
Thermoplastic polyacetals: chemistry from the past for a sustainable future?
Andrea
Hufendiek
 *,
Sophie
Lingier
and
Filip E.
Du Prez
*
*,
Sophie
Lingier
and
Filip E.
Du Prez
*
Polymer Chemistry Research Group, Centre of Macromolecular Chemistry (CMaC), Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4-bis, B-9000 Ghent, Belgium. E-mail: Filip.DuPrez@UGent.be; andrea.hufendiek@nottingham.ac.uk
First published on 9th October 2018
Abstract
Synthetic thermoplastic polyacetals have a long history dating back to 1912. While polymers with non-cyclic acetal repeat units are typically well soluble and degrade easily at biologically relevant pH values, polycycloacetals have a rigid polymer backbone, resulting in favorable thermal and mechanical properties but are often insoluble and thus challenging to process. In recent years, the degradation behavior and the availability of many building blocks from renewable resources have sparked renewed interest in poly(cyclo)acetals. This review provides a critical overview over the synthetic routes to polyacetals, highlighting where possible the material properties. Direct polyacetalization and polytransacetalization techniques are discussed first, followed by the polymerization of acetal containing monomers and recent ring opening polymerization approaches. Thermoplastic polyacetals show promise in delivery applications but also as bulk materials, where a combination of excellent material properties, a potential for renewable sourcing and degradation as an end-of-life option are of ever increasing importance.
 Andrea Hufendiek | Andrea Hufendiek received her PhD in chemistry from the Karlsruhe Institute of Technology (KIT) in 2015 working on the synthesis of cellulose copolymers in homogeneous media. She joined the Polymer Research Group at Ghent University in 2016 focusing on polycondensation techniques for renewable polymers. Since 2018 she is a Senior Research Officer in the Polymers and Supercritical Fluids Group at the University of Nottingham. Her research interests include the synthesis of renewable monomers and polymers, and sustainable reaction media. |
 Sophie Lingier | Sophie Lingier graduated with a bachelor's and master's degree from Ghent University, Belgium. She completed her master's thesis in the Polymer Chemistry Research Group in 2013 working on amphiphilic graft copolymers based on renewable resources. In December 2017, she obtained her Ph.D. degree at Ghent University, in which she focused on polycycloacetals from new renewable resources. Since January 2018, Sophie is working as a researcher for Beaulieu International Group. |
 Filip E. Du Prez | Filip Du Prez is since 1999 heading the Polymer Chemistry Research Group (http://www.PCR.UGent.be) at Ghent University in which about 25 researchers are dealing with the development of new polymer structures, biosourced polymers, exploration of powerful functionalisation methods and the design of dynamic/renewable polymer materials for specific applications. In 2008, he had a Visiting Professor position at the CAMD Research Center (UNSW, Sydney). He is author of more than 280 peer-reviewed publications, 12 patents, 10 book chapters and (co-)chairman of 10 (inter)national conferences on polymer chemistry related topics. In 2018, he became associate editor of Polymer Chemistry. |
Introduction: polyacetals
The first report on a polyacetal in 1912 dates back prior to the time of the macromolecular hypothesis in the 1920s. John Read reports on the condensation of glyoxal and pentaerythritol and describes the reaction product as exhibiting “all the characteristics of a substance of very high molecular weight”.1 Despite the existence of industrially produced polyoxymethylene (POM, Chart 1, top left) – a linear polyacetal – and polyvinyl butyral (Chart 1, top middle) – a cyclic polyvinyl acetal – synthetic man-made acetal containing polymers had a lower impact on our daily lives than polycondensation products, such as polyesters or polyamides.2–5 POM is used as an engineering thermoplastic in precision parts, which require high stiffness, low friction and excellent dimensional stability. As a tough, solvent and fuel resistant plastic, it is used for example in gears, zips, lighters, aerosol valves, fasteners and furniture components, whereas polyvinyl butyral is mainly produced for safety glass applications. However, when taking naturally occurring polymers such as cellulose (Chart 1, top right) and starch into account as well, the acetal bond is one of the most common bonds in (bio)polymers. In addition, the instability of the acetal bond against hydrolysis in media of low pH resulted for example in its use as a protecting group for polymer chemistry and in the development of many acetal based crosslinkers for degradable materials.6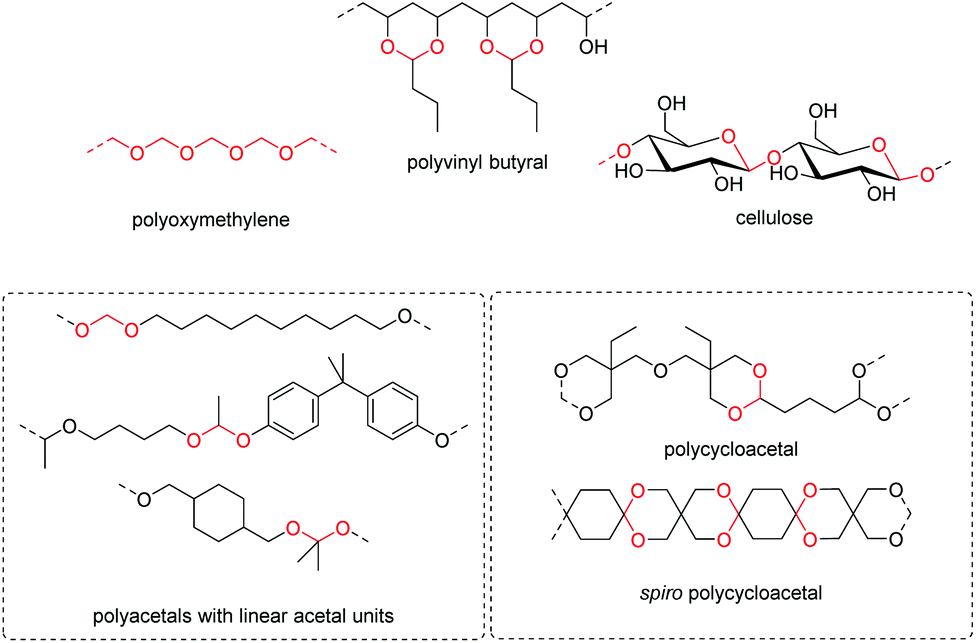 | ||
| Chart 1 Examples of polymers that contain acetals in the repeat unit. The acetal units are highlighted in red. Polyoxymethylene (top left) and polyvinyl acetals (mainly polyvinyl butyral, top middle) are polymers of commercial relevance. Cellulose (top right) is an example of a polysaccharide. This review discusses polyacetals with linear acetals units from formaldehyde, other aldehydes and ketones (bottom left), such as a polyacetal formally derived from formaldehyde and 1,10-decanediol (top; vide infra: acetal metathesis polymerization, AMP), a mixed polyacetal formally derived from acetaldehyde, butanediol and bisphenol A (middle; vide infra: polyaddition of diols to divinylethers), and a polyacetal formally derived from acetone and 1,4-cyclohexanedimethanol (bottom; vide infra: polytransacetalization). Polycycloacetals (bottom right) are also discussed, such as a polycycloacetal derived from glutaraldehyde and di-trimethylolpropane (di-TMP) (top) and a spiro polycycloacetal from pentaerythritol and 1,4-cyclohexanedione (bottom; vide infra: polyacetalization). | ||
Since the hydrolysis of especially non-cyclic acetal units can already take place at biologically relevant pH values ≤7.4, depending on the substituents of the acetal group, degradable networks based on acetal crosslinkers have been investigated mostly in the context of biomaterials and delivery systems and have been reviewed numerous times.7–16 Degradable acetal crosslinkers were also used in lithography applications.17 Very recently, the reversibility of acetal formation was used for the first time for reshapable crosslinked materials.18 This review however, focuses on thermoplastic, thus non-crosslinked polyacetals. In the field of (bio)medical applications, these polymers are mostly investigated as drug delivery vehicles. For example, semi-synthetic polyacetals derived from polysaccharides have been commercialized under the trade name Fleximer® for drug conjugates.9,19,20
In recent years, there has also been renewed interest in polyacetals in the context of renewable and degradable polymers because they have a high oxygen to carbon ratio, and many of the carbonyl and polyol monomers for polyacetals are already available from nature or have the potential for renewable sourcing as a result of the abundance of oxygen rich biomass. In this context, polymers with cyclic acetal units such as polycycloacetals (Chart 1, bottom left) or polyvinyl butyrate, typically show better thermal and mechanical properties, and are less sensitive towards hydrolytic degradation (vide infra) compared to polyacetals with linear acetal units (Chart 1, bottom right). Thus, their properties may constitute a promising compromise between good material properties and stability during their use and the potential for (bio)degradation as the end-of-life option. The field of renewable polyacetals is not only progressing by using established polymerization techniques for new carbonyl and/or polyol (derived) monomers, but new polymerization procedures, such as acetal metathesis polymerization (AMP), or new monomer classes, such as 1,3-dioxolan-4-ones (DOXs) for ring opening polymerizations (ROP), are being developed as well.
This review will mainly focus on the synthetic approaches toward polyacetals, highlighting in particular the different monomers for polyacetals and discussing the resulting material properties where possible. After some general considerations, direct polyacetalization and polytransacetalization are critically discussed, followed by the polymerization of acetal containing monomers and polyacetals via ROP. Finally, we also highlight the challenges in the synthesis of polyacetals, such as the synthesis of high molecular weight polymers, solubility, and the compromise between stability and tailored degradability, which all relate to the still developing understanding of the structure–property relationships for this emerging class of polymers.
Synthesis of polyacetals
This review focuses on thermoplastic polyacetals that contain acetals in the repeat unit in the backbone, excluding for the most part polyacetals based on formaldehyde (such as POM) except for a few selected, more recent examples. With the term acetal we refer to all diethers of geminal diols without distinguishing between acetals derived from aldehydes and acetals derived from ketones, which are often also referred to as ketals.21 We will also briefly discuss related structures such as hemiacetal esters (vide infra). In the simplest case, acetals are obtained from the reaction of a carbonyl, i.e. an aldehyde or a ketone, with two hydroxyl groups. This reaction, but also the reverse reaction (degradation) proceeds via acid catalyzed equilibria and a less stable hemiacetal intermediate (Scheme 1).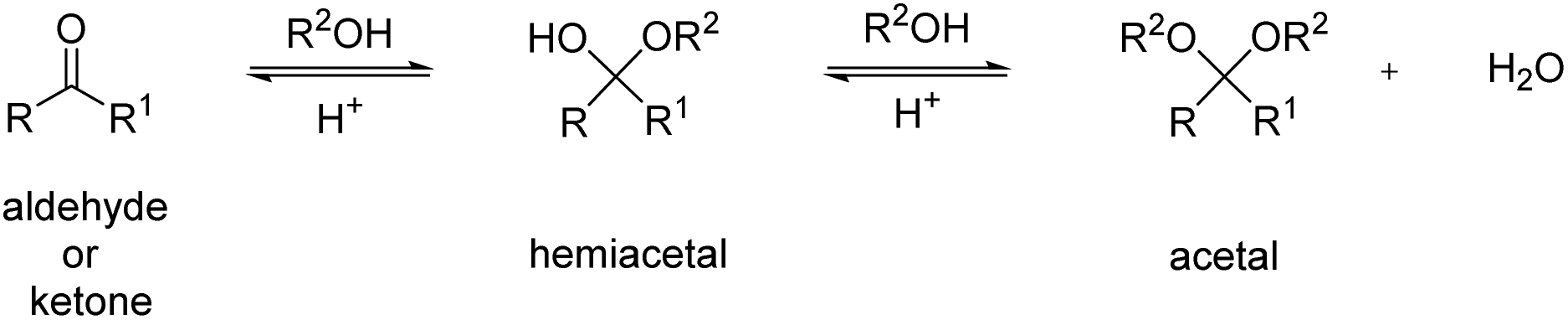 | ||
| Scheme 1 Synthesis of an acetal by reaction of an aldehyde or ketone with an alcohol. | ||
However, acetals are stable against bases. When combining a carbonyl with a diol, a cyclic acetal is usually formed. Cyclic acetals are typically more stable against hydrolysis than their linear counterparts, since their hydrolytic degradation is less entropically favored. Five and six membered cyclic acetals are particularly stable. While aldehydes tend to favor the formation of the six membered acetals (e.g. in a reaction with glycerol), ketones tend to favor the formation of five membered acetals, but in fact all combinations are possible.22,23 Typically, acetals of ketones are more susceptible to hydrolysis than acetals of aldehydes, which are in turn more susceptible to hydrolysis than the acetals of formaldehyde. This is attributed to the relative stability of the carboxonium type intermediates occurring during hydrolysis, which decreases in the order ketones > aldehydes > formaldehyde.24
There are several methods for synthesizing polymers containing acetal groups. We have structured the synthetic approaches into four groups. (i) Polymerization by formation of acetal bonds, which includes the reaction of polyols with carbonyl compounds, acetal exchange reactions (=transacetalization), and the reaction of diols with vinyl ethers. The second group (ii) comprises polymerizations of acetal containing monomers by other polymerization techniques, which do not involve the acetal moiety. The third group (iii) is only discussed briefly and comprises recent approaches on (re)polymerization of paraformaldehyde, and finally the fourth group (iv) comprises ROP, where the focus will also be on recent approaches.
Polyacetalization
By reacting equimolar amounts of an aldehyde or ketone and a diol in the presence of an acid catalyst (most commonly a Brønsted acid) while removing the condensate, water, polyacetals can in theory be formed in a polycondensation reaction (Scheme 2). A commonly employed method to remove water from the reaction mixture is azeotropic distillation, performed with a Dean–Stark apparatus. However, a serious limitation to this technique is the tendency to form cyclic acetals. Advantage can be taken of this tendency to form cyclic acetals by reacting tetraols with dialdehydes or diketones to form polycycloacetals (Scheme 3).1,26–32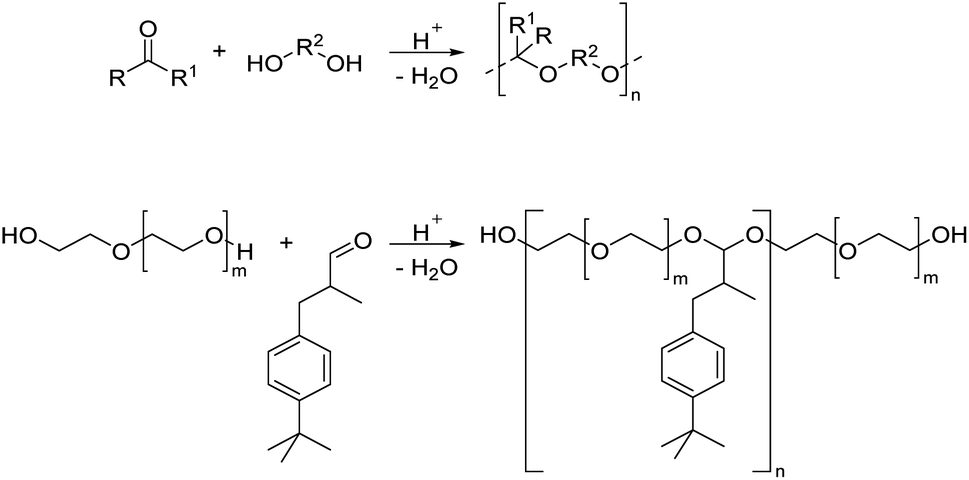 | ||
| Scheme 2 Synthesis of polyacetals from an aldehyde or ketone and a diol (top) and an example from the literature25 (bottom) showing the synthesis of poly(ether acetals) from PEG and lilial (3-(4-tert-butylphenyl)-2-methyl propanal). | ||
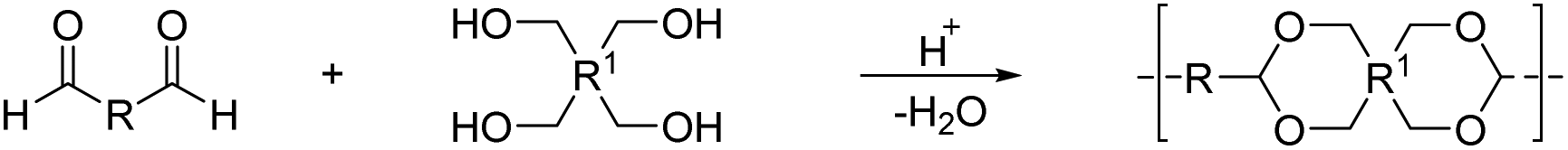 | ||
| Scheme 3 Synthesis of a polycycloacetal from a tetraol and a dialdehyde. | ||
In fact, the vast majority of reports on direct polyacetalizations use the formation of cyclic acetal units by a combination of tetraol and dicarbonyl monomers (vide infra). Linear polyacetals by direct polyacetalization of a linear diol and an aldehyde were reported for poly(ethylene glycol) (PEG) diols and lilial (Scheme 2), where the chain length of the PEG diols prevents the formation of cyclic acetals.25 Yet, this approach resulted in only about 13 or fewer acetal units in the polymer backbone, depending on the molecular weight of the PEG diol. All polymers prepared in this way were susceptible to hydrolytic degradation at pH <5–7.
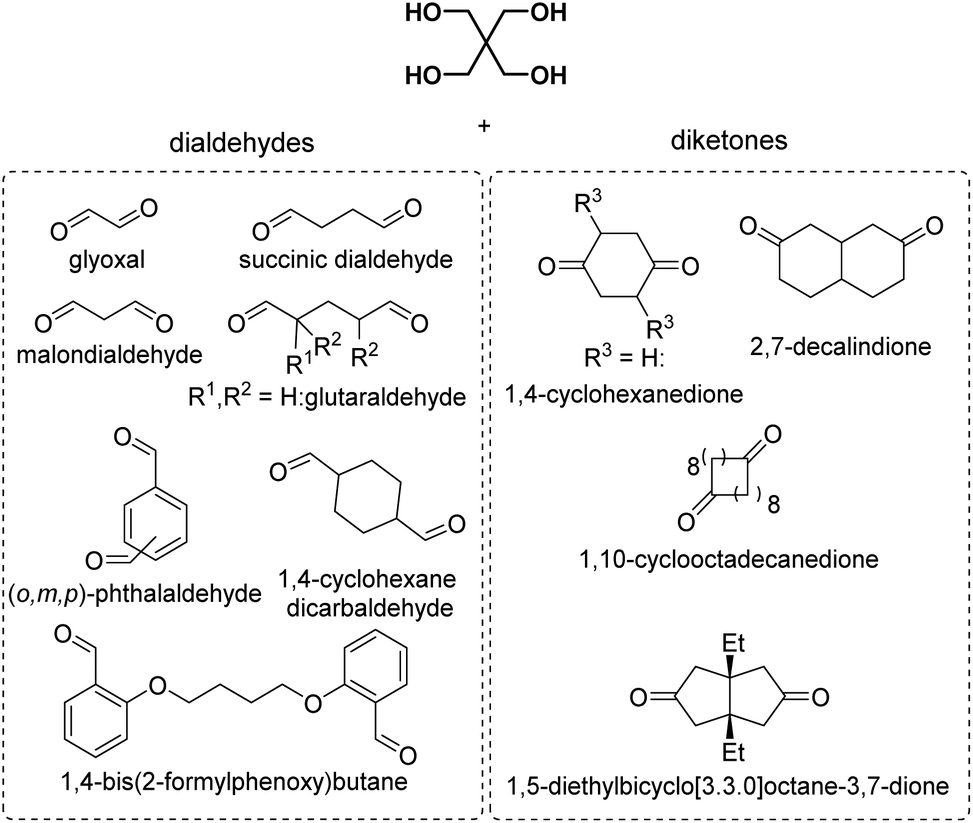 | ||
| Chart 2 Overview of the reported dialdehydes and diketones for polyacetalization with pentaerythritol (R1 = CH3OCH2, R2 = CH3: 2-methoxymethyl-2,4-dimethylpentanedial; R1 = CH3CH2OCH2, R2 = CH3: 2-ethoxymethyl-2,4-dimethylpentanedial; R3 = (CH2)11CH3: 2,5-didodecylcyclohexane-1,4-dione). 2,7-Decalindione was polymerized with 1,2,4,5-tetramethylolcyclohexane (Chart 4). | ||
Subsequently, pentaerythritol was also reacted with glutaraldehyde, 3-methylglutaraldehyde, and terephthalaldehyde, yielding low to moderate molecular weight polymers (Mn = 0.3–8.4 kg mol−1).27,28 High melting points (Tm = 148–221 °C) are reported for these polymers, although most polycycloacetals seem to exhibit competing melting and degradation. Replacing the pentaerythritol in part by dipentaerythritol resulted in products with improved adhesion and film formation, and crosslinking was always observed for dipentaerythritol contents exceeding 25%.27,28 Pentaerythritol was also reacted with 1,4-bis(2-formylphenoxy) butane (Chart 2) resulting in a polycycloacetal with a low Mn of 3.1 kg mol−1 (Đ = 1.30). The authors also investigated an alternative approach to the same polymer: the corresponding spiroacetal diol was synthesized first and reacted with 1,4 dibromobutane in a polyetherification. This second approach resulted in polycycloacetals of slightly higher molecular weights (Mn = 5.0 kg mol−1, Đ = 1.65), but elimination and substitution reactions were a limitation.38 The polymer was amorphous (Tg = 66 °C) and its acetal units were oxidized to yield a poly(ether ester) using N-bromosuccinimide.
More recently, Miller and co-workers investigated polycycloacetals from pentaerythritol and dicarbonyl monomers obtained from 4-hydroxybenzaldehyde, vanillin, and syringaldehyde, which can be derived from lignin, as well as ethylvanillin (Chart 3).39 The authors pointed out that pentaerythritol has the potential for sourcing from biomass, since it is obtained from acetaldehyde and formaldehyde by an aldol reaction and subsequent Cannizzaro reaction.40,41 Pentaerythritol from renewable resources is available from Perstorp under the tradename Voxtar™ M100.42 A physical drying agent (MgSO4), which was placed between the reaction vessel containing halogenated solvents and the reflux condenser, was used to remove the water from the reaction mixture. Thermally stable (temperature at which a 5% weight loss occurs (Td,5%) >300 °C) polycycloacetals of high molecular weight (Mn = 10.6–22.2 kg mol−1) were obtained, except for the 4-hydroxybenzaldehyde derived polymers, which were poorly soluble. The authors also used di(trimethylolpropane) (di-TMP, Chart 3) as a tetraol and compared the influence of the tetraol component on the thermal properties of the polycycloacetals (Table 1). Di-TMP is currently obtained as a by-product in trimethylolpropane synthesis from butanal and formaldehyde, which are starting materials that could be sourced from renewable feedstock in the future.39 When using di-TMP as the tetraol monomer, the polycycloacetals were more soluble as compared to pentaerythritol based polycycloacetals. However, the combination of di-TMP with 4-hydroxybenzaldehyde resulted in a polymer that was very sensitive to acid induced degradation, which hampered its characterization. The pentaerythritol derived polymers generally showed higher glass transition temperatures (Tg = 108–129 °C) than the di-TMP derived polymers (Tg = 68–98 °C, Table 1). All polycycloacetals except for those based on 4-hydroxybenzaldehyde were amorphous. The higher Tgs of the pentaerythritol based polycycloacetals are attributed to the more rigid backbone that results from the presence of spirocyclic units. 4-Hydroxybenzaldehyde based polycycloacetals did not exhibit Tgs but a high Tm of 259 °C when di-TMP was used as the tetraol, whereas neither a Tg nor Tm could be detected when using pentaerythritol. For the different aromatic substitution patterns of the dicarbonyl monomers, a general increase in Tg was observed in the series ethylvanillin < vanillin < syringaldehyde, which was explained by free volume effects and conformational restraints. Substantial hydrolytic degradation was observed for the vanillin based polycycloacetals in concentrated and dilute (2 M) hydrochloric acid within 24 or 48 h respectively, where polymers synthesized with di-TMP showed faster degradation than polymers synthesized with pentaerythritol.
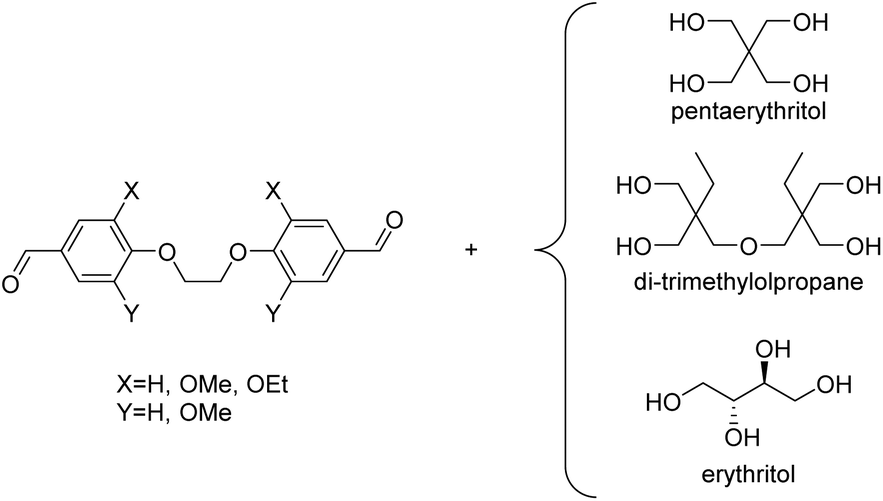 | ||
| Chart 3 Dialdehydes from vanillin (X = OMe, Y = H; VV), ethylvanillin (X = OEt, Y = H; EE), syringaldehyde (X = Y = OMe; SS) and 4-hydroxybenzaldehyde (X = Y = H; BB) and their combination with tetraols for the synthesis of polycycloacetals. | ||
| Tetraol | Dicarbonyl | T g [°C] | T m [°C] | ΔHm [J g−1] | E [GPa] | Ref. |
|---|---|---|---|---|---|---|
| Pentaerythritol | 1,4-Bis(2-formylphenoxy) butane | 66 | — | — | / | 38 |
| Pentaerythritol | BB | — | — | — | / | 39 |
| Pentaerythritol | VV | 129 | — | — | / | 39 |
| Pentaerythritol | EE | 108 | — | — | / | 39 |
| Pentaerythritol | SS | 152 | — | — | / | 39 |
| Pentaerythritol | 1,5Diethylbicyclo[3.3.0]octane-3,7-dione | 180 | — | — | / | 49 |
| Pentaerythritol | 2,5-Didodecylcyclohexane-1,4-dione | 45 | — | — | / | 49 |
| Pentaerythritol | 2,5-Hexanedione | / | 225–238 | / | / | 27 |
| di-TMP | Glutaraldehyde | 30 | — | — | 0.5 | 44 |
| di-TMP | o-Phthalaldehyde | 105 | — | — | / | 44 |
| di-TMP | m-Phthalaldehyde | 90 | — | — | / | 44 |
| di-TMP | p-Phthalaldehyde | 115 | — | — | / | 44 |
| di-TMP | BB | — | 259 | 0.19 | / | 39 |
| di-TMP | VV | 80 | — | — | / | 39 |
| di-TMP | EE | 68 | — | — | / | 39 |
| di-TMP | SS | 98 | — | — | / | 39 |
| di-TMP | 1,4-Cyclohexanedione | 97 | 139 | 22 | / | 69 |
| Diglycerol | 1,4-Cyclohexanedione | 48 | 210 | 44 | / | 69 |
| Diglycerol | 4,4′-Bicyclohexanone | 65 | 241 | 33 | / | 69 |
| Erythritol | VV | 74 | — | — | / | 43 |
| Erythritol | SS | 135 | — | — | / | 43 |
| 1,4-Di-O-methyl-myo-inositol | 1,4-Cyclohexanedione | 130 | 250 | — | / | 51 |
| 1,4-Di-O-allyl-myo-inositol | 1,4-Cyclohexanedione | 90 | — | — | / | 51 |
| 1,4-Di-O-benzyl-myo-inositol | 1,4-Cyclohexanedione | 123 | — | — | / | 51 |
| Pentaerythritol/erythritol: 91/9 | VV | 110 | — | — | / | 43 |
| Pentaerythritol/erythritol: 57/43 | VV | 97 | — | — | / | 43 |
| Pentaerythritol/erythritol: 29/71 | VV | 58 | 159 | — | / | 43 |
| Pentaerythritol/erythritol: 84/16 | SS | 85 | — | — | / | 43 |
| Pentaerythritol/erythritol: 66/34 | SS | 121 | — | — | / | 43 |
| Pentaerythritol/erythritol: 24/76 | SS | 98 | — | — | / | 43 |
| di-TMP/diglycerol: 24/76 | 1,4-Cyclohexanedione | 60 | — | — | 1.4 | 69 |
| di-TMP/diglycerol: 48/52 | 1,4-Cyclohexanedione | 71 | — | — | 1.1 | 69 |
| di-TMP/erythritol: 94/6 | VV | 73 | — | — | / | 43 |
| di-TMP/erythritol: 67/33 | VV | 57 | — | — | / | 43 |
| di-TMP/erythritol: 27/73 | VV | 75 | — | — | / | 43 |
| di-TMP/erythritol: 94/6 | SS | 91 | — | — | / | 43 |
| di-TMP/erythritol: 76/24 | SS | 101 | — | — | / | 43 |
| di-TMP/erythritol: 14/86 | SS | 71 | — | — | / | 43 |
The same authors investigated the vanillin and syringaldehyde based dicarbonyl monomers in combination with either pentaerythritol and biogenic erythritol (Chart 3), or di-TMP and erythritol in a later report.43 Erythritol was less reactive than pentaerythritol and di-TMP, which is ascribed to the presence of two secondary hydroxyl groups in the molecule. Generally, the erythritol ratio in the polycycloacetal was lower than the erythritol feed ratio and polymerizations with a high erythritol feed ratio resulted in lower yields and molecular weights than the polymerizations with a low erythritol feed ratio. The effect of erythritol incorporation on the Tgs of the polymers varied (Table 1), but a tendency towards higher Tgs was observed for both very high (>90%) and very low (<10%) erythritol feed ratios. In most cases however, a high erythritol feed ratio resulted in a lower thermal stability as evidenced from a decrease in the degradation temperature. A typical decrease in Td,5% when increasing the erythritol feed ratio from 0–100% was 30–40 °C. A long-term degradation study on a polycycloacetal synthesized with an erythritol/di-TMP feed ratio of 50/50 and the syringaldehyde based dicarbonyl revealed that the polycycloacetal was stable in sea water, deionized water, tap water, and a buffer solution of pH = 5 for one year, whereas a decrease of 40% in molecular weight was observed at pH = 1.
We recently investigated polycycloacetals from glutaraldehyde and o, m, and p-phthalaldehyde in combination with di-TMP, employing either MgSO4 as a physical drying agent or a Dean–Stark set-up for the removal of water from the polycondensation.44 For glutaraldehyde as the dicarbonyl monomer, high molecular weights (Mn = 34–38 kg mol−1) and broad molecular weight distributions (Đ = 2.6–4.0) were obtained for both polycondensation set-ups, and the polymer was thermally stable with Td,5% >350 °C. This polycycloacetal has a low Tg (Tg,DSC = 30 °C, Tg,DMA = 35 °C); but nevertheless mechanical properties could be determined by tensile testing. The polymer showed ductile behavior, a Young's modulus E of ca. 0.5 GPa and a tensile strength of ca. 15–16 MPa. Terephthalaldehyde as a monomer resulted in an insoluble polymer that could not be processed. For m-phthalaldehyde and o-phthalaldehyde, the combination of DCM with MgSO4 gave slightly higher molecular weights (Mn = 18–20 kg mol−1, Đ = 1.4) than the Dean–Stark set-up (Mn = 15–16 kg mol−1, Đ = 1.7–1.9), yet it must be noted that the Dean–Stark set-up generally allowed for shorter reaction times (6–8 h vs. 65–90 h). The polycycloacetals based on m- and o-phthalaldehyde were thermally stable but could not be characterized by tensile testing since the samples were too brittle. The substitution pattern of the phthalaldehyde monomers had a pronounced influence on the thermal properties of the polycycloacetals as a result of free volume effects: m-phthalaldehyde resulted in the lowest Tg of 90 °C, o-phthalaldehyde resulted in a Tg of 105 °C, while terephthalaldehyde resulted in a Tg of 115 °C (Table 1). The polycycloacetals from glutaraldehyde and di-TMP were stable against hydrolysis in a range in pH = 3–10 for one month at a temperature of 50 °C.
Polycycloacetal oligomers for subsequent use in epoxy resins for powder coatings were also synthesized from di-TMP, 2,2,6,6-tetramethylolcyclohexanol (Chart 4), or diglycerol and an aqueous solutions of glutaraldehyde and glyoxal using hypophosphorous acid as the catalyst (Mn = 1.0–2.2 kg mol−1, Đ = 1.8–2.3).31 Intrigued by the high thermal and chemical stability of ladder polymers, Wudl and co-workers investigated four different tetraols (2,2,6,6-tetramethylolcyclohexanol, 2,2,6,6-tetramethylolcyclohexanone, 2,2,5,5-tetramethylolcyclopentanone, and 1,1,4,4-tetramethylolcyclohexane, Chart 4) for the condensation with terephthalaldehyde, m-phthalaldehyde and 1,4-cyclohexanedicarbaldehyde.31,32 Thermally stable products were obtained (200 °C < Td,5% < 300 °C) but information on the molecular weight of the products was not provided. The polymers were insoluble in most common solvents, but well soluble in hexafluoroisopropanol (HFIP) and N-methy-2-pyrrolidone (NMP), or slightly soluble upon heating in polar aprotic solvents in some cases. The polycycloacetals based on m-phthalaldehyde and 1,4-cyclohexanedicarbaldehyde were more likely to show a wider range in solubility.
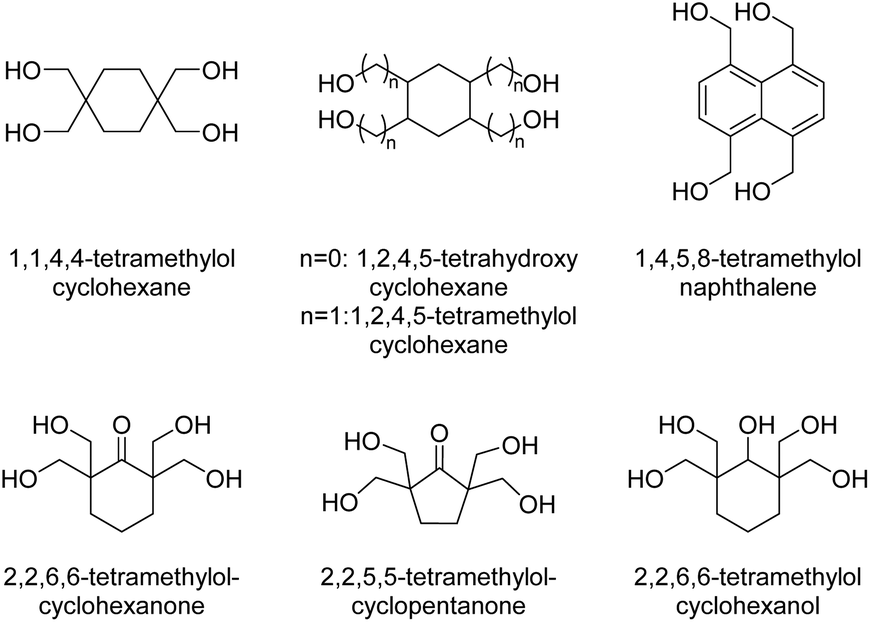 | ||
| Chart 4 Tetraol monomers synthesized for polyacetalization. | ||
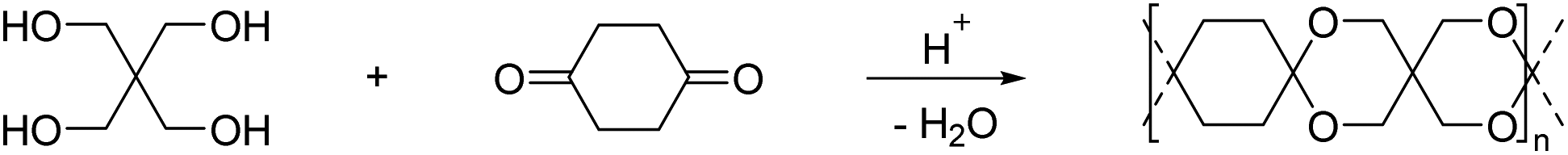 | ||
| Scheme 4 Synthesis of a spiro polycycloacetal from 1,4-cyclohexanedione and pentaerythritol. | ||
A set of spiro polymers synthesized by combining 1,4-cyclohexanedione with 1,1,4,4-tetramethylolcyclohexane, 2,2,6,6-tetramethylolcyclohexanone, 2,2,5,5-tetramethylolcyclopentanone, 1,2,4,5-tetrahydroxy-cyclohexane, 1,2,4,5-tetramethylolcyclohexane, and 1,4,5,8-tetramethylolnaphthalene (Chart 4) was later reported. 1,2,4,5-tetramethylolcyclohexane was also reacted with 2,7-decalindione, and self-condensation of dihydroxyacetone was performed as well. In all cases poorly soluble products were obtained and this insolubility results in a lack of reliable characterization data, such as molecular weights. Only the polycycloacetals based on 2,2,6,6-tetramethylolcyclohexanone and 2,2,5,5-tetramethylolcyclopentanone appeared to soften or melt (Tm = 255–290 °C or 370–395 °C, respectively).47 Self-condensation of dihydroxyacetone was later attempted again and reported to result in a polymer that was slightly soluble in DMSO.48 McKeown and co-workers addressed the poor solubility of these polycycloacetals by introducing alternative diketone building blocks for the polyacetalization with pentaerythritol, which either bear solubilizing groups or result in a contorted structure of the polycycloacetals.49 Functionalizing 1,4-cyclohexanedione yielded 2,5-didodecylcyclohexane-1,4-dione (Chart 2; R3 = (CH2)11CH3), and the subsequent polymerization with pentaerythritol resulted in a product that was well soluble in DCM and THF. The polymer showed a moderate Tg of 45 °C, but SEC analysis revealed a low molecular weight material (Mn = 5.0–6.0 kg mol−1, Đ = 1.6–1.8), most likely due to steric hindrance in the acetal formation. However, the polymerization of pentaerythritol with 1,5-diethylbicyclo[3.3.0]octane-3,7-dione (Chart 2), resulted in polycycloacetals with high molecular weights (Mn = 12–14 kg mol−1, Đ = 1.8–2.5), which were nevertheless well soluble in common organic solvents. The latter polymer was amorphous (Tg = 180 °C) and stable up to 280 °C, but sensitive to hydrolysis. Oishi and co-workers later investigated the polycycloacetal for polymer electrolyte applications.50
Spiro oligocycloacetals were also synthesized from 1,4-cyclohexanedione and a disubstituted derivative of myo-inositol, a naturally occurring cyclic hexaol (Scheme 5), resulting in oligomers (Mn = 1.4–2.1 kg mol−1, Đ = 1.9).51 Mass spectrometry confirmed the typical end group distribution of a polycondensation (i.e. AA-, BB-, and AB-monomer end-capped chains) from which the authors concluded that the low molecular weights should be ascribed to an incomplete removal of water from the equilibrium reaction rather than to side reactions. The spiro oligocycloacetals were soluble in polar aprotic solvents, thermally stable (Td,5% = 320–330 °C) and showed moderate to high Tgs (Table 1, Scheme 5). The oligomers based on diallyl, and dibenzyl substituted myo-inositol were amorphous, whereas the oligomer based on dimethyl substituted myo-inositol was semi-crystalline (Tm = 250 °C). Post-polymerization modification by radical thiol–ene chemistry was successfully applied on the spiro oligocycloacetal bearing allyl substituents.
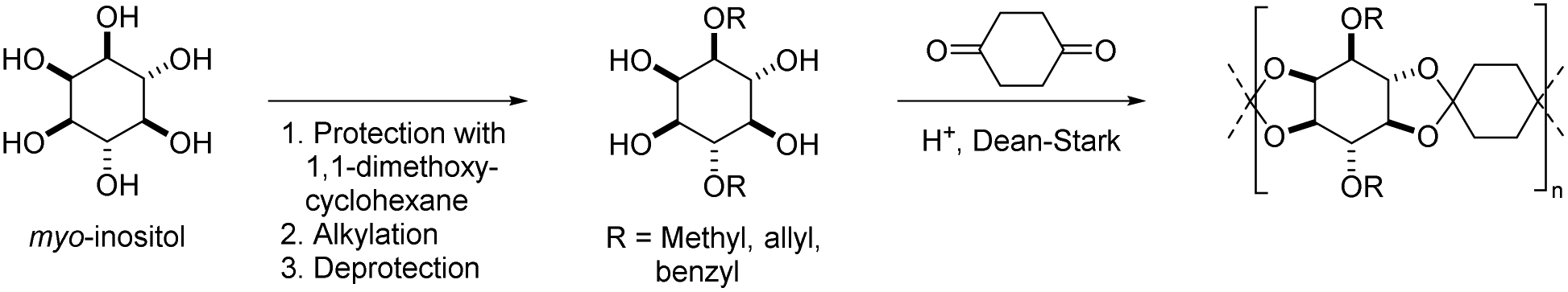 | ||
| Scheme 5 The synthesis of spiro oligocycloacetals from disubstituted myo-inositol and 1,4-cyclohexanedione. | ||
In addition to the synthesis of linear polycycloacetals by direct polyacetalization, an approach towards hyperbranched polymers and dendrimers by polyacetalization was presented by Taton and co-workers in 2014.52 4-Hydroxymethylbenzaldehyde was used as an AB2 monomer yielding non-cyclic acetal repeat units by polyacetalization in bulk, which resulted in high molecular weights (Mn = 12 kg mol−1, Đ = 1.7) in relatively short reaction times (2 h). A degree of branching (DB) equal to 1 was observed, which was explained by the high reactivity of the intermediate hemiacetal species. Interestingly, using the dimethylacetal of hydroxybenzaldehyde resulted in hyperbranched polymers with a DB of ca. 0.5 instead (Mn = 4.8 kg mol−1, Đ = 2.9), since the reaction does not proceed via the reactive hemiacetal species but via mixed acetals. The latter approach to hyperbranched polyacetals was also reported before by Ramakrishnan and co-workers, as a route toward degradable scaffolds (Mn = 7.0 kg mol−1, Đ = 3.0).53 The authors also investigated the synthesis of hyperbranched polymers from the dibutyl and dihexyl acetal of 4-hydroxymethyl benzaldehyde. As compared to the dimethyl acetal, the acetals from higher alcohols were more susceptible towards hydrolysis of the acetal groups during synthesis. Using the dibutyl and dihexyl acetal of hydroxybenzaldehyde however, results in the same polymer structure along the chains, but more hydrophobic end-groups. Whereas the hyperbranched polymers bearing methyl and butyl end-groups readily degraded in water at pH = 4, the hexyl terminated hyperbranched polymer did not degrade within 3 days. On the other hand, in aprotic deuterated chloroform containing trifluoroacetic acid, all polymers degraded within hours, with faster degradation rates for increasing lengths of the alkyl rests of the end groups.53 These last examples of polycondensations leading to hyperbranched polyacetals are in fact polytransacetalizations, which will be discussed in more detail in the next section.
Polytransacetalization
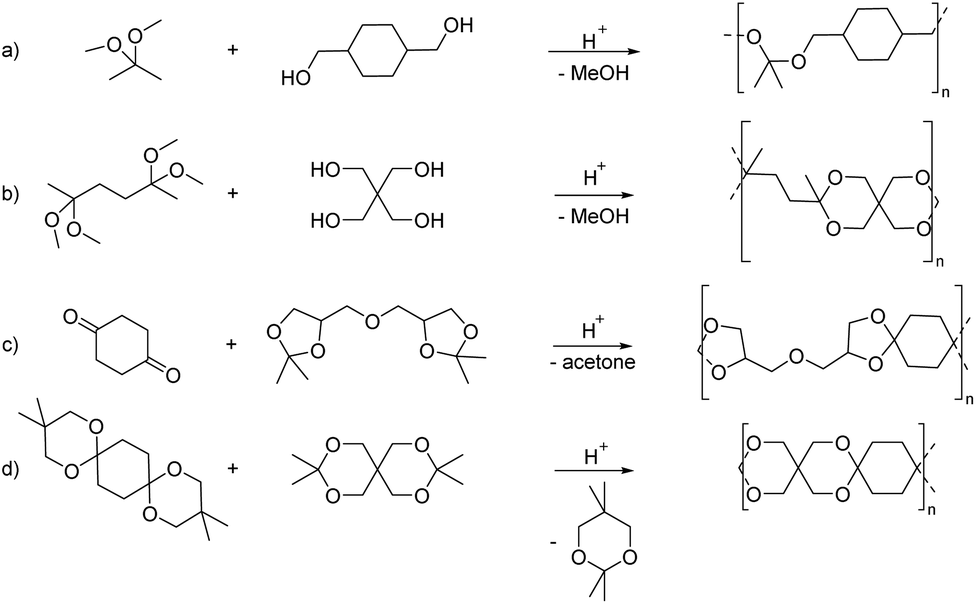 | ||
| Scheme 6 Approaches towards transacetalization, illustrated using examples from the literature. | ||
In particular the polyacetals obtained from reacting 2,2-dimethoxypropane with 1,4-benzenedimethanol, 1,4-cyclohexanediol or a mixture of 1,4-cyclohexanediol and a linear aliphatic diol in the presence of p-TsOH as the catalyst (Scheme 6(a)) have more recently attracted increasing interest for drug delivery applications.57–65 These examples illustrate one of the advantages of using the polytransacetalization approach, i.e. the removal of condensates with a relatively low boiling point (bp). 2,2-Dimethoxypropane for example has a bp of 80 °C (compare acetone: bp = 56 °C), which allows for the removal of methanol (bp = 65 °C) as the condensate from the reaction mixture. Nevertheless, additional doses of 2,2-dimethoxypropane are added to the reaction mixture to account for the loss of the volatile monomer. The polyacetals obtained in this way typically have low molecular weights (Mn = 2.1–4.1 kg mol−1, Đ = 1.43–1.79) and are already very sensitive towards hydrolysis at pH = 7.4 resulting in 30% hydrolysis within 72 h. The fast hydrolytic degradation at biologically relevant pH-values renders these polyacetals interesting for drug delivery applications, where the polyacetals can be used in the form of nanoparticles as delivery vehicles for the encapsulation of drugs. However, this sensitivity towards hydrolytic degradation, even at neutral pH-values, prevents the application of these polyacetals in polymer bulk materials. Transacetalization was also investigated for the synthesis of polycycloacetal dendrimers from the tris(dimethyl acetal) of 1,3,5-benzene-tricarbaldehyde.67 The tris(dimethyl acetal) was reacted as the core with the tris(pentaerythritol acetal) of the same tricarbaldehyde (1,3,5-benzene-tricarbaldehyde), thereby releasing methanol as the condensate and resulting in a sparsely soluble dendrimer with 12 terminal hydroxy groups.
Polytransketalization resulting in the release of acetone as the condensate for the synthesis of poly(ester acetal)s was also described. In this case, diesters of a diacid (e.g. maleic acid for subsequent crosslinking, Scheme 7) and the glycerol acetal of acetone (1,2-isopropylideneglycerol, solketal) were prepared as a tetraol-type monomer.68 Diesters were also prepared from butanediol for example, with two equivalents of levulinic acid, a renewable keto acid, yielding the corresponding dicarbonyl monomer. The same polymer was also prepared by direct polyacetalization using the corresponding tetraol monomer, but it is not clear which polycondensation approach was more successful or resulted in higher molecular weight material. Eventually, the resulting poly(ester acetal)s were crosslinked with styrene using radical polymerization. Structurally similar monomers were also used for the synthesis of polymers from acetal containing monomers using other step-growth polymerization techniques (vide infra).
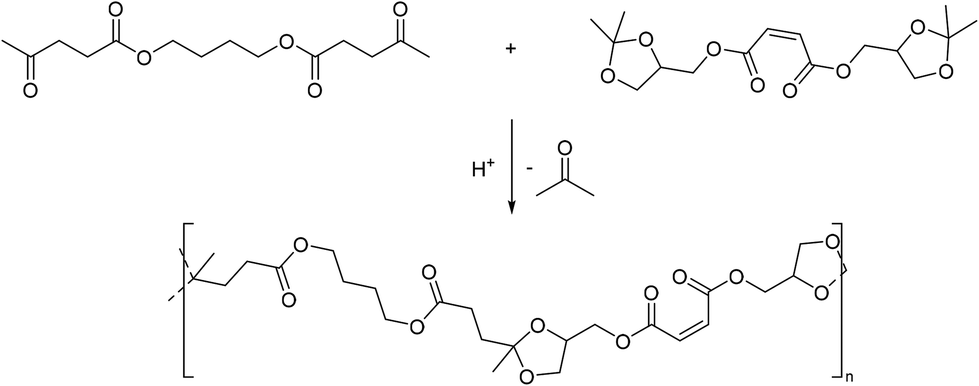 | ||
| Scheme 7 Polytransketalization of 1,4-butanediol dilevulinate and disolketal maleate. | ||
Very recently we employed polytransacetalization for the synthesis of polycycloacetals from diglycerol bisacetonide and 1,4-cyclohexanedione (Scheme 6(c)).69 Diglycerol is obtained from the condensation of glycerol, which is a waste product of the biodiesel industry, at high temperatures. However, diglycerol is commercially only available in approximately 90% purity, rendering commercially available samples unsuitable for polycondensation reactions. Yet, the bisacetonide was easily synthesized from diglycerol, purified by vacuum distillation and subsequently used in the polytransacetalization with 1,4-cyclohexanedione and 4,4′-bicyclohexanone. High molecular weight polycycloacetals, which were soluble in chloroform, were obtained (Mn = 28.1–22.6 kg mol−1; Đ = 2.5–4.0). The high dispersities and the formation of a small amount of insoluble gel-like material indicate branching/crosslinking as a side-reaction, most likely via the formation of linear acetal units at high conversions. The polycycloacetals were semi-crystalline with high melting points (Tg = 48 and 65 °C, Tm = 210 and 241 °C, where 4,4′-bicyclohexanone as a monomer resulted in the polymers with the thermal transitions at higher temperatures, Table 1). The high melting points made processing by compression molding impossible because of sample degradation. Thus, copolycycloacetals were synthesized by partly replacing the diglycerol bisacetonide by di-TMP in the polymerization with 1,4-cyclohexanedione. The incorporation of di-TMP decreased the melting temperature (10% di-TMP: Tm = 172 °C) and crystallinity, whereas the glass transition temperature increased (10% di-TMP: Tg = 55 °C). Completely amorphous polymers were obtained at a di-TMP content of 24% and 48% (Tg(24% di-TMP) = 60 °C; Tg(48% di-TMP) = 71 °C). All polycycloacetals were thermally stable (Td,5% >300 °C) and have renewable carbon contents in the range of 33–100%. For the completely amorphous copolycycloacetals, Young's moduli of 1.1 and 1.4 GPa, elongations at break of 2.6 and 1.2% and tensile strengths at break of 24 and 7 GPa were determined by tensile testing, where the copolycycloacetal with a lower di-TMP content of 24% showed the better mechanical properties. The semi-crystalline polymers were too brittle for mechanical characterization. Interestingly, the di-TMP content also influenced the hydrolytic degradation of the copolycycloacetals. In general, the copolycycloacetals were stable at pH values greater than 3 and showed increasing hydrolytic degradation with increasing diglycerol bisacetonide content at pH = 1. At this pH value, the copolycycloacetals were recovered in more than 65% of the original weight with only moderate reductions in molecular weight depending on the di-TMP content, whereas the polycycloacetal from diglycerol bisacetonide and 1,4-cyclohexanedione showed a weight loss of 87% and a reduction in molecular weight from Mn = 28.1 kg mol−1 to Mn = 8.4 kg mol−1 for a powder sample. A processed sample of the latter polymer degraded to a lesser extend at pH = 1. Finally, the polycycloacetal from 1,4-cyclohexanedione and di-TMP showed a weight loss of 17% and a reduction in molecular weight by 28% at pH = 1. This polymer showed an intriguing thermal behavior, with a Tm close to the Tg (Tm = 139 °C, Tg = 97 °C) in the first heating scan in DSC. It is this close temperature range in which both the Tg and the Tm occur that is thought to prevent crystallization from the melt, resulting in an amorphous sample.
Alder et al. reported an attempt to obtain the spiro polycycloacetals based on 1,4-cyclohexanedione and pentaerythritol via transketalization (Scheme 6(d)).66 The bisacetonide of pentaerythritol was reacted with the bis(dimethyl-1,3-propanediol) acetal of 1,4-cyclohexanedione or the bis(ethylene glycol) acetal of 1,4-cyclohexanedione under acidic conditions, releasing 2,2,5,5-tetramethyl-1,3-dioxane or acetone ethylene acetal as the condensate and insoluble oligomers were obtained in all cases.66 While highly interesting from a synthetic perspective, this polytransacetalization approach suffered from the insolubility of the resulting spiro polycycloacetal, which was previously reported for the same polycycloacetal synthesized by direct polyacetalization (vide supra). In particular, the polymer was not soluble in the reaction mixture (in toluene), which most likely prevented the formation of high molecular weight polymers. The authors also report on a polycycloacetal, which is formally a self-condensate of dihydroxyacetone. This polymer was soluble in DMSO, but molecular weights are not reported.
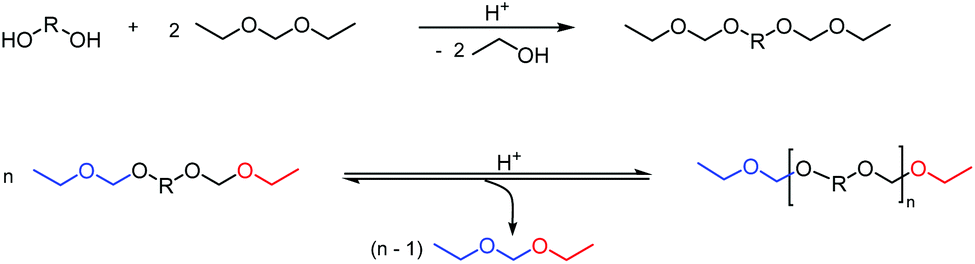 | ||
| Scheme 8 Brønsted acid catalyzed synthesis and subsequent acetal metathesis polymerization of a bis(ethyl acetal). | ||
So far, biobased linear aliphatic α,ω-diols and the isohexides isomannide, isosorbide and isoidide were employed as diol building blocks in AMP. However, for the less reactive secondary hydroxy groups of the isohexides, the bisacetal monomers had to be synthesized using the highly carcinogenic chloromethyl methyl ether.74 Finally, the isohexide bis(methyl acetals) were combined with long-chain aliphatic bis(methyl acetals), which were synthesized from the corresponding diol and dimethoxymethane.75 For polyacetals based on linear aliphatic diols, mostly high molecular weights in the range of Mn = 7.8–38.8 kg mol−1 (SEC in THF) were obtained with the expected broad molecular weight distributions, and the polyacetals exhibited moderate melting points (Tm = 28–64 °C), whereas Tgs are not reported. The polyacetals were susceptible to Brønsted acid catalyzed hydrolytic degradation in THF/water.70 Isohexide based polyacetals on the other hand were obtained with molecular weights in the range of Mn = 3.2–27.6 kg mol−1 (NMR) depending on the type of isohexide. The semi-crystalline polymers with Tgs in the range of 38–66 °C and Tms in the range of 103–156 °C were susceptible to acid catalyzed hydrolytic degradation as well. Interestingly, the copolymerization of isohexide bis(methyl acetals) with bis(methyl acetals) of long chain aliphatic α,ω-diols resulted in polyacetals that were much less sensitive to hydrolytic degradation, probably owing to the hydrophobic nature of the long aliphatic chains. For example, treatment with 9 M HCl solution over a period of 15 days led to a weight loss of only 30%.75 In a later report, Miller an co-workers transferred the concept of AMP to silicon acetals.76 Here, bis(silicon acetal) monomers were synthesized from a variety of diols and dimethoxydimethylsilane. The obtained polymers were of similar molecular weights as previously reported for AMP (Mn = 6.6–26.6 kg mol−1). For some of the poly(silicon acetals) Tgs could be detected (Tg = −54 to −45 °C) and all poly(silicon acetals) were semi-crystalline with Tm in the range of 42–104 °C depending on the nature of the employed diols. The poly(silicon acetals) based on diols containing aromatic units showed the highest Tms of approximately 100 °C.
Polyaddition of diols to divinylethers
Polyacetals can also be prepared by an entirely different route, namely the acid catalyzed stepwise polyaddition of diols to divinyl ethers (Scheme 9), yielding high molar mass polymers containing non-cyclic acetaldehyde acetal repeat units. Polymers of this type have been investigated mostly in the context of degradable polymers for controlled drug release.9 Heller et al. synthesized polyacetals from diethyleneglycol divinyl ether or 1,4-divinyloxybutane and 1,4-cyclohexanediol, 1,4-cyclohexanedimethanol, or bisphenol A.77,78 The reaction proceeded by Brønsted acid catalysis (p-TsOH) in THF at room temperature under an inert atmosphere, and within 1 h the formation of polymers with high molecular weights (Mw = 33–200 kg mol−1 (light scattering), Đ = 1.56–2.93) was reported.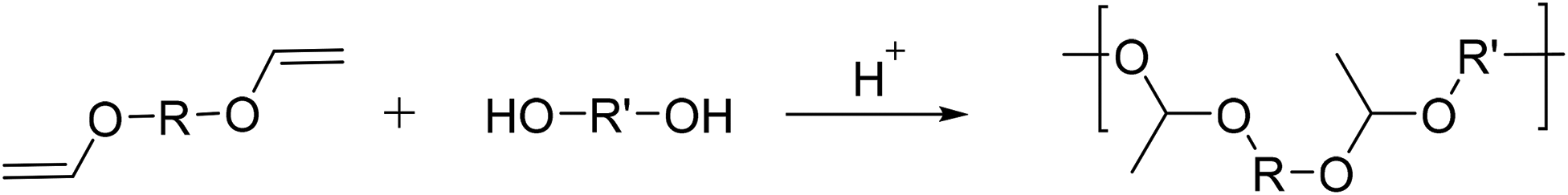 | ||
| Scheme 9 Acid-catalyzed step-growth addition polymerization of diols and divinyl ethers. | ||
Tomlinson et al. investigated the polymerization of tri(ethylene glycol) divinyl ether in combination with a poly(ethylene glycol) diol and 9-fluorenylmethyloxycarbonyl (FMOC) protected 2-amino-1,3-propanediol (serinol) for subsequent drug conjugation.79,80 The authors highlighted that the removal of the catalyst after polymerization (by precipitation) was crucial to prevent polymer degradation during storage. The polyacetals showed pronounced degradation at pH = 7.4 (35% molecular weight loss after 500 h), and 90% molecular weight loss within 500 h at pH = 5. These polyacetals were used to prepare a conjugate with doxorubicin, which was liberated during main chain degradation and biologically active. The amino functionality of serinol was also used to prepare polyacetal-graft-PEG copolymers, and the resulting thermogels exhibited a lower critical solution temperature and were investigated in hydrolysis and release studies.81
The polyaddition reaction with divinylethers was also extended to dicarboxylic acids instead of diols, resulting in the formation of poly(hemiacetal esters).82 Polymers with Mn = 6.0–14.1 kg mol−1 (Đ = 1.5–3.0) were obtained within 24 h from ethylene glycol divinyl ether and various dicarboxylic acids. For a series of linear aliphatic dicarboxylic acids, the thermal stability increased with increasing length of the aliphatic spacer. For example, malonic acid resulted in a poly(hemiacetal ester), which degraded at T <150 °C whereas succinic acid, adipic acid, and sebacic acid resulted in polymers that were stable above 200 °C. Thermal degradation in the range of 145–200 °C was observed for polymers from diacids that stabilize carboxylate anions by mesomeric effects (terephthalic acid, maleic acid, fumaric acid) whereas a poly(hemiacetal ester) containing an isocyanuric acid moiety in the repeat unit degraded at 150 °C, which was explained by enhanced dissociation due to hydrogen bonding between the isocyanuric acid groups and the carbonyl groups of the hemiacetal esters.82 The potential of polyacetals prepared in this way for application in biomaterials has been reviewed elsewhere.9
In addition to the polycondensation and polyaddition methods, which build up the polymer backbone by the formation or exchange of acetal bonds, the polymerization of monomers that already contain the acetal unit in their structure has been intensively investigated in the literature.
Polymerization of monomers containing the acetal functional group
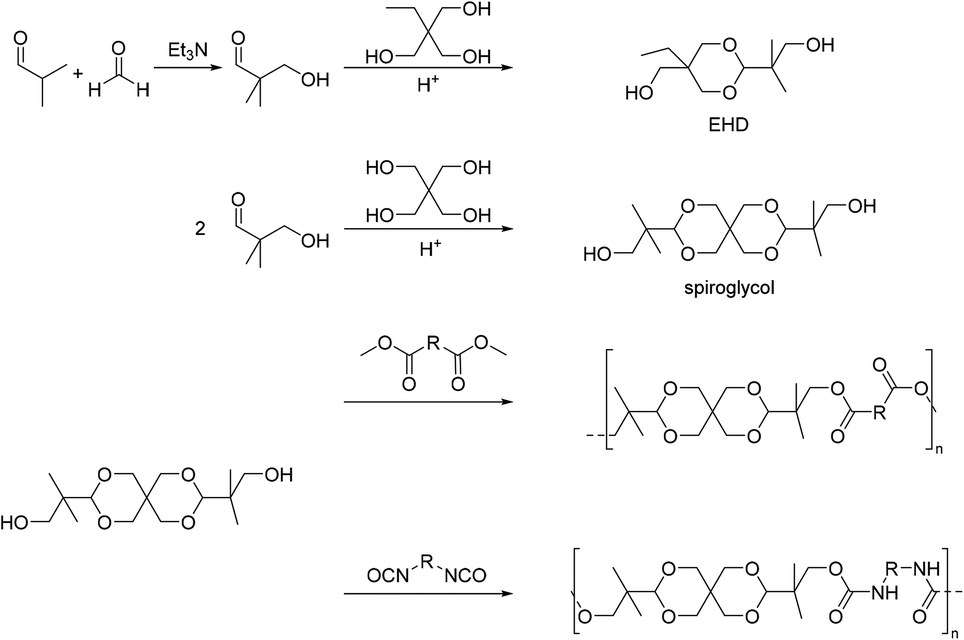 | ||
| Scheme 10 Synthesis of acetal containing diols EHD and spiroglycol (top) and schematic representation of polycondensation/addition reactions of spiroglycol to obtain polyesters or polyurethanes (bottom). | ||
Pentaerythritol was also reacted with 2- or 4-hydroxybenzaldehyde in order to obtain a diol.38,91–93 The diol based on 4-hydroxybenzaldehyde was polymerized with 2,5-alkoxyl substituted terephthaloyl dichlorides. The molecular weight of the poly(ester acetal)s decreased with increasing length of the alkoxy side chains. For example, for dipropyloxy substituted terephthaloyl dicholoride as the monomer Mw = 15 kg mol−1 was obtained, whereas the terephthaloyl dicholoride with two pentadecyloxy substituents yielded a poly(ester acetal) with a molecular weight of only Mw = 7.6 kg mol−1. The poly(ester acetals) were soluble in a range of solvents. The polymer with propyloxy substituents was amorphous with a Tg = 117 °C whereas all other polymers showed three melting transitions. The two melting transitions at low temperatures were attributed to the side chains, while the third melting transition, associated with the polymer backbone, occurred at decreasing temperatures with increasing length of the alkoxy substituents (Tm = 182–228 °C). All poly(ester acetals) were thermally stable (Td,5% = 350–370 °C).91 The same authors used the diol obtained from 4-hydroxybenzaldehyde and pentaerythritol also in polyester synthesis with bis(4-chlorocarbonylphenyl)dimethylsilane and bis(4-chlorocarbonylphenyl)diphenylsilane. Molecular weights were not reported but the thermal stability of the polymers was excellent (Td,10% = 365–410 °C); the samples were amorphous with high Tg = 161–203 °C, and soluble in polar aprotic solvents.93
We investigated the bisacrolein acetal of pentaerythritol (3,9-divinyl-2,4,8,10-tetraoxaspiro[5.5]undecane), for the synthesis of a diol monomer by radical thiol–ene addition of 2-mercaptoethanol (Scheme 11).94 Later a diamine monomer was synthesized in the same way (vide infra). The bisacrolein acetal can potentially be sourced from renewable resources and was readily soluble in common solvents. Linear thermoplastic poly(urethane acetal)s could be synthesized from the diol monomer and six different diisocyanate monomers. As expected, linear aliphatic diisocyanates, such as hexamethylene diisocyanate (HDI), resulted in polymers of lower Tgs (Tg = 24–26 °C) as compared to cycloaliphatic diisocyanates (e.g. 4,4′-methylenebis(cyclohexyl isocyanate) (hMDI)) and aromatic diisocyanates (e.g. 4,4′-methylene diphenyl diisocyanate (MDI)) (both Tg = 85 °C). The degradation temperature (Td,5% = 260–300 °C) of the poly(urethane acetal)s depended on the diisocyanate: the use of aromatic diisocyanates resulted in lower degradation temperatures than the use of aliphatic diisocyanates. Since the poly(urethane acetal)s were obtained with moderate to excellent molecular weights (Mn = 8.0–45.0 kg mol−1), the mechanical properties could be established for most poly(urethane acetal)s and Young's moduli in the range from 0.1–1.4 GPa were observed, which is in good agreement with commercially available thermoplastic polyurethanes.95 Hydrolytic degradation test were performed at pH = 3, 7 and 10 for selected poly(urethane acetal)s and the polymers proved to be stable against degradation under these conditions.
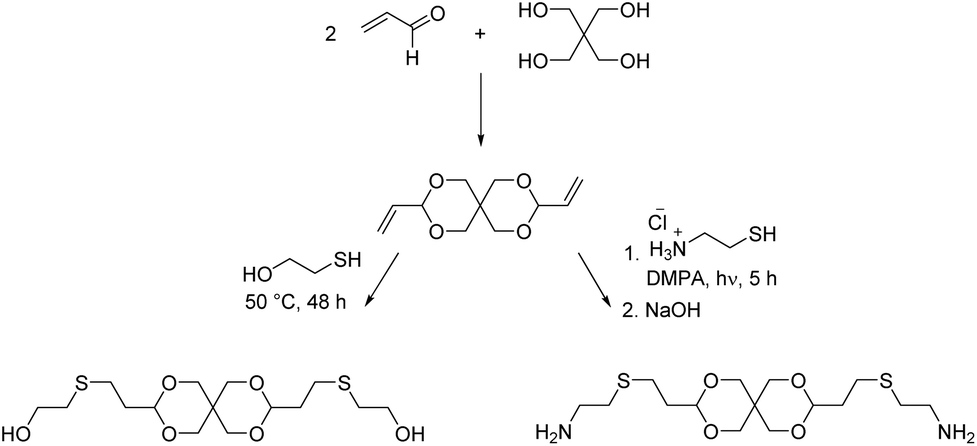 | ||
| Scheme 11 Synthesis of a spiroacetal based diol and diamine monomer via radical thiol–ene addition to pentaerythritol bis(crotonaldehyde) acetal. The diol was synthesized at elevated temperatures whereas the diamine was synthesized using the photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA). Adapted from ref. 94 and 96. | ||
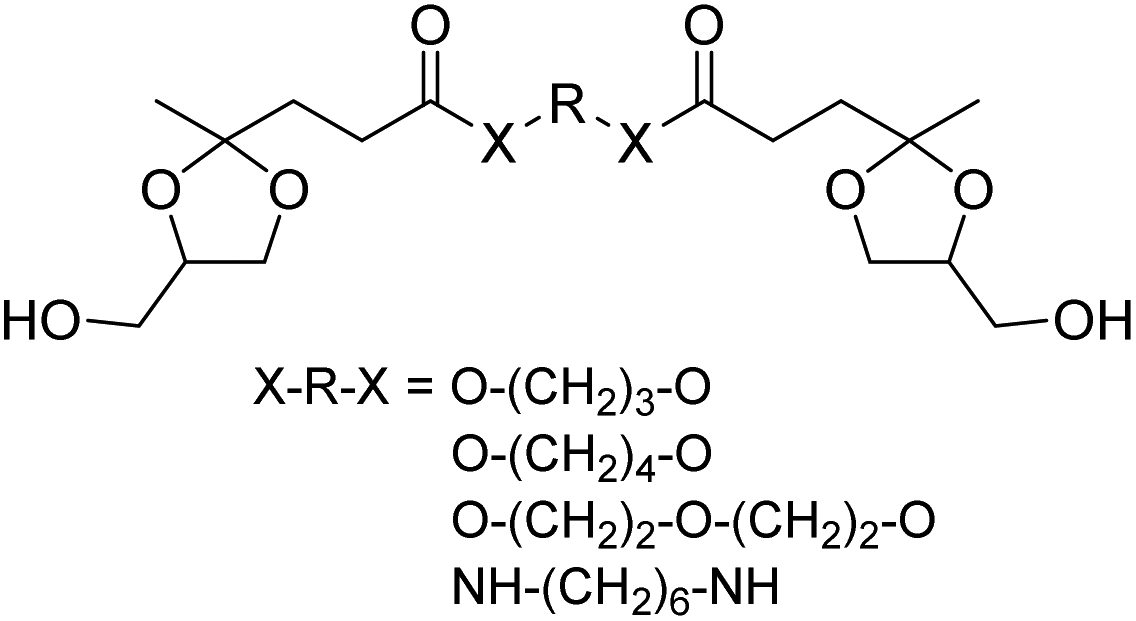 | ||
| Chart 5 Diol monomers from two equivalents of the glycerol acetal of ethyl levulinate and diols or diamines. | ||
Suh and co-workers reported on a diol monomer from the bis(glycerol) acetal of camphorquinone (CaG, Scheme 12).99,100 CaG was first investigated in (co)polycarbonate synthesis with diphenyl carbonate (DPC) and bisphenol A (BPA). The molecular weight of the obtained poly(carbonate acetal)s was moderate to high (Mn = 8.2–18.0 kg mol−1, Đ = 2.0–4.9) and increased with increasing BPA content (0–80%). Similarly, the Tg of the amorphous poly(carbonate acetals) increased with increasing BPA content (Tg = 128–151 °C) as did the thermal stability of the polymers (Td,5% = 276–330 °C), with the exception of the poly(carbonate acetal) that did not contain any BPA (Td,5% = 289 °C). Overall, the thermal stability was thus comparable to the industrially produced polycarbonate based on 100% BPA (Td,5% = 333 °C). Subsequently, CaG was also investigated in copoly(ester acetal) synthesis with dimethyl terephthalate (DMT) and ethylene glycol (0–100%). High molecular weight polymers (Mn = 10.0–21.0 kg mol−1, Đ = 1.9–3.3) with good thermal stability were obtained (Td,5% = 323–377 °C). Only the polymers with the highest ethylene glycol contents (>90%) were semi-crystalline, (Tg = 78 °C, Tm = 209 °C, 90% ethylene glycol), whereas all other polymers were amorphous with increasing Tg for higher CaG content (Tg = 78–129 °C). In alkaline medium at pH = 10, the poly(ester acetal)s were stable (less than 10% reduction in molecular weight), whereas at pH = 2.0 (citric acid buffer) a reduction in molecular weight was observed within 12 days. The degradation was more pronounced for higher CaG contents (e.g. ethylene glycol/CaG: 50/50, reduction in molecular weight by 50%). While these observations already indicate that degradation takes place via acetal hydrolysis rather than hydrolysis of the ester bonds, this was also supported by a small molecule model compound NMR study.
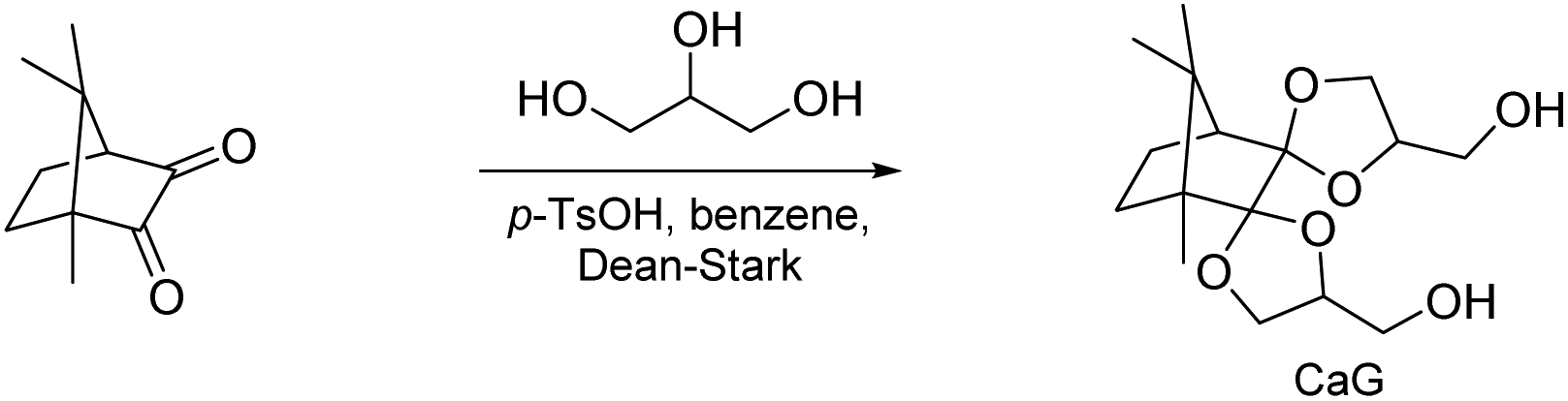 | ||
| Scheme 12 Synthesis of CaG from camphorquinone and glycerol. | ||
Diol monomers from bis(glycerol acetal)s of the diketones 1,4-cyclohexanedione (CHD) and 4,4′-bicyclohexanone (BCD) were also prepared and employed for the synthesis of polyesters, polyurethanes, and polycarbonates (Scheme 13).101 The diol prepared in this way from 1,4-cyclohexanedione is completely renewable. This diol was also employed for the synthesis of a degradable crosslinker for nanoimprint lithography.102 On the on the other hand, the BCD diol from 4,4′-bicyclohexanone, which is not a renewable compound, can easily be purified by recrystallization, which allows for straightforward scaling of the monomer synthesis.
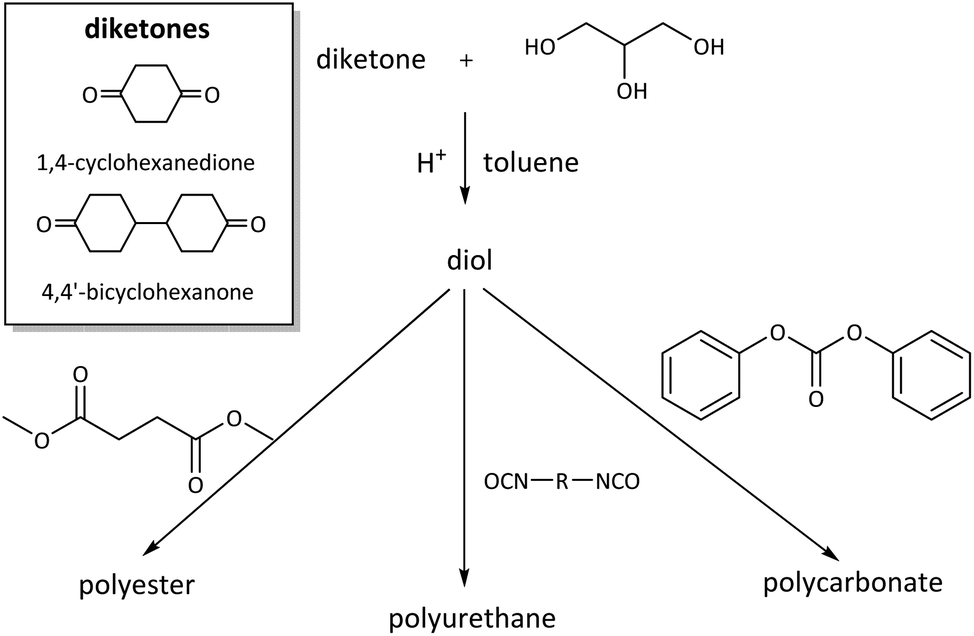 | ||
| Scheme 13 Synthesis of CHD and BCD diol monomers from glycerol and 1,4-cyclohexanedione or 4,4′-bicyclohexanone and their use in polyester, polyurethane, and polycarbonate synthesis. Reprinted with permission from ref. 101. Copyright 2017 American Chemical Society. | ||
Both CHD and BCD diols were successfully used for the synthesis of poly(carbonate acetal)s of high molecular weight (Mn = 24–33 kg mol−1, Đ = 2.8–3.2). The polymers showed lower Tgs and a lower thermal stability (Tg = 72 °C (CHD), 100 °C (BCD), Td,5% = 350 °C (CHD), 320 °C (BCD)) than polycarbonates from BPA. For the BCD based poly(carbonate acetal), tensile measurements could be performed, revealing a promising ductile behavior of the polymer, although the mechanical properties of BPA based polycarbonates were not fully matched. The synthesis of poly(ester acetal)s proved more challenging, but polymers were obtained for both BCD and CHD as the diol monomer (Mn = 17–19 kg mol−1, Đ = 2.3–3.8) when using dibutyltin oxide as the catalyst. However, the obtained molecular weights were too low for processing and the determination of the mechanical properties of the polymers. In an attempt to increase the moderate Tgs of the poly(ester acetal)s from dimethyl succinate (Tg = 40 °C (CHD), 50 °C (BCD)), poly(ester acetals) with dimethyl furandicarboxylate were prepared as well. Although this approach did indeed lead to an increase in Tg (Tg = 96 °C (CHD), 65 °C (BCD)), the molecular weights of the polyesters based on furandicarboxylic acid were low (Mn = 8.6–6.0 kg mol−1, Đ = 2.3–4.0). In all cases, the poly(ester acetal)s were thermally stable (Td,5% = 310–320 °C). Finally, CHD and BCD were tested in polyaddition reactions with four different diisocyanates each (isophorone diisocyanate (IPDI), toluene diisocyanate (TDI), MDI, hMDI). Poly(urethane acetal)s of high molecular weight were obtained for most combinations of diisocyanate/diol (Mn = 17–50 kg mol−1, Đ = 1.7–2.3). Thermally stable poly(urethane acetal)s (typically Td,5%ca. 300 °C) with high Tgs were obtained (e.g. MDI: Tg = 150 °C (CHD), 145 °C (BCD)), depending on the diisocyanate. Importantly, the materials could be processed by compression molding and were characterized by tensile testing. Although the materials were brittle, as expected from the absence of soft segments, they had excellent Young's moduli in the range of commercially available (rigid) thermoplastic polyurethanes (E = 1.10–1.42 GPa, ultimate tensile strength = 16–75 MPa). For selected examples of polycarbonates, polyesters and polyurethanes, hydrolysis studies were performed as well, and interestingly all polymers were stable against hydrolysis for two weeks at 50 °C and pH = 1, 3, 5 and 7, which has been related to the hydrophobicity of the polymer backbones. Together with the previous discussion on spiroglycol based polyesters, as well as polyurethanes based on the bisacrolein acetal of pentaerythritol, and in contrast to other reports discussed previously, this observation shows that the susceptibility of poly(cyclo)acetals to hydrolytic degradation is not self-evident and in fact highly dependent on the precise structure of the polymers.
 | ||
| Scheme 14 Synthesis of a diol from 1,3-dihydroxy acetone. | ||
Muñoz-Guerra and co-workers have intensely investigated diol monomers based on formaldehyde acetals of alditols in the past years, with the aim to obtain partly renewable copolyesters, mostly terephthalate copolyesters, with high Tgs.104–107 Although, strictly speaking, the resulting polymers do not contain the acetal group in their backbone, but as a side group, the formation of acetals enables the use of alditol based structures as diol monomers.
These efforts and related reports were recently reviewed by the authors in 2014.107 Therefore, only the latest contributions to this field are included here. For completion however, the alditol based diol monomers reported so far are summarized (Table 2).
| Alditol | Chemical structure diol | Chemical structure diester |
|---|---|---|
| Galacitol |
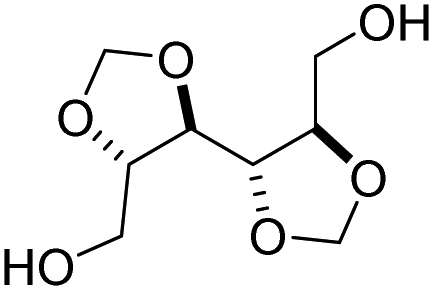
|
|
| D-Mannitol |
|
|
| D-Glucitol |
|
|
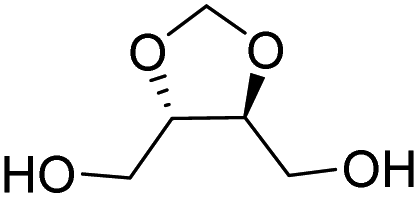
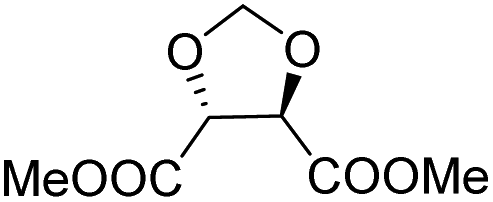
| L-Threitol |
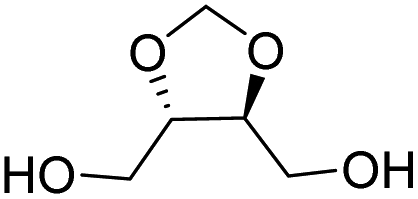
|
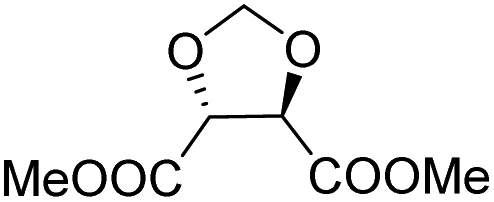
|
Bernaerts, de Wildeman and co-workers recently investigated polyamides from galactarate acetals.111,112 For 2,3:4,5-di-O-methylene-galactarate and 2,3:4,5-di-O-isopropylidene-galactarate different polymerization techniques, such as melt-condensation of the galactarate/diamine salt or of the diethyl galactarate and the diamine, and direct polymerization using a phosphorylation technique (for aromatic diamines), were investigated with a series of diamines. While the melt polymerization of the salt was hampered by the high temperatures required to melt the salts, which lead to degradation, low to high molecular weight polyamides were obtained for both galactarate monomers when using the diethyl galactarate monomers in the melt (Mn = 2.1–24 kg mol−1, Đ = 1.72–5.94). Direct polycondensation using a phosphorylation technique resulted in high molecular weights for the acetone acetal of galactaric acid (Mn = 26–36 kg mol−1, Đ = 2.15–2.52), but in moderate molecular weights for the formaldehyde acetal of galactaric acid (Mn = 6.7–17 kg mol−1, Đ = 1.9–2.32).
The latter monomer typically polymerized faster but also resulted in higher dispersities during polymerization in the melt, probably due to branching or crosslinking caused by ring opening of the acetal, which results in pendant hydroxyl groups. As acknowledged by the authors this observation is contrary to what is expected for the stability of formaldehyde acetals. All polyamides showed high Tgs when aromatic or cycloaliphatic amines were used (Tg = 119–224 °C) and were stable at temperatures greater than 300 °C. Polyamides bearing isopropylidene groups were soluble in a wider range of solvents than polyamides bearing methylene groups. While the polymer backbones were stable in acidic medium, such as 5% citric acid or a commercially available cleaning agent, 95% of the isopropylidene groups were removed within 60 days, whereas the methylene groups were much less sensitive to hydrolysis. The authors also investigated the water uptake of the polyamides, which revealed that the polyamides based on 2,3:4,5-di-O-isopropylidene-galactarate show a reduced water uptake as compared to 2,3:4,5-di-O-methylene-galactarate based polyamides. Subsequently, the monomer range was expanded further to the cyclohexanone, benzaldehyde and p-fluorobenzaldehyde acetal of galactaric acid.112 The polymers showed Tgs in the range of 73–97 °C, which increased with increasing bulkiness of the acetal group. The highest Tgs were obtained for polymers with benzaldehyde acetals although these polymers also showed the lowest molecular weights (Mn ≤10 kg mol−1, Đ = 2.2–3.8), which was attributed to a slower polymerization. The latter polymers showed the lowest water uptake (1.20%), whereas the polyamide based on 2,3:4,5-di-O-methylene-galactarate showed the highest water uptake (4.25%). Thus, the general hydrophilicity of the acetal and amide functionality can be reduced by choosing sterically demanding hydrophobic carbonyls for the formation of the acetal side group. All polyamides were thermally stable (Td,10% >340 °C), and thermal degradation could be adjusted further by varying the acetal substituent. For example, polyamides bearing a p-fluorobenzyl substituent degraded at a higher temperature (Td,10% = 366 °C) than polyamides bearing a benzyl substituent (Td,10% = 344 °C).
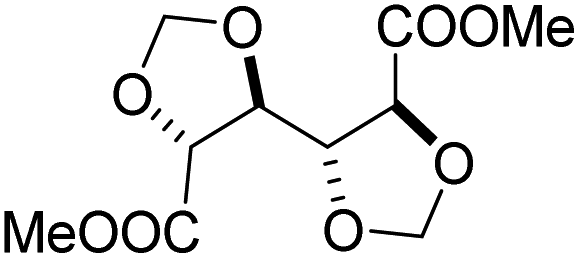
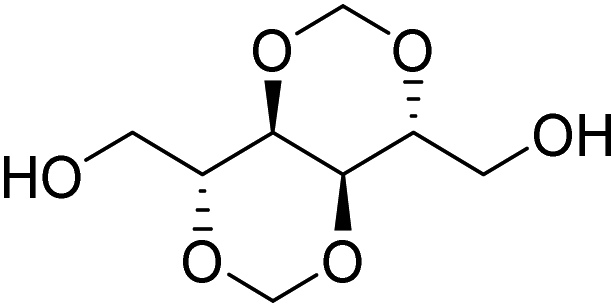
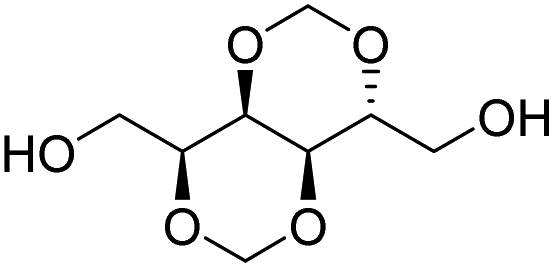
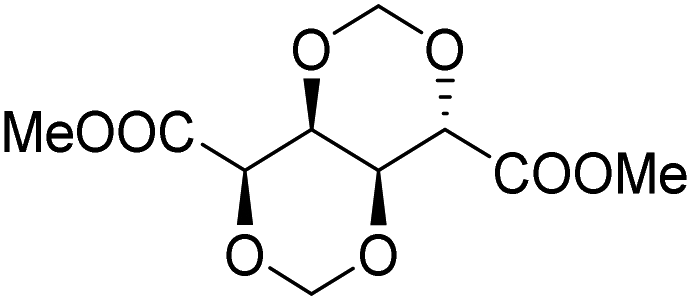
Muñoz-Guerra and co-workers, and Jacob and co-workers also investigated di(carboxylic acid) monomers based on cyclic acetals of tartaric acid derivatives. The corresponding diol is formally a L-threitol based acetal (Table 2).108–110 The formaldehyde acetals of L-threitol and dimethyl tartrate were used in copolymers of poly(ethylene terephthalate) (PET) and poly(butylene succinate) (PBS) and the copolyesters were typically obtained with molecular weights in the range of Mn = 10–15 kg mol−1 (Đ >2.0). For PET copolyesters, an increasing content of the L-threitol based monomer or the dimethyl tartrate based monomer reduced the melt enthalpy and the Tm, yielding amorphous polymers at comonomer contents of 20–30 wt% (Table 3). While the L-threitol based monomer did not affect the Tg, the dimethyl tartrate monomer caused a gradual decrease in Tg. Similarly, the thermal stability of the polyesters was not affected by varying the ratio of the diol monomers, whereas the dimethyl tartrate based monomer lead to a decrease in Td,10% from 410 °C to 322 °C. These results indicate that the decrease in terephthalate content has the most pronounced effect on the thermal properties of the copolyesters. For copolyesters of PBS, neither the incorporation of the di(carboxylic acid) monomer nor the incorporation of the diol monomer had a pronounced influence on the thermal stability of the polyesters. The Tg on the other hand, gradually increased with increasing acetal functional monomer content for both poly(2,3-di-O-methylene-L-threitol succinate) and poly(butylene 2,3-di-O-methylene-L-threarate) (Table 3). All PBS (co)polyesters were semi-crystalline, except for 2,3-di-O-methylene-L-threitol contents above 80%. While 2,3-di-O-methylene-L-threitol decreased both the Tm and the melt enthalpy of PBS, 2,3-di-O-methylene-L-threarate first decreased (up to comonomer content of 64%) and subsequently increased the Tm and the melt enthalpy of PBS copolyesters (Table 3). The copolyesters from acetal containing monomers showed enhanced hydrophilic degradation at pH = 7.4 in both the presence and absence of lipase.
| Polyester | Comonomer | Comonomer content [%] | T g [°C] | T m [°C] |
|---|---|---|---|---|
| PET |
|
0 | 81 | 250 |
| 8 | 82 | 235 | ||
| 23 | 84 | 190 | ||
| 57 | 84 | — | ||
| 100 | 83 | — | ||
|
7 | 75 | 238 | |
| 20 | 63 | 207 | ||
| 30 | 59 | 178 | ||
| 100 | 7 | — | ||
| PBS |
|
0 | −37 | 117 |
| 21 | −24 | 90 | ||
| 40 | −13 | 35 | ||
| 78 | 7 | — | ||
| 100 | 13 | — | ||
|
22 | −30 | 90 | |
| 42 | −18 | 61 | ||
| 83 | −6 | 53 | ||
| 100 | −3 | 104 | ||
|
100 | −9 | — | |
|
100 | 6 | — |
Jacobs and co-workers investigated copolyesters of the acetone acetal of dimethyl tartrate and L-threitol with aliphatic diols and aliphatic di(carboxylic acids) instead. In all cases, this resulted in amorphous polymers with low Tgs in the range of −36 °C to −8 °C and varying thermal stability (Td,max = 298–415 °C). The molecular weights of the polyesters were typically low (Mn <10 kg mol−1, Đ = 1.7–2.3), except for poly(hexamethylene 2,3-O-isopropylidene-L-threarate (Mn = 15.7 kg mol−1, Đ = 2.1). The isopropylidene side group could easily be removed by acid catalyzed hydrolysis, resulting in hydroxy functional polymers, which typically showed an increase in Tg, by 10–20 °C as compared to the acetal functionalized copolyesters.
Sudo and co-workers reported diol monomers based on diacetals of myo-inositol in an approach to polycondensates that is essentially the reverse to their approach previously discussed in the polyacetalization section (vide supra).113,114 In a first step, a diacetal was synthesized from myo-inositol and either 1,1-dimethoxy cyclohexane or a combination of butanedione and trimethyl orthoformate, resulting in diol monomers (Scheme 15), which were subsequently employed in the synthesis of polyurethanes with HMDI, IPDI, TDI and MDI as diisocyanates. The polyurethanes from the di(cyclohexanone acetal) of myo-inositol (myo-inositol diol 1) were obtained with high molecular weights (Mn = 19.3–61.6 kg mol−1, Đ = 1.53–2.53) as semi-crystalline polymers with high Tms in the range of 199–265 °C. The thermal stability of these polyurethanes (Td,5% = 283–296 °C) was 30–90 °C higher than the stability of the corresponding polyurethanes from 1,4-cyclohexanediol. The Tgs of myo-inositol based polyurethanes (Tg = 143–250 °C) were typically 30–40 °C higher than the Tgs of polyurethanes based on 1,4-cyclohexanediol, although Tgs were only observed for 1,4-cyclohexanediol in combination with IPDI and TDI. For the myo-inositol based diol, which was acetalized with butanedione (myo-inositol diol 2), polyurethanes were synthesized with HMDI, IPDI, MDI and 3-bis(isocyanatomethyl)cyclohexane (BICMC). All polyurethanes synthesized in this way were amorphous, and for IPDI and HMDI as diisocyanates higher Tgs (50 °C or 20 °C higher Tg) were observed using myo-inositol diol 2 than when using myo-inositol diol 1. In case of MDI based polyurethanes however, myo-inositol diol 2 resulted in a reduction in Tgs by 35 °C as compared to myo-inositol diol 1. Generally, myo-inositol diol 2 also resulted in lower molecular weights of the polyurethanes (Mn = 8.0–13.7 kg mol−1). In a ternary polyaddition with neopentyl glycol and HMDI, an increasing myo-inositol diol 2 content efficiently increased the Tgs from 39 °C (0% myo-inositol diol 2) to 132 °C (75% myo-inositol diol 2). Importantly, both myo-inositol diol 1 and 2 are obtained as a mixture of regioisomers upon acetalization, but the symmetric diacetals can be obtained in pure form by recrystallization, albeit in a low overall yield (20–21%).
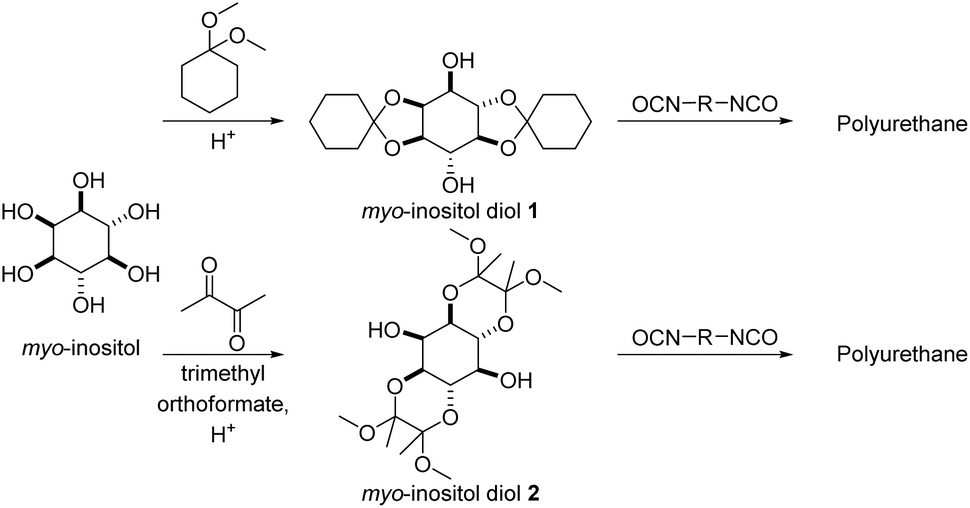 | ||
| Scheme 15 Polyacetals and polyurethanes from myo-inositol derived diols. | ||
 | ||
| Chart 6 Pentaerythritol based diamines from p-substituted benzaldehyde derivatives. | ||
A series of poly(amide acetal)s was synthesized by Bernaerts and coworkers from succinic acid, adipic acid, suberic acid, sebacic acid, terephthalic acid, isophthalic acid and a diamine obtained from radical thiol–ene addition of cysteamine hydrochloride to 3,9-divinyl-2,4,8,10-tetraoxaspiro[5.5]undecane (Scheme 11).96 Using terephthalic acid and isophthalic acid as monomers resulted in low molecular weights and broad dispersities (Mn = 2.6 and 6.0 kg mol−1, Đ = 5.9 and 5.4, respectively), while succinic acid as a monomer resulted in end-capping via succinimide formation and consequently in low molecular weights as well (Mn = 2.5 kg mol−1, Đ = 2.0). On the other hand, the polymerization with adipic acid, suberic and sebacic acid as monomers resulted in high molecular weights of Mn = 13.5–20.0 kg mol−1 (Đ = 2.3–2.9). The thermal stability was typically good (Td,10% = 295–350 °C). The poly(amide acetal)s from aromatic diacids were amorphous and showed the highest Tgs (Tg = 82–84 °C), whereas all other poly(amide acetal)s were semi-crystalline. For the poly(amide acetal)s from adipic acid, suberic acid and sebacic acid, the Tg and the Tm decreased with increasing length of the alkyl chain of the dicarboxylic acid (Tg = 48, 42, 36 °C, Tm = 144, 133, 132 °C). The mechanical properties of the latter three poly(amide acetal)s were investigated as well, and as one might expect, both the Young's modulus (E = 0.98–1.52 GPa) and the yield stress (23.0–31.2 MPa) increased with decreasing length of the alkyl chain of the diacid, whereas the strain at break decreased (43.5–52.3%). Hydrolytic degradation studies in water at pH = 1, 3, 5, and 7 for two weeks showed water induced cold crystallization after one day, but no weight loss was observed. However, at pH = 1, SEC analysis revealed a decrease in molecular weight. Nevertheless, it could be concluded that the poly(amide acetal)s were stable at pH >3.
A diamine monomer was also obtained by hydrogenation using RANEY® Nickel as the catalyst from the corresponding bisnitrile acetal of pentaerythritol (Scheme 16).118 This diamine was used for example for the curing of epoxy resins.119,120 Moreover, it was combined with benzophenone-3,3,4,4-tetracarboxylic acid, or its ester or dianhydride, resulting in poly(acetal imide)s for electrically insulating varnishes.121 Poly(amide acetal)s of low molecular weight were also reported.122
 | ||
| Scheme 16 Synthesis of an acetal containing diamine monomer by hydrogenation of the corresponding bisnitrile. | ||
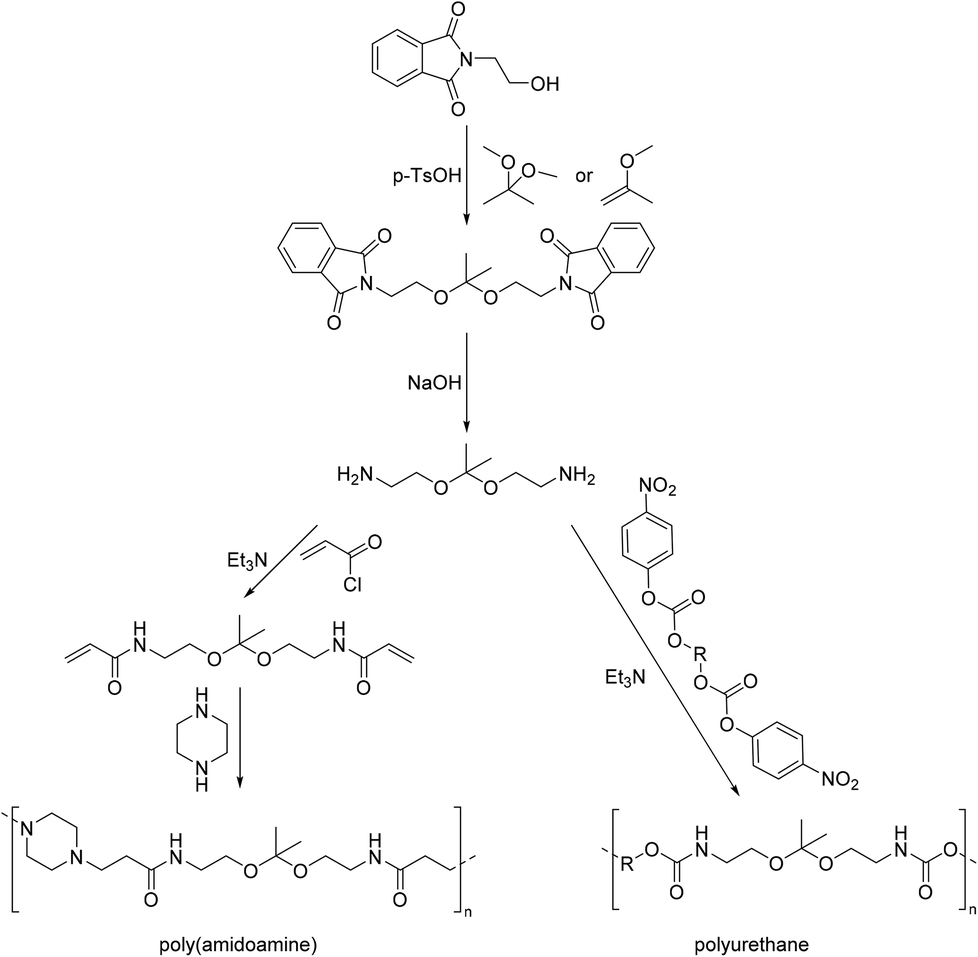 | ||
| Scheme 17 Exemplary synthesis of poly(amido amine)s and polyurethanes from linear acetal diamines. | ||
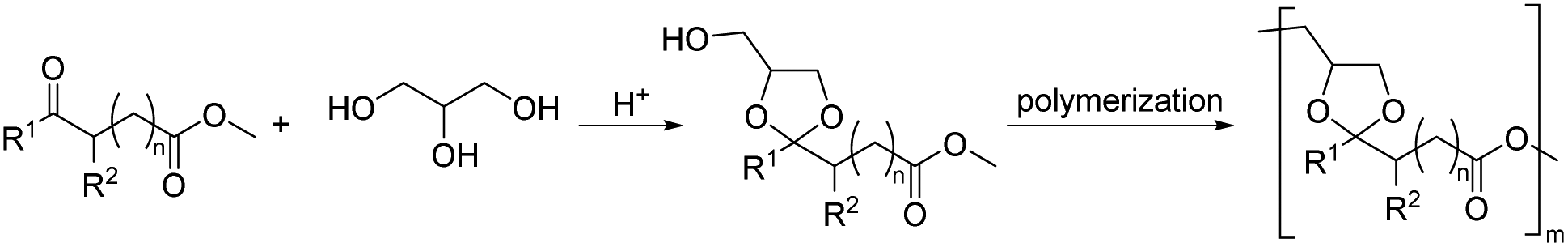 | ||
| Scheme 18 Polyesters from AB monomers containing cyclic acetal moieties synthesized from glycerol and (a) methyl azelaaldehydate (R1, R2 = H, n = 6) or (b) methyl levulinate (R1 = Me, R2 = H, n = 1) or (c) methyl 9- or 10-formylstearate (R1 = H, R2 = H3C(CH2)k, where n + k = 15 and n = 7 is methyl 9-formylstearate and n = 8 is methyl 10-formylstearate). | ||
For poly(ester acetals) from the glycerol acetal of methyl azelaaldehydate, several methods to synthesize the monomers and polymers were investigated.127,128 Reacting glycerol with the dimethyl acetal of methyl azelaaldehydate resulted in quantitative yield, whereas the direct acetalization of methyl azelaaldehydate with glycerol resulted in 65% yield of the AB monomer. This monomer was in fact a mixture of the five and six-membered cyclic acetal of glycerol. Lime as the catalyst resulted in the polymers with the highest molecular weights (Mn = 6 kg mol−1) as determined by osmometry. The semi-crystalline poly(ester acetal) showed a Tg of −20 to −25 °C, a Tm of 45–50 °C and good thermal stability (Td,10% = 350–380 °C). Excessive heating during the polymerization or using zinc acetate as the catalyst resulted in crosslinked material. The reverse synthetic approach was also investigated, where the solketal ester of azelaaldehydate dimethyl acetal was prepared first, which was then polymerized by transacetalization. However, this approach resulted in low molecular weight oligomers (Mn <1.5 kg mol−1). Other catalysts and reaction conditions were not investigated. However, interestingly, the monomer for the latter approach could not only be prepared by transesterification of solketal with methyl azelaaldehydate dimethyl acetal, but also by ozonolysis of glycerol oleate yielding the glycerol azelaaldehydate ester, followed by reaction with 2,2-dimethoxy propane to yield the solketal azelaaldehydate dimethyl acetal monomer.127,128
AB monomers from methyl azelaaldehydate were also synthesized with 1,2,6-hexanetriol, trimethylolethane, and trimethylolpropane.129 Interestingly, the five membered and the six membered cyclic acetals of glycerol and methyl azelaaldehydate were prepared selectively. Both the five membered cyclic acetal and the six membered cyclic acetal exist in a cis and a trans form, but purification on a preparative scale by column chromatography or fractional distillation and crystallization achieved separation of the cis and trans isomers only for the six membered cyclic acetal.130 Typical molecular weights after polycondensation were between 4 kg mol−1 and 10 kg mol−1 and most poly(ester acetal)s were semi-crystalline. The investigation of the thermal properties (Table 4) revealed that the poly(ester acetal)s from glycerol containing a five membered cyclic acetal linkage exhibit lower Tg and Tm than the poly(ester acetal) containing a six membered cyclic acetal of glycerol in the repeat unit. The latter poly(ester acetal) showed slightly different thermal properties depending on whether the cis- or the trans isomer of the AB monomer were polymerized and the trans isomers resulted in higher Tg and Tm. The poly(ester acetal)s based on trimethylolethane showed an intermediate Tg and Tm (Table 4), whereas the trimethylolethane based poly(ester acetal) was amorphous (Tg not reported).
| Triol | Chemical structure repeat unit | T g [°C] | T m [°C] |
|---|---|---|---|
| Glycerol |
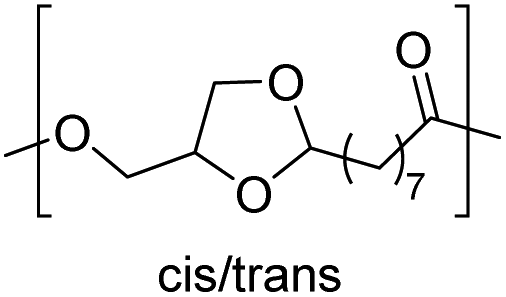
|
−35 | 54 |
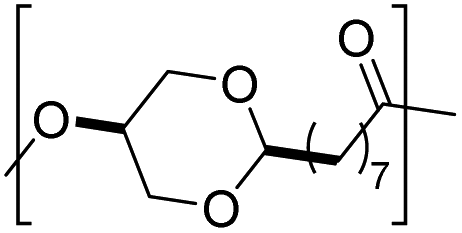
|
2 | 122 | |
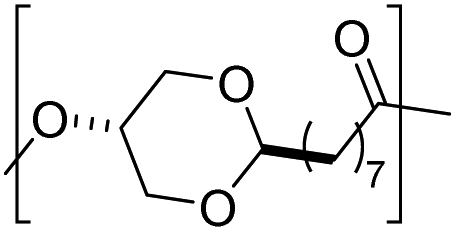
|
7 | 150 | |
| Trimethylol-ethane |
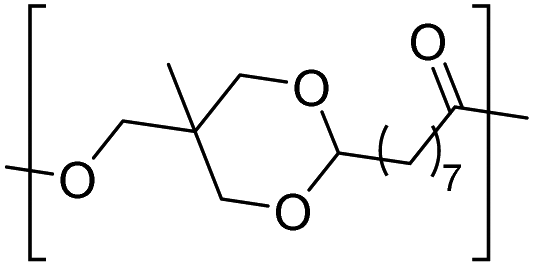
|
−26 | 81 |
Methyl azelaaldehydate was also exploited for the synthesis of dicarboxylic acid monomers by reaction with tetraols, such as diglycerol, pentaerythritol and 1,2,4,5-tetrakis(hydroxymethyl)benzene.129 These dicarboxylic acid monomers were subsequently used to synthesize poly(ester acetal)s with ethylene glycol or poly(amide acetal)s with hexamethylene diamine. In general, the diglycerol based monomer resulted in the lowest Tms of the polycondensates (Tm = 19 or 145 °C, respectively). The pentaerythritol based monomer resulted in the highest Tm for poly(ester acetal)s (Tm = 85 °C, Tm(poly(acetal amide)) = 186 °C), whereas the 1,2,4,5-tetrakis(hydroxymethyl)benzene based monomer resulted in the highest Tm for poly(acetal amide)s (Tm = 243 °C, Tm(poly(ester acetal)) = 42 °C). Several copolymers with PET and Nylon 66 were prepared as well. Finally, the poly(ester acetal)s were crosslinked by curing the polymers at T >200 °C through acetal interchange reactions, resulting in ring-opening of the cyclic acetal moieties. Lewis acid catalysts proved particularly active for crosslinking.131,132 Crosslinked copoly(ester acetal)s from the pentaerythritol acetal of methyl azelaaldehydate, diethylene glycol and dimethyl 1,4-cyclohexanedicarboxylate in combination with an acidic siliceous support were investigated as stationary phases for gas chromatography.133
The dimethyl acetal of a mixture of methyl 9- and 10-formylstearate was converted into an AB monomer with glycerol (Scheme 18), and a dicarboxylic acid monomer with pentaerythritol (Chart 7) in the same way as reported previously for the methyl azelaaldehydate.134 The glycerol acetal was polymerized by self-condensation (Mn = 1.5 kg mol−1) and by copolymerization with caprolactam (Mn = 2.3 kg mol−1), whereas the pentaerythritol based acetal was used in polycondensations with ethylene glycol (Mn = 7.2 kg mol−1), ethylene diamine (Mn = 6.2–11.6 kg mol−1), or hexamethylene diamine (Mn = 7.2–10.2 kg mol−1). The poly(ester acetal) from ethylene glycol and the poly(amide acetal) from ethylene diamine were also synthesized by a different approach. First the enol ether methyl methoxymethylstearate was prepared, which was then reacted with either ethylene glycol or ethylene diamine, followed by polyacetalization with pentaerythritol, but this method generally resulted in lower molecular weight of the final polymers. For the poly(ester acetal) with ethylene glycol, only unexpectedly low melting temperatures are reported (Tm = −34 to −40 °C). However, as expected, poly(amide acetal)s showed higher melting points irrespective of the diamine monomer (Tm = 48–56 °C) but low Tgs (Tg = −12 to −2 °C). For the self-condensate of the glycerol acetal of methyl formylstearate, a low Tm (thermomechanical analysis) was reported as well (Tm = −53 °C), whereas the caprolactam copolymer showed a higher Tm of 58 °C but again a low Tg of −34 °C. All polymers were thermally stable, and the 10% weight loss occurred in the range from 320–420 °C. Compared to the polymers from methyl azelaaldehydate, the polymers from methyl formylstearate showed better solubility. The latter polymers were also investigated for applications in chromatographic stationary phases.135 Another series of dicarboxylic acid ester monomers based on pentaerythritol acetals were reported as well and used, for example, in low molecular weight soluble poly(amide acetal)s for application as binders in printing inks (Chart 7).122
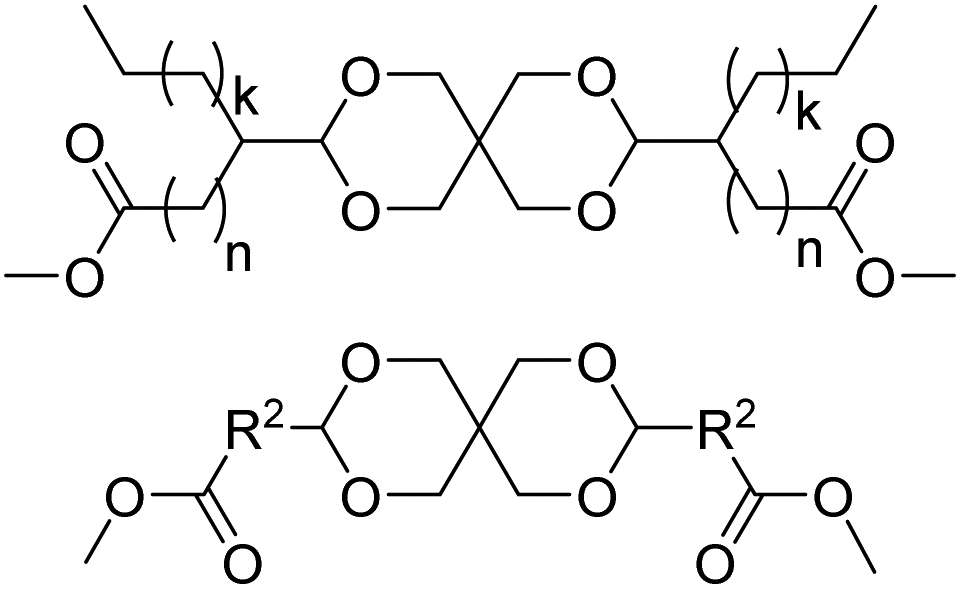 | ||
| Chart 7 Diester monomer from pentaerythritol and methyl 9(10) formylstearate (top, where n + k = 15, n = 7 is methyl 9-formylstearate, and n = 8 is methyl 10-formylstearate) and a series of other diester monomers (R2 = –(CH2)2–, –(CH(CH3))–, –(CH(CH3)CH2)–, –(C(CH3)2)–, –(CH2)2C(CH3)2–). | ||
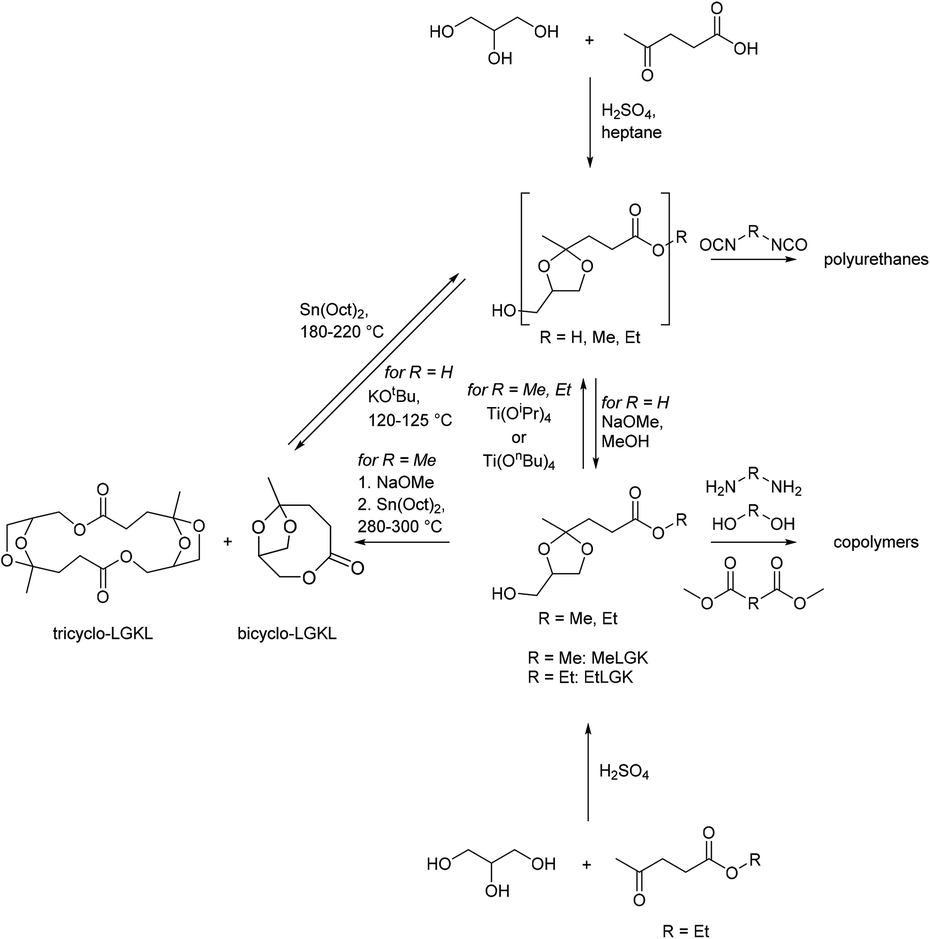 | ||
| Scheme 19 The monomer and polymer platform based on levulinic acid and glycerol. | ||
Other monomers based on acetals of polyols and keto carboxylic acids that have been reported can be grouped into bisacetals of two equivalents of levulinic acid and a tetraol (diglycerol (EtBLDK), pentaerythritol (EtBLPK), erythritol (EtBLEK), Chart 8), bisacetals of erythritol and a keto carboxylic acid ester (ethyl pyruvate (EtBPEK), ethyl acetoacetate (EtBAEK), Chart 8) and acetals of acetonides of sorbitol and keto carboxylic acids (diethyl 4-oxo-pimelate (EtPmSK), ethyl levulinate (EtBLSK), Chart 8).138–141 For bisacetals of two equivalents of levulinic acid and a tetraol, EtBLEK was most intensely investigated. For EtBLDK and EtBLPK poly(ester acetal)s with ethylene glycol and poly(acetal amide)s with hexamethylene diamine are reported.139 Amorphous polymers with Tgs in the range of 35–45 °C were obtained. Similar to EtLGK, EtBLEK was polymerized in combination with dicarboxylic acid monomers (DMT, dimethyl isophthalate, dimethyl adipate), aliphatic diols, aliphatic diamines, and hydroxy amines. The copolymers with DMT resulted in the highest Tgs, being 97 °C for a copoly(acetal amide) with DMT and hexamethylene diamine. EtBPEK and EtBAEK were polymerized with a combination of ethylene glycol, and hexamethylene diamine in a step-wise polymerization.141 An ethylene glycol poly(ester acetal) prepolymer was synthesized in the first step, which was then reacted with hexamethylene diamine in the second step. Low to moderate Tgs were reported (for EtBPEK: Tg = 7 °C, for EtBAEK: Tg = 37 °C). In all cases similar trends as previously discussed for EtLGK were observed regarding the molecular weights, i.e. for Mn >5–7.5 kg mol−1 unusually broad dispersities were observed. For example, for a poly(ester acetal) of EtBLEK and ethylene glycol, synthesized using Ti(OiPr)4 as the catalyst a dispersity of 15 was determined for Mn = 10.9 kg mol−1. For poly(acetal amide)s based on hexamethylene diamine and EtPmSK and EtBLSK, Tgs in the range of 65–80 °C were reported, whereas for the poly(ester acetal) from EtBLSK and ethylene glycol, a Tg of 17 °C was reported. Interestingly, the poly(amide acetal) from EtBLSK and hexamethylene diamine, was obtained with a comparatively high molecular weight (Mn = 12.6 kg mol−1) but a low dispersity was observed in this case (Đ = 1.61).140 In general, the acetal containing polymers based on polyols and keto carboxylic acids were investigated as plasticizers for commodity polymers (e.g. poly(vinyl chloride)), as prepolymers for polyurethane synthesis, and as compatibilizers. Although there is not yet a lot of information on the structure–property relationships and the polymerization procedure is not fully understood, these polymers may comprise a versatile and promising platform for the future based on renewable feedstock, especially since the starting materials are often already commercially available on a large scale.
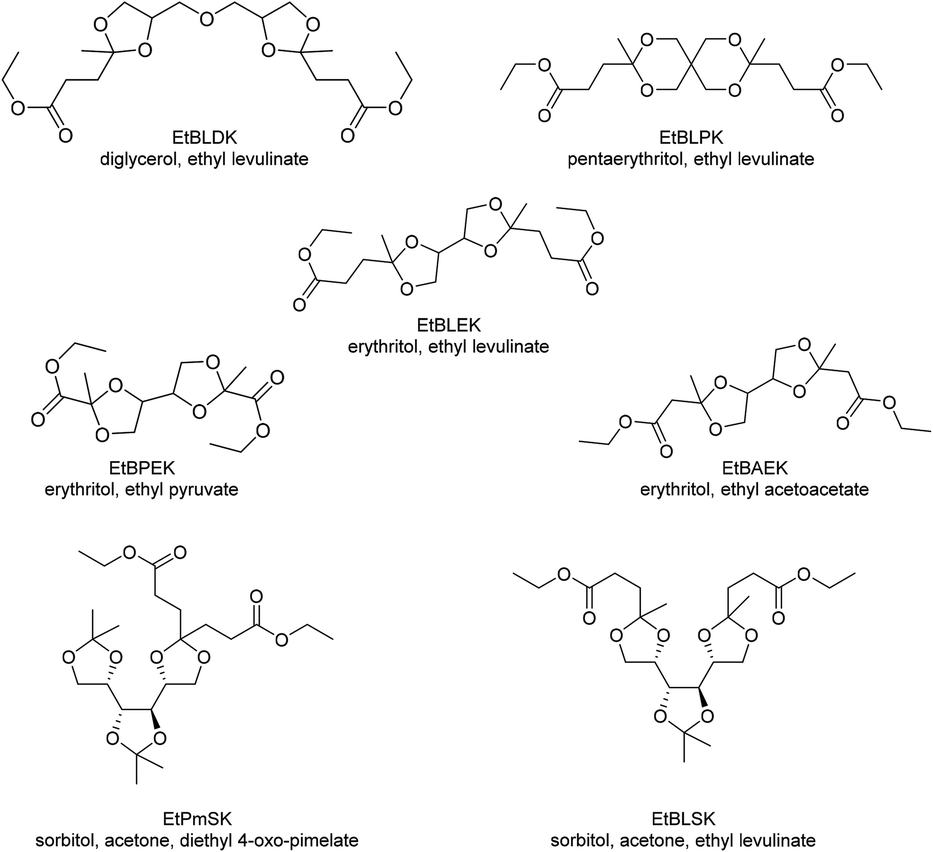 | ||
| Chart 8 Dicarboxylic acid ester monomers based on the combination of polyols with keto carboxylic acids. | ||
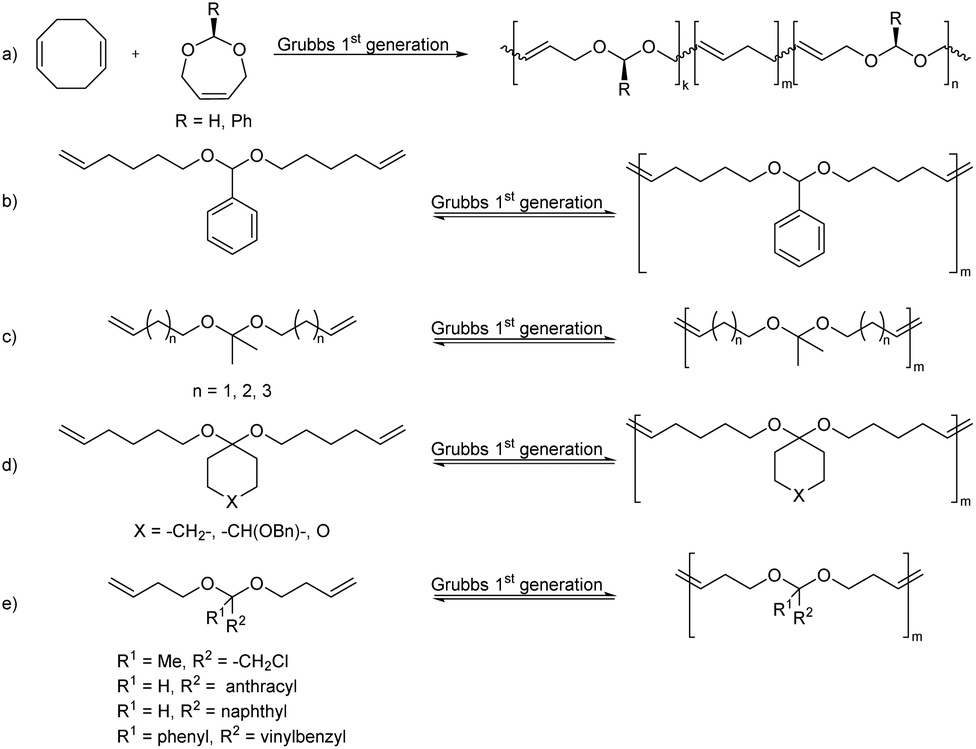 | ||
| Scheme 20 Polyacetal (co)polymers via ROMP (a) and ADMET (b–e) polymerization. | ||
Thus, 3,9-bischloromethyl-2,4,8,10-tetraoxaspiro[5.5]undecane was synthesized by transacetalization of two equivalents of chloroacetaldehyde dimethyl acetal with pentaerythritol (Scheme 21, R = H). A similar monomer was also synthesized from α-chloropropionaldehyde (Scheme 21, R = Me) and pentaerythritol.
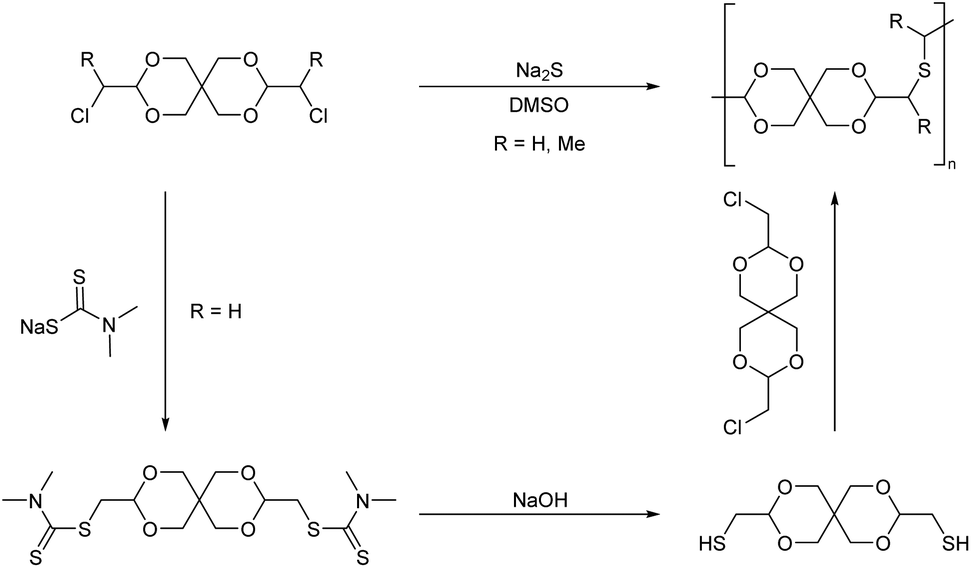 | ||
| Scheme 21 Synthesis of poly(thioether acetal)s via nucleophilic substitution reactions of pentaerythritol based dichloro and dithiol monomers. | ||
The polymerizations were performed by nucleophilic substitution reaction with sodium sulfide in DMSO at T >120 °C, for comparatively short reaction times (25 min–2 h). This resulted in oligomers (Mn = 1.3 kg mol−1) for the α-chloropropionaldehyde derived monomer. For the chloroacetaldehyde derived monomer, an alternative polymerization resulting in the same polymer backbone was investigated as well. The chloromethyl units of the monomer were first reacted with sodium N,N-dimethyldithiocarbamate, after which the resulting bis(dimethyldithiocarbamate) was hydrolyzed with sodium hydroxide to yield a bis(mercaptomethyl) monomer, which was then combined with the bis(chloromethyl) monomer in the presence of sodium hydroxide in DMSO. This polymer, prepared from the chloroacetaldehyde derived monomer, was soluble in chloroform and DCM at ambient temperature and gave solutions of higher viscosity than the polymer derived from α-chloropropionaldehyde, irrespective of the synthesis route, which could be explained by a lower reactivity due to increased steric hindrance of the latter monomer.
Wessig, Schlaad and co-workers also reported on poly(thioether acetal)s in the context of their work on oligospiroketal rods.146,147 However, in this case the authors pursued a thiol–ene addition polymerization strategy (Scheme 22) aiming at increasing the usually poor solubility of the rod-like structures by connecting them through structurally more flexible joints. Thus, a rod-like rigid dithiol monomer was combined with bismaleimides of varying flexibility by nucleophilic thiol–ene reactions. Interestingly, in all cases regardless of the flexibility of the bismaleimide, cyclic oligomers were formed as the main product as determined by NMR and Raman spectroscopy. The authors postulate that this strong tendency to cyclize, results from a folding of the rod-like units onto each other, thereby bringing the reactive end-groups into close proximity of each other. The formation of cyclic oligomers in polycycloacetal synthesis was not investigated previously but the observed phenomenon could in part explain the often low molecular weights that are reported for polyacetals comprising very rigid moieties.
 | ||
| Scheme 22 Nucleophilic thiol–ene polymerization of rigid dithiol monomers based on pentaerythritol bisacetals and bismaleimides. | ||
A nucleophilic Michael-type polyaddition reaction was also recently reported for the synthesis of dendritic polyacetals with a β-cyclodextrin core (Scheme 23).148 The cyclodextrin was first functionalized with methacryloyl units, followed by thia-Michael addition of cysteamine to these methacryloyl groups. The amino groups of cysteamine subsequently underwent aza-Michael additions with the acryloyl unit of an acryloyl/methacryloyl functional acetal of acetaldehyde. These dendritic units containing linear acetal units showed degradation at pH = 5 within a few hours and thus resulted in pH sensitive charge-reversible supramolecular amphiphiles based on host–guest chemistry for drug delivery applications.
 | ||
| Scheme 23 Synthesis of dendritic polyacetals by sequential thia-Michael and aza-Michael additions using an acryloyl/methacryloyl functional acetal of acetaldehyde. | ||
Polyacetals from paraformaldehyde
Following the pioneering reports from the 1960s, the utilization of (para)formaldehyde for the synthesis of copolymers has recently awakened again, since formaldehyde can be considered as a renewable C1 feedstock.149–152 Wolf and co-workers reported on the synthesis of polyols using a double metal cyanide catalyst to polymerize propylene oxide or ethylene oxide using paraformaldehyde as the initiator and in both the presence and absence of CO2.153Paraformaldehyde as the initiator results in ABA block-copolymer structures of poly(ether acetal)s or poly(ether carbonate acetal)s. The obtained block copolymers exhibited molecular weights in the range of Mn = 0.7–2.7 kg mol−1. The poly(ether acetal)s were stable against degradation up to a temperature of 310 °C, whereas the poly(ether carbonate acetal)s degraded at a lower temperature of 225 °C. For the paraformaldehyde-polypropylene oxide copolymer, a Tg of −60 °C is reported. Müller and co-workers employed a combination of Lewis acid and Cs2CO3 as a catalytic system for the repolymerization of paraformaldehyde in the presence of CO2 (30 bar).154 Incorporation of the solvent, 1,4-dioxane, in the anticipated poly(acetal carbonate)s structure was observed. The obtained oligomers (Mn <1.0 kg mol−1) were sensitive to thermal degradation, which occurred in two steps at 119 °C and 302 °C. However, acetylation of the hydroxyl end-groups improved the thermal stability of the oligomers, and the onset of degradation was observed at 232 °C for acetate terminated poly(acetal carbonate)s. While a low Tg = −60 °C was observed, the poly(carbonate acetal)s were semi-crystalline with two Tm at 35 and 120 °C, indicating different types of crystallites, which was attributed to the presence of long oxymethylene segments. The reactivity of the hydroxyl end-groups of the poly(carbonate acetal)s makes them potentially interesting for applications in polyol chemistry.
Ring opening polymerization
Cationic ROP of cyclic formaldehyde or acetaldehyde acetals was investigated intensely in the 1970s and 1980s using Lewis or Brønsted acid catalysis.155,156 The most commonly investigated monomers are 1,3-dioxolanes, but other cyclic monomers such as 1,3-dioxepanes or 1,3,6-trioxacyclooctane were studied as well.157,158 Since this process was reviewed elsewhere, especially with respect to the polymerization mechanism, we refrain from a more detailed discussion here.159–161 However, the drive toward (degradable) polymers from renewable monomers has resulted in an interest in ROP of 1,3-dioxolan-4-one (DOX) monomers in recent years (Scheme 24), which result in hemiacetal ester repeat units. The simplest DOX monomer is obtained from formaldehyde and glycolic acid, which can potentially be sourced from renewable C1 feedstock.162 Miller and co-workers reported the copolymerization of DOX with L-lactide via ROP. The main interest of the authors was the degradability of the resulting copolymers, and indeed the polymers were labile in both distilled water and in seawater. Hillmyer and co-workers investigated a similar monomer, the cyclic esteracetal 2-methyl-1,3-dioxane-4-one (MDO, Scheme 24), which can be obtained by reacting 3-hydroxypropionic acid with acetaldehyde, which both can be obtained from biomass.163 Alternatively, the MDO monomer is available by Baeyer–Villiger oxidation of 2-methyltetrahydrofuran-3-one.163 MDO was polymerized to yield the corresponding poly(hemiacetal ester) (PMDO) using diethylzinc as the catalyst at low catalyst/initiator ratios ([Zn(Et)2]/[BnOH] = 0.02/1). Interestingly, the authors observed that acetaldehyde could also be expelled during the polymerization, yielding a polyester (PHA) when using higher catalyst/initiator ratios ([Zn(Et)2]/[BnOH] = 2/1) and the polymerizations were studied in detail. This approach to polyesters was exploited further by Shaver and co-workers.164 For PDMO, molecular weights in the range of Mn = 7.6–30 kg mol−1 as determined by NMR are reported and the poly(hemiacetal ester) is an amorphous polymer with a low Tg, of −25 to −32 °C. On the other hand, PHA is a semi-crystalline polymer with a Tg in the same range as for PMDO (Tg(PHA) = −28 to −34 °C) and a Tm in the range of 59–77 °C. The molecular weights obtained for PHA were in the range of Mn = 8–43 kg mol−1. Whereas the PMDO poly(hemiacetal ester) is susceptible to hydrolytic degradation, especially at low pH values, the PHA polyester did not show a reduction in molecular weight during degradation studies (10.5 days). | ||
| Scheme 24 ROP approaches from 1,3-dioxolan-4-one (DOX) monomers to polyesters and poly(hemiacetal ester)s. | ||
Subsequently, the same authors investigated ROP of MDO using Brønsted acid catalysis.165 In this context, the authors also attempted to reproduce the results reported by Miller and co-workers162 on the co-polymerization of DOX and L-lactide. However, Hillmyer and co-workers concluded that 1H and 13C NMR data on the DOX monomer and the polymer in fact suggest that formaldehyde is expelled during the polymerization of DOX and L-lactide, resulting in a copolymer of glycolic acid L-lactide instead. It was highlighted that for ROP of MDO or DOX monomers, a careful investigation of the reaction conditions is crucial to avoid the elimination of the volatile carbonyl compounds. For ROP of MDO, using Brønsted acid catalysis by diphenylphosphoric acid, a higher activity of the catalyst as compared to diethyl zinc as the catalyst was observed. It was shown that the polymerization can proceed via both an activated monomer mechanism and an activated chain end mechanism, depending on the concentration of the initiator. High concentrations of the initiator (benzyl alcohol) favor polymerization via the activated monomer mechanism, whereas polymerization by the activated chain end mechanism occurs as well at low initiator concentrations (Scheme 24). Although the PDMO was thermally unstable (Td,5% = 121 °C), esterification of the polymer end-group increased the Td,5% to 146 °C. Based on the comparatively low degradation temperatures the author proposed that PDMO may be interesting for applications in lithography.
In another approach, ROP of bicyclic acetal monomers, which are available from epoxy ketones, was performed (Scheme 25).166,167 Similar polymerizations were reported in the 1970 in attempts to synthesize polysaccharide mimics.168–171 The epoxy ketone starting materials can be polymerized directly in the presence of Lewis acids, but this approach resulted in low to moderate molecular weight polymers (Mw ≤14.8 kg mol−1, Đ = 1.3–1.7).167 ROP of the bicyclic acetal with BF3·OEt2 as the catalyst however, resulted in polyacetals in very short reaction times (5–30 min) and moderate to good yields (24–71%).166 The highest molecular weights (Mn = 110–190 kg mol−1, Đ = 1.3–2.0) were obtained for the 1-methyl-2,7-dioxabicyclo[2.2.1]-heptane monomer (Scheme 24), but 1,4-dimethyl-2,7-dioxabicyclo[2.2.1]-heptane and 1-phenyl-2,7-dioxabicyclo[2.2.1]-heptane were successfully polymerized as well. The polymers showed Tgs of 23, 37 and 47 °C, respectively. For the polymer with phenyl substituents, a Tm of 144 °C was observed, but only in the first heating scan.
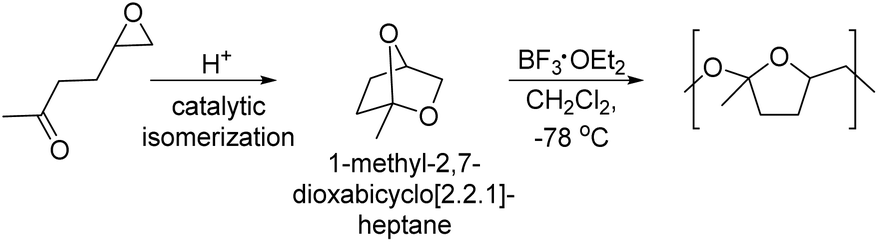 | ||
| Scheme 25 Polyacetals from epoxyketones via ROP of dioxabicyclo[2.2.1]-heptanes. | ||
Conclusions
Despite the long timeframe in which polyacetals have been discussed (>100 years) and the often-promising properties that have been observed depending on their exact structure, one can argue that polyacetals are a class of polymers that was not yet extensively investigated compared to other polycondensates. For example, a high thermal stability and good mechanical properties are often observed for polycycloacetals, whereas linear polyacetals show great promise in easily degradable materials. Yet, there is a limited number of reports focusing on a more detailed study regarding the understanding of how the structure–property relationships of these polymers affect their synthesis but also their properties. This includes for example the effect of isomerism (regioisomers, cis/trans isomers), the intra- and intermolecular interactions (such as folding), the stability or structural integrity of the acetal unit during polymerization with certain catalyst systems or at high temperatures, but also the stability or instability of the acetal backbone in the final materials against degradation.In recent years, there has been an increased interest in polyacetals, mainly because of their degradable nature and the high oxygen to carbon ratio, which often allows their synthesis from renewable building blocks. Despite the intensely investigated hydrolytic degradation of polyacetals, there is still a lack of information on their biodegradation. The interest in renewable polymers and the versatility of the polyacetals that we aimed to demonstrate in this review, in combination with an evolving understanding of their structure–property relationships, may well provide the basis for an increased impact of these polymers on our daily lives in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. L. acknowledges the Flanders Innovation & Entrepreneurship agency (VLAIO) for funding her PhD fellowship.References
- J. Read, J. Chem. Soc. Trans., 1912, 101, 2090 RSC.
- Polyoxymethylene Handbook: Structure, Properties, Applications and their Nanocomposites, ed. S. Lüftl, P. M. Visakh and S. Chandran, Scrivener Publishing LLC, Beverly, MA, 2014 Search PubMed.
- M. Haubs, K. Kurz and G. Sextro, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000 Search PubMed.
- M. L. Hallensleben, R. Fuss and F. Mummy, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2015 Search PubMed.
- M. Rostagno, S. Shen, I. Ghiviriga and S. A. Miller, Polym. Chem., 2017, 8, 5049 RSC.
- E. R. Gillies and J. M. J. Fréchet, Chem. Commun., 2003, 1640 RSC.
- E. E. Falco, M. Patel and J. P. Fisher, Pharm. Res., 2008, 25, 2348 CrossRef CAS.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082 RSC.
- S. Carmali and S. Brocchini, in Natural and Synthetic Biomedical Polymers, ed. S. Kumbar, C. Laurencin and M. Deng, Elsevier Inc., Burlington, MA, 1st edn, 2014, pp. 219–233 Search PubMed.
- G. Tillet, B. Boutevin and B. Ameduri, Prog. Polym. Sci., 2011, 36, 191 CrossRef CAS.
- S. Hou, D. M. Hoyle, C. J. Blackwell, K. Haernvall, V. Perz, G. M. Guebitz and E. Khosravi, Green Chem., 2016, 18, 5190 RSC.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Fréchet, J. Am. Chem. Soc., 2002, 124, 12398 CrossRef CAS.
- L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromol. Rapid Commun., 2008, 29, 123 CrossRef CAS.
- S. J. Lee, K. H. Min, H. J. Lee, A. N. Koo, H. P. Rim, B. J. Jeon, S. Y. Jeong, J. S. Heo and S. C. Lee, Biomacromolecules, 2011, 12, 1224 CrossRef CAS.
- H. Noh, H. J. Kim and S. K. Yang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3804 CrossRef CAS.
- D. N. Amato, D. V. Amato, O. V. Mavrodi, W. B. Martin, S. N. Swilley, K. H. Parsons, D. V. Mavrodi and D. L. Patton, ACS Macro Lett., 2017, 6, 171 CrossRef CAS.
- X. Hu, T. Yang, R. Gu, Y. Cui, C. Yuan, H. Ge, W. Wu, W. Li and Y. Chen, J. Mater. Chem. C, 2014, 2, 1836 RSC.
- H. Matsukizono and T. Endo, J. Am. Chem. Soc., 2018, 140, 884 CrossRef CAS.
- M. I. Papisov, A. Hiller, A. Yurkovetskiy, M. Yin, M. Barzana, S. Hillier and A. J. Fischman, Biomacromolecules, 2005, 6, 2659 CrossRef CAS.
- Mersana therapeutics, http://www.mersana.com/our-technology (01/08/2018).
- IUPAC. Compendium of Chemical Terminology, ed. A. D. McNaught and A. Wilkinson, Blackwell Scientific Publications, Oxford, 2nd edn, 1997 Search PubMed.
- Science of Synthesis: Houben−Weyl Methods of Molecular Transformations, ed. S. L. Warriner, Georg Thieme Verlag, Stuttgart, 2007 Search PubMed.
- G. J. F. Chittenden, Carbohydr. Res., 1983, 121, 316 CrossRef CAS.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306 CrossRef CAS.
- Y. Wang, H. Morinaga, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 596 CrossRef CAS.
- E. L. Kropa and W. M. Thomas, US Pat, 2643236, 1953 Search PubMed.
- S. M. Cohen and E. Lavin, US Pat, 2963464, 1960 Search PubMed.
- S. M. Cohen and E. Lavin, J. Appl. Polym. Sci., 1962, 6, 503–507 CrossRef CAS.
- S. M. Cohen, C. F. Hunt, R. E. Kass and A. H. Markhart, J. Appl. Polym. Sci., 1962, VI, 508–517 CrossRef.
- J. Cotting and A. Renner, EP Pat, 0297030, 1988 Search PubMed.
- H. B. Sonmez, F. G. Kuloglu, K. Karadag and F. Wudl, Polym. J., 2012, 44, 217–223 CrossRef CAS.
- G. Akbulut, H. B. Sonmez and F. Wudl, J. Polym. Res., 2013, 97 CrossRef.
- R. Kass, A. Markhart and E. Lavin, US Pat, 3063955, 1962 Search PubMed.
- A. Markhart, C. Hunt and S. Cohen, US Pat, 3048560, 1962 Search PubMed.
- A. Markhart, C. Hunt and E. Lavin, US Pat, 3015643, 1962 Search PubMed.
- C. E. Hollis, D. Faulkner and L. S. Abbott, GB Pat, 718502, 1954 Search PubMed.
- L. S. Abbott, D. Faulkner and C. E. Hollis, US Pat, 2739972, 1956 Search PubMed.
- J. Maślińska-Solich and S. Kukowka, Polym. Int., 2003, 52, 1633–1640 CrossRef.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214 RSC.
- B. Tollens and P. Wigand, Justus Liebigs Ann. Chem., 1891, 316–340 CrossRef CAS.
- H. B. J. Schurink, Org. Synth., 1925, 4, 53 CrossRef.
- Perstorp, VoxtarTM M100, https://www.perstorp.com/products/voxtar_m100 (01/08/2018).
- M. Rostagno, E. J. Price, A. G. Pemba, I. Ghiviriga, K. A. Abboud and S. A. Miller, J. Appl. Polym. Sci., 2016, 133, 44089 CrossRef.
- S. Lingier, S. Nevejans, P. Espeel, S. De Wildeman and F. E. Du Prez, Polymer, 2016, 103, 98 CrossRef CAS.
- W. J. Bailey and A. A. Volpe, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 2109 CrossRef CAS.
- A. T. Nielsen and W. R. Carpenter, Org. Synth., 1965, 45, 25 CrossRef CAS.
- W. J. Bailey, C. F. Beam, E. D. Cappuccilli, I. Haddad and A. A. Volpe, ACS Symp. Ser., 1982, 195, 391 CrossRef CAS.
- A. Akar and N. Talinli, Macromol. Rapid Commun., 1989, 10, 127 CrossRef CAS.
- S. Makhseed and N. B. McKeown, Chem. Commun., 1999, 88, 255 RSC.
- H. Tsutsumi, R. Shirotani, K. Onimura and T. Oishi, Electrochem. Solid-State Lett., 2001, 4, A195 CrossRef CAS.
- A. Sudo, T. Sano, M. Harada and D. Ishida, ACS Macro Lett., 2014, 3, 808 CrossRef CAS.
- N. Liu, J. Vignolle, J. M. Vincent, F. Robert, Y. Landais, H. Cramail and D. Taton, Macromolecules, 2014, 47, 1532 CrossRef CAS.
- S. Chatterjee and S. Ramakrishnan, Macromolecules, 2011, 44, 4658 CrossRef CAS.
- Société european research associates, BE Pat, 568181, 1958 Search PubMed.
- D. Capps, US Pat, 2889290, 1959 Search PubMed.
- Chemische Werke Hüls AG, GB Pat., 1041084, 1966 Search PubMed.
- M. J. Heffernan and N. Murthy, Bioconjugate Chem., 2005, 16, 1340 CrossRef CAS.
- Modified-release Drug Delivery Technology, ed. M. J. Rathbone, J. Hadgraft and M. S. Roberts, CRC Press, New York, 2003 Search PubMed.
- V. F. Fiore, M. C. Lofton, S. Roser-Page, S. C. Yang, J. Roman, N. Murthy and T. H. Barker, Biomaterials, 2010, 31, 810 CrossRef CAS PubMed.
- G. Seshadri, J. C. Sy, M. Brown, S. Dikalov, S. C. Yang, N. Murthy and M. E. Davis, Biomaterials, 2010, 31, 1372 CrossRef CAS.
- D. Chen and H. Wang, J. Nanosci. Nanotechnol., 2014, 14, 983 CrossRef CAS.
- S. Yang, M. Bhide, I. Crispe, R. Pierce and N. Murphy, Bioconjugate Chem., 2009, 19, 1164 CrossRef.
- M. J. Heffernan, S. P. Kasturi, S. C. Yang, B. Pulendran and N. Murthy, Biomaterials, 2009, 30, 910 CrossRef CAS.
- S. Lee, S. C. Yang, M. J. Heffernan, W. R. Taylor and N. Murthy, Bioconjugate Chem., 2007, 18, 4 CrossRef CAS.
- N. Murphy, B. Pulendran and R. Pierce, US Pat, 8252846, 2012 Search PubMed.
- R. W. Alder and B. S. R. Reddy, Polymer, 1994, 35, 5765 CrossRef CAS.
- N. G. Lemcoff and B. Fuchs, Org. Lett., 2002, 4, 731 CrossRef CAS.
- B. D. Mullen and E. J. Molitor, US Pat, 20150291721A1, 2015 Search PubMed.
- A. Hufendiek, S. Lingier, P. Espeel, S. De Wildeman and F. E. Du Prez, Polym. Chem., 2018, 9, 4789–4797 RSC.
- A. G. Pemba, J. A. Flores and S. A. Miller, Green Chem., 2013, 15, 325 RSC.
- S. A. Miller, ACS Macro Lett., 2013, 2, 550 CrossRef CAS.
- S. A. Miller and A. G. Pemba, US Pat, 9217058, 2015 Search PubMed.
- U.-A. Schaper, Synthesis, 1981, 794 CrossRef CAS.
- B. S. Rajput, S. R. Gaikwad, S. K. Menon and S. H. Chikkali, Green Chem., 2014, 16, 3810 RSC.
- B. S. Rajput, U. Chander, K. Arole, F. Stempfle, S. Menon, S. Mecking and S. H. Chikkali, Macromol. Chem. Phys., 2016, 217, 1396 CrossRef CAS.
- E. Sahmetlioglu, H. T. H. Nguyen, O. Nsengiyumva, E. Göktürk and S. A. Miller, ACS Macro Lett., 2016, 5, 466 CrossRef CAS.
- University of Strathclyde, FR Pat, 2336936, 1977 Search PubMed.
- J. Heller, D. W. H. Penhale and R. F. Helwing, J. Polym. Sci., Polym. Lett. Ed., 2018, 18, 293 CrossRef.
- R. Tomlinson, M. Klee, S. Garrett, J. Heller, R. Duncan and S. Brocchini, Macromolecules, 2002, 35, 473 CrossRef CAS.
- R. Tomlinson, J. Heller, S. Brocchini and R. Duncan, Bioconjugate Chem., 2003, 14, 1096 CrossRef CAS.
- E. Schacht, V. Toncheva, K. Vandertaelen and J. Heller, J. Controlled Release, 2006, 116, 219 CrossRef CAS.
- H. Otsuka and T. Endo, Macromolecules, 1999, 32, 9059 CrossRef CAS.
- J. R. Caldwell, R. Gilkey and B. S. Meeks, US Pat, 2945008, 1960 Search PubMed.
- Y. Masaoka and M. Kubo, GB Pat, 2162841, 1987 Search PubMed.
- H. Tanaka, S. Nishino and H. Takeuchi, US Pat, 6593419, 2003 Search PubMed.
- T. Hirokane, T. Oyama and S. Kuwahara, US Pat, 7179869, 2007 Search PubMed.
- M. Watanabe, J. Amemiya and I. Kuzuhara, US Pat, 9067950, 2015 Search PubMed.
- Mitsubishi Gas Chemical Company, Altester, https://www.mgc.co.jp/eng/products/nc/altester.html (01/08/2018).
- G. M. Rodriguez and S. Atsumi, Microb. Cell Fact., 2012, 11, 90 CrossRef CAS.
- S. Kaihara, S. Matsumura and J. P. Fisher, Macromolecules, 2007, 40, 7625 CrossRef CAS.
- K.-S. Lee, H. M. Kim and J. M. Rhee, Makromol. Chem., 1991, 192, 1033 CrossRef CAS.
- K.-S. Lee, J. M. Rhee, K.-Y. Choi, H.-M. Kim and S.-M. Lee, in Frontiers of Polymer Research, ed. P. N. Prasad and J. K. Nigam, Springer Science+Business Media, LLC, 1991, pp. 529–540 Search PubMed.
- K. S. Kim, S. M. Lee, K. C. Ryu and K. S. Lee, Polym. Bull., 1995, 35, 57 CrossRef CAS.
- S. Lingier, P. Espeel, S. S. Suarez, O. Türünç, S. De Wildeman and F. E. Du Prez, Eur. Polym. J., 2015, 70, 232 CrossRef CAS.
- BASF, Thermoplastic Polyurethane Elastomers, http://www.polyurethanes.basf.de/pu/solutions/en/function/conversions:/publish/content/group/Arbeitsgebiete_und_Produkte/Thermoplastische_Spezialelastomere/Infomaterial/elastollan_material_uk.pdf (01/08/2018).
- A. A. Wróblewska, S. Lingier, J. Noordijk, F. E. Du Prez, S. M. A. De Wildeman and K. V. Bernaerts, Eur. Polym. J., 2017, 96, 221 CrossRef.
- S. Selifonov, S. D. Rothstein and B. D. Mullen, US Pat, 8604223, 2013 Search PubMed.
- B. D. Mullen, M. D. Scholten, D. J. Yontz, C. M. Leibig and M. J. Tjosaas, US Pat, 9187598, 2015 Search PubMed.
- G. H. Choi, D. Y. Hwang and D. H. Suh, Macromolecules, 2015, 48, 6839 CrossRef CAS.
- J. E. Park, D. Y. Hwang, G. H. Choi, K. H. Choi and D. H. Suh, Biomacromolecules, 2017, 18, 2633 CrossRef CAS.
- S. Lingier, Y. Spiesschaert, B. Dhanis, S. De Wildeman and F. E. Du Prez, Macromolecules, 2017, 50, 5346 CrossRef CAS.
- X. Hu, T. Yang, R. Gu, Y. Cui, C. Yuan, H. Ge, W. Wu, W. Li and Y. Chen, J. Mater. Chem. C, 2014, 2, 1836 RSC.
- A. N. Zelikin and D. Putnam, Macromolecules, 2005, 38, 5532 CrossRef CAS.
- C. Lavilla, E. Gubbels, A. Mart, B. A. J. Noordover and C. E. Koning, Macromolecules, 2013, 46, 4335 CrossRef CAS.
- E. Gubbels, C. Lavilla, A. M. De Ilarduya, B. A. J. Noordover, C. E. Koning and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 164 CrossRef CAS.
- E. Zakharova, A. M. De Ilarduya, S. León and S. Muñoz-Guerra, Des. Monomers Polym., 2017, 20, 157 CrossRef CAS.
- S. Muñoz-Guerra, C. Lavilla, C. Japu and A. Martínez de Ilarduya, Green Chem., 2014, 16, 1716 RSC.
- S. Dhamaniya and J. Jacob, Polymer, 2010, 51, 5392 CrossRef CAS.
- E. Zakharova, C. Lavilla, A. Alla, A. Martínez De Ilarduya and S. Muñoz-Guerra, Eur. Polym. J., 2014, 61, 263 CrossRef CAS.
- C. Japu, A. Martínez De Ilarduya, A. Alla and S. Muñoz-Guerra, Polymer, 2014, 55, 2294 CrossRef CAS.
- A. A. Wróblewska, K. V. Bernaerts and S. M. A. De Wildeman, Polymer, 2017, 124, 252 CrossRef.
- A. A. Wróblewska, J. Noordijk, N. Das, C. Gerards, S. M. A. De Wildeman and K. V. Bernaerts, Macromol. Rapid Commun., 2018, 39 DOI:10.1002/marc.201800077.
- A. Sudo, Y. Shibata and A. Miyamoto, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3956 CrossRef CAS.
- A. Yoshida and A. Sudo, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3798 CrossRef CAS.
- M. M. Koton, T. M. Kiseleva, N. P. Kuznetsov, S. N. Nikolaeva and Y. N. Sazanov, Vysokomol. Soedin., Ser. B, 1976, 18, 456 CAS.
- J.-B. Lee, M.-S. Bang, K.-S. Kim, K.-S. Lee and S.-M. Lee, Polymer, 1996, 20, 701 CAS.
- S.-D. Han, B.-K. So, S.-H. Lee and S.-M. Lee, Polymer, 1997, 21, 913 CAS.
- K. Iijima, Y. Komachiya and E. Negoro, US Pat, 3609169, 1971 Search PubMed.
- I. Yoshimura, N. Fukue, H. Sakamoto, H. Murofushi and T. Hiyama, US Pat, 3679707, 1972 Search PubMed.
- I. Yoshimura, N. Fukue, H. Sakamoto, H. Murofushi and T. Hiyama, US Pat, 3773790, 1973 Search PubMed.
- M. Suzuki, E. Hosokawa and M. Waki, US Pat, 3634304, 1972 Search PubMed.
- H. Tashiro, US Pat, 3597376, 1971 Search PubMed.
- R. Jain, S. M. Standley and J. M. J. Fréchet, Macromolecules, 2007, 40, 452 CrossRef CAS.
- S. E. Paramonov, E. M. Bachelder, T. T. Beaudette, S. M. Standley, C. C. Lee, J. Dashe and J. M. J. Fréchet, Bioconjugate Chem., 2008, 19, 911 CrossRef CAS.
- E. M. Bachelder, T. T. Beaudette, K. E. Broaders, S. E. Paramonov, J. Dashe and J. M. J. Fréchet, Mol. Pharm., 2008, 5, 876 CrossRef CAS.
- J. L. Jensen, L. R. Herold, P. A. Lenz, S. Trusty, V. Sergi, K. Bell and P. Rogers, J. Am. Chem. Soc., 1979, 101, 4672 CrossRef CAS.
- W. R. Miller, J. C. Cowan and E. H. Pryde, US Pat, 3287326, 1966 Search PubMed.
- W. R. Miller, R. A. Awl, E. H. Pryde and J. C. Cowan, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 415 CrossRef CAS.
- R. W. Lenz, Macromolecules, 1969, 2, 129 CrossRef CAS.
- R. W. Lenz, R. A. Awl, W. R. Miller and E. H. Pryde, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 429 CrossRef CAS.
- E. H. Pryde, US Pat, 3183215, 1965 Search PubMed.
- E. H. Pryde, US Pat, 3223683, 1965 Search PubMed.
- W. E. Neff, E. H. Pryde, E. Selke and J. C. Cowan, J. Chromatogr. Sci., 1972, 512–517 CrossRef CAS.
- R. A. Awl, W. E. Neff, D. Welsleder and E. H. Pryde, J. Am. Oil Chem. Soc., 1976, 9, 20–26 CrossRef.
- W. E. Neff, R. A. Awl, E. H. Pryde and J. C. Cowan, J. Am. Oil Chem. Soc., 1973, 50, 235–239 CrossRef CAS.
- S. Selifonov, US Pat, 8178701, 2012 Search PubMed.
- A. S. Amarasekara and S. A. Hawkins, Eur. Polym. J., 2011, 47, 2451–2457 CrossRef CAS.
- S. Selifonov, S. D. Rothstein, D. A. Wicks, B. D. Mullen, T. J. Mullen, J. D. Pratt, C. T. Williams and C. Wu, CA Pat, 2674778, 2009 Search PubMed.
- S. Selifonov, S. D. Rothstein, D. A. Wicks, B. D. Mullen, T. J. Mullen, J. D. Pratt, C. T. Williams, C. Wu and N. Zhou, US Pat, 8546519, 2013 Search PubMed.
- S. Selifonov, A. E. Goetz and F. Jing, US Pat, 8653223, 2014 Search PubMed.
- S. Selifonov, A. E. Goetz, M. D. Scholten and N. Zhou, US8969559, 2015 Search PubMed.
- P. S. Wolfe and K. B. Wagener, Macromol. Rapid Commun., 1998, 19, 305 CrossRef CAS.
- S. D. Khaja, S. Lee and N. Murthy, Biomacromolecules, 2007, 8, 1391 CrossRef CAS.
- C. Fraser, M. A. Hillmyer, E. Gutierrez and R. H. Grubbs, Macromolecules, 1995, 28, 7256 CrossRef CAS.
- F. V. Zalar, Macromolecules, 1972, 5, 539 CrossRef CAS.
- P. Wessig and K. Möllnitz, J. Org. Chem., 2012, 77, 3907 CrossRef CAS.
- P. Wessig, T. Schulze, A. Pfennig, S. M. Weidner, S. Prentzel and H. Schlaad, Polym. Chem., 2017, 8, 6879 RSC.
- D. Huang, Y. Wang, F. Yang, H. Shen, Z. Weng and D. Wu, Polym. Chem., 2017, 8, 6675 RSC.
- W. R. Grace & Co., GB Pat., 960744, 1962 Search PubMed.
- K. Noro, H. Kawazura, T. Moriyama and S. Yoshioka, Makromol. Chem., 1965, 83, 35 CrossRef CAS.
- W. E. Smith, F. R. Galiano, D. Rankin and G. J. Mantell, J. Appl. Polym. Sci., 1966, 10, 1659 CrossRef CAS.
- T. Kagiya, K. Narita and M. Kondo, Bull. Chem. Soc. Jpn., 1969, 42, 3211–3215 CrossRef CAS.
- J. Langanke, J. Hofmann, C. Gürtler and A. Wolf, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2071 CrossRef CAS.
- C. Bizzarri, H. Vogt, G. Baráth, C. Gürtler, W. Leitner and T. E. Müller, Green Chem., 2016, 18, 5160 RSC.
- Z. Jedliński, J. Lukaszczyk, J. Dudek and M. Gibas, Macromolecules, 1975, 9, 622 CrossRef.
- Z. Jedliński and J. Lukaszczyk, Macromolecules, 1975, 8, 700 CrossRef.
- L. C. Reibel, C. P. Durand and E. Franta, Can. J. Chem., 1985, 63, 264 CrossRef CAS.
- R. S. Velichkova, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 2561–2568 CrossRef CAS.
- S. Penczek, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1919 CrossRef CAS.
- R. Szymański, P. Kubisa and S. Penczek, Macromolecules, 1983, 16, 1000 CrossRef.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Słomkowski, in Controlled and Living Polymerizations, ed. A. H. E. Müller and K. Matyjaszewski, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009, pp. 241–296 Search PubMed.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768 RSC.
- A. E. Neitzel, M. A. Petersen, E. Kokkoli and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 1156 CrossRef CAS.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990 RSC.
- A. E. Neitzel, T. J. Haversang and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11747 CrossRef CAS.
- B. T. Whiting and G. W. Coates, J. Am. Chem. Soc., 2013, 135, 10974 CrossRef CAS.
- M. E. Benz and L. L. Luo, US Pat, 7741375, 2007 Search PubMed.
- C. Schuerch, Acc. Chem. Res., 1973, 6, 184 CrossRef CAS.
- H. K. Hall, L. J. Carr, R. Kellman and F. De Blauwe, J. Am. Chem. Soc., 1974, 96, 7265 CrossRef CAS.
- H. K. Hall and F. De Blauwe, J. Am. Chem. Soc., 1975, 97, 655 CrossRef CAS.
- S. Penczek, Ed., in Models of Biopolymers by Ring-Opening Polymerization, CRC Press, Inc., Boca Raton, Florida, 2000 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |