (Chart 18). However, azacorranulene 3-18 should have an open-shell electronic configuration in its neutral state. Peripheral modification of 3-18 by five fused benzo units should provide crossed-shell nitrogen-doped corannulene 3-19. However, the synthesis of this molecule was also difficult.
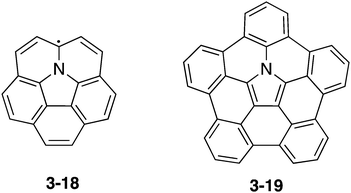 |
| | Chart 18 Azacorannulene 3-18 and pentabenzoazacorannulene 3-19. | |
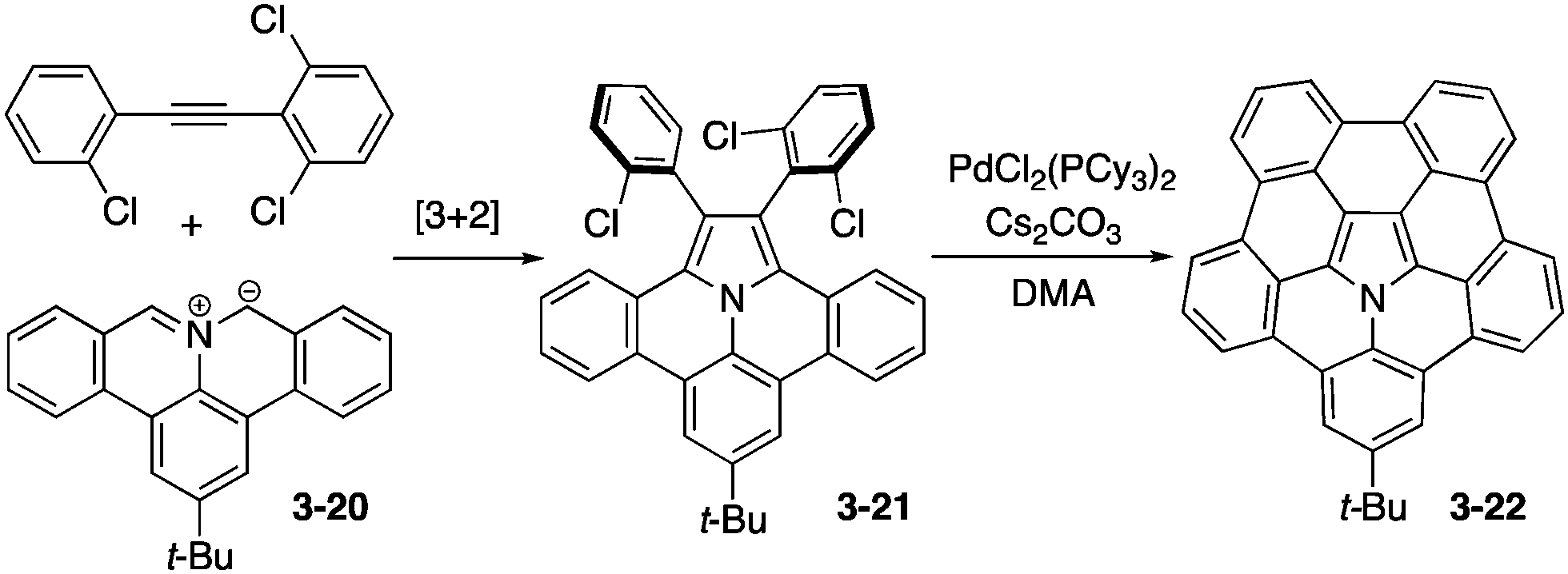
Ito and Nozaki reported the first synthesis of pentabenzoazacorannulene 3-22 through expeditious use of [3+2] dipolar cycloaddition of an azomethine ylide 3-20 with an alkyne followed by a palladium-catalysed fusion reaction of 3-21 (Scheme 6).164 X-ray diffraction analysis of the tert-butylpentabenzoazacorannulene 3-22 elucidated its beautiful bowl-shaped conformation as well as its unique packing structure, in which the molecule exhibited a one-directional concave–convex π–π stacking, which may be suitable to exhibit high charge carrier mobility (Fig. 30). In addition, the bowl-to-bowl inversion energy of azacorranulene 3-22 was investigated by DFT calculations.
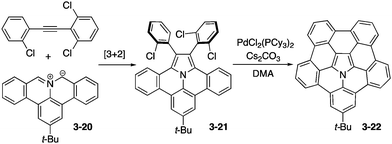 |
| | Scheme 6 Synthesis of pentabenzoazacorannulene 3-22. | |
 |
| | Fig. 30 Packing structure of pentabenzoazacorannulene 3-22. | |
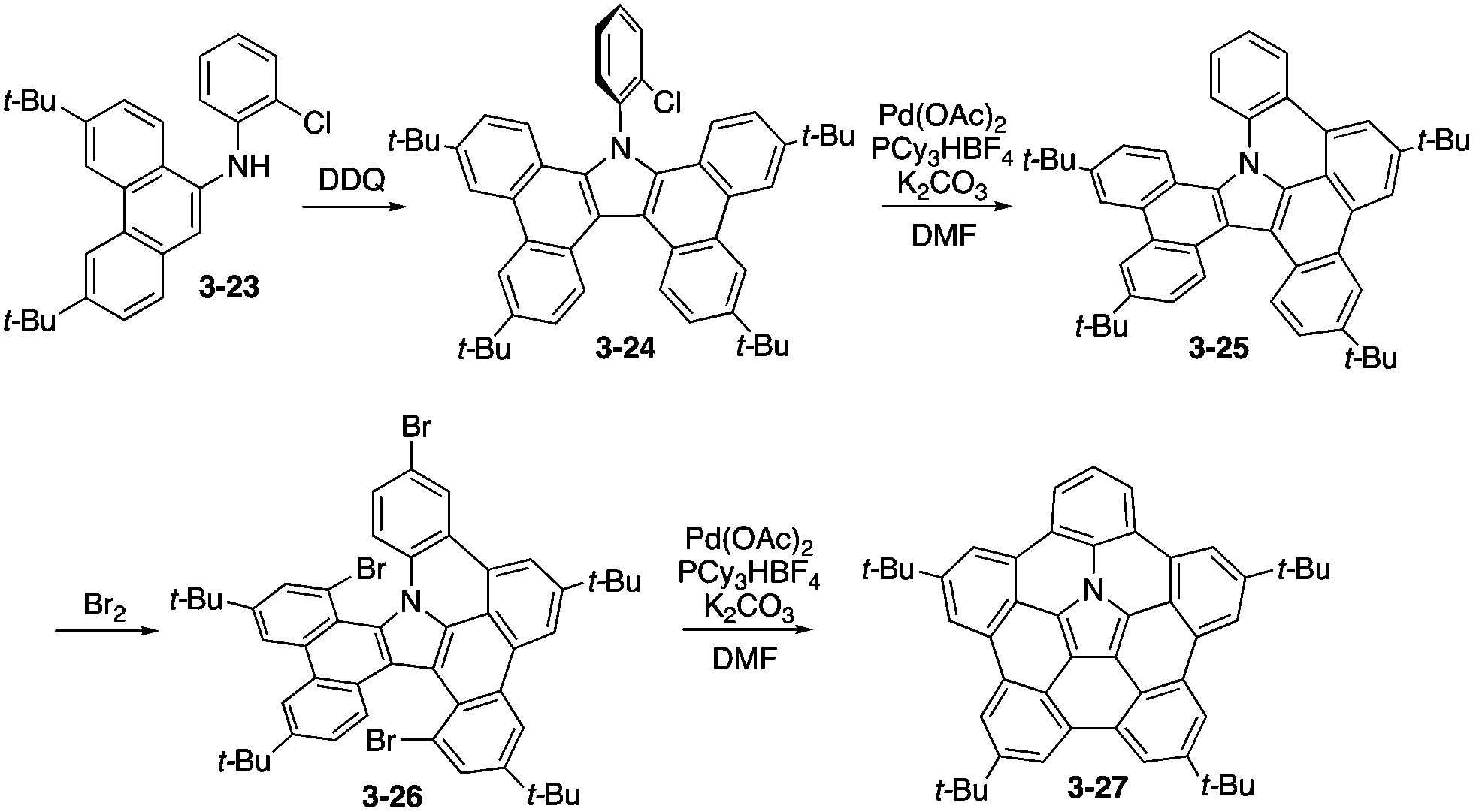
The synthesis of analogous tetra(tert-butyl)pentabenzoazacorranulene 3-27 was then developed on the basis of efficient oxidative dimerization of aminophenanthrene 3-23 to tetrabenzocarbazole 3-24 as the key step (Scheme 7).165 For the ring-fusion steps from 3-24 to 3-25 and 3-26 to 3-27, palladium-mediated C–H/C–Br coupling was employed. In this care, the bowl-to-bowl inversion barrier of 3-27 was experimentally evaluated by the 2D NMR technique. In addition, highly electron-donating nature of azacorranulene 3-27 was revealed through electrochemical analysis by cyclic voltammogram.
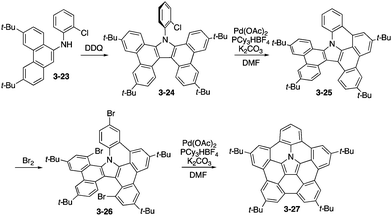 |
| | Scheme 7 Synthesis of pentabenzoazacorannulene 3-27. | |
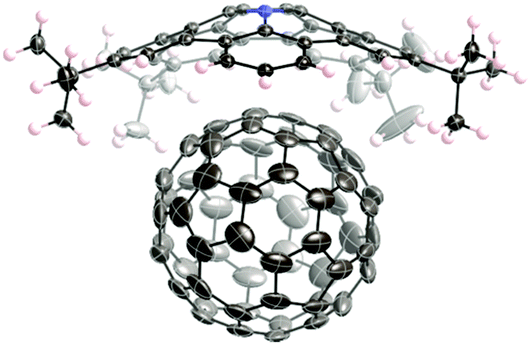
Some of the bowl-shaped π-conjugated molecules are effective at providing inclusion complexes with fullerenes but their binding constants are not very high due to weak electronic interactions between them. Electron-donating bowl-shaped π-conjugated molecules should exhibit enhanced association behaviour. In fact, pentabenzoazacorranulene 3-27 exhibits particularly strong binding with C60 due to the formation of the charge-transfer complex. The association constant of azacorranulene 3-27 with C60 was determined to be 3800 M−1 by titration in 1,2-dichlorobenzene. The structure of the inclusion complex of azacorranulene 3-27 with C60 was elucidated by the X-ray diffraction analysis (Fig. 31). Both C60 and azacorranulene 3-27 construct one-dimensional chain alignments in the co-crystal. Furthermore, the co-crystal showed high charge mobility (0.17 cm2 V−1 s−1) determined by flash-photolysis time-resolved microwave conductivity (FP-TRMC) measurements.
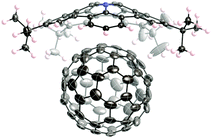 |
| | Fig. 31 Crystal structure of pentabenzoazacorannulene 3-27 with C60. | |
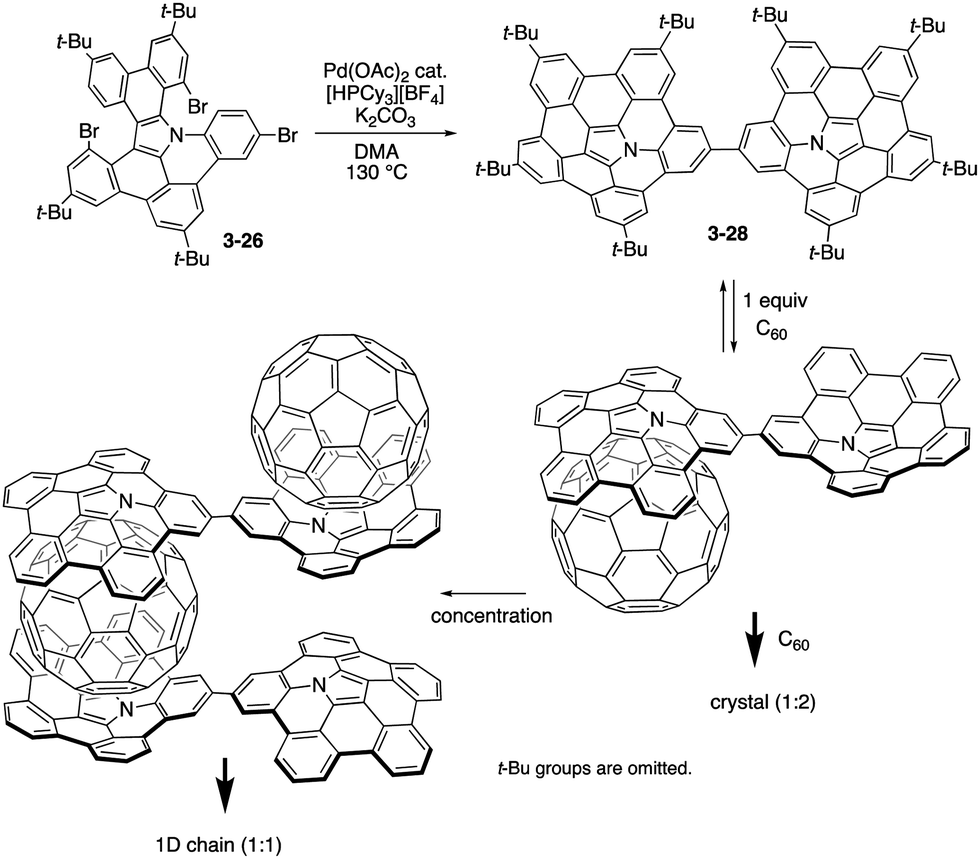
Recently, Shinokubo prepared a directly linked pentabenzoazacorranulene dimer 3-28 through the similar palladium-catalysed C–H/C–Br coupling of the precursor 3-26 (Scheme 8).166 The azabuckybowl dimer 3-28 exhibited intriguing association behaviour with pristine C60. In the presence of 2 equiv. of C60, the dimer 3-28 afforded crystals with a 1:2 stoichiometry. Segregated stacks of the dimer and C60 were detected in the crystal packing, suggesting efficient charge-carrier mobility. In contrast, dimer 3-28 forms a 1:1 inclusion complex with C60 in solution. Furthermore, upon concentrating the solution containing the 1:1 complex, 1D chain supramolecular assemblies were generated to provide fibre-like aggregates upon increasing the concentration.
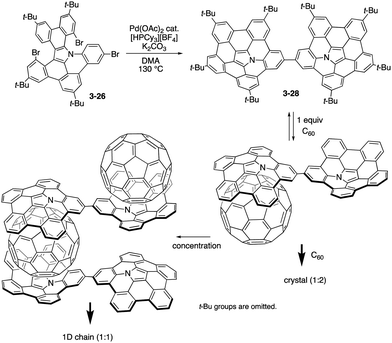 |
| | Scheme 8 Synthesis of pentabenzoazacorannulene dimer 3-28 and its association behaviour with C60. | |
3.5 Other nitrogen-embedded buckybowls
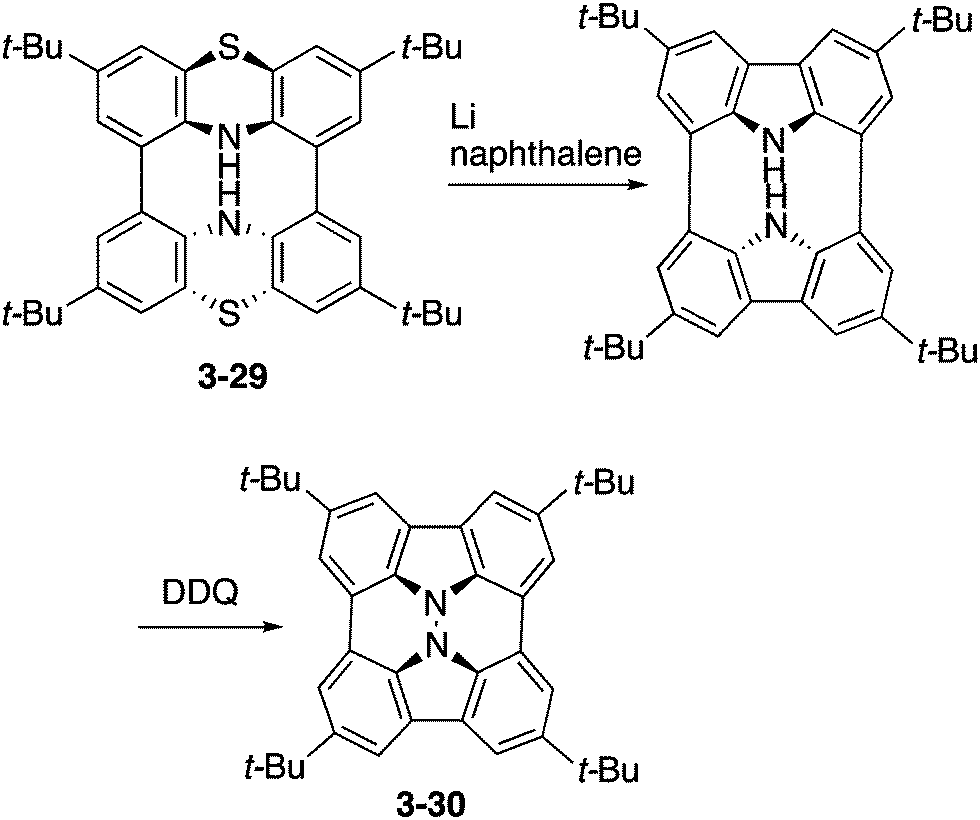
Higashibayashi and co-workers have recently reported the synthesis of carbazole dimer 3-30 as a “hydrazinobuckybowl” (Scheme 9).167 This molecule 3-30 adopted a twisted nonplanar conformation in the crystal, but DFT calculations suggested the bowl-shaped structure as the most stable conformation. Due to its electron-rich nature, hydrazinobuckybowl underwent facile oxidation to provide isolable radical cation and dication species. In addition, the electron-donating nature of the carbazole moieties would enable some application of this molecule as an optoelectronic material.
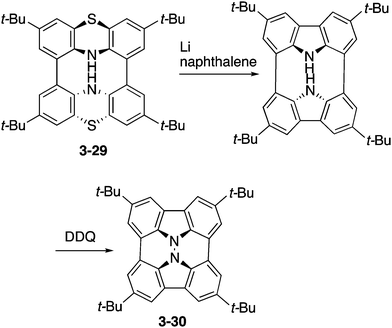 |
| | Scheme 9 Synthesis of hydrazinobuckybowl 3-30. | |
3.6 Azasumanenes
Among possible nitrogen-embedded sumanene derivatives, only triazasumanene has been prepared. One of the intriguing features of triazasumanenes is its bowl chirality. Because of the C3 symmetric structure and bowl-shaped conformation, triazasumanenes should have two enantiomers, which are potentially separable if the bowl-to-bowl inversion barrier is sufficiently large (Chart 19).
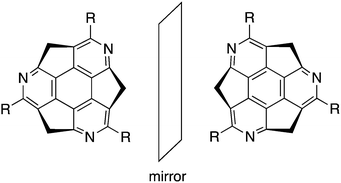 |
| | Chart 19 Chirality of triazasumanene. | |
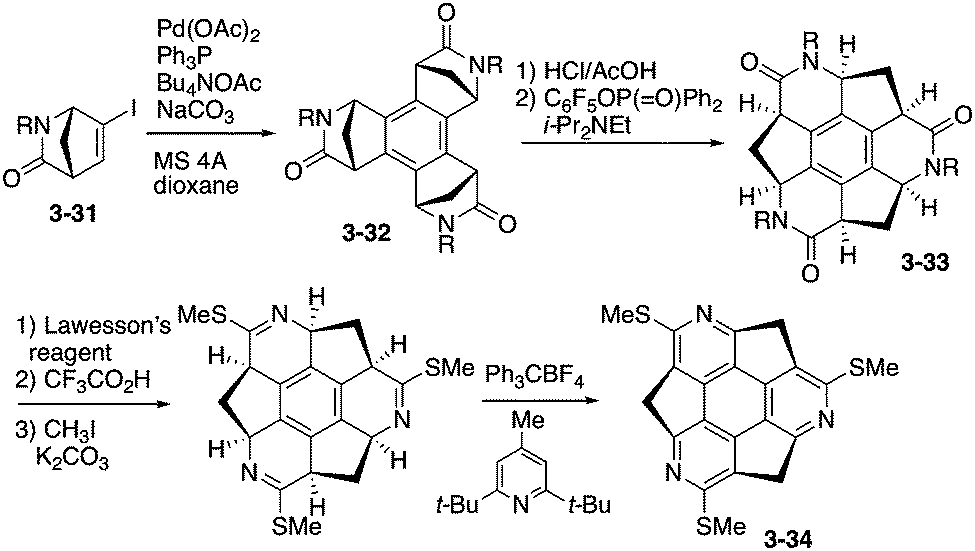
The enantioselective synthesis of triazasumanenes was achieved for the first time by Sakurai, Higashibayashi and co-workers.168 They prepared tris(methylthio)triazasumanene 3-34 through stereoselective trimerization of bicyclic amide 3-31 and transamidation from 3-32 to 3-33 as the key steps (Scheme 10). Due to the high inversion barrier (42.2 kcal mol−1), triazasumanene 3-34 has rather stable chirality. No racemization of 3-34 was observed. This is due to the deeper bowl structure than that of the pristine sumanene, created by the incorporation of the nitrogen atoms.
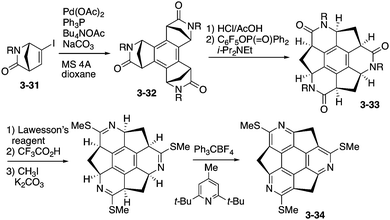 |
| | Scheme 10 Synthesis of tris(methylthio)triazasumanene 3-34. | |
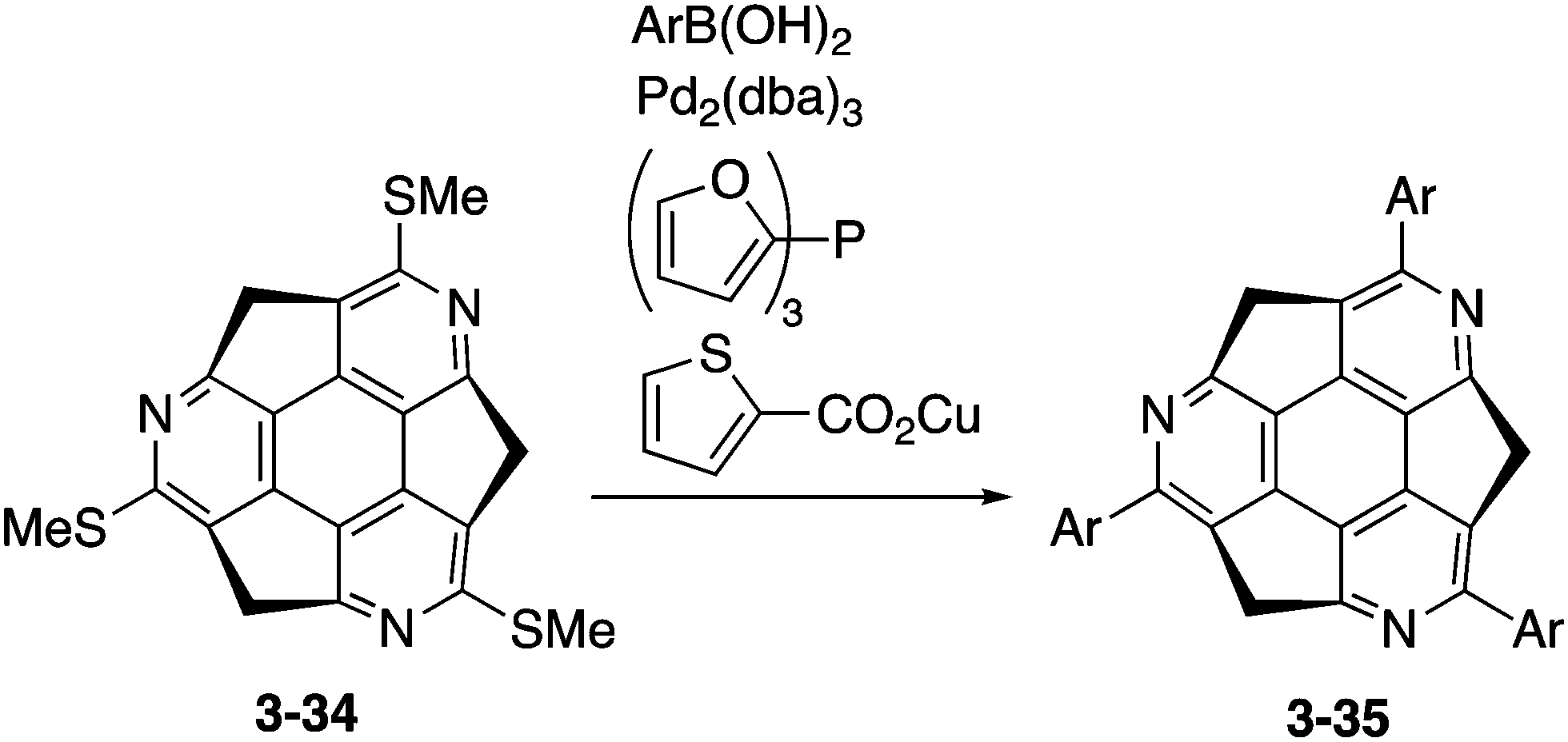
Optically active triarylated triazasumanenes 3-35 were prepared by Liebeskind's palladium/copper-catalysed cross-coupling of tris(methylthio)triazasumanene 3-34 with various arylboronic acids (Scheme 11).169 During the coupling reaction, no racemization of the chiral bowl was observed, indicating the highly stable chirality of triazasumanenes. Now the synthetic methodology for a variety of chiral substituted triazasumanenes has been established. Chemists are ready for exploration of applications of azasumanene derivatives in various fields.
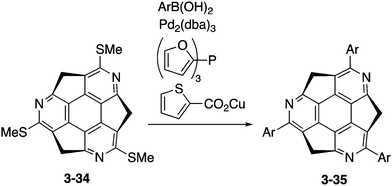 |
| | Scheme 11 Synthesis of arylated triazasumanene 3-35. | |
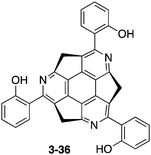
The transformation through the coupling reaction of methylthio derivatives 3-34 with arylboronic acids allows efficient access to various functionalized triazasumanenes. For instance, cross-coupling of 3-34 with 2-hydroxyphenylboronic acid provided triazasumanene 3-36 bearing phenol moieties (Chart 20).170 In the crystal structure of 3-36, intramolecular hydrogen bonding interactions between hydroxy groups and nitrogen atoms were observed. Interestingly, triazasumanene 3-36 showed a single peak emission in solution but its single crystals exhibited a dual emission with a large Stokes shift due to the excited state intramolecular proton transfer (ESIPT) process. The addition of a poor solvent (hexane) in the CH2Cl2 solution of 3-36 resulted in the formation of supramolecular aggregates, which exhibit enhanced emission due to ESIPT.
 |
| | Chart 20 Tri(2-hydroxyphenyl)triazasumanene. | |
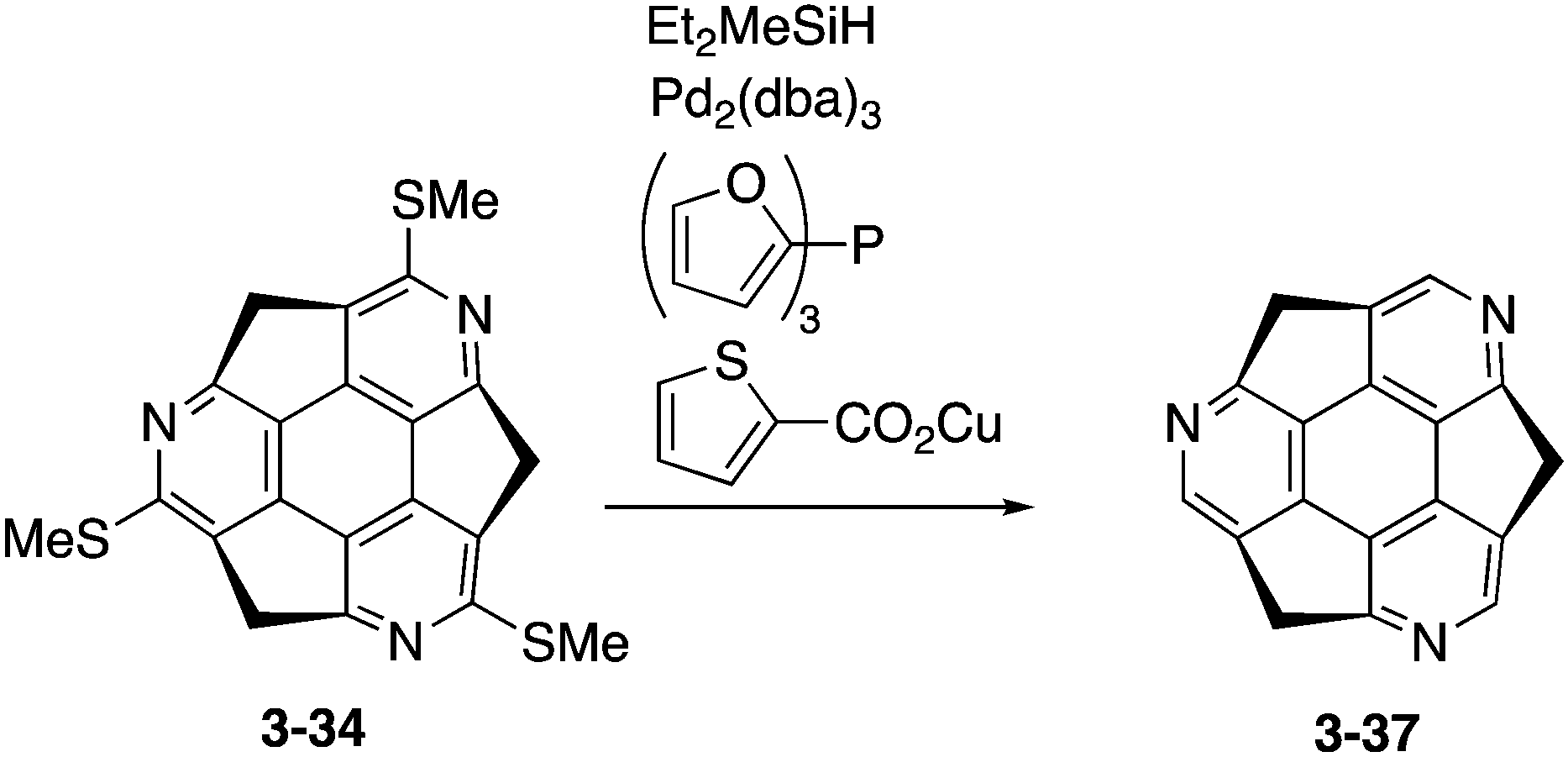
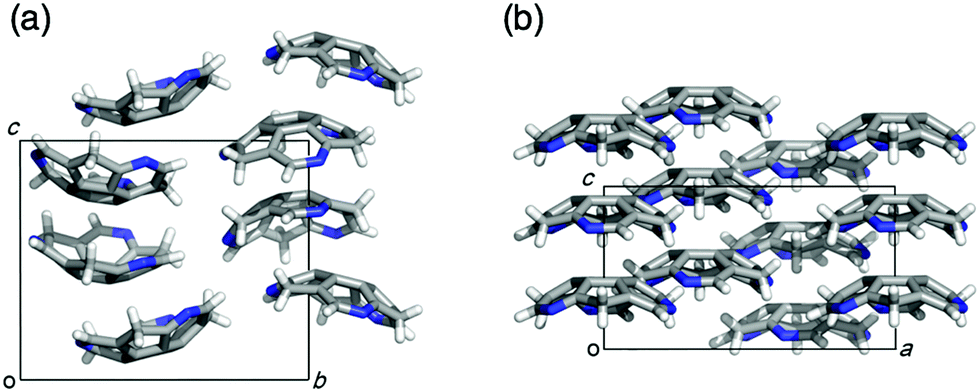
Eventually, pristine triazasumanene 3-37 has been successfully synthesized in a enantiomerically pure form through the palladium/copper-catalysed reduction of tris(methylthio)triazasumanene 3-34 with hydrosilanes (Scheme 12).171 Similar to tris(methylthio)triazasumanene 3-34, unsubstituted triazasumanene 3-37 underwent no racemization of the bowl chirality due to its high bowl inversion barrier. The racemic form of 3-37 was prepared by mixing both enantiomers in equal amounts. Interestingly, the racemic mixture provided crystals, which exhibited a totally different packing structure from the enantiomerically pure form of 3-37 (Fig. 32). While the helical columnar packing with slipped π–π stacking was observed in the crystal of the single enantiomer 3-37, the racemic compound exhibited the unidirectional columnar packing, in which both enantiomers stacked alternatively. The different arrangement of the triazasumanene molecules in the solid state should affect various bulk properties. Hydrolysis of the peripheral imine moiety in acidic aqueous media was also investigated.
 |
| | Scheme 12 Synthesis of pristine triazasumanene. | |
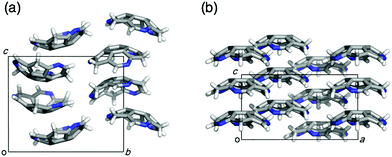 |
| | Fig. 32 Packing structures of chiral (a) and racemic (b) crystals of pristine triazasumanene. | |
4. Recent advances in the chemistry of bowl-shaped heterasumanenes
4.1 Historical background on heterasumanenes
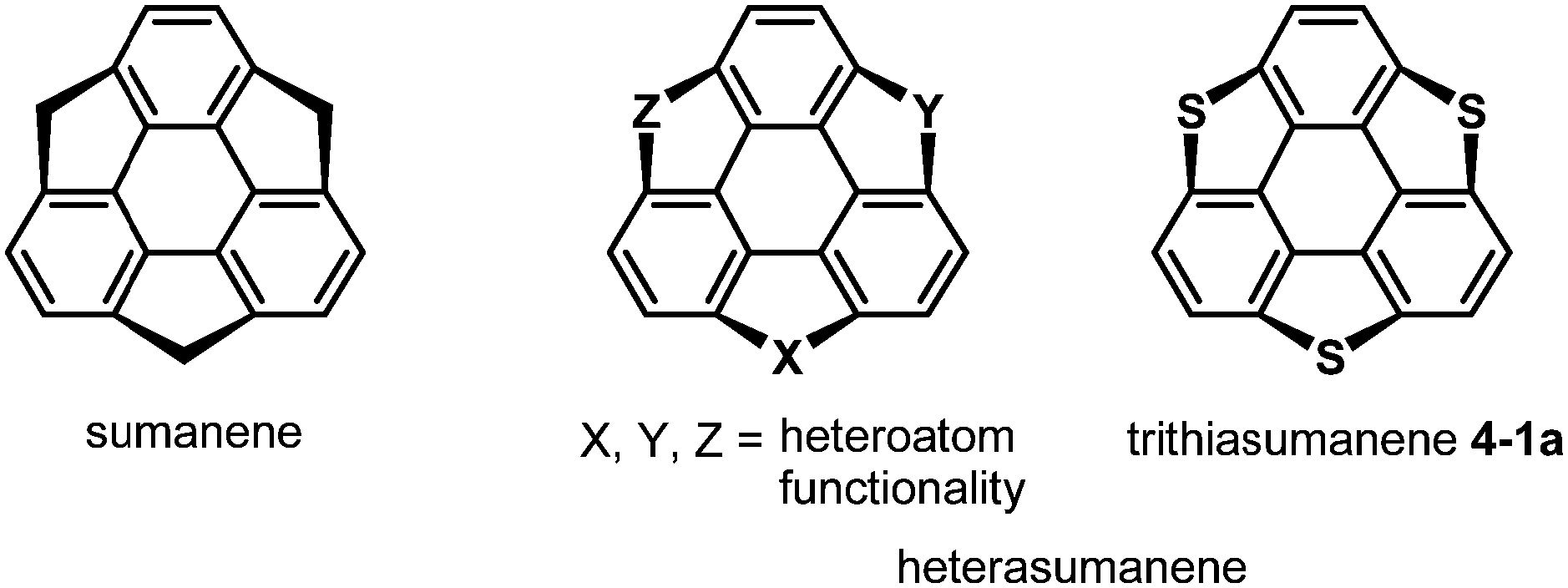
After the first synthesis of sumanene,7 attention has next focused on the synthesis of bowl-shaped compounds containing heteroatoms. One possible method for the introduction of heteroatoms into the sumanene skeleton is the substitution of benzylic carbon atoms by heteroatoms, and the resulting compounds are now called heterasumanenes (Chart 21).17 In contrast to the bowl-shaped structure of sumanene,25 according to theoretical calculations, the structures of heterasumanenes are highly dependent on heteroatoms introduced at the benzylic positions.172 The calculated bowl depth of trioxasumanene (1.49 Å) is larger than the experimentally determined one of sumanene (1.11 Å),25 whereas trisilasumanene was predicted to have a highly planar π-framework172 and experimental studies indeed justified the theoretical prediction.173–177
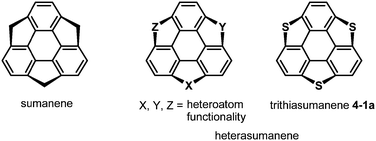 |
| | Chart 21 Sumanene and heterasumanenes. | |
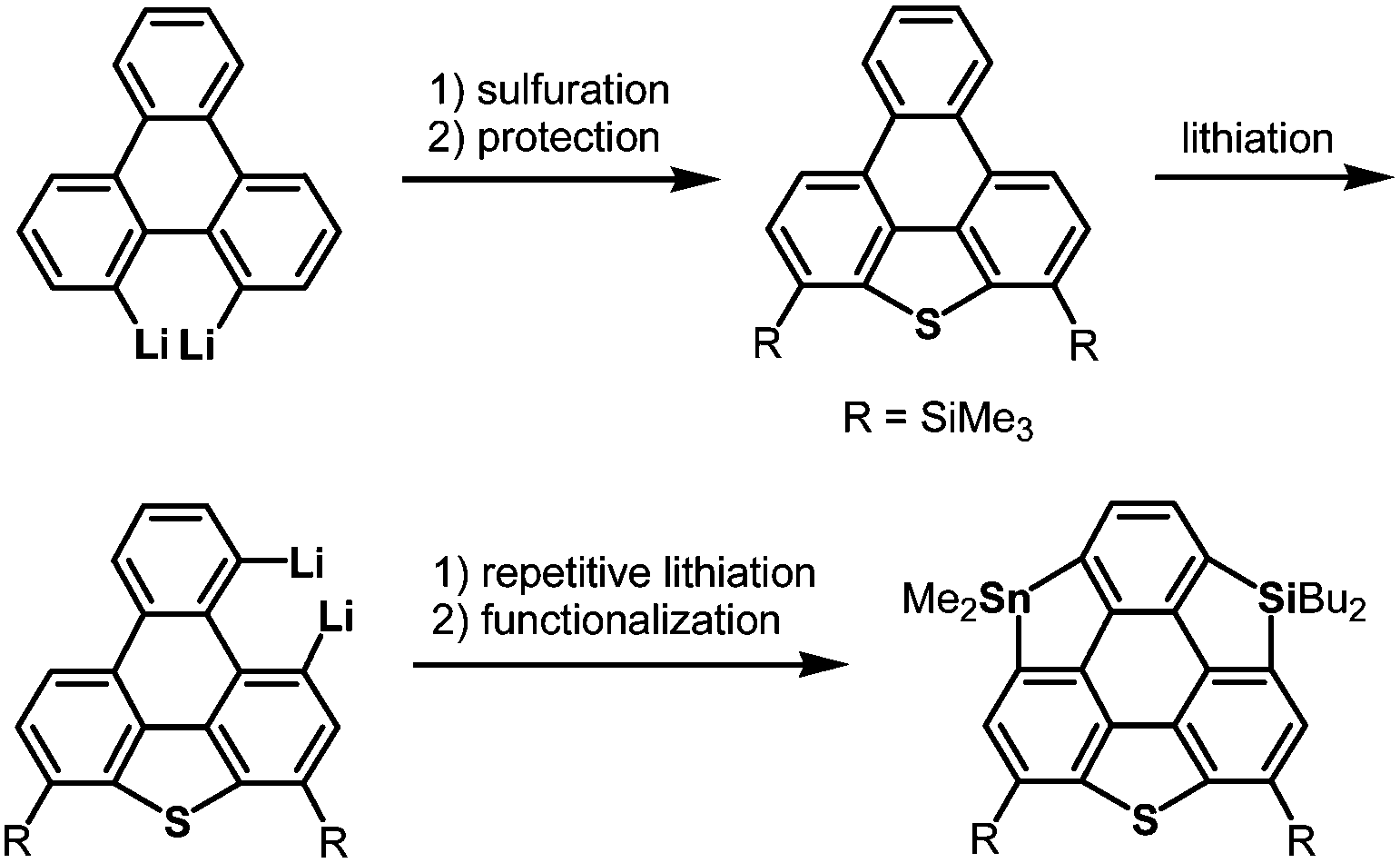
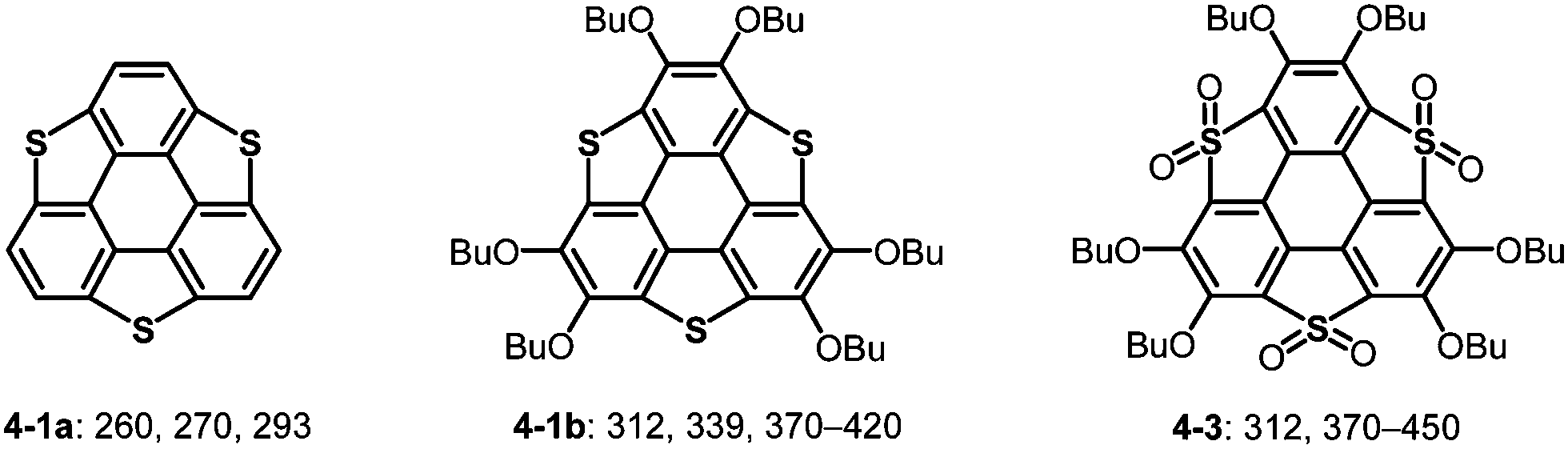
The heterasumanene with the largest bowl depth among those already prepared is pristine trithiasumanene 4-1a. The first synthesis of pristine trithiasumanene 4-1a was achieved under severe reaction conditions of flash vacuum pyrolysis, and its bowl depth (0.65 Å) shallower than that of sumanene was determined by X-ray diffraction analysis.178 After a silence of 10 years, the synthesis of heterasumanenes containing three silicon atoms or three different heteroatoms was accomplished by sila-Friedel–Crafts reactions173 (Scheme 13) and stepwise lithiation of triphenylene (Scheme 14),179 respectively.
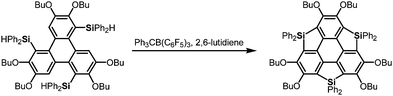 |
| | Scheme 13 Synthesis of a trisilasumanene by sila-Friedel–Crafts reactions. | |
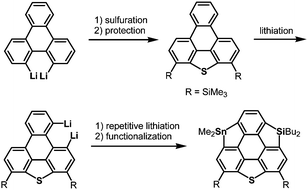 |
| | Scheme 14 A heterasumanene containing three different heteroatoms synthesized by stepwise lithiation of triphenylene. | |
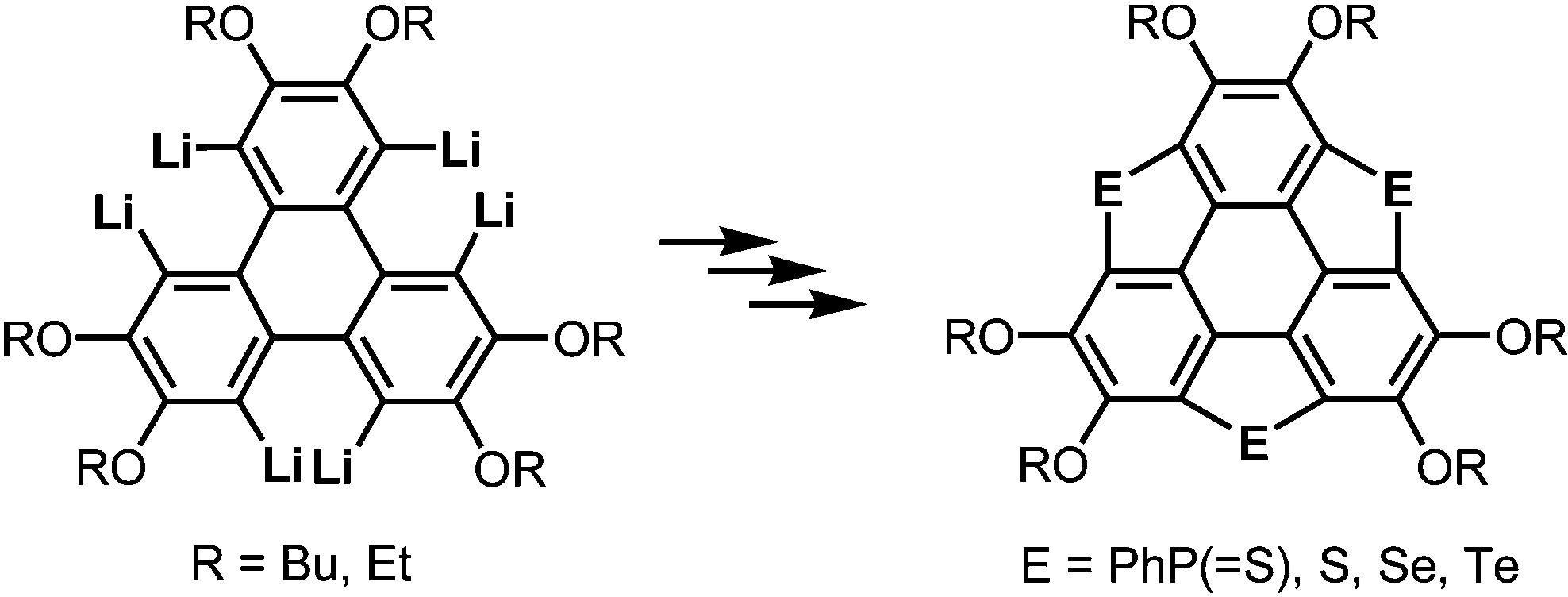
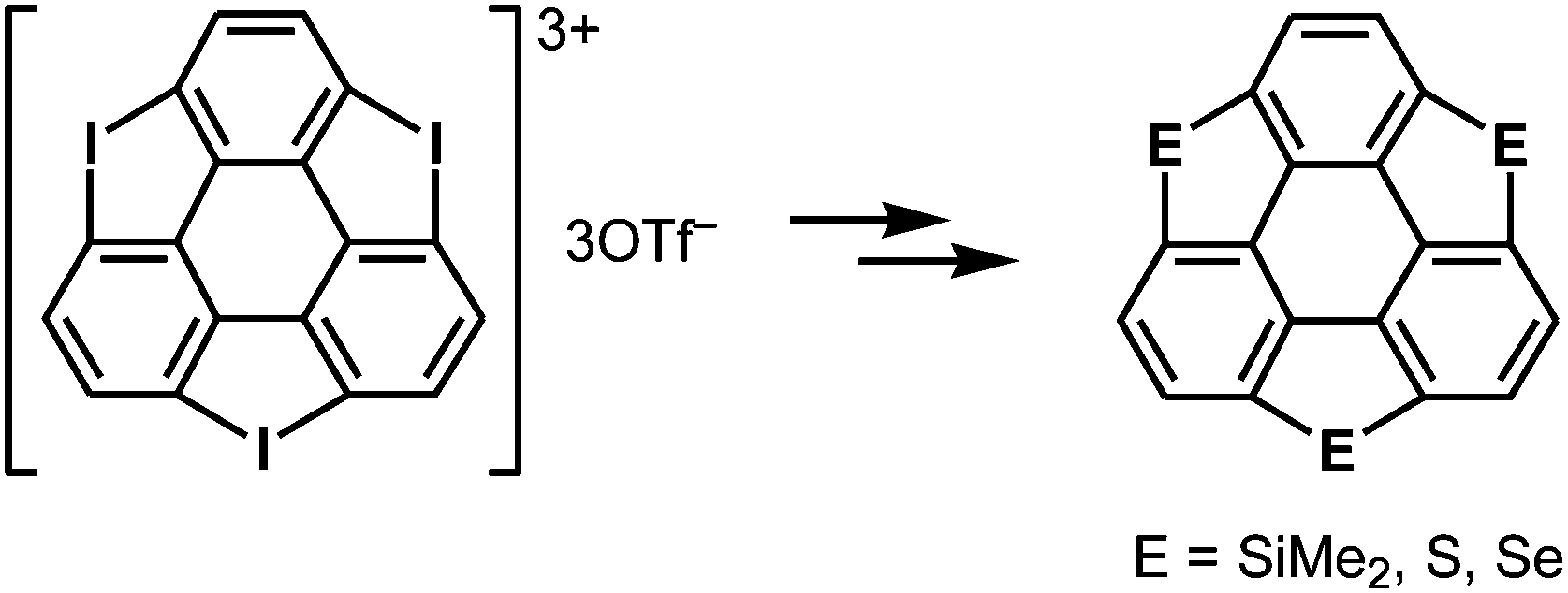
More recently, a large-scale synthesis of trithia- and triselena-sumanenes was accomplished through hexalithiation of hexa-alkoxytriphenylenes,180,181 and the bowl depth of triselenasumanene was determined to be 0.47 Å (Scheme 15). The hexalithiation method was also applied for the preparation of a triphosphasumanene trisulfide and tritellurasumanenes with a bowl depth of 0.46 Å182 and with a highly planar π-framework,183 respectively (Scheme 15). Quite recently, an iodine-bridged sumanene was developed. Each of the bay regions of triphenylene was bridged by an iodine cation to afford the tricationic iodine-bridged sumanene, which functioned as a key intermediate for the preparation of trisila-, trithia- and triselena-sumanenes (Schemes 16).177 Intramolecular cyclization reactions via Si–H and Ge–H activation176 using a Rh catalyst (Kuninobu reaction) provided trisila- and trigerma-sumanenes (Scheme 17).184 These facile synthetic methods opened chemistry of heterasumanenes to investigate their electronic properties because heterasumanenes are composed of three 9-heteraflurorene units, which have long received considerable attention in terms of their fluorescent185,186 and semiconductive nature.187,188
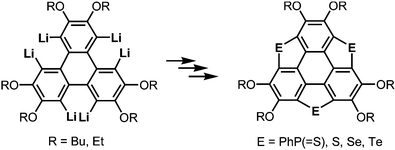 |
| | Scheme 15 Synthesis of trichalcogenasumanenes (E = S, Se, Te) and triphosphasumanene trisulfides (E = PhP(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)) via hexalithiation of hexa-alkoxytriphenylenes. S)) via hexalithiation of hexa-alkoxytriphenylenes. | |
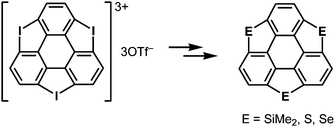 |
| | Scheme 16 Synthesis of heterasumanenes via a tricationic iodine-bridged sumanene. | |
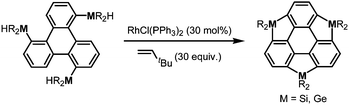 |
| | Scheme 17 Synthesis of trimetallasumanenes via Si–H and Ge–H activation using a Rh catalyst. | |
4.2 Reactivity of heterasumanenes
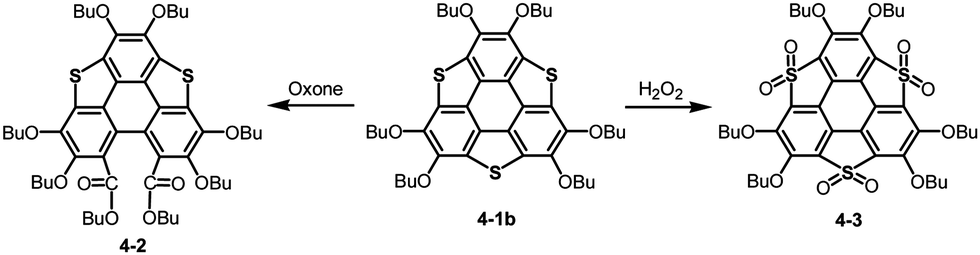
Bowl-shaped molecules are envisaged to exhibit reactivities different from those of planar molecules because of their highly strained structures. A remarkable example is the collapse of a benzene ring found in the oxidation of trithiasumanene 4-1b. The reaction of trithiasumanene 4-1b with Oxone® brought about ring opening of a flanking benzene ring to provide triphenylenodithiophene diester 4-2, whereas oxidation of the same trithiasumanene with hydrogen peroxide provided the expected tris(S,S-dioxide) 4-3 (Scheme 18),189 as was observed in the oxidation of the parent thiophene to afford 1,1-dioxide.190
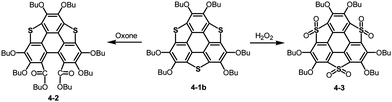 |
| | Scheme 18 Oxidation of trithiasumanene 4-1b. | |
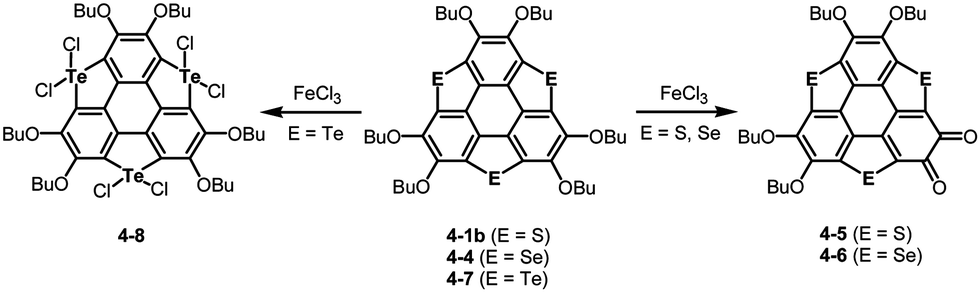
Another striking reactivity was also found in the oxidation of trichalcogenasumanenes. Trithia-4-1b and triselenasumanenes 4-4 reacted with FeCl3 to undergo selective transformation of butoxy groups to provide trichalcogenasumanene ortho-quinones, 4-5 and 4-6 (Scheme 19).191 Remarkably, the direct transformation of 1,2-dibuthoxybenzene to ortho-quinone by FeCl3 has never been reported. Quantum-chemical calculations of model compounds predicted that the trithiasumanene ortho-quinone should have a bowl-depth (0.56 Å) shallower than that of the trithiasumanene (0.67 Å), even though X-ray diffraction analysis of both the compounds was not successful. In contrast, using tritellurasumanene 4-7 brought about a different reaction to produce the corresponding hexachloro adduct 4-8 (Scheme 19),23 which is similar to a hexabromo adduct obtained by the reaction of the tritellurasumanene with bromine.183
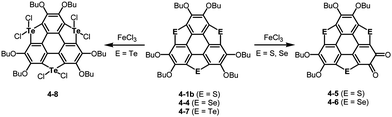 |
| | Scheme 19 Oxidation of trichalcogenasumanenes with FeCl3. | |
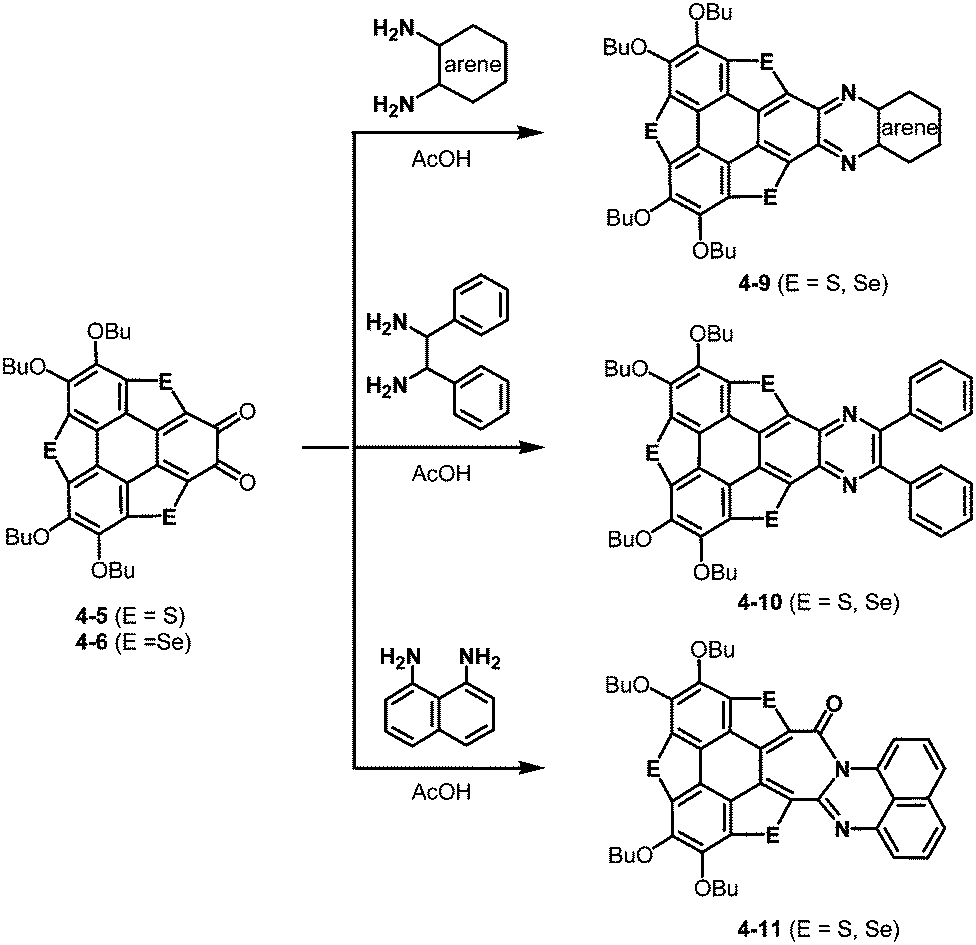
The resulting ortho-quinones were applied for the preparation of new pyrazine derivatives, 4-9 and 4-10 using ortho-aryldiamines (Scheme 20).191 Interestingly, an aryl moiety is not essential for this transformation. The use of 1,2-diphenylethane-1,2-diamine afforded the corresponding pyrazine derivative 4-10 (Scheme 20). Moreover, the treatment with naphthalene-1,8-diamine resulted in the formation of the [7-6-6]-fused system 4-11 (Scheme 20). Reflecting a bowl-shaped structure of the trithiasumanene, the pyrazine derivatives consist of bowl-shaped trithiasumanene and planar pyrazine moieties, while the π-frameworks derived from the triselenasumanene and pyrazine derivatives are almost planar.
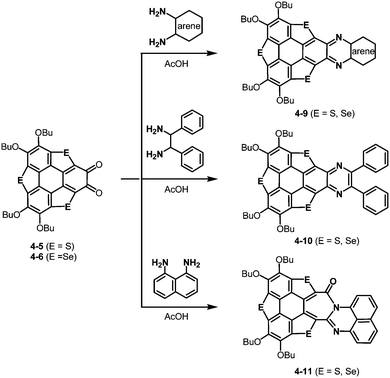 |
| | Scheme 20 Formation of pyrazine rings from trichalcogenasumanene ortho-quinones 4-5 and 4-6. | |
4.3 Optical properties of heterasumanenes
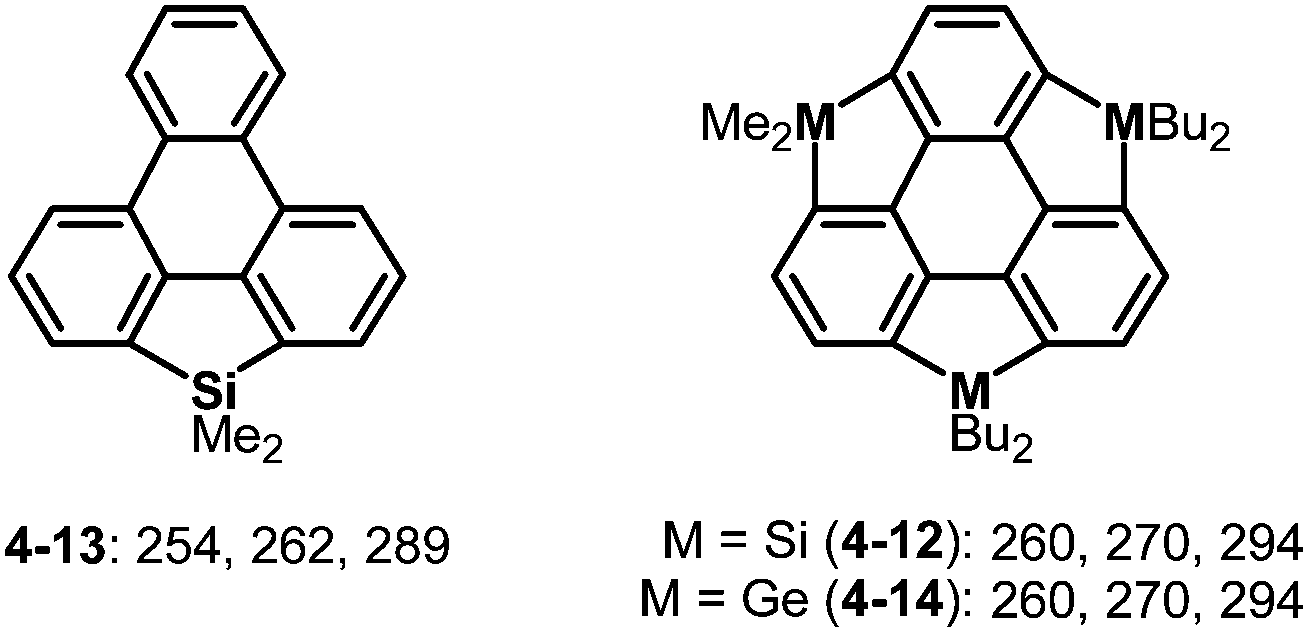
Trisilasumanenes exhibit fluorescent nature derived from their silafluorence units. The HOMO of trisilasumanene 4-12 is mainly composed of a triphenylene π-framework, while the LUMO consists of a σ*(Si–C)–π*(triphenylene) interaction distributed in the π-framework, and lowering the LUMO level causes electronic absorptions that are red-shifted compared to those of the triphenylenosilole 4-13 (Chart 22).175,184 Similar tendencies were observed in the electronic absorption and fluorescence spectra of trigermasumanene 4-14 and therefore group 14 atoms introduced little effect on the electronic properties of the resulting heterasumanenes (Chart 22).176,184 Likewise, the UV-Vis absorption spectra of trithia- and triselena-sumanenes bearing similar substituents on the π-frameworks are almost identical.177,180
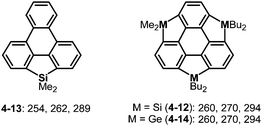 |
| | Chart 22 UV-Vis absorption maxima (nm) of trimetallasumanenes and their related compounds. | |
On the other hand, substituents on the carbon atoms of the triphenylene skeleton significantly affect the optical properties of heterasumanenes. The lone pair of oxygen atoms of alkoxy groups significantly contributes to the increase of the HOMO level, resulting in the HOMO–LUMO gap becoming narrower than those of the corresponding derivatives without alkoxy groups (Chart 23).174–176,184 Valence of the heteroatoms also affects the electronic absorption of trichalcogenasumanenes (Chart 23). Both HOMO and LUMO levels of trithiasumanene S,S-dioxide 4-3 were lower than those of trithiasumanane 4-1b, and the extent of lowering of the LUMO level was substantially large because of electron-withdrawing SO2 units. Consequently, the longest absorption of S,S-dioxide 4-3 is red-shifted, compared to that of the unoxidized congener 4-1b.
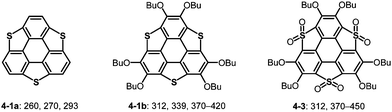 |
| | Chart 23 UV-Vis absorption maxima (nm) of trithiasumanenes. | |
4.4 Application of heterasumanenes as potential precursors for materials
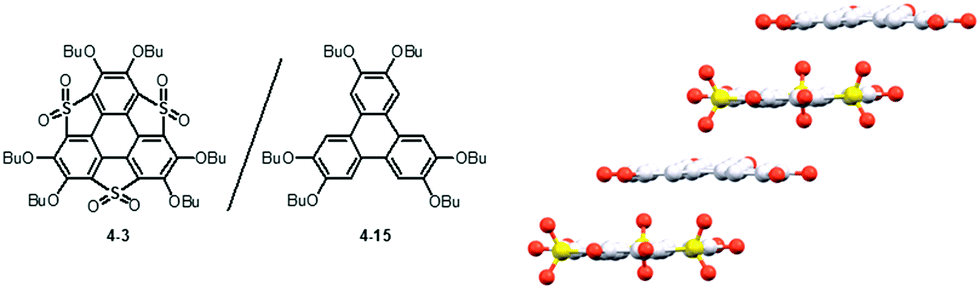
Heteroatom functionalities in heterasumanenes cause fascinating properties, which could be applied as potential precursors for future materials. Trithiasumanene S,S-dioxide 4-3 has an electron-deficient triphenylene π-framework because of electron-withdrawing SO2 groups, whereas that of the starting hexabutoxy triphenylene (HBT) 4-15 is more electron-rich because of electron-donating butoxy groups. The electrostatic interactions arising from these complementary electronic structures resulted in the formation of co-crystals composed of S,S-dioxide 4-3 and HBT 4-15 (Fig. 33).189 In the co-crystals, both components stack alternately to form columnar structures with a distance of about 3.58 Å (av.). The solid-state absorption spectrum of the co-crystals dispersed on KBr pellets revealed a HOMO–LUMO gap narrower than that of the S,S-dioxide, which causes a bathochromic shift of the emission (500–700 nm), compared to those of the two components (398 nm and 479 nm for HBT and the S,S-dioxide, respectively). Another interesting feature of the co-crystal is its noncentrosymmetric nature with the space group P1, which was indeed confirmed by circular dichroism spectroscopy. It is concluded that the combination of two components that have distinct electronic structures can produce new properties that are not demonstrated by each of the two components.
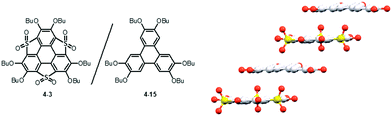 |
| | Fig. 33 A packing structure of a co-crystal composed of trithiasumanene S,S-dioxide 3-3 and hexabutoxytriphenylene 4-15. | |
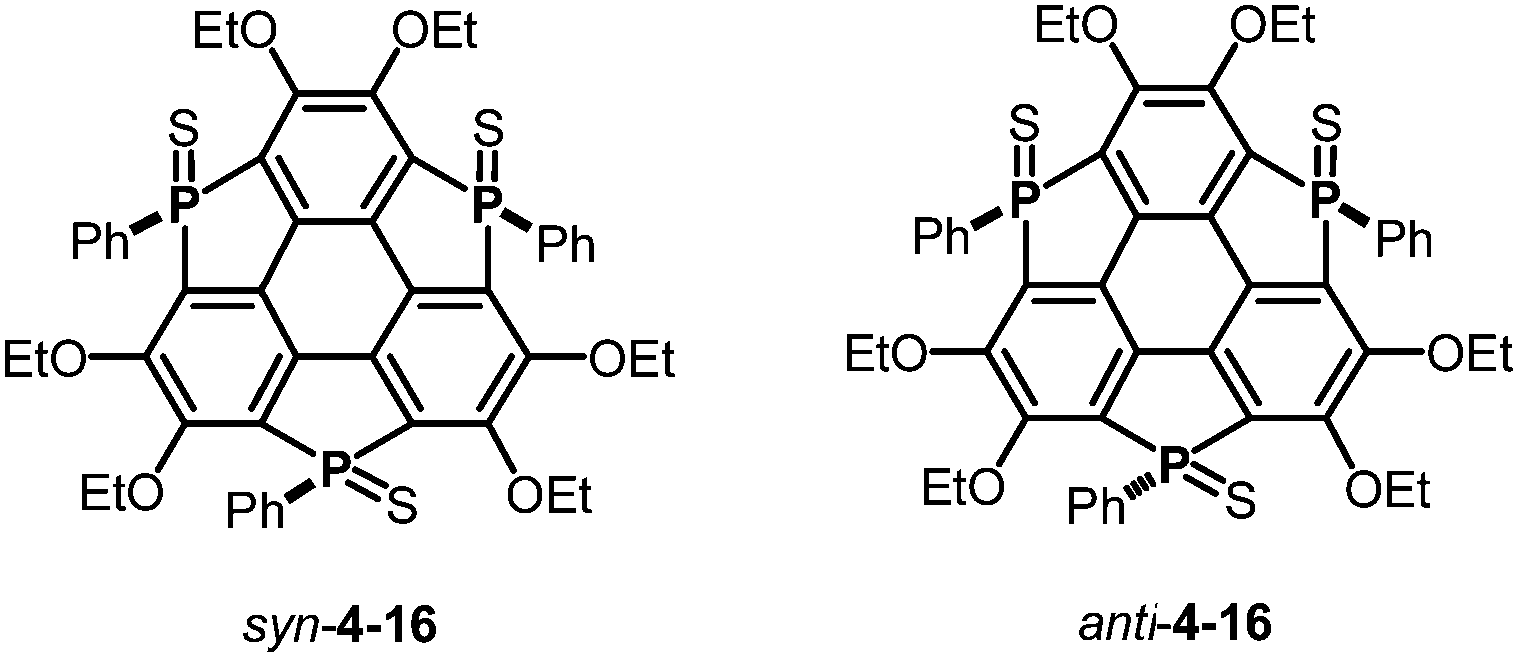
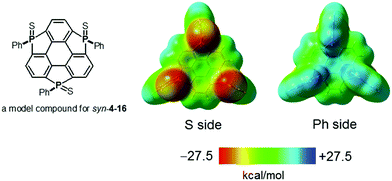
The choice of heteroatom functionalities enables making two faces of a π-framework with distinct electronic features; the resulting π-framework can possess out-of-plane anisotropy.182 Triphosphasumanene trisulfide 4-16 has two isomers; one has three PS moieties on the same side, denoted as a syn form, while one of the three PS moieties is on an opposite side of the π-framework, denoted as an anti form (Chart 24). In the syn form, the side bearing PS moieties possess a negative electrostatic potential surface due to electro-negative sulfur atoms, whereas the opposite side exhibits an electrostatic potential surface, allowing a differentiation between the front and back of the π-surface (a Janus-type π-surface), as evidenced by quantum-chemical calculations of the model compound for syn-4-16 shown in Fig. 34. On account of its Janus nature, the syn form is calculated to have a large dipole moment of 13.7 D, which is much larger than those of boron subphthalocyanine (5.40 D)192 and titanium oxide phthalocyanine (3.73 D).193 The two-face character was experimentally elucidated by the adsorption of a molecule on the Au(111) surface. The X-ray photoelectron spectra of the syn-form on Au(111) revealed that the sulfur atoms of the syn-form bind to the Au(111) surface. According to quantum-chemical calculations, the adsorption energy of the configuration where three sulfur atoms equally interact with the Au surface is 93.5 kcal mol−1 more stable than that of the overturned configuration where the phenyl groups are oriented to the Au(111) surface (Fig. 35).
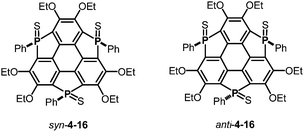 |
| | Chart 24 Triphosphasumanene trisulfides 4-16 (left: syn form; right: anti form). | |
 |
| | Fig. 34 Calculated electrostatic potential map of the model compound for syn-4-16. | |
 |
| | Fig. 35 Optimized adsorption modes of syn-form of triphosphasumanene trisulfide 4-16. The three sulfur atoms equally contact the Au(111) surface (left), while the phenyl side contacts the Au(111) surface. | |
A dense columnar π–π stacking structure is of crucial importance to achieve high charge mobility in organic semiconductors.194 Considerable efforts have long been made for the modification of the chemical structures of planar polyaromatic hydrocarbons to enhance π–π stacking by introducing heteroacenes, envisaging the increased charge mobility.195 During the course of the study to explore π–π stacking structures efficient for organic semiconductor devices, triselenasumanene 4-6 was investigated. Organic field-effect transistors (OFETs) based on single-crystal nanoribbons of the triselenasumanene exhibited a hole mobility of 0.37 cm2 V−1 s−1,196 which is the highest among OFETs composed of corannulene and sumanene derivatives.197–199
4.5 Phosphorus-based concave molecules
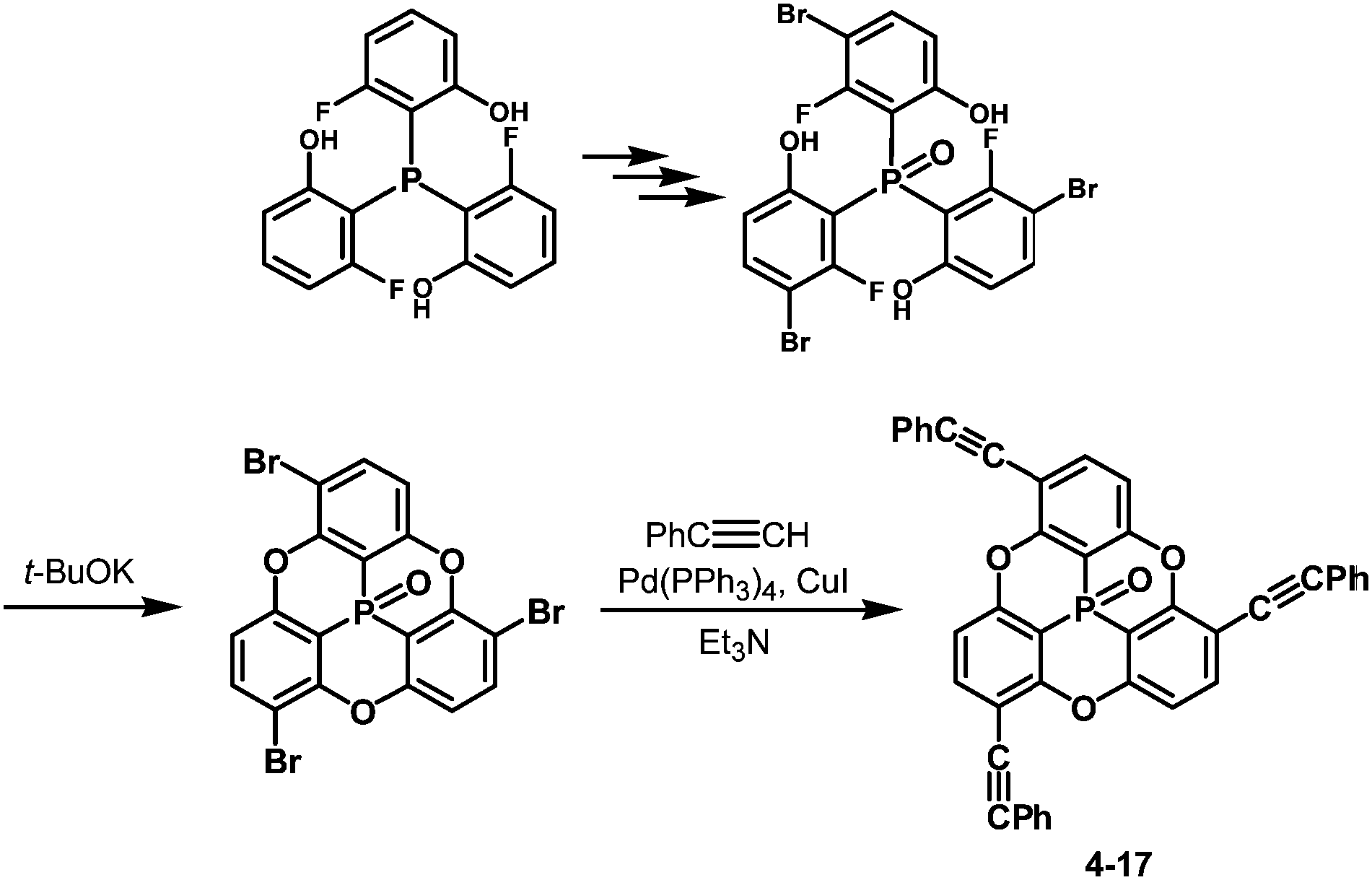
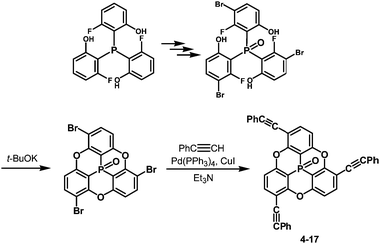
One of the challenging tasks in using bowl-shaped molecules is molecular recognition of fullerene C60, permitting its purification, nano-scale organization and perturbation of its electronic and photochemical properties. The C60 recognition is usually achieved by interactions between the convex surface of C60 and the concave surface of a bowl-shaped molecule. As bowl-shaped host molecules of C60, corannulene and sumanene are promising because they are regarded to possess partial structures of C60, and therefore their concave surfaces can precisely fit into the convex surface of C60. Indeed, the interaction between corannulene and C60 has already been observed in the solid state.95 However, it was not effective in solution due to rapid bowl-to-bowl inversion of corannulene in solution at room temperature. To avoid rapid bowl-to-bowl inversion, in other words, to construct a rigid concave structure, a phosphorus atom would be one of the choices as the bottom of a concave molecule because planar tertiary phosphines have never been reported, meaning that concave molecules bearing phosphorus atoms at the bottom would have high bowl-to-bowl inversion barriers. With this in mind, phosphangulenes, which were firstly prepared by Krebs,200 were utilized as hosts of C60. Yamamura and his co-workers prepared phosphangulene 4-17 with a C3 symmetry bearing three phenylethynyl groups (Scheme 21).201
 |
| | Scheme 21 Preparation of phosphangulene 4-17 with a C3 symmetry bearing three phenylethynyl groups. | |
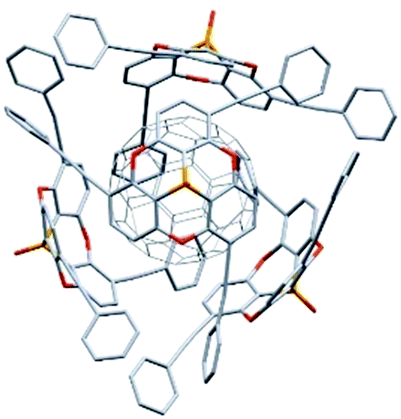
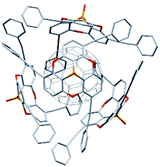
Remarkably, phosphangulene 4-17 formed co-crystals with pristine C60, wrapped by four phosphangulene molecules (Fig. 36). On the other hand, MALDI-TOF spectroscopy showed an ion peak that was assigned to be a 1:1 complex of phosphangulene 4-17 and a C60 molecule, revealing that the concave/convex interaction is strong and sufficient to maintain the encapsulated complex even during ionization, even though the ratio of concave/convex molecules is different from that found in the solid state. More importantly, the 1H NMR analysis of a mixture containing phosphangulene 4-17 with a functionalized C60 (Chart 25), which is more soluble than pristine C60, indicated that the 1:1 complex was formed in solution. In contrast, no interactions were observed between Kreb's phosphangulene (Chart 26) and the same functionalized C60 in solution. It is concluded that the phenylethynyl groups play an essential role in the interactions with C60.
 |
| | Fig. 36 The structure of a co-crystal composed of one C60 and four phosphangulene molecules. | |
 |
| | Chart 25 The functionalized C60 utilized for the NMR analysis. | |
 |
| | Chart 26 Kreb's phosphangulene. | |
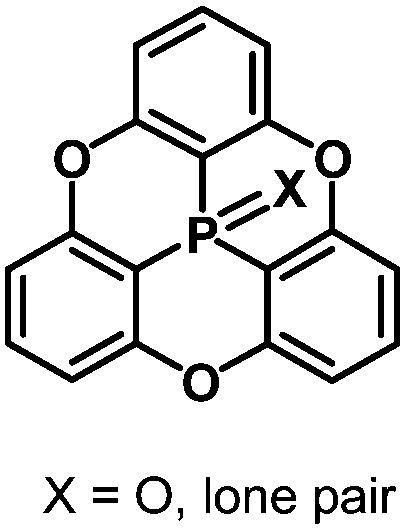
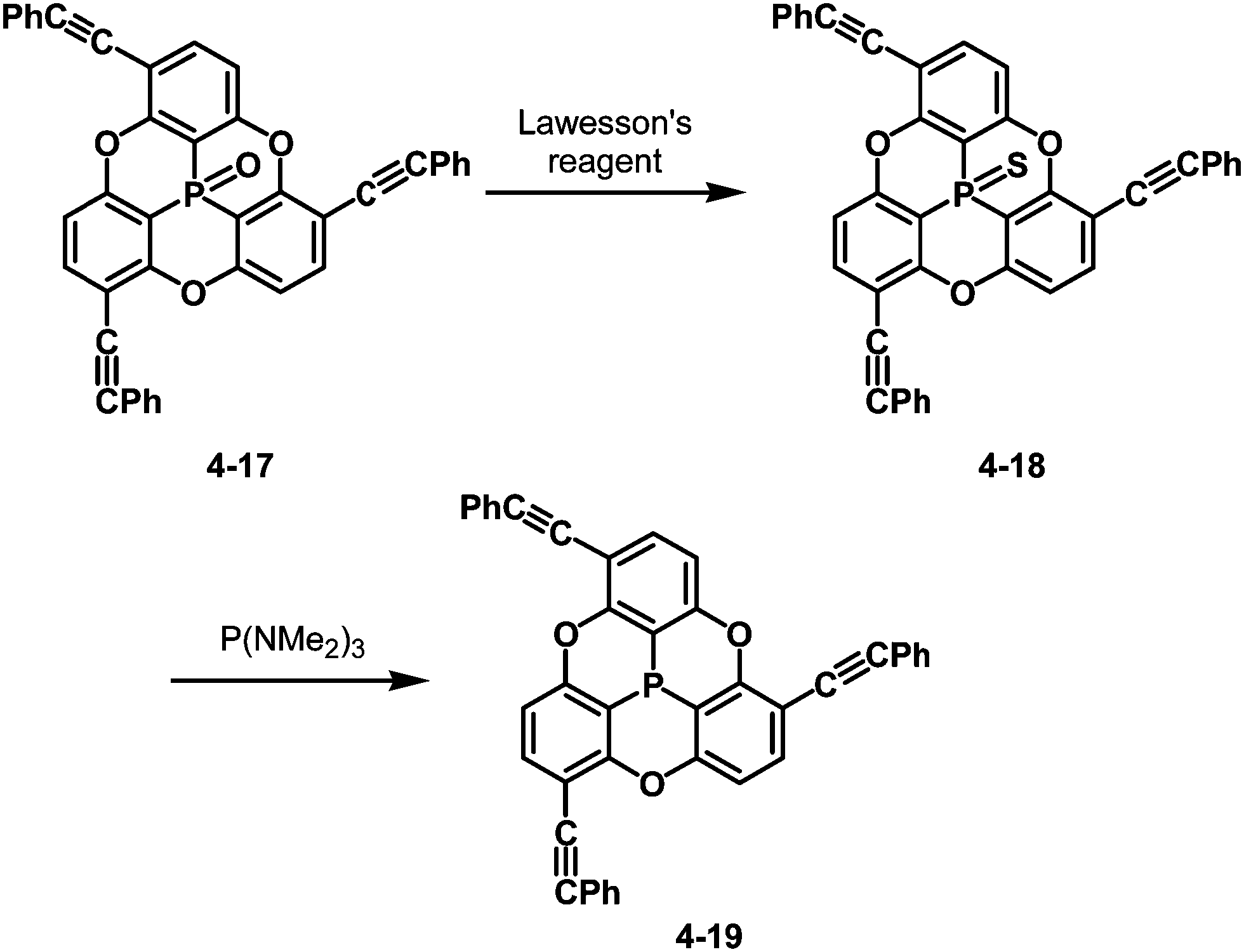
The bowl-depth of phosphangulene derivatives can be controlled by cone angles around the phosphorus atoms, dependent on the electronic states of the phosphorus atoms. The phosphangulene bearing a PO moiety was converted into the corresponding PS derivative by the use of Lawesson's reagent, and subsequent desulfuration provided the phosphine derivative (Scheme 22).202
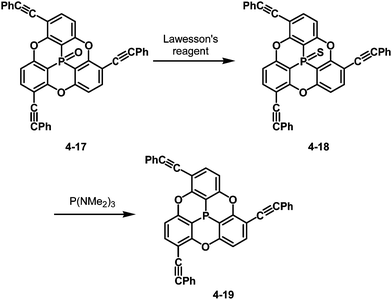 |
| | Scheme 22 Transformation of PO phosphangulene 4-17 into PS one 4-18 and phosphine derivative 4-19. | |
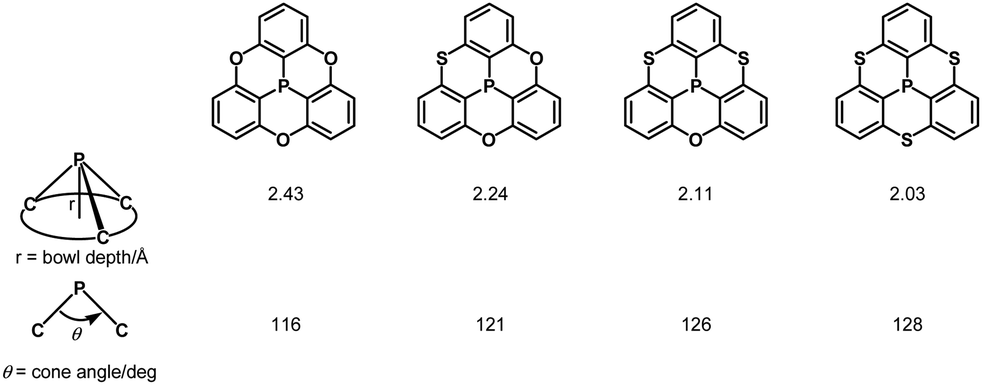
Quantum-chemical calculations revealed that a cone angle of the phosphine derivative (115°) is smaller than those of the PO and PS derivatives (about 121°), which causes a bowl depth of 2.46 Å, deeper than those of the PO and PS derivatives (about 2.23 Å) (Fig. 37).
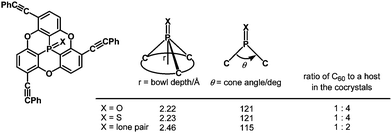 |
| | Fig. 37 Correlation between bowl depths and cone angles in phosphangulenes. | |
The bowl depths and cone angles of phosphangulenes affected the structures of encapsulated complexes containing C60. The PO and PS derivatives formed co-crystals composed of a 1:4 mixture with pristine C60, whereas two molecules of the phosphine derivative wrapped pristine C60 in the co-crystals, indicating that even small differences in the cone angle (6°) and in the bowl depth (0.23 Å) can significantly affect encapsulation.
Inspired by this sophisticated crystal engineering, the control of the bowl depth of a phosphangulene was investigated, and the bowl depth was found to depend on bridging heteroatoms.203 Trithiaphosphangulene 4-20 prepared according to Scheme 23 has the smallest bowl depth (2.03 Å) among the prepared congeners (Fig. 38), reflecting a P–S bond longer than a P–O bond. These sophisticated controls of differences would inspire a new strategy for crystal engineering.
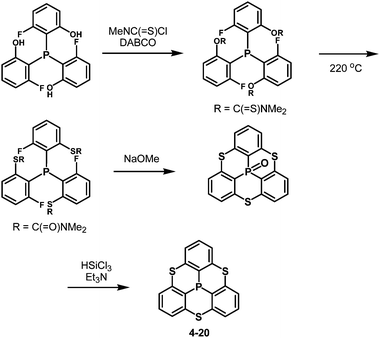 |
| | Scheme 23 Preparation of trithiaphosphangulene 4-20. | |
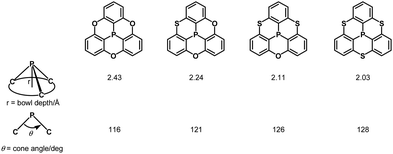 |
| | Fig. 38 Bowl depths and cone angles of phosphangulenes. | |
Conclusions
The chemistry of bowl-shaped π-conjugated molecules is quite unique, rich and promising. Among them, corannulene and its derivatives have already advanced well, mainly because of its longer history and ready availability. In particular, the progress in recent years in its application to materials science is magnificent as is obvious from the number of publications. Further excellent outputs are highly expected in the coming decade. On the other hand, the chemistry of sumanene, started more than ten years later than that of corannulene, seems still on the way of development, and more time is needed for studying its application in materials chemistry. However, the difference in the substitution pattern of sumanene is attractive, and fundamental studies have revealed the characteristics of sumanene against corannulene. The commercialization of sumanene from 2017 will trigger the explosive development of sumanene chemistry.
With regard to the heteroatom-doped systems, the research in this field has just started, therefore, the development of efficient synthetic routes and bulk-scale preparation are still urgent issues. However, without doubt, this field is becoming more and more important because the doping of heteroatoms into bowl-shaped molecules has been found to be particularly effective in modulating their optoelectronic properties. Such molecules would contribute to the development of materials science.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Area “π-System Figuration” from the Ministry of Education, Culture, Sports, Science and Technology of Japan (JP26102002, JP26102003 and JP26102006).
Notes and references
- H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl and R. D. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380–381 CrossRef CAS.
- L. T. Scott, M. M. Hashemi and M. S. Bratcher, J. Am. Chem. Soc., 1992, 114, 1920–1921 CrossRef CAS.
- A. Borchardt, A. Fuchicello, K. V. Kilway, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 1921–1923 CrossRef CAS.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664–676 CrossRef CAS.
- H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed.
- C.-N. Feng, Y.-C. Hsieh and Y.-T. Wu, Chem. Rec., 2015, 15, 266–279 CrossRef CAS PubMed.
- For example, K. Y. Amsharov, M. A. Kabdulov and M. Jansen, Angew. Chem., Int. Ed., 2012, 51, 4594–4597 CrossRef CAS PubMed.
- P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235–242 CrossRef CAS.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS PubMed.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- S. Higashibayashi and H. Sakurai, Chem. Lett., 2011, 40, 122–128 CrossRef CAS.
- M. Stępień, Synlett, 2013, 1316–1321 CrossRef.
- X. Li and X. Shao, Synlett, 2014, 1795–1798 CAS.
- T. Amaya and T. Hirao, Chem. Rec., 2015, 15, 310–321 CrossRef CAS PubMed.
- M. Saito, S. Furukawa, J. Kobayashi and T. Kawashima, Chem. Rec., 2016, 16, 64–72 CrossRef CAS PubMed.
- X. Li, F. Kang and M. Inagaki, Small, 2016, 12, 3206–3223 CrossRef CAS PubMed.
- X.-Q. Hou, Y.-T. Sun, L. Liu, S.-T. Wang, R.-L. Geng and X.-F. Shao, Chin. Chem. Lett., 2016, 27, 1166–1174 CrossRef CAS.
- S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed.
- R. A. Pascal, Jr., Chem. Rev., 2006, 106, 4809–4819 CrossRef PubMed.
- S. Higashibayashi, S. Onogi, H. K. Srivastava, G. N. Sastry, Y.-T. Wu and H. Sakurai, Angew. Chem., Int. Ed., 2013, 52, 7314–7316 CrossRef CAS PubMed.
- J. C. Hanson and C. E. Nordman, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, B32, 1147–1153 CrossRef CAS.
- H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS PubMed.
- S. Mebs, M. Weber, P. Luger, B. M. Schmidt, H. Sakurai, S. Higashibayashi, S. Onogi and D. Lentz, Org. Biomol. Chem., 2012, 10, 2218–2222 CAS.
- T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef CAS PubMed.
- B. B. Shrestha, S. Karanjit, S. Higashibayashi and H. Sakurai, Pure Appl. Chem., 2014, 86, 747–753 CrossRef CAS.
- T.-C. Wu, M.-K. Chen, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 1289–1293 CrossRef CAS PubMed.
- R.-F. Chen, C. Zheng, Q.-L. Fan and W. Huang, J. Comput. Chem., 2007, 28, 2091–2101 CrossRef CAS PubMed.
- S. Yamaguchi and K. Tamao, J. Chem. Soc., Dalton Trans., 1998, 3693–3702 RSC.
- J. C. Sanchez, S. A. Urbas, S. J. Toal, A. G. DiPasquale, A. L. Rheingold and W. C. Trogler, Macromolecules, 2008, 41, 1237–1245 CrossRef CAS.
- K. Tamura, M. Shiotsuki, N. Kobayashi, T. Masuda and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3506–3517 CrossRef CAS.
- S. M. King, I. I. Perepichka, I. F. Perepichka, F. B. Dias, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 586–591 CrossRef CAS.
- J. You, T. Yan, H. Zhao, Z. Sun, L. Xia, Y. Suo and Y. Li, Anal. Chim. Acta, 2009, 636, 95–104 CrossRef CAS PubMed.
- J. C. Sanchez, A. G. DiPasquale, A. A. Mrse and W. C. Trogler, Anal. Bioanal. Chem., 2009, 395, 387–392 CrossRef CAS PubMed.
- B. M. Schmidt, B. Topolinski, M. Yamada, S. Higashibayashi, M. Shionoya, H. Sakurai and D. Lentz, Chem. – Eur. J., 2013, 19, 13872–13880 CrossRef CAS PubMed.
- B. M. Schmidt, B. Topolinski, S. Higashibayashi, T. Kojima, M. Kawano, D. Lentz and H. Sakurai, Chem. – Eur. J., 2013, 19, 3282–3286 CrossRef CAS PubMed.
- I. V. Kuvychko, S. N. Spisak, Y.-S. Chen, A. A. Popov, M. A. Petrukhina, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2012, 51, 4939–4942 CrossRef CAS PubMed.
- B. M. Schmidt, S. Seki, B. Topolinski, K. Ohkubo, S. Fukuzumi, H. Sakurai and D. Lentz, Angew. Chem., Int. Ed., 2012, 51, 11385–11388 CrossRef CAS PubMed.
- I. V. Kuvychko, C. Dubceac, S. H. M. Deng, X.-B. Wang, A. A. Granovsky, A. A. Popov, M. A. Petrukhina, S. H. Strauss and O. V. Boltalina, Angew. Chem., Int. Ed., 2013, 52, 7505–7508 CrossRef CAS PubMed.
- K. Shoyama, D. Schmidt, M. Mahl and F. Würthner, Org. Lett., 2017, 19, 5328–5331 CrossRef CAS PubMed.
- Y.-L. Wu, M. C. Stuparu, C. Boudon, J.-P. Gisselbrecht, W. B. Schweizer, K. K. Baldridge, J. S. Siegel and F. Diederich, J. Org. Chem., 2012, 77, 11014–11026 CrossRef CAS PubMed.
- J. Li, A. Terec, Y. Wang, H. Joshi, Y. Lu, H. Sun and M. C. Stuparu, J. Am. Chem. Soc., 2017, 139, 3089–3094 CrossRef CAS PubMed.
- C. S. Jones, E. Elliott and J. S. Siegel, Synlett, 2004, 187–191 CAS.
- Y.-T. Wu, D. Bandera, R. Maag, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2008, 130, 10729–10739 CrossRef CAS PubMed.
- J. Mack, P. Vogel, D. Jones, N. Kaval and A. Sutton, Org. Biomol. Chem., 2007, 5, 2448–2452 CAS.
- J. Li, A. Terec, Y. Wang, H. Joshi, Y. Lu, H. Sun and M. Stuparu, J. Am. Chem. Soc., 2017, 139, 3089–3094 CrossRef CAS PubMed.
- H. Joshi, S. Sreejith, R. Dey and M. Stuparu, RSC Adv., 2016, 6, 110001–110003 RSC.
- S. Sreejith, N. V. Menon, Y. Wang, H. Joshi, S. Liu, K. C. Chong, Y. Kang, H. Sun and M. C. Stuparu, Mater. Chem. Front., 2017, 1, 831–837 RSC.
- D. Wu, T. Shao, J. Men, X. Chen and G. Gao, Dalton Trans., 2014, 43, 1753–1761 RSC.
- T. Amaya, M. Hifumi, M. Okada, Y. Shimizu, T. Moriuchi, K. Segawa, Y. Ando and T. Hirao, J. Org. Chem., 2011, 76, 8049–8052 CrossRef CAS PubMed.
- B. B. Shrestha, Y. Morita, T. Kojima, M. Kawano, S. Higashibayashi and H. Sakurai, Chem. Lett., 2014, 43, 1294–1296 CrossRef CAS.
- S. Higashibayashi, B. B. Shrestha, Y. Morita, M. Ehara, K. Ohkubo, S. Fukuzumi and H. Sakurai, Chem. Lett., 2014, 43, 1297–1299 CrossRef CAS.
- K. Kanahara, M. D. M. R. Badal, S. Hatano, M. Abe, S. Higashibayashi, N. Takashina and H. Sakurai, Bull. Chem. Soc. Jpn., 2015, 88, 1612–1617 CrossRef CAS.
- Y. Morita, S. Nakao, S. Haesuwannakij, S. Higashibayashi and H. Sakurai, Chem. Commun., 2012, 48, 9050–9052 RSC.
- R. Temirov, S. Soubatch, A. Luican and F. S. Tautz, Nature, 2006, 444, 350–353 CrossRef CAS PubMed.
- M. Feng, J. Zhao and H. Petek, Science, 2008, 320, 359–362 CrossRef CAS PubMed.
- J. Zhao, M. Feng, J. Yang and H. Petek, ACS Nano, 2009, 3, 853–864 CrossRef CAS PubMed.
- M. Feng, J. Zhao, T. Huang, X. Zhu and H. Petek, Acc. Chem. Res., 2011, 44, 360–368 CrossRef CAS PubMed.
- N. Pastukhova, L. M. Samos, L. Zoppi, E. Pavlica, J. Mathew, G. Bratina, J. S. Siegel and K. K. Baldridge, RSC Adv., 2017, 7, 45601–45606 RSC.
- T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408–409 CrossRef CAS PubMed.
- S. Higashibayashi, R. Tsuruoka, Y. Soujanya, U. Purushotham, G. N. Sastry, S. Seki, T. Ishikawa, S. Toyota and H. Sakurai, Bull. Chem. Soc. Jpn., 2012, 85, 450–467 CrossRef CAS.
- S. Sanyal, A. K. Manna and S. K. Pati, ChemPhysChem, 2014, 15, 885–893 CrossRef CAS PubMed.
- B.-T. Wang, M. A. Petrukhina and E. R. Margine, Carbon, 2015, 94, 174–180 CrossRef CAS.
- R. Chen, R.-Q. Lu, P.-C. Shi and X.-Y. Cao, Chin. Chem. Lett., 2016, 27, 1175–1183 CrossRef CAS.
- K. Shi, T. Lei, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Sci., 2014, 5, 1041–1045 RSC.
- R. Chen, R.-Q. Lu, K. Shi, F. Wu, H.-X. Fang, Z.-X. Niu, X.-Y. Yan, M. Luo, X.-C. Wang, C.-Y. Yang, X.-Y. Wang, B. Xu, H. Xia, J. Pei and X.-Y. Cao, Chem. Commun., 2015, 51, 13768–13771 RSC.
- R.-Q. Lu, Y.-N. Zhou, X.-Y. Yan, K. Shi, Y.-Q. Zheng, M. Luo, X.-C. Wang, J. Pei, H. Xia, L. Zoppi, K. K. Baldridge, J. S. Siegel and X.-Y. Cao, Chem. Commun., 2015, 51, 1681–1684 RSC.
- R.-Q. Lu, W. Xuan, Y.-Q. Zheng, Y.-N. Zhou, X.-Y. Yan, J.-H. Dou, R. Chen, J. Pei, W. Weng and X.-Y. Cao, RSC Adv., 2014, 4, 56749–56755 RSC.
- R.-Q. Lu, Y.-Q. Zheng, Y.-N. Zhou, X.-Y. Yan, T. Lei, K. Shi, Y. Zhou, J. Pei, L. Zoppi, K. K. Baldridge and J. S. Siegel, J. Mater. Chem. A, 2014, 2, 20515–20519 CAS.
- Y. Deng, B. Xu, E. Castro, O. Fernandez-Delgado, L. Echegoyen, K. K. Baldridge and J. S. Siegel, Eur. J. Org. Chem., 2017, 4338–4342 CrossRef CAS.
- A. Steinauer, A. M. Butterfield, A. Linden, A. Molina-Ontario, D. C. Buck, R. W. Cotta, L. Echegoyen, K. K. Baldridge and J. S. Siegel, J. Braz. Chem. Soc., 2016, 27, 1866–1871 CAS.
- A. Mishra, M. Ulaganathan, E. Edison, P. Borah, A. Mishra, S. Sreejith, S. Madhavi and M. C. Stuparu, ACS Macro Lett., 2017, 6, 1212–1216 CrossRef CAS.
- Y. Inada, T. Amaya, Y. Shimizu, A. Saeki, T. Otsuka, R. Tsuji, S. Seki and T. Hirao, Chem. – Asian J., 2013, 8, 2569–2574 CrossRef CAS PubMed.
- T. Amaya, Y. Inada, Y. Shimizu, A. Saeki, R. Tsuji, S. Seki and T. Hirao, Chem. – Asian J., 2014, 9, 2568–2575 CrossRef CAS PubMed.
- Y. Inada, T. Amaya, A. Saeki, S. Seki and T. Hirao, Bull. Chem. Soc. Jpn., 2015, 88, 330–332 CrossRef.
- J. Fujita, M. Tachi, K. Murakami, H. Sakurai, Y. Morita, S. Higashibayashi and M. Takeguchi, Appl. Phys. Lett., 2014, 104, 043107 Search PubMed.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS PubMed.
- V. H. Le, M. Yanney, M. McGuire, A. Sygula and E. A. Lewis, J. Phys. Chem. B, 2014, 118, 11956–11964 CrossRef CAS PubMed.
- M. Yanney and A. Sygula, Tetrahedron Lett., 2013, 54, 2604–2607 CrossRef CAS.
- M. Yanney, F. R. Fronczek and A. Sygula, Angew. Chem., Int. Ed., 2015, 54, 11153–11156 CrossRef CAS PubMed.
- P. L. Abeyratne Kuragama, F. R. Fronczek and A. Sygula, Org. Lett., 2015, 17, 5292–5295 CrossRef CAS PubMed.
- K. G. U. R. Kumarasinghe, F. R. Fronczek, H. U. Valle and A. Sygula, Org. Lett., 2016, 18, 3054–3057 CrossRef CAS PubMed.
- C. M. Álvarez, L. A. García-Escudero, R. García-Rodríguez, J. M. Martín-Álvarez, D. Miguel and V. M. Rayón, Dalton Trans., 2014, 43, 15693–15696 RSC.
- C. M. Álvarez, G. Aullón, H. Barbero, L. A. García-Escudero, C. Martínez-Pérez, J. M. Martín-Álvarez and D. Miguel, Org. Lett., 2015, 17, 2578–2581 CrossRef PubMed.
- H. Barbero, S. Ferrero, L. Álvarez-Miguel, P. Gómez-Iglesias, D. Miguel and C. M. Álvarez, Chem. Commun., 2016, 52, 12964–12967 RSC.
- C. M. Álvarez, H. Barbero, S. Ferrero and D. Miguel, J. Org. Chem., 2016, 81, 6081–6086 CrossRef PubMed.
- M. C. Stuparu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2641–2649 CrossRef CAS.
- M. C. Stuparu, Angew. Chem., Int. Ed., 2013, 52, 7786–7790 CrossRef CAS PubMed.
- S. Mizyed, P. E. Georghiou, M. Bancu, B. Cuadra, A. K. Rai, P. Cheng and L. T. Scott, J. Am. Chem. Soc., 2001, 123, 12770–12774 CrossRef CAS PubMed.
- P. E. Georghiou, A. H. Tran, S. Mizyed, M. Bancu and L. T. Scott, J. Org. Chem., 2005, 70, 6158–6163 CrossRef CAS PubMed.
- H. Becker, G. Javahery, S. Petrie, P.-C. Cheng, H. Schwarz, L. T. Scott and D. K. Bohme, J. Am. Chem. Soc., 1993, 115, 11636–11637 CrossRef CAS.
- W. Xiao, D. Passerone, P. Ruffieux, K. Aït-Mansour, O. Gröning, E. Tosatti, J. S. Siegel and R. Fasel, J. Am. Chem. Soc., 2008, 130, 4767–4771 CrossRef CAS PubMed.
- L. N. Dawe, T. A. AlHujran, H.-A. Tran, J. I. Mercer, E. A. Jackson, L. T. Scott and P. E. Georghiou, Chem. Commun., 2012, 48, 5563–5565 RSC.
- Y. Wang, Y. Li, W. Zhu, J. Liu, X. Zhang, R. Li, Y. Zhen, H. Dong and W. Hu, Nanoscale, 2016, 8, 14920–14924 RSC.
- A. S. Filatov, M. V. Ferguson, S. N. Spisak, B. Li, C. F. Campana and M. A. Petrukhina, Cryst. Growth Des., 2014, 14, 756–762 CAS.
- L. M. Roch, L. Zoppi, J. S. Siegel and K. K. Baldridge, J. Phys. Chem. C, 2017, 121, 1220–1234 CAS.
- Y.-Z. Liu, K. Yuan, Z. Yuan, Y.-C. Zhu, S.-D. Zhao and L.-L. Lv, RSC Adv., 2017, 7, 27960–27968 RSC.
- Y. Shoji, T. Kajitani, F. Ishiwari, Q. Ding, H. Sato, H. Anetai, T. Akutagawa, H. Sakurai and T. Fukushima, Chem. Sci., 2017, 8, 8405–8410 RSC.
- M. Yamada, K. Ohkubo, M. Shionoya and S. Fukuzumi, J. Am. Chem. Soc., 2014, 136, 13240–13248 CrossRef CAS PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, V. M. Grigoryants and M. A. Petrukhina, Chem. – Eur. J., 2012, 18, 6476–6484 CrossRef CAS PubMed.
- A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Organometallics, 2012, 31, 5541–5545 CrossRef CAS.
- A. V. Zabula, S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Angew. Chem., Int. Ed., 2012, 51, 12194–12198 CrossRef CAS PubMed.
- S. N. Spisak, A. V. Zabula, M. V. Ferguson, A. S. Filatov and M. A. Petrukhina, Organometallics, 2013, 32, 538–543 CrossRef CAS.
- T. Bauert, L. Zoppi, G. Koller, J. S. Siegel, K. K. Baldridge and K.-H. Ernst, J. Am. Chem. Soc., 2013, 135, 12857–12860 CrossRef CAS PubMed.
- A. S. Filatov, A. V. Zabula, S. N. Spisak, A. Y. Rogachev and M. A. Petrukhina, Angew. Chem., Int. Ed., 2014, 53, 140–145 CrossRef CAS PubMed.
- A. S. Filatov, S. N. Spisak, A. V. Zabula, J. McNeely, A. Y. Rogachev and M. A. Petrukhina, Chem. Sci., 2015, 6, 1959–1966 RSC.
- A. V. Zabula, S. N. Spisak, A. S. Filatov, A. Y. Rogachev, R. Clérac and M. A. Petrukhina, Chem. Sci., 2016, 7, 1954–1961 RSC.
- S. N. Spisak, Z. Wei, N. J. O’Neil, A. Y. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, J. Am. Chem. Soc., 2015, 137, 9768–9771 CrossRef CAS PubMed.
- S. N. Spisak, Z. Wei, A. Y. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, Angew. Chem., Int. Ed., 2017, 56, 2582–2587 CrossRef CAS PubMed.
- R. M. Chin, M. S. Jarosh, J. D. Russell and R. J. Lachicotte, Organometallics, 2002, 21, 2027–2029 CrossRef CAS.
- T. Amaya, H. Sakane and T. Hirao, Angew. Chem., Int. Ed., 2007, 46, 8376–8379 CrossRef CAS PubMed.
- H. Sakane, T. Amaya, T. Moriuchi and T. Hirao, Angew. Chem., Int. Ed., 2009, 48, 1640–1643 CrossRef CAS PubMed.
- T. Amaya, W.-Z. Wang, H. Sakane, T. Moriuchi and T. Hirao, Angew. Chem., Int. Ed., 2010, 49, 403–406 CrossRef CAS PubMed.
- T. Amaya, Y. Takahashi, T. Moriuchi and T. Hirao, J. Am. Chem. Soc., 2014, 136, 12794–12798 CrossRef CAS PubMed.
- B. Topolinski, B. M. Schmidt, M. Kathan, S. I. Troyanov and D. Lentz, Chem. Commun., 2012, 48, 6298–6300 RSC.
- B. Topolinski, B. M. Schmidt, S. Higashibayashi, H. Sakurai and D. Lentz, Dalton Trans., 2013, 42, 13809–13812 RSC.
- B. Topolinski, B. M. Schmidt, S. Schwagerus, M. Kathan and D. Lentz, Eur. J. Inorg. Chem., 2014, 5391–5405 CrossRef CAS.
- M. Yamada, S. Tashiro, R. Miyake and M. Shionoya, Dalton Trans., 2013, 42, 3300–3303 RSC.
- J. Gao and F. Ding, J. Phys. Chem. C, 2015, 119, 11086–11093 CAS.
- M. Parschau, R. Fasel, K.-H. Ernst, O. Gröning, L. Brandenberger, R. Schillinger, T. Greber, A. P. Seitsonen, Y.-T. Wu and J. S. Siegel, Angew. Chem., Int. Ed., 2007, 46, 8258–8261 CrossRef CAS PubMed.
- L. Merz, M. Parschau, L. Zoppi, K. K. Baldridge, J. S. Siegel and K.-H. Ernst, Angew. Chem., Int. Ed., 2009, 48, 1966–1969 CrossRef CAS PubMed.
- T. Bauert, K. K. Baldridge, J. S. Siegel and K.-H. Ernst, Chem. Commun., 2011, 47, 7995–7997 RSC.
- P. Angelova, E. Solel, G. Parvari, A. Turchanin, M. Botoshansky, A. Gölzhäuser and E. Keinan, Langmuir, 2013, 29, 2217–2223 CrossRef CAS PubMed.
- Q. S. Stöckl, Y.-C. Hsieh, A. Mairena, Y.-T. Wu and K.-H. Ernst, J. Am. Chem. Soc., 2016, 138, 6111–6114 CrossRef PubMed.
- R. Jaafar, C. A. Pignedoli, G. Bussi, K. Aït-Mansour, O. Groening, T. Amaya, T. Hirao, R. Fasel and P. Ruffieux, J. Am. Chem. Soc., 2014, 136, 13666–13671 CrossRef CAS PubMed.
- S. Fujii, M. Ziatdinov, S. Higashibayashi, H. Sakurai and M. Kiguchi, J. Am. Chem. Soc., 2016, 138, 12142–12149 CrossRef CAS PubMed.
- O. Guillermet, E. Niemi, S. Nagarajan, X. Bouju, D. Martrou, A. Gourdon and S. Gauthier, Angew. Chem., Int. Ed., 2009, 48, 1970–1973 CrossRef CAS PubMed.
- L. Zoppi, T. Bauert, J. S. Siegel, K. K. Baldridge and K.-H. Ernst, Phys. Chem. Chem. Phys., 2012, 14, 13365–13369 RSC.
- Q. Stöckl, D. Bandera, C. S. Kaplan, K.-H. Ernst and J. S. Siegel, J. Am. Chem. Soc., 2013, 136, 606–609 CrossRef PubMed.
- W. Xiao, K.-H. Ernst, K. Palotas, Y. Zhang, E. Bruyer, L. Peng, T. Greber, W. A. Hofer, L. T. Scott and R. Fasel, Nat. Chem., 2016, 8, 326–330 CrossRef CAS PubMed.
- D. Miyajima, K. Tashiro, F. Araoka, H. Takezoe, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2009, 131, 44–45 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- S. H. Mahadevegowda and M. Stuparu, Eur. J. Org. Chem., 2017, 570–576 CrossRef CAS.
- S. H. Mahadevegowda and M. Stuparu, ACS Omega, 2017, 2, 4964–4971 CrossRef CAS.
- M. Mattarella, J. M. Haberl, J. Ruokolainen, E. M. Landau, R. Mezzenga and J. S. Siegel, Chem. Commun., 2013, 49, 7204–7206 RSC.
- M. Mattarella, L. Berstis, K. K. Baldridge and J. S. Siegel, Bioconjugate Chem., 2014, 25, 115–128 CrossRef CAS PubMed.
- N. Kishi, Z. Li, Y. Sei, M. Akita, K. Yoza, J. S. Siegel and M. Yoshizawa, Chem. – Eur. J., 2013, 19, 6313–6320 CrossRef CAS PubMed.
- B. M. Schmidt, T. Osuga, T. Sawada, M. Hoshino and M. Fujita, Angew. Chem., Int. Ed., 2016, 55, 1561–1564 CrossRef CAS PubMed.
- M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed.
- Y. Kim and M. Lee, Chem. – Eur. J., 2015, 21, 5736–5740 CrossRef CAS PubMed.
- W. B. Fellows, A. M. Rice, D. E. Williams, E. A. Dolgopolova, A. K. Vannucci, P. J. Pellechia, M. D. Smith, J. A. Krause and N. B. Shustova, Angew. Chem., Int. Ed., 2016, 55, 2195–2199 CrossRef CAS PubMed.
- A. M. Rice, W. B. Fellows, E. A. Dolgopolova, A. B. Greytak, A. K. Vannucci, M. D. Smith, S. G. Karakalos, J. A. Krause, S. M. Avdoshenko, A. A. Popov and N. B. Shustova, Angew. Chem., Int. Ed., 2017, 56, 4525–4529 CrossRef CAS PubMed.
- I. Hisaki, H. Toda, H. Sato, N. Tohnai and H. Sakurai, Angew. Chem., Int. Ed., 2017, 56, 15294–15298 CrossRef CAS PubMed.
- A. Meller and A. Ossko, Monatsh. Chem., 1972, 103, 150–155 CrossRef CAS.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed.
- C. G. Claessens, D. González-Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed.
- S. Shimizu and N. Kobayashi, Chem. Commun., 2014, 50, 6949–6966 RSC.
- Y. Inokuma, J. H. Kwon, T. K. Ahn, M.-C. Yoo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 961–964 CrossRef CAS PubMed.
- N. Kobayashi, Y. Takeuchi and A. Matsuda, Angew. Chem., Int. Ed., 2007, 46, 758–760 CrossRef CAS PubMed.
- Y. Takeuchi, A. Matsuda and N. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8271–8281 CrossRef CAS PubMed.
- Y. Inokuma, Z. S. Yoon, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 4747–4761 CrossRef CAS PubMed.
- T. Torres, Angew. Chem., Int. Ed., 2006, 45, 2834–2837 CrossRef CAS PubMed.
- S. Shimizu, Chem. Rev., 2017, 117, 2730–2784 CrossRef CAS PubMed.
- Y. Inokuma and A. Osuka, Dalton Trans., 2008, 2517–2526 RSC.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542–8545 CrossRef CAS PubMed.
- J.-Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 3096–3101 CrossRef CAS PubMed.
- S. Fujii, S. Marqués-González, J.-Y. Shin, H. Shinokubo, T. Masuda, T. Nishino, N. P. Arasu, H. Vázquez and M. Kiguchi, Nat. Commun., 2017, 8, 15984 CrossRef CAS PubMed.
- T. Yonezawa, S. A. Shafie, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2017, 56, 11822–11825 CrossRef CAS PubMed.
- V. M. Tsefrikas, A. K. Greene and L. T. Scott, Org. Chem. Front., 2017, 4, 688–698 RSC.
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082–7084 CrossRef CAS.
- V. M. Tsefrikas, S. Arns, P. M. Merner, C. C. Warford, B. L. Merner, L. T. Scott and G. J. Bodwell, Org. Lett., 2006, 8, 5195–5198 CrossRef CAS PubMed.
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed.
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed.
- H. Yokoi, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Sci., 2018, 9, 819–824 RSC.
- S. Higashibayashi, P. Pandit, R. Haruki, S.-I. Adachi and R. Kumai, Angew. Chem., Int. Ed., 2016, 55, 10830–10834 CrossRef CAS PubMed.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed.
- P. Kaewmati, Q. Tan, S. Higashibayashi, Y. Yakiyama and H. Sakurai, Chem. Lett., 2016, 46, 146–148 CrossRef.
- P. Kaewmati, Y. Yakiyama, H. Ohtsu, M. Kawano, S. Haesuwannakij, S. Higashibayashi and H. Sakurai, Mater. Chem. Front., 2018 10.1039/C7QM00530J.
- Q. Tan, P. Kaewmati, S. Higashibayashi, M. Kawano, Y. Yakiyama and H. Sakurai, Bull. Chem. Soc. Jpn., 2018 DOI:10.1246/bcsj.20170384.
- U. D. Priyakumar and G. N. Sastry, J. Org. Chem., 2001, 66, 6523 CrossRef CAS PubMed.
- S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192 CrossRef CAS PubMed.
- S. Furukawa, J. Kobayashi and T. Kawashima, Dalton Trans., 2010, 39, 9329 RSC.
- T. Tanikawa, M. Saito, J. D. Guo and S. Nagase, Org. Biomol. Chem., 2011, 9, 1731 CAS.
- D. Zhou, Y. Gao, B. Liu, Q. Tan and B. Xu, Org. Lett., 2017, 19, 4628 CrossRef CAS PubMed.
- Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279 RSC.
- K. Imamura, K. Takimiya, T. Otsubo and Y. Aso, Chem. Commun., 1999, 1859 RSC.
- M. Saito, T. Tanikawa, T. Tajima, J. D. Guo and S. Nagase, Tetrahedron Lett., 2010, 51, 672 CrossRef CAS.
- X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535 CrossRef CAS PubMed.
- H. Zeng, X. Zhang, Y. Deng, Y. Chang, X. Shao, X. Xu and Z. Li, Synlett, 2016, 442 CAS.
- S. Furukawa, Y. Suda, J. Kobayashi, T. Kawashima, T. Tada, S. Fujii, M. Kiguchi and M. Saito, J. Am. Chem. Soc., 2017, 139, 5787 CrossRef CAS PubMed.
- S. Wang, X. Li, X. Hou, Y. Sun and X. Shao, Chem. Commun., 2016, 52, 14486 RSC.
- T. Tanikawa, M. Saito, J. D. Guo, S. Nagase and M. Minoura, Eur. J. Org. Chem., 2012, 7135 CrossRef CAS.
- J. C. Sanchez, A. G. DiPasquale, A. L. Rheingold and W. C. Trogler, Chem. Mater., 2007, 19, 6459 CrossRef CAS.
- J. C. Sanchez and W. C. Trogler, J. Mater. Chem., 2008, 18, 3143 RSC.
- H. Sirringhaus, R. H. Friend, C. Wang, J. Leuninger and K. Müllen, J. Mater. Chem., 1999, 9, 2095 RSC.
- J. Gao, L. Li, Q. Meng, R. Li, H. Jiang, H. Li and W. Hu, J. Mater. Chem., 2007, 17, 1421 RSC.
- X. Hou, Y. Zhu, Y. Qin, L. Chen, X. Li, H.-L. Zhang, W. Xu, D. Zhu and X. Shao, Chem. Commun., 2017, 53, 1546 RSC.
- J. Nakayama, H. Nagasawa, Y. Sugihara and A. Ishii, J. Am. Chem. Soc., 1997, 119, 9077 CrossRef CAS.
- Y. Sun, X. Li, C. Sun, H. Shen, X. Hou, D. Lin, H.-L. Zhang, C.-A. Di, D. Zhu and X. Shao, Angew. Chem., Int. Ed., 2017, 56, 13470 CrossRef CAS PubMed.
- B. del Rey, U. Keller, T. Torres, G. Rojo, F. Agulló-López, S. Nonell, C. Martí, S. Brasselet, I. Ledoux and J. Zyss, J. Am. Chem. Soc., 1998, 120, 12808 CrossRef CAS.
- H. Fukagawa, H. Yamane, S. Kera, K. K. Okudaira and N. Ueno, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 041302 CrossRef.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971 CrossRef PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed.
- B. Fu, X. Hou, C. Wang, Y. Wang, X. Zhang, R. Li, X. Shao and W. Hu, Chem. Commun., 2017, 53, 11407–11409 RSC.
- K. Shi, T. Lei, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Sci., 2014, 5, 1041 RSC.
- R.-Q. Lu, Y.-Q. Zheng, Y.-N. Zhou, X.-Y. Yan, T. Lei, K. Shi, Y. Zhou, J. Pei, L. Zoppi, K. K. Baldridge, J. S. Siegel and X.-Y. Cao, J. Mater. Chem. A, 2014, 2, 20515 CAS.
- R.-Q. Lu, Y.-N. Zhou, X.-Y. Yan, K. Shi, Y.-Q. Zheng, M. Luo, X.-C. Wang, J. Pei, H. Xia, L. Zoppi, K. K. Baldridge, J. S. Siegel and X.-Y. Cao, Chem. Commun., 2015, 51, 1681 RSC.
- F. C. Krebs, P. S. Larsen, J. Larsen, C. S. Jacobsen, C. Boutton and N. Thorup, J. Am. Chem. Soc., 1997, 119, 1208 CrossRef CAS.
- M. Yamamura, T. Saito and T. Nabeshima, J. Am. Chem. Soc., 2014, 136, 14299 CrossRef CAS PubMed.
- M. Yamamura, K. Sukegawa and T. Nabeshima, Chem. Commun., 2015, 51, 12080 RSC.
- M. Yamamura, T. Hasegawa and T. Nabeshima, Org. Lett., 2016, 18, 816 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Hiroshi
Shinokubo
*a,
Hiroshi
Shinokubo