DOI:
10.1039/C8QI01150H
(Research Article)
Inorg. Chem. Front., 2019,
6, 317-325
The design of a novel and resistant Zn(PZDC)(ATZ) MOF catalyst for the chemical fixation of CO2 under solvent-free conditions†
Received
24th October 2018
, Accepted 28th November 2018
First published on 29th November 2018
Abstract
A well-designed 3D metal–organic framework with accessible Lewis acid–base sites, [Zn2(PZDC)(1/2ATZ)2(H2O)2·2.5H2O] (H2PZDC = 3,5-pyrazoledicarboxylic acid, 5-ATZ = 5-aminoterazole) abbreviated as Zn(PZDC)(ATZ), was successfully prepared by incorporating zinc(II) ions, 3,5-pyrazoledicarboxylic acid and nitrogen-rich 5-aminoterazole. The existence of abundant Lewis bases can increase the chemical affinity to carbon dioxide, thus resulting in their high CO2 capture ability. Additionally, the heterogeneous Zn(PZDC)(ATZ) catalyst facilitated the CO2 coupling reaction of various epoxides into cyclic carbonates with the aid of Bu4NBr under solvent-free conditions (90 °C, 1 MPa, 6 h); besides, a high TON (131.0) and TOF (21.8 h−1) of propylene carbonates were obtained. Furthermore, the Zn(PZDC)(ATZ) catalyst also showed good chemical stability and recyclability for seven cycles without a significant catalytic loss. What's more, a plausible catalytic mechanism of synergistic effects derived from Zn(PZDC)(ATZ)/Bu4NBr catalysts for CO2 conversion to cyclic carbonate was proposed.
Introduction
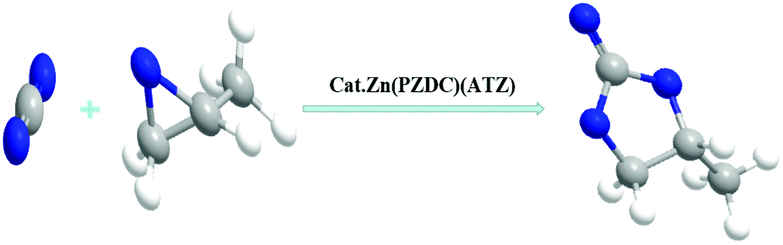
The greenhouse effect has already become an intractable environmental problem due to the excessive emission of carbon dioxide caused by human activities from the large-scale use of fossil energy.1,2 Carbon capture and utilization (CCU) could transform CO2 as carbon feedstock into high value-added chemicals so as to limit CO2 destabilization;3 therefore, the development of the capture and conversion of CO2 has drawn considerable interest. The main challenge for CO2 utilization is the high kinetic and thermodynamic stability of the CO2 molecule, which requires a large amount of energy to be activated. The effective and industrial application method for CO2 utilization is the conversion of CO2 into cyclic carbonates (Scheme 1), which is attractive for its 100% atom-economical reaction in terms of sustainable and green chemistry.4 Furthermore, the cyclic carbonates can be employed as reaction intermediates for chemicals, precursors of polymers, electrolytes and so on.5
 |
| | Scheme 1 Synthesis of cyclic carbonates from CO2 and epoxide. | |
Numerous catalysts for coupling CO2 and epoxides into cyclic carbonates have been reported in the past few decades. For example, homogeneous catalysts showed exceptional properties under mild conditions, such as ionic liquids (ILs),6 metal complexes7 and organic bases;8 however, the problems of product separation and recyclability decreased the demand of these homogeneous catalysts and limited industrial production. Therefore, more and more attention has been focused on the development of heterogeneous catalysts like functional zeolites9 and porous organic polymers.10 In particular, a series of self-assembled MOFs with an ordered structure, tunable architecture, high adsorption capacity and modifiable groups have emerged as novel catalysts for the fixation of CO2. For instance, MIL-68(In)-NH2,11 ZIF-78(Zn),12 and BIT-103(Zn)13 have been developed and presented high catalytic performance at high temperature. Particularly, in order to meet the needs of activity and selectivity, organic solvents were added into the reaction system and the preparation of catalysts was complex and expensive.14,15 Even some MOFs such as MOF-892(Zr) exhibited catalytic performance at ambient CO2 pressure over 60 h.16 Moreover, only a few studies have considerably concentrated on the stability of catalysts, for example, MOFs based on Zn ions and organic carboxylates are usually subjected to hydrolysis in the presence of moisture.17,18 Obviously, these disadvantages restrict their applications in the industrial production. For the CO2 cycloaddition reactions, it is generally regarded that the synergistic effects of Lewis acid–base sites and the auxiliary effect of nucleophilic reagents from the catalysts are favorable to CO2 cycloaddition to epoxide smoothly.19,20 Furthermore, the amino group and water existed in MOFs as hydrogen bond donors (HBD) are conducive to the activation of propylene oxide (PO) and the abundant nitrogen units in MOFs can improve the affinity of CO2 molecules.21 Taking these into account, it is imperative to design the metal organic framework catalyst with Lewis acid–base sites that are inexpensive, efficient, stable, and highly resistant to the synthesis of cyclic carbonates.
Herein, a novel zinc-based MOF catalyst constructed from the mixed ligands of 3,5-pyrazoledicarboxylic acid and 5-aminoterazole has been reported, the developed Zn(PZDC)(ATZ) not only contains Lewis acid–base sites with a three-dimensional framework in crystals but also shows great chemical affinity to CO2 due to the existing abundant N atoms. What's more, this Zn(PZDC)(ATZ) with unique structures displays fascinating catalytic properties for the cycloaddition reaction of epoxides and CO2, and can be easily recycled for seven runs. Remarkably, the Zn(PZDC)(ATZ) catalyst possesses excellent hydrothermal and structural stability in a wide range of pH values. Finally, a possible synergistic mechanism catalyzed by Zn(PZDC)(ATZ)/Bu4NBr catalysts under solvent-free conditions was proposed. The developed Zn(PZDC)(ATZ) can both capture and transform carbon dioxide, and its performance is comparable or superior to the reported MOFs in the literature (as shown in Table 3).
Results and discussion
Zn(PZDC)(ATZ) catalyst characterization
Crystal structure description.
The compound Zn(PZDC)(ATZ) crystallizes in an orthorhombic system with the space group Pnnm and displays a three-dimensional (3D) framework with one-dimensional (1D) channels. The asymmetric unit of Zn(PZDC)(ATZ) consists of two crystallographically independent Zn(II) centers, one PZDC2− anion, two halves of ATZ− anions and two coordinated water molecules (Fig. 1a). Both zinc centers locate in a slightly distorted octahedral coordination geometry and are coordinated by three carboxylate oxygen atoms from three different PZDC2− anions, two nitrogen atoms from PZDC2− and ATZ− anions, respectively, and one coordinated with water molecules. The bond distances range for Zn–O from 2.035(3) to 2.347(2) Å and for Zn–N from 2.062(3) to 2.104(3) Å. The H2PZDC ligand in Zn(PZDC)(ATZ) loses three protons to form the PZDC2− anion, in which two protons arise from carboxyl groups and one proton from the pyrazole ring. The PZDC2− anion adopts a μ6-κN,O:κO:κO′:κO′′:κO′′′:κO′′′,N′ coordination mode, in which both carboxylate groups coordinated with three zinc centers by the η1:η2:μ3 coordination mode (Fig. 1b). Thus, the zinc centers are connected by the PZDC2− anion into a 2D sheet in the ab plane (Fig. 2a). The ATZ− anions coordinated to the Zn center of adjacent 2D sheets with a Zn⋯Zn distance of 6.3358(2) Å. Thus, the 2D sheets are further pillared by the ATZ− anions to construct the 3D framework. A feature of Zn(PZDC)(ATZ) is that there are 1D rectangular tunnels with dimensions of ca. 6.34 Å × 7.84 Å, which are filled by the lattice water molecules (Fig. 2b). Furthermore, the 1D tunnels in the neighboring sheets are interlaced with each other at an angle of ca. 68.12 degrees.
 |
| | Fig. 1 (a) A view of the asymmetric unit and some symmetry-related atoms in Zn(PZDC)(ATZ). [Symmetry codes: (i) x, y, −z; (ii) −x, −y, z; (iii) −0.5 − x, 0.5 + y, 0.5 − z; (iv) x, y, 1 − z]. (b) The coordination mode of the PZDC2− anion. | |
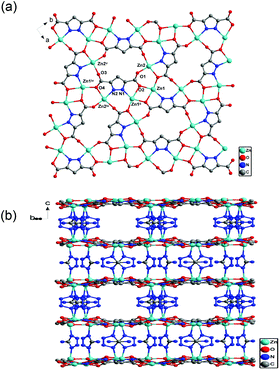 |
| | Fig. 2 (a) View down the c axis, showing the 2D sheet structure built up of Zn centers connected by the PZDC2− anion. (b) A view of the 3D framework of Zn(PZDC)(ATZ). | |
PXRD and FT-IR characterization.
The probed powder diffraction patterns had good agreement between the simulated and measured samples in Fig. 3a, which confirmed the structural integrity and phase homogeneity.
 |
| | Fig. 3 (a) PXRD patterns of the as-synthesized Zn(PZDC)(ATZ) and simulated, (b) FT-IR spectra of Zn(PZDC)(ATZ). | |
Furthermore, the stretching vibration of the primary amine was observed at 3458 and 3453 cm−1 in the FT-IR spectrum (Fig. 3b). The absorption bands at 792 and 755 cm−1 were attributed to the N–H flexural vibrations of the primary amine,22 and a broad band that appeared at around 3180 cm−1 was ascribed to the O–H stretching vibration,23 coming from the water molecules contained in Zn(PZDC)(ATZ). Moreover, the absorption band at around 1652 cm−1 belonged to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration.24 The possibility of the involvement of the tetrazolium and pyrazole carboxylic acid ligands in the coordination was confirmed in Zn(PZDC)(ATZ).
O stretching vibration.24 The possibility of the involvement of the tetrazolium and pyrazole carboxylic acid ligands in the coordination was confirmed in Zn(PZDC)(ATZ).
XPS analysis.
XPS was assigned to investigate the surface photo-electron states of elements in the Zn(PZDC)(ATZ) sample as shown in Fig. 4. The survey spectrum indicated that it consisted of C, N, O, Zn signals. The Zn 2p XPS high resolution spectrum in Fig. 4b was deconvoluted into four peaks, two peaks at 1021.4 and 1026.7 eV attributed to Zn 2p3/2, while the Zn 2p1/2 region contained the peaks at 1044.8 eV and 1049.7 eV.
 |
| | Fig. 4 XPS spectra of (a) the survey spectrum and (b) the Zn 2p XPS core level spectrum of Zn(PZDC)(ATZ). | |
Thermal analysis.
The TG-DSC revealed the thermal stability and decomposition of the skeleton construction of Zn(PZDC)(ATZ). As shown in Fig. 5, the weight loss could be decomposed into four steps, the initial 10.2% weight loss before 170 °C was attributed to the loss of existing H2O in Zn(PZDC)(ATZ). From 170 °C to 300 °C, a small weight loss of 10.3% came from the coordinated water with Zn atoms. Whereas, about 11.7% of Zn(PZDC)(ATZ) that disintegrated within 300–450 °C was assigned to the subsequent decomposition of organic ligands under an N2 atmosphere. Ultimately, the residual mass of 48.5% was caused by a complete collapse. Thus, the Zn(PZDC)(ATZ) sample was dried at 120 °C before the catalytic test and Zn(PZDC)(ATZ) possessed thermostability during the process of CO2 fixation reactions conducted at 90 °C in the following.
 |
| | Fig. 5 TG-DSC curves of the as-synthesized Zn(PZDC)(ATZ). | |
Gas adsorption properties.
The incorporation of nitrogen-rich groups into Zn(PZDC)(ATZ) was expected to result in high CO2 sorption behaviors. As illustrated in Fig. 6, its CO2 adsorption capacity reached 119 mg g−1 at 273 K and 80 mg g−1 at 298 K under 1 atm. The CO2 uptake over Zn(PZDC)(ATZ) at 298 K was comparable to USTC-253(Al) which possessed adsorption sites (79 mg g−1, 1 atm)25 and higher than several reported MOFs such as MOF-892(Zr) (38 mg g−1, 0.8 atm),16 MOF-177(Zn) (35 mg g−1, 1 atm),26 and ZnGlu (5 mg g−1, 1 atm).27 Based on the adsorption isotherms at 273 and 298 K, the isosteric heat of adsorption (Qst) was calculated to be ∼19.8 kJ mol−1 (ESI, Fig. S1†),15 which suggested the ease of desorption and adsorbent regeneration. Meanwhile, Zn(PZDC)(ATZ) showed a slightly higher Qst value than Zn4O(BDC-NH2)3 (IRMOF-3, 19 kJ mol−1);28 but less than those of [Zn2(NH2BDC)2(dpNDI)]n (46.5 kJ mol−1)29 and [{Cd2(L-glu)2(bpe)3(H2O)}·2H2O] (40.8 kJ mol−1).30 However, on the basis of the Zeo++ calculation,31 it has been suggested that the aperture window size and maximum pore diameter were 2.23 Å and 4.90 Å, since the corresponding pore window size was smaller than the kinetic diameter of N2 (3.64 Å), and therefore, no significant N2 adsorption isotherms could be obtained. In contrast, the Zn(PZDC)(ATZ) framework pores decorated with highly polar NH2 groups were favourable to the quadrupole moment of CO2, and thus Zn(PZDC)(ATZ) showed selective adsorption properties of CO2 over N2.
 |
| | Fig. 6 CO2 adsorption isotherms over Zn(PZDC)(ATZ) at 273 K and 298 K. | |
CO2 and epoxide cycloaddition reaction
The cocatalyst screening for CO2 cycloaddition to propylene oxide.
CO2 and propylene oxide (PO) were selected as the model substrates for the optimization of the reaction conditions (Table 1). It was obviously observed that both Zn(PZDC)(ATZ) and Bu4NBr showed low activities when used alone (entries 1 and 2). Noticeably, Zn(PZDC)(ATZ) combined with tetrabutyl ammonium bromide (Bu4NBr) exhibited a significant catalytic activity at 94% propylene carbonate (PC) yield and 99% selectivity at 90 °C and under 1.0 MPa for 6 h without any additional organic solvent (entry 3). The enhanced activity was attributed to the existence of anion nucleophilicity from Bu4NBr, which was conducive to the ring-opening step in the synthesis of PC. This will be discussed in the tentative reaction mechanism section. Simultaneously, two kinds of common cocatalyst halides were investigated including tetrabutylammonium halides (Bu4NX with X = Cl, Br, I) and potassium halides (KX with X = Cl, Br, I). To our delight, Zn(PZDC)(ATZ) with a Bu4NBr cocatalyst displayed a much better synergistic effect than Bu4NI and Bu4NCl (entries 3, 5 and 7). This result was ascribed to the higher nucleophilicity of bromide than iodide and to its better leaving ability in comparison with chloride.32 However, only a small number of target products were detected by GC with KX surveyed (entries 4, 6 and 8). During the catalytic reaction, Bu4NBr was decomposed into tributylamine, which could activate CO2, thus both the anion and cation from Bu4NBr played vital roles in the CO2 cycloaddition reaction.33 Therefore, all of the following investigations were carried out with Bu4NBr as the cocatalyst. Even at a low temperature of 50 °C, Zn(PZDC)(ATZ)/Bu4NBr still showed a moderate 74% catalytic activity (entry 9). Furthermore, the concentration of CO2 in industrial waste gas is generally lower than 15%,34 and it is important to study the cycloaddition reaction of CO2 at a lower concentration. The result indicated that the yield of PC only reached 7% at room temperature and 1 atm pressure with mixed gases of 15% CO2 and 85% N2 (entry 11).
Table 1 Screening of the cocatalyst for the CO2 cycloaddition reactiona
| Entry |
Catalyst |
Cocatalyst |
T (°C) |
Time (h) |
Y
(%) |
S
(%) |
|
Reaction conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), cocatalyst (100 mg), 1.0 MPa.
Based on the GC analysis.
Bu4NBr (400 mg), 1.0 MPa.
Bu4NBr (400 mg), CO2 concentration of 15% at 1 atm.
|
| 1 |
Zn(PZDC)(ATZ) |
— |
90 |
6 |
5 |
99 |
| 2 |
— |
Bu4NBr |
90 |
6 |
28 |
98 |
| 3 |
Zn(PZDC)(ATZ) |
Bu4NBr |
90 |
6 |
94 |
99 |
| 4 |
Zn(PZDC)(ATZ) |
KBr |
90 |
6 |
2 |
99 |
| 5 |
Zn(PZDC)(ATZ) |
Bu4NI |
90 |
6 |
86 |
96 |
| 6 |
Zn(PZDC)(ATZ) |
KI |
90 |
6 |
16 |
99 |
| 7 |
Zn(PZDC)(ATZ) |
Bu4NCl |
90 |
6 |
59 |
98 |
| 8 |
Zn(PZDC)(ATZ) |
KCl |
90 |
6 |
4 |
99 |
| 9c |
Zn(PZDC)(ATZ) |
Bu4NBr |
50 |
24 |
74 |
98 |
| 10c |
Zn(PZDC)(ATZ) |
Bu4NBr |
30 |
24 |
35 |
99 |
| 11d |
Zn(PZDC)(ATZ) |
Bu4NBr |
25 |
24 |
7 |
97 |
Effects of reaction parameters.
It was worth pointing out that temperature, cocatalyst loadings, CO2 pressure, and time variables had effects on the catalytic activities. Obviously, the temperature was a vital factor in the cycloaddition reaction. When the temperature rose from 60 °C to 100 °C (Fig. 7a), PC was obtained with 97% yield, and the selectivity was excellent. This result was due to that the high temperature promoted the effective collisions between the reaction substrate and the active centers of the catalyst. In consideration of energy utilization, the following reactions were performed at 90 °C.
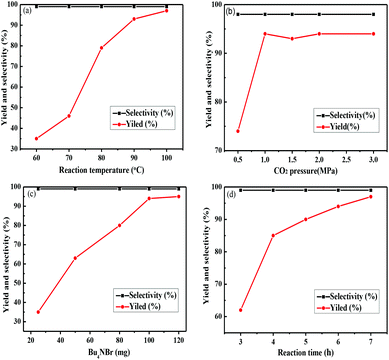 |
| | Fig. 7 Effects of different reaction condition parameters on the PC synthesis. (a) Effect of reaction temperature, conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), Bu4NBr (100 mg), 1.0 MPa, 6.0 h; (b) effect of CO2 pressure, conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), Bu4NBr (100 mg), 90 °C, 6.0 h; (c) effect of Bu4NBr loading, conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), 90 °C, 1.0 MPa, 6.0 h; (d) effect of reaction time, conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), Bu4NBr (100 mg), 90 °C, 1.0 MPa. | |
Remarkably, when the pressure was increased from 0.5 to 1.0 MPa at 90 °C, the yield of PC had a dramatic increase (Fig. 7b); however, the PC yield had little changes in the pressure range of 1–3 MPa, which was attributed to CO2 saturation in the reactive phase with the pressure increasing. What's more, as shown in Fig. 7c, when 100 mg Bu4NBr was used, the PC yield was up to 94%. However, the yield increased slightly with the further addition of the cocatalyst.
Additionally, on the basis of the above optimization conditions, the effect on the reaction time was investigated (Fig. 7d). The yield of PC increased notably in the first 4 h. When the reaction time was further extended and PC increased steadily, apparently, PO was almost completely converted into a PC product with good selectivity.
Recyclability and stability
Encouraged by the superior catalytic performance of Zn(PZDC)(ATZ), its recyclability was also investigated. As shown in Fig. 8a, Zn(PZDC)(ATZ) could be recycled for seven cycles without any significant catalytic deactivation and the selectivity was still above 99%. From the XRD pattern (Fig. 8b), the spent catalyst was identical to the fresh sample, suggesting that Zn(PZDC)(ATZ) possessed remarkable stability. Additionally, a major difference in the peak intensity suggested that the crystallinity of the spent sample was poor through the reaction runs. Meanwhile, it was pointed out that the structure and active sites of the catalyst were not destroyed. Thus, the good catalytic performance of the catalyst could be mainly attributed to the synergistic effects of the active metal sites/amino of the frameworks exposed for interactions with the substrates. In order to further confirm the structural stability, SEM images of the fresh and used Zn(PZDC)(ATZ) catalysts are shown in Fig. S2,† and no obvious morphological changes were observed after seven runs. Simultaneously, the structural stability had been systematically assessed by immersing Zn(PZDC)(ATZ) at different pH values in Fig. 9a. To our surprise, powder X-ray diffraction patterns had no changes, and the unusual chemical stability of Zn(PZDC)(ATZ) might have resulted from the unique structure composed of a high six-coordinated Zn(II) ion subunit and Zn–N coordination interaction, which was stronger than Zn–O cluster interaction,18 thus the high strength of the metal–nitrogen bond prevented its complete destruction under acidic–alkaline conditions. Generally, MOFs based on Zn ions and organic carboxylates are usually sensitive to moisture, only a few known MOFs show such an excellent chemical stability. So the synthesized Zn(PZDC)(ATZ) was immersed in water for 24 h at different temperatures (100 °C, 120 °C, 150 °C), and then dried at room temperature. Fortunately, no significant peak changes could be observed from the PXRD patterns in Fig. 9b, which indicates the excellent hydrothermal stability of Zn(PZDC)(ATZ).
 |
| | Fig. 8 (a) Recycling experiments of the Zn(PZDC)(ATZ) catalyst. Reaction conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), Bu4NBr (100 mg), 90 °C, 1.0 MPa CO2, 6.0 h, (b) PXRD patterns of the Zn(PZDC)(ATZ) catalyst for fresh and reuses for seven runs. | |
 |
| | Fig. 9 PXRD patterns of Zn(PZDC)(ATZ) (a) after being immersed under acid and base conditions for 24 h, (b) treatment in boiling water. | |
Catalytic cycloaddition of CO2 to various epoxides
In order to study the generality of Zn(PZDC)(ATZ)/Bu4NBr catalysts, a series of substituted epoxides were examined, as shown in Table 2, and all could be successfully transformed into the corresponding cyclic carbonates with high yields and excellent selectivity under solvent-free conditions. The yields of cycloaddition products decreased to 94%, 91%, and 58% for 1,2-epoxy propane, 1,2-epoxybutane, and 1,2-epoxyhexane (entries 1–3), respectively. The remarkably lowered catalytic conversion of 1,2-epoxyhexane was mainly ascribed to the steric hindrance of the substituent groups; besides, on account of the large size of 1,2-epoxyhexane (∼8.198 × 3.39 Å2),15 its interaction with the active sites inside the pore was difficult. However, the yield of Cl-substituted 1,2-epoxybutane reached the highest, because the existence of an electron withdrawing group (–Cl) allowed the breakage of the C–O bond, thereby facilitating the formation of the corresponding carbonate. Nevertheless, for the cycloadditions of styrene oxide and cyclohexene with CO2, the catalytic activities were remarkably affected (entries 5 and 6), which resulted from the steric hindrance of the substituent that makes the nucleophilic attack of the Br− reaction rate reduce significantly. Fortunately, a satisfactory yield of cyclohexene carbonate could be obtained by increasing the reaction temperature or reaction time.
Table 2 Coupling reactions of CO2 with various epoxidesa
| Entry |
Epoxide |
Product |
T(°C) |
Time (h) |
Reaction resultsb |
|
Y (%) |
S (%) |
|
Reaction conditions: PO (34.5 mmol), Zn(PZDC)(ATZ) (0.25 mmol), Bu4NBr (100 mg), 1.0 MPa CO2.
Based on the GC analysis.
|
| 1 |

|

|
90 |
6 |
94 |
99 |
| 2 |

|

|
90 |
6 |
91 |
99 |
| 3 |

|

|
90 |
6 |
58 |
89 |
| 4 |

|
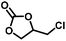
|
90 |
4 |
98 |
99 |
| 5 |

|

|
100 |
6 |
87 |
98 |
| 6 |
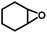
|

|
100 |
12 |
43 |
69 |
Zn(PZDC)(ATZ)/Bu4NBr catalysts with abundant nitrogen units and Lewis acid sites exhibited high performance for coupling CO2 into cyclic carbonates under mild conditions (90 °C, 1.0 MPa). It was worthwhile to compare the catalytic activity of our developed Zn-based MOF catalyst with other reported porous materials in the relevant CO2 cycloaddition reaction (Table 3). Notably, Zn(PZDC)(ATZ)/Bu4NBr systems displayed comparable activities to some reported catalysts, and it could be concluded that the Lewis acid–base sites in Zn(PZDC)(ATZ) are beneficial for the synthesis of cyclic carbonates.
Table 3 Comparisons of the catalysts of CO2 with epoxides in previous reports
| Type |
Catalyst (mmol) |
Cocatalyst (mmol) |
Epoxide (mmol) |
Reaction conditions |
TONd/TOFe (h−1) |
Ref. |
| Yield (%) |
|
SO, styrene oxide.
PO, propylene oxide.
ECH, epichlorohydrin.
TON (turnover number) = product (mmol)/catalyst (mmol)).
TOF (turnover frequency) = moles of product formed per mole of catalyst per hour.
|
| MOF |
Cu(HIP)2(BPY) (0.30) |
— |
POb (18.6) |
120 °C, 1.2 MPa, 6 h |
38/6.4 |
35
|
| 62 |
| CuTrp (0.21) |
Bu4NBr (0.21) |
ECHc (25.5) |
100 °C, 1.2 MPa, 9 h |
118/13.1 |
36
|
| 98 |
| Ni(btzip)(H2btzip) (0.20) |
Bu4NBr (2.0) |
SOa (20.0) |
80 °C, 2.0 MPa, 4 h |
40/10.0 |
37
|
| 40 |
| Sm(BTB)(H2O) (0.10) |
Bu4NBr (0.10) |
SO (20.0) |
80 °C, 1.0 atm, 15 h |
182/12.1 |
38
|
| 93 |
| Co(muco)(bpa)(2H2O) (0.10) |
Bu4NBr (0.10) |
SO (20.0) |
100 °C, 1.0 atm, 12 h |
171/14.2 |
39
|
| 86 |
| Zn(PZDC)(ATZ) (0.25) |
Bu4NBr (0.31) |
ECH (34.5) |
90 °C, 1.0 MPa, 4 h |
135/33.8 |
This work |
| 98 |
| Porous polymers |
PS-HEIMBr (1.60) |
— |
PO (100) |
120 °C, 2.5 MPa, 4 h |
61/15.3 |
40
|
| 98 |
| CBAP-1 (EDA) (0.40) |
Bu4NBr (0.36) |
ECH (20.0) |
80 °C, 1.0 MPa, 8 h |
49/6.1 |
41
|
| 97 |
| IT-POP-1 (0.042) |
— |
PO (41.6) |
130 °C, 1.0 MPa, 10 h |
950/95.0 |
42
|
| 96 |
| Other porous materials |
P-C3N4 (9.0 wt%) |
Bu4NBr (0.69) |
PO (28.6) |
100 °C, 2.0 MPa, 3 h |
38/12.7 |
43
|
| 91 |
| NH2–Zn/SBA-15 (5.0 wt%) |
— |
PO (34.5) |
150 °C, 3.0 MPa, 12 h |
1020/85 |
44
|
| 86 |
| AA-850 (0.27 wt%) |
— |
ECH (20.0) |
150 °C, 4.0 MPa, 16 h |
8848/553 |
45
|
| 54 |
Tentative reaction mechanism
Previous studies have shown that the coupling reaction of CO2 with epoxides can be promoted by the activation of both CO2 and epoxides. Epoxides are usually activated by forming hydrogen bonds through functional groups such as NH2 and coordinated water, or the Lewis acid metal sites such as Zn2+ could also form M–O adducts to activate epoxides, which give rise to the polarization of C–O of epoxide, availing the subsequent of ring-opening process.46,47 Moreover, Lewis bases such as the nitrogenous sites can activate CO2. Likewise, when the developed Zn(PZDC)(ATZ) catalyst was treated with CO2 at room temperature and 1 MPa, a new absorption peak at 1785 cm−1 was found as shown in Fig. 10, which belonged to the asymmetric stretching CO vibration of the carbamate salt,48 and it indicated that the CO2 molecule was activated by basic sites with the formation of a carbamate salt (Scheme 2). Besides, the physical absorption of CO2 appeared as a peak at 2334 cm−1.49 Therefore, a tentative mechanism for the chemical fixation reaction of CO2 is proposed in Scheme 2. First, the cycloaddition reaction was triggered by the activation of the oxygen atoms of epoxides through the Zn(PZDC)(ATZ) catalyst, which made the ring-opening procedure of the epoxide and the subsequent CO2 insertion easy. Meanwhile, the Br− nucleophile from Bu4NBr attacked the less hindered β-carbon atom of the epoxides to assist in the ring-opening process. Finally, the intermediate was formed rapidly by the interaction of activated CO2 with the oxygen anions of the opened epoxy ring, which was further converted into the corresponding cyclic carbonates through the ring-closing step and the catalysts were regenerated for the next run.
 |
| | Fig. 10 FT-IR spectra of fresh Zn(PZDC)(ATZ) and its adsorption of CO2 treated at room temperature and 1 MPa. | |
 |
| | Scheme 2 Possible reaction mechanism for CO2 coupling over Zn(PZDC)(ATZ)/Bu4NBr catalysts. HBD stands for NH2 and water in the Zn(PZDC)(ATZ) catalyst. | |
Conclusions
A novel Lewis acid–base 3D zinc-based MOF was successfully prepared under hydrothermal conditions, and the structure was composed of Zn2+ and functional NH2 groups for the purpose of achieving high activity and adsorption. The abundant nitrogen-containing groups presented excellent adsorption to CO2 molecules at 1 bar (119 mg g−1 at 273 K and 80 mg g−1 at 298 K). By screening the effect of a halide salt cocatalyst, the catalytic performance of Zn(PZDC)(ATZ) was optimized, and organic halides gave a higher activity than the corresponding inorganic halides. Remarkably, Zn(PZDC)(ATZ) showed an excellent activity cooperating with Bu4NBr for fixing CO2 to cyclic carbonates under solvent-free and mild conditions. In addition, the scope versatility, reusability and chemical stability were also exhibited. Zn(PZDC)(ATZ)/Bu4NBr as effective catalysts showed great significance in the practical conversion of CO2 to valuable chemicals under mild conditions. Also, the attractive advantages such as the better skeleton stability and abundant N atoms of the Zn(PZDC)(ATZ) catalyst might be further extended to other application fields.
Experimental section
Preparation of the Zn-MOF catalyst
The synthesis of [Zn2(PZDC)(1/2ATZ)2(H2O)2·2.5H2O] (abbreviated as Zn(PZDC)(ATZ)): a mixture of (0.2 mmol) Zn(NO3)2·6H2O, (0.1 mmol) 3,5-pyrazoledicarboxylic acid, and (0.2 mmol) 5-aminoterazole was dissolved in mixed solvents of H2O (7.5 mL) and DMF (2.5 mL), and then heated at 120 °C for 33 h, and after the completion of the reaction, the mixture was cooled down to room temperature. The crystals were obtained and dried at 120 °C overnight before use.
Materials and characterization
All reagents and solvents were purchased commercially and used without further purification. Single crystal X-ray diffraction analysis was operated on an Agilent Technology Super Nova Eos Dual system with Mo Kα radiation (λ = 1.54184 Å) at 293 K and processed using CrysAlisPro.50 The structures were solved and refined by means of SHELXS-97 and SHELXL-97.51 The patterns of powder X-ray diffraction were conducted on a PANalytical X'Pert PRO with Cu Kα radiation (40 mA, 40 kV) for phase identification. Fourier transform infrared (FT-IR) analysis was conducted on a PerkinElmer Spectrum 100 with KBr pellets in the 4000–450 cm−1 region. Thermogravimetric analysis (TGA) was performed using a NETZSCH STA 449F3 thermal analyzer from 25 to 700 °C at a rate of 10 °C min−1 under a N2 atmosphere. The sample was pre-treated at 120 °C for 12 h, and then the CO2 adsorption capacities at 273 and 298 K were measured using a Micromeritics ASAP 2020 system. GC analysis was performed on an Agilent GC-7890A equipped with a capillary column (Agilent 19091J-413) using a flame ionization detector.
Catalytic conversion of CO2 into cyclic carbonates
All of the catalytic reactions were conducted in a 50 mL stainless-steel autoclave with magnetic stirring. In a typical run, the autoclave was first purged with CO2 to replace the air in the autoclave, then 100 mg Zn(PZDC)(ATZ) catalyst (0.25 mmol), 100 mg cocatalyst Bu4NBr, and propylene oxide (34.5 mmol) were added, respectively. The autoclave was heated to 90 °C followed by introducing CO2 to 1 MPa and stirring for a certain period of time. Upon completion, the mixtures were cooled down to room temperature slowly. The products were quantitatively analyzed by GC. Furthermore, the catalysts could be recycled by centrifugation, washed with diethyl ether, dried under vacuum, and then reused for the next run.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We sincerely acknowledge the financial support from the National Natural Science Foundation of China (21673060) and the State Key Lab of Urban Water Resource and Environment of Harbin Institute of Technology (HIT2017DX10).
Notes and references
- Z. J. Zhang, Z. Z. Yao, S. C. Xiang and B. L. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 RSC
 .
.
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef CAS PubMed .
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC .
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC .
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS .
- S. Yue, P. P. Wang, X. J. Hao and S. L. Zang, J. CO2 Util., 2017, 21, 238–246 CrossRef CAS .
- C. K. Karan and M. Bhattacharjee, Inorg. Chem., 2018, 57, 4649–4656 CrossRef CAS PubMed .
- K. R. Roshan, R. A. Palissery, A. C. Kathalikkattil, R. Babu, G. Mathai, H. Lee and D. A. Park, Catal. Sci. Technol., 2016, 6, 3997–4004 RSC .
- C. G. Li, L. Xu, P. Wu, H. H. Wu and M. Y. He, Chem. Commun., 2014, 50, 15764–15767 RSC .
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960–1967 CrossRef .
- T. Lescouet, C. Chizallet and D. Farrusseng, ChemCatChem, 2012, 4, 1725–1728 CrossRef CAS .
- Y. F. Lin, K. W. Huang, B. T. Ko and K. Y. Lin, J. CO2 Util., 2017, 178–182 CrossRef .
- X. Q. Huang, Y. F. Chen, Z. G. Lin, X. Q. Ren, Y. N. Song, Z. Z. Xu, X. M. Dong, X. G. Li, C. W. Hu and B. Wang, Chem. Commun., 2014, 50, 2624–2627 RSC .
- H. F. Zhou, B. Liu, L. Hou, W. Y. Zhang and Y. Y. Wang, Chem. Commun., 2018, 54, 456–459 RSC .
- N. Sharma, S. S. Dhankhar, S. Kumar, T. J. D. Kumar and C. M. Nagaraja, Chem. – Eur. J., 2018, 24, 16662–16669 CrossRef CAS PubMed .
- P. T. K. Nguyen, H. T. D. Nguyen, H. N. Nguyen, C. A. Trickett, Q. T. Ton, E. Gutiérrez-Puebla, M. Á. Monge, K. E. Cordova and F. Gándara, ACS Appl. Mater. Interfaces, 2018, 10, 733–744 CrossRef CAS .
- L. F. Liang, C. P. Liu, F. L. Jiang, Q. H. Chen, L. J. Zhang, H. Xue, H. L. Jiang, J. J. Qian, D. Q. Yuan and M. C. Hong, Nat. Commun., 2017, 8, 1233–1243 CrossRef PubMed .
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed .
- H. M. He, J. A. Perman, G. S. Zhu and S. Q. Ma, Small, 2016, 12, 6309–6324 CrossRef CAS PubMed .
- R. Babu, A. C. Kathalikkattil, R. Roshan, J. Tharun, D. Kim and D. Park, Green Chem., 2016, 18, 232–242 RSC .
- M. S. Liu, L. Liang, X. Li, X. X. Gao and J. M. Sun, Green Chem., 2016, 18, 2851–2863 RSC .
- S. Nagata, H. Sato, K. Sugikawa, K. Kokado and K. Sada, CrystEngComm, 2012, 14, 4137–4141 RSC .
- D. Müller, R. Hochleitner and K. T. Fehr, J. Raman Spectrosc., 2015, 47, 602–606 CrossRef .
- V. Krishnakumar, V. Balachandran and T. Chithambarathanu, Spectrochim. Acta, Part A, 2005, 62, 918–925 CrossRef CAS .
- Z. Jiang, H. Wang, Y. Hu, J. Lu and H. Jiang, ChemSusChem, 2015, 8, 878–885 CrossRef CAS .
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998–17999 CrossRef CAS PubMed .
- A. C. Kathalikkattil, R. Roshan, J. Tharun, R. Babu, G. S. Jeong, D. W. Kim, S. J. Cho and D. W. Park, Chem. Commun., 2016, 52, 280–283 RSC .
- R. Sabouni, H. Kazemian and S. Rohani, Environ. Sci. Pollut. Res., 2014, 21, 5427–5449 CrossRef CAS .
- S. S. Dhankhar, N. Sharma, S. Kumar, T. J. D. Kumar and C. M. Nagaraja, Chem. – Eur. J., 2017, 23, 16204–16212 CrossRef .
- B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Inorg. Chem. Front., 2017, 4, 348–359 RSC .
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS .
- M. Taherimehr, B. Van de Voorde, L. H. Wee, J. A. Martens, D. E. De Vos and P. P. Pescarmona, ChemSusChem, 2017, 10, 1283–1291 CrossRef CAS PubMed .
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed .
- Z. F. Dai, Q. Sun, X. L. Liu, C. Q. Bian, Q. M. Wu, S. X. Pan, L. Wang, X. J. Meng, F. Deng and F. S. Xiao, J. Catal., 2016, 338, 202–209 CrossRef CAS .
- A. C. Kathalikkattil, D. W. Kim, J. Tharun, H. G. Soek, R. Roshan and D. W. Park, Green Chem., 2014, 16, 1607–1616 RSC .
- G. S. Jeong, A. C. Kathalikkattil, R. Babu, Y. G. Chung and D. W. Park, Chin. J. Catal., 2018, 39, 63–70 CrossRef CAS .
- Y. Z. Li, H. H. Wang, H. Y. Yang, L. Hou, Y. Y. Wang and Z. H. Zhu, Chem. – Eur. J., 2018, 24, 865–871 CrossRef CAS PubMed .
- B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Cryst. Growth Des., 2018, 18, 2432–2440 CrossRef CAS .
- B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Cryst. Growth Des., 2017, 17, 3295–3305 CrossRef CAS .
- J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS .
- S. Ravi, P. Puthiaraj and W. S. Ahn, J. CO2 Util., 2017, 21, 450–458 CrossRef CAS .
- H. Zhong, Y. Q. Su, X. W. Chen, X. J. Li and R. H. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed .
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS .
- M. S. Liu, B. Liu, L. Liang, F. X. Wang, L. Shi and J. M. Sun, J. Mol. Catal. A: Chem., 2016, 418, 78–85 CrossRef .
- X. Ma, B. Zou, M. Cao, S. L. Chen and C. Hu, J. Mater. Chem. A, 2014, 2, 18360–18366 RSC .
- Y. H. Han, Z. Y. Zhou, C. B. Tian and S. W. Du, Green Chem., 2016, 18, 4086–4091 RSC .
- H. Zhong, Y. Q. Su, X. W. Chen, X. J. Li and R. H. Wang, ChemSusChem, 2017, 10, 4855–4863 CrossRef CAS PubMed .
- F. Adam and M. S. Batagarawa, Appl. Catal., A, 2013, 454, 164–171 CrossRef CAS .
- A. Saad, S. Atwa, N. Yasuda and M. Fujii, Radiat. Meas., 2001, 34, 51–54 CrossRef CAS .
- A. Rothkirch, G. D. Gatta, M. Meyer, S. Merkel, M. Merlini and H. P. Liermann, J. Synchrotron Radiat., 2013, 20, 711–720 CrossRef CAS PubMed .
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1872616. For the ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c8qi01150h |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Zimin
Jiang
and
Jianmin
Sun
,
Zimin
Jiang
and
Jianmin
Sun