
Secondary-amine-functionalized isoreticular metal–organic frameworks for controllable and selective dye capture†
Jing
Tan‡
,
Beibei
Zhou‡
,
Congcong
Liang
 ,
Hannah
Zinky
,
Hao-Long
Zhou
and
Yue-Biao
Zhang
*
,
Hannah
Zinky
,
Hao-Long
Zhou
and
Yue-Biao
Zhang
*
School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: zhangyb@shanghaitech.edu.cn
First published on 3rd November 2017
Abstract
By adding 4,4′-bipyridine (BPY) to cross-link the paddle-wheel clusters M2(–COO)4L2, isoreticular metal–organic frameworks (MOFs) have been constructed from a flexible tritopic linker, 4,4′,4′′-s-triazine-1,3,5-triyltri-p-aminobenzoic acid (H3TATAB), termed as ST-14 [Cu6(TATAB)4(BPY)3] and ST-15 [Zn6(TATAB)4(BPY)3]. With a secondary-amine group, these MOFs have shown exculsive cationic dye capture properties with charge selectivity, metal-dependent adsorption kinetics, and solvent-dependent dye release. This work sheds light on further designing smart porous materials for drug delivery, molecular inclusion, sensing and so on.
 Yue-Biao Zhang | He obtained his PhD degree in 2011 from Sun Yat-Sen University at Guangzhou under the supervision of Prof. Xiao-Ming Chen. He was a postdoc researcher in the Yaghi lab at UCLA, LBNL and UC Berkeley from 2011 to 2015. He is now an Assistant Professor at the School of Physical Science & Technology, ShanghaiTech University. He is awarded by the “1000 Young Talent Program”. His research interests include the discovery of metal–organic frameworks and design of covalent organic frameworks for gas storage and separation. |
Introduction
Metal–organic frameworks (MOFs), as a new class of crystalline porous materials constructed from inorganic and organic building units, have attracted great interest due to their regular pore structures, designable frameworks and bright potential applications.1 Recently, due to their diverse structural topologies, tunable porosity including pore sizes and pore surface, and unique host–guest interactions, MOFs have emerged as a promising adsorbent for liquid mixture purification, drug delivery, large molecular inclusion, dye capture and so on.2–22 Although extensive effort has been devoted to exploring the use of MOFs in the adsorption and separation of dyes,6–11,23–25 examples of controllable dye adsorption behaviours and studies of the related mechanism are still scarce.12–15 To investigate the effect of framework composition on the dye adsorption behaviour, it is beneficial to obtain isostructural MOFs for dye adsorption experiments thereby avoiding the uncertainty imposed by different structures.26–29Constructing a highly porous framework is a prerequisite for accommodating dye molecules.2,3 As the most common 3-connected organic unit, trigonal tricarboxylate linkers are very attractive for highly porous crystal engineering.2,30–34 For example, Zn4O(BTB)2 (MOF-177, H3BTB = benzene-1,3,5-trisbenzoic acid)2 has extra-large pores capable of large polycyclic organic molecule inclusion. In addition, by choosing between coplanar or noncoplanar trigonal tricarboxylate linkers, two kinds of ultraporous coordination frameworks with tbo or pto nets can be controllably assembled based on binuclear paddle-wheel nodes.35,36 Therefore, the trigonal tricarboxylate linkers are ideal for rationally designing highly porous frameworks. 4,4′,4′′-s-triazine-1,3,5-triyltri-p-aminobenzoic acid (H3TATAB) has been considered a favorable functionalized linker to make MOFs with secondary-amine groups decorating the pore surface.37–42 Previously, Zhou et al. reported two TATAB-based mesoporous frameworks, Cu3(TATAB)2 (meso-PCN)37 with a tbo net and Zn4O(TATAB)2 (PCN-100)38 with a pyr net (Fig. 1). In other words, the different coordination behaviours of metal ions hinder the formation of isostructural TATAB-based highly porous MOFs. Recently, by mixing H3TATAB with various ditopic linkers, a series of ultraporous MOFs with fine-tuned mesoporosity and high capacity methane storage have been systematically constructed.42
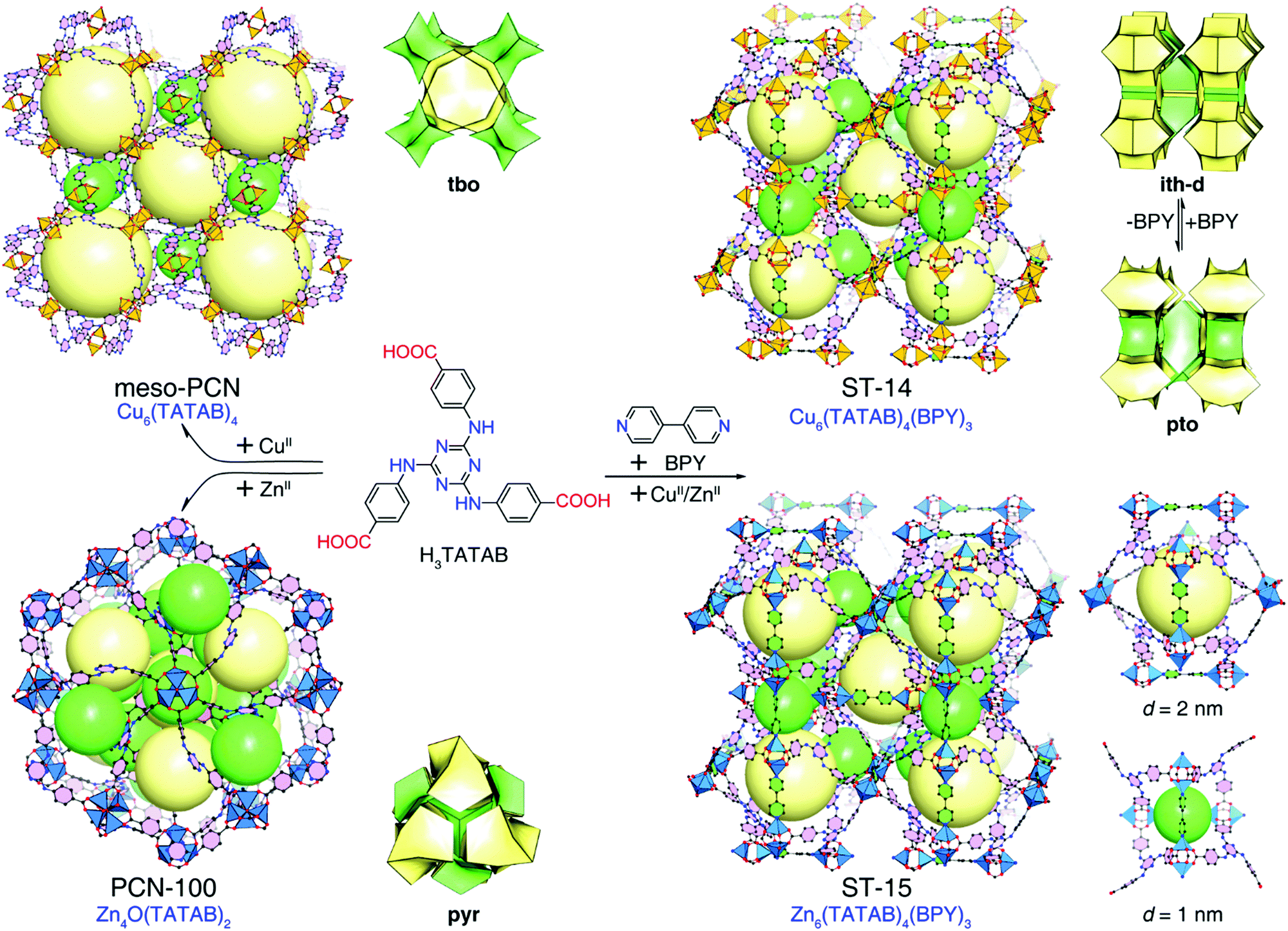 | ||
| Fig. 1 Illustration of the ligand-directed strategy to construct isostructural TATAB-based MOFs ST-14 and ST-15 and their single-crystal structures, topologies, and pore diameters. | ||
Herein, we adopted a ligand-directed strategy to successfully construct isostructural TATAB-based MOFs with Zn(II) and Cu(II) ions by introducing the ditopic neutral ligand 4,4′-bipyridine (BPY). The newly synthesized MOFs, ST-14 [Cu6(TATAB)4(BPY)3] and ST-15 [Zn6(TATAB)4(BPY)3] (ST = ShanghaiTech University), adopt crosslinked-pto (also simplified as ith-d) topology, comprising two kinds of polyhedral nanocages with diameters of 2.0 nm and 1.0 nm (Fig. 1). Selective dye capture behaviours, metal-ion-dependent adsorption kinetics, and controllable dye release behaviours were investigated for ST-14 and ST-15.
Experimental section
Materials and methods
All of the chemicals were purchased from commercial sources and used as received without further purification unless otherwise stated. Infrared (IR) spectra were recorded in the range of 4000–400 cm−1 with a Perkin-Elmer Fourier Transform FT-IR spectrometer using KBr pellets. Elemental analyses (EA) were performed on a Vario EL Cube CHNS elemental analyser. Thermogravimetric analyses (TGA) were performed on a TGA 8000 in flowing N2 at a heating rate of 10 °C min−1. Powder X-ray diffraction (PXRD) patterns were collected on a Bruker D8 ADVANCE X-ray diffractometer with CuKα radiation (λ = 1.54184 Å). NMR spectra were recorded on a Bruker AVANCE III HD 500 MHz NMR spectrometer.Synthesis of ST-14, Cu6(TATAB)4(BPY)3
A mixture of Cu(NO3)2·3H2O (39 mg, 0.16 mmol), BPY (7.5 mg, 0.05 mmol), and H3TATAB (15 mg, 0.03 mmol) was dissolved in a 20 mL vial containing 4 mL of DMF/ethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixed solvent and 2 drops of trifluoroacetic acid. The reaction mixture was then capped tightly, sonicated for 5 minutes, and heated at 65 °C for 36 hours in an isothermal oven. After cooling to room temperature, green block crystals of ST-14 were obtained with a yield of 30% based on H3TATAB. EA of the activated sample of ST-14: calcd for C126H132N30O48Cu6: C, 47.06; H, 4.14; N, 13.07%. Found: C, 46.77; H, 3.56; N, 13.59%. FTIR (4000–400 cm−1) (Fig. S1, ESI†): 1653(m), 1582(m), 1550(w), 1482(s), 1373(s), 1346(s), 1310(s), 1239(s), 1223(m), 1218(m), 1176(m), 1094(m), 1065(w), 1013(w), 858(m), 805(m),780(m), 732(w), 701(m), 659(m), 621(m), 572(w), 499(m), 472(m).
:1) mixed solvent and 2 drops of trifluoroacetic acid. The reaction mixture was then capped tightly, sonicated for 5 minutes, and heated at 65 °C for 36 hours in an isothermal oven. After cooling to room temperature, green block crystals of ST-14 were obtained with a yield of 30% based on H3TATAB. EA of the activated sample of ST-14: calcd for C126H132N30O48Cu6: C, 47.06; H, 4.14; N, 13.07%. Found: C, 46.77; H, 3.56; N, 13.59%. FTIR (4000–400 cm−1) (Fig. S1, ESI†): 1653(m), 1582(m), 1550(w), 1482(s), 1373(s), 1346(s), 1310(s), 1239(s), 1223(m), 1218(m), 1176(m), 1094(m), 1065(w), 1013(w), 858(m), 805(m),780(m), 732(w), 701(m), 659(m), 621(m), 572(w), 499(m), 472(m).
Synthesis of ST-15, Zn6(TATAB)4(BPY)3
A mixture of Zn(NO3)2·4H2O (42 mg, 0.16 mmol), BPY (7.5 mg, 0.05 mmol) and H3TATAB (15 mg, 0.03 mmol) was dissolved in a 20 mL vial containing 4 mL of DMF/ethanol (v/v = 2.33:1) mixed solvent. The reaction mixture was then capped tightly, sonicated for 5 minutes, and heated at 65 °C for 48 hours in an isothermal oven. After cooling to room temperature, colourless block crystals of ST-15 were obtained with a yield of 30% based on H3TATAB. EA of the activated sample of ST-15: calcd for C126H108N30O36Zn6: C, 50.27; H, 3.62; N, 13.96%. Found: C, 50.37; H, 3.63; N, 13.91%. FTIR (4000–400 cm−1) (Fig. S1, ESI†): 1654(m), 1585(m), 1550(w), 1483(s), 1373(s), 1342(s), 1310(s), 1240(s), 1223(m), 1218(m), 1176(m), 1096(m), 1065(w), 1013(w), 859(m), 806(m), 783(m), 732(w), 701(m), 659(m), 621(m), 571(w), 500(m), 466(m).
Single crystal X-ray diffraction measurement and crystallographic data refinement
Single-crystal X-ray diffraction data were collected on a Bruker D8 Venture diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 173(2) K. Multi-scan absorption corrections were performed with a Bruker APEX III. The initial structure models were solved by direct methods, and all the non-H atoms were refined anisotropically using the SHELXTL-2014 software package. All hydrogen atoms were generated geometrically and refined with a riding model. Due to the serious disorder of guests, the solvent accessible void electron density was treated by using the Olex2 solvent mask for ST-14 and ST-15. Crystallographic data and structural refinement details are summarized in Table S1 (ESI†).Dye adsorption/separation experiments
Solutions of different dyes with concentrations from 5 ppm to 40 ppm in ethanol were prepared for the adsorption experiments. 50 mg of freshly prepared ST-14/15 was weighed out into a 20 ml vial, exchanged with ethanol 3 times, and then the ethanol was replaced with 5 ml of dye-containing ethanol solution. The dye content was analysed by UV-Vis absorption spectroscopy. The intensity value at the maximum absorbance wavelength was chosen to calculate the dye content and the absorbance value at 0 h was normalized as 100% dye content in the UV-Vis spectra. The UV-Vis spectra were recorded on a Cary 5000 UV-Vis-NIR spectrophotometer after 0, 0.5, 1, 2, 3, 4, 5, 6, 7, and 8 hours.Results and discussion
Synthesis and crystal structure analysis
The solvothermal reaction of Cu(NO3)2·3H2O and Zn(NO3)2·4H2O, and H3TATAB in a mixture of DMF and ethanol at 65 °C can give single crystals of meso-PCN37 based on the Cu2(–COO)4 clusters and PCN-10038 based on the Zn4O(–COO)6 clusters, respectively. Considering that the BPY ligand can induce both Cu(II) and Zn(II) ions to form the M2(–COO)4(–Py)2 clusters,43–45 various mole ratios of H3TATAB and BPY in the presence of excess Cu(NO3)2·3H2O and Zn(NO3)2·4H2O were heated under the same conditions, and the crystalline phases of solid products were monitored by PXRD (Fig. 2). By adding H3TATAB and BPY in the ratio of 8:2, a new phase was observed, while by adding H3TATAB and BPY in the ratio of 4:6, the pure new phase could be achieved in the bulk sample. Indicated by the similar PXRD patterns, the reactions of both Cu(NO3)2·3H2O and Zn(NO3)2·4H2O could give isostructural products.
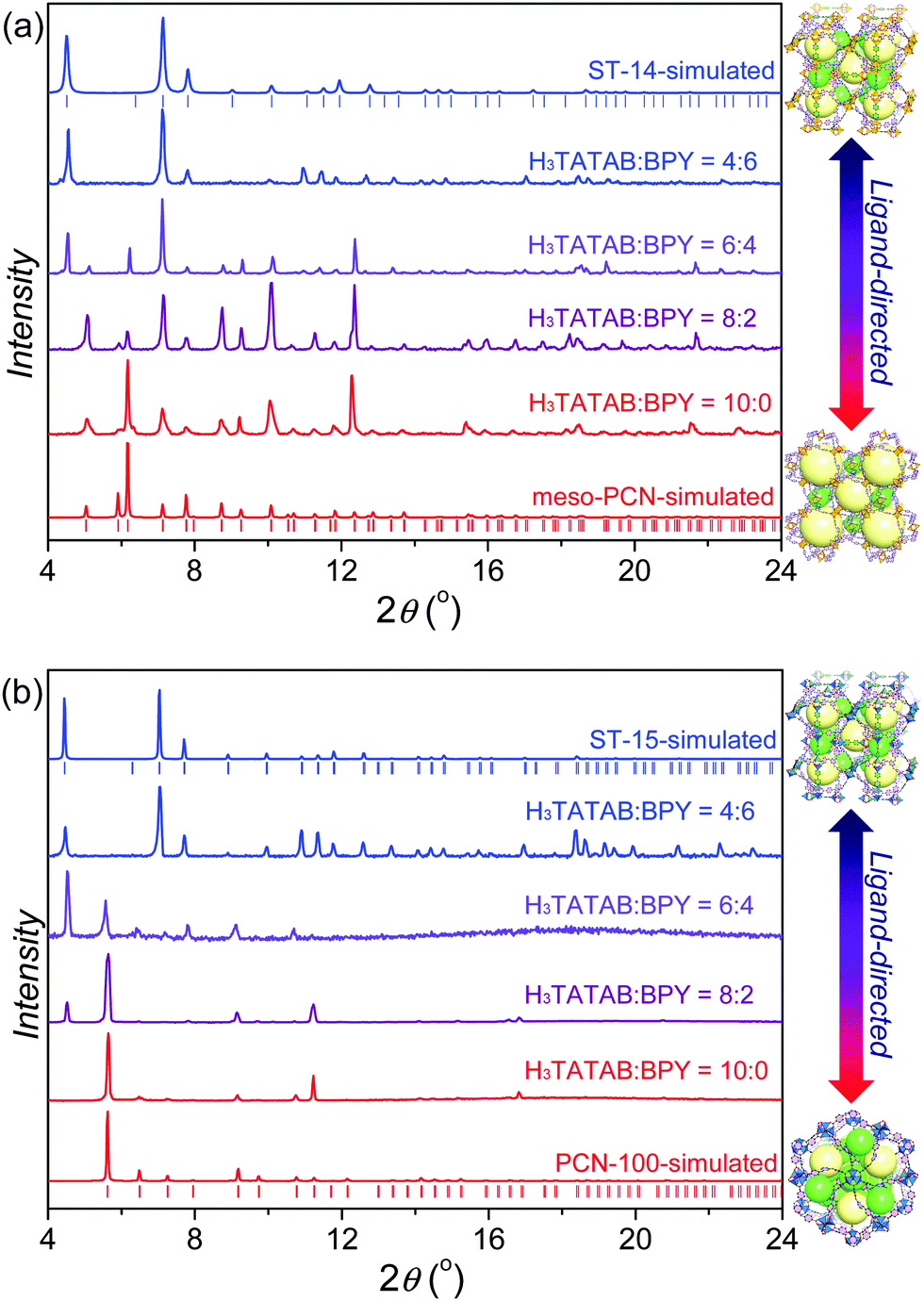 | ||
| Fig. 2 PXRD patterns of the products with various ratios of H3TATAB and BPY to show the ligand-directed syntheses (a) from meso-PCN to ST-14 and (b) from PCN-100 to ST-15. | ||
Single-crystal X-ray diffraction analysis confirmed that the obtained crystals for Cu6(TATAB)4(BPY)3 (called ST-14) and Zn6(TATAB)4(BPY)3 (called ST-15) are isostructural. For simplicity, we use ST-14 as an example to introduce their structure. ST-14 crystallizes in the centrosymmetric cubic space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3n, containing half a metal ion, one third of a TATAB ligand and one quarter of a BPY ligand in its asymmetric unit (Fig. S2, ESI†). As expected, the metal ion adopts a five-coordinated configuration and is bridged to another one by four carboxylate groups to form a typical M2(–COO)4 paddle-wheel cluster. Regarding the paddle-wheel cluster as a 4-connected node and the TATAB ligand as a 3-connected node, the coordination framework without the BPY ligands can be simplified as a (3,4)-connected pto net. The M2(–COO)4 clusters are pillared by the BPY ligands leading to the overall framework of ST-14 becoming a crosslinked pto net (also known as a (3,6)-connected ith-d net)46–48 (Fig. 1).
3n, containing half a metal ion, one third of a TATAB ligand and one quarter of a BPY ligand in its asymmetric unit (Fig. S2, ESI†). As expected, the metal ion adopts a five-coordinated configuration and is bridged to another one by four carboxylate groups to form a typical M2(–COO)4 paddle-wheel cluster. Regarding the paddle-wheel cluster as a 4-connected node and the TATAB ligand as a 3-connected node, the coordination framework without the BPY ligands can be simplified as a (3,4)-connected pto net. The M2(–COO)4 clusters are pillared by the BPY ligands leading to the overall framework of ST-14 becoming a crosslinked pto net (also known as a (3,6)-connected ith-d net)46–48 (Fig. 1).
It is worth noting that the trigonal tricarboxylate ligands need to be twisted in the pto structure. Therefore, though the trigonal TATAB ligand normally adopts a coplanar configuration in reported structures,37 in ST-14, it is twisted along the C–N single bond of the secondary-amine groups to adopt the noncoplanar conformation (Fig. S2, ESI†). The addition of BPY ligands effectively changes the coordination behaviour of the metal ions and triggers the adaptability of TATAB, thus inducing the formation of the underlying pto structure. While the optimal addition of BPY is far from excessive (twice equivalent) to bypass phase separation, the single crystal X-ray diffraction data and the digested-solution 1H NMR spectrum (Fig. S3, ESI†) confirmed that the ratio of TATAB and BPY in the crosslinked pto structure is 4:3. This phenomenon suggests that in the ligand-directed synthesis strategy, an excess of auxiliary ligands is essential for ensuring the full formation of such a target structure.
ST-14 possesses two kinds of polyhedral nanocages with pore sizes of 2.0 nm and 1.0 nm, and a solvent-accessible volume void of ca. 80%. The phase purity of as-synthesized ST-14 was confirmed by the good match between the experimental PXRD patterns and the simulated ones (Fig. 2). Despite no obvious platforms appearing in the 25–700 °C range, the TGA curve of as-synthesized ST-14 revealed a rapid weight loss of more than 50% before 150 °C revealing its high porosity (Fig. S4, ESI†).
Charge-dependent selective dye capture
Considering synthetic organic dyes have been widely used and sometimes act as pollutants,23–25,49 the dye adsorption behaviours of ST-14 and ST-15 were investigated. Seven organic dyes with different charges and sizes were selected (Fig. 3 and Table S2, ESI†): methylene blue (MLB, +1), rhodamine B (RHB, +1), crystal violet (CV, +1), solvent yellow 2 (SY2, 0), acid red 88 (AR88, −1), orange G (OG, −2) and new coccine (NC, −3).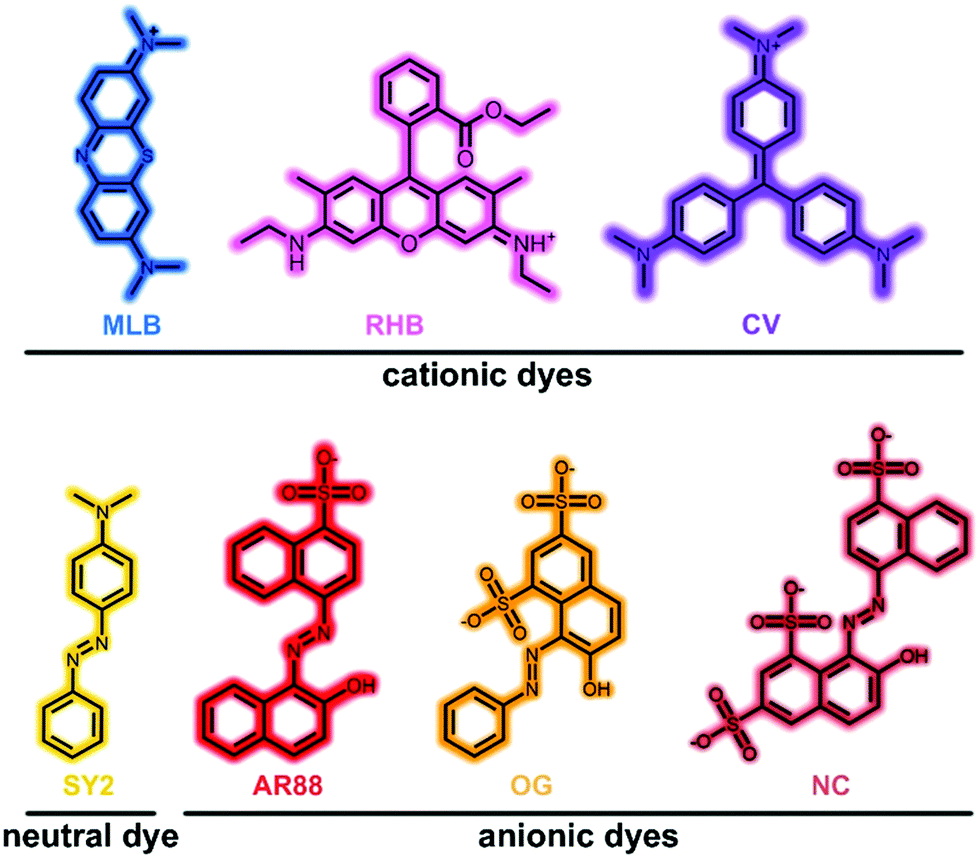 | ||
| Fig. 3 Chemical structures of seven organic dyes discussed in this work. | ||
First, the ST-15 crystals with sizes larger than 0.1 × 0.1 × 0.1 mm3 were soaked in 5 mL of EtOH solution containing a single dye and the dye content was determined by UV-Vis spectroscopy monitoring the adsorption behaviours with time. Interestingly, although ST-15 is an electrically neutral coordination framework, it can selectively adsorb cationic dyes (Fig. 4 and Fig. S5, S6, ESI†). While almost no adsorption behaviour was observed for neutral and anionic dyes after 480 min, the content of cationic dyes MLB, RHB and CV decreased by 99, 93, and 85%, respectively. As a consequence, the crystals became blue, pink and purple after adsorption of cationic dyes MLB, RHB and CV, respectively. PXRD patterns of the resulting sample showed that ST-15 still kept its crystallinity after the dye adsorption experiments (Fig. S7, ESI†). Although the nanocages (up to 2.0 nm) of ST-15 can accommodate these dyes with large molecular sizes, the relatively smaller cage windows (<0.8 nm) could limit the entry and exit of these dyes. The adsorption rate of the cationic dyes exhibits a trend of MLB > RHB > CV, which showed a negative correlation with the dye sizes (MLB < RHB < CV). This size-dependent adsorption rate indicates that ST-15 can adsorb the dye in the pore channels with the assistance of the dynamic behaviours and dissociation of the ligand,14,50 otherwise surface adsorption is not selective in general. Subsequently, the selective adsorption capacity of ST-15 to the cationic dye was verified by performing a separation experiment of mixed dyes (Fig. 5). Time-dependent UV-Vis spectroscopy indicated that ST-15 can selectively and almost completely capture the cationic dye MLB from the mixed dye system even with a ratio of only 1:10 of MLB to the other dyes. In other words, even in the presence of neutral and anionic dyes, its cationic dye capture selectivity will not be disturbed, illustrating its utility for dye separation.
 | ||
| Fig. 4 (a) Photographs of ST-15 samples after dye-adsorption for 480 minutes; (b) differently charged dye contents in solution in the presence of ST-15 monitored with time. | ||
 | ||
| Fig. 5 UV-Vis spectroscopy changes of mixed dye solutions in the presence of ST-15. The photographs show the colours of the dye solutions, their mixture before and after adsorption. | ||
A similar selective capture of cationic dyes has also been reported in other neutral frameworks.6,7 Actually, according to previous reports, MOFs can show higher selectivity and adsorption capacity for cationic dyes after modification with amino groups.27,28 In our case, such excellent selectivity may be attributed to the secondary-amine groups of their pore surfaces. In order to confirm our hypothesis, qualitative dye adsorption experiments were performed on neutral PCN-100 and meso-PCN based on the TATAB ligand,37,38 which also display selective cationic dye capture behaviour (Fig. S5, ESI†). It can be seen that the TATAB ligand plays an important role in selective adsorption behaviours.
Metal-ion dependence of dye adsorption kinetics
ST-14 and ST-15 are isostructural, which is beneficial for studying the relationship between framework composition and framework adsorption behaviour. Therefore, the dye adsorption behaviour of ST-14 was also characterized for comparison (Fig. S5 and S8–S11, ESI†). As a general observation, the adsorption rate of ST-14 was much slower than that of ST-15, although both ST-14 and ST-15 showed a similar charge and size selectivity to adsorb cationic dyes (Fig. 6). With the zinc metal centres, ST-15 can rapidly take up MLB, RHB and CV dyes in 480 minutes as much as 99, 93, and 85%, respectively. In comparison, ST-14 using the same time can only adsorb MLB, RHB and CV dyes up to 54, 43, and 25%, respectively. Since sufficiently large sized crystals are used in the experiments, such a difference in adsorption kinetics must be caused by the intrinsic properties of the frameworks. | ||
| Fig. 6 The difference in adsorption kinetics between ST-14 and ST-15 for the cationic dyes (a) MLB, (b) RHB and (c) CV. | ||
Although there are many possibilities that can affect the adsorption kinetics, the major difference in this pair of isostructural MOFs is the metal centres. In the context of coordination chemistry, 5-coordinated square-pyramidal geometry is stable for a copper centre, while a zinc ion prefers to have 4-coordinated tetrahedral geometry or 6-coordinated octahedral geometry. Here, we attribute this phenomena to the dissociation mechanism.14 That is, the copper centre in ST-14 preferred a square-pyramidal geometry leading to slower ligand dissociation, while the zinc centre in ST-15 is relatively dynamic for faster ligand dissociation. This observation is also supported by the trend of metal exchange process in several MOFs previously reported.48
Controllable dye release behaviours
Reversible adsorption and release behaviours for dyes receive high attention in practical applications, especially for drug delivery. In general, charged dyes can be controllably released in a salt solution rather than in a blank solution. However, no release behaviours were observed after the sample of MLB-loaded ST-15 was soaked in either blank or NaNO3-containing EtOH solution (Fig. 7), meaning that the dye release for ST-15 is not simply dependent on an ion-exchange process. Interestingly, when the sample was soaked into NaNO3-containing DMF solution, the dye could be released rapidly. At the same time, the dye cannot be released in a blank DMF solution. This indicates that the dye release will be triggered only by the cooperation of the salt and the appropriate solvent molecules. To the best of our knowledge, ST-15 is the first case of a MOF material exhibiting solvent-dependent dye release behaviour, which may be caused by the unique host–guest interaction in the MOF system. PXRD revealed that ST-15 retained its framework integrity after the dye release experiments (Fig. S12, ESI†). Although the mechanism is unclear and remains to be explored, this phenomenon suggests that MOFs have great potential as candidates for designing and obtaining dye adsorbents and drug capsules.51,52 | ||
| Fig. 7 The change of dye content in the release experiments of ST-15 in the blank EtOH, NaNO3-containing EtOH, blank DMF and NaNO3-containing DMF solutions. | ||
Conclusions
In summary, by virtue of the ligand-directed strategy and the adaptability of the trigonal tricarboxylate linker H3TATAB, we have successfully constructed crosslinked-pto type MOFs based on Zn(II) and Cu(II) metal centres. These two highly porous MOFs, with secondary-amine-groups decorating their nanocages exhibited exceptional ability to selectively capture cationic dyes, and being isostructural, the pair provided an ideal opportunity to study the composition dependence of dye adsorption. Exploration of the kinetics highlights that dye capture by MOFs can be rationally fine-tuned through the choice of metal ion. This first report of solvent-dependent dye release gives a new direction for further studies on the mechanism of molecule inclusion and release in the liquid phase.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the National Natural Science Foundation of China (No. 21522105), the 1000 Young Talents Plan, and the ShanghaiTech University Research Startup Fund. The National Postdoctoral Program for Innovative Talents (BX201700272) is acknowledged by H. L. Z. We thank the staff from the BL-17B Beamline of the National Center for Protein Sciences Shanghai (NCPSS) at the Shanghai Synchrotron Radiation Facility (SSRF) for assistance during data collection.Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- L. Q. Ma, J. M. Falkowski, C. Abney and W. B. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- Q.-R. Fang, G.-S. Zhu, Z. Jin, Y.-Y. Ji, J.-W. Ye, M. Xue, H. Yang, Y. Wang and S.-L. Qiu, Angew. Chem., Int. Ed., 2007, 119, 6758–6762 CrossRef.
- H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed.
- Y. Q. Lan, H. L. Jiang, S. L. Li and Q. Xu, Adv. Mater., 2011, 23, 5015–5020 CrossRef CAS PubMed.
- B. Q. Song, X. L. Wang, Y. T. Zhang, X. S. Wu, H. S. Liu, K. Z. Shao and Z. M. Su, Chem. Commun., 2015, 51, 9515–9518 RSC.
- S. R. Zhang, J. Li, D. Y. Du, J. S. Qin, S. L. Li, W. W. He, Z. M. Su and Y. Q. Lan, J. Mater. Chem. A, 2015, 3, 23426–23434 CAS.
- X. S. Wang, J. Liang, L. Li, Z. J. Lin, P. P. Bag, S. Y. Gao, Y. B. Huang and R. Cao, Inorg. Chem., 2016, 55, 2641–2649 CrossRef CAS PubMed.
- Z. Zhu, Y. L. Bai, L. Zhang, D. Sun, J. Fang and S. Zhu, Chem. Commun., 2014, 50, 14674–14677 RSC.
- S. B. Han, Y. H. Wei, C. Valente, I. Lagzi, J. J. Gassensmith, A. Coskun, J. F. Stoddart and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 16358–16361 CrossRef CAS PubMed.
- X. Zhao, X. Bu, T. Wu, S. T. Zheng, L. Wang and P. Feng, Nat. Commun., 2013, 4, 2344 Search PubMed.
- J. V. Morabito, L. Y. Chou, Z. H. Li, C. M. Manna, C. A. Petroff, R. J. Kyada, J. M. Palomba, J. A. Byers and C. K. Tsung, J. Am. Chem. Soc., 2014, 136, 12540–12543 CrossRef CAS PubMed.
- Y. Han, S. Sheng, F. Yang, Y. Xie, M. Zhao and J.-R. Li, J. Mater. Chem. A, 2015, 3, 12804–12809 CAS.
- K. Williams, L. Meng, S. Lee, L. Lux, W. Y. Gao and S. Q. Ma, Inorg. Chem. Front., 2016, 3, 393–396 RSC.
- S. Wang, Q. Wang, X. Feng, B. Wang and L. Yang, Adv. Mater., 2017, 29, 1701898 CrossRef PubMed.
- J. Yu, Y. Cui, H. Xu, Y. Yang, Z. Wang, B. Chen and G. Qian, Nat. Commun., 2013, 4, 2719 Search PubMed.
- B. Tu, Q. Pang, D. Wu, Y. Song, L. Weng and Q. Li, J. Am. Chem. Soc., 2014, 136, 14465–14471 CrossRef CAS PubMed.
- J. W. Ye, H. L. Zhou, S. Y. Liu, X. N. Cheng, R. B. Lin, X. L. Qi, J. P. Zhang and X. M. Chen, Chem. Mater., 2015, 27, 8255–8260 CrossRef CAS.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- Y. J. Cui, B. L. Chen and G. D. Qian, Coord. Chem. Rev., 2014, 273, 76–86 CrossRef.
- A. A. Adeyemo, I. O. Adeoye and O. S. Bello, Toxicol. Environ. Chem., 2012, 94, 1846–1863 CrossRef CAS.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244-245, 444–456 CrossRef CAS PubMed.
- I. Ahmed and S. H. Jhung, J. Hazard. Mater., 2016, 301, 259–276 CrossRef CAS PubMed.
- M. Tong, D. Liu, Q. Yang, S. Devautour-Vinot, G. Maurin and C. Zhong, J. Mater. Chem. A, 2013, 1, 8534 CAS.
- Q. Chen, Q. He, M. Lv, Y. Xu, H. Yang, X. Liu and F. Wei, Appl. Surf. Sci., 2015, 327, 77–85 CrossRef CAS.
- C. Li, Z. Xiong, J. Zhang and C. Wu, J. Chem. Eng. Data, 2015, 60, 3414–3422 CrossRef CAS.
- K. E. Cordova and O. M. Yaghi, Mater. Chem. Front., 2017, 1, 1304–1309 RSC.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- Y. B. Zhang, H. Furukawa, N. Ko, W. X. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. X. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed.
- P. Li, N. A. Vermeulen, C. D. Malliakas, D. A. Gomez-Gualdron, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624–627 CrossRef CAS PubMed.
- E. Virmani, O. Beyer, U. Luning, U. Ruschewitz and S. Wuttke, Mater. Chem. Front., 2017, 1, 1965–1974 RSC.
- H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe-Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef CAS PubMed.
- P. Müller, R. Grünker, V. Bon, M. Pfeffermann, I. Senkovska, M. S. Weiss, X. Feng and S. Kaskel, CrystEngComm, 2016, 18, 8164–8171 RSC.
- X. S. Wang, S. Q. Ma, D. F. Sun, S. Parkin and H. C. Zhou, J. Am. Chem. Soc., 2006, 128, 16474–16475 CrossRef CAS PubMed.
- Q. R. Fang, D. Q. Yuan, J. Sculley, J. R. Li, Z. B. Han and H. C. Zhou, Inorg. Chem., 2010, 49, 11637–11642 CrossRef CAS PubMed.
- M. Du, X. Wang, M. Chen, C. P. Li, J. Y. Tian, Z. W. Wang and C. S. Liu, Chem. – Eur. J., 2015, 21, 9713–9719 CrossRef CAS PubMed.
- H. Zhang, D. Chen, H. Ma and P. Cheng, Chem. – Eur. J., 2015, 21, 15854–15859 CrossRef CAS PubMed.
- T. Xia, T. Song, Y. Cui, Y. Yang and G. Qian, Dalton Trans., 2016, 45, 18689–18695 RSC.
- C. Liang, Z. Shi, C.-T. He, J. Tan, H. Zhou, H.-L. Zhou, Y. Lee and Y.-B. Zhang, J. Am. Chem. Soc., 2017, 139, 13300–13303 CrossRef CAS PubMed.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- B. Q. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912–4914 CrossRef CAS PubMed.
- S. Bureekaew, H. Sato, R. Matsuda, Y. Kubota, R. Hirose, J. Kim, K. Kato, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 7660–7664 CrossRef CAS PubMed.
- N. Klein, I. Senkovska, I. A. Baburin, R. Grunker, U. Stoeck, M. Schlichtenmayer, B. Streppel, U. Mueller, S. Leoni, M. Hirscher and S. Kaskel, Chem. – Eur. J., 2011, 17, 13007–13016 CrossRef CAS PubMed.
- D. Han, F. L. Jiang, M. Y. Wu, L. Chen, Q. H. Chen and M. C. Hong, Chem. Commun., 2011, 47, 9861–9863 RSC.
- X. Song, T. K. Kim, H. Kim, D. Kim, S. Jeong, H. R. Moon and M. S. Lah, Chem. Mater., 2012, 24, 3065–3073 CrossRef CAS.
- P. Samanta, P. Chandra, A. V. Desai and S. K. Ghosh, Mater. Chem. Front., 2017, 1, 1384–1388 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- J. Della Rocca, D. M. Liu and W. B. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic tables, PXRD patterns, TGA curves, NMR and dye UV-vis absorbance spectroscopy. CCDC 1573691 and 1573692. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00414a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |