
Synthesis of sheet-coil-helix and coil-sheet-helix triblock copolymers by combining ROMP with palladium-mediated isocyanide polymerization†

Abstract
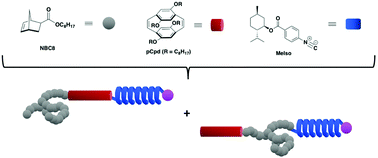
We report a synthetic route to construct architecturally well-defined triblock copolymers, in particular copolymers featuring secondary structures that form sheet-coil-helix and coil-sheet-helix motifs, through a macroinitiation strategy that combines sequential ring-opening metathesis polymerization (ROMP) with palladium-mediated isocyanide polymerization. Throughout the triblock copolymer fabrication, the individual blocks retained their secondary structures as evidenced by circular dicroism (CD) and fluorescence spectroscopies. Our strategy introduces a facile way to create complex ABC triblock copolymers that contain architecturally-diverse backbones, whilst retaining the ability to control sequence: sheet-coil-helix or coil-sheet-helix.
 Please wait while we load your content...
Please wait while we load your content...

