
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Giant polymersomes from non-assisted film hydration of phosphate-based block copolymers†
Emeline
Rideau
 ,
Frederik R.
Wurm
* and
Katharina
Landfester
*
,
Frederik R.
Wurm
* and
Katharina
Landfester
*
Max-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
First published on 11th October 2018
Abstract
The self-assembly of amphiphilic block copolymers is a fast way to prepare chemically versatile and stable “protocells” that can act as a reactor or a confinement. However, controlling their self-assembly into giant unilamellar vesicles (GUVs) with diameters of several micrometers is challenging. Electroformation has been used to generate GUVs from amphiphilic block copolymers, which can be studied by light microscopy and resemble cell-like entities. However, a mild film hydration protocol for GUV preparation would be desirable in order to prepare libraries of protocells for further applications. Here, we present the self-assembly of novel amphiphilic polybutadiene-block-polyphosphoester block copolymers into GUVs by simple film hydration. These amphiphiles are synthetic analogs of phospholipids and possess the hydrophilic poly(ethylene ethyl phosphate) (PEEP) block. The GUVs (with diameters of ca. 10–40 μm) were formed in high yields by simple non-assisted film hydration requiring no external forces and with no need of the commonly applied electroformation. PEEP-based block copolymers with a lamellar bulk morphology produced GUVs in high yields and outperformed commonly used block copolymers (e.g. with poly(ethylene oxide) as a hydrophilic segment). We quantified their respective yield (number of GUVs formed) and diameters and monitored their stability over time. In addition, we proved their encapsulation capacity and permeability to hydrophobic and hydrophilic fluorescent cargo. Due to their high performance, these phosphate-based amphiphilic block copolymers are promising candidates for the generation of protocells and self-assembled microreactors.
Introduction
Compartmentalization of cells is a key feature of life.1,2 The plasma membrane is composed of a complex, balanced ratio of lipids, proteins, and small molecules and has many functions.3 Cell membrane mimicking has become an important quest for simplifying the understanding of the membrane's inherent properties, functions and behaviors4–6 as well as for using their biocompatibility to expand drugs’ bioavailability, medical imaging and diagnostics7–9 or to compartmentalize incompatible entities in the synthesis.2,10Cell membrane mimicking was originally achieved with liposomes, and despite their resemblance to cell membranes, these vesicles are difficult to use and specialize, as they are unstable, fluid, and permeable.10–13 More recently, polymeric vesicles (polymersomes) have gained in popularity, as block copolymers are chemically more versatile, malleable, and tougher than lipids, resulting in easily functionalizable, more stable vesicles.11,13,14 Classically, polymersomes are generated from commercially available block copolymers and almost always using poly(ethylene oxide) (PEO) as the hydrophilic block.15 A shortcoming of polymersomes is their lower biocompatibility and mimicry of cell membranes compared to liposomes as they are constituted entirely of synthetic entities.13 In this study, we propose a novel block copolymer, namely polybutadiene-b-poly(ethylene ethyl phosphate) (PB-b-PEEP). The EEP block is interesting as its phosphate moiety resembles natural phospholipids and is biodegradable, bridging the gap between liposomes and polymersomes.16,17 Despite this advantage, polymersomes bearing phosphate moieties are rare.18,19 PB is also commonly used as the hydrophobic block in polymersomes as it has a low glass transition temperature (Tg) (Tg ≈ −21 °C for Mn = 105k; Tg ≈ −77 °C for Mn = 50k).20 Low Tg materials are desirable, as they are flexible under the self-assembly conditions (room temperature or above) and thus are able to mimic the fluidity of bio-membranes contrary to more rigid hydrophobic blocks like polystyrene.13,21,22
The vast majority of studies on polymersomes focus on small vesicles of ca. 100 nm diameters, the so-called small or large unilamellar vesicles (SUVs and LUVs, respectively) as they are readily achievable by multiple methodologies.23 However, cells are much larger (∼10–100 μm) and giant unilamellar vesicles (GUVs) (>1 μm) are thus better mimics than SUVs.24 As increasing evidence suggests that factors such as the membrane curvature, effective encapsulated volume, stability, and permeability differ depending on size, efficient formation of GUVs becomes necessary.25,26
Polymeric-GUVs are more challenging to obtain than SUVs and are often generated with microfluidic devices. This controlled water-in-oil-in-water double emulsion technique selectively forms vesicles of any size in very low polydispersity.20,27–29 However, this solvent displacement method requires complex mixtures of additives, which can be difficult to adapt to new conditions and contaminate the vesicles (e.g. the remaining solvent, surfactants, and additives in the membrane or in the lumen), significantly changing the membrane properties.27,30–32 Solvent- and additive-free methodologies generating GUVs are still desirable for the robust and high-yield formation and encapsulation. Film hydration methods are based on the initial formation of a thin layer of the amphiphile on a surface by solvent evaporation followed by hydration of this solvent-free film.14,20,23 It is generally accepted that simple hydration of amphiphilic block copolymer films does not result in polymersome formation, and especially no GUVs are obtained by this procedure.24,31,33,34 Water cannot penetrate the dry polymer film to induce the self-assembly. Forces are required to enhance the film hydration of amphiphilic block copolymers or lipids. Commonly, shear forces like sonicating or stirring lead to SUVs or LUVs and alternative current (AC) or the use of swelling hydrogel substrates to GUVs.31,34–37
The exact mechanism behind the effect of AC on vesicle formation is not well understood. Despite the success of the so-called electroformation, that is, the AC-aided film hydration, for liposomes, this technique has been used only in a few studies for assembling polymeric-GUVs.35,38–41 In the case of hydrogel-mediated hydration, the amphiphilic film is formed on a pre-dehydrated hydrophilic polymer or gel (such as poly(vinyl alcohol) or agarose). Hydration of the gel causes deformation of the amphiphilic film as a driving force for the formation of vesicles. Hydrogel-mediated polymersome formation has only been rarely described42 and can also cause undesired membrane alteration.31 Therefore, since the initial report of polymeric GUVs in 1999,35 there is a clear niche for methods to form solvent and additive-free polymeric GUVs.
In this study, we first generated a library of amphiphilic PB-b-PEEP by sequential anionic polymerization. Then, we describe how with the appropriate block ratio PB-b-PEEP can generate GUVs by electroformation and even by spontaneous non-assisted direct hydration of their film within only 1 h. We quantified their yield and mean diameter, examine their stability in terms of number and size evolution over a month, and finally their encapsulation capacity to hydrophobic and hydrophilic fluorescent dyes. All these factors proved the polyphosphate-based block copolymers to be efficient amphiphiles for polymersome formation and encapsulation, superior to the commonly used block copolymers.
Results and discussion
PB-b-PEEP synthesis
PB-b-PEEP block copolymers were synthesized by sequential anionic polymerization (Scheme 1). The first step was the anionic polymerization of 1,3-butadiene, initialized by organolithium reagents and end-capped with ethylene oxide (EO) to yield a hydroxyl-functionalized PB-macroinitiator (PB-OH) (a).43,44 Preferential 1,2- or 1,4-polymerization can be achieved by using THF or cyclohexane, respectively. With cyclohexane, we obtained PB-OH with 92% 1,4-microstructure (PB(1,4)-OH) (Fig. 1ii) with a low molar mass dispersity (Đ = 1.06) (Fig. 1i). The degree of polymerization of PB-OH was determined by NMR with reference to the methyl end-groups at 0.87 ppm (Fig. 1ii).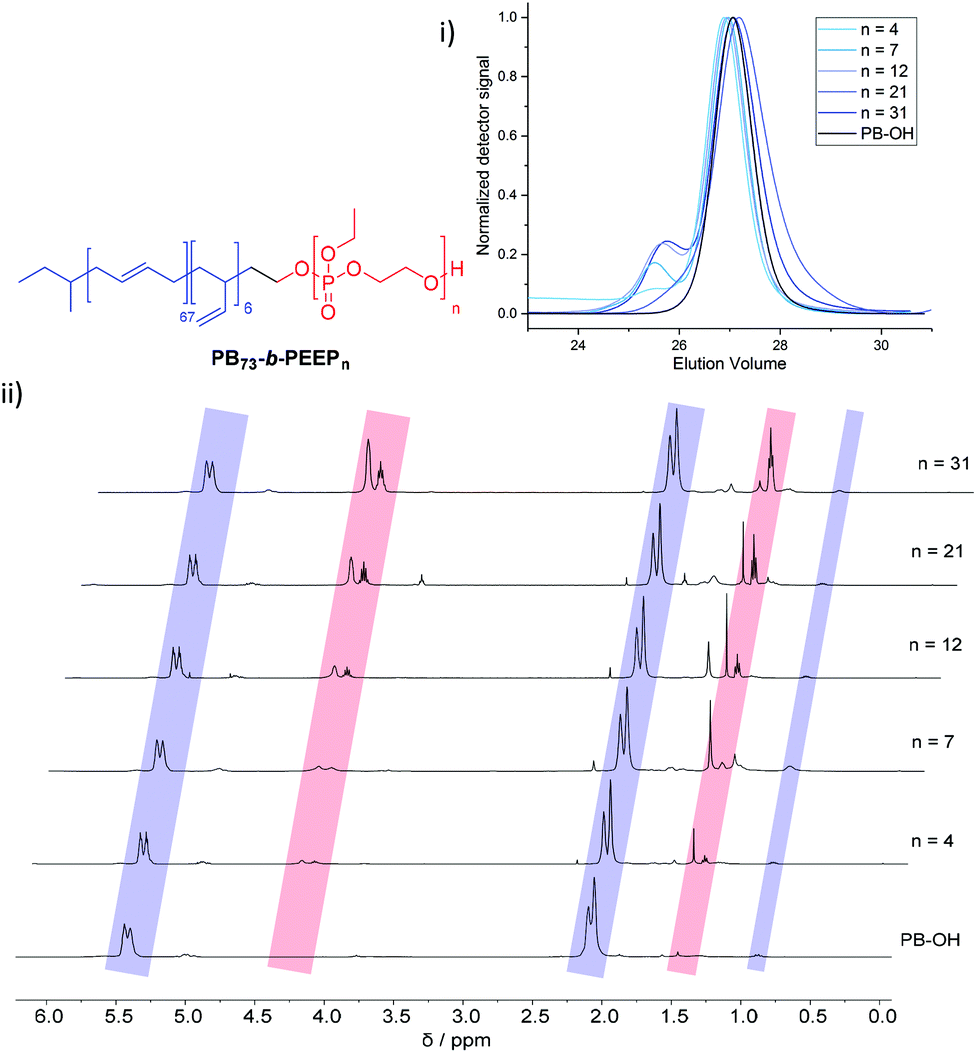 | ||
| Fig. 1 Stacked GPC curves: (i) 1H NMR spectra and (ii) of PB-OH and PB73-b-PEEPn with the corresponding signal assignments. | ||
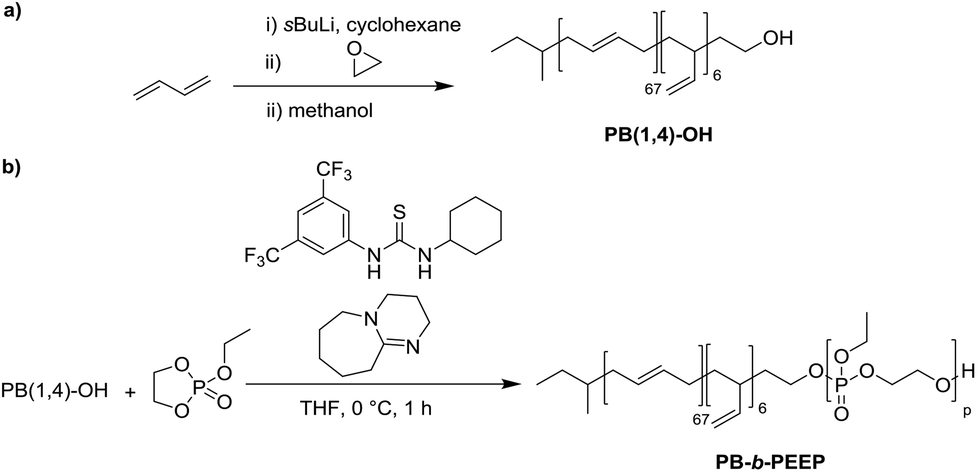 | ||
| Scheme 1 Synthesis of polybutadiene-b-poly(ethylene ethyl phosphate) (PB-b-PEEP). (a) Initial living anionic polymerization of butadiene to generate hydroxyl terminated 1,4-rich polybutadiene (PB(1,4)-OH). (b) Organocatalyzed anionic ring-opening polymerization of ethylene ethyl phosphate to the amphiphilic block copolymers PB-b-PEEP. | ||
PB(1,4)-OH was then used to polymerize ethyl ethylene phosphate (EEP) in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base to activate the macroinitiator and a thiourea cocatalyst (b).16 These additives reduce side reactions such as the transesterification of the EEP moiety.16 Transesterification could still be observed as a small shoulder in the GPC curves at lower elution volume than the desired block copolymer (Fig. 1i). The shoulder appeared more prominent when targeting a higher degree of polymerization. Despite the transesterification side-reaction, all block copolymers were obtained with a narrow molar mass dispersity Đ (Đ < 1.2) (Fig. 1i).
In comparison, classically used amphiphilic block copolymers consisting of a hydrophilic PEO block are synthesized by anionic ring-opening polymerization of the gas ethylene oxide (EO) at elevated temperature, high pressure, overnight onto the hydrophobic macroinitiator, such as PB-OH for PB-b-PEO.43 However, EO is a carcinogenic, colorless, flammable gas and special care has to be taken when handled in the lab.45 Therefore, the synthesis of PB-b-PEEPs is simpler, faster, and less toxic than that of PB-b-PEOs.
We generated a library of PB-b-PEEPs with a range of hydrophilic fractions f (f = Mn(hydrophilic block)/Mn(block copolymer)) (Fig. 1ii B–F and Table 1). The degree of hydrophilicity f of amphiphilic block copolymers is an important property as it determines which macromolecular self-assembly is entropically favored.10,15,46,47 The nature of the polymer blocks and f determines their self-assembly morphologies (micelles, cylindrical micelles (worms), reverse micelles, lamellae, vesicles, etc.). As a general rule 0.25 < f < 0.45 yields polymersomes11,48 and our library is well within these boundaries. The f values of each block copolymer were determined by comparing their PB(1,4) signal at 5.39 ppm (Fig. 1ii) already established for the PB-OH macroinitiator with the CH2–O signals at 4.34–4.10 ppm (Fig. 1ii) of the PEEP block. These values were then used to determine the respective Mn of each block copolymer.
| Entry | Polymera | f |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Đ |
|---|---|---|---|---|
| a Degree of polymerization and Mn were determined by NMR. b Hydrophilic fraction defined as f = Mn(hydrophilic block)/Mn(block copolymer). c Đ, the molar mass dispersity, was determined by GPC. | ||||
| 1 | PB(1,4)73-b-PEEP4 | 0.13 | 5000 | 1.07 |
| 2 | PB(1,4)73-b-PEEP7 | 0.21 | 5000 | 1.13 |
| 3 | PB(1,4)73-b-PEEP12 | 0.32 | 6000 | 1.14 |
| 4 | PB(1,4)73-b-PEEP21 | 0.45 | 7000 | 1.19 |
| 5 | PB(1,4)73-b-PEEP31 | 0.54 | 9000 | 1.17 |
PB-b-PEEP self-assembly into GUVs by electroformation
Using our homemade electro-chamber with Pt wires (Fig. 2) we tested our library of PB-b-PEEPs for GUV formation by electroformation (EF) following a modified method of Discher (see ESI† pS22 for details).35 PB73-b-PEEP12A (entry 3) and PB73-b-PEEP21B (entry 4) gave GUVs in high yields. Other ratios outside these boundaries yielded no vesicles. Therefore, PB-b-PEEPs behave similarly to other classical amphiphilic block copolymers, although they appear to favor slightly above average f for vesicles as PB73-b-PEEP7 (f = 0.21) did not self-assemble into GUVs while PB73-b-PEEP21B (f = 0.45) did.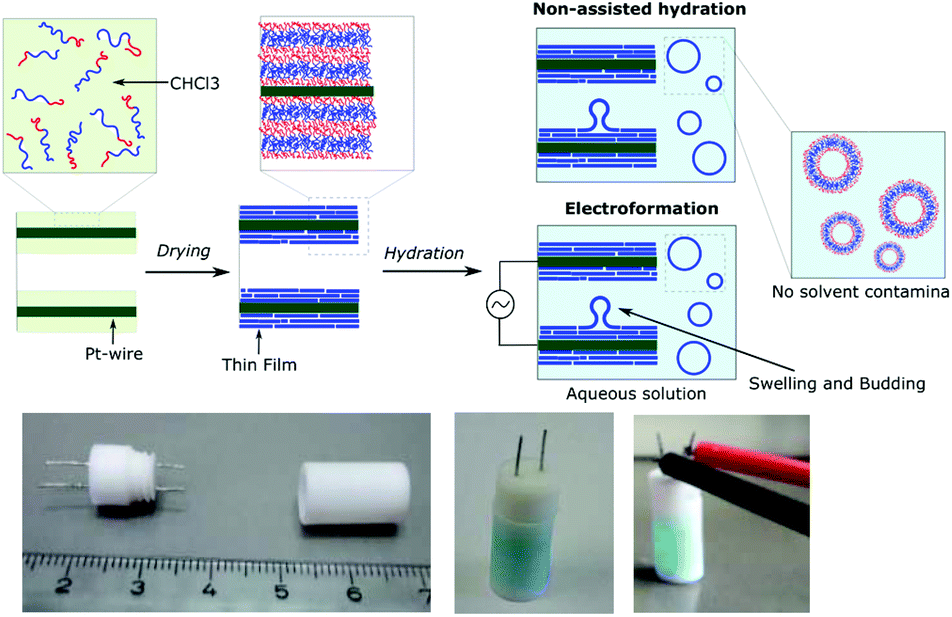 | ||
| Fig. 2 Electrochambers and electroformation methodology. (a) Scheme of non-assisted film hydration and electroformation methods. (b) Picture of the open homemade electro-chamber (left), the closed chamber after filling here with aqueous sucrose solution doped with AF647 as used in the non-assisted film hydration method (middle) and then connected to the generator for electroformation (right). | ||
Most surprisingly, control experiments showed that the same polymers (PB73-b-PEEP12A and PB73-b-PEEP21B) could also spontaneously self-assemble into GUVs in the absence of an alternating current within the same time period (1 h) on Pt-wires and on glass slides. All the other PB-b-PEEPs did not self-assemble into GUVs under these conditions. Non-assisted film hydration in such a fast timescale to form GUVs has never been reported before. Even in the case of lipidic GUVs, gentle hydration has only rarely been described as it requires long swelling times (typically several hours to days), is highly sensitive to any form of agitation, is unsuccessful for many amphiphiles and forms multilamellar deformed vesicles.38,49,50 For polymersomes, reports even state that they have high energy requirements towards their self-assembly.51 Control experiments for the formation of GUVs involving electroformation have not been explicitly described in previous studies.35,40,41 Dimova et al. reported that the time required to form GUVs is much longer (3 h) at lower voltage (800 mV) and this resulted in smaller vesicles than those for 15 min at 9 V yielding GUVs of 40 μm radius on average.38 The authors also showed that simple swelling, on Teflon surfaces, of PB-b-PEO and PEE-b-PEO took 3 days and resulted in smaller vesicles. Therefore, our fast non-assisted film hydration of PB-b-PEEPs into GUVs is unprecedented.
In order to compare the GUV formation between non-assisted film hydration (na-FH) and electroformation (EF), we quantified the yield (the number of GUVs formed) and their mean diameters. In analogy to the well-established standard mammalian cell counting methods using a hemocytometer,52 we manually counted the vesicles present in a number of random locations at the bottom of the well from microscopy images at a magnification of 20×. A magnification of 20× allows the counting to be done on an area of 6.4 × 105 μm2 (divided into 16 squares of 200 μm length to ease counting – ESI† pS23) and was found it to be optimal for evaluating the vesicles formed. The number of vesicles at each location was then averaged out and back calculated to the vesicular yield obtained in the electro-chamber in GUVs per μL. Similarly, to the cell counting method, our yield estimation is prone to errors. For example, despite the density difference used between the inner phase (sucrose) and the outer phase (glucose), not all vesicles settled to the bottom where we counted them. We controlled that only a small proportion of vesicles could be found floating in the wells and no significant discrepancy was observed between settling times as long as a short 10 min latent period was given. Most importantly, for polymers that showed little to no vesicles, no vesicles were also observed floating and no changes in the results were reported at later times that could account for slower settling of the vesicles. Thus, it seems that the assumption that the majority of vesicle settle at the bottom rapidly does not have a significant impact on the estimated yield. Other parameters such as the number of vesicles transferred to the well, the location analyzed in the well, and counting errors, as well as experimental parameters such as film formation, the electro-chamber used and room conditions (temperature and humidity) could also affect the number of vesicles observed. The effect of these parameters can be minimized by systematically repeating the same protocol. In order to obtain a realistic yield estimation, we counted the cells at many different locations in the well (>5), replicated the experiments at least in triplicates and calculated the standard deviation between replicates. We also measured the diameter of each vesicle in order to determine the mean diameter of the vesicles per replicate. We then calculated the average of the mean diameters over the triplicate and their respective standard deviation. Finally, in analogy to dynamic light scattering (DLS) analysis of nanosized particles,53,54 we calculated the polydispersity (PDI) as PDI = (standard deviation/mean diameter)2 for each replicate and then calculated the average PDI. The average yield and their standard deviation, the mean diameter and their standard deviation and their respective average PDI are summarized in Table 2.
| Entry | Polymer | Yield (GUVs per μL)a | Mean Øb (μm) | PDIc |
|---|---|---|---|---|
| a Determined by phase contrast optical microscopy. b The mean diameter. c Polydispersity index defined as the average of the (standard deviation/mean)2. For more details, the GUV yields for each replicate can be found in ESI Table S1 and their diameter and PDI in Tables S11–25, including frequency diagrams for their size distributions (Fig. S10–12). | ||||
| 1 | PB73-b-PEEP12 | 355 ± 186 | 20 ± 2 | 0.78 |
| 161 ± 47 | 16 ± 4 | 0.80 | ||
| 2 | PB73-b-PEEP21 | 452 ± 144 | 14 ± 2 | 0.59 |
| 181 ± 70 | 16 ± 2 | 0.67 | ||
| 3 | PB46-b-PEO23 | 0.00 ± 0.00 | — | - |
| 3.54 ± 3.23 | 37 ± 11 | 0.35 | ||
| 4 | PDMS67-b-PEO48 | 0.00 ± 0.00 | — | — |
| 0.00 ± 0.00 | ||||
| 5 | PDMS60-b-PMOXA21 | 4.77 ± 4.61 | 23 ± 4 | 0.20 |
| 4.00 ± 2.72 | 27 ± 17 | 0.33 | ||
| 6 | PMOXA22-b-PDMS119-b-PMOXA22 | 0.00 ± 0.00 | — | — |
| 0.00 ± 0.00 | ||||
We observed that PB73-b-PEEP12 (entry 1) and PB73-b-PEEP21 (entry 2) clearly outperform the commonly used PB46-b-PEO23 (f = 0.29) (entry 3),55–58 PDMS67-b-PEO48 (f = 0.30) (entry 4),59,60 PDMS60-b-PMOXA21 (f = 0.29) (entry 5),61–63 and PMOXA22-b-PDMS119-b-PMOXA22 (f = 0.31) (entry 6)36,64–67 in both EF and na-FH (ESI Table S1† for details of the replicated GUV yield). PB73-b-PEEP12 and PB73-b-PEEP21 gave similar high yields for both na-FH and EF (400 GUVs per μL and 175 GUVs per μL, respectively), typically giving a phase contrast image as seen in Fig. 3a. On the other hand, PB46-b-PEO23 (entry 3), PDMS67-b-PEO48 (entry 4) and PMOXA22-b-PDMS119-b-PMOXA22 (entry 6) did not yield any GUVs by na-FH while PDMS60-b-PMOXA21 (entry 5) gave a low yield (4.77 ± 4.61 GUVs per μL). PB46-b-PEO23 (entry 3) and PDMS60-b-PMOXA21 (entry 5) gave a few GUVs using EF (<5 GUV per μL) contrary to PDMS67-b-PEO48 and PMOXA22-b-PDMS119-b-PMOXA22 (Fig. 3b–d). Similar PB-b-PEOs with a variety of properties tested such as 0.2 ≤ f ≤ 0.4 and 3000 ≤ Mn ≤ 17000 also failed to produce GUVs by EF (ESI Table S1†) despite the frequent recurrence of PB-b-PEO in the formation of SUV and even GUV by various other methods.37,42,55,58 In the same perspective, Mingotaud and coworkers also expressed difficulties in obtaining polymersomes with PB-b-PEO by EF on ITO plates and its narrow hydrophilic ratio range37 as well as Greene et al. by EF on Pt wires.42 Interestingly, despite the common assumption that EF improves the vesicular self-assembly,12,68 we did not observe such an improvement for any of the block copolymers used and na-FH performed even slightly better for PB-b-PEEPs. We hypothesized that EF results in a smaller number of GUVs than na-FH as the electrical current might catalyze the degradation of GUVs perhaps by altering the polymer structure, in parallel with the previously studied degradation of polyunsaturated phospholipids.69–71
 | ||
| Fig. 3 Typical phase contrast microscopy image of (a) PB73-b-PEEP12, (b) PB46-b-PEO23, (c) PDMS67-b-PEO48, and (d) PDMS60-b-PMOXA21 by film hydration. Scale bar: 100 μm. | ||
In terms of size, all samples had a large size distribution (PDI > 0.2); nonetheless, the replicates consistently gave the same mean diameters. The mean diameter was 20 ± 2 μm for PB73-b-PEEP12A and 14 ± 2 μm for PB73-b-PEEP21B during na-FH (the error representing the mean diameter uncertainty between replicates). Tuning the GUVs’ size by EF to larger monodisperse vesicles was not observed, giving identical sizes and PDI to na-FH. In the case of PB46-b-PEO23, the mean diameter was 37 ± 11 μm, a significantly larger diameter than that for PB-b-PEEPs and PDMS60-b-PMOXA21. PB-b-PEEPs gave an apparent Gaussian distribution with a maximum at 5 μm (ESI Fig. S10–12†). Smaller vesicles than 1 μm were probably also formed but cannot be accounted for on the optical microscope. Experimentally, any object below 1 μm could not be definitely distinguished between vesicles or impurities by optical microscopy or SUVs be assessed by DLS due to the presence of GUVs, altering the scattering's statistical average.
In order to determine how long the GUVs self-assemble by na-FH, we conducted a kinetic study in triplicate with PB73-b-PEEP12A (Fig. 4). We observed that the optimal vesicle number is achieved within 2 h, already achieving a large number of vesicles within 1 h (ESI Table S3†). The mean diameter of the vesicles decreased slightly over time from 23 ± 3 μm to 14 ± 1 μm, exemplifying that larger GUVs seem to be formed first (ESI Table S4†). Further na-FH experiments were thus carried out for 1 h in order to directly correlate EF and na-FH over the same timescale.
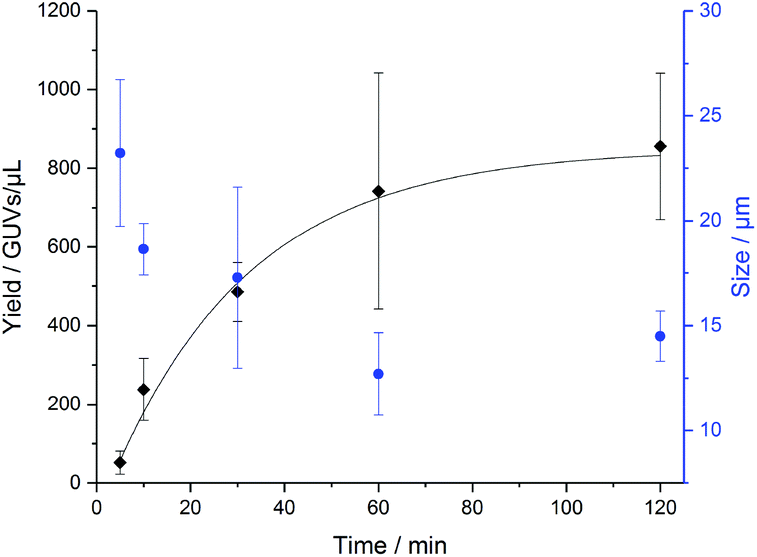 | ||
| Fig. 4 Formation of PB73-b-PEEP12 GUVs over time by non-assisted film hydration (na-FH) measuring the resulting yield in GUVs per μL (black) and the mean diameter of the vesicles in μm (blue). Details of the GUV yields and diameter for each replicate can be found in ESI Tables S3 and S4.† | ||
In the last decade, polymersomes have been increasingly used because of their inherent stability compared to liposomes.24 Block copolymers are much less prone to chemical degradation than lipids and as they are larger molecules, entanglement in the bilayer can be greater, resulting in higher mechanical stability than that of liposomes.14,47,67 We analyzed the size (ESI Tables S7 and S8† for more details) and yield evolution (Tables S5 and S6† for more details) of our PB-b-PEEP GUVs under no special storing conditions (kept in aqueous dispersion at room temperature). We observed for our PB-b-PEEPs that the vesicle yield slowly decreased (Fig. 5). After 1 month, 56 ± 10 GUVs per μL of vesicles were still present for PB73-b-PEEP12, thus effectively losing 63% in yield. In contrast, only 5% of PB73-b-PEEP21 remained. In terms of size, the mean diameter and size distribution of PB73-b-PEEP12 polymersomes over one month were similar to the freshly prepared GUVs, while the vast majority of PB73-b-PEEP21 were much smaller. For PB73-b-PEEP21, >80% of vesicle size was between 1 and 10 μm compared to 50% at the formation and with a mean diameter dropping to 6 ± 1 μm. Thus, it appears that PB73-b-PEEP12 GUVs are more stable than PB73-b-PEEP21 GUVs, influenced by a favored hydrophilic/hydrophobic block ratio.
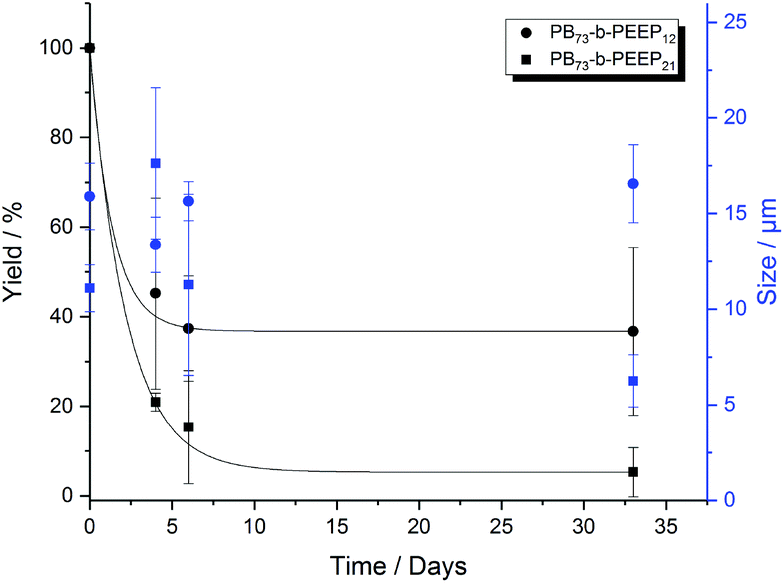 | ||
| Fig. 5 Yield (black) and size (blue) evolution over a period of 1 month of PB73-b-PEEP12 A and PB73-b-PEEP21 B. Details of the GUV yields and diameter for each replicate can be found in ESI Tables S5–8.† | ||
Scaling up the film hydration protocol in a round bottom flask using 4 mg of polymer in 5 mL of aqueous sucrose solution (100 mM) was also successful. A similarly high number of GUVs for both PB-b-PEEP A and B was obtained in a round bottom flask, even whilst vigorously stirring, than in our small 350 μL-capacity reactors. These agitated film hydration protocols are most frequently used to obtain a large amount of polydispersed multilamellar vesicles (MLV), usually <1 μm, which are then extruded through a polycarbonate membrane with small pores to obtain a homogeneous SUV population.20,72,73 In the case of PB-b-PEEPs, many GUVs were obtained with diameters >25 μm, whilst PDMS-b-PEO and PMOXA-b-PDMS-b-PMOXA did not yield any GUVs, PB-b-PEO formed only a few GUVs (with smaller diameters of ca. 5 μm) and PDMS-b-PMOXA formed a small number of GUVs (20 μm).
The polymers’ physical properties
We wanted to determine why the PB-b-PEEP block copolymers formed a much higher number of GUVs than the classically used block copolymers. By analysis of the block themselves, we concluded that the hydrophobic block has a limited impact on the GUV yield as PB74-b-PEO45 with a similar degree of polymerization of PB, Mn (6000 g mol−1) and f (0.33) to the successful PB73-b-PEEP12 yielded no vesicles. Thus, we believe that the hydrophilic block has the largest influence on vesicle formation.During EF and na-FH, one of the crucial steps is polymeric film formation on Pt-electrodes; thus the block copolymers are in their neat state at ambient temperature before hydration. Depending on their inherent properties, polymers would stack and adhere to the surface differently, which would influence their subsequent self-assembly into vesicles. In order to assess these variations in the block copolymers, we ran differential scanning calorimetry (DSC). For vesicle formation, the block copolymers are first dissolved in CHCl3 and then dried under reduced pressure once coated onto the Pt-wires. When treating our neat commercial PB-b-PEO in a similar way (dissolved in CHCl3 at 4.0 mg mL−1 and subsequently rapidly dried under reduced pressure), the DSC curve (ESI Fig. S7†) was similar to the second DSC heating curve of the neat polymer (Fig. S6†) and thus was analyzed as such for simplification.
Both PB73-b-PEEP12A and PB73-b-PEEP21B showed similar behavior: two Tg at low values for both phase-separated blocks were determined (−97 °C and −59 °C for A (Fig. S4†) and −96 °C and −45 °C for B (Fig. S5†)) (Table 3). The lower Tg at −90 °C corresponds to the PB block20,74 while the Tg around −50 °C corresponds to PEEP.75 PB-b-PEEP are thus fully amorphous. By contrast, PB-b-PEO (Fig. S6†) and PDMS-b-PEO (Fig. S8†) both exhibited a melting temperature (Tm) of 33 °C and 51 °C, respectively (corresponding to PEO).20 PB-b-PEO also has a single detectable Tg at −60 °C. PDMS-b-PMOXA exhibited two Tm at −41 °C and 60 °C (Fig. S9†). PB-b-PEO, PDMS-b-PEO, and PDMS-b-PMOXA are all thus semi-crystalline. At ambient temperature, at which the films are formed, all the commonly used block copolymers are below their Tm and thus exhibit a crystalline-like packing.
| Block copolymer | f | σ (mN m−1) | Thermal analysisb (°C) |
|---|---|---|---|
| a Interfacial tension σ measured by spinning drop tensiometry between CHCl3 and H2O at a concentration of 1.0 mg mL−1. b Measured by differential scanning calorimetry (DSC) between −100 °C and 100 °C. c The solution of PDMS-b-PMOXA in CHCl3 could not be run as despite full dissolution the block copolymer crashes out during spinning in the tensiometer. Filtering with a PTFE 0.45 μm filter prior to σ measurement did not limit that effect. The DSC curve for the thermal analysis can be found in ESI Fig. S4–9. | |||
| PB73-b-PEEP12 | 0.32 | 8.96 ± 0.34 | T g = −97 |
| T g = −59 | |||
| PB73-b-PEEP21 | 0.45 | 9.16 ± 0.15 | T g = −96 |
| T g = −45 | |||
| PB46-b-PEO23 | 0.29 | 19.82 ± 0.49 | T g = −60 |
| T m = 33 (30 J g−1) | |||
| PDMS67-b-PEO48 | 0.30 | 19.80 ± 0.70 | T m = 51 (92 J g−1) |
| PDMS60-b-PMOXA21 | 0.29 | —c | T m = −41 (8.0 J g−1) |
| T m = 60 | |||
The majority of EF studies rely on the operator's manual expertise in drop-cast film formation rather than automated methods like spin coating. Recently, Stein et al. suggested that non-uniform films (by manual drop-casting) might behave better than homogeneous films (for example, by spin-coating) as defects would allow a better water influx through the films, leading to facilitated GUV self-assembly.71 The same assumption can be drawn to ordered partly crystalline block copolymers. The lack of defects present in the crystalline-like films in comparison with their disordered amorphous counterpart could alter the water infiltration within the films and consequently the formation of vesicles. The partly crystalline block copolymers would thus require a higher input of energy than the amorphous PB-b-PEEP to achieve similar levels of self-assembly into GUVs.14,20,23,51 PB46-b-PEO23 could, therefore, form a few GUVs by EF but not by the milder na-FH methodology (Table 2, entry 3).
In the case of liposomes, self-assembly is always carried out at a temperature above the Tm of the lipids.68,76 We carried out the EF of PB-b-PEO at 50 °C, above its Tm, and obtained on average 1.7 ± 0.6 GUVs per μL, a yield similar to room temperature. Thus, contrary to lipids, modifying the temperature conditions does not change the behavior of PB-b-PEO in a manner that facilitates their self-assembly. PEO is known to have an inverse solubility–temperature relationship in aqueous solution with a lower critical solution temperature (LCST) typically >100 °C due to changes in its hydrogen bonding network with water.77,78 It would therefore not be surprising that at elevated temperature, PB-b-PEO exhibits modified hydrophilicity behaviors inhibiting vesicular self-assembly.
We further analyzed the bulk morphology of PB-b-PEEP by transmission electron microscopy (TEM). Staining the double bonds of PB with RuO4 was carried to observe the contrast between the hydrophilic and hydrophobic blocks. We observed that polymers A and B (f = 0.32 and 0.45, respectively) exhibited extensive lamellar morphologies (Fig. 6 and ESI Fig. S13 and S14†). Interestingly, while the lamellar thickness was similar, the phase separation obtained with these polymers was inverted: PB73-b-PEEP12A yielded PEEP thick lamellar structures (10.4 ± 1.3 nm for PEEP and 3.5 ± 0.4 nm for PB) whilst PB73-b-PEEP21B yielded PB thick lamellar structures (11.0 ± 1.5 nm for PB and 4.4 ± 0.7 nm for PEEP). In contrast, PB73-b-PEEP31 (f = 0.54) formed lamellar bulk structures (ESI Fig. S15†) but did not assemble into GUV. PB73-b-PEEP4 (f = 0.13) and PB73-b-PEEP7 (f = 0.21) did not form lamellar structures as expected from block copolymer phase separation theory (ESI Fig. S16 and S17†).46 PB46-b-PEO23 (f = 0.29), which is well within the 0.25 < f < 0.45 polymersome-forming range11,48 but only yielded a small number of GUVs (Table 2, entry 3), did not form lamellar structures in the bulk (Fig. S18†). Thus, the presence of amorphous lamellar films in the bulk structure might ease the formation of GUVs.
 | ||
| Fig. 6 Transmission electron microscopy (TEM) of PB73-b-PEEP21 films stained with RuO4 exhibiting extensive lamellar networks. Scale bar: 100 nm. | ||
Furthermore, we measured the interfacial tension σ between CHCl3 and H2O at a polymeric concentration of 1.0 mg mL−1.The interfacial tension of CHCl3 was determined to be 26.64 ± 0.67 mN m−1. We observed that PB-b-PEEP lowers σ to 8.96 ± 0.34 mN m−1 for PB73-b-PEEP12A and 9.16 ± 0.15 mN m−1 for PB73-b-PEEP21B while PB46-b-PEO23 and PDMS67-b-PEO48 gave 19.82 ± 0.49 mN m−1 and 19.80 ± 0.70 mN m−1, respectively. In comparison, the phospholipid POPC gave 3.90 ± 0.11 mN m−1. POPC and, in general, other phospholipids are known to readily self-assemble into lipidic GUVs. Thus, a low interfacial tension might be an indication that amphiphiles (lipids and polymers) are more likely to form vesicles.
In addition, other properties of the block copolymers influence their self-assembly into vesicles such as hydrogen bonding. EEP is composed of four oxygen atoms, and thus it has eight available lone pairs that could form extensive hydrogen bonding networks. In contrast, PEO and PMOXA only have two and three available lone pairs, respectively, from oxygen or nitrogen atoms. This difference in hydrogen-bonding potential might also contribute to the success of PB-b-PEEPs. Moreover, PEEP and PMOXA allow delocalization of electrons onto O to form charged species that can also modify their adherence to Pt or glass surfaces and aid their self-assembly in aqueous solution.
Encapsulation of hydrophilic and hydrophobic dyes
Ultimately, vesicles are interesting because of their compartmentalization, whether for chemical synthesis or biological mimicking. They are especially versatile compared to other carriers as they can encapsulate both hydrophilic and hydrophobic cargos with increasing complexity such as transmembrane proteins and even living cells.32,79–81 These advanced encapsulations are almost exclusively carried out by microfluidics. Because of the nature of the methodology, encapsulation of hydrophilic moieties by film hydration techniques (EF, gel-assisted hydration) has been reported to be challenging even for liposomes.14,20,76 Only sparse examples of passive encapsulation of hydrophilic cargos have been described for GUVs by film hydration. In the case of hydrophobic cargo, entrapment in the membrane has been well reported for polymersomes and liposomes.14,40 Encapsulation of hydrophobic dyes is easier as the cargo is mixed with the amphiphilic agent prior to swelling and its entrapment in the membrane is entropically favored in aqueous media.We first tested the encapsulation of the hydrophobic dye Nile Red (NR) (Table 4 and ESI Table S2† for more details). We define the encapsulation efficiency as the number of vesicles exhibiting fluorescence compared to the total number of vesicles observed in phase contrast (ESI† pS24). For both PB73-b-PEEPs A and B (entries 1 and 2), using NR did not disrupt the vesicular yield and was even significantly improved for PB73-b-PEEP21B to a high 1579 ± 279 GUVs per μL. Na-FH was again better at performing than EF, giving yields >580 GUVs per μL compared to 100–200 GUVs per μL for EF (Table 4, entries 1 and 2). Results for EF were similar to those in the absence of hydrophobic cargo. In the case of PB46-b-PEO23 (entry 3), similar low yields were obtained to those in the absence of dye: no GUVs were formed by na-FH and only a small number by EF (0.21 ± 0.36 GUVs per μL).
| Entry | Polymer | Additive | Yield (GUVs per μL) | eea (%) |
|---|---|---|---|---|
| a Encapsulation efficiency. Defined as the number of vesicles expressing fluorescence over the total number of vesicles observed by phase contrast. Details of the GUV yields and ee for each replicate can be found in ESI Table S2. | ||||
| 1 | PB73-b-PEEP12 | NR | 583 ± 101 | >99 |
| 100 ± 36 | >99 | |||
| 2 | PB73-b-PEEP21 | 1579 ± 279 | >99 | |
| 211 ± 106 | >99 | |||
| 3 | PB46-b-PEO23 | 0.00 ± 0.00 | — | |
| 0.21 ± 0.36 | >99 | |||
| 4 | PB73-b-PEEP12 | AF647 | 49 ± 11 | 46 ± 8 |
| 117 ± 72 | 55 ± 15 | |||
| 5 | PB73-b-PEEP21 | 74 ± 36 | 8 ± 3 | |
| 145 ± 128 | 23 ± 5 | |||
For all polymers, the encapsulation efficiency (ee) was optimal (all vesicles formed have encapsulated the hydrophobic dye in their membrane – Fig. 6 left). Thus, regardless of the block copolymer used, the encapsulation of NR has no effect or is improving the yield of vesicles obtained and the cargo can be efficiently encapsulated.
Encapsulation of the hydrophilic dye Alexa Fluor 647 (AF647) was carried out by doping the aqueous sucrose medium with AF647. Following hydration, the electro-chamber medium was then diluted into an AF647-free glucose solution for observation. Thus any AF647 in the extra-vesicular media is diluted by a factor of five and allows a contrast to be observed if AF647 is encapsulated (Fig. 7 – right). When using AF647 for passive encapsulation into PB-b-PEEP GUVs during na-FH, the yield significantly decreased: PB73-b-PEEP12A produced only 49 ± 11 GUVs per μL and PB73-b-PEEP21B 74 ± 36 GUVs per μL compared to typically 400 GUVs per μL (Table 4 and ESI Table S2† for more details). Thus hydrophilic cargo can have a strong negative influence on the self-assembly of the GUVs themselves. Small quantities of additives such as ions or salts have been shown to affect the self-assembly processes of SUVs to yield aggregates of various morphologies.82,83 Thus it is not surprising that charged dyes like AF647 would disturb GUV formation.
 | ||
| Fig. 7 Typical fluorescence microscopy image of PB-b-PEEPs of NR (left) and AF647 (right) encapsulation. | ||
For EF, the yield of PB-b-PEEP GUVs in the presence of AF647 was two times higher than for na-FH, giving similar values to the standard experiments in the absence of hydrophilic cargo (∼100 GUVs per μL). In other EF studies, large frequencies (500 Hz) have been reported to compensate for ionic strength, such as charged lipids36 or physiological buffers.84,85 It is thus reasonable to conclude that the use of a moderate frequency (10 Hz) in our system is enough to compensate for the ionic strength of AF647, impairing the spontaneous swelling of the polymeric films. This observation can also be correlated to the encapsulation of hydrophilic cargos by electroporation, a technique first tested on living cells.51 We thus observed for the first time the beneficial effect of EF compared to na-FH when using PB-b-PEEPs.
The ee was higher than expected for PB73-b-PEEP12A with a decent ∼50% ee for both EF and na-FH, although generally, the fluorescence was weak. In the case of PB73-b-PEEP21B, only low encapsulation (8 ± 3%) was obtained by na-FH. EF slightly improved the encapsulation to 23 ± 5%. Therefore, hydrophilic dyes are indeed harder to encapsulate than hydrophobic dyes; nonetheless, we were able to obtain a decent encapsulation efficiency when using PB73-b-PEEP12A.
Polymersomes have been described to be less permeable than liposomes due to a much lower membrane fluidity.24,86–88 This allows polymersomes to retain hydrophilic cargo and thus makes them promising candidates for protocells. In order to assess the permeability of the PB-b-PEEP vesicles, we analyzed the evolution over time of the intravesicular AF647 fluorescence (Fig. 8, ESI Tables S9 and 10†). Vesicles of PB73-b-PEEP12A appeared to be relatively hermetic with their fluorescence oscillating around 100% over 100 min, retaining AF647 in the polymersome's lumen. PB73-b-PEEP21B GUVs are more permeable, losing 85% fluorescence during the first 100 min. These results might also explain why the ee of AF647 in GUVs of B was significantly lower compared to GUVs prepared from A, rendering A a promising candidate for generating protocells.
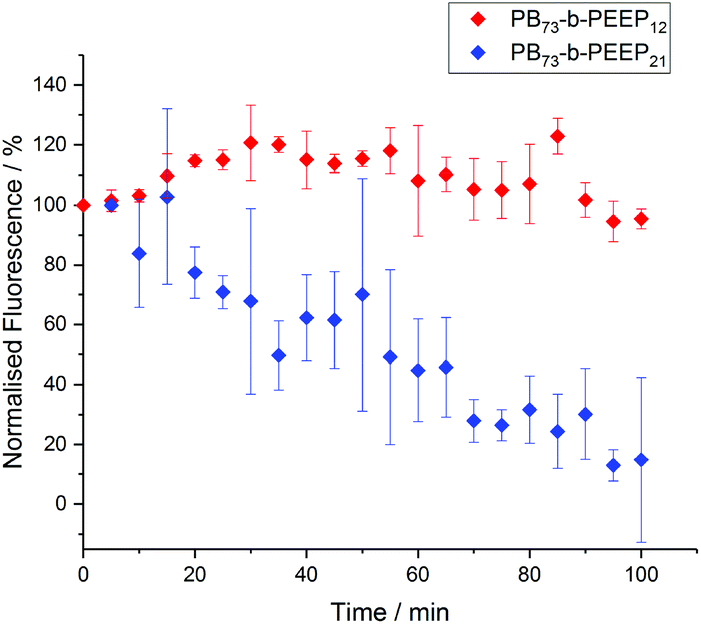 | ||
| Fig. 8 AF647 retention over time in GUVs based on the difference between the intravesicular emitted fluorescence and the background fluorescence. The fluorescence was normalized to 100% at t = 0. Details of normalized fluorescence for each replicate can be found in ESI Tables S9 and S10.† | ||
Summary
We successfully synthesized a library of novel amphiphilic block copolymers (polybutadiene-block-poly(ethyl ethylene phosphate) (PB-b-PEEP)), with a polyphosphoester as the hydrophilic segment, resembling phospholipid-like structures for protocell assembly. PB-b-PEEPs with hydrophilic ratios of 0.32 and 0.45 successfully self-assembled into solvent- and additive-free GUVs with high yields by electroformation and non-assisted direct film hydration, i.e. in the absence of an alternating current or any other energy forces. In contrast to classically used block copolymers for polymersome formation, which are PB-b-PEO, PDMS-b-PEO, and PDMS-b-PMOXA block copolymers, we observed that polyphosphoester-based amphiphiles produced GUVs by spontaneous film hydration or electroformation very efficiently. Stability experiments proved that PB-b-PEEP GUVs could be stored at room temperature for several weeks. Furthermore, we proved that hydrophobic and hydrophilic cargos were encapsulated into the GUVs. Hydrophobic dyes were efficiently encapsulated by non-assisted film hydration. Hydrophilic dyes tested with AF647 were more challenging to encapsulate into the GUVs. Nevertheless, 50% encapsulation efficiency could be achieved in PB73-b-PEEP12 GUVs and could be efficiently retained in the polymersomes for at least 2 h.The straightforward synthesis of well-defined PB-b-PEEP block copolymers, their structural similarities to phospholipids, and the ease of producing loaded GUVs by simple film hydration make them promising new materials for the generation of protocells and microreactors.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the MaxSynBio consortium, which is jointly funded by the Federal Ministry of Education and Research of Germany and the Max Planck Society. We would like to thank Kathrin Kirchhoff and Dr Ingo Lieberwirth (MPIP) for TEM measurements. Open Access funding provided by the Max Planck Society.Notes and references
- M. Li, X. Huang, T. Y. D. Tang and S. Mann, Curr. Opin. Chem. Biol., 2014, 22, 1–11 CrossRef CAS PubMed.
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, Curr. Opin. Biotechnol., 2014, 28, 10–16 CrossRef CAS PubMed.
- A. I. Lamond, Nature, 2002, 417, 383–383 CrossRef CAS.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS PubMed.
- B. C. Buddingh and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769–777 CrossRef CAS PubMed.
- K. Gopfrich, I. Platzman and J. P. Spatz, Trends Biotechnol., 2018, 36, 938–951 CrossRef PubMed.
- L. K. Müller and K. Landfester, Biochem. Biophys. Res. Commun., 2015, 468, 411–418 CrossRef PubMed.
- V. Balasubramanian, B. Herranz-Blanco, P. V. Almeida, J. Hirvonen and H. A. Santos, Prog. Polym. Sci., 2016, 60, 51–85 CrossRef CAS.
- X. L. Hu, Y. G. Zhang, Z. G. Xie, X. B. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- H. L. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- A. Jesorka and O. Orwar, Annu. Rev. Anal. Chem., 2008, 1, 801–832 CrossRef CAS PubMed.
- J. F. Le Meins, C. Schatz, S. Lecommandoux and O. Sandre, Mater. Today, 2013, 16, 397–402 CrossRef CAS.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Curr. Opin. Pharmacol., 2014, 18, 104–111 CrossRef CAS PubMed.
- V. Malinova, S. Belegrinou, D. D. Ouboter and W. P. Meier, Adv. Polym. Sci., 2010, 224, 113–165 CAS.
- B. Clément, B. Grignard, L. Koole, C. Jérôme and P. Lecomte, Macromolecules, 2012, 45, 4476–4486 CrossRef.
- K. N. Bauer, H. T. Tee, M. M. Velencoso and F. R. Wurm, Prog. Polym. Sci., 2017, 73, 61–122 CrossRef CAS.
- F. Wang, Y.-C. Wang, L.-F. Yan and J. Wang, Polymer, 2009, 50, 5048–5054 CrossRef CAS.
- T. Wolf, T. Rheinberger, J. Simon and F. R. Wurm, J. Am. Chem. Soc., 2017, 139, 11064–11072 CrossRef CAS PubMed.
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS PubMed.
- S. So, L. J. Yao and T. P. Lodge, J. Phys. Chem. B, 2015, 119, 15054–15062 CrossRef CAS PubMed.
- R. S. M. Rikken, H. Engelkamp, R. J. M. Nolte, J. C. Maan, J. C. M. van Hest, D. A. Wilson and P. C. M. Christianen, Nat. Commun., 2016, 7 CAS , 12606.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3176 RSC.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018 10.1039/c8cs00162f.
- M. Chemin, P. M. Brun, S. Lecommandoux, O. Sandre and J. F. Le Meins, Soft Matter, 2012, 8, 2867–2874 RSC.
- T. P. T. Dao, F. Fernandes, M. Er-Rafik, R. Salva, M. Schmutz, A. Brulet, M. Prieto, O. Sandre and J. F. Le Meins, ACS Macro Lett., 2015, 4, 182–186 CrossRef CAS.
- C. Martino and A. J. deMello, Interface Focus, 2016, 6, 20160011 CrossRef PubMed.
- J. Petit, I. Polenz, J. C. Baret, S. Herminghaus and O. Baumchen, Eur. Phys. J. E: Soft Matter Biol. Phys., 2016, 39, 59 CrossRef PubMed.
- J. Petit, L. Thomi, J. Schultze, M. Makowski, I. Negwer, K. Koynov, S. Herminghaus, F. R. Wurm, O. Baumchen and K. Landfester, Soft Matter, 2018, 14, 894–900 RSC.
- D. van Swaay and A. deMello, Lab Chip, 2013, 13, 752–767 RSC.
- T. P. T. Dao, M. Fauquignon, F. Fernandes, E. Ibarboure, A. Vax, M. Prieto and J. F. Le Meins, Colloids Surf., A, 2017, 533, 347–353 CrossRef CAS.
- M. Weiss, J. P. Frohnmayer, L. T. Benk, B. Haller, J. W. Janiesch, T. Heitkamp, M. Borsch, R. B. Lira, R. Dimova, R. Lipowsky, E. Bodenschatz, J. C. Baret, T. Vidakovic-Koch, K. Sundmacher, I. Platzman and J. P. Spatz, Nat. Mater., 2018, 17(1), 89–96 CrossRef CAS PubMed.
- K. Tsumoto, H. Matsuo, M. Tomita and T. Yoshimura, Colloids Surf., B, 2009, 68, 98–105 CrossRef CAS PubMed.
- T. J. Politano, V. E. Froude, B. Jing and Y. Zhu, Colloids Surf., B, 2010, 79, 75–82 CrossRef CAS PubMed.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- A. Weinberger, F. C. Tsai, G. H. Koenderink, T. F. Schmidt, R. Itri, W. Meier, T. Schmatko, A. Schroder and C. Marques, Biophys. J., 2013, 105, 154–164 CrossRef CAS PubMed.
- M. Dionzou, A. Morere, C. Roux, B. Lonetti, J. D. Marty, C. Mingotaud, P. Joseph, D. Goudouneche, B. Payre, M. Leonetti and A. F. Mingotaud, Soft Matter, 2016, 12, 2166–2176 RSC.
- R. Dimova, U. Seifert, B. Pouligny, S. Forster and H. G. Dobereiner, Eur. Phys. J. E, 2002, 7, 241–250 CAS.
- L. Theogarajan, S. Desai, M. Baldo and C. Scholz, Polym. Int., 2008, 57, 660–667 CrossRef CAS.
- A. M. Eissa, M. J. P. Smith, A. Kubilis, J. A. Mosely and N. R. Cameron, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5184–5193 CrossRef CAS.
- A. Kubilis, A. Abdulkarim, A. M. Eissa and N. R. Cameron, Sci. Rep., 2016, 6 CAS , 32414.
- A. C. Greene, I. M. Henderson, A. Gomez, W. F. Paxton, V. VanDelinder and G. D. Bachand, PLoS One, 2016, 11, e0158729 CrossRef PubMed.
- M. A. Hillmyer and F. S. Bates, Macromolecules, 1996, 29, 6994–7002 CrossRef CAS.
- J. M. Chen, Z. J. Lu, G. Q. Pan, Y. X. Qi, J. J. Yi and H. J. Bai, Chin. J. Polym. Sci., 2010, 28, 715–720 CrossRef CAS.
- P. Patnaik, A Comprehensive Guide to the Hazardous Properties of Chemical Substances, Wiley, 2007 Search PubMed.
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- X. Y. Zhang, P. Tanner, A. Graff, C. G. Palivan and W. Meier, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2293–2318 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- J. P. Reeves and R. M. Dowben, J. Cell Physiol., 1969, 73, 49–60 CrossRef CAS PubMed.
- N. Rodriguez, F. Pincet and S. Cribier, Colloids Surf., B, 2005, 42, 125–130 CrossRef CAS PubMed.
- L. G. Wang, L. Chierico, D. Little, N. Patikarnmonthon, Z. Yang, M. Azzouz, J. Madsen, S. P. Armes and G. Battaglia, Angew. Chem., Int. Ed., 2012, 51, 11122–11125 CrossRef CAS PubMed.
- M. Absher, in Tissue Culture, Academic Press, 1973, pp. 395–397, DOI:10.1016/B978-0-12-427150-0.50098-X.
- Z. T. Qin, J. Joo, L. Gu and M. J. Sailor, Part. Part. Syst. Charact., 2014, 31, 252–256 CrossRef CAS.
- N. K. Kozlov, U. A. Natashina, K. P. Tamarov, M. B. Gongalsky, V. V. Solovyev, A. A. Kudryavtsev, V. Sivakov and L. A. Osminkina, Mater. Res. Express, 2017, 4 CAS , 095026.
- J. Gaspard, L. M. Casey, M. Rozin, D. J. Munoz-Pinto, J. A. Silas and M. S. Hahn, Sensors, 2016, 16(3), 390 CrossRef PubMed.
- H. L. Che, B. C. Buddingh’ and J. C. M. van Hest, Angew. Chem., Int. Ed., 2017, 56, 12581–12585 CrossRef CAS PubMed.
- A. Peyret, E. Ibarboure, N. Pippa and S. Lecommandoux, Langmuir, 2017, 33, 7079–7085 CrossRef CAS PubMed.
- A. Peyret, E. Ibarboure, J. F. Le Meins and S. Lecommandoux, Adv. Sci., 2018, 5, 1700453 CrossRef PubMed.
- G. Kickelbick, J. Bauer, N. Huesing, M. Andersson and K. Holmberg, Langmuir, 2003, 19, 10073–10076 CrossRef CAS.
- G. Kickelbick, J. Bauer, N. Husing, M. Andersson and A. Palmqvist, Langmuir, 2003, 19, 3198–3201 CrossRef CAS.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- S. Winzen, M. Bernhardt, D. Schaeffel, A. Koch, M. Kappl, K. Koynov, K. Landfester and A. Kroeger, Soft Matter, 2013, 9, 5883–5890 RSC.
- K. Kiene, S. H. Schenk, F. Porta, A. Ernst, D. Witzigmann, P. Grossen and J. Huwyler, Eur. J. Pharm. Biopharm., 2017, 119, 322–332 CrossRef CAS PubMed.
- Y. R. Kim, S. Jung, H. Ryu, Y. E. Yoo, S. M. Kim and T. J. Jeon, Sensors, 2012, 12, 9530–9550 CrossRef CAS PubMed.
- A. Najer, S. Thamboo, C. G. Palivan, H. P. Beck and W. Meier, Chimia, 2016, 70, 288–291 CrossRef CAS PubMed.
- L. Klermund, S. T. Poschenrieder and K. Castiglione, ACS Catal., 2017, 7, 3900–3904 CrossRef CAS.
- S. T. Poschenrieder, S. K. Schiebel and K. Castiglione, Eng. Life Sci., 2018, 18, 101–113 CrossRef CAS.
- M. I. Angelova and D. S. Dimitrov, Faraday Discuss., 1986, 81, 303–311 RSC.
- Y. Zhou, C. K. Berry, P. A. Storer and R. M. Raphael, Biomaterials, 2007, 28, 1298–1306 CrossRef CAS PubMed.
- M. Breton, M. Amirkavei and L. M. Mir, J. Membr. Biol., 2015, 248, 827–835 CrossRef CAS PubMed.
- H. Stein, S. Spindler, N. Bonakdar, C. Wang and V. Sandoghdar, Front. Physiol., 2017, 8(63) DOI:10.3389/fphys.2017.00063.
- L. D. Mayer, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, 1986, 858, 161–168 CrossRef CAS.
- S. Vemuri and C. T. Rhodes, Pharm. Acta Helv., 1995, 70, 95–111 CrossRef CAS PubMed.
- G. Janowska and L. Slusarski, J. Therm. Anal. Calorim., 2001, 65, 205–212 CrossRef CAS.
- S. Zhang, J. Zou, F. Zhang, M. Elsabahy, S. E. Felder, J. Zhu, D. J. Pochan and K. L. Wooley, J. Am. Chem. Soc., 2012, 134, 18467–18474 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102–111 CrossRef PubMed.
- R. Kjellander and E. Florin, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2053–2077 RSC.
- F. Kafer, F. Liu, U. Stahlschmidt, V. Jerome, R. Freitag, M. Karg and S. Agarwal, Langmuir, 2015, 31, 8940–8946 CrossRef PubMed.
- R. Chandrawati and F. Caruso, Langmuir, 2012, 28, 13798–13807 CrossRef CAS PubMed.
- F. Fernandez-Trillo, L. M. Grover, A. Stephenson-Brown, P. Harrison and P. M. Mendes, Angew. Chem., Int. Ed., 2017, 56, 3142–3160 CrossRef CAS PubMed.
- Y. Elani, T. Trantidou, D. Wylie, L. Dekker, K. Polizzi, R. V. Law and O. Ces, Sci. Rep., 2018, 8 CAS , 4564.
- P. Bucher, A. Fischer, P. L. Luisi, T. Oberholzer and P. Walde, Langmuir, 1998, 14, 2712–2721 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef CAS.
- S. Aimon, J. Manzi, D. Schmidt, J. A. Poveda Larrosa, P. Bassereau and G. E. Toombes, PLoS One, 2011, 6, e25529 CrossRef CAS PubMed.
- W. Zong, S. H. Ma, X. N. Zhang, X. J. Wang, Q. C. Li and X. J. Han, J. Am. Chem. Soc., 2017, 139, 9955–9960 CrossRef CAS PubMed.
- J. F. Le Meins, O. Sandre and S. Lecommandoux, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 14 CrossRef PubMed.
- R. Dimova, in Advances in Planar Lipid Bilayers and Liposomes, ed. A. Iglič, Academic Press, 2012, vol. 16, pp. 1–50 Search PubMed.
- R. Dimova, Adv. Colloid Interface Sci., 2014, 208, 225–234 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py00992a |
| This journal is © The Royal Society of Chemistry 2018 |