DOI:
10.1039/C7QM00536A
(Research Article)
Mater. Chem. Front., 2018,
2, 796-806
Photocatalytic activity of SnO2–α-Fe2O3 composite mixtures: exploration of number of active sites, turnover number and turnover frequency†
Received
20th November 2017
, Accepted 27th January 2018
First published on 29th January 2018
Introduction
The concept of combining two or more semiconductors with varying band gaps to form a heterojunction has attracted the attention of many researchers. Such composites show several unique properties that are otherwise not shown by their individual counterparts. Researchers have reported the preparation of several composite mixtures by taking two or more semiconductors in different proportions, so that the resultant mixture is expected to show unique properties especially aiming to reduce the rate of recombination of photogenerated charge carriers and also to extend the photoresponse to the visible region.1 The other goals to achieve efficiency are high photo-oxidative capacity, photo-stability, non-toxicity and reproducibility. Such materials are expected to be used for solar energy conversion, H2-production and for environmental remediation processes. In the past decade, efforts were made to improve the efficiency of SnO2 by combining it with various other metal oxides like TiO2, ZnO etc.2 SnO2 is a direct wide band gap semiconductor that does not show any absorption in the visible region and shows a comparatively lower quantum efficiency in the UV region. While designing a catalytic reaction system, it is of great importance to establish both the activities and stabilities of the studied catalysts.3 SnO2 and α-Fe2O3 are two metal oxides possessing high and low band gap energies respectively, which are amphoteric in nature when they are used individually as photocatalysts. One of the limitations of the use of these metal oxides is the high rate of recombination of photogenerated charge carriers.4 In the case of α-Fe2O3, the diffusion path length of holes is very short (2–4 nm) and its oxidation ability is low.5 Other drawbacks of α-Fe2O3 include the low energy conversion efficiency and small optical absorption coefficient.6 However, when SnO2 is coupled with α-Fe2O3, it is observed that the composite possesses unique catalytic, optical and electronic properties. In the present research work, an attempt has been made to show that an extremely small amount of catalyst was capable of providing a considerable increase in the rate of the degradation reaction. Emphasis should be made here that no specific definition can be given for a catalyst that acts at a very low concentration. However, the effectiveness of the catalyst at very low concentrations can be provided by terms referred to as turn-over-number (TON) and turn-over-frequency (TOF). Both these factors can vary with several reaction parameters like temperature, concentration etc.
Experimental
Chemicals used
Tin chloride (SnCl2·2H2O) was obtained from Merck Chemicals Limited. Ferric chloride (FeCl3·6H2O), ammonia (NH3) and phenol (C6H5OH) were obtained from SD Fine Chem. Limited. Hydrogen peroxide (H2O2) (30%, w/v) was obtained from Sisco-Chemical Industries. Double distilled water was used in all the experiments.
Preparation of the SnO2 photocatalyst
The adopted method for the preparation of SnO2 was that reported by K. Melghit et al.7 5 g of tin(II) dichloride (SnCl2·2H2O) was dissolved in 1 L of double distilled water. The solution was continuously stirred for 42 hours. In this extended period of stirring, the solution converted to a white cloudy mixture. During this process, a gelatinous milky white precipitate of tin hydroxide was formed. The mixture was allowed to settle. The gel at the bottom of the beaker can be easily separated from the solution by a decantation process. It was then washed several times with distilled water to completely remove all the chloride ions. The obtained precipitate was dried in an oven at 100 °C and further calcined at 400 °C for 4 h to get the crystalline sample of SnO2.| |  | (1) |
Preparation of α-Fe2O3 photocatalyst
The procedure adopted for the preparation of the α-Fe2O3 photocatalyst was that reported by N. C. Pramanik et al.8 5 g of FeCl3·6H2O was dissolved in 1 L of double distilled water. The solution was continuously stirred. Then, 10% aqueous NH3 solution was slowly added to the above solution until the pH condition changed to 2–3. A dark brown coloured metal hydroxide precipitate was formed and this precipitate was allowed to age in the solution for five days under ambient conditions. Excess double distilled water was added to the above suspension and stirred vigorously for 3 h. The precipitate was first washed thoroughly with a dilute NH3 solution and later with double distilled water until all the chloride ions were removed. The solid thus obtained was then dried in an oven at 100 °C followed by calcination at 400 °C for 4 h to get the crystalline α-Fe2O3 sample.| |  | (2) |
Preparation of mixed metal oxide composite samples
Individual metal oxides in various proportions were mixed to get the composite samples. During the process of the preparation of composites, calculated amounts of the catalysts were taken in a pestle and mortar and they were grinded for 2 h. Homogenization of the mixture was achieved by adding a small quantity of a volatile organic liquid like acetone or alcohol to form a paste. During the process of grinding and mixing, the organic liquid gradually volatilized and evaporated completely. For example, 0.75 g of SnO2 along with 0.25 g of α-Fe2O3 was grinded using a pestle and mortar for two hours and the sample was designated as Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe (0.75:0.25). The same procedure was adopted for the preparation of 0.5 g of SnO2 with 0.5 g of α-Fe2O3 and 0.25 g of SnO2 with 0.75 g of α-Fe2O3 and the prepared samples were designated as Sn:Fe (0.5:0.5) and Sn:Fe (0.25:0.75), respectively.
:Fe (0.75:0.25). The same procedure was adopted for the preparation of 0.5 g of SnO2 with 0.5 g of α-Fe2O3 and 0.25 g of SnO2 with 0.75 g of α-Fe2O3 and the prepared samples were designated as Sn:Fe (0.5:0.5) and Sn:Fe (0.25:0.75), respectively.
Characterization
Powder X-ray diffraction (PXRD) patterns were obtained by using an Analytical X’Pert Pro MPD: PW3204 diffractometer at a scan rate of 2° per minute in the 2θ range of 20–80° at room temperature using Cu Kα radiation as a source with a Ni filter. To study the light absorption characteristics of the prepared photocatalyst, the absorption spectra were recorded using a Shimadzu UV 3101 PC UV-VIS-NIR UV-Vis spectrophotometer in the range of 200–800 nm. The baseline correction and calibration were done by using a BaSO4 sample. The obtained absorbance (A) data were transformed into reflectance values (R∞ = 10−A). The Kubelka–Munk method was used for the band gap calculation. The surface morphology was analyzed by scanning electron microscopy (SEM) coupled with EDAX using a TESCAN Vega 3 LMU SEM, operating at 25 kV on the specimen upon which a thin layer of platinum had been evaporated. The nitrogen adsorption–desorption isotherms were recorded on a Quantachrome® ASiQwin™ – automated gas sorption analyzer at 77.4 K liquid nitrogen temperature. The samples were degassed at 300 °C prior to the measurement. The surface area was measured by the Brunauer–Emmett–Teller (BET) method and the pore size distribution was calculated from the adsorption branch of the isotherm by the Barret–Joyner Halenda (BJH) model. The photoluminescence (PL) spectra were measured on a Hitachi F-7000 fluorescence spectrophotometer. A multi N/C 2100 analyzer (analytic Jena, Germany) was used to determine the total organic carbon (TOC).
Experimental setup for photochemical measurements
A 125 W medium-pressure mercury vapour lamp was used as the UV light source whose emitting wavelength is in the range of 250–400 nm. The photon flux of the light source was found to be 7.68 mW cm−2 (as determined by ferrioxalate actinometry). A Pyrex glass reactor of 1 L capacity whose surface area is 176 cm2 was used for the experiment. The entire photoreactor system was maintained at 25 °C using a thermostat. The degradation experiment under solar light irradiation was carried out in an open air environment from 11 a.m. to 2 p.m. during bright sunny days. The latitude and longitude of Bangalore, India was found to be 12.58 N and 77.38 E, respectively. The intensity of sunlight was found to be around 1270 W m−2. A convex lens was used for focusing and concentrating the solar light intensity and the reaction mixture was exposed to this concentrated sunlight. In a typical experiment, 10 mg of the catalyst was finely dispersed in 250 mL of a 20 ppm phenol solution. The experiment involving an additional oxidant was performed with an optimized concentration of 10 ppm H2O2 solution. The test samples were taken out at different time intervals and the solution was centrifuged at 1400 rpm for 2 min and then filtered to remove the catalyst particles. The residual phenol concentration was estimated using a UV-vis spectrophotometer. The photocatalytic degradation of phenol was found to obey the first-order decay kinetics according to the following formula:| |  | (3) |
where C0 is the initial concentration of phenol (mole L−1), C is the concentration of the phenol at various specified time intervals (mole L−1), t is the time period of illumination (min) and k is the reaction rate constant.
Results and discussion
PXRD studies
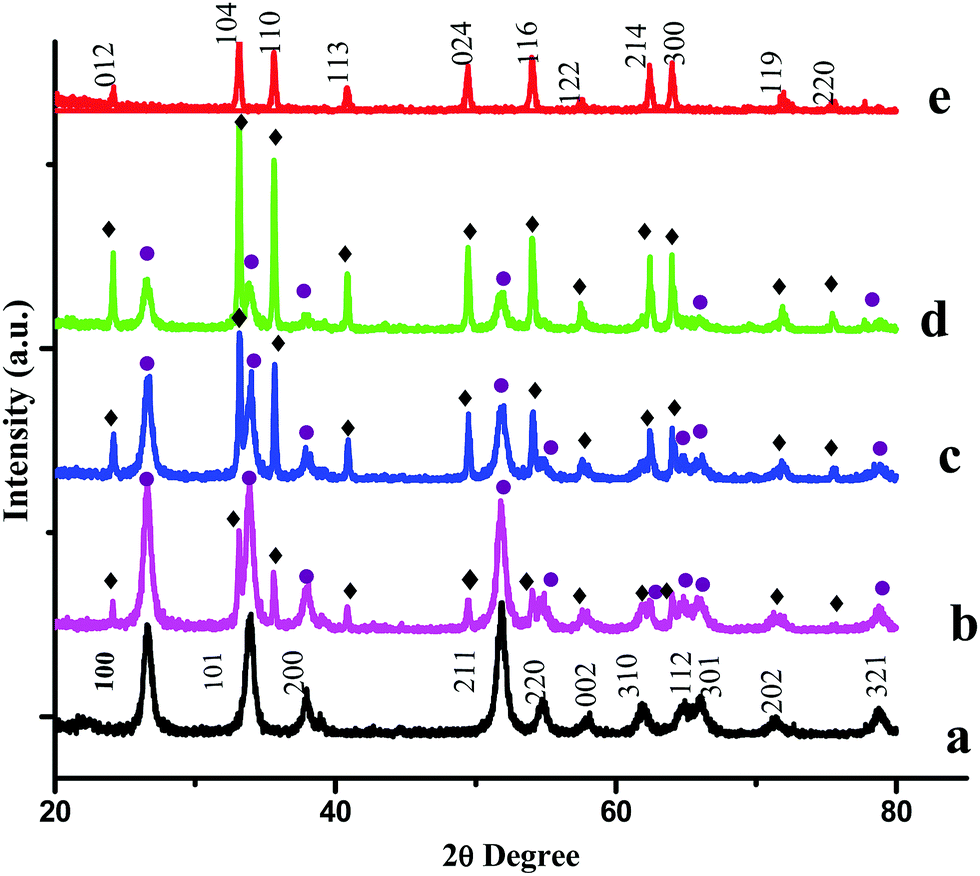
PXRD measurements were carried out to characterize the phases and crystal structures of the prepared metal oxides (Fig. 1). Fig. 1(a) shows the diffraction peaks of SnO2, which can be indexed to the tetragonal rutile structure (JCPDS card of SnO2, No. 41-1445) and the 2θ values (along with the hkl values) are: 26.611° (110), 33.893° (101), 37.950° (200), 51.781° (211), 54.759° (220), 57.820° (002), 61.872° (310), 64.719° (112), 65.939° (301), 71.278° (202), and 78.714° (321).9 The calculated lattice parameters for SnO2 were found to be a = 0.4738 nm and c = 0.3187 nm. α-Fe2O3 exhibits a rhombohedral structure (JCPDS card of α-Fe2O3, No. 33-0664) with 2θ diffraction peaks (along with corresponding hkl values) at 24.1° (012), 33.1° (104), 35.6° (110), 40.9° (113), 49.5° (024), 54.1° (116), 62.4° (214) and 64.0° (300).10 The calculated lattice parameters for α-Fe2O3 were found to be a = 0.5035 nm and c = 1.375 nm. The PXRD results confirmed the existence of both SnO2 and α-Fe2O3 in the composites. The PXRD peak intensities of the composites depend on the concentration of each constituent. The higher the concentration, the higher the corresponding peak intensity. Besides the qualitative analysis, a quantitative estimation of either the SnO2 or α-Fe2O3 content in the composites can be estimated from the following semi quantitative formula:11| |  | (4) |
Ci in the above equation represents the percentage concentration of either SnO2 or α-Fe2O3. Hi denotes the strongest characteristic peak intensity of either SnO2 or α-Fe2O3 in the SnO2–α-Fe2O3 composite. ∑Hi represents the summation of all the strongest characteristic peak intensities observed in the PXRD patterns of the composite (Table 1).
 |
| | Fig. 1 PXRD patterns of (a) SnO2, (b) Sn:Fe (0.75:0.25), (c) Sn:Fe (0.5:0.5), (d) Sn:Fe (0.25:0.75), and (e) α-Fe2O3 catalysts. The numbers on the peaks denote the standard (hkl) values. The SnO2 and Fe2O3 peaks are represented by ● and ♦ respectively. | |
Table 1 Calculated percentage phase composition of the prepared photocatalyst samples
| Photocatalysts |
% of SnO2 |
% of Fe2O3 |
| SnO2 |
100 |
— |
| Fe2O3 |
— |
100 |
| Sn:Fe (0.25:0.75) |
28.51 |
71.49 |
| Sn:Fe (0.5:0.5) |
52.93 |
47.07 |
| Sn:Fe (0.75:0.25) |
71.59 |
28.41 |
UV-visible absorbance studies
The optical absorption spectra in the range of 190–900 nm for the SnO2, α-Fe2O3 and SnO2–α-Fe2O3 composite mixture photocatalyst samples are shown in Fig. 2. The α-Fe2O3 and SnO2–α-Fe2O3 composite mixtures show strong absorption peaks in both the UV and visible regions, while SnO2 shows an absorption peak only in the UV region. The reddish brown colour of α-Fe2O3 indicates that the metal oxide is most probably hydrated. The hydrolysed α-Fe2O3 is known to show charge-transfer bands (CT) in the UV region. The edges of these CT bands appear in the ultraviolet region and are extended to the visible region, and the d–d transitions are also expected to show peaks in the visible region.12 The complete analysis of the CT bands and d–d transition bands is rather difficult. Sn4+ has a completely filled d-orbital electronic configuration of d10. The total spin quantum number in the z-direction can be represented as Ms and it is the summation of all individual ‘m’ values, which is equal to zero (S = 0) and the multiplicity 2S + 1 = 1. The total orbital angular momentum quantum number in the z-direction is ML = ∑m = 0 hence L = 0, which corresponds to the S state. The electrons of a closed shell always produce the singlet S state (1S).
 |
| | Fig. 2 UV-visible absorbance spectra of the α-Fe2O3, SnO2 and SnO2–α-Fe2O3 composites. | |
Fe3+ has a d5 electronic configuration. There are five unpaired electrons with parallel spins. Any electronic transition within the d orbital must involve a reversal of spin and such spin forbidden transitions should appear as a weak band in the absorption spectra. The ground state term symbol is 6S. Eleven excited states can be expected, if a reversing of the spin state of the electron occurs. The probability of such transitions is extremely small but still such forbidden transitions do occur. The eleven excited states are 4G, 4F, 4D, 4P, 2I, 2H, 2G, 2F, 2D, 2P and 2S. Among these states, the four quartets 4G, 4F, 4D and 4P involve the reversal of only one spin. The other seven states are doublets and are doubly spin forbidden and are still less probable. In an octahedral field, these four states split into ten states and extremely weak absorption bands can be observed. Many of the bands are broad and they are spin-allowed. The ground state 6S is not split and transforms to the 6A1g state. 4Eg(G), 4A1g, 4Eg(D) and 4A2g(F) energies are independent of the crystal field. Due to the continuous vibration, the crystal field strength varies about a mean value and hence broad absorption peaks are observed. The transition to the four states mentioned above will give rise to distinct peaks, whereas 4T1g(G) and 4T2g(G) give broader bands. They can be assigned as follows:13
| 6A1g → 4Eg and 6A1g → 4A1g both appear between 400.48–395.25 nm, |
| 6A1g → 4T2g(D) (357.14 nm) and 6A1g → 4Eg(D) (336.7 nm) |
The band gap energy (Eg) of each catalyst was determined by the Kubelka–Munk method, where, [F(R∞)hν]½ is plotted versus hν (expressed in eV as per eqn (7)) (Fig. S1) (ESI†). The Kubelka–Munk function F(R∞) and the photon energy (hν) in eV can be calculated by the following eqn (5)–(7):14
| | 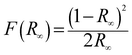 | (5) |
where
| |  | (7) |
where
R∞ is the reflection coefficient of the sample,
A is the absorbance intensity of the sample and
λ is the absorption wavelength. The calculated band gaps of α-Fe
2O
3, SnO
2, Sn
:
Fe (0.75
:
0.25), Sn
:
Fe (0.5
:
0.5) and Sn
:
Fe (0.25
:
0.75) samples were found to be 2.06 eV, 3.6eV, 2.29 eV, 2.22 eV and 2.06 eV, respectively. The obtained band gap values for SnO
2 and α-Fe
2O
3 are in good agreement with the reported values in the literature.
15–17
SEM and EDAX analysis
The SEM images of the SnO2 and Sn:Fe (0.75:0.25) composites are shown in Fig. 3a and b. The SEM image of SnO2 shows the morphology of the aggregated microspheres. The microspheres observed in the Sn:Fe (0.75:0.25) sample were larger. The EDAX analysis confirmed the presence of O and Sn in the SnO2 sample (Fig. S2(a)) (ESI†) and Fe, O and Sn in the Sn:Fe (0.75:0.25) composite sample (Fig. S2(b)) (refer ESI†) along with the weight percentage and atom percentage of each element (Table 2).
 |
| | Fig. 3 SEM images for (a) SnO2 and (b) the Sn:Fe (0.75:0.25) composite. | |
Table 2 The results of the EDX analysis of the SnO2 and Sn:Fe (0.75:0.25) composites
| Photocatalyst |
Element |
Weight (%) |
Atom (%) |
| SnO2 |
Sn |
86.59 |
53.46 |
| O |
13.41 |
46.54 |
|
|
| Sn:Fe (0.75:0.25) |
Sn |
26.02 |
56.73 |
| Fe |
8.88 |
2.61 |
| O |
65.11 |
40.66 |
BET studies
The nitrogen adsorption–desorption isotherms and the pore size distribution curves of the SnO2, α-Fe2O3 and SnO2–α-Fe2O3 samples are shown in Fig. 4a and b respectively. The surface areas of the catalysts were measured using the BET technique. The SnO2 and SnO2 rich composite samples exhibited a typical characteristic mesoporous structure, which was confirmed by the N2 gas sorption, which shows a type-IV adsorption isotherm with an elongated S-type hysteresis loop, according to the IUPAC classification. α-Fe2O3 also shows a type-IV adsorption isotherm, but shows comparatively less mesoporosity than SnO2.18 The BET surface area, pore volume and pore diameter were calculated by the BJH method for all the catalysts and the values are presented in Table 3. In the present investigation, the surface areas of the photocatalysts varied from 26 to13 m2 g−1. The SnO2 sample shows the highest surface area of 26.53 m2 g−1. Among all the prepared mixed composite photocatalysts, the BET surface area of the Sn:Fe (0.75:0.25) catalyst is maximum and is found to be 21.21 m2 g−1 with a pore volume and an average pore diameter of 0.06 cc g−1 and 10.41 nm, respectively. The Sn:Fe (0.5:0.5) and Sn:Fe (0.25:0.75) samples seem to be relatively less mesoporous with pore diameters of 2.5 nm and 1.6 nm, respectively.19 Based on the above surface morphology results, it can be clearly concluded that the SnO2, Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) samples are highly mesoporous with higher surface areas. The Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) composites form an organized aggregation. However, such an organized aggregation is not found in the Sn:Fe (0.5:0.5) composite and it does not show any intermediate characteristics between the other two mentioned composites, which may be due to the weak coordination interaction when the concentration of each component is increased. Preservation of the mesoporous structure naturally takes place for the Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) composites with distinct junction/interface properties. The mesoporosity of the α-Fe2O3 sample is much less compared to all the other samples and the pore diameter is found to be 2–3 nm.
 |
| | Fig. 4 (a) The N2 adsorption–desorption isotherms of the α-Fe2O3, SnO2 and SnO2–α-Fe2O3 photocatalysts. (b) The BJH pore size distribution curves of the α-Fe2O3, SnO2 and SnO2–α-Fe2O3 photocatalysts. | |
Table 3 BET surface areas, pore volumes and pore diameters of the SnO2, Fe2O3 and SnO2/α-Fe2O3 photocatalysts
| Catalysts |
Surface area (m2 g−1) |
Pore volume (cc g−1) |
Pore diameter (nm) |
| SnO2 |
26.53 |
0.09 |
14.64 |
| Fe2O3 |
18.11 |
0.02 |
2.36 |
| Sn:Fe (0.25:0.75) |
15.27 |
0.03 |
1.62 |
| Sn:Fe (0.5:0.5) |
13.63 |
0.02 |
2.49 |
| Sn:Fe (0.75:0.25) |
21.21 |
0.06 |
10.41 |
Photoluminescence studies
Photoluminescence spectra have been used to study the various charge recombination processes involving the photogenerated electron–hole pairs and in turn the mobility of the charge carriers. It gives a glimpse into the separation and recombination of photogenerated charge carriers for various transitions.10 PL emission results from the radiative recombination of excited electrons and holes. In other words, it is an essential requirement for a good photocatalyst to have a minimum electron–hole recombination and a low PL intensity spectrum.20 The PL spectrum of the SnO2 sample shows a strong band in the UV region due to the recombination of the photogenerated charge carriers from the valence band to the conduction band (Fig. 5 and the inset (A)). The origin of luminescence in the visible region for the SnO2 sample is due to the presence of interstitials and non-stoichiometric intrinsic defects (Fig. 5). The oxygen vacancies in the SnO2 sample that act as the radiative centres for luminescence in the visible region can be categorised into three different defect types: neutral oxygen vacancy V0, singly ionised oxygen vacancy  and doubly ionised oxygen vacancy
and doubly ionised oxygen vacancy  .21 V0 is a very shallow donor, which means it can donate and accept charge carriers more easily. These vacancies are usually associated with metal ions (Sn3+–V0) and are expected to be located below the SnO2 conduction band (∼0.3–0.4 eV). Excitation of an electron from the valence band to these states may result in recombination, which is responsible for the peaks observed in the visible region. The PL intensities of α-Fe2O3 and the composites containing α-Fe2O3 are shown in the inset (B) of Fig. 5. The PL intensity of pure α-Fe2O3 is high when compared to those of the composites containing α-Fe2O3. The recombination reactions are reduced considerably in the Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) composites. The PL intensities are found to be high for the pure SnO2 and α-Fe2O3 samples. But the intensities completely change when they are mixed in different proportions due to the synergistic effect of the band energy positions of these materials with respect to one another for the smooth transfer of charge carriers.
.21 V0 is a very shallow donor, which means it can donate and accept charge carriers more easily. These vacancies are usually associated with metal ions (Sn3+–V0) and are expected to be located below the SnO2 conduction band (∼0.3–0.4 eV). Excitation of an electron from the valence band to these states may result in recombination, which is responsible for the peaks observed in the visible region. The PL intensities of α-Fe2O3 and the composites containing α-Fe2O3 are shown in the inset (B) of Fig. 5. The PL intensity of pure α-Fe2O3 is high when compared to those of the composites containing α-Fe2O3. The recombination reactions are reduced considerably in the Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) composites. The PL intensities are found to be high for the pure SnO2 and α-Fe2O3 samples. But the intensities completely change when they are mixed in different proportions due to the synergistic effect of the band energy positions of these materials with respect to one another for the smooth transfer of charge carriers.
 |
| | Fig. 5 PL spectra of the SnO2, α-Fe2O3 and SnO2–α-Fe2O3 composites. Inset (A) PL spectrum of SnO2 in the UV region. Inset (B) Magnified PL spectra of α-Fe2O3 and the SnO2–α-Fe2O3 composites. | |
Photocatalytic degradation studies
Active sites, turnover number and turnover frequency.
The term active site is often applied to those sites for adsorption that are the effective sites for a particular heterogeneous catalytic reaction. The knowledge of the number of active sites can be used to describe an ensemble of sites at/on which a catalytic reaction takes place.22 The number of active sites depends on the concentration (expressed in moles) of metal ions in the specified amount of the catalyst (10 mg) taken for a particular reaction. For example, 10 mg of the SnO2 sample contains 6.57 × 10−5 moles of Sn, which is approximated to the number of active sites.
An extremely small amount of catalyst (10 mg) brings about a considerable increase in the rate of the reaction. The effectiveness of the catalyst is expressed in this reaction system as “turnover number” (TON), which is merely a number that does not have any units and can be defined as:3,23,24
| |  | (8) |
Percentage conversion (%) actually means the extent of the degradation of a substrate molecule, which can be calculated using the formula:  where, C0 and C have the same significance as defined in eqn (3). The detailed calculations of the number of moles of the substrate and catalyst are given in S3 (ESI†). The term turnover frequency (TOF) is expressed as a time inverse unit, which is actually the number of reaction cycles taking place in a given time period and is usually employed to study the reaction rate, which can be defined as:
where, C0 and C have the same significance as defined in eqn (3). The detailed calculations of the number of moles of the substrate and catalyst are given in S3 (ESI†). The term turnover frequency (TOF) is expressed as a time inverse unit, which is actually the number of reaction cycles taking place in a given time period and is usually employed to study the reaction rate, which can be defined as:
| |  | (9) |
Evaluation of photocatalytic activity
The photocatalytic activities of the SnO2, α-Fe2O3, Sn:Fe (0.25:0.75), Sn:Fe (0.5:0.5), and Sn:Fe (0.75:0.25) samples were studied in the degradation of phenol as a model pollutant under both UV and solar light irradiation. The pH was measured initially before the catalysis and also at the end of the experiment and it was found to be 6. No buffers or acid/bases were used to maintain the pH. The natural pH value of the reaction mixture was found to be around 6–7. A blank degradation experiment involving only phenol in the absence of any photocatalyst under UV/solar illumination did not show any contribution from direct photolysis. The adsorption characteristics of all the catalysts were studied by taking 10 mg of the catalyst dispersed in 250 mL of a 20 ppm phenol solution and the reaction mixture was stirred for 30 minutes before illumination to ensure the establishment of the adsorption/desorption equilibrium. The extent of adsorption qe (mg g−1) was calculated from the following equation:25| |  | (10) |
where C0 and C in the above eqn (10) are the phenol concentrations (mg L−1) before and after adsorption, V is the volume (250 mL) of the reaction mixture and W is the amount of catalyst used (10 mg). The residual concentration of phenol was determined by a UV-vis spectroscopic technique. The extent of adsorption was higher for the Sn:Fe (0.75:0.25) and Sn:Fe (0.25:0.75) catalysts and the detailed results are given in Fig. S4 (Table S5) (see the ESI†).
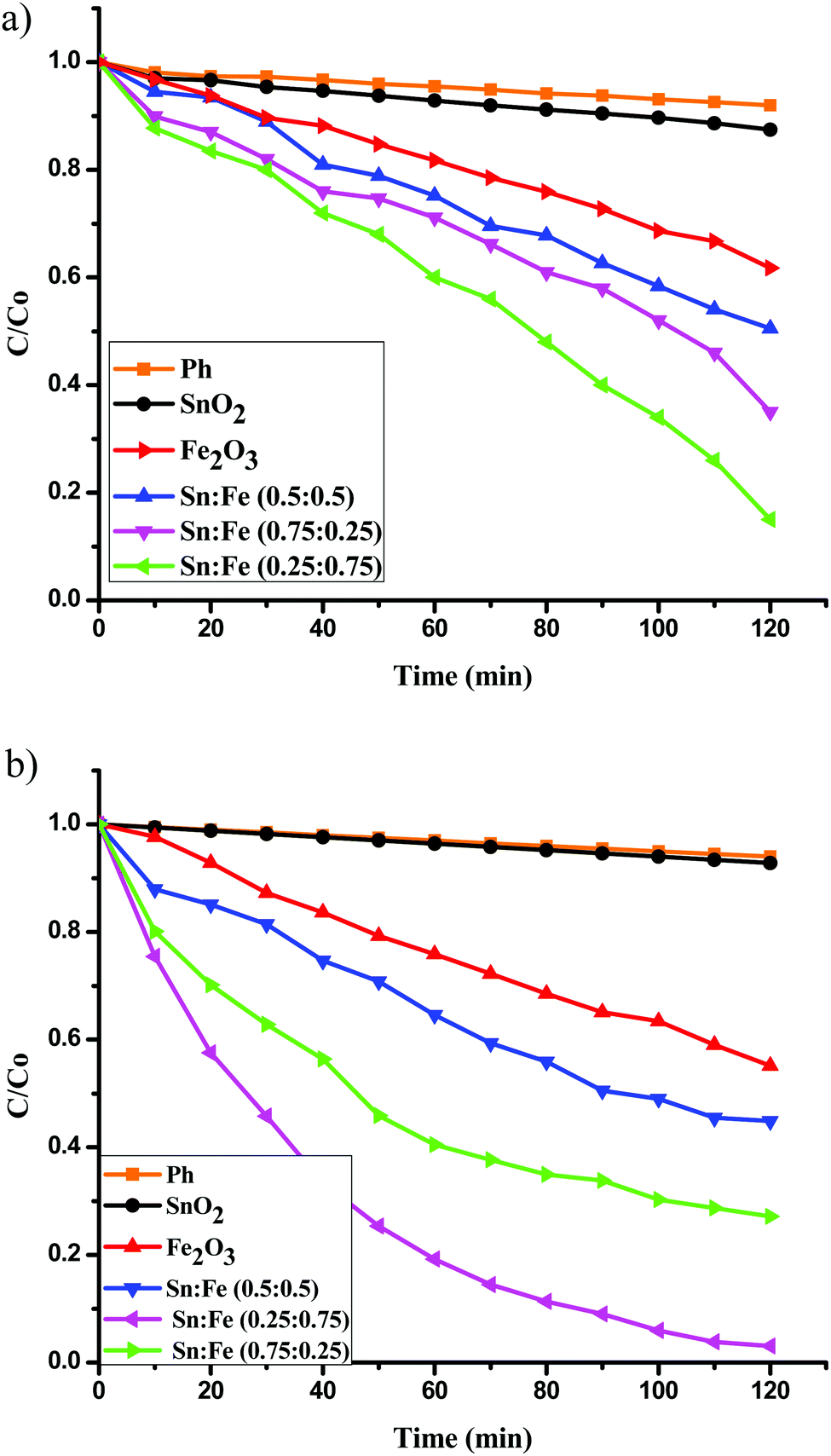
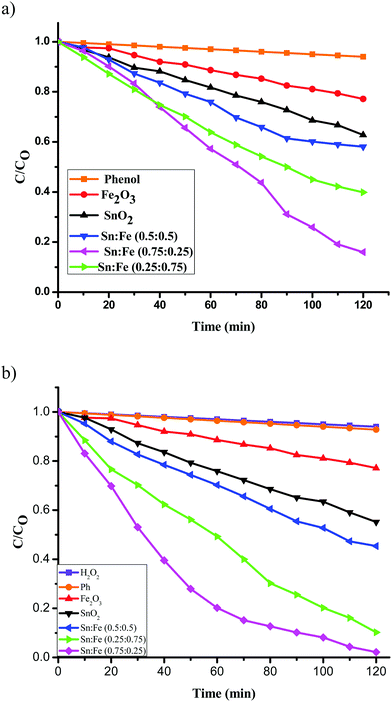
The photocatalytic activities of the above mentioned catalysts were studied under UV illumination. The observed results show the following decreasing order; Sn:Fe (0.75:0.25) > Sn:Fe (0.25:0.75) > Sn:Fe (0.5:0.5) > SnO2 > α-Fe2O3 and the activities under solar illumination were found to be in the order of Sn:Fe (0.25:0.75) > Sn:Fe (0.75:0.25) > Sn:Fe (0.5:0.5) >α-Fe2O3 > SnO2. The order of reactivity of these catalysts was found to be same, with higher quantum efficiency, when the above experiments were performed in the presence of H2O2 as an oxidant under UV/solar light irradiation. The results suggest that the composite systems showed better activity when compared to the pristine individual photocatalysts (Fig. 6(a), (b) and 7(a), (b)). The rate constant values are presented in Tables 4 and 5. The results suggest a higher activity of the Sn:Fe (0.75:0.25) catalyst under UV/visible light and the Sn:Fe (0.25:0.75) catalyst under solar light irradiation. The interfaces in the composites play a definite role in aiding the overall photocatalytic activity. The band gap values of SnO2 and α-Fe2O3 are found to be 3.6 and 2.2 eV, respectively as mentioned earlier (Fig. S1, ESI†). These values suggest that each of them will have a different electron affinity and band configuration. The CB edge of SnO2 is 0.2 eV lower than the CB edge of α-Fe2O3, leading to the staggered type band edge positions in the composite. A tentative charge transfer mechanism in these energy levels of composites at the interface is proposed. Fig. 8 shows the schematic energy band gap diagram and the charge transfer mechanism in the composite semiconductor system.
 |
| | Fig. 6 Plot of C/C0versus time for the degradation of phenol under UV light irradiation (a) in the absence of oxidant, (b) along with H2O2 as an oxidant. | |
 |
| | Fig. 7 Plot of C/C0versus time for the degradation of phenol under solar light irradiation (a) in the absence of oxidant, (b) along with H2O2 as an oxidant. | |
Table 4 Rate constants and percentage degradation of phenol with the mentioned photocatalysts under UV light illumination and along with H2O2 as an oxidising agent
| Photocatalysts |
UV light (without H2O2) |
UV light (with H2O2) |
| Rate constant (k) in 10−3 min−1 |
Percentage degradation in 120 min |
Rate constant (k) in 10−3 min−1 |
Percentage degradation in 120 min |
| SnO2 |
3.84 |
35 |
4.97 |
45 |
| Fe2O3 |
2.16 |
23 |
3.27 |
32 |
| Sn:Fe (0.25:0.75) |
7.93 |
60 |
23.9 |
96 |
| Sn:Fe (0.5:0.5) |
5.04 |
41 |
6.67 |
56 |
| Sn:Fe (0.75:0.25) |
15.56 |
84 |
40.96 |
99 |
Table 5 Rate constants and percentage degradation of phenol with the mentioned photocatalysts under solar light illumination and along with H2O2 as an oxidising agent
| Photocatalysts |
Solar light (without H2O2) |
Solar light (with H2O2) |
| Rate constant (k) in 10−3 min−1 |
Percentage degradation in 120 min |
Rate constant (k) in 10−3 min−1 |
Percentage degradation in 120 min |
| SnO2 |
0.99 |
13 |
0.62 |
17 |
| Fe2O3 |
3.84 |
38 |
4.97 |
45 |
| Sn:Fe (0.25:0.75) |
13.27 |
85 |
28.82 |
97 |
| Sn:Fe (0.5:0.5) |
5.66 |
50 |
1.83 |
56 |
| Sn:Fe (0.75:0.25) |
7.83 |
65 |
10.62 |
73 |
 |
| | Fig. 8 Schematic diagram showing the energy band edge positions, vectorial charge transfer process and electron hole separation process in the SnO2–α-Fe2O3 composite under UV/visible light irradiation. | |
Estimation of the rate constant, TON and TOF values
The activity of the Sn:Fe composites was higher compared to bare SnO2 and α-Fe2O3 (Fig. 6(a) and Table 4). This can be explained based on the band configuration of the composites, which prevents the electron hole recombination, thereby increasing the photocatalytic activity. Further, the catalytic activity of the Sn:Fe (0.75:0.25) catalyst was found to be maximum when compared to the other composites. These results are concordant with the PL studies and the calculated TON and TOF values given in Table 6. This observed enhancement in the degradation rate can be attributed to the higher concentration of SnO2, which can efficiently absorb photons under UV light compared to α-Fe2O3. The number of active sites per 10 mg of Sn:Fe (0.75:0.25) composite is found to be 5.04 × 10−5 moles (Table 6). Though the number of active sites is higher for the SnO2, α-Fe2O3 and Sn:Fe (0.25:0.75) samples, these sites probably act as recombination centres and hence the TON and TOF values are lower for these catalysts. The recombination of photogenerated charge carriers is high for pure compounds, while in the composites, the recombination centres are fewer because of the synergistic effects of the band edge positions of the semiconductors. The electron hole separation efficiency is improved by tuning the energy levels of the valence and conduction bands of the semiconductors. The spatial separation of the charge carriers (excitons) on photoexcitation leads to a higher activity. When the above photocatalytic reaction was carried out in the presence of H2O2 as an oxidant, there was a remarkable increase in the values of rate constant, TON and TOF compared to the values obtained for the experiments in the absence of H2O2. The highest TON value was found to be 1.04 for the Sn:Fe (0.75:0.25) composite. This can be attributed to the synergistic effects of the photocatalytic process and photo-Fenton process. The photo-Fenton process is expected to take place because of the presence of Fe3+ ions in the composite, which have the ability to be oxidised and then reduced continuously leading to a cyclic process. In the photocatalytic reaction pathway, the photogenerated hole interacts with the H2O/OH− groups present on the surface of the catalyst to generate the hydroxyl free radicals (OH˙). The transfer of the photogenerated electrons from the conduction band to the molecular oxygen is a minor reaction pathway due to the mismatch of the energy levels. The presence of H2O2 in the reaction medium accelerates the rate of generation of OH˙ radicals. The rate is further increased by the cyclic photo-Fenton process (Fig. 6(b)). This synergistic effect of both photocatalysis and the photo-Fenton process enhances the degradation rate by 63%.
Table 6 Determination of the active sites, TON and TOF in the UV system for all the photocatalysts
| Photocatalyst |
Number of active sites in 10 mg (10−5 moles) |
UV light (without H2O2) |
UV light (with H2O2) |
| TON (10−3 min−1) |
TOF (10−3 min−1) |
TON (10−3 min−1) |
TOF (10−3 min−1) |
| SnO2 |
6.57 |
0.37 |
2.56 |
0.36 |
3.03 |
| Fe2O3 |
6.26 |
0.19 |
1.61 |
0.26 |
2.24 |
| Sn:Fe (0.25:0.75) |
6.37 |
0.50 |
4.17 |
0.80 |
6.70 |
| Sn:Fe (0.5:0.5) |
3.98 |
0.43 |
3.58 |
0.74 |
6.20 |
| Sn:Fe (0.75:0.25) |
5.04 |
0.88 |
7.38 |
1.04 |
8.68 |
The pure SnO2 sample does not show any photocatalytic activity under solar light irradiation, due to its wide band gap energy (Fig. 7(a)). The band gap of α-Fe2O3 was more favourable for visible light absorption (Fig. 8). However, the bare α-Fe2O3 exhibits a lower photocatalytic activity compared to the Sn:Fe composites. This is because of its rapid charge carrier recombination and low absorption coefficient. The significant activity of the composites can be attributed to the effective charge carrier separation and migration to the surface to produce highly reactive free radicals that in turn oxidize phenol. The higher activity of the Sn:Fe (0.25:0.75) sample under visible light can be attributed to the higher concentration of α-Fe2O3. Further increases in the rate constant, TON and TOF values (Table 7) in the presence of H2O2 can be attributed to the simultaneous process of photocatalysis and photo-Fenton reactions (Fig. 7(b)).
Table 7 Determination of the active sites, TON and TOF in the solar system for all the photocatalysts
| Photocatalyst |
Number of active sites in 10 mg (10−5 moles) |
Solar light (without H2O2) |
Solar light (with H2O2) |
| TON (10−3 min−1) |
TOF (10−3 min−1) |
TON (10−3 min−1) |
TOF (10−3 min−1) |
| SnO2 |
6.57 |
0.10 |
0.80 |
0.14 |
1.14 |
| Fe2O3 |
6.26 |
0.32 |
2.70 |
0.41 |
3.39 |
| Sn:Fe (0.25:0.75) |
6.37 |
0.71 |
5.90 |
0.81 |
6.73 |
| Sn:Fe (0.5:0.5) |
3.98 |
0.67 |
5.56 |
0.74 |
6.20 |
| Sn:Fe (0.75:0.25) |
5.03 |
0.69 |
5.71 |
0.78 |
6.46 |
Effect of the oxidant
Oxidation of phenol, an organic pollutant, by the combination of UV/solar light with H2O2 implies a higher magnitude of generation of hydroxyl free radicals. The oxidation potential of hydrogen peroxide is 1.78 V and that of hydroxyl radical is 2.8 V. The most powerful oxidizing agent is a short lived hydroxyl radical, which is capable of oxidizing most organic compounds by (i) hydrogen abstraction, (ii) electrophilic addition and (iii) electron transfer reaction. These reactions generate several free radicals and additionally these radicals actively participate in the oxidative degradation. The intermediates formed during the reaction initiate several chain reactions. Besides, hydrogen abstraction and electrophilic addition, electron transfer to hydroxyl radicals constitutes another mechanism of oxidation degradation.| | | H2O2 → 2HO˙ (only under UV irradiation) | (11) |
| | | HO˙ + C6H5OH → C6H5O˙ + H2O (hydrogen abstraction) | (12) |
| | | HO˙ + C6H5OH → HOC6H5OH (electrophilic attack) | (13) |
| | | HO˙ + C6H5OH → C6H5OH˙ + HO− (electron transfer reaction) | (14) |
The use of H2O2 as an oxidant has a number of advantages in comparison to the other methods of degradation. Extensive commercial availability, thermal stability, infinite solubility in water, and no associated mass transfer problems make H2O2 a more appropriate oxidant. The production of two hydroxyl radicals under UV-irradiation and the generation of peroxy radicals on reaction with hydroxyl free radicals or organic free radicals lead to the subsequent oxidation reactions. The use of H2O2 as an oxidant under UV irradiation leads to minimal capital investment, a very cost effective source of hydroxyl radical and involves simple operational procedures. In the presence of solar light, the Fe3+ ions of α-Fe2O3 can react with H2O2 to generate Fe2+ ions and hydroxy peroxyl radicals. Fe2+ thus formed in the reaction will actively participate in the photo-Fenton reaction.
| | | Fe3+ + H2O2 → Fe2+ + ˙O2H + H+ | (15) |
Further, the Fe2+ ions can also undergo the following reactions.
| | | Fe2+ + H2O2 → Fe3+ + 2OH˙ | (16) |
| | | Fe3+ + OOH˙ → Fe2+ + O2 + H+ | (17) |
| | | Fe2+ + OOH˙ + H+ → Fe3+ + H2O2 | (18) |
| | | Fe2+ + OH˙ → Fe3+ + OH− | (19) |
| | | H2O2 + OH˙ → OOH˙ + H2O | (20) |
Thus, the formation of the Fe2+ and Fe3+ ions continuously takes place and leads to the cyclic photo-Fenton process.
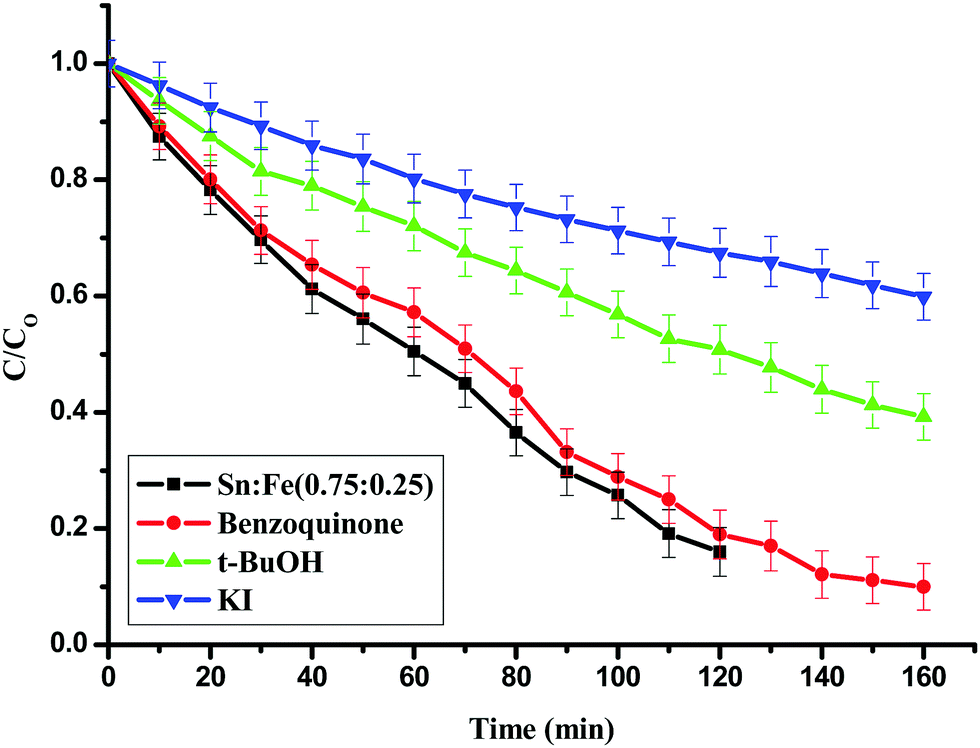
Effect of addition of scavengers like KI, t-BuOH and Benzoquinone on the photocatalytic activity of Sn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Fe (0.75:0.25)
:Fe (0.75:0.25)
The degradation reaction was carried out in the presence of KI (surface hydroxyl radical scavenger), t-BuOH (bulk hydroxyl radical scavenger) and benzoquinone (super oxygen radical scavenger) with the Sn:Fe (0.75:0.25) photocatalyst.26,27 Iodide ions are an excellent scavenger of VB holes and their presence hinders the formation of surface hydroxyl radicals.28 It was observed that the extent of degradation decreased by 70% upon addition of KI and 50% upon addition of t-BuoH, which are surface and bulk hydroxyl radical scavengers. The extent of degradation did not change upon addition of benzoquinone, which is more effective in scavenging super oxide radicals (Fig. 9). This implies the prominent role of surface and bulk hydroxide radicals compared to super oxide radicals.
 |
| | Fig. 9 Effect of KI, t-BuOH and benzoquinone, which are hydroxyl and super oxide radical scavengers for the degradation of phenol using the Sn:Fe (0.75:0.25) catalyst under UV light irradiation. | |
Energy band configuration and electron–hole separation
The enhanced photocatalytic activity of the SnO2–α-Fe2O3 composite under both UV and solar light illumination could be attributed to: (i) the favourable electronic energy levels of SnO2 and α-Fe2O3. The staggered positions of the conduction band edges with respect to one another facilitate the electron transfer process, (ii) the synergetic effects between photocatalysis and photo-Fenton processes, (iii) the oxidation potential of the oxidant (H2O2) suitably matches with the valence band edge positions to generate highly potent hydroxyl free radicals, (iv) the photoexcitation of SnO2 takes place only under UV light illumination, whereas α-Fe2O3 readily absorbs solar light and the extent of excitation of each is based on the weight proportion of these photocatalysts in the composite. Fig. 8 shows the schematic representation of various reaction pathways taking place at the interface of the composite catalyst. The photocatalytic degradation mainly takes place through the valence band holes. The transfer of electrons from the conduction band to the oxygen molecule is limited due to the mismatch of the conduction band energy level positions with the oxygen redox potential.
TOC analysis
The plot of TOC/TOC0versus time for the degradation of phenol (20 ppm) in the presence of the Sn:Fe (0.75:0.25) (10 mg) catalyst along with H2O2 (10 ppm) under UV illumination is shown in Fig. 10, where TOC0 corresponds to the initial phenol concentration and TOC is the concentration of phenol at different time intervals. The complete degradation of phenol is confirmed by TOC analysis.
 |
| | Fig. 10 The plot of TOC/TOC0versus time for the degradation of phenol (20 ppm) in the presence of the Sn:Fe (0.75:0.25) (10 mg) catalyst along with H2O2 (10 ppm) under UV illumination. | |
Recyclability of Sn:Fe (0.75:0.25)
The stability and reusability of the prepared Sn:Fe (0.75:0.25) composite photocatalyst was checked by using 10 mg of the catalyst for three consecutive cycles (Fig. 11) and in the third cycle, the percentage of degradation was found to be 88.12% due to the loss of some amount of catalyst while washing and filtrating. The structural stability of the catalyst after three cycles was confirmed by PXRD and UV-visible absorbance spectroscopic techniques (Fig. S6) (ESI†). The results unambiguously demonstrate that the Sn:Fe (0.75:0.25) photocatalyst is stable, efficient, and can be reused for up to three cycles.
 |
| | Fig. 11 Catalytic recycling of Sn:Fe (0.75:0.25) in the degradation of phenol. | |
Conclusions
SnO2 and α-Fe2O3 were synthesized by a sol–gel method and the composite systems were prepared by mixing and grinding the required proportions of each individual metal oxide. The photocatalytic activities were studied using phenol as the model compound. The material composition, optical properties, morphologies and surface areas as well as luminescence characteristics were studied. The reduction in the band gap takes place for the composite systems and these values suggest that each of them will have a different electron affinity and band configuration. The composites show a higher extent of absorption in the visible region due to the larger red shift of the band gap values. The effectiveness of the catalyst at very low concentrations is suggested by the calculation of the number of active sites, turnover number (TON) and turnover frequency (TOF) for all the systems under both UV/solar light illumination. Sn:Fe (0.75:0.25) shows higher activity both under UV and visible light illumination and Sn:Fe (0.25:0.75) shows higher activity only under visible light due to the higher proportion of α-Fe2O3. The activities were further enhanced when the degradation reaction was carried out in the presence of an oxidant (H2O2). The degradation reaction mainly takes place by the surface and bulk hydroxyl free radicals generated by the valence band holes rather than super oxide radicals. The photocatalytic oxidation process dominates the degradation reaction rather than the photocatalytic reduction.
Conflicts of interest
There are no conflicts of interest.
References
- S. Zhang, H. Yang, H. Huang, H. Gao, X. Wang, R. Cao, J. Li, X. Xu and X. Wang, J. Mater. Chem. A, 2017, 5, 15913–15922 Search PubMed; S. Zhanga, H. Gao, J. Li, Y. Huang, A. Alsaedi, T. Hayat, X. Xu and X. Wang, J. Hazard. Mater., 2017, 321, 92–102 CrossRef CAS PubMed; S. Zhang, H. Yang, H. Gao, R. Cao, J. Huang and X. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 23635–23646 Search PubMed.
- B. Palanisamya, C. M. Babua, B. Sundaravela, S. Anandanb and V. Murugesana, J. Hazard. Mater., 2013, 252–253, 233–242 CrossRef CAS PubMed; A. Hernandez, L. Maya, E. Sanchez-Mora and E. M. Sanchez, J. Sol Gel. Sci. Technol., 2007, 42, 71–78 CrossRef.
- S. Kozuch and J. M. L. Martin, ACS Catal., 2012, 2, 2787–2794 CrossRef CAS.
- S. Huang, Z. Xiao, J. Li, J. Zhong, W. Hu and J. He, Adv. Mater. Res., 2013, 734–737, 2278–2281 CrossRef.
- J. Kang, Q. Kuang, Z.-X. Xie and L.-S. Zheng, J. Phys. Chem. C, 2011, 115, 7874–7879 CAS.
- M. Niu, F. Huang, L. Cui, P. Huang, Y. Yu and Y. Wang, ACS Nano, 2010, 4, 681–688 CrossRef CAS PubMed.
- K. Melghita, A. Kabir Mohammed and I. Al-Amri, Mater. Sci. Eng., B, 2005, 117, 302–306 CrossRef.
- N. C. Pramanik, T. I. Bhuiyan, M. Nakanishi, T. Fujii, J. Takada and S. I. L. Seok, Mater. Lett., 2005, 59, 3783–3787 CrossRef CAS.
- P. Sun, C. Wang, J. Liu, X. Zhou, X. Li, X. Hu and G. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 19119–19125 CAS.
- X. Wang, J. Wang, Z. Cui, S. Wang and M. Cao, RSC Adv., 2014, 4, 34387–34394 RSC.
- Y. Liao, H. Li, Y. Liu, Z. Zou, D. Zeng and C. Xie, J. Comb. Chem., 2010, 12, 883–889 CrossRef CAS PubMed.
- J. Liu, C. Liang, H. Zhang, Z. Tian and S. Zhang, J. Phys. Chem. C, 2012, 116, 4986–4992 CAS.
-
J. D. Lee, Concise, Inorganic Chemistry, Wiley Indian edition, 5th edn, 2007 Search PubMed.
- R. Kavitha and L. Gomathi Devi, J. Environ. Chem. Eng., 2014, 2, 857–867 CrossRef CAS.
- Y. R. Smith, K. Joseph Antony Raj, V. (Ravi) Subramanian and B. Viswanathan, Colloids Surf., A, 2010, 367, 140–147 CrossRef CAS.
- W.-K. Jo and N. Clament Sagaya Selvam, Dalton Trans., 2015, 44, 16024–16035 RSC.
- J. Kang, Q. Kuang, Z.-X. Xie and L.-S. Zheng, J. Phys. Chem. C, 2011, 115, 7874–7879 CAS.
- Y. Mi, Y. Hong Cao, X. Li Liu, J. Bao Yi, H. Ru Tan, P. Ma, H. Hao, X. Zhang and H. Ming Fan, Mater. Chem. Phys., 2013, 143, 311–321 CrossRef CAS.
- T. Wang, G. Yanga, J. Liu, B. Yang, S. Dingc, Z. Yan and T. Xiao, Appl. Surf. Sci., 2014, 311, 314–323 CrossRef CAS.
- G. Kumar Pradhan, D. Kumar Padhi and K. M. Parida, ACS Appl. Mater. Interfaces, 2013, 5, 9101–9110 Search PubMed.
- A. Kar, S. Kundu and A. Patra, RSC Adv., 2012, 2, 10222–10230 RSC.
- L. Robert and J. R. Burwell, Pure Appl. Chem., 1976, 46, 71–90 Search PubMed.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
-
O. Verho, Transition metal catalyzed redox reactions, Stockholm University, 2012, ISBN 978-91-7447-828-0 Search PubMed.
- L. Gomathi Devi and K. Mohan Reddy, Appl. Surf. Sci., 2010, 256, 3116–3121 CrossRef CAS.
- L. Gomathi Devi and R. Kavitha, Mater. Chem. Phys., 2014, 143, 1300–1308 CrossRef.
- W. Gu, F. Teng, Z. Liu, Z. Liu, J. Yang and Y. Teng, J. Photochem. Photobiol., A, 2018, 353, 395–400 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00536a |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
R.
Shyamala
* and
R.
Shyamala









 and doubly ionised oxygen vacancy
and doubly ionised oxygen vacancy  .21 V0 is a very shallow donor, which means it can donate and accept charge carriers more easily. These vacancies are usually associated with metal ions (Sn3+–V0) and are expected to be located below the SnO2 conduction band (∼0.3–0.4 eV). Excitation of an electron from the valence band to these states may result in recombination, which is responsible for the peaks observed in the visible region. The PL intensities of α-Fe2O3 and the composites containing α-Fe2O3 are shown in the inset (B) of Fig. 5. The PL intensity of pure α-Fe2O3 is high when compared to those of the composites containing α-Fe2O3. The recombination reactions are reduced considerably in the Sn
.21 V0 is a very shallow donor, which means it can donate and accept charge carriers more easily. These vacancies are usually associated with metal ions (Sn3+–V0) and are expected to be located below the SnO2 conduction band (∼0.3–0.4 eV). Excitation of an electron from the valence band to these states may result in recombination, which is responsible for the peaks observed in the visible region. The PL intensities of α-Fe2O3 and the composites containing α-Fe2O3 are shown in the inset (B) of Fig. 5. The PL intensity of pure α-Fe2O3 is high when compared to those of the composites containing α-Fe2O3. The recombination reactions are reduced considerably in the Sn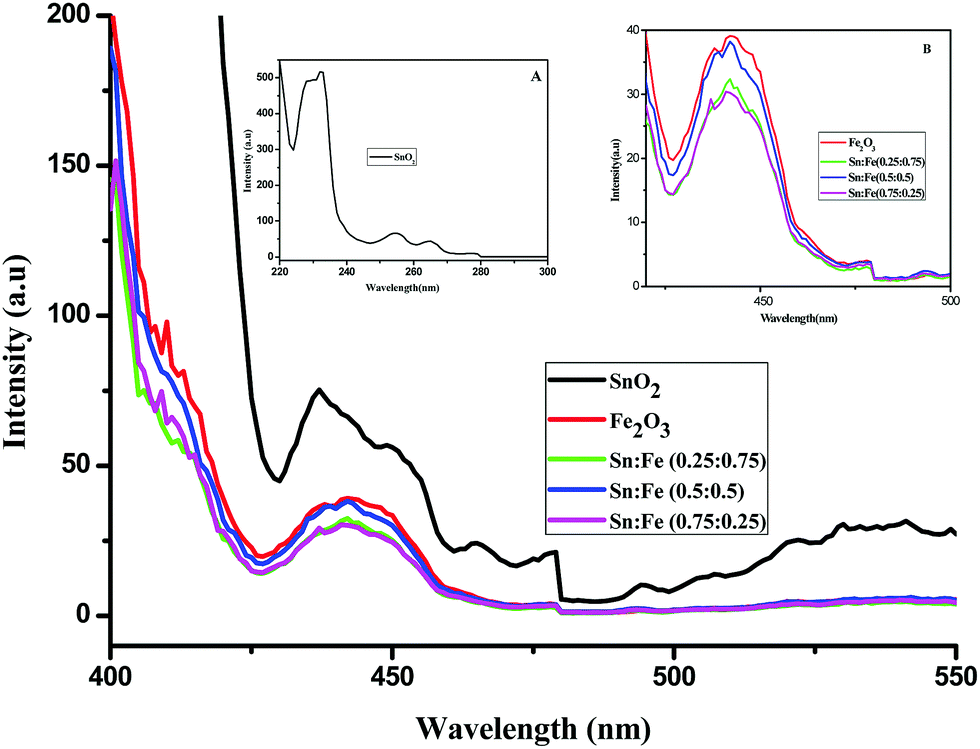
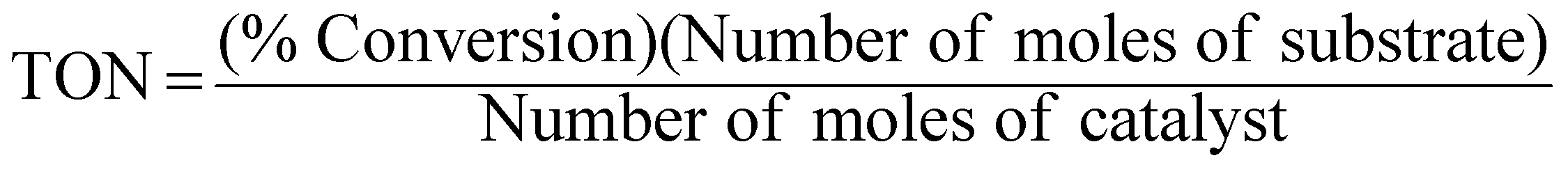
 where, C0 and C have the same significance as defined in eqn (3). The detailed calculations of the number of moles of the substrate and catalyst are given in S3 (ESI†). The term turnover frequency (TOF) is expressed as a time inverse unit, which is actually the number of reaction cycles taking place in a given time period and is usually employed to study the reaction rate, which can be defined as:
where, C0 and C have the same significance as defined in eqn (3). The detailed calculations of the number of moles of the substrate and catalyst are given in S3 (ESI†). The term turnover frequency (TOF) is expressed as a time inverse unit, which is actually the number of reaction cycles taking place in a given time period and is usually employed to study the reaction rate, which can be defined as: