
Tris(2-hydroxyphenyl)triazasumanene: bowl-shaped excited-state intramolecular proton transfer (ESIPT) fluorophore coupled with aggregation-induced enhanced emission (AIEE)†
Patcharin
Kaewmati
a,
Yumi
Yakiyama
 a,
Hiroyoshi
Ohtsu
b,
Masaki
Kawano
b,
Setsiri
Haesuwannakij
c,
Shuhei
Higashibayashi
d and
Hidehiro
Sakurai
*a
a,
Hiroyoshi
Ohtsu
b,
Masaki
Kawano
b,
Setsiri
Haesuwannakij
c,
Shuhei
Higashibayashi
d and
Hidehiro
Sakurai
*a
aDivision of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: hsakurai@chem.eng.osaka-u.ac.jp
bDepartment of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8551, Japan
cSchool of Molecular Science & Engineering, Vidyasirimedhi Instituteof Science and Technology (VISTEC), 555 Moo 1 Payupnai, Wangchan, Rayong 21210, Thailand
dFaculty of Pharmacy, Keio University, 1-5-30 Shibakoen, Minato-ku, Tokyo 105-8512, Japan
First published on 15th January 2018
Abstract
Tris(2-hydroxyphenyl)triazasumanene ((A)-(+)-1), a bowl-shaped molecule, which possesses 2-(2′-hydroxyphenyl)pyridine moieties, was successfully synthesised. (A)-(+)-1 showed a single peak emission in CH2Cl2, which overlapped with the emission of the triazasumanene skeleton and diminished at a high concentration, corresponding to the formation of the excited enol form. In contrast, single crystals of (A)-(+)-1 exhibited a dual emission with a large Stokes shift, indicating the presence of the excited keto form by the excited state intramolecular proton transfer (ESIPT) process. In the solution of (A)-(+)-1 containing a mixture of a large amount of a poor solvent (hexane or MeOH) and a small amount of CH2Cl2, colloidal aggregates emerged with the continuous increment of emission intensity by further addition of the poor solvent, demonstrating aggregation-induced enhanced emission (AIEE). The analysis of the morphology and the structure of the aggregates using a scanning electron microscope (SEM) and powder X-ray diffraction (PXRD) revealed the well-ordered structure of the aggregates, which possessed a molecular packing pattern similar to that of an (A)-(+)-1 single crystal.
Introduction
In the molecular design of organic emissive materials, which are receiving a lot of attention due to their wide application, one of the most important strategies is to introduce a key structure that exhibits a unique optical property. Excited state intramolecular proton transfer (ESIPT)1 is a widely studied phenomenon in terms of its potential application for laser dyes,2 ultraviolet (UV) photostabilizers,3 photoswitches4 and electroluminescent materials.5 ESIPT occurs via an ultrafast reaction process to create a large Stokes shift (6000–12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1),1a which often shows dual emission. In general, dual emission consists of a shorter wavelength one derived from the excited state enol form (E*) and a longer wavelength one due to the excited state keto form (K*). ESIPT is also well known to be very sensitive to the surrounding environment, such as the solvent system and acidic conditions. As a minimum requirement to realise this unique property of ESIPT, a preformed intramolecular hydrogen bond (H-bond) between the proton donor (–OH, –NH2) and the proton acceptor (–C
000 cm−1),1a which often shows dual emission. In general, dual emission consists of a shorter wavelength one derived from the excited state enol form (E*) and a longer wavelength one due to the excited state keto form (K*). ESIPT is also well known to be very sensitive to the surrounding environment, such as the solvent system and acidic conditions. As a minimum requirement to realise this unique property of ESIPT, a preformed intramolecular hydrogen bond (H-bond) between the proton donor (–OH, –NH2) and the proton acceptor (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –N) groups must be in close proximity to each other in a molecule.
O, –N) groups must be in close proximity to each other in a molecule.
Triazasumanene (TAS) is a C3-symmetric chiral azabuckybowl, a nitrogen analogue of sumanene,6 and it possesses three peripheral pyridine skeletons (Fig. 1).7 Previously, we reported on the enantioselective synthesis of trisubstituted C3-symmetric TAS derivatives with MeS (2), MeSO2 and various aryl groups.7 These were found to have unique properties, such as a deeper bowl depth (1.30 Å for tri-MeSO2; 1.27–1.28 Å for tri-(4-CF3Ph) derivatives) than that of pristine sumanene (1.11 Å), and high bowl inversion energy (ca. 40 kcal mol−1 for tri-MeS derivative), which enables the successful separation of two enantiomers defined by the inherent bowl chirality of TAS (cf. 20.3 kcal mol−1 for pristine sumanene).6c These features are attributable to nitrogen doping on the sumanene skeleton. These findings encouraged us to further study the functionalisation of the TAS skeleton with the expectation of identifying additional aspects. In the present study, we synthesised three 2-hydroxyphenyl group-introduced TAS as a single enantiomer (A)-(+)-tris(2-hydroxyphenyl)triazasumanene ((A)-(+)-1). Spectroscopic analyses revealed that single crystals of (A)-(+)-1 can exhibit the dual emission caused by ESIPT. This finding was also supported by the single crystal X-ray analysis, which showed the formation of strong OH⋯N type hydrogen bonds. Further investigation using solvent- and concentration-dependent emission spectra led us to find that (A)-(+)-1 can also exhibit aggregation-induced enhanced emission (AIEE),8 which is one of the effective methods used to obtain highly emissive solid materials. An AIEE-active system coupled with ESIPT9 is still rare, and this paper presents the first example of the curved-π molecule-based AIEE-active system coupled with ESIPT.10
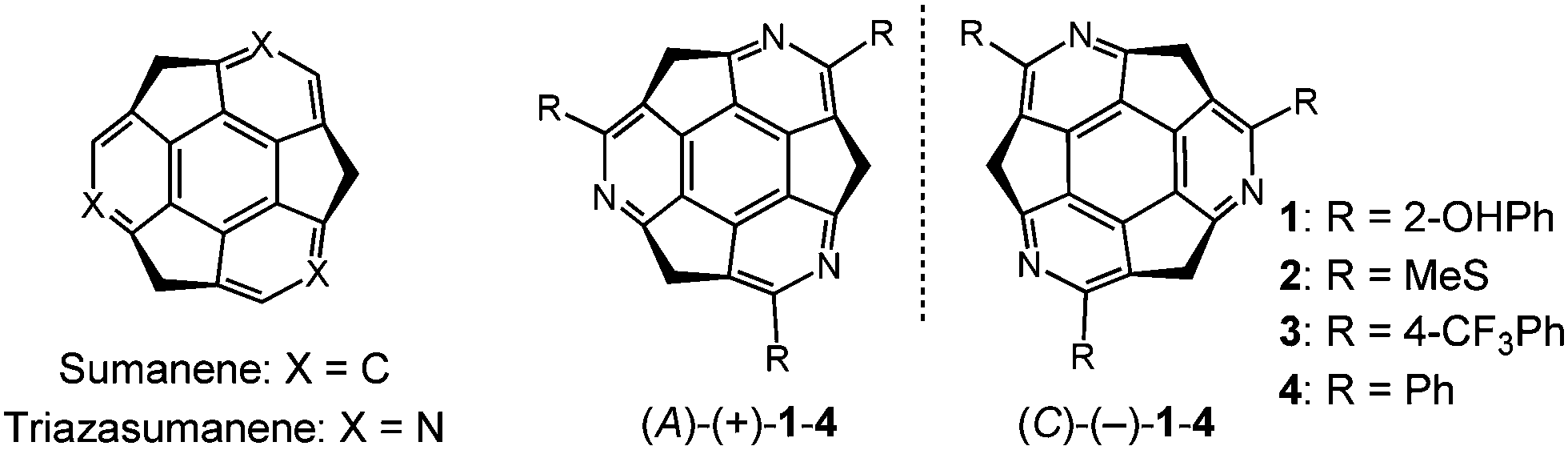 | ||
| Fig. 1 Molecular structures of sumanene and triazasumanenes. | ||
Results and discussions
Synthesis and crystal structure of (A)-(+)-1
The synthesis of (A)-(+)-1 was performed using the previously reported cross-coupling reaction of (A)-(+)-tris(methylthio)triazasumanene ((A)-(+)-2) with 2-hydroxyphenylboronic acid.7 A tetrahydrofuran (THF) solution of (A)-(+)-2, Pd2(dba)3 (20 mol%), 2-hydroxyphenylboronic acid (500 mol%), tri(2-furyl)phosphine (TFP, 100 mol%) and Cu(I)thiophene carboxylate (CuTC, 500 mol%) were heated at 50 °C to afford (A)-(+)-1 in 80% yield as a yellow solid (Scheme 1). In 1H NMR (in CDCl3), the peak assignable to phenol hydroxyl proton appeared with significant downfield shift at 13.4 ppm, indicating the formation of stable intramolecular hydrogen bond (ESI†).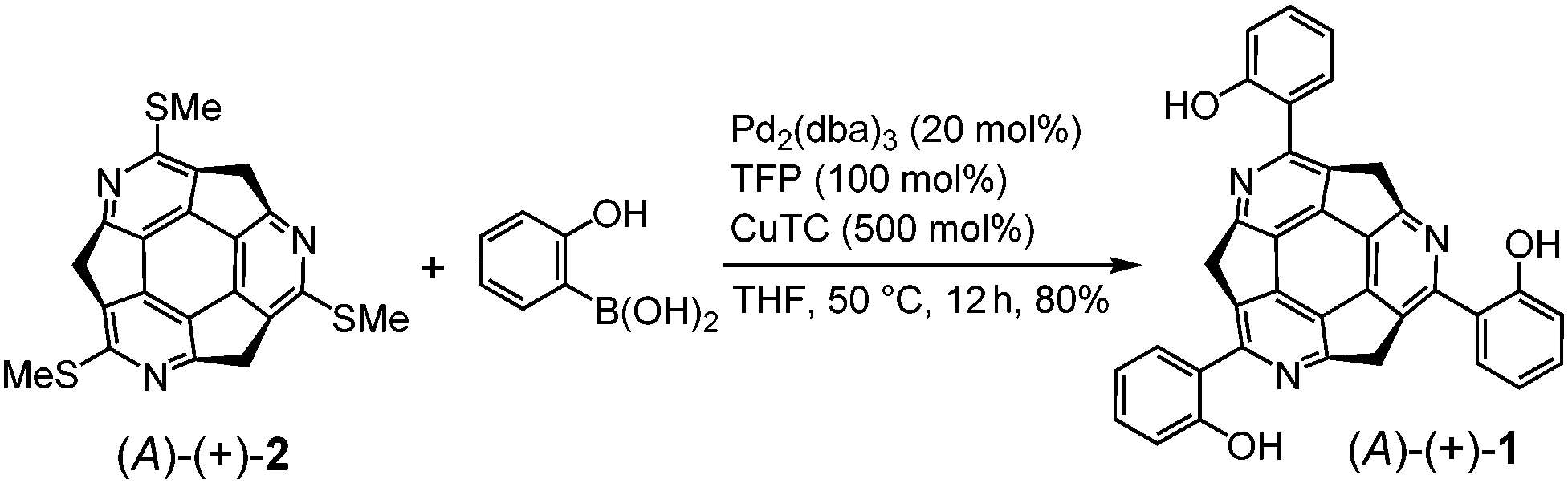 | ||
| Scheme 1 Synthesis of (A)-(+)-1. | ||
Single crystal X-ray analysis revealed the unique packing structure of (A)-(+)-1 in its crystalline state. In the structure, there is only one crystallographically independent (A)-(+)-1 because the three sets of the dihedral angles around the three peripheral pyridine rings on the TAS skeleton and the three phenol rings connecting to them were slightly different (Fig. 2a). On the (A)-(+)-1 skeleton, three intramolecular OH⋯N type hydrogen bonds were observed between the phenol hydroxy group and the N atom on the peripheral pyridine skeleton. Their O⋯N lengths (2.554(2)–2.588(2) Å) were shorter than the general OH⋯N type hydrogen bond distance (2.7–2.8 Å),11 indicating the presence of a relatively strong attractive force within the three hydrogen bonds. The bowl depth, which can be defined by the distance between the centre of the hexagonal ring at the bottom of the TAS skeleton (blue part in Fig. 2a) and the plane, consisting of three N atoms (N1, N2 and N3), was 1.34 Å (Fig. 2b). This bowl depth was slightly deeper than the values that have been previously reported (A)-(+)-2 (1.30 Å) and tris(p-trifluoromethylphenyl)triazasumanene (3) (1.27–1.28 Å).7 Each (A)-(+)-1 formed a CH⋯π and π–π interaction at the 2-(2′-hydroxyphenyl)pyridine moieties to give a slippery-stacked columnar structure along the b axis (Fig. 2c); hence, the TAS skeleton did not form the direct overlap, which is often found in sumanene derivatives due to the convex-concave-type stacking structure.6 These stacking columns resulted in additional CH⋯π interactions among the neighbouring columns to construct a one-dimensional channel structure along the b axis with the size of 5.8 Å × 3.5 Å, in which CH2Cl2 (0.25 Occ.) was encapsulated as the crystalline solvent (Fig. 2d).
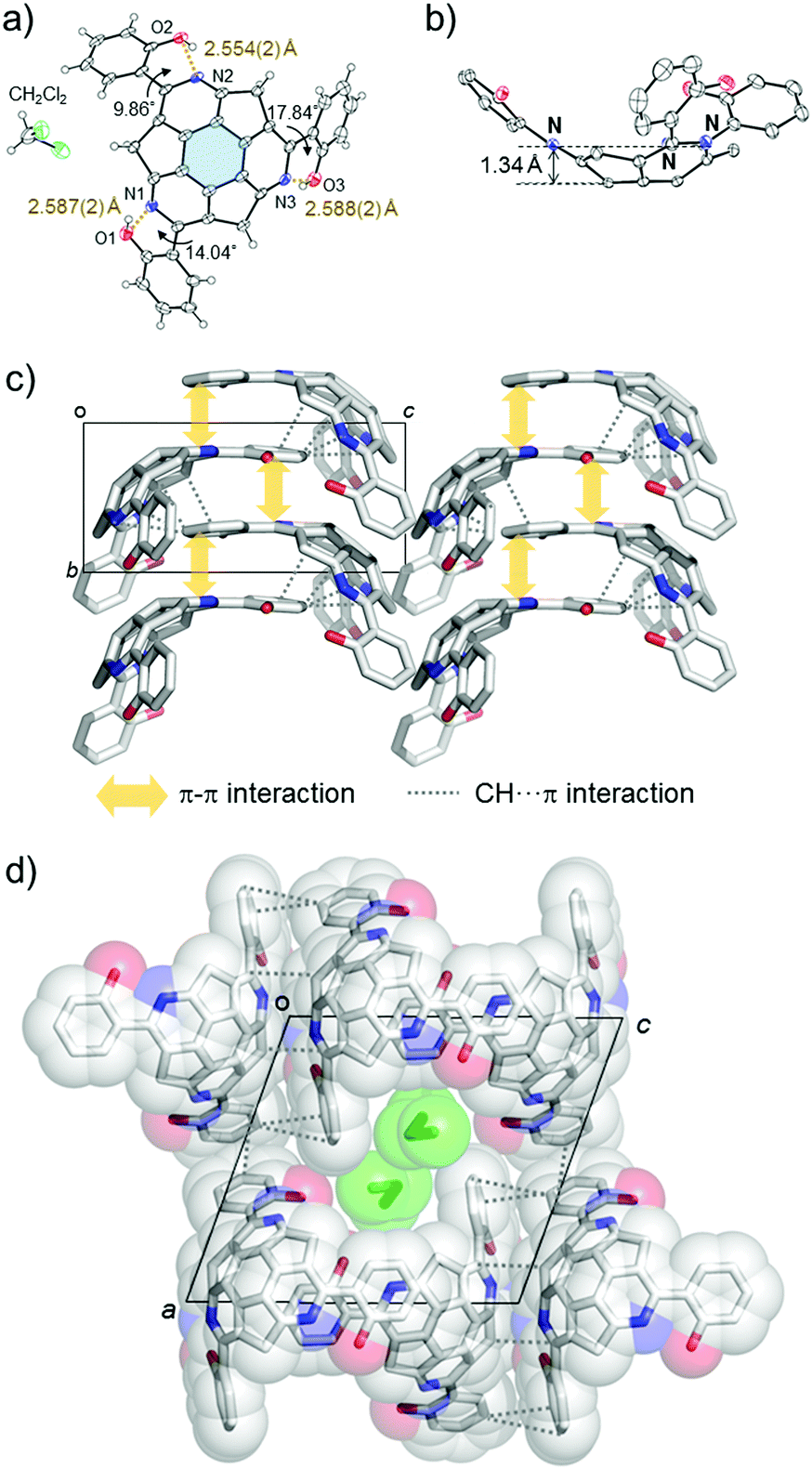 | ||
| Fig. 2 Crystal structure of (A)-(+)-1. (a) ORTEP model of (A)-(+)-1. The brown dotted lines show the intramolecular hydrogen bonds. (b) The deepest position of the bowl depth. In (a and b), the ellipsoids are shown in 50% probability. (c) Stacking structure viewed from the b axis with intermolecular π–π (yellow solid arrow) and CH⋯π (grey dotted line) interactions. (d) One-dimensional channel structure with intercolumnar CH⋯π interactions. Green molecules correspond to the encapsulated CH2Cl2. The colour codes in each of the images in this figure are: C, grey; N, blue; O, red; Cl, light green; however, (e) in (a), the hydrogen atoms are omitted for clarity. | ||
Photophysical property of (A)-(+)-1
The ultraviolet-visible (UV-vis) spectra of (A)-(+)-1 showed absorption maxima at 370 nm in the CH2Cl2 solution, which slightly red shifted to 382 nm in the single crystals of (A)-(+)-1 (Fig. 3). The small UV peak shift may be simply attributed to the intermolecular interactions involved in the J-aggregation-like stacking structure of 2-(2′-hydroxyphenyl)pyridine moiety in the crystalline state (Fig. 2 and Fig. S1, ESI†). The emission spectrum of single crystals of (A)-(+)-1 was significantly different from the spectrum measured in the CH2Cl2 solution. On excitation at 370 nm, the 4.0 × 10−5 M CH2Cl2 solution of (A)-(+)-1 showed a single emission peak at 447 nm, and its intensity significantly reduced at a higher concentration due to the concentration quenching (Fig. 3 and Fig. S2, ESI†). In contrast, a large shift (294 nm) in the additional emission band (λmax = 631 nm) was observed for the single crystals of (A)-(+)-1. Consequently, it was expected that (A)-(+)-1 possesses the ESIPT-active 2-(2′-hydroxyphenyl)pyridine moiety,12 which forms strong OH⋯N type hydrogen bonds in the crystal structure (Fig. 2a). This dual emission can be explained by the ESIPT effect at the 2-(2′-hydroxyphenyl)pyridine moiety of (A)-(+)-1. Therefore, the lower energy emission at 631 nm can be assigned to the K* form. The structured higher energy emission at around 410–500 nm was well overlapped with the single emission observed in the CH2Cl2 solution. Therefore, it might be easy to assign this to the E* form of emission. However, a similar emission was also observed in non-ESIPT-active triphenyltriazasumanene (4) and 3 together with another broad band around 540–590 nm (Fig. S3, ESI†).7 All these peaks were attributed to the TAS-skeleton, and therefore, the structured emission should be assigned to the mixture of the TAS skeleton-based structures rather than the E* form.13 In the CH2Cl2 solution, the contribution of the E* form might be larger than it is in the solid state because the fast rotation of the 2-hydroxyphenyl ring in CH2Cl2 restricts the ESIPT process via the formation of the OH⋯N type hydrogen bond.14 It would be interesting to know if these three OH⋯N hydrogen bonds contribute to the ESIPT process of (A)-(+)-1 in a concerted process or in a stepwise process.13,15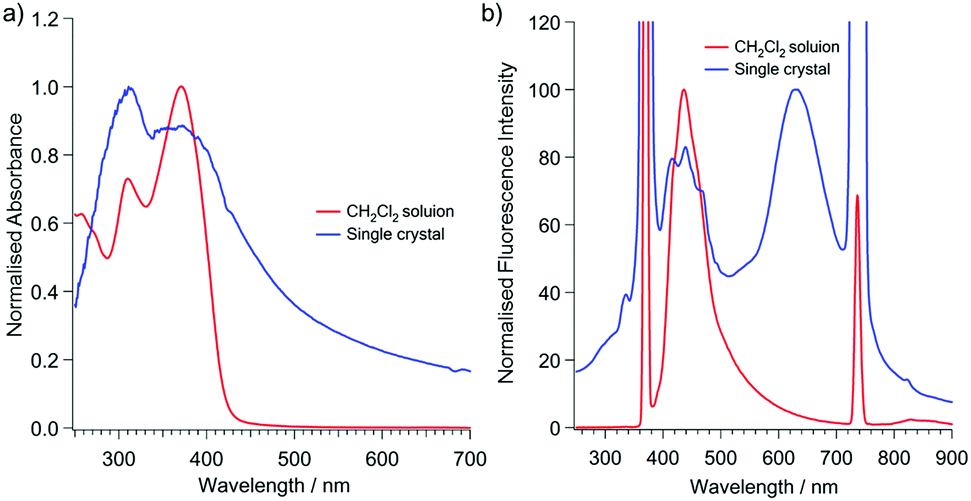 | ||
| Fig. 3 Absorption (a) and emission (b) spectra of (A)-(+)-1 at room temperature. Red: 4.0 × 10−5 M CH2Cl2 solution. Blue: single crystalline solid. λex = 370 nm. | ||
The emission spectra of (A)-(+)-1 were also recorded for different compositions of the CH2Cl2–hexane or MeOH mixtures (100:0 to 2:98; v/v) (Fig. 4 and Fig. S4, S5, ESI†). In the process of adding hexane, the emission intensity at around 430 nm, derived from both the TAS skeleton and the E* form, did not change much until the volumetric ratio was 80:20 (hexane:CH2Cl2). However, after the ratio was 90:10, the emission peak intensity at 430 nm suddenly increased and a new emission peak appeared at 634 nm, which was attributable to the K* form of (A)-(+)-1. Furthermore, at the same hexane:CH2Cl2 (90:10) ratio, we confirmed in the UV cell that the colloidal aggregates of (A)-(+)-1a dispersed homogeneously just after the appropriate amount of hexane was added to the CH2Cl2 stock solution. The amount of aggregates and the emission peak intensities at both 430 nm and 634 nm continuously increased as the ratio of hexane increased, indicating that (A)-(+)-1 is AIEE active. The relative ratio of both emission intensities was completely different from those of the crystalline sample (Fig. 3). This can be explained by the fact that the emission at around 430 nm includes the emission from the aggregates as well as the emission from the saturated CH2Cl2 solution of (A)-(+)-1. In the case in which MeOH was added, we also confirmed that the AIEE phenomenon showed a continuous increment of the emission peaks at both 430 nm and 634 nm after the MeOH:CH2Cl2 volumetric ratio was 90:10. However, the emission from the K* form at 634 nm in the MeOH–CH2Cl2 mixture was much weaker than that of the hexane–CH2Cl2 mixture, and the magnitude of the intensity increment was very small even in the 98:2 MeOH–CH2Cl2 solution. This can simply be explained by the high polarity of MeOH, which disturbs the formation of the K* form by covering the hydrogen bonding moiety to exhibit the emission from the E* form dominantly after photo irradiation.16
 | ||
| Fig. 4 Aggregate formation study of (A)-(+)-1 in (a) hexane/CH2Cl2, (b) MeOH/CH2Cl2 systems. λex = 370 nm. The annotation indicates the volumetric ratio of CH2Cl2 in each mixture. Dotted lines (y axis: right side) indicate the dried aggregates. | ||
Fig. 4 also shows the emission spectra of the aggregates after the removal of the solvent (dotted lines). Both dried aggregates exhibited dual emission spectra similar to that of the single crystals. In the emission spectra of both dried aggregates, the emission peak corresponding to the K* form was also observed at around 625 nm. Their relative intensity against the emission at around 430 nm was much stronger than in the case of non-dried aggregates. Meanwhile, the higher energy emission bands of the dried aggregates at around 430 nm were broader than those of the non-dried aggregates, also indicating that the intermolecular interaction between (A)-(+)-1 and the solvents has a strong impact on their emission properties. In addition, these results indicate that both of the dried aggregates, wherever they formed, constructed specific molecular arrangements, inducing AIEE.
To obtain further insights about the structure of the aggregated products of (A)-(+)-1, scanning electron microscope (SEM) measurements and powder X-ray diffraction (PXRD) studies were performed (Fig. 5 and Fig. S6, ESI†). The SEM images of the aggregates prepared from both the MeOH–CH2Cl2 and hexane–CH2Cl2 mixed solutions of (A)-(+)-1 revealed the formation of microcrystals. The PXRD analysis of all the dried aggregates, which showed diffraction patterns indicating the presence of the crystalline species, also supported the SEM observations. Their diffraction patterns were similar to that of the simulated pattern obtained from the single crystal data, indicating that all the aggregates possess a molecular packing structure similar to that of the single crystal of (A)-(+)-1, which realised the solid-state emission.
 | ||
| Fig. 5 SEM images of the dried-aggregates of (A)-(+)-1. (a) From the hexane/CH2Cl2 mixture. (b) From the MeOH/CH2Cl2 mixture. | ||
Conclusion
As described in this paper, we successfully synthesised tris(2-hydroxyphenyl)triazasumanene (A)-(+)-1 as a new non-planar ESIPT fluorophore model of a curved-π molecule-based AIEE-active system coupled with ESIPT. The single crystal analysis revealed the J-aggregation-like packing pattern of 2-hydroxyphenyl moieties on (A)-(+)-1 to prevent the formation of a strong π–π stacking column in the TAS skeleton. This single crystalline (A)-(+)-1 showed dual emission due to the ESIPT process. This solvent- and concentration-dependent emission study exhibited that (A)-(+)-1 achieved AIEE at the large volumetric ratio of the poor solvent. The SEM and PXRD measurements of the aggregates of (A)-(+)-1 revealed the formation of crystalline materials with a molecular arrangement similar to that of single crystalline state, which exhibits a solid-state emission.Experimental section
General information
Chemical reagents and solvents were commercially purchased and purified according to the standard methods, if necessary. Air- and moisture-sensitive reactions were carried out using commercially available anhydrous solvents under an inert atmosphere of nitrogen. 1H and 13C NMR spectra were recorded on 400 MHz JEOL JNM-ECS400 NMR spectrometer (1H: 400 MHz and 13C: 100 MHz). Chemical shifts (δ) are expressed relative to the resonances of the residual non-deuterated solvent for 1H (CDCl3: 1H(δ) = 7.26 ppm) and for 13C (CDCl3: 13C(δ) = 77.0 ppm). Solution state UV-vis absorption and emission spectra were recorded on a JASCO V-670 spectrophotometer and a FP-6500 spectrofluorometer using 1 cm−1 path-length quartz cells, respectively. Solid state UV-vis absorption spectra of the single crystals of (A)-(+)-1 dispersed in BaSO4 powder was recorded on a JASCO V-670 spectrophotometer with an integrating sphere. High-resolution fast atom bombardment (FAB) mass spectra were measured on a JEOL JMS-700 spectrometer. Melting points were determined on a Stanford Research Systems Optimelt MPA100 and were uncorrected. The SEM images were measured on a JEOL JSM-7610F field emission scanning electron microscope. Powder diffraction X-ray analysis (PXRD) data were recorded by using Rigaku RINT-2000 using graphite-monochromitized Cu-Kα radiation (λ = 1.54187 Å) at room temperature. The preparative TLC (PTLC) purification was conducted using Wakogel B-5F PTLC plates. The quantum chemical calculation was performed at the CAM-B3LYP/6-31G+(d,p) level17 with Gaussian 09 program.18Synthesis of tris(2-hydroxyphenyl)triazasumaene (A)-(+)-1
A solution of Pd2(dba)3 (2.3 mg, 0.0025 mmol) and tris(2-furyl)phosphine (2.8 mg, 0.012 mmol) in THF (200 µL) was added to a mixture of tris(methylthio)triazasumanene ((A)-(+)-1)4 (5.0 mg, 0.012 mmol), 2-hydroxyphenylboronic acid (8.5 mg, 0.062 mmol) and CuTC (11.8 mg, 0.0617 mmol) in THF (3 mL) under N2 atmosphere. The mixture was stirred at 50 °C under N2 condition for 12 h, then cooled to room temperature and quenched with 28% NH3 aqueous solution in brine (1:1 (v/v), 10 mL). The mixture was then stirred for 10 min and extracted with CH2Cl2 for 3 times. The combined organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel preparative TLC using CH2Cl2 as eluent to afford (A)-(+)-1 (5.3 mg, 80% yield) as a yellow solid. Single crystals for X-ray structure analysis were obtained by slow evaporation method from CH2Cl2.
R f = 0.60 (CH2Cl2); m.p.: 139 °C (dec.); [α]20D = +278 (c = 0.12, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ (ppm) 13.42 (s, 3H), 7.80 (dd, J = 7.6, 1.6 Hz, 3H), 7.33 (ddd, J = 16.8, 8.4, 1.6 Hz, 3H), 7.03 (ddd, 16.0, 8.4, 1.6, 3H), 6.99 (dd, J = 7.6, 1.6 Hz, 3H), 5.32 (d, J = 19.2 Hz, 3H), 3.59 (d, J = 20.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ (ppm) 172.3, 160.7, 160.0, 158.2, 140.4, 135.6, 132.2, 130.1, 119.6, 119.1, 118.5, 45.5; IR (KBr): ν (cm−1) 2956, 1616, 1577, 1548, 1463, 1396, 1363, 1295, 1249, HRMS (FAB) m/z calcd for C36H27N3 [M + H+]: 544.1661. Found: 544.1679.
Crystal data for (A)-(+)-1
The diffraction data for (A)-(+)-1 was recorded on a ADSC Q210 CCD area detector with a synchrotron radiation (λ = 0.70000 Å) at 2D beamline in Pohang Accelerator Laboratory (PAL) at 100 K. The diffraction images were processed by using HKL3000. The structures were solved by direct methods (SHELXS) and refined by full-matrix least squares calculations on F2 (SHELXL) using the Olex2 program package.C36H21N3O3(CH2Cl2)0.24, Mw = 564.36, crystal dimensions 0.04 × 0.03 × 0.02 mm3, monoclinic, space group = P21, a = 14.1631(1) Å, b = 6.8184(1) Å, c = 15.4813(1) Å, β = 109.465(3)°, V = 1409.58(3) Å3, T = −173 °C, Z = 2, ρcalcd = 1.330 g cm−3, μ = 0.91 cm−1, 7287 unique reflections out of 8036 with I > 2σ(I), 410 parameters, 2.657° < θ < 29.523°, R1 = 0.0403, wR2 = 0.1087, GOF = 1.063. Flack = −0.02(4). CCDC 1552563.†
Solid UV-vis and fluorescence spectra
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research on Innovative Area ‘π Space Figuration’ from MEXT (No. JP26102002), and JSPS KAKENHI (JP26288020). S. H. acknowledges ACT-C, JST (JPMJCR12YZ), for the support. Y. Y. acknowledges Ogasawara Foundation for the Promotion of Science & Engineering and Iketani Science and Technology Foundation for the support. We thank Prof. Norimitu Tohnai for the help of PXRD measurement. The X-ray diffraction study of (A)-(+)-1 with synchrotron radiation was performed at the Pohang Accelerator Laboratory (Beamline 2D) supported by POSTECH.Notes and references
- Recent review of ESIPT, see; (a) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed; (b) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- K. Sakai, T. Tsuzuki, Y. Itoh, M. Ichikawa and Y. Taniguchi, Appl. Phys. Lett., 2005, 86, 081103 CrossRef.
- M. J. Paterson, M. A. Robb and L. Blancafort, J. Phys. Chem. A, 2005, 109, 7527–7537 CrossRef CAS PubMed.
- A. L. Sobolewski, Phys. Chem. Chem. Phys., 2008, 10, 1243–1247 RSC.
- S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed.
- (a) H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS PubMed; (b) H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS PubMed; (c) T. Amaya, H. Sakane, T. Muneishi and T. Hirao, Chem. Commun., 2008, 765–767 RSC; (d) T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408–409 CrossRef CAS PubMed; (e) S. Higashibayashi, R. B. N. Baig, Y. Morita and H. Sakurai, Chem. Lett., 2012, 41, 84–86 CrossRef CAS; (f) S. Mebs, M. Weber, P. Luger, B. M. Schmidt, H. Sakurai, S. Higashibayashi, S. Onogi and D. Lentz, Org. Biomol. Chem., 2012, 10, 2218–2222 RSC.
- (a) Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed; (b) P. Kaewmati, Q. Tan, S. Higashibayashi, Y. Yakiyama and H. Sakurai, Chem. Lett., 2016, 46, 146–148 CrossRef.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- (a) Y. Qian, S. Li, G. Zhang, Q. Wang, S. Wang, H. Xu, C. Li, Y. Li and G. Yang, J. Phys. Chem. B, 2007, 111, 5861–5868 CrossRef CAS PubMed; (b) R. Hu, S. Li, J. Chen, S. Wang, Y. Li and G. Yang, Phys. Chem. Chem. Phys., 2011, 13, 2044–2051 RSC; (c) A. Kindu, P. S. Harihasan, K. Prabakaran, D. Moon and S. P. Anthony, Cryst. Growth Des., 2016, 116, 3400–3408 CrossRef.
- Recently, aggregation induced emission (AIE) active curved-π molecule has been reported. See; S. H. Mahadevegowda and M. C. Stuparu, Eur. J. Org. Chem., 2017, 570–576 CrossRef CAS.
- Crystal Engineering, ed. G. R. Desiraju, J. J. Vittal and A. Ramanan, World Scientific, 2011 Search PubMed.
- (a) D. LeGourriérec, V. Kharlanov, R. G. Brown and W. Rettig, J. Photochem. Photobiol., A, 1998, 117, 209–216 CrossRef; (b) N. Basaric and P. Wan, Photochem. Photobiol. Sci., 2006, 5, 656–664 RSC.
- A time-dependent density functional theory (TD-DFT) calculation-based detailed discussion will be reported as an independent paper.
- R. Hu, S. Li, Y. Zeng, J. Chen, S. Wang, Y. Li and G. Yang, Phys. Chem. Chem. Phys., 2011, 13, 2044–2051 RSC.
- R. Pradhan, A. K. Harshan, G. S. K. Chandrika, A. Srinivasan and U. Lourderaj, J. Phys. Chem. A, 2016, 120, 9894–9906 CrossRef CAS PubMed.
- F. Rodriguez-Prieto, J. C. Penedo and M. Mosquera, J. Chem. Soc., Faraday Trans., 1998, 94, 2775–2782 RSC.
- (a) D. Ghosh, S. Batuta, N. A. Begum and D. Mandal, J. Lumin., 2017, 184, 64–73 CrossRef CAS; (b) Y. Peng, Y. Ye, X. Xiu and S. Sun, J. Phys. Chem. A, 2017, 121, 5625–5634 CrossRef CAS PubMed; (c) A. Szemik-Hojniak, Ł. Wiśniewski, I. Deperasińska, A. Makarewicz, L. Jerzykiewicz, A. Puszko, Y. Erez and D. Huppert, Phys. Chem. Chem. Phys., 2012, 14, 8147–8159 RSC; (d) M. Li, W. Ren, Z. He and Y. Zhu, J. Cluster Sci., 2017, 28, 2111–2122 CrossRef CAS; (e) D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- The fluorescence intensity continued to increase even more than 6 h after the sample preparation (Fig. S7, ESI†). For data collection, we stirred the mixtures for 5 min and quickly applied to measure emission spectra.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1552563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00530j |
| This journal is © the Partner Organisations 2018 |