
New perspectives on aryl azide noncanonical amino acid use in yeast†
Eric M.
Tippmann
 *,
Seja
Culpepper
,
Wade
Bunnel
and
Nolan
Appel
*,
Seja
Culpepper
,
Wade
Bunnel
and
Nolan
Appel
Purdue University, 2101 E. Coliseum Blvd., Fort Wayne, IN 46805, USA. E-mail: tippmane@pfw.edu
First published on 9th October 2018
Abstract
A photochemically chemically active noncanonical amino acid para-azido-L-phenylalanine widely used in biology was found to be metabolized by Saccharomyces cerevisiae. Contrary to multiple reports, the azide moiety is not reduced to the corresponding amine. The amino acid's concentration was found to decline somewhat with time which was due, at least in part, to modification of the amino acid side chain. The metabolite was found to be photochemically active and further characterization concluded the azide moiety was still intact. This work also goes onto highlight paramount areas of concern with regards to (photo)chemical compatibility, handling, and fidelity in genetically encoding aryl azide amino acids.
The ability to site-specifically incorporate noncanonical amino acids (ncAAs) into proteins using an in vivo host is a well-established methodology to expand the chemical functionality of a given protein.1–3 The incorporation of ncAAs has been demonstrated across a variety of cell types, including: bacteria,4 yeast,5,6Xenopus oocytes,7Drosophila melanogaster,8 and other cell lines.9,10 In any given host, the ncAA should be: efficiently transported across the cell membrane, incorporated with high fidelity by a cognate aminoacyl tRNA synthetase (AARS),11 and not be detrimentally toxic or unstable.12 One widely used amino acid that meets these criteria is para-azidophenylalanine 1 (AzF; Fig. 1).5,13,14 Since the biological chemistry of disparate hosts may be as different as the (photo)chemistry of the ncAAs themselves there is a need to better understand the fate and/or impact of such ncAAs on the metabolic level. To this end, the metabolic profile of various amino acids were examined in yeast with a special emphasis on AzF.
 | ||
| Fig. 1 Noncanonical amino acids analyzed in yeast cultures. | ||
A photo- and chemically active amino acid, AzF has been introduced into a number of proteins. Although it enjoys widespread use,15–17 there are discrepancies observed across reports of AzF related to fidelity of incorporation but also the calculated and observed mass spectra for whole and digested peptide fragments harboring this amino acid.15–19 Still others have reported that aryl azides are efficiently reduced by nothing more than Saccharomyces cerevisiae (S. cerevisiae).20,21 With regards to the latter, if this organism efficiently reduced aryl azides, then this would undermine efforts to efficiently incorporate this amino acid – primarily because the amino acid is generally desired for the azido group's utility in bioconjugate18,19 and photochemical applications.22
The ncAA AzF was added to cultures of S. cerevisiae and the organic and aqueous extracts monitored by reverse phase HPLC. The concentration of AzF does decline somewhat with time (Fig. 2 and S1†). A new peak, with a longer retention time (rt) was observed (Fig. 3). The new peak was not observed in cultures of other ncAAs nor observed in control cultures in the absence of AzF (Fig. S2†) The longer rt of the product suggested a more nonpolar compound. The new metabolite was initially suspected to be 4-azidophenylpropanoic acid by deamination (Scheme 1; inset). Such a process appeared consistent with the literature: deamination at the amino acid side chain is known to precede decarboxylation during catabolism of phenylalanine,23 and deamination of phenylalanine leads to formation of 4-hydroxycinnamic acid and other products.24 To this end, 4-azidophenylpropanoic acid, 2, as well as 4-azidophenylpropenoic acid, 3, were synthesized using the procedure of Liu and Tor.25 HPLC analysis indicated that the rt of the metabolite product was different from the rt of both the synthesized azides (Fig. S3†). The metabolite was photolyzed alone as well as alongside azide 2; upon photolysis at 302 nm, the peaks for both compounds diminished, suggesting the possibility the metabolite retained the azide moiety (Fig. S4†). Prep-HPLC was used to isolate the metabolite and 1H NMR and FT-IR spectra were recorded and compared with an authentic sample (Fig. S5–S7†). The IR data suggested an intact aryl azide; the NMR is consistent with known compound: 2-(4-azidophenyl)ethanol.26,27 The metabolism seems general for other ncAAs considered here. Cultures were also separately supplemented with p-bromophenylalanine. 5, and p-benzoylphenylalanine, 6, and new metabolite peaks appeared with similar, but unique retention times approximating 2-(4-azidophenyl)ethanol. Furthermore, no aminophenylalanine (AmF) could be accounted for in AzF-supplemented cultures (Fig. S13†). Further identification of the metabolites derived from ncAAs 5 or 6 was not pursued.
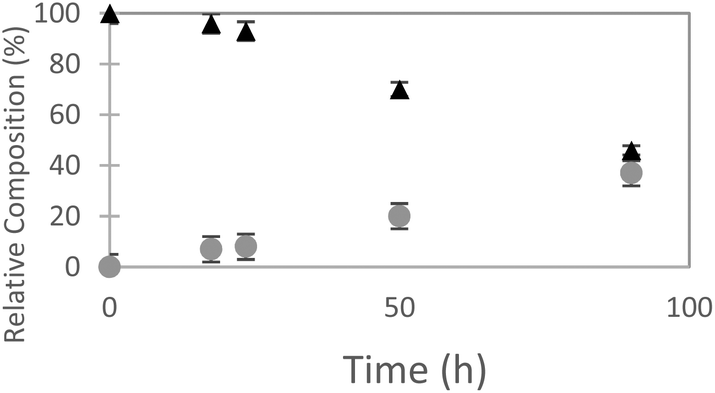 | ||
| Fig. 2 Relative composition of 4-azidophenylalanine (black triangles) to 2-(4-azidophenyl)ethanol (gray circles) monitored in S. cerevisiae cultures (orbital incubator, 30 °C. | ||
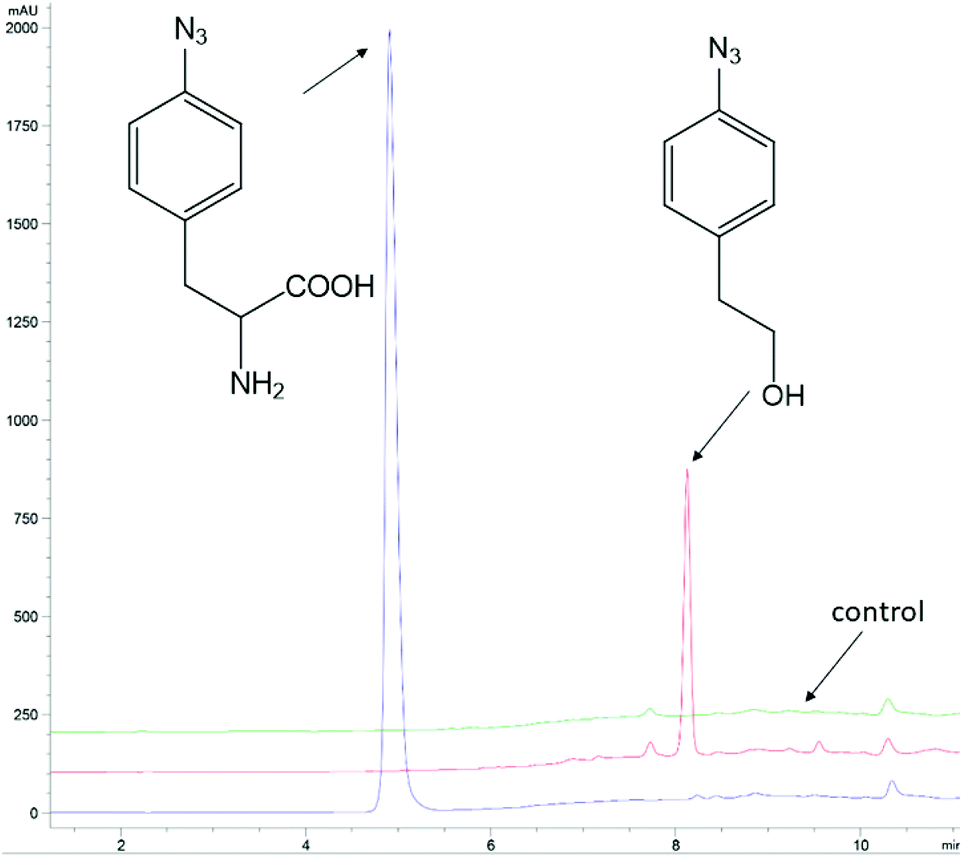 | ||
| Fig. 3 Analysis of 4-azidophenylalanine (AzF) in baker's yeast cultures. The blue trace is an AzF commercial solution. The red trace is HPLC analysis of a CH2Cl2 extract of a yeast culture (+) AzF at 2 h. The green trace is an HPLC analysis of a CH2Cl2 extract of a yeast culture (−) AzF at 2 h. Note: AzF remains in the aqueous growth media rather than partition with 4-azidophenylethanol into the organic CH2Cl2 layer. The HPLC traces are offset by 10% on the absorbance axis; the time axis has not been offset. The HPLC peaks were monitored at 254 nm. HPLC was performed with Method A (ESI†). | ||
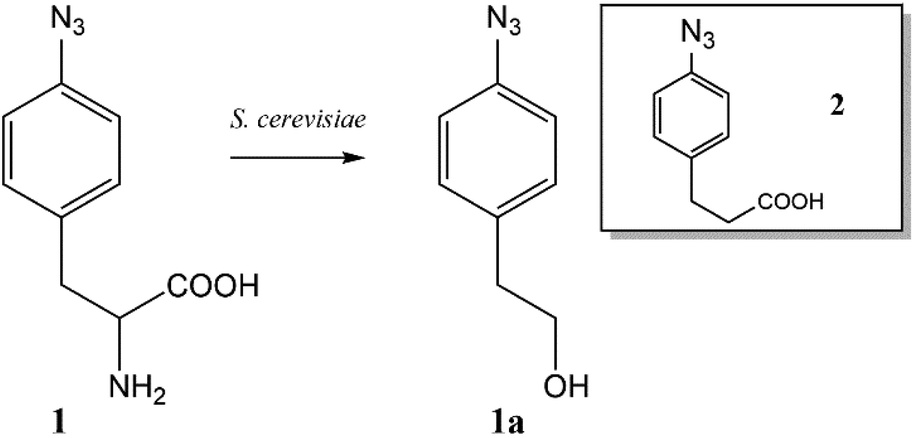 | ||
| Scheme 1 Metabolite 2-(4-azidophenyl)ethanol formed from azidophenylalanine supplemented yeast cultures. Inset: (4-Azido)phenylpropanoic acid as another suspected metabolite. | ||
With a variety of growth media conditions known, it is reasonable to expect differences in amino acid up-take, intracellular concentration, and metabolism.28 From a practical consideration though, the typical end-user is likely unconcerned by most metabolic processes. This is because the losses incurred here, combined with the examined culturing times – would not greatly imperil a typical genetic ncAA incorporation experiment which can succeed at sub-millimolar concentrations or may only require culturing overnight.29 Furthermore, accumulation of 2-(4-azidophenyl)ethanol is not of great concern to most users since this compound is an unlikely substrate to compete with AzF for tRNA aminoacylation. The latter scenario would not impact the fidelity of AzF incorporation, which is a paramount end-user concern (vide infra).
Finally, the notion of aryl azide reduction in yeast was revisited. It begs to reason that the many successful reports of AzF incorporation would not be possible if yeast efficiently reduced the azido group to an amino group in vivo. This is because each successful report utilizing AzF in yeast not only incorporates the ncAA, but typically has an application contingent upon an intact azide (i.e. photocrosslinking, 3 + 2 cycloadditions, infrared analysis).22 The end applications would not be impossible if the proteins harbored AmF, the reduced product of AzF (as well as AARS recognition would be impacted). Thus, it is interesting that is has been reported that yeast reduces a number of aryl azides of the general structure X-phenyl-N3 where X might be a 3-, or 4-substituted: –OMe, Cl, Br, Me, etc. In one report, the reduction was reported to be quick (i.e. 2–3 h) and proceeding in high yields (generally >80%).21 Using a commercial solution of 4-bromophenyl azide, we could not reproduce the reduction of this simple aryl azide by yeast using the reported conditions or by employing more rigorous conditions. Our culturing, extraction, and HPLC methodology afforded a detection limit on the order of nanomoles for 4-bromoaniline detection (Fig. S8–S10†). Multiple attempts ranging from the reported 2 h to even 4 days, or even using rich media, etc. routinely failed to improve upon the nanomolar amounts of 4-bromoaniline already present in commercial solutions of 1-azido-4-bromobenzene (Fig. 4). And although the previous studies reported high yields of amine from other aryl azides, we did not pursue the reduction of any of the other aryl azides listed in those reports.
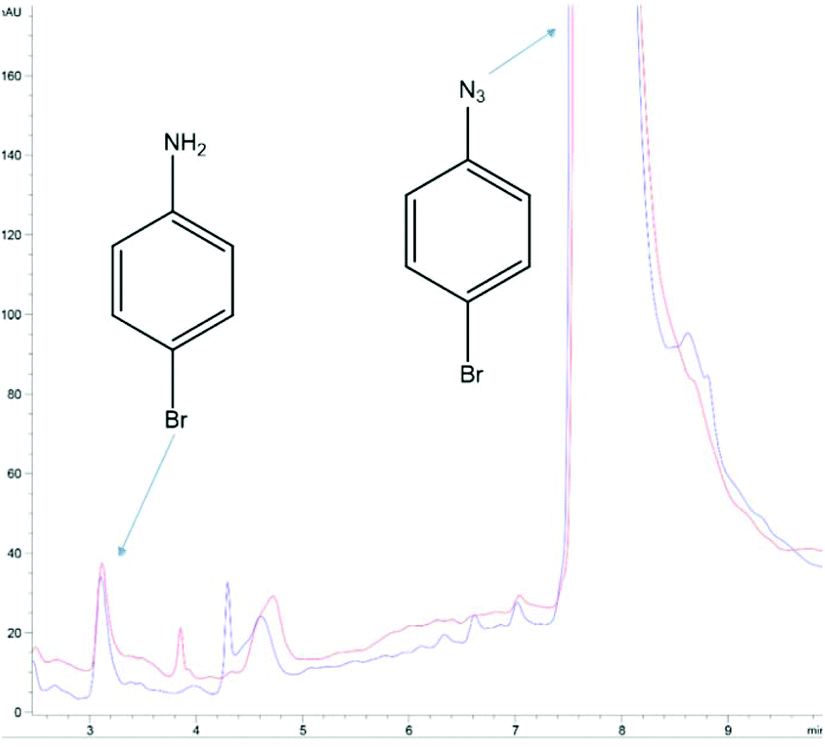 | ||
| Fig. 4 Null result determined by HPLC analysis of 1-azido-4-bromobenzene in yeast cultures. The blue trace is at t = 0 h; 125 μmole azide was added to a yeast culture (2 g Baker's yeast, 6 mL water, 2.5 mL methanol) and 500 μL of the yeast culture was immediately withdrawn, combined with 500 μL CH2Cl2, vortexed, and the organic layer separated for analysis. The red trace followed the same extraction protocol 5.5 h later. The area of the 4-bromoaniline peak at t = 0 suggests the commercial product contains ∼0.4% of the amine (from which the azide is manufactured). The analysis was performed with HPLC Method B (ESI†). The results were typical of >12 cultures of varying duration and conditions, including: (1) those reported by Baruah et al. (ref. 21) and (2) with glucose yeast extract rich media. | ||
Our observed lack of aryl azide reduction in yeast, coupled with successful reports in yeast requiring an intact AzF, suggests other explanations are required when AmF is observed or invoked in ncAA studies of AzF use in yeast. Since AzF is: (1) chemically, (2) photochemically and (3) thermally labile, the analysis techniques post-production of the ncAA-protein should be scrutinized. Prior to mass spectrometry or SDS-PAGE analysis, it is common to incubate protein solutions to reduce disulfide bonds with the reducing agent DTT. The laser used to vaporize samples in MALDI-TOF experiments would be expected to active some azides as is observed for photoactive ncAA functional groups like diazirines.30 Finally, electrospray MS techniques may employ injector temperatures of 150–300 °C, more than enough to decompose an aryl azide thermally. The actual (photo)chemistry of aryl azides has historically been convoluted due to the propensity of aryl azides to primarily form tar upon treatment with light or heat where a synthetic organic chemist might remark, “…phenylnitrene as a reagent is never described with complimentary adjectives”.31
Scheme 2 summarizes the reactivity of a phenylazide-phenylnitrene system modelled onto AzF. For a simple aryl azide, the main azide decomposition product, a singlet nitrene ring expands to a ketenimine – which is the crosslinking species on the singlet manifold.32 The AmF, which is so commonly invoked in studies utilizing AzF, may only form after intersystem crossing of the singlet nitrene to the triplet nitrene – a slower process for most simple aryl azides. Furthermore, H-abstraction to form aniline is also then unfavorable.33,34 If the AzF protein sample has not been reduced by DTT (Fig. S11†), then it is more likely that in some of the instances where an AmF mass is invoked it is actually an azepine, dehydroazepine or other intermediate from a nitrene (Scheme 2).
 | ||
| Scheme 2 Mechanism for the thermal or photochemical activation of aryl azide modelled onto azidophenylalanine, AzF. Intersystem crossing (isc) indicated for known singlet to triplet nitrene states. | ||
Indeed, as these azides become more widely used in biological applications, a variety of spectroscopic and molecular details have emerged to support the aforementioned nitrene chemistry. For example, the Jones’ group provided crystallographic evidence for a dehydroazepine-like side chain from in crystallo irradiation of a superfolder GFP148AzF mutant.35 In our own work, we have proposed the formation of AmF from AzF based on low temperature EPR.22 However, we made no claims as to the total yield of AmF, and we could not rule out products from the singlet nitrene manifold, which would be invisible to EPR. Our GFP66AzF study also differed from Jones’ GFP148AzF study in that position 66 incorporates AzF into the GFP chromophore.36 The conditions of low temperature, highly polar environment, and increased resonance energy from being in the chromophore would all affect nitrene singlet–triplet intersystem crossing.37 Relevant to most studies though is the fact that azepine, dehydrozepine, or even nitrene insertion reactions would give observed masses within about 1 Da of a putative AmF.
A closer look at AzF usage showed that many reports typically employ at least one of the three practices (i.e. reducing agent, ultraviolet light, or heat) which could activate or reduce an aryl azide. Thus, for some applications it may be that 4-azidomethyl phenylalanine (AzMF) would suffice. This ncAA removes the azide from conjugating the ring, increasing resilience to reduction, ESI-Q-TOF and UV light.38,39 TheAzMF amino acid is also apparently a more robust partner for cycloaddition reactions.40
Finally, the AmF vs. AzF dispute is also a concern for the fidelity of incorporation. Here, other observations may be helpful about the putative presence of AzF, AmF, dehydroazepine or even tyrosine. There is a noticeable decrease in fidelity from the initial report of AzF incorporated by the evolved E. coli aaRSs in yeast. The original report claimed high to excellent fidelity for the incorporation of AzF in S. cerevisiae.5 Subsequent reports have also reassessed the fidelity of this system in various ways.16,17,41 Fidelity in the use of orthogonal AARS-tRNA pairs is typically a measure of how well the mutant AARS has “forgotten” about its canonical amino acid. The standard experiment to ascertain fidelity is protein production in the presence (+) and absence (−) of the ncAA. In the absence of ncAA ribosomal synthesis should cease at UAG (in a high-fidelity result). In the original yeast AzF report, the fidelity experiment indicated a robust AARS for AzF (termed an AzRS) and ideal fidelity. Indeed, full length protein was only observed in the presence (+) of AzF, whereas no detectable protein in the absence (−) was reported. Moreover, a claim of up to 99.8% incorporation was made. The same fidelity was indirectly determined in the Sohn lab. They incorporated AzF into eight mutants of Aha1 using a AzRS provided from the original study.17 Their fidelity across these eight experiments was poor and produced significant protein in the absence (−) of AzF (Fig. S12†). Sohn and coworkers had previously16 observed similar marginal fidelity for the same AzRS when working in S. cerevisiae and Candida albicans with other amber mutants (although one should hesitate to draw strong conclusions with Candida albicans because little ncAA work has been performed in this model organism).
The work by Sohn and coworkers appeared shortly after a focused analysis of fidelity was published on the same E. coli AzRSs.41 A performance analysis of the original AzRSs indicated these enzymes did not incorporate appreciable amounts AzF, and in in vitro aminoacylation assays the AzRSs activated tyrosine but not AzF. This activation of tyrosine is consistent with Sohn's experiments in the absence of AzF (although mass spectrometry was not performed on the (−) AzF samples, the most likely contaminating amino acid is tyrosine for an evolved tyrosyl AARS. It is worth mentioning here the oft overlooked, or subtle feature that any given AARS may not truly be dedicated to one nonnatural amino acid, but rather tend to be promiscuous even with other non-natural amino acids. It should also be noted that this discussion only applies to a limited set of results, specifically those results using AzF with E. coli AARSs in yeast.29 The use of ncAAs in yeast is identical in concept to, but should not be confused with the robust use of the same AzF with evolved Methanocaldococcus jannaschii AzRS's in E. coli.14,42
Conclusions
The utility of genetically incorporating noncanonical amino acids allows one to surmount biological limitations using solutions rooted in synthetic and physical organic chemistry. However, there can be implications with the biological chemistry and certain organic functional groups.43,44 A good example is an aryl azide amino acid which is not appreciably reduced in yeast, and yet may be partially metabolized, and still possess chemical, thermal, and photochemical concerns. Thus, specific considerations may need to be given to synthetic and/or physical organic chemistry – perhaps not the primary concern for most cell biologists, for example. Failure to consider nitrene chemistry for example (or indeed consider any noncanonical functional group's chemistry), at a given stage of use may result, in the worst case – in an explosion,45 but more often than that in a failed application or inconclusive or convoluted spectra.Experimental
Experimental procedures, chromatograms and spectra are available in the ESI.†Culturing of S. cerevisiae/cell extracts
A rich media consisted of yeast extract (0.5 g L−1) and glucose (0.5 g L−1) which were autoclaved and filter sterilized, respectively. S. cerevisiae was incubated in water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH ratios as reported or in the rich media in an orbital shaker at RT or 30 °C. The target amino acid was dissolved in 1 mL water:NaOH (∼1 eq. NaOH was necessary for most amino acids to dissolve – with the exception AmF). 1-Azido-bromobenzene was purchased as a 0.5 M solution in tert-butyl methyl ether. Cells were extracted, vortexed and the aqueous layer was separated from CH2Cl2 by centrifuge (13000 rpm, 1 min). The resulting bottom organic layer was used to determine 4-bromoaniline: 1-azido-bromobenzene ratios.
:MeOH ratios as reported or in the rich media in an orbital shaker at RT or 30 °C. The target amino acid was dissolved in 1 mL water:NaOH (∼1 eq. NaOH was necessary for most amino acids to dissolve – with the exception AmF). 1-Azido-bromobenzene was purchased as a 0.5 M solution in tert-butyl methyl ether. Cells were extracted, vortexed and the aqueous layer was separated from CH2Cl2 by centrifuge (13000 rpm, 1 min). The resulting bottom organic layer was used to determine 4-bromoaniline: 1-azido-bromobenzene ratios.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author appreciates Indiana-Purdue University Fort Wayne for financial assistance. The author also thanks Kelsie Hilarie, and Carol Goss for technical assistance and Michael Hall, Madison Sido, and Jimmy Nguyen for a critical reading of manuscript drafts.Notes and references
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8650–8655 CrossRef CAS PubMed.
- S. Lepthien, L. Merkel and N. Budisa, Angew. Chem., Int. Ed., 2010, 49, 5446–5450 CrossRef CAS PubMed.
- R. Furter, Protein Sci., 1998, 7, 419–426 CrossRef CAS PubMed.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. W. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS PubMed.
- B. Wiltschi, W. Wenger, S. Nehring and N. Budisa, Yeast, 2008, 25, 775–786 CrossRef CAS PubMed.
- M. W. Nowak, P. C. Kearney, J. R. Sampson, M. E. Saks, C. G. Labarca, S. K. Silverman, W. Zhong, J. Thorson, J. N. Abelson and N. Davidson, et al. , Science, 1995, 268, 439–442 CrossRef CAS PubMed.
- T. Mukai, M. Wakiyama, K. Sakamoto and S. Yokoyama, Protein Sci., 2010, 19, 440–448 CAS.
- W. Liu, A. Brock, S. Chen and P. G. Schultz, Nat. Methods, 2007, 4, 239–244 CrossRef CAS PubMed.
- B. Shen, Z. Xiang, B. Miller, G. Louie, W. Wang, J. P. Noel, F. H. Gage and L. Wang, Stem Cells, 2011, 29, 1231–1240 CrossRef CAS PubMed.
- A. K. Antonczak, Z. Simova, I. T. Yonemoto, M. Bochtler, A. Piasecka, H. Czapinska, A. Brancale and E. M. Tippmann, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1320–1325 CrossRef CAS PubMed.
- D. Liu and P. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4780–4785 CrossRef CAS.
- S. Ye, T. Huber, R. Vogel and T. P. Sakmar, Nat. Chem. Biol., 2009, 5, 397–399 CrossRef CAS PubMed.
- J. Chin, S. Santoro, A. Martin, D. King, L. Wang and P. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- N. Shao, N. S. Singh, S. E. Slade, A. M. Jones and M. K. Balasubramanian, Sci. Rep., 2015, 5, 17196 CrossRef CAS PubMed.
- S. Palzer, Y. Bantel, F. Kazenwadel, M. Berg, S. Rupp and K. Sohn, Eukaryotic Cell, 2013, 12, 816–827 CrossRef CAS PubMed.
- M. Berg, A. Michalowski, S. Palzer, S. Rupp and K. Sohn, PLoS One, 2014, 9, e89436 CrossRef PubMed.
- A. Deiters, T. Cropp, D. Summerer, M. Mukherji and P. Schultz, Bioorg. Med. Chem. Lett., 2004, 14, 5743–5745 CrossRef CAS PubMed.
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782–11783 CrossRef CAS PubMed.
- A. Kamal, Y. Damayanthi, B. S. Narayan Reddy, B. Lakminarayana and B. S. Praveen Reddy, Chem. Commun., 1997, 1015–1016 RSC.
- M. Baruah, A. Boruah, D. Prajapati and J. S. Sandhu, Synlett, 1996, 1193–1194 CrossRef CAS.
- J. L. Morris, S. C. Reddington, D. M. Murphy, D. D. Jones, J. A. Platts and E. M. Tippmann, Org. Lett., 2013, 15, 728–731 CrossRef CAS PubMed.
- J. R. Dickinson, L. E. Salgado and M. J. Hewlins, J. Biol. Chem., 2003, 278, 8028–8034 CrossRef CAS PubMed.
- T. Vannelli, W. Wei Qi, J. Sweigard, A. A. Gatenby and F. S. Sariaslani, Metab. Eng., 2007, 9, 142–151 CrossRef CAS PubMed.
- Q. Liu and Y. Tor, Org. Lett., 2003, 5, 2571–2572 CrossRef CAS PubMed.
- J. R. Dickinson, L. Eshantha, J. Salgado and M. J. E. Hewlins, J. Biol. Chem., 2003, 278, 8028–8034 CrossRef CAS PubMed.
- Y. Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146–153 CrossRef CAS PubMed.
- J. A. Barnett, Yeast, 2008, 25, 689–731 CrossRef CAS PubMed.
- B. Wiltschi, Fungal Genet. Biol., 2016, 89, 137–156 CrossRef CAS PubMed.
- E. M. Tippmann, W. Liu, D. Summerer, A. V. Mack and P. G. Schultz, ChemBioChem, 2007, 8, 2210–2214 CrossRef CAS PubMed.
- M. S. Platz, Acc. Chem. Res., 1995, 28, 487–492 CrossRef CAS.
- K. L. Buchmueller, B. T. Hill, M. S. Platz and K. M. Weeks, J. Am. Chem. Soc., 2003, 125, 10850–10861 CrossRef CAS PubMed.
- W. T. Borden, N. P. Gritsan, C. M. Hadad, W. L. Karney, C. R. Kemnitz and M. S. Platz, Acc. Chem. Res., 2000, 33, 765–771 CrossRef CAS PubMed.
- J. Sankaranarayanan, S. Rajam, C. M. Hadad and A. D. Gudmundsdottir, J. Phys. Org. Chem., 2010, 23, 370–375 CrossRef CAS.
- A. M. Hartley, H. L. Worthy, S. C. Reddington, P. J. Rizkallah and D. D. Jones, Chem. Sci., 2016, 7, 6484–6491 RSC.
- L. Wang, J. Xie, A. A. Deniz and P. G. Schultz, J. Org. Chem., 2003, 68, 174–176 CrossRef CAS PubMed.
- N. J. Rau, E. A. Welles and P. G. Wenthold, J. Am. Chem. Soc., 2013, 135, 683–690 CrossRef CAS PubMed.
- T. Hosoya, T. Hiramatsu, T. Ikemoto, M. Nakanishi, H. Aoyama, A. Hosoya, T. Iwata, K. Maruyama, M. Endo and M. Suzuki, Org. Biol. Chem., 2004, 2, 637–641 RSC.
- C. G. Bazewicz, M. T. Liskov, K. J. Hines and S. H. Brewer, J. Phys. Chem. B, 2013, 117, 8987–8993 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS PubMed.
- S. Nehring, N. Budisa and B. Wiltschi, PLoS One, 2012, 7, e31992 CrossRef CAS PubMed.
- M. Amiram, A. D. Haimovich, C. Fan, Y. S. Wang, H. R. Aerni, I. Ntai, D. W. Moonan, N. J. Ma, A. J. Rovner, S. H. Hong, N. L. Kelleher, A. L. Goodman, M. C. Jewett, D. Soll, J. Rinehart and F. J. Isaacs, Nat. Biotechnol., 2015, 33, 1272–1279 CrossRef CAS PubMed.
- C. J. Pickens, S. N. Johnson, M. M. Pressnall, M. A. Leon and C. J. Berkland, Bioconjugate Chem., 2018, 29, 686–701 CrossRef CAS PubMed.
- A. K. Antonczak, Z. Simova and E. M. Tippmann, J. Biol. Chem., 2009, 284, 28795–28800 CrossRef CAS PubMed.
- M. B. Richardson, D. B. Brown, C. A. Vasquez, J. W. Ziller, K. M. Johnston and G. A. Weiss, J. Org. Chem., 2018, 83, 4525–4536 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8pp00243f |
| This journal is © The Royal Society of Chemistry and Owner Societies 2019 |