
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic polypyrrole-poly(Schiff base) copolymers with poly(ethylene glycol) side chains: synthesis, properties and applications†
Brenda G.
Molina
 ab,
Luminita
Cianga
c,
Anca-Dana
Bendrea
c,
Ioan
Cianga
*c,
Luis J.
del Valle
ab,
Francesc
Estrany
ab,
Carlos
Alemán
*ab and
Elaine
Armelin
*ab
ab,
Luminita
Cianga
c,
Anca-Dana
Bendrea
c,
Ioan
Cianga
*c,
Luis J.
del Valle
ab,
Francesc
Estrany
ab,
Carlos
Alemán
*ab and
Elaine
Armelin
*ab
aDepartament d'Enginyeria Química, EEBE, Universitat Politècnica de Catalunya, C/ Eduard Maristany, 10-14, Ed. I.2, 08019, Barcelona, Spain
bBarcelona Research Center for Multiscale Science and Engineering, Universitat Politècnica de Catalunya, C/ Eduard Maristany, 10-14, Ed. I.S, 08019, Barcelona, Spain. E-mail: carlos.aleman@upc.edu; elaine.armelin@upc.edu
c“Petru Poni” Institute of Macromolecular Chemistry, 41A, Grigore –Ghica Voda Alley, 700487, Iasi, Romania. E-mail: ioanc@icmpp.ro
First published on 11th July 2018
Abstract
New amphiphilic poly(ethylene glycol) (PEG)-grafted random intrinsically conducting copolymers which combine three different functionalities have been engineered, prepared and characterized. Specifically, these “rod-coil” type copolymers bear conducting polypyrrole (PPy) and poly(Schiff base) (PSB) sequences randomly distributed in their backbones; hydrophilic grafted side chains consisting of well-defined PEG chains are attached to the PSB units. Basically, the synthesis of the copolymers is conducted sequentially by employing the “macromonomer” technique via electrochemical co-polymerization of a bis (pyrrole) benzoic Schiff base-containing PEG macromonomer with pyrrole monomers. After investigation of the chemical and electrochemical properties of the synthesized copolymers, their advantages of multi-functionality in terms of biomedical applications have been demonstrated. More specifically, the conjugated PPy and PSB sequences enabled the grafted copolymers to exhibit great ability to catalyse the oxidation of serotonin, an important neurotransmitter found in blood platelets and in the central nervous systems of animals and humans. On the other hand, the enhanced biocompatibility in comparison with bare PPy is due to the presence of PEG side chains, while bacteriostatic activity against both Gram-negative and Gram-positive bacteria is imparted by the synergistic combination of the polycationic character of the PPy main chain with the benzoic Schiff base functional groups and with the bent-shaped architecture of the facially amphiphilic PSB sequences, respectively. Accordingly, these grafted copolymers are promising materials for developing implantable electrodes for serotonin detection which present the abovementioned characteristics.
Introduction
Since their discovery, electroactive conjugated polymers, also known as intrinsically conducting polymers (ICPs), have been foreseen to be prospective materials with applications in many fields despite several obvious drawbacks related to their processing (innate rigidity, insolubility, and infusibility). Therefore, the discovery in the late 1980s1 of the concept of flexible side chains as a “chemically bound solvent” for solubilizing the rod-like ICPs was welcome; this discovery enabled the development of currently well-known solution processing technologies, although it was demonstrated that the introduction of alkyl or alkoxy side chains alters the planarity of the conjugated backbone by their steric interaction, thus negatively affecting the electrical conductivity and decisively influencing the spectral features and specific phase behaviour.2However, in recent years, ICPs-based graft copolymers with linear biocompatible thermoplastics as side chains have emerged as a new class of useful hybrid materials. The advantage of the synergistic combination of very different characteristics and properties is due to the presence of the semiconducting main chain and grafted segments in a single structure.3 The high conductivity, good capacitive behavior and wide range of unique electromechanical and electrochromic characteristics4–8 of ICPs differentiate them from thermoplastics, thermosets and elastomers. Moreover, these properties can be reversibly tuned through electrochemical reactions; thus, this class of materials are ideal biomimetic candidates for the development of biosensing devices,9–13 bioactuators,14–16 regulated drug-delivery systems,17–21 and bioelectrodes, among other biomedical applications.22–26
The design of new biocompatible conducting polymers using biodegradable thermoplastics is an exciting area of research. Ma and co-workers recently reported a review27 compiling the main studies on bioactive scaffolds for tissue regeneration that employ electrically conducting polymers, such as polyaniline (PAni) and oligomers, polypyrrole (PPy) and polythiophene (PTh) combined with poly(ethylene glycol) (PEG),28,29 polylactide (PLA),30 and polycaprolactone (PCL).31 Although amphiphilic conducting polymers for biomedical functions have not been yet extensively addressed, systems derived from polypyrrole (PPy) and polythiophene (PTh) are among the most well studied. For example, pH responsive PTh-g-poly(N,N-dimethylaminoethylmethacrylate) (PTh-g-PDMA) was proposed as an aqueous sensor for biological applications,32 an amphiphilic copolymer consisting of a PTh backbone and sparsely attached oligo-ε-caprolactone side chains (PTh-g-PCL) was proven effective as a dopamine biosensor,33 and PPy-g-poly(styrenesulfonic acid) (PPy-g-PSSA) was used for the electrocatalytic biosensing of hypoxanthine.34 Also, functionalized PTh-graft copolymers have been designed to effectively induce specific adsorption of cells and proteins.35,36 In the last few years, most of our research interest in PTh-graft copolymers for biomedical applications has been focused on systems containing PEG side chains anchored to a PTh backbone (PTh-g-PEG).1,37–41 For example, we used pentathiophene-PEG macromonomers (Th5-PEGM, where M refers to the molecular weight of the PEG side chain, with Mn = 1000 or 2000 g mol−1) to produce graft copolymers, denoted as PTh5-g-PEGM (Scheme 1).1,38,40 These materials were successfully employed as active surfaces for the selective adsorption of proteins and as cellular matrices for tissue engineering.
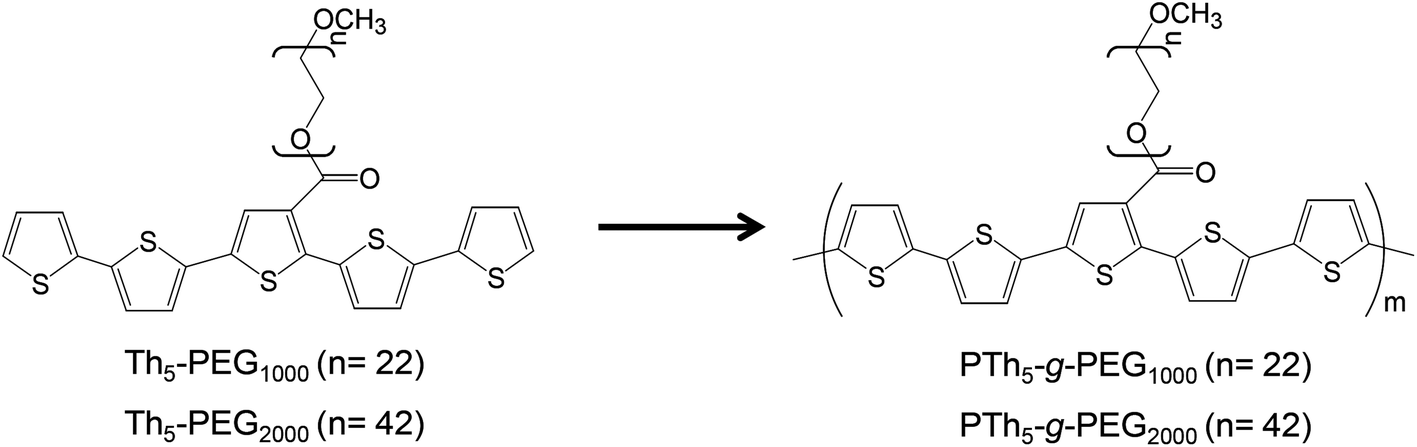 | ||
| Scheme 1 Synthesis of PTh5-g-PEGM.38 | ||
More recently, we synthesized PTh3-g-PEG2000 copolymers (Scheme 2) via a macromonomer approach using a terthiophene-PEG macromonomer (Th3-PEG2000) copolymerized with terthiophene (Th3) using different Th3-PEG2000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Th3 ratios.40 These materials showed excellent behavior as bioactive platforms; they were more biocompatible in terms of cellular adhesion and proliferation and more electroactive than PTh5-g-PEGM (Scheme 1).
:Th3 ratios.40 These materials showed excellent behavior as bioactive platforms; they were more biocompatible in terms of cellular adhesion and proliferation and more electroactive than PTh5-g-PEGM (Scheme 1).
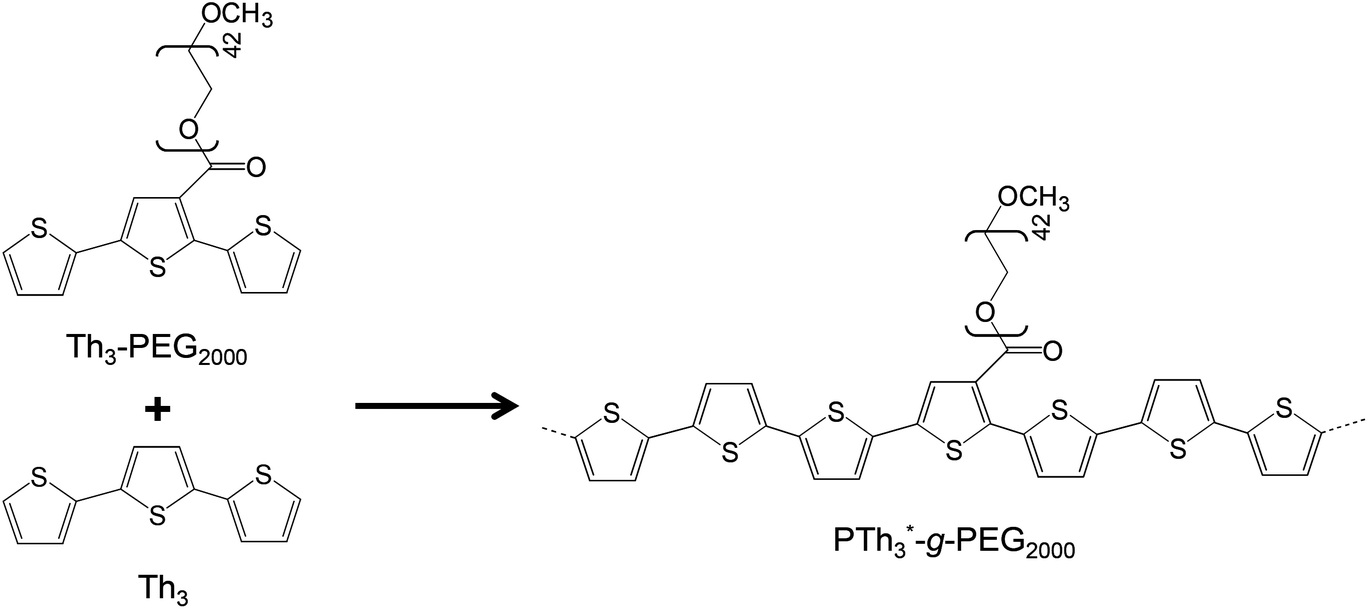 | ||
| Scheme 2 Synthesis of PTh3-g-PEG2000 copolymers.40 | ||
On the other hand, PSB are recognized ICPs42,43 with potential application areas ranging from LED devices,44 organic photovoltaics (OPV),45 and electrochromic devices (ECD)46 to organic field effect transistors (OFET)47 and chemosensors.48
In a series of reported studies on chemical and electrochemical syntheses of several bis(pyrrole) Schiff bases,49,50 investigations of their properties revealed that their electrical conductivities,49 redox properties51 and behaviour in electrochemical polymerization52 are similar to those of polypyrrole. In another approach, a polystyrene-containing bis(pyrrole) Schiff base macromonomer was prepared,53 and its copolymer with pyrrole showed interesting electrochromic properties.46
When biomedical applications are considered, even if PSB are well-recognized pharmacophores, their application as excellent plasmid DNA damage protecting agents54 or as well-performing nonviral vectors for efficient cellular DNA transfection has also been reported.55 Also, in a recent study, we prepared new functional synthetic biomaterials with similar optical properties to those displayed by PTh by combining Schiff base functions with Th units in the polymeric main chain.56 More specifically, a Schiff base-containing bis-thienyl monomer, (AzbT), was synthesized and electrochemically copolymerized with Th3 using different AzbT:Th3 ratios (Scheme 3). The resulting P(AzbT-co-Th3) copolymers behaved as bioactive substrates that promoted cellular viability; this was attributed to the presence of hydrophilic pendant carboxyl groups. Moreover, the antibacterial activity, usually associated with the presence of the Schiff base functionality, suggested that the P(AzbT-co-Th3) construction can be considered as a dual-functionalized platform for advanced biomedical applications. The introduction of inherent antibacterial ability into materials for tissue engineering scaffolds, cell therapy or other bio-applications brings benefits, such as decreased infection risk.
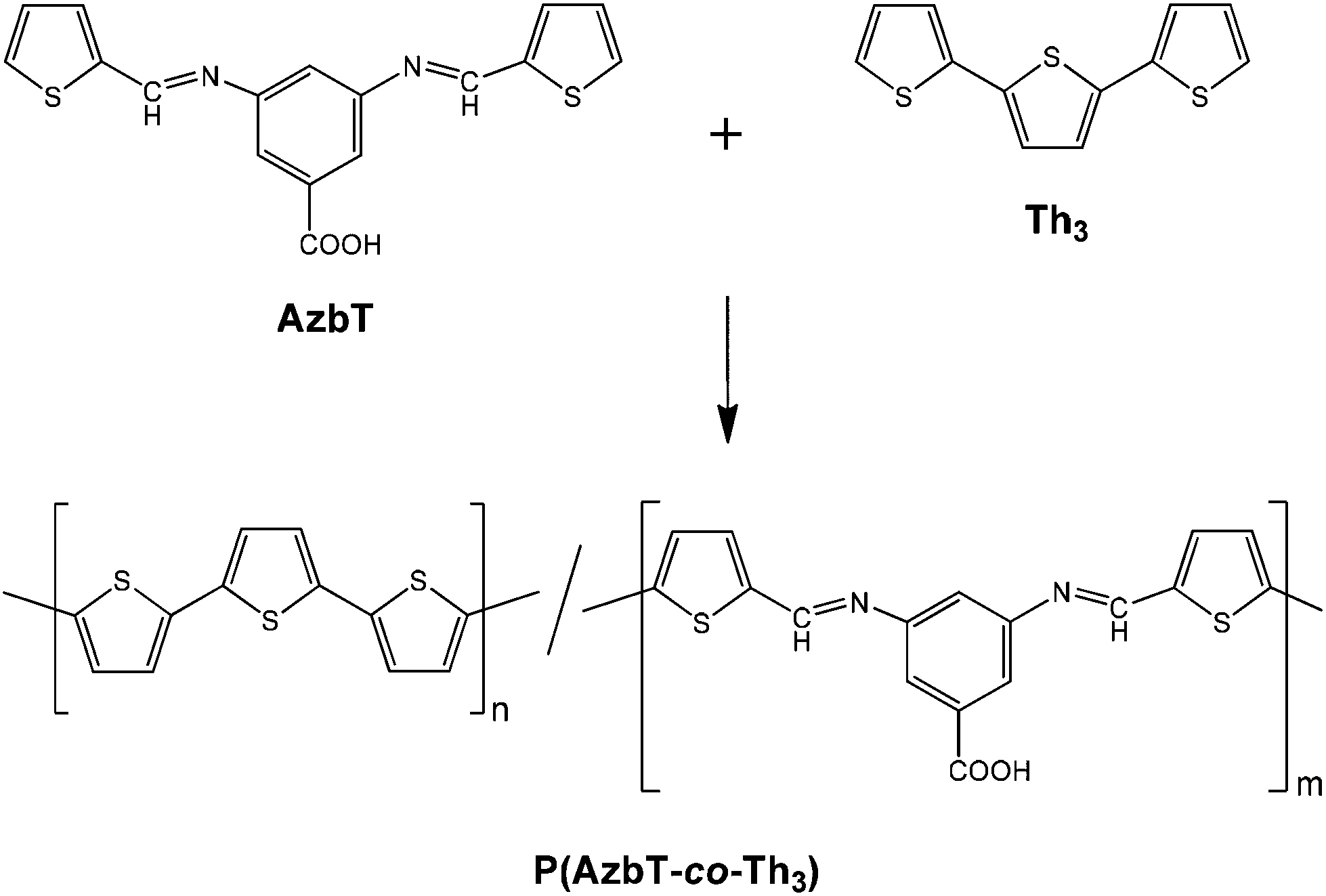 | ||
| Scheme 3 Synthesis of P(AzbT-co-Th3) copolymers.56 | ||
In the present work, new amphiphilic random copolymers are reported that combine the benefits associated with both the graft PEG chains and the presence of a benzoic Schiff base in a polyheterocyclic backbone. For this purpose, in analogy with AzbT reported in our previous study,56 a new macromonomer has been designed and subsequently synthesized in two reaction steps. More specifically, in this new compound, the end-thienyl moieties of AzbT have been replaced by pyrrole (Py) units, while the carboxyl moiety of the aromatic phenyl ring has been used to attach PEG2000 side chains. After synthesizing and characterizing the macromonomer, hereafter denoted AzbPy-g-PEG, it was electrochemically copolymerized with Py, and the chemical and physical properties of the resulting graft copolymers, P(Py-co-AzbPy-g-PEG), were determined. Finally, the suitability of P(Py-co-AzbPy-g-PEG) in potential bioactive and biosensing platforms has been considered. Thus, studies on the protein adhesion, cellular proliferation, antimicrobial activity, and neurotransmitter detection biocapacities of P(Py-co-AzbPy-g-PEG) are also discussed.
Experimental section
Materials
Pyrrole (Py; from Aldrich, 98%) was freshly distilled and 2-pyrrole-carboxaldehyde (from Fluka, 97%) was recrystallized from petroleum ether before use. p-Toluenesulfonic acid (PTSA; from Aldrich, 99%) and 3,5-diaminobenzoic acid (DABA; from Aldrich, 98%), poly(ethylene glycol) methyl ether with Mn = 2000 g mol−1 (PEG2000; from Aldrich), N,N′-dicyclohexylcarbodiimide (DCCI; from Merck), 4-dimethylamino pyridine (DMAP; from Aldrich) and acetonitrile (ACN; from Panreac S.A., PA) were used as received. Meanwhile, anhydrous lithium perchlorate (from Sigma-Aldrich, 95%) was stored in an oven at 70 °C before its use in the electrochemical trials.Synthesis of the azomethine-containing bis-pyrrole monomer: 3,5-bis(((E)-(1H-pyrrol-2-yl)methylene)amino)benzoic acid (AzbPyBA)
A 250 mL three-neck round bottom flask equipped with a condenser, a Dean–Stark trap, a nitrogen inlet–outlet and a magnetic stirrer was charged with 2-pyrrole-carboxaldehyde (5.89 g; 0.062 mol), DABA (4.56 g, 0.03 mol), PTSA (0.015 g) and toluene (150 mL). Nitrogen was purged through the reaction mixture for 15 minutes (Scheme 4). | ||
| Scheme 4 Synthesis of the AzbPy-g-PEG macromonomer. | ||
The mixture was heated to reflux with stirring for 20 hours while continuously removing the toluene–water azeotrope. After cooling the reaction mixture, the formed solid product was filtered and purified by column chromatography (aluminum oxide-Fluka Type 507c), first with dichloromethane as eluent and then with THF. The second fraction was concentrated by evaporation under reduced pressure, and the resulting dark-brown solid was dried. A dark-brownish solid was obtained. Yield: 62%.
Synthesis of pyrrole-g-polyethylene glycol macromonomer functionalized with azomethine-containing bis-pyrrole moieties (AzbPy-g-PEG)
2 g (0.001 mol) of PEG2000 and 0.46 g (0.0015 mol) of AzbPyBA were placed in a three-neck round-bottom flask equipped with a dropping funnel under nitrogen. 17 mL of THF and 0.01815 g (0.00015 mol) of DMAP were added to the flask. 0.3078 g (0.0015 mol) of DCCI in 1.5 mL THF were placed in the dropping funnel and added over about 15 min. The mixture was stirred at room temperature for three days (Scheme 4). The resulting solution was filtered and precipitated in cold diethyl ether to remove the catalyst and the unreacted reagents. After filtration and drying, a white solid was obtained. The product was passed through a silica gel-filled column and re-precipitated in cold diethyl ether using CH2Cl2 as solvent.Copolymerization of pyrrole monomer and pyrrole-g-polyethylene glycol macromonomer to obtain P(Py-co-AzbPy-g-PEG)
All electrochemical assays, including anodic polymerizations, were carried out in a standard 50 mL three-electrode cell under nitrogen atmosphere (99.995% purity) at room temperature. The anodic compartment was filled with 10 mL of a 1 mM 1:1 Py:AzbPy-g-PEG in ACN solution with 0.1 M LiClO4 as the supporting electrolyte; meanwhile, the cathodic compartment was filled with 10 mL of the same electrolyte solution. Stainless steel (SS) AISI 316 sheets with areas of 0.5 cm2 (surface roughness determined by AFM, Rq = 11.4 Å) were employed as working electrodes (WE) and counter electrodes (CE), respectively. The reference electrode (RE) was an Ag|AgCl electrode containing a saturated aqueous KCl solution (E° = 0.222 V at 25 °C) which was connected to the working compartment through a salt bridge containing the electrolyte solution. P(Py-co-AzbPy-g-PEG) polymeric films were prepared by chronoamperometry (CA) using a constant potential of +1.60 V and considering three different polymerization times (θ = 300, 500 and 1000 s) (Fig. 1). The stainless steel WE surfaces were covered by the polymers after the current pass and polymerization times were established. The final compositions of the monomer units in the copolymer films were determined by determining the ratios of the FTIR absorption bands for the N–H stretching in the Py units (at 3430 cm−1) and the ether deformation (at 1108 cm−1) in the PEG units. For calculations, the Py:PEG molar ratio (2:1) and the molecular weight of the macromonomer (2313.4 g mol−1) were taken into account.
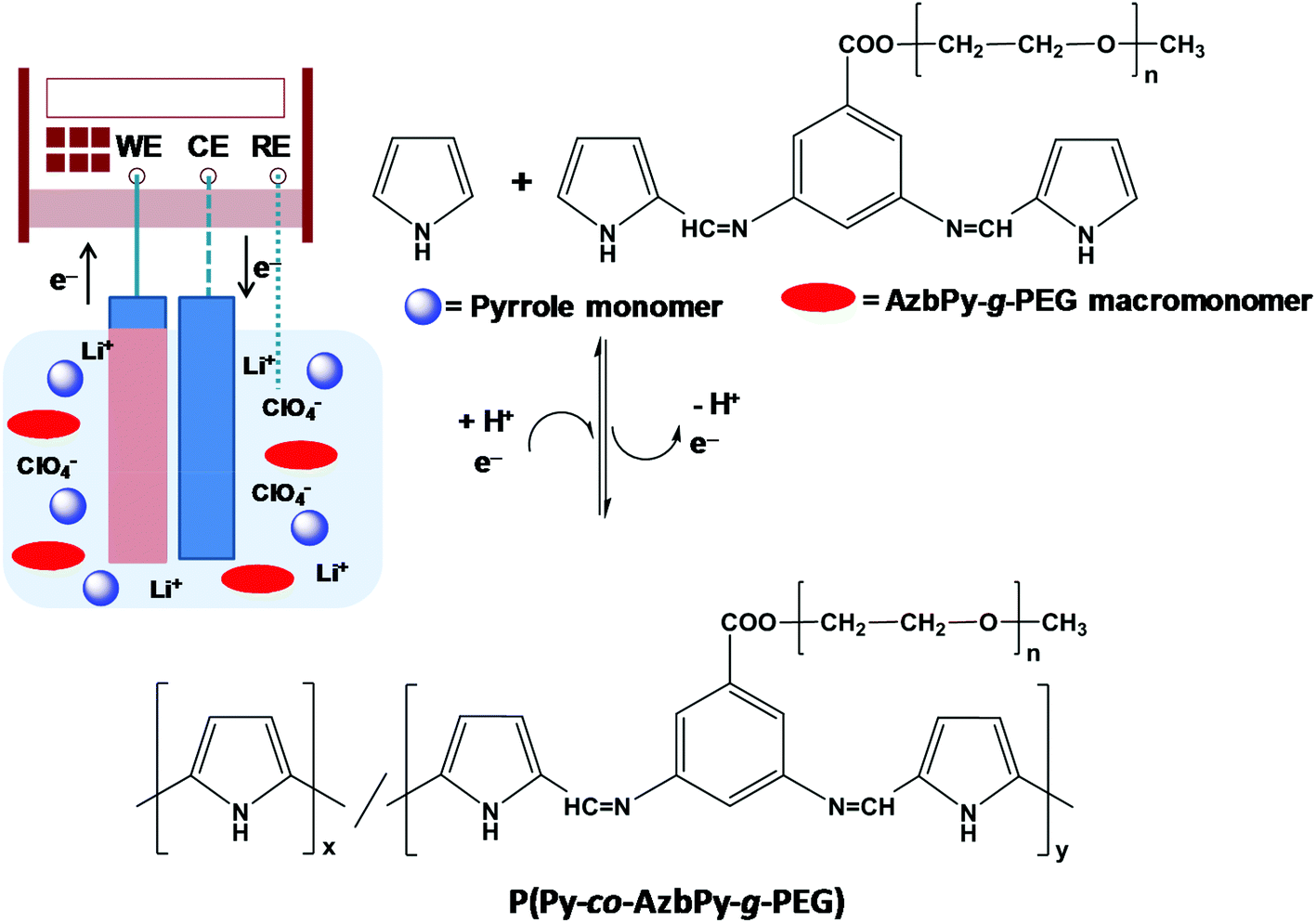 | ||
| Fig. 1 Scheme illustrating the anodic polymerization of P(Py-co-AzbPy-g-PEG). The experimental conditions are described in the Methods section. | ||
PPy homopolymer (PPy) was also prepared and characterized as a standard for comparison of the properties of the copolymers. The generation medium was 1 mM of Py in acetonitrile solvent, with 0.1 M LiClO4 solution as the supporting electrolyte, and the anodic polymerization was performed using a constant potential of +1.70 V for θ = 300 s.
Characterization of the macromonomer and copolymers
The AzbPy-g-PEG macromonomer was characterized by 1H- and 13C-NMR spectroscopy, FTIR, UV-vis and fluorescence spectroscopies and thermal analyses. The experimental methods of all the characterization techniques for all the monomers and graft copolymers are detailed in the ESI.†Biological studies
In order to evaluate the ability of P(Py-co-AzbPy-g-PEG) to interact with different proteins, adsorption assays were conducted out using albumin (BSA) and lysozyme (Lyz) proteins. The cytotoxicity was evaluated using the COS-1 and Vero cell lines. Finally, the antimicrobial properties of AzbPy-g-PEG macromonomer, PPy and P(Py-co-AzbPy-g-PEG) were investigated using Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus), which are Gram-negative and Gram-positive bacteria, respectively. The complete experimental procedures are described in the ESI.†Electrochemical detection of serotonin neurotransmitter
Differential pulse voltammetry (DPV) was applied for the electrochemical detection of serotonin. The assays were carried out in a tree electrode cell according to the procedure described in the ESI.†Results and discussion
Design, synthesis and characterization of AzbPy-g-PEG macromonomer
In a previous study, an electrochemically active bis-thienyl-containing monomer, AzbT, with preformed Schiff base linkages was synthesized starting from 2-thiophene carboxaldehyde and the versatile bis-aminoacid DABA, the presence of which enabled the preparation of P(AzbT-co-Th3) copolymers with free carboxyl functionality.56In the present work, a similar approach was used; however, with the aim of encoding multifunctionality by design, several essential modifications were performed (Scheme 4). Firstly, the thienyl-end groups were replaced by pyrrole rings. As with thiophene, these moieties maintained their electrochemical activity; however, the obtained PPy, in its oxidized form, is a biocompatible polycation with recognized antibacterial activity in all its forms (powder, particles, nanotubes and thin film surfaces).57,58 The benzoic Schiff base bonds were retained for their potential antibacterial activity and also as desirable and easy-to-use functionalities for the construction of pH-responsive systems. Benzoic Schiff bases are differentiated from other types of Schiff bases by their stability at normal physiological pH (7.4) and their ability to undergo hydrolysis under very mild acidic conditions (pH 6.5–6.8). Due to this behavior, materials containing these functional bonds can find various practical applications, including transient scaffolds and transient bioelectronic devices.59 The presence of free carboxyl groups in AzbPyBA (Scheme 4) enabled the attachment of the non-ionic PEG by esterification in the second step of synthesis; thus, the structure of the new macromonomer AzbPy-g-PEG was amphiphilic, with high hydrophilicity, water dispersibility and the ability to complex with metal ions.60 Also, it is known that incorporation of hydrophilic moieties such as PEG can dramatically improve the biocompatibility of polycations without significant loss of antimicrobial activity. These modifications can be used to improve the selectivity of the antimicrobial agent towards bacteria over mammalian cells.61 Moreover, although they represent a nascent technique in the field, phase-separated amphiphilic copolymer coatings have significant potential to prevent bacterial infections on implanted medical devices.62 These coatings phase-separate on the length scale of biomacromolecules and disrupt their adhesion mechanisms. Last but not least in the importance order of the chemical-structural features of AzbPy-g-PEG is the bent-shaped geometry imposed by the 3,5-substituted phenyl ring from the DABA moiety, as well as the attachment of PEG side chains in the first position of this phenyl ring. Due to these structural peculiarities, the new macromonomer is a facially amphiphilic structure (FA). The importance of this aspect is related to the widespread FA in nature, from steroids to antimicrobial peptides, such as magainin, cecropin and defensins. For host defense peptides, it has been shown that this architecture is imperative for antibacterial activity and selectivity, rather than the precise chemical composition or amino acid sequence.63 Thus, synthetic polymers that mimic the essential physicochemical features of these specialized natural macromolecules can provide fast access to less expensive materials for medical coatings, antimicrobial tubing, and other applications.63
After the synthesis, the structures of the new compounds were unambiguously elucidated by combining FTIR, 1H-NMR and 13C-NMR spectroscopy.
Fig. 2 compares the FTIR spectra of DABA, AzbPyBA and AzbPy-g-PEG. The spectrum of DABA presents the characteristic absorption bands of the primary aromatic amine functionality at 3427 cm−1 (νasim NH2), 3339 cm−1 (νsim NH2), and 1289 cm−1 and 1267 cm−1 (ν C–N of NH2); these are absent from the spectrum of AzbPyBA. The successful synthesis of the latter is corroborated by the new band at 1669 cm−1 (ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), which is due to the formation of azomethine bonds, as well as by the appearance of bands typically associated with the Py ring. These are the broad band centered at 3344 cm−1 (ν NH), the shoulder at 3124 cm−1 (ν C–H), and other bands at 1089 cm−1 (β NH), 1042 cm−1, and 960 cm−1 (β C–H).
N), which is due to the formation of azomethine bonds, as well as by the appearance of bands typically associated with the Py ring. These are the broad band centered at 3344 cm−1 (ν NH), the shoulder at 3124 cm−1 (ν C–H), and other bands at 1089 cm−1 (β NH), 1042 cm−1, and 960 cm−1 (β C–H).
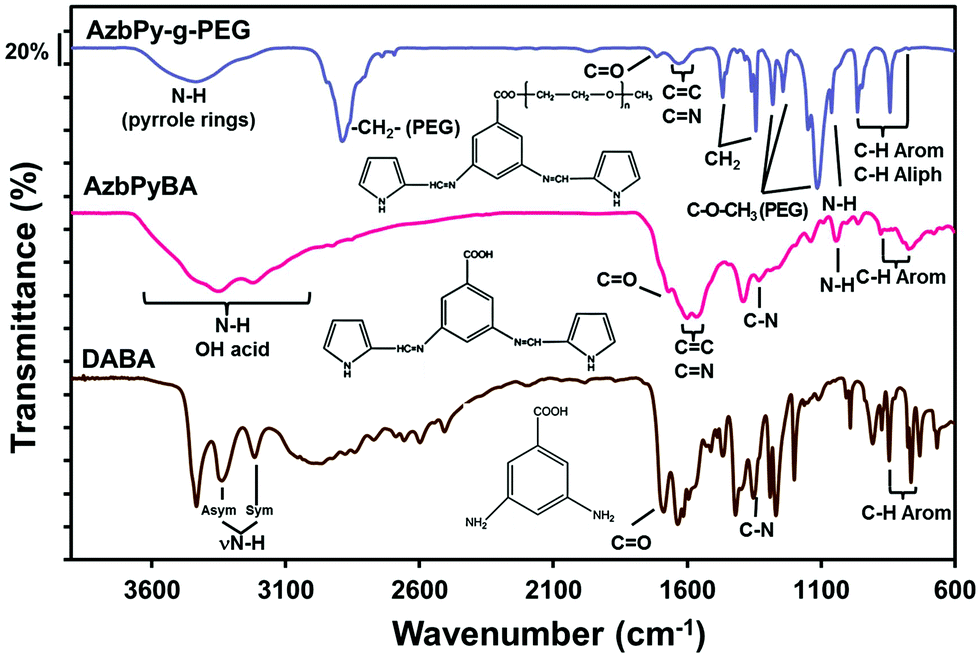 | ||
| Fig. 2 FTIR spectra of AzbPy-g-PEG, AzbPyBA and DABA. The latter two compounds are the precursors used for the synthesis of the AzbPy-g-PEG macromonomer. | ||
On the other hand, the FTIR spectra of DABA and AzbPyBA reveal that both aromatic acids are in a dimerized form. Thus, the absorption bands due to the carbonyls of the carboxylic functionalities are centered at values (1686 cm−1 and 1701 cm−1 for DABA and AzbPyBA, respectively) lower than that typically found for the monomeric form (∼1760 cm−1). These bands are accompanied by other bands at 3212 cm−1 (DABA) and 3221 cm−1 (AzbPyBA), which are also specific for dimeric forms of aromatic acids. On further analysis, it can be observed that in the FTIR spectrum of AzbPy-g-PEG, the absorption band at 3221 cm−1 disappeared concomitantly with the appearance of a broad band centered at approximately 1715 cm−1 due to the formation of the ester group. The band at 1632 cm−1 in this spectrum was attributed to the azomethine bond. The presence of the PEG in the structure of AzbPy-g-PEG is confirmed by additional bands at 1360, 1278 and 1059 cm−1 (common vibrations of crystallized PEG in a 7/2 helical structure) in addition to those attributed to the trans zig-zag conformation at 1241 and 963 cm−1, supporting the assumption that PEG in AzbPy-g-PEG is in the crystallized form.64
The expected structures were also confirmed by NMR spectroscopy (1H-NMR, 13C-NMR). The solvents used to record the spectra were carefully chosen; on the one hand, both the structural peculiarities of the compounds and their possible particular interactions during the solution preparation and NMR recording were taken into account. On the other hand, care was taken to avoid eventual solvent interference with the signals of the investigated compounds. Thus, the spectrum of the precursor AzbPyBA, containing highly polar functional groups (carboxyl, imine, pyrrole ring), was recorded in polar DMSO-d6 for solubility reasons, despite the presence of an appreciable amount of water that can be associated with DMSO due to its hygroscopic nature. This remnant water can be detrimental to the Schiff base linkages, as previously described.56 In the case of the AzbPy-g-PEG macromonomer, a propensity for self-assembly (SA) in PEG-selective solvents such as DMSO (ε = 47, δt = 26.7 MPa1/2) can be intuitively envisioned, driven by the synergetic combination of hydrophobic and π–π stacking interactions.31 Thus, the less polar acetone-d6 (ε = 21, δt = 19.9 MPa1/2) was employed for the NMR recording in order to decrease the solvent selectivity toward PEG (δt = 24 MPa1/2) as much as possible and, thus, to avoid decreased intensity of the peaks for the hydrophobic and aromatic moieties in the spectrum due to SA.65 However, some water is present in acetone as well; consequently, the values of the integrals were weakly affected for both hydrophobic (slightly decreased) and hydrophilic moieties (slightly increased), as can be seen in the spectrum in Fig. 3a.
 | ||
| Fig. 3 1H-NMR spectra of (a) AzbPy-g-PEG in acetone-d6 and (b) AzbPyBA solid compound in DMSO-d6. | ||
Fig. 3 also compares the 1H-NMR spectra of AzbPy-g-PEG and AzbPyBA. It is well known that polar solvents induce downfield shifting of signals. As the spectrum of the AzbPy-g-PEG macromonomer was recorded in acetone-d6 (Fig. 3a), an upfield shifting for the peaks is expected in comparison with the spectrum of AzbPyBA, for which the more polar DMSO-d6 solvent was used (Fig. 3b). In this case, this shifting was only appreciable for the N–H proton of the Py ring (h type in the 1H-NMR spectrum in Fig. 3a). This phenomenon has been attributed to the higher sensitivity of the N–H moiety towards solvent polarity in comparison with the other aromatic protons and to the enhanced electron density in the structure of AzbPy-g-PEG due to the presence of PEG. On the other hand, the 1H-NMR spectra displayed in Fig. 3 for the two compounds indicate that the azomethine functionality is stable in the registration conditions and that it is present in both the syn and anti-isomeric forms.56 The increased amount of syn-conformer in the case of AzbPy-g-PEG compared to the AzbPyBA organic molecule has been associated with the effect of the PEG chain grafted at the phenylene ring, which can modify its positions with respect to the azomethine linkage.66 Therefore, the presence of the hydrophilic PEG molecules increases the deviation from planarity about the azomethine bond due to steric interactions.67 The 13C-NMR spectra of AzbPyBA and AzbPy-g-PEG molecules are shown in Fig. S1.†
The photochemical and thermal properties of the new macromonomer were also investigated and compared to those previously reported for the AzbT monomer (Scheme 3).56 Comparison of the UV-vis spectra indicates that the λmax position does not vary when the thiophene rings in AzbT are replaced by Py rings in AzbPyBA. However, a notable shift was obtained when PEG was incorporated into the macromonomer (Fig. S2a†); the band gap decreased by a factor of 20% with respect to the AzbPyBA precursor (Table 1). This behavior was attributed to the electron-donor character of the PEG graft chain. The optical band gap was estimated from the onset wavelength of optical absorption in DMSO solution (Eg = 1240/λonset). The molar extinction coefficients (ε) were calculated by applying the Lambert–Beer law at the values of λmax in the 340 to 347 nm range.
| Sample |
T
d,0b (°C) |
Char yieldb (%) |
λ
maxc (nm) |
λ
maxd (nm) |
Stokes shift (nm) | ε (×104 M−1 cm−1) |
E
gf (eV) |
|---|---|---|---|---|---|---|---|
| a Data from ref. 51. b Initial decomposition temperatures and char yields as obtained by TGA. c Maximum absorption bands obtained by UV-vis spectroscopy in DMSO. d Fluorescence spectra were recorded in DMSO using the values of λmax derived from UV-vis spectroscopy as λex. e The molar extinction coefficients (ε) were calculated by applying the Lambert–Beer law at the values of λmax obtained by UV-vis spectroscopy in the range of 340 to 347 nm. f Optical band gap estimated from the onset wavelength of optical absorption in solution (Eg = 1240/λonset) by UV-visible spectroscopy. nd – not determined. | |||||||
| AzbTa | nd | nd | 289, 340 | 360; 490 (289) | 71 | 0.340 | 3.06 |
| AzbPyBA | 220 | 5.4 | 290, 344 | 412 (290) | 68, 66 | 10.5 | 3.10 |
| 410 (344) | |||||||
| AzbPy-g-PEG | 354 | 6.7 | 347, 458 | 422 (347) | 75, 68 | 8.00 | 2.48 |
| 526 (458) | |||||||
In the case of the fluorescence studies, both the shape and the λmax of AzbPy-g-PEG reflect changes with respect to AzbT.56 The fluorescence curve of AzbT was reported to be bimodal, whereas AzbPy-g-PEG shows a non-symmetric monomodal shape (Fig. S2b†) and a blue shift of λmax (Table 1). The solutions of AzbPy-g-PEG are highly fluorescent at different concentrations under 365 nm of irradiation and also in daylight. This optoelectronic property has been attributed to the extended conjugation, which is induced through the self-assembly between neighboring molecules forming complementary hydrogen bonds.
The thermal properties of AzbPyBA and AzbPy-g-PEG macromonomer were evaluated by DSC. Fig. S3a† displays a broad exothermic peak in the range of 210 °C to 150 °C that was registered during the cooling cycle. This exothermic peak can be assigned to crystallization of the compound but also to other structural modifications that can take place due to the complex structural re-organization of this compound. In the second heating curve, only the first shallow transition corresponding to the beginning of the exothermic peak was revealed. This crystallization temperature (206 °C) was further confirmed in a second DSC experiment, which was conducted with a very low scan rate (2 °C min−1) for the second heating cycle (Fig. S3b†). On the other hand, the AzbPy-g-PEG macromonomer shows only the melting temperature (54 °C) and crystallization temperature (28 °C) corresponding to the PEG side chains (Fig. S3c†). From the TGA analyses, it was demonstrated that the AzbPy-g-PEG precursor molecule, AzbPyBA, starts to decompose at around 220 °C, whereas the macromonomer starts at ∼354 °C (Td,0) with a maximum decomposition temperature of 405 °C (Td,max) (Fig. S3d†). The optical and thermal data are displayed in Table 1, and a detailed discussion of these results is included in the ESI.†
Synthesis and characterization of P(Py-co-AzbPy-g-PEG) graft copolymers
It is widely known that the oxidative chemical polymerization of PPy is limited by the tendency of this ICP to over-oxidize in oxygen-rich environments, and very low yields are usually obtained. Therefore, electrochemical synthesis has become the most versatile method for the generation of Py-containing polymers and copolymers;46,50–52,68,69 this procedure is also widely employed for the synthesis of other heterocyclic ICPs.70–73 However, the success of anodic polymerization processes, like those used in this work, largely depends on the mobility of the monomers that must reach the electrode surface, i.e. small, lightweight monomers move more quickly than heavy, bulky macromonomers. This may explain the unsuccessful homopolymerization of the AzbPy-g-PEG macromonomer, which due to its large size presents reduced mobility in organic solutions. This assumption is also sustained by the fact that other bis-pyrrole Schiff bases with similar structures but lower molecular weights were successfully homo-electropolymerized.50In order to overcome the limitations associated with the low mobility of the macromonomer, P(Py-co-AzbPy-g-PEG) copolymers were prepared. Long polymerization times (θ = 300, 500 and 1000 s) were required to obtain homogenous films when the ratio of the comonomers in the feed (Py:AzbPy-g-PEG macromonomer) was 1:1. However, the real comonomer ratios in the hybrid material, as determined by FTIR, were approximately 521:1, 208:1, and 125:1 (Py:macromonomer) with 300 s, 500 s and 1000 s of electrochemical polymerization, respectively. According to the previous discussion, the excessively low content of the macromonomer units in the copolymer films is probably due to the lower mobility of the AzbPy-g-PEG macromonomer compared to the higher mobility of Py molecules. These results are in accordance with our previous studies with thiophene derivatives.56 As stated, the poor solubility of ICPs in organic solvents prevents the use of more accurate techniques, such as NMR and GPC, for precise evaluation of the comonomer ratio. Moreover, solid techniques such as mass spectrometry and solid 13C-NMR spectroscopy are also complicated to employ due to the charges present in the doped film.
Furthermore, bare PPy films were prepared using θ = 300 s under identical experimental conditions for comparison. The SEM micrographs and AFM images displayed in Fig. 4a and b, c, respectively, reveal a cauliflower-like morphology for the P(Py-co-AzbPy-g-PEG) films independently of θ, which is similar to the results for PPy.74,75 The film thickness (L) of P(Py-co-AzbPy-g-PEG) increases with θ, as was demonstrated by optical profilometry measurements. Thus, values of L = 1.4 ± 0.4 nm, 2.6 ± 0.5 nm, and 4.1 ± 0.5 nm were obtained for films generated using θ = 300, 500, and 1000 s, respectively. It can be assumed that the bent-shaped geometry and the presence of long PEG side chains in the AzbPy-g-PEG comonomer hinder the intermolecular chain packing of the copolymers, resulting in increased film thickness. Similarly, the roughness (Rq) increased from 91 ± 5 nm to 123 ± 11 nm when θ was increased from 300 s to 1000 s. The L and Rq values of the PPy films prepared using θ = 300 s (2.2 ± 0.1 nm and 112 ± 9.9 nm, respectively) are higher than those of the graft copolymer obtained using the same θ value. A possible explanation for these experimental findings is that the anodic polymerization of PPy is dominated by the crosslinks in the β- and β′-ring positions,76 allowing non-regular growth of the polymer chains. The formation of multidirectional branches results in increments of both L and Rq compared to other ICPs.75
 | ||
| Fig. 4 (A) SEM micrographs, (B) 2D height AFM images (5 × 5 μm2) and (C) 3D topographic AFM images (5 × 5 μm2) of PPy obtained by CA using θ = 300 s (a) and P(Py-co-AzbPy-g-PEG) prepared by CA using θ = 300, 500 and 1000 s (b, c and d, respectively). All samples for AFM analyses were supported on stainless steel electrodes. | ||
As part of the surface characterization, the wettability of the copolymer was determined by measuring the water contact angle (WCA). As expected, the wettability of the copolymers increased with the roughness of the films and the content of hydrophilic grafted PEG chains. Thus, the values of WCA determined for the P(Py-co-AzbPy-g-PEG) films obtained at θ = 300, 500 and 1000 s were 71.0° ± 0.4°, 62.6° ± 0.3°, and 48.5° ± 0.3°, respectively, demonstrating that the hydrophilicity increases with θ. The WCA exhibited by PPy, (58.9° ± 0.1°), was intermediate between those obtained for the copolymers prepared using θ = 300 and 1000 s, which is fully consistent with previous evidence of the influence of Rq on the wettability of PPy.77
The increased wettability observed for the P(Py-co-AzbPy-g-PEG) films synthesized using the largest θ value can also be attributed to a potential higher content of PEG on the copolymer film surface. Due to the insolubility of the as-prepared films in deuterated solvents, unfortunately, determination of the ratios between the Py and AzbPy-g-PEG comonomers in the final copolymers using NMR spectroscopy was not possible. However, the presence of both PEG and AzbPy groups in the copolymers was assessed by comparing the FTIR spectra of the monomers with those of the final copolymers. Thus, the Py monomer shows remarkable bands centered at 3125 and 720 cm−1, which correspond to the Cα–H stretching and out-of-plane vibration modes, respectively (Fig. 5a). The absence of these two absorption bands in the PPy homopolymer spectrum indicates that the hydrogen atoms at the Cα-position were removed during the polymerization process. The characteristic absorption bands of the PPy samples are the fundamental vibrations of the Py rings at 1525 and 1596 cm−1, the C–N stretching vibrations at 1464 and 1413 cm−1, the C–H in-plane vibration at 1072 and 1040 cm−1, and the N–H in-plane vibration at 1010 cm−1.78 Although the Py monomer was distilled before use, the appearance of the CO band at 1714 cm−1 indicates some overoxidation after the electropolymerization in acetonitrile.68
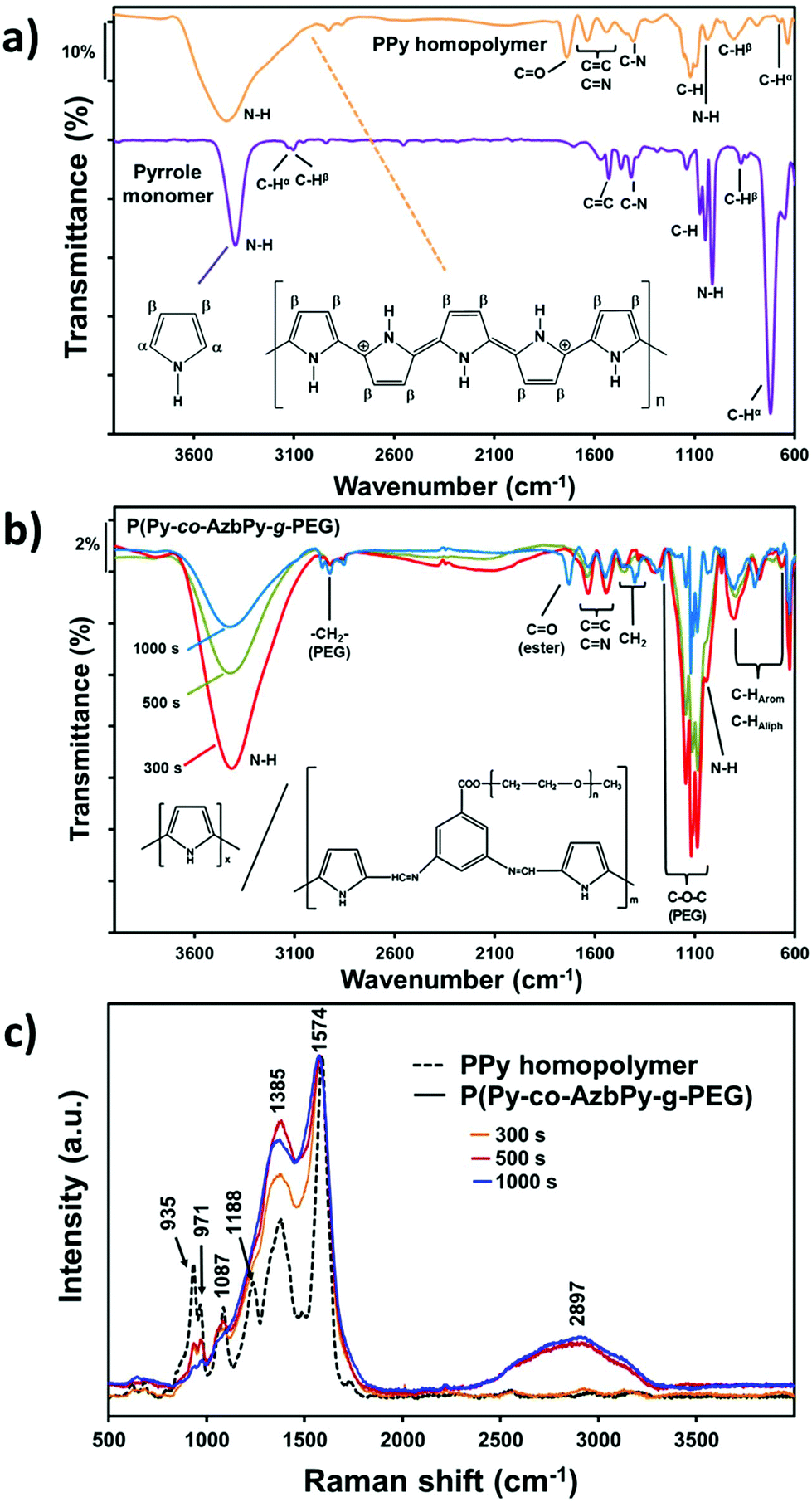 | ||
| Fig. 5 FTIR spectra of: (a) Py monomer and PPy prepared by CA using θ = 300 s; and (b) P(Py-co-AzbPy-g-PEG) synthesized by CA using θ = 300, 500 and 1000 s. (c) Raman spectra of P(Py-co-AzbPy-g-PEG) synthesized by CA using θ = 300, 500 and 1000 s. All films were generated on stainless steel electrodes as working electrodes. | ||
After copolymerization, the most relevant bands of the PPy homopolymer remained unaltered, and new absorption bands from the PEG side chains appeared (Fig. 5b). The absorption band at 2921 cm−1 is usually attributed to the large methylene units (–CH2–) from PEG chains; the bands at 1395 and 1446 cm−1 (not present in PPy) corroborate this assignment. The incorporation of AzbPy-g-PEG macromonomer into the copolymer formulation was particularly demonstrated when the time of electropolymerization increased from 300 s to 1000 s, due to the appearance of the sharp peak at 1727 cm−1, which is characteristic of ester linkages. Table S1† in summarizes the main absorption bands obtained for the monomers and polymers.
The structure of P(Py-co-AzbPy-g-PEG) was also corroborated by Raman spectroscopy, as reflected in Fig. 5c. The spectrum of PPy presents peaks at 1574 cm−1 and 1367 cm−1, which arise from the conjugated polymer backbone (CC stretching mode) and Py ring, respectively, and three bands at around 1090 cm−1, 971 cm−1 and 935 cm−1 that are related to the in-plane and out-of-plane deformations of N–H and C–H groups.79,80 The graft copolymers generated using θ = 500 and 1000 s also present broad bands at around 2890 to 2940 cm−1, which are attributed to the symmetric stretching vibrations of the methylene groups of the PEG chains.37,41
The grafting of hydrophilic, polar PEG chains to the conjugated polymer backbone is expected to affect its electrochemical activity and doping level. Control cyclic voltammograms of PPy deposited onto stainless steel showed the highest electroactivity with a voltammetric charge of 5.2 × 10−2 C (Fig. 6a). However, the control homopolymer displayed the highest loss of electrochemical activity (LEA = 48%) after 50 consecutive cycles, which has been attributed to the dense network of chemical crosslinks formed during the anodic polymerization process.76 Thus, the formation of cross-links, which is also promoted by oxidation–reduction cycles, favors the organization of polymer chains in compact structures, precluding the diffusion of the dopant ions through the polymer/electrolyte interface.
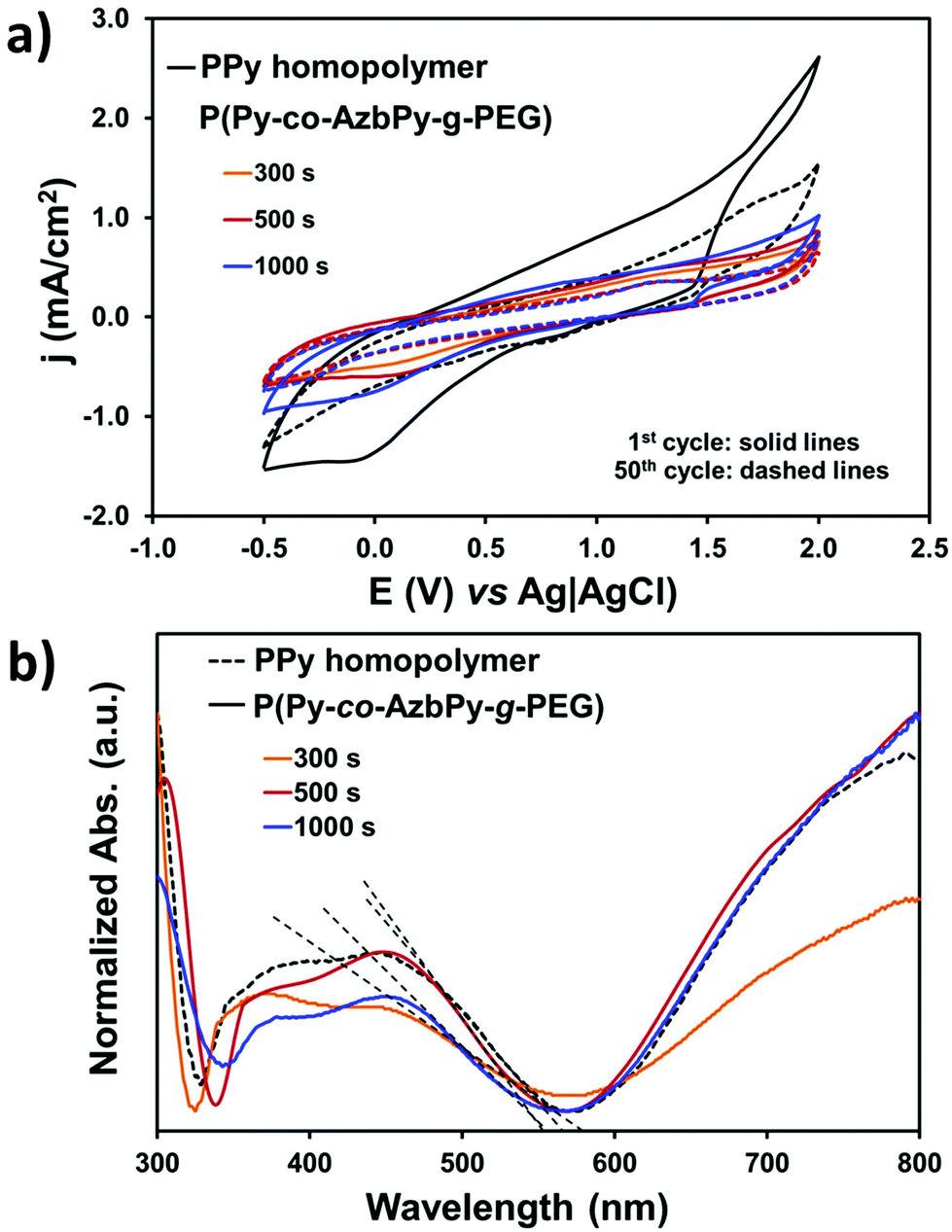 | ||
| Fig. 6 (a) Cyclic voltammograms (scan rate: 50 mV s−1) recorded in a 0.1 M LiClO4 acetonitrile solution for PPy generated using θ = 300 s, and P(Py-co-AzbPy-g-PEG) copolymers obtained using θ = 300, 500 and 1000 s: 2nd cycle (solid lines) and 50th cycle (dashed lines). (b) UV-vis spectra of the homopolymer and the copolymers freshly generated by CA onto ITO glass working electrodes. The dotted straight lines represent the best fit for the tangent of the band used to determine the εg for each system. | ||
On the other hand, the electroactivity of the P(Py-co-AzbPy-g-PEG) copolymers was less than half of that determined for the PPy homopolymer, even when θ = 1000 s. More specifically, the electrochemical activity experienced a decrease of 54% to 65% (2.8 to 3.4 × 10−2 C) upon incorporation of the AzbPy-g-PEG macromonomer. This is consistent with the well-known detrimental effect on heterocyclic ring conjugation caused by electron-donating substituents.81 However, the LEA was significantly lower for the grafted copolymers than for the homopolymer after fifty oxidation–reduction cycles, demonstrating that the new semiconducting copolymers present good electrochemical stability when consecutive redox processes are applied.
The UV-vis spectra displayed in Fig. 6b indicate that the electronic properties of P(Py-co-AzbPy-g-PEG) and PPy are quite similar; no detrimental effects due to the presence of the electron-donating groups were detected. The UV-vis spectra show an absorption band in the visible region (∼300 nm), which has been attributed to the π–π* transition in the tri-substituted benzene ring of AzbPy units. On the other hand, both the PPy and P(Py-co-AzbPy-g-PEG) copolymers present two broad absorption transitions starting at 320 nm and ending at ∼500 nm, which have been associated with the polaron and bipolaron bands of the conductive quinoid and aromatic forms of the heterocycle conjugated rings and Schiff base linkages.51 Additionally, another high absorption phenomenon starts at 600 nm, extending to values higher than 800 nm. This corresponds to the interaction of the dopant molecules with the polymer chains, reflecting the complete oxidation of the latter.82 This latter transition is practically identical for all copolymers, with the exception of P(Py-co-AzbPy-g-PEG) generated at θ = 300 s, which shows a slight reduction; this demonstrates that the doping levels are quite similar for all systems.
The band gap energy (εg) was calculated for each sample from the intersection of the dotted line, which corresponds to the tangent to the absorption band, and the absorption wavelength edge, as shown in Fig. 6b. The absorption maxima (λmax) and the εg values of the grafted copolymers and PPy are listed in Table S2.† The band gaps obtained for our systems are between 2.25 (PPy) and 2.30 eV (P(Py-co-AzbPy-g-PEG) at θ = 300 s). These values are in accordance with those reported by Brooke et al.83 for films of PPy with PEG-PP-PEG triblock copolymers which were prepared by vapor phase deposition.
Protein adsorption
In addition to their material properties, several features of proteins, such as their molecular weights, structures, shapes and electrostatic charges, can influence their non-specific interactions with a material surface, in particular with ICPs.84In the present study, bovine serum albumin (BSA) and lysozyme (Lyz) were used as model proteins for adsorption experiments. These proteins, which contain free cysteine residues (which can be exploited in conjugation studies or in chemical adsorption with other substances), have considerably different molecular weights (66.5 kDa for BSA and 14.3 kDa for Lyz). Furthermore, Lyz exhibits an ellipsoidal shape as a quaternary structure, in which both ordered (30% α-helix, 27% β-turn and 13% β-sheet secondary structures) and unordered (30%) regions coexist;85 meanwhile, the “heart”-shaped86 secondary structure of BSA is predominantly α-helical, with the remaining polypeptide occurring in turns and extended or flexible regions (i.e. with no β-sheets).87
The adsorptions of BSA and Lyz onto the surface of steel, PPy and P(Py-co-AzbPy-g-PEG) generated at θ = 1000 s are shown in Fig. 7a. As can be seen, the adsorptions of both BSA and Lyz were significantly higher on the graft copolymer film than on the PPy homopolymer. This has been attributed to the presence of PEG groups, which facilitates the interaction with the hydrophilic residues of the proteins [Referee #2].
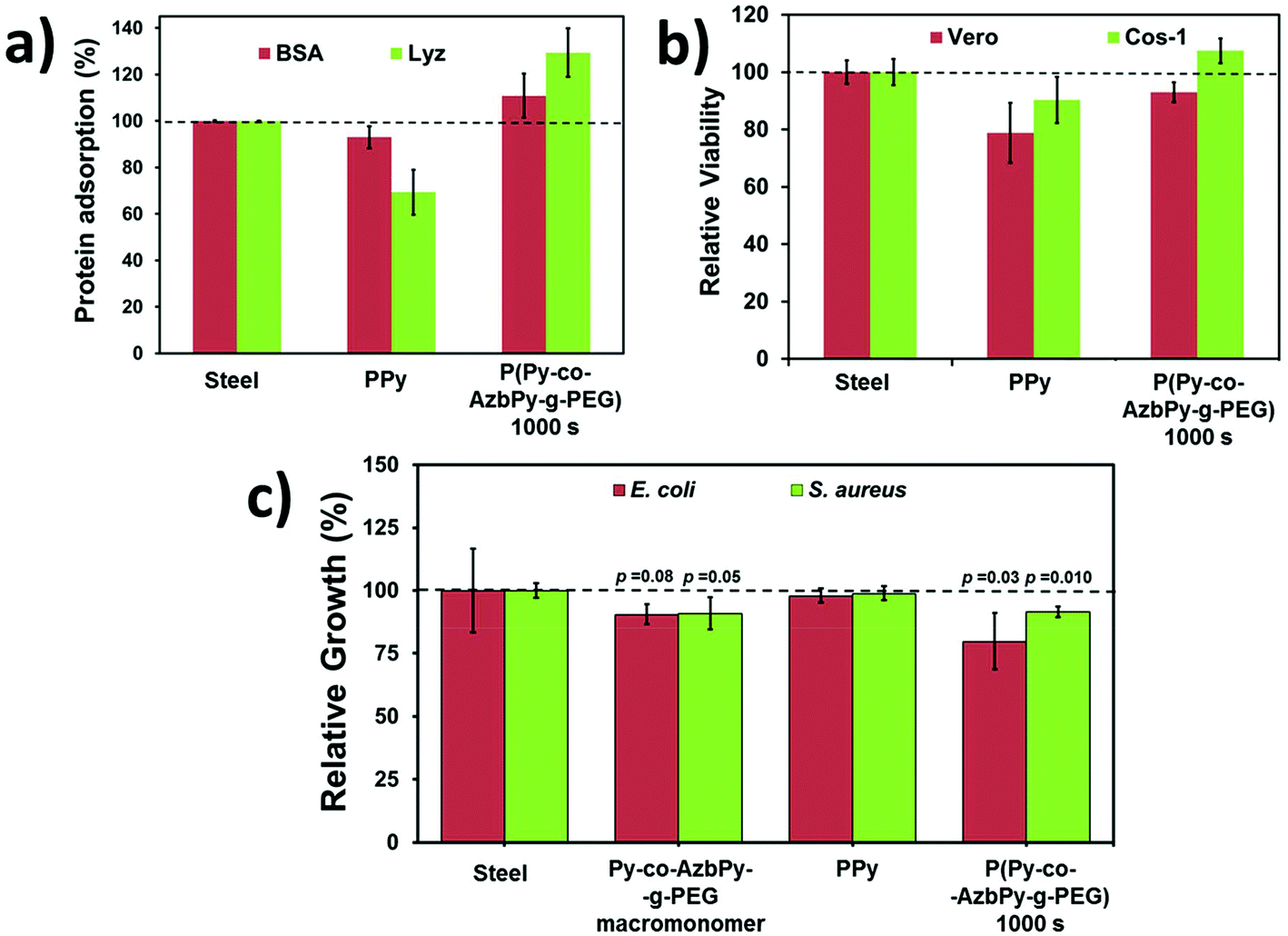 | ||
| Fig. 7 For PPy and P(Py-co-AzbPy-g-PEG) obtained using θ = 1000 s: (a) protein adsorption using BSA and Lyz; (b) cytotoxicity using Vero and COS-1 cultured cells; and (c) antimicrobial response using Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). The antimicrobial response of the AzbPy-g-PEG macromonomer is also evaluated in (c). Steel was used as a control substrate in all cases. Statistical analyses were performed with a confidence level of 95% (p < 0.05) using Student's T-test. | ||
In contrast, the N–H groups of PPy are expected to be sterically hindered; thus, they cannot readily interact with the polar residues of the proteins. Indeed, the adsorption of proteins on PPy is even lower than that observed on the steel substrate. These results are fully consistent with the hydrophilicity ranking determined by WCA measurements (i.e. 48.5° ± 0.3° and 58.9° ± 0.1° for P(Py-co-AzbPy-g-PEG) and PPy, respectively). Also, the low protein adsorption observed for PPy in comparison with P(Py-co-AzbPy-g-PEG) indicates that in the latter, the PEG groups play the most important role in the interactions with Lyz and BSA.
On the other hand, P(Py-co-AzbPy-g-PEG) exhibits higher affinity for Lyz than for BSA, even though the amount of charged residues is significantly higher in the latter than in the former. This result indicates the preference of the copolymer towards small ellipsoid proteins rather than bulky proteins organized in helical bundle domains, in which the attractive electrostatic interactions induced by many charged residues are shielded by residues located at neighboring secondary motifs.
On the other hand, comparison with previous results indicates that P(Py-co-AzbPy-g-PEG) exhibits higher affinity towards proteins than PTh5-g-PEG2000 and PTh5-g-PEG1000.3 More specifically, the adsorptions of BSA with respect to steel (100%) were reported to be 113% and 106% for PTh5-g-PEG2000 and PTh5-g-PEG1000, respectively, while the adsorption of collagen was ∼100% for these copolymers.3 These values are lower than those achieved by P(Py-co-AzbPy-g-PEG) for BSA (118%) and, especially, Lyz (137%) (Fig. 7a).
In vitro cytotoxicity and antimicrobial activity
The potential cytotoxicities of P(Py-co-AzbPy-g-PEG), PPy and steel were evaluated using the MTT assay for cell viability determination, which was conducted after 72 h of culturing Vero and COS-1 cells. No evidence of cytotoxicity was found for PPy or P(Py-co-AzbPy-g-PEG). However, differences in the number of cells adhering to the films were detected (Fig. 7b); the cell viabilities were 10% to 20% higher for P(Py-co-AzbPy-g-PEG) than for PPy. This behaviour has been attributed to the well-known biocompatibility of grafted PEG chains, which improves the cellular adhesion behaviour of the semiconducting polymer backbone. The influences of the steel, PPy and P(Py-co-AzbPy-g-PEG) films on Cos-1 cell morphology were qualitatively evaluated by SEM (Fig. S4†).On the other hand, it has been reported that materials with azomethine exhibit antimicrobial activity.88,89 The antimicrobial responses of PPy, P(Py-co-AzbPy-g-PEG) and AzbPy-g-PEG are compared in Fig. 7c. As can be seen, both the macromonomer and the graft copolymer exhibit some antibacterial response against Escherichia coli and Staphylococcus aureus, while the antimicrobial activities of steel and PPy are null. Previous studies indicated that the response of PPy to bacteria strongly depends on the dopant and redox state of the ICP and the characteristics of the bacterial strain.58,90,91
Although this effect may be due to the action of the Schiff base moiety, other explanations are also possible because the concentration at which the antibacterial test was performed (4.3 mM) is in approximately the same range as that (6 mM) in which “T-shaped” septithiophene oligomers grafted with PEG2000 formed self-assembled “core–shell” type micellar structures.92 In that case, the access to the Schiff base groups was hindered by the PEG shell of the micelles; therefore, the observed inhibitory effect may be due to the ability of PEG to complex metal cations, particularly Ca2+.93 Thus, displacement of Ca2+ ions from the outer membrane of bacterial cells toward AzbPy-g-PEG self-assembled micelles can disrupt the integrity of the bacterial cells,94 resulting in inhibition of their growth.
These results, combined with the observed cell viability, suggest that P(Py-co-AzbPy-g-PEG) is a promising candidate for the development of biomedical applications, such as implantable integrated devices for release of neurotransmitters under real-time monitoring. In the next section, we explore the potential of P(Py-co-AzbPy-g-PEG) for the sensitive detection of serotonin (5-hydroxytryptamine), which is a monoamine neurotransmitter biochemically derived from tryptophan that is primarily found in blood platelets and the central nervous systems of animals and humans.
Specific electrochemical detection capability of serotonin
Py is a well-known π-conjugated building block that is typically employed for engineering planar anionic receptors, acting as both a metal-coordination ligand (N sites) and a hydrogen-bonding donor (N–H moiety).95 Additionally, the complementarity between the electron-donor and electron-acceptor behavior of the Py ring and the serotonin neurotransmitter, respectively, is beneficial for the design of sensitive sensors.Fig. 8a and b compare the differential pulse voltammograms recorded in solutions with different concentrations of serotonin (from 0 to 20 μM) in 0.1 M PBS (pH 7.4) for PPy (θ = 300 s) and P(Py-co-AzbPy-g-PEG) (θ = 1000 s), respectively. As can be seen, both polymers are able to catalyze the oxidation of serotonin molecules in a low potential range. Although the anodic peak current decreases with decreasing serotonin concentration, the position of the peak potential in the copolymer film remains almost constant at 0.29 to 0.31 V for all the tested concentrations. In a very recent study, Cristea and co-workers96 prepared detectors of serotonin using electrochemically generated PPy nanoparticles which were decorated with catalytic gold nanoparticles. However, this P(Py-co-AzbPy-g-PEG) sensor presents important advantages with respect to the latter, such as the facile preparation of polymeric films without the need of activation with metal nanoparticles.
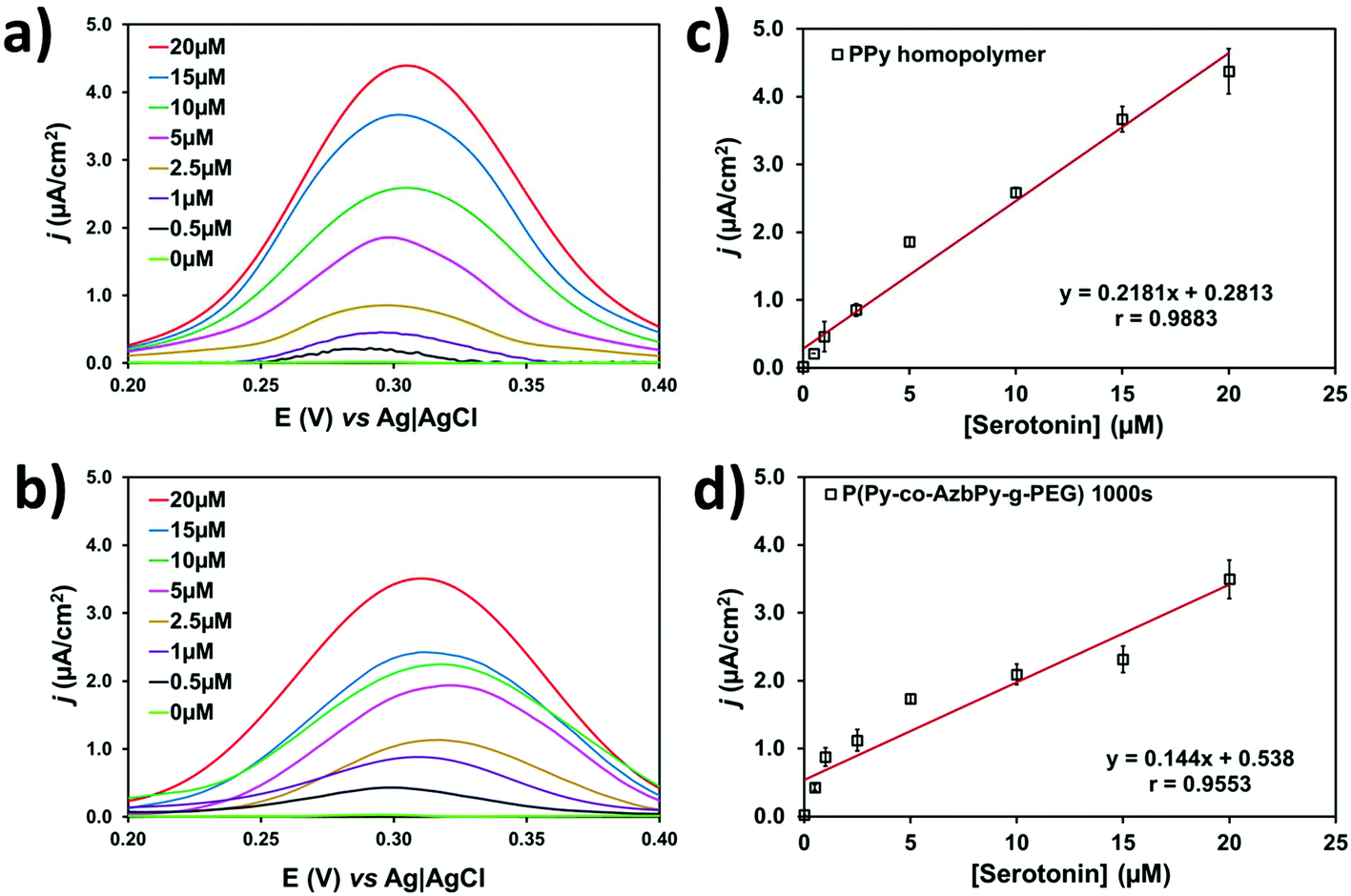 | ||
| Fig. 8 Differential pulse voltammograms in 0.1 M PBS (pH 7.2) with serotonin concentrations ranging from 0.5 to 20 μM for: (a) PPy and (b) P(Py-co-AzbPy-g-PEG) generated on glassy carbon electrode using θ = 300 and 1000 s, respectively. Calibration curves for serotonin detection in a concentration range from 0 to 20 μM for (c) PPy and (d) P(Py-co-AzbPy-g-PEG). | ||
The serotonin detection limits of the PPy and P(Py-co-AzbPy-g-PEG) sensors were derived from the variations of the maximum peak current density (j) against the serotonin concentration, as determined by DPV. The resulting calibration curves (Fig. 8c and d) show linear behavior in the whole interval of the examined serotonin concentrations. The detection limit is expressed as 3.3·σ/S, where σ and S are the standard deviation of the response and the slope of the calibration curve for serotonin concentrations ranging from 0.5 to 20 μM; the limits are 0.04 and 0.07 μM for PPy and P(Py-co-AzbPy-g-PEG), respectively.
In summary, the structural characteristics of P(Py-co-AzbPy-g-PEG) are suitable for complex biomedical applications in which several functions must be fulfilled simultaneously, such as implantable electrodes to monitor the level of serotonin. This type of electrode requires not only a high detection capacity, such as that imparted by the conjugated main chain of the copolymer, but also must present biocompatibility, provided by the PEG grafted chains, to facilitate its integration in the body, and antimicrobial activity, induced by the synergistic combination of the chemical and architectural peculiarities of the copolymer, to prevent infections after implantation. Additionally, the modified electrode obtained in this study is easy to prepare compared to other hybrid materials being developed, such as those combining metallic nanoparticles and carbon nanotubes97 or complex electrodes.98–100
Conclusions
In this work, chemically and architecturally complex copolymers containing Py ring and Schiff base functionalities in their backbones and PEG as grafted chains have been engineered and characterized. The properties of the obtained P(Py-co-AzbPy-g-PEG) have been compared with those of bare PPy homopolymer and other related copolymers from which they were inspired, such as (i) P(AzbT-co-Th3), in which an azomethine-containing Schiff base was copolymerized with Th3; and (ii) PTh-g-PEG, a family of grafted copolymers without Schiff base functionalities in which PEG side chains were anchored to the PTh backbone. The antimicrobial activity of the novel graft copolymer was enhanced compared to that of previously studied copolymers.Our results show that this synthetic approach, which sequentially combines the “macromonomer technique” with electrochemical polymerization, can be a useful alternative that enables the combination of otherwise incompatible building blocks to obtain multifunctional materials capable of encompassing diverse applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MINECO (MAT2015-69367-R) and the Agència de Gestió d'Ajuts Universitaris i de Recerca (2017SGR359). Support for the research by C. A. was received through the prize “ICREA Academia 2015” for excellence in research, funded by the Generalitat de Catalunya (Catalonia-Spain). B. G. M. is thankful to CONACYT agency for their financial support through a postgraduate scholarship (328467 CVU 621314). The authors are grateful to Prof. G. Lisa from the Faculty of Chemical Engineering and Environmental Protection of “Gheorghe Asachi” Technical University, Iasi, Romania for TGA and DSC measurements. The Romanian authors dedicate this paper to Professor Bogdan C. Simionescu on the occasion of his 70th birthday.References
- M. Rehahn and A. D. Schluter, Polymer, 1989, 30, 1054–1059 CrossRef.
- W. A. Goedel, N. S. Somanathan, V. Ekelman and G. Wegner, Makromol. Chem., 1992, 193, 1195–1206 CrossRef.
- A.-D. Bendrea, G. Fabregat, L. Cianga, F. Estrany, L. J. del Valle, I. Cianga and C. Alemán, Polym. Chem., 2013, 4, 2709–2723 RSC.
- T. F. Otero, Electrochim. Acta, 2016, 212, 440–457 CrossRef.
- M. E. Abdelhamid, A. P. O'Mullane and G. A. Snook, RSC Adv., 2015, 5, 11611–11626 RSC.
- T. F. Otero and J. G. Martinez, J. Mater. Chem. B, 2016, 4, 2069–2085 RSC.
- Y.-Z. Long, M.-M. Li, C. Gu, M. Wan, J.-L. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef.
- T.-H. Le, Y. Kim and H. Yoon, Polymers, 2017, 9, 150 CrossRef.
- S. Soylemez, S. O. Hacioglu, M. Kesik, H. Unay, A. Cirpan and L. Toppare, ACS Appl. Mater. Interfaces, 2014, 6, 18290–18300 CrossRef PubMed.
- G. Fabregat, E. Córdova-Mateo, E. Armelin, O. Bertran and C. Alemán, J. Phys. Chem. C, 2011, 115, 14933–14941 CrossRef.
- G. Fabregat, E. Armelin and C. Alemán, J. Phys. Chem. B, 2014, 118, 4669–4682 CrossRef PubMed.
- L. D. Sappia, E. Piccinini, W. Marmisollé, N. Santilli, E. Maza, S. Moya, F. Battaglini, R. E. Madrid and O. Azzaroni, Adv. Mater. Interfaces, 2017, 4, 1700502 CrossRef.
- N. Aydemir, J. Malmström and J. Travas-Sejdic, Phys. Chem. Chem. Phys., 2016, 18, 8264–8277 RSC.
- S. B. Thourson and C. K. Payne, Sci. Rep., 2017, 7, 10402 CrossRef PubMed.
- T. F. Otero and J. G. Martinez, Prog. Polym. Sci., 2015, 44, 62–78 CrossRef.
- C. Plesse, F. Vidal, D. Teyssié and C. Chevrot, Chem. Commun., 2010, 46, 2910 RSC.
- A. Puiggalí-Jou, P. Micheletti, F. Estrany, L. J. del Valle and C. Alemán, Adv. Healthcare Mater., 2017, 6, 1700453 CrossRef PubMed.
- D. Samanta, N. Hosseini-Nassab and R. N. Zare, Nanoscale, 2016, 8, 9310–9317 RSC.
- D. Samanta, J. L. Meiser and R. N. Zare, Nanoscale, 2015, 7, 9497–9504 RSC.
- Y. Wang, Y. Xiao and R. Tang, Chem. – Eur. J., 2014, 20, 11826–11834 CrossRef PubMed.
- D. Park, Y. Cho, S.-H. Goh and Y. Choi, Chem. Commun., 2014, 50, 15014–15017 RSC.
- C. Zhong, D. Ke, L. Wang, Y. Lu and L. Wang, Electrochem. Commun., 2017, 79, 59–62 CrossRef.
- J. Goding, A. Gilmour, P. Martens, L. Poole-Warren and R. Green, Adv. Healthcare Mater., 2017, 6, 1601177 CrossRef PubMed.
- C. Yu, C. Wang, X. Liu, X. Jia, S. Naficy, K. Shu, M. Forsyth and G. G. Wallace, Adv. Mater., 2016, 28, 9349–9355 CrossRef PubMed.
- A. Jonsson, S. Inal, L. Uguz, A. J. Williamson, L. Kergoat, J. Rivnay, D. Khodagholy, M. Berggren, C. Bernard, G. G. Malliaras and D. T. Simon, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9440–9445 CrossRef PubMed.
- R. Green and M. R. Abidian, Adv. Mater., 2015, 27, 7620–7637 CrossRef PubMed.
- B. Guo and P. X. Ma, Biomacromolecules, 2018, 19, 1764–1782 CrossRef PubMed.
- Y. Wu, B. Guo and P. X. Ma, ACS Macro Lett., 2014, 3, 1145–1150 CrossRef.
- R. Dong, X. Zhao, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 17138–17150 CrossRef PubMed.
- M. Xie, L. Wang, J. Ge, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 6772–6781 CrossRef PubMed.
- X. Zhao, B. Guo and P. X. Ma, J. Mater. Chem. B, 2015, 3, 8459–8468 RSC.
- M. Wang, S. Zou, G. Guerin, L. Shen, K. Deng, M. Jones, G. C. Walker, G. D. Scholes and M. A. Winnik, Macromolecules, 2008, 41, 6993–7002 CrossRef.
- B. G. Molina, A. D. Bendrea, L. Cianga, E. Armelin, L. J. del Valle, I. Cianga and C. Alemán, Polym. Chem., 2017, 8, 6112–6122 RSC.
- J. Zhang, J. Lei, R. Pan, Y. Xue and H. Ju, Biosens. Bioelectron., 2010, 26, 371–376 CrossRef PubMed.
- H. Zhao, B. Zhu, S.-C. Luo, H.-A. Lin, A. Nakao, Y. Yamashita and H. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 4536–4543 CrossRef PubMed.
- M. Waugaman, B. Sannigrahi, P. McGeady and I. M. Khan, Eur. Polym. J., 2007, 39, 1405–1412 CrossRef.
- A.-D. Bendrea, G. Fabregat, L. Cianga, F. Estrany, L. J. del Valle, I. Cianga and C. Alemán, Polym. Chem., 2013, 4, 2709–2723 RSC.
- A.-D. Bendrea, G. Fabregat, J. Torras, S. Maione, L. Cianga, L. J. del Valle, I. Cianga and C. Alemán, J. Mater. Chem. B, 2013, 1, 4135–4145 RSC.
- A.-D. Bendrea, L. Cianga, E. Hitruc, I. Titorencu and I. Cianga, Mater. Plast., 2013, 50, 71–78 Search PubMed.
- S. Maione, G. Fabregat, L. J. del Valle, A.-D. Bendrea, L. Cianga, I. Cianga, F. Estrany and C. Alemán, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 239–252 CrossRef.
- L. Cianga, A.-D. Bendrea, N. Fifere, L. E. Nita, F. Doroftei, D. Ag, M. Seleci, S. Timur and I. Cianga, RSC Adv., 2014, 4, 56385–56405 RSC.
- A. Iwan and D. Sek, Prog. Polym. Sci., 2008, 33, 289–345 CrossRef.
- C. I. Simionescu, I. Cianga, M. Ivanoiu, A. Duca, I. Cocarla and M. Grigoras, Eur. Polym. J., 1999, 35, 587–599 CrossRef.
- M. Grucela-Zajac, K. Bijak, E. Zaleckas, S. Grigalevicius, M. Wiacek, H. Janeczek and E. Schab-Balcerzak, Opt. Mater, 2014, 37, 543–551 CrossRef.
- A. Bolduc, S. Barik, M. R. Lenze, K. Meerholz and W. G. Skene, J. Mater. Chem. A, 2014, 2, 15620–15626 RSC.
- S. Tarkuc, E. Sahin, L. Toppare, D. Colak, I. Cianga and Y. Yagci, Polymer, 2006, 47, 2001–2009 CrossRef.
- D. Isik, C. Santato, S. Barik and W. G. Skene, Org. Electron., 2012, 13, 3022–3031 CrossRef.
- C. Mallet, M. Le Borgne, M. Starck and W. G. Skene, Polym. Chem., 2013, 4, 250–254 RSC.
- C. I. Simionescu, M. Grigoras, I. Cianga, I. Diaconu and A. Farcas, Polym. Bull., 1994, 32, 257–264 CrossRef.
- C. I. Simionescu, M. Grovu-Ivanoiu, M. Grigoras and I. Cianga, Angew. Makromol. Chem., 1994, 221, 103–115 CrossRef.
- C. I. Simionescu, M. Grovu-Ivanoiu, I. Cianga, M. Grigoras, A. Duca and I. Cocarla, Angew. Makromol. Chem., 1996, 239, 1–12 CrossRef.
- C. I. Simionescu, I. Cianga, M. Ivanoiu, A. Airinei, M. Grigoras and I. Radu, Eur. Polym. J., 1998, 35, 1895–1905 CrossRef.
- D. Colak, I. Cianga, A. E. Muftuoglu and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 727–743 CrossRef.
- A. Gul, Z. Akhter, M. Siddiq, S. Sarfraz and B. Mirza, Macromolecules, 2013, 46, 2800–2807 CrossRef.
- L. Marin, D. Ailincai, M. Calin, D. Stan, C. A. Constantinescu, L. Ursu, F. Doroftei, M. Pinteala, B. C. Simionescu and M. Barboiu, ACS Biomater. Sci. Eng., 2016, 2, 104–111 CrossRef.
- M. M. Pérez-Madrigal, L. Cianga, L. J. del Valle, I. Cianga and C. Alemán, Polym. Chem., 2015, 6, 4319–4335 RSC.
- F. A. G. da Silva Jr., J. C. Queiroz, E. R. Macedo, A. W. C. Fernandes, N. B. Freire, M. M. da Costa and H. P. de Oliveira, Mater. Sci. Eng., C, 2016, 62, 317–322 CrossRef PubMed.
- M. Golabi, A. P. F. Turner and E. W. H. Jager, Macromol. Chem. Phys., 2016, 217, 1128–1135 CrossRef.
- X. Qu and Z. Yang, Chem. – Asian J., 2016, 11, 2633–2641 CrossRef PubMed.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, Chem. Mater., 2018, 30, 2945–2953 CrossRef PubMed.
- M. S. Ganewatta, M. A. Rahman, L. Mercado, T. Shokfai, A. W. Decho, T. M. Reineke and C. Tang, Bioact. Mat., 2018, 3, 186–193, DOI:10.1016/j.bioactmat.2018.02.001.
- J. L. Kerstetter and W. Gramlich, J. Mater. Chem. B, 2014, 2, 8043–8052 RSC.
- L. Arnt, K. Nusslein and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3860–3864 CrossRef.
- D. G. Colak, I. Cianga, L. Cianga and Y. Yagci, Des. Monomers Polym., 2016, 19, 508–534 CrossRef.
- The values for Hansen solubility parameters, δt (in MPa1/2) of DMSO and acetone were taken by accessing the address http://www.accudynetest.com CrossRef PubMed, while that for PEG was considered from K. Adamska, A. Voelkel and A. Berlinska, J. Pharm. Biomed. Anal., 2016, 127, 202–206 CrossRef PubMed.
- W. G. Herkstroeter, J. Am. Chem. Soc., 1973, 95, 8686–8691 CrossRef.
- P. Douglas, C. Couture, D. Clarke, D. Reed, I. H. Sadler and T. Wear, J. Chem. Soc., Perkin Trans. 2, 1994, 1295–1298 RSC.
- B. Teixeira-Dias, C. Alemán, F. Estrany, D. S. Azambuja and E. Armelin, Electrochim. Acta, 2011, 56, 5836–5843 CrossRef.
- D. Aradilla, F. Estrany, E. Armelin, R. Oliver, J. I. Iribarren and C. Alemán, Macromol. Chem. Phys., 2010, 211, 1663–1672 CrossRef.
- G. Fabregat, E. Córdova-Mateo, E. Armelin, O. Bertran and C. Alemán, J. Phys. Chem. C, 2011, 115, 14933–14941 CrossRef.
- C. Ocampo, E. Armelin, F. Estrany, L. J. Del Valle, R. Oliver, F. Sépulcre and C. Alemán, Macromol. Mater. Eng., 2007, 292, 85–94 CrossRef.
- C. Alemán, F. Estrany, E. Armelin, R. Oliver and J. Casanovas, Polymer, 2007, 48, 6162–6169 CrossRef.
- D. Aradilla, F. Estrany, E. Armelin and C. Alemán, Thin Solid Films, 2012, 520, 4402–4409 CrossRef.
- F. Estrany, D. Aradilla, R. Oliver, E. Armelin and C. Alemán, Eur. Polym. J., 2008, 44, 1323–1330 CrossRef.
- D. Aradilla, F. Estrany, E. Armelin and C. Alemán, Thin Solid Films, 2010, 518, 4203–4210 CrossRef.
- C. Alemán, J. Casanovas, J. Torras, O. Bertrán, E. Armelin, R. Oliver and F. Estrany, Polymer, 2008, 49, 1066–1075 CrossRef.
- K. S. Teh, Y. Takahashi, Z. Yao and Y. W. Lu, Sens. Actuators, A, 2009, 155, 113–119 CrossRef.
- M. Omastova, M. Trchova, J. Kovarova and J. Stejskal, Synth. Met., 2003, 138, 447–445 CrossRef.
- V. Chandra and K. S. Kim, Chem. Commun., 2011, 47, 3942–3944 RSC.
- X. Fan, Z. Yang and N. He, RSC Adv., 2015, 5, 15096–15102 RSC.
- A. R. Murphy, J. Liu, C. Luscombe, D. Kavulak, J. M. J. Fréchet, R. J. Kline and M. D. McGehee, Chem. Mater., 2005, 17, 4892–4899 CrossRef.
- J. Arjomandi, A.-U.-H. A. Shah, S. Bilal, H. Van Hoang and R. Holze, Spectrochim. Acta, Part A, 2011, 78, 1–6 CrossRef PubMed.
- R. Brooke, D. Evans, P. Hojati-Talemi, P. Murphy and M. Fabretto, Eur. Polym. J., 2014, 51, 28–36 CrossRef.
- M. J. Higgins, P. J. Molino, Z. Yue and G. G. Wallace, Chem. Mater., 2012, 24, 828–839 CrossRef.
- M. S. Weiss, G. J. Palm and R. Hilgenfeld, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 952–958 CrossRef.
- M. L. Ferrer, R. Duchowicz, B. Carrasco, J. G. de la Torre and A. U. Acuna, Biophys. J., 2001, 80, 2422–2430 CrossRef PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef PubMed.
- N. Yilmaz Baran, M. Karakişla, H. Ö. Demir and M. Saçak, J. Mol. Struct., 2016, 1123, 153–161 CrossRef.
- C. M. Da Silva, D. L. Da Silva, L. V. Modolo, R. B. Alves, M. A. De Resende, C. V. B. Martins and A. De Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- M. Golabi, A. P. F. Turner and E. W. H. Jager, Sens. Actuators, B, 2016, 222, 839–848 CrossRef.
- W. Zhou, L. Lu, D. Chen, Z. Wang, J. Zhai, R. Wang, G. Tan, J. Mao, P. Yu and C. Ning, J. Mater. Chem. B, 2018, 6, 3128–3135 RSC.
- A.-D. Bendrea, L. Cianga, E. G. Hitruc and I. Cianga, Int. J. Polym. Anal. Charact., 2013, 18, 189–198 CrossRef.
- M. Pannuzzo, D. H. De Jong, A. Raudino and S. J. Marrink, J. Chem. Phys., 2014, 140, 124905, DOI:10.1063/1.4869176.
- L. A. Clifton, M. W. A. Skoda, A. P. Le Brun, F. Ciesielski, I. Kuzmenko, S. A. Holt and J. H. Lakey, Langmuir, 2015, 31, 404–412 CrossRef PubMed.
- Q. Li, Intelligent stimuli-responsive materials: from well-defined nanostructures to applications, John Wiley & Sons, ch. 12, 2013 Search PubMed.
- M. Tertiş, A. Cernat, D. Lacatiş, A. Florea, D. Bogdan, M. Suciu, R. Săndulescu and C. Cristea, Electrochem. Commun., 2017, 75, 43–47 CrossRef.
- I. Cesarino, H. V. Galesco and S. A. S. Machado, Mater. Sci. Eng., C, 2014, 40, 49–54 CrossRef PubMed.
- X. Jiang and X. Lin, Anal. Chim. Acta, 2005, 537, 145–151 CrossRef.
- J. Li and X. Lin, Sens. Actuators, B, 2007, 124, 486–493 CrossRef.
- S. Sharmaa, N. Singha, V. Tomara and R. Chandra, Biosens. Bioelectron., 2018, 107, 76–93 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8py00762d |
| This journal is © The Royal Society of Chemistry 2018 |