
Triphenylsulfonium topophotochemistry†
E.
Despagnet-Ayoub
 *a,
W. W.
Kramer
a,
W.
Sattler
b,
A.
Sattler
a,
P. J.
LaBeaume
c,
J. W.
Thackeray
c,
J. F.
Cameron
c,
T.
Cardolaccia
c,
A. A.
Rachford
c,
J. R.
Winkler
*a and
H. B.
Gray
*a
*a,
W. W.
Kramer
a,
W.
Sattler
b,
A.
Sattler
a,
P. J.
LaBeaume
c,
J. W.
Thackeray
c,
J. F.
Cameron
c,
T.
Cardolaccia
c,
A. A.
Rachford
c,
J. R.
Winkler
*a and
H. B.
Gray
*a
aBeckman Institute, California Institute of Technology, Pasadena, California 91125, USA. E-mail: despagne@caltech.edu
bThe Dow Chemical Company, Formulation Science, Core R&D, 400 Arcola Road, Collegeville, Pennsylvania 19426, USA
cDow Electronic Materials, 455 Forest Street, Marlborough, Massachusetts 01752, USA
First published on 3rd November 2017
Abstract
The products from the 193 nm irradiation of triphenylsulfonium nonaflate (TPS) embedded in a poly(methyl methacrylate) (PMMA) film have been characterized. The analysis of the photoproduct formation was performed using chromatographic techniques including HPLC, GPC and GC-MS as well as UV-vis and NMR spectroscopic methods. Two previously unreported TPS photoproducts, triphenylene and dibenzothiophene, were detected; additionally, GPC and DOSY-NMR spectroscopic analyses after irradiation suggested that TPS fragments had been incorporated into the polymer film. The irradiation of acetonitrile solutions containing 10% w/v PMMA and 1% w/v TPS in a 1 cm-path-length cuvette showed only a trace amount of triphenylene or dibenzothiophene, indicating that topochemical factors were important for the formation of these molecules. The accumulated evidence indicates that both products were formed by in-cage, secondary photochemical reactions: 2-(phenylthio)biphenyl to triphenylene, and diphenylsulfide to dibenzothiophene.
Introduction
Rigid media restrict the motion of embedded molecules, greatly reducing the number of allowed reaction pathways. Importantly, investigations of related solution and solid state photoreactions have established that differences in product distributions can be exploited in stereo- and regioselective syntheses.1We studied the photochemistry of the triphenylsulfonium (TPS) cation, a photoacid generator employed in photolithography,2 in both solution and rigid media employing 193 nm irradiation as well as other excitation sources. In earlier work on TPS reactions in solution, it was established that both in-cage and cage-escape products3–5 are formed, most likely by the heterolysis or homolysis of sulfur–carbon bonds. The heterolysis pathway produces a phenyl cation and diphenylsulfide, whereas a diphenylsulfinyl radical cation6 and a phenyl radical are the expected products from homolysis.3,7 Recombination reactions of the different fragments yielded 2-, 3- and 4-(phenylthio)biphenyl with one equivalent of acid. After cage-escape, the fragments reacted with the solvent or with fragments from another TPS molecule or other contaminants, generating diphenylsulfide, biphenyl, and acid (Scheme 1). An investigation of crystalline or finely ground solid onium salt photochemistry revealed decomposition pathways similar to those found in solution, but with increased amounts of in-cage products, likely owing to the greatly reduced fragment diffusion.8,9 There have been few prior reports of the photochemistry of TPS in polymeric matrices,10 only one of which with results from experiments involving 193 nm irradiation.11
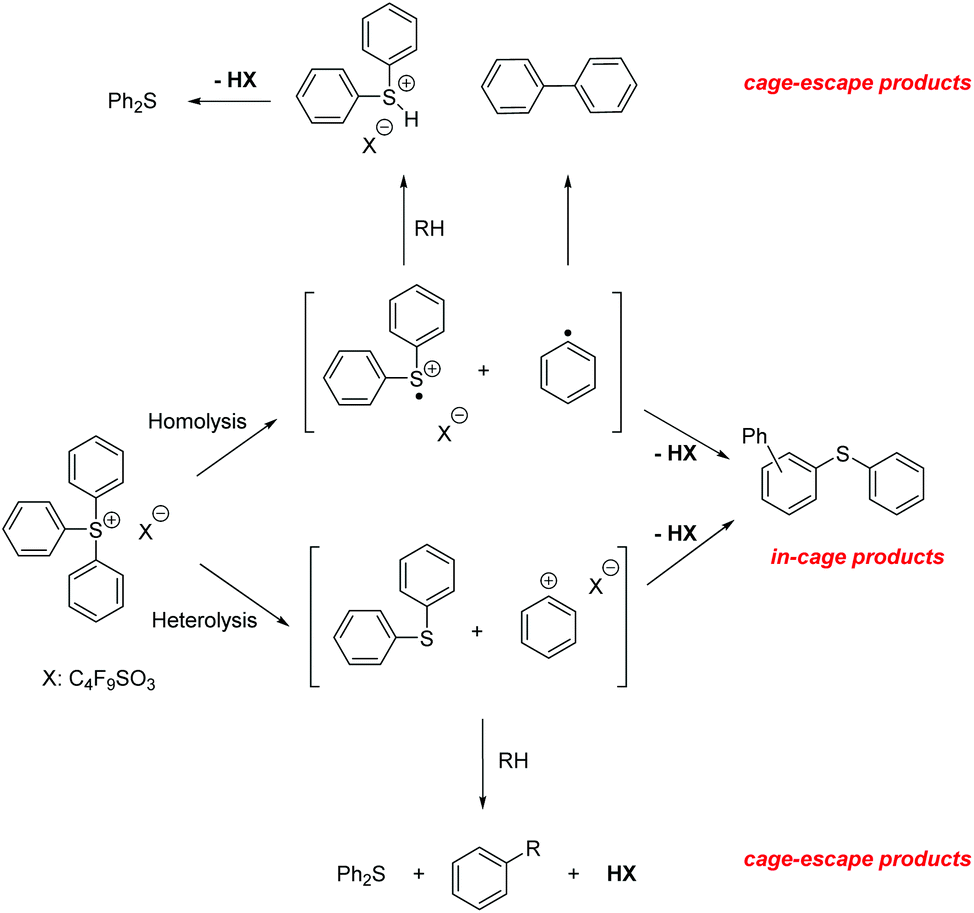 | ||
| Scheme 1 193 nm photochemistry of the triphenylsulfonium cation. | ||
As TPS is normally embedded in polymers in the photolithographic process, it is of importance to elucidate the photodecomposition pathways of this photoacid generator in confined environments, with the goal of establishing whether or not these pathways are qualitatively different from those observed in solution.
Notably, in our work on the 193 nm photochemistry of triphenylsulfonium nonaflate in multiple polymeric matrices, we found two products that were not observed in liquid phase experiments: the new topochemistry12 product is triphenylene, a desulfurized TPS derivative. Such discovery will also have a great impact on organic electronics; the introduction of TPS in polymer light emitting devices allowed the improvement of conductivity by charge injection. Therefore, the formation of polycyclic aromatic compounds during the irradiation process is an attractive characteristic as it will certainly favor even more the conductivity of these polymer films.13
Experimental
General methods
Poly(methyl methacrylate) (PMMA, average Mw ∼ 15 kDa by GPC, cas # 9011-14-7), poly(ethylene glycol) methyl ether (PEGME, Mw = 2000), 4-(phenylthio)biphenyl and tris(4-tert-butylphenyl)sulfonium nonaflate (tBu-TPS) were used as received from Aldrich. 2- and 3-(Phenylthio)biphenyl were synthesized according to a reported procedure.14 Triphenylsulfonium nonaflate (TPS) and poly((tert-butylmethacrylate)-co-(2-hydroxyethylmethacrylate)-co-(gamma-butyrolactonemethacrylate)) (poly(tBMA-co-HEMA-co-GBLMA)) with different weight-average molecular weights (Mw = 4600; 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; 5300 and 8600) were provided by The Dow Chemical Company. The reported weight-average molecular weights (Mw) were determined using narrow poly(styrene) calibration standards. Organic solvents (acetonitrile, DMSO, pentane and THF) were used as received from VWR. UV-vis absorption spectra were recorded on an HP 8453 spectrometer. In situ UV-vis spectra were recorded using an HP spectrometer (254 nm) and a B&W Tek spectrometer (193 nm). NMR spectra were recorded on a Bruker 400 with a Prodigy broadband cryoprobe and a broadband Smart probe (103Rh-31P plus 19F/1H) using Shigemi NMR tubes in MeOH-d4.
000; 5300 and 8600) were provided by The Dow Chemical Company. The reported weight-average molecular weights (Mw) were determined using narrow poly(styrene) calibration standards. Organic solvents (acetonitrile, DMSO, pentane and THF) were used as received from VWR. UV-vis absorption spectra were recorded on an HP 8453 spectrometer. In situ UV-vis spectra were recorded using an HP spectrometer (254 nm) and a B&W Tek spectrometer (193 nm). NMR spectra were recorded on a Bruker 400 with a Prodigy broadband cryoprobe and a broadband Smart probe (103Rh-31P plus 19F/1H) using Shigemi NMR tubes in MeOH-d4.
General film preparation and irradiation of TPS-PMMA
A solution of 10% w/v PMMA (1 g) and 1% w/v triphenylsulfonium nonaflate (TPS) (0.1 g) in tetrahydrofuran (10 mL) was spin-coated on a 1.5 in diameter fused-silica disc at a rotation rate of 3000 rpm for 30 s. The film thickness was determined using a Dektak XT Stylus profilometer and found to be 0.8 μm.Irradiation was performed using one of three different light sources. The 193 nm irradiation employed a home-built nitrogen-purged exposure chamber equipped with a D2-lamp and a 193 nm interference filter (20 ± 5 nm FWHM, Tmax = 15%) and a 1 cm water filter (emission spectrum: see Fig. S1 in the ESI,† dose <10 μW as measured by using a high-sensitivity thermal power sensor (ThorLabs S401C)). 254 nm radiation was produced using a krypton fluoride excimer mirror to direct the output from a Xe arc lamp through two 1 cm-path-length water infrared filters and a 254 nm interference filter. The irradiation power measured with a photodiode (ThorLabs Standard Photodiode Power Sensor (S120C), Si, 200–1100 nm) was 360 μW (360 μJ s−1) (emission spectrum: see Fig. S2 in the ESI†). Coated discs were placed in front of the light source for 3 h (Xe-lamp) or 3 days (3 d) (D2-lamp). Pulsed laser excitation (266 nm, 8 ns) was from the fourth harmonic of a Q-switched Nd:YAG laser (Spectra-Physics Quanta-Ray PRO-Series) operating at 10 Hz. A mechanical shutter was used to select individual pulses at a reduced repetition rate (1 Hz).
Crossover experiment in a tBu-TPS/TPS-PMMA film
A solution of 10% w/v PMMA, 0.5% w/v TPS, and 0.5% w/v tBu-TPS in THF was spin-coated on a 1.5-inch diameter fused-silica disc using the conditions described above. The disc was irradiated at 193 nm for 3 d after which the film was dissolved in acetone and evaporated to dryness. 1H NMR (400 MHz, MeOH-d4): δ 8.74 (m, triphenylene), 8.66 (d, 3JHH = 5.6 Hz, tBu-triphenylene), 8.63 (s, tBu-triphenylene); other signals were hidden under TPS peaks.Irradiation of solution samples
Solution phase experiments were conducted with 193 and 254 nm irradiation under the same conditions described for the TPS-PMMA films. Experiments were performed in a 1 cm fused-silica cuvette; solutions of 10% w/v PMMA and 1% w/v TPS (17.8 mM TPS concentration) in CH3CN were first purged with an inert gas and cooled in an ice bath to limit evaporation. The samples were then irradiated with constant stirring. After irradiation, 100 μL of the reaction mixture was diluted with approximately 2 mL DMSO. The resulting solution was filtered through a Celite plug and further diluted to a final volume of 10 mL with DMSO to obtain samples for HPLC analysis.Irradiation of wafers in a semiconductor fabrication facility at Dow Electronic Materials
A solution of poly(methyl methacrylate) (PMMA, 5 g) and triphenylsulfonium nonaflate (TPS, 0.5 g) in propylene glycol monomethyl ether acetate (PGMEA, 50 g) was prepared by mixing at room temperature and then rolling overnight. Six different 200 mm silicon wafers were first primed with hexamethyldisilazane (HMDS) at 120 °C for 30 seconds. AF40A bottom antireflective coating (BARC) was then spin-coated and baked at 205 °C for 60 seconds to give a thickness of 80 nm on each wafer. The PMMA/TPS/PGMEA formulation was then spin-coated onto each wafer at 1500 rpm, and baked at 95 °C for 60 seconds to give film thicknesses of approximately 320 nm. An exposure ladder was accomplished using an ASM-L/1100 lithography tool. The center doses were set to 0, 10, 20, 30, 40 and 50 mJ cm−2. A center portion of each wafer (ca. 1.5 × 3 inches) was then cleaved, which was extracted with either THF (GPC analysis) or DMSO (HPLC analysis).The absorbance k-value was determined using a 320 nm thick film, soft-baked at 95 °C for 60 s on 200 mm silicon wafers, and a J. A. Woollam WVASE32 vacuum ultraviolet variable angle spectroscopic ellipsometer using polarized light. Changes to the polarized light phase and amplitude after it passed through the coated film were observed to cover 180–900 nm using a range of 1.4 eV to 6.875 eV in increments of 0.0375 eV and three incident angles of 65, 70, and 75 degrees. These raw data were fit to a model using J. A. Woollam software to determine the real and imaginary optical constants of the film at each wavelength.
Chromatography
After irradiation, the film was dissolved in 1 mL DMSO and filtered through Celite. The resulting solution was then analyzed by high performance liquid chromatography (Agilent 1100 series preparative HPLC) and eluted through an InertSustain® Phenyl column (GL Sciences, 5 μm, 4.6 × 250 mm) at a flow rate of 0.7 mL min−1 using a UV detector at 254 nm (Agilent 1100 Variable Wavelength Detector). A gradient elution was used starting from 70:30 (v/v) H2O/CH3CN up to 30:70 (v/v) H2O/CH3CN over a period of 60 min, and then held at 30:70 (v/v) H2O/CH3CN for 10 min. 50 μL samples were injected by using an auto-sampler. Due to the poor separation of the three (phenylthio)biphenyl isomers, a MATLAB program was written to fit the HPLC data and quantify each isomer based on reference solutions. Each product was identified by the retention time and spiking with pure compounds. Calibration curves were generated for the quantification of all photoproducts.
The detection of the remaining TPS in the reaction mixture employed a Zorbax SB-C18 column (5 μm, 4.6 × 150 mm) with UV-detection at 214 nm. The eluent used was 50:50 (v/v) CH3CN/(H2O + 0.1% TFA) for 15 min, and then held at 100% CH3CN for 10 min to wash the column.
Gas chromatography-mass spectrometry (GC-MS) was conducted on a MSD5972/GC5890 with an HP6890 autosampler and an electron ionization (EI) method. The column was a DB-5 30 m × 0.25 mm × 0.25 μm film. The injector was set at 250 °C and the detector at 280 °C. The procedure: 50 °C for 2 min; 15 °C for 15 min; and then 270 °C for 3 min.
Gel permeation chromatography (GPC) employed a Waters Alliance system with an e2695 Separations Module using differential refractive index and photodiode array detection and THF (1.2 mL min−1) as the elution solvent (30 °C).
DOSY-NMR performed in DMSO-d6 was a DgcsteSL (DOSY gradient compensated stimulated echo Spin Lock) experiment15 with a diffusion delay of 50 ms, a difference in gradient pulse of 5 ms and an array of 15 gradient values.
Results and discussion
Detection of secondary photoproducts
Fused-silica discs spin-coated with a solution of 10% w/v PMMA and 1% w/v TPS in tetrahydrofuran afforded 0.8 μm films (measured by profilometry). UV-vis spectroscopy of the as-prepared films revealed absorption bands at 268 and 276 nm that are characteristic of the TPS starting material (Fig. 1).16 PMMA films containing 1% w/v TPS were then irradiated at 193 nm for 3 d under an N2 atmosphere. The UV-vis spectra of irradiated films revealed the presence of a new feature at 260 nm that displayed a prominent vibronic structure (Fig. 1). This feature indicates that there is minor structural distortion in the electronic excited state, consistent with a molecule containing fused/multiple aromatic rings.17 This signal did not match the UV-vis spectrum of the starting PMMA-TPS film, or of the spin-coated PMMA films containing previously reported photoproducts (Scheme 1). In situ UV-vis spectroscopy showed that the formation of this photoproduct increased over the course of 3 d irradiation (Fig. S3 in the ESI†).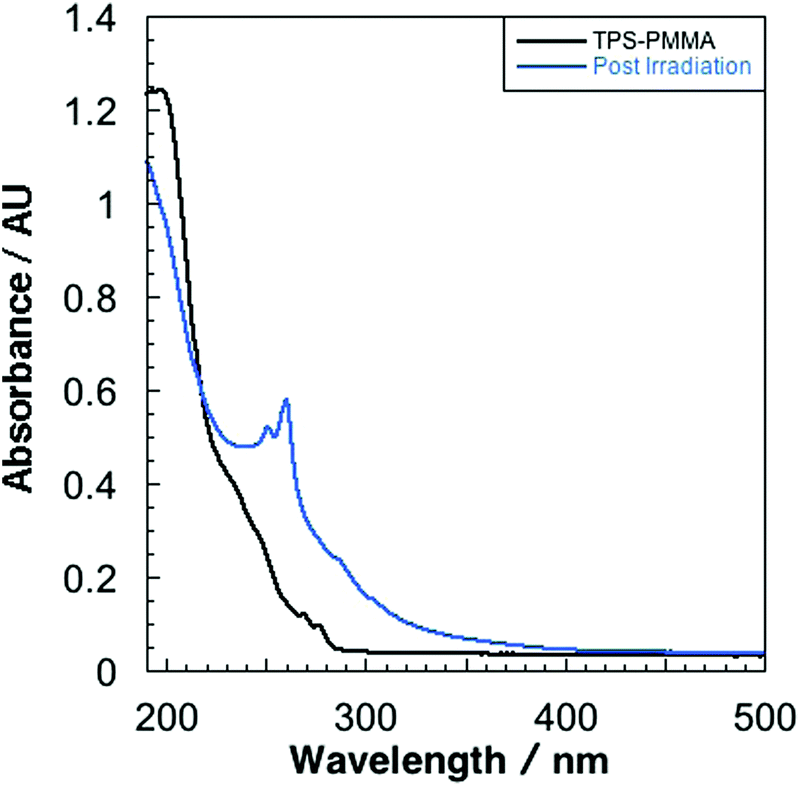 | ||
| Fig. 1 UV-vis spectra of PMMA/TPS films: before and after (3 d) 193 nm irradiation. | ||
HPLC was employed to separate and identify the different photoproducts present in the irradiated TPS-PMMA films. The HPLC trace (Fig. 2) shows the presence of several compounds in the reaction mixture. Many of these peaks were assigned by comparing their retention times in HPLC and GC-MS with those of previously reported photoproducts 2-, 3- and 4-(phenylthio)biphenyl (2-BiPhSPh, 3-BiPhSPh, and 4-BiPhSPh), biphenyl (PhPh), and diphenylsulfide (PhSPh).3,5,18 However, two peaks with retention times of ∼43 and ∼51 min did not match any known photoproducts. The product with the retention time of 43 min was identified as dibenzothiophene by GC-MS and by spiking an HPLC sample with a small amount of the pure substance. The product with retention time of 51 min was isolated by prep-HPLC and characterized by NMR spectroscopy. The 1H NMR spectrum of the product showed only two downfield signals (8.87 and 7.70 ppm), consistent with a highly symmetric aromatic ring system. GC-MS of the product gave a molecular weight of 228 m/z. Based on the 1H NMR and GC-MS analyses, we concluded that the photoproduct was triphenylene. We further confirmed this assignment by comparing the UV-vis spectrum, HPLC retention time, and 1H NMR spectrum with those of pure triphenylene.
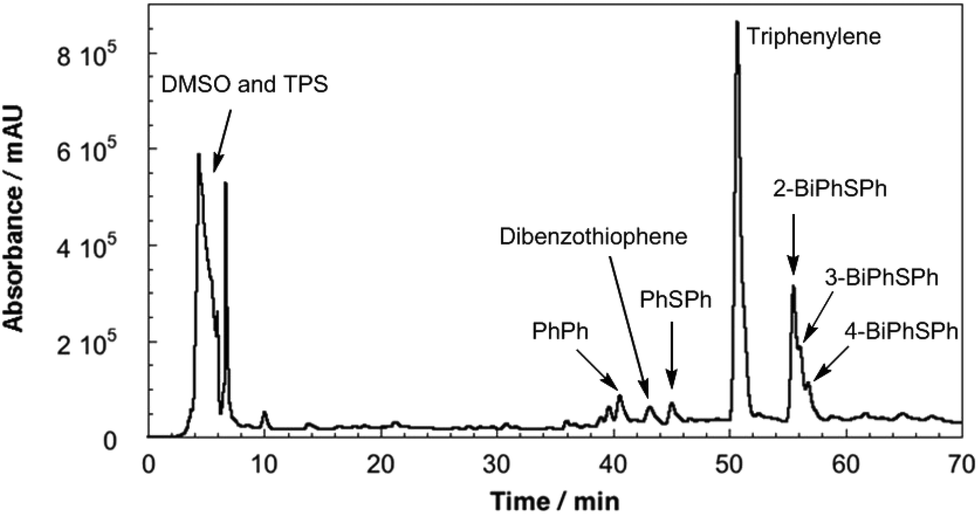 | ||
| Fig. 2 HPLC chromatogram (absorbance at 254 nm) of PMMA/TPS film after 193 nm irradiation for 3 d. | ||
As neither dibenzothiophene nor triphenylene has been previously identified as a product of TPS photolysis, additional control experiments were conducted in order to validate the assignment. The influence of the coating solvent as well as the identity of the polymer and irradiation wavelength were investigated by monitoring the appearance of triphenylene by UV-vis spectroscopy of the irradiated films. Triphenylene formation was not affected by the use of PGMEA or cyclohexanone as the coating solvent in place of THF, and the use of polymers other than PMMA had no effect on triphenylene formation. Indeed, the UV-vis spectra of the irradiated films of 1% w/v TPS in PEGME and poly(tBMA-co-HEMA-co-GBLMA) with different molecular weights (Mw = 4600; 14000; 5300 and 8600) (Fig. S4 and S5 in the ESI†) clearly demonstrated the presence of triphenylene, as evidenced by the sharp vibronic feature at 260 nm. Additionally, we investigated the effect of the irradiation wavelength by testing the TPS-PMMA film after exposure to 254 nm light from a Xe-lamp. The in situ UV-vis spectra of these films under 254 nm irradiation (Fig. S6 in the ESI†) show triphenylene generation (260 nm signal), reaching a maximum at 3 h exposure followed by a small decrease in band intensity. Triphenylene was observed in all cases.
The photoproducts from the 193 and 254 nm irradiation of TPS-PMMA films were quantified by HPLC. Irradiation for 3 h with 254 nm radiation led to quantitative conversion of TPS, whereas irradiation for 3 d with 193 nm radiation resulted in 90% conversion (Table 1). In each case the main product was the in-cage molecule, 2-(phenylthio)biphenyl (2-BiPhSPh), in addition to smaller amounts of triphenylene; substantial amounts of cage-escape products (diphenylsulfide (PhSPh), dibenzothiophene) are observed. At both irradiation wavelengths, the photoproduct distribution favored in-cage products over cage-escape ones (77% in both cases), as expected due to limited diffusion through the polymer film.9
The material recovered only accounts for 47% (or 27%) of the initial TPS after the 193 (or 254) nm exposure despite ≥90% consumption of the starting material. This finding suggests that many TPS fragments reacted with PMMA. Indeed, GPC analysis of the PMMA-TPS film after irradiation showed increased near-UV absorption (260 nm) for irradiated samples compared to PMMA itself (Fig. 3), indicating that aromatic TPS fragments were incorporated in the polymer matrix. Furthermore, a DOSY-NMR experiment was performed on an irradiated film showing aromatic signals matching the diffusion rate of the PMMA polymer (Fig. S7 in the ESI†).
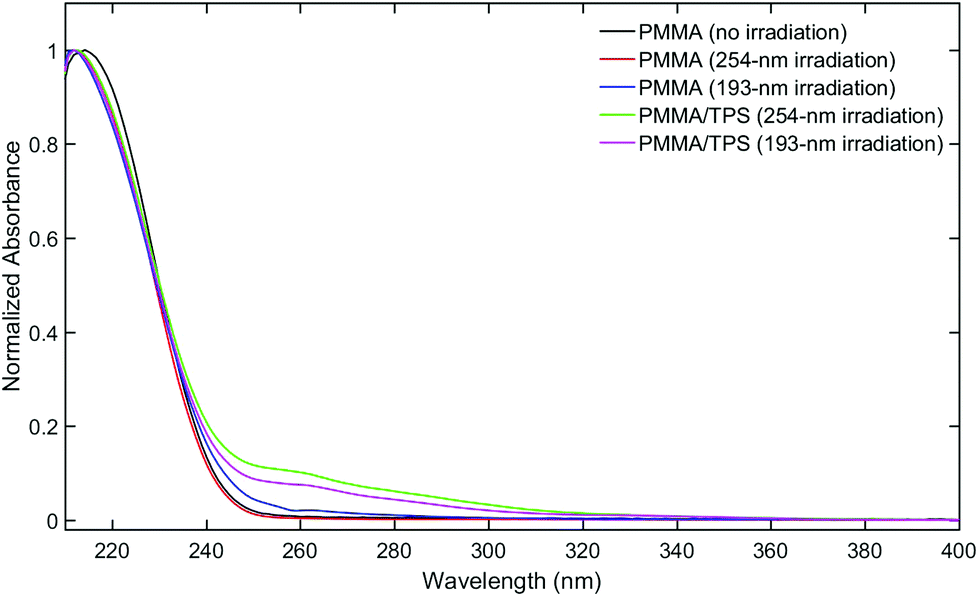 | ||
| Fig. 3 UV-vis spectra of high MW GPC fractions of films before and after irradiation with and without TPS. | ||
TPS solution photoproducts
Solution phase experiments were conducted in order to evaluate whether the formation of triphenylene as a TPS photoproduct was dependent on sulfonium salt immobilization in the polymer. Acetonitrile solutions containing 10% w/v PMMA and 1% TPS w/v were irradiated for 3 d (193 nm) and 3 h (254 nm). The UV-vis spectra of these solutions after irradiation showed no evidence of the formation of triphenylene (Fig. S8 in the ESI†). Irrespective of the irradiation wavelength, only 4 photoproducts were detected by HPLC: acetanilide, PhSPh, 2-BiPhSPh, and 4-BiPhSPh (Fig. S9, S10 and Table S1 in the ESI†). Neither triphenylene nor dibenzothiophene could be detected. The variation in the irradiation wavelength had no effect on the distribution of these photoproducts. The ratio of the in-cage products 2-BiPhSPh and 4-BiPhSPh to the cage-escape product PhSPh was 1.5:1 for both the 193 and 254 nm irradiated samples. Photoproduct concentrations were much greater in samples irradiated at 254 nm, in good agreement with the TPS consumption data. Note that there was 25% loss of initial TPS concentration after 3 h of the 254 nm irradiation, but there was much less TPS loss (7%) after 3 d of the 193 nm irradiation, a difference attributable to the lower power of the 193 nm source.
Solution experiments showed lower TPS conversion compared to in-film experiments (3 d under 193 nm irradiation: 7% vs. 90%; 3 h under 254 nm irradiation: 25% vs. 100%). To confirm that detectable quantities of triphenylene or dibenzothiophene are not produced in solution at higher TPS conversion, longer irradiation was conducted at 254 nm. The TPS consumption was monitored by HPLC and the reaction was stopped after 48 h as the TPS consumption had reached 88%. Shown in Fig. 4 are the time traces of the consumption of TPS and the yields of the various photoproducts over the course of the reaction. Again, acetanilide, PhSPh, 2-BiPhSPh, and 4-BiPhSPh, but biphenyl also appeared at longer irradiation times. The ratio of in-cage to cage-escape products remained constant throughout the reaction. After 24 hours of irradiation (72% TPS consumption), benzene, dibenzothiophene, and triphenylene could be detected, but only in trace amounts (<1% yields). The total material recovered after 48 hours of irradiation accounts for 73% of the starting TPS, suggesting that reactions between TPS fragments and PMMA occur in solution as well.
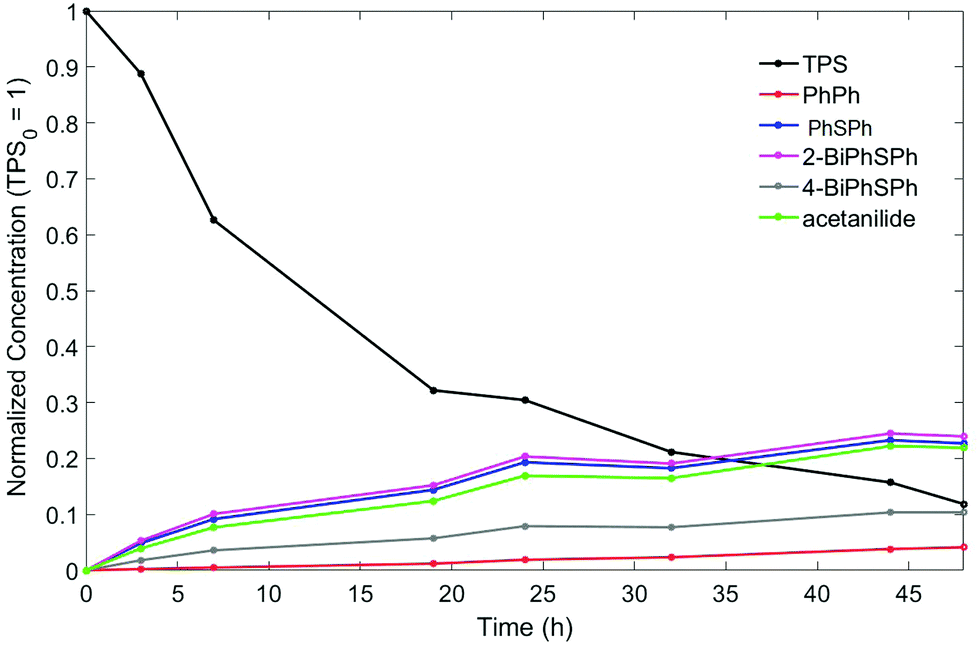 | ||
| Fig. 4 Consumption of TPS and the formation of photoproducts during 48 h irradiation of 1% TPS and 10% PMMA acetonitrile solutions at 254 nm. The concentration has been normalized versus TPS, which is set to 1 at an exposure dose of 0 mJ cm−2. | ||
TPS photoproducts determined in a semiconductor fabrication facility
The triphenylsulfonium cation embedded in polymer films is a key molecule in photolithography for the generation of an acid. To determine the relevance of the film studies in industrial processes, additional experiments were performed in a semiconductor fabrication facility. Specifically, we used an ASM-L/1100 lithographic scanner that produced monochromatic 193 nm radiation. 200 mm silicon wafers were first primed with hexamethyldisilazane (HMDS) after which a bottom antireflective coating (BARC) was applied (coated and cured) in order to prevent reflection from the silicon wafer back into the films. Under these conditions, precise measurement of the exposure dose can be carried out. The absorbance k-value at 193 nm for the resist film unexposed is 0.0496 which is primarily attributed to the TPS cation. A run sequence was developed that resulted in complete wafer exposure. A ladder study was conducted which used doses of 0, 10, 20, 30, 40 and 50 mJ cm−2. Following the exposure, the wafers were cleaved and extracted with a solvent in order to perform GPC and HPLC analyses. Both chromatography experiments showed definitive evidence of TPS consumption in addition to the production of the same photoproducts observed with conventional irradiation sources.Fig. 5 shows the GPC traces obtained for each of the films. Due to the small differences in the areas of cleaved wafers, the GPC traces were normalized at a retention time of 12 minutes corresponding to the PMMA fraction. As expected, TPS (ca. retention time = 14.8 minutes) decreases with increasing exposure, whereas the photoproducts increase (retention times between 15 and 16 minutes) with increasing exposure dose. The use of a photodiode array detector on the GPC allowed for the definite determination of photoproducts. Fig. 6 shows the UV-vis spectra obtained at a retention time of 15.8 minutes, corresponding to the elution of triphenylene (Fig. S11: UV-vis spectra obtained at a retention time of 15.1 minutes, corresponding to the elution of 2-BiPhSPh; see the ESI†). As was observed in the studies described above, triphenylene is produced with increasing exposure dose. Notably, triphenylene is produced at doses as low as 20 mJ cm−2. Significant production of triphenylene occurs with increasing exposure as clearly demonstrated by the UV-vis spectra. In order to quantify the photoproducts, we applied the same HPLC method as described above. The relative amounts of photoproducts are plotted in Fig. 7, demonstrating the rapid production of 2-BiPhSPh with later production of secondary photoproducts such as triphenylene. As these data were obtained in a DOW semiconductor fabrication facility, with quantified exposure doses, they demonstrate the relevance of this topophotochemistry to the lithographic world. At doses in the range of common exposure regimes, triphenylene is produced with a loss of sulfur.
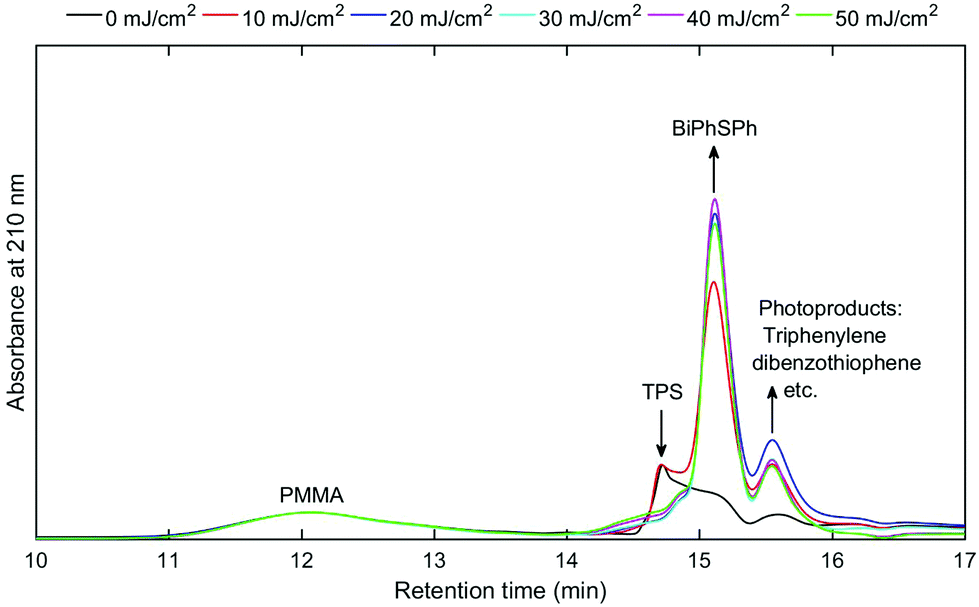 | ||
| Fig. 5 GPC traces (absorbance at 210 nm, normalized at 12 minute retention time) of extracted films from 0, 10, 20, 30, 40 and 50 mJ cm−2 exposure ladder study. | ||
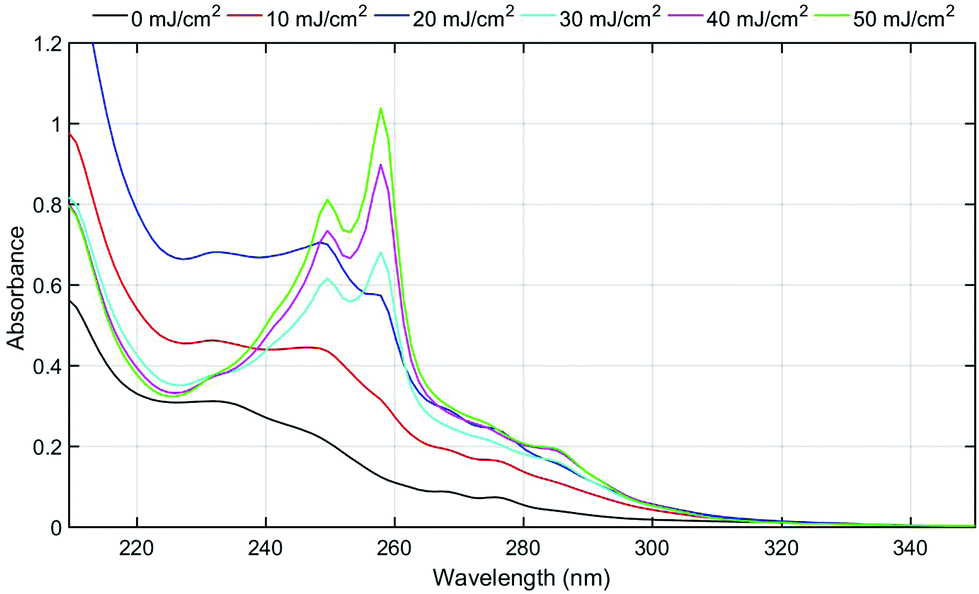 | ||
| Fig. 6 UV-vis spectra (corrected via the normalization factor taken in Fig. 5) obtained at 15.8 minute retention time from GPC traces, corresponding to the triphenylene fraction. | ||
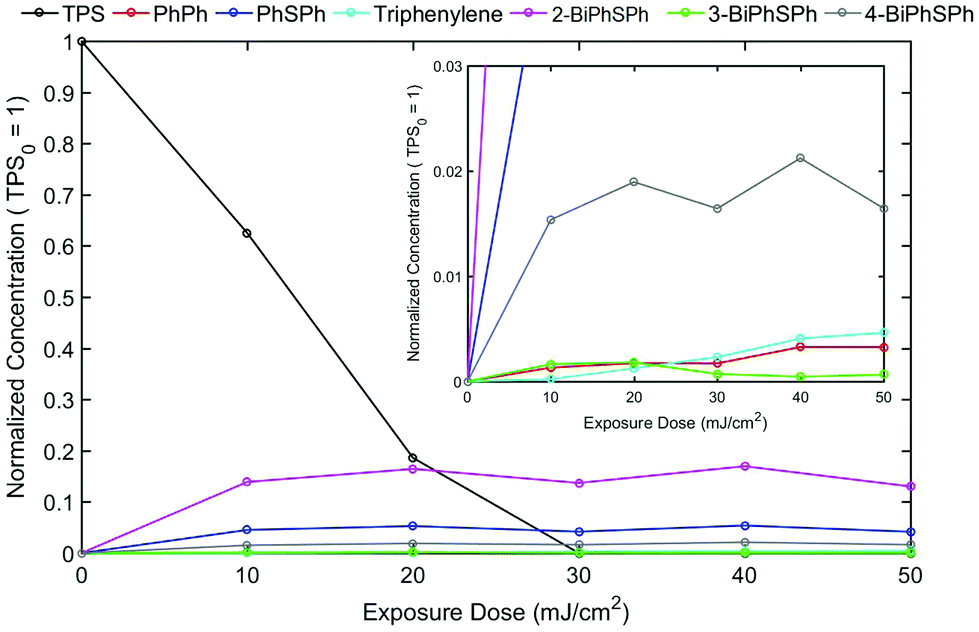 | ||
| Fig. 7 Photoproduct quantification by HPLC analysis. Concentrations have been normalized to the initial TPS consumption, which is set to 1 at an exposure dose of 0 mJ cm−2. | ||
The Dill C-parameter is a commonly used term to quantify the rate of a photochemical reaction of a photoacid generator (PAG) in a photoresist, and is defined as the first-order photodecomposition decay constant (i.e., M = M0 e−CE, where M = PAG concentration, M0 = initial PAG concentration, C = Dill C-parameter and E = exposure energy).19 Assuming that the destruction of TPS is first order and that each photochemical event leads to one photoacid produced, then the C-parameter can be determined by fitting the equation [H+] = 1 − e−CE, where [H+] is the normalized acid concentration (which is assumed to be equal to 1 − [TPS]/[TPS]0), C is the C-parameter and E is the exposure energy. From the data shown in Fig. 7, from exposure energies of 0 to 20 mJ cm−2, C is determined to be approximately 0.06 cm2 mJ−1, which is consistent with previous literature values.20
tBu-TPS/TPS-PMMA crossover experiment
A crossover experiment was conducted using TPS and (tris(4-tert-butylphenyl)sulfonium nonaflate: tBu-TPS) to probe the pathway for the formation of triphenylene in the polymer film. The UV-vis spectrum recorded after the irradiation of a 1:1 mixture of TPS and tBu-TPS in PMMA at 193 nm exhibited the characteristic vibronic absorption signature of triphenylene (Fig. S12 in the ESI†). NMR analysis of the photoproducts (Fig. S13 and S14 in the ESI†) revealed only non-substituted triphenylene and tris-tert-butyltriphenylene. No cross-products were observed, indicating that triphenylene was formed by an in-cage process, likely owing to the sluggish fragment diffusion through the polymer matrix. Moreover, the main isomer of tris-tert-butyltriphenylene (by NMR) was C3 symmetric 2,6,11-tris-tert-butyltriphenylene, establishing the connectivity of three aromatic rings (Scheme 2).
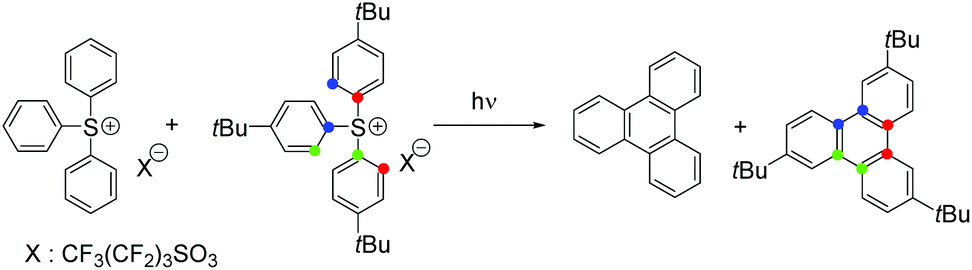 | ||
| Scheme 2 Products of the crossover experiment between TPS and tBu-TPS. | ||
Dose dependence of TPS photoproduct formation
In order to have better control of the exposure of the PMMA/TPS discs and to control the amount of secondary/tertiary photoproducts, the discs were irradiated with a 266 nm, 8 ns pulsed laser at a dose of 2.2 mJ per cm2 per pulse. Different exposures were used with each disc, and the photoproducts were analyzed by HPLC to quantify the amount of TPS consumed. The results for the dose dependence of TPS consumption and photoproduct formation are shown in Fig. 8.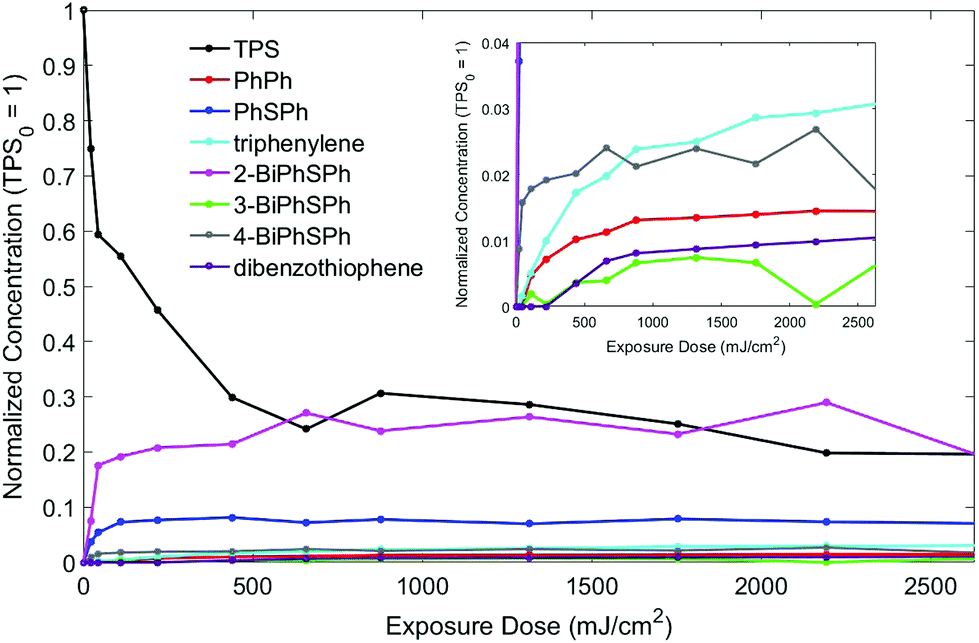 | ||
| Fig. 8 Reaction profiles: PMMA/TPS film exposed to 266 nm pulsed laser irradiation. The concentration has been normalized versus TPS, which is set to 1 at an exposure dose of 0 mJ cm−2. | ||
The consumption of TPS plateaued after exposure to 658 mJ cm−2 (300 shots) at 83% conversion.21 The major photoproduct at all doses was the ortho-isomer, 2-(phenylthio)biphenyl (19.6%), as expected given the diffusion constraints in the polymer matrix. A small amount of the para-isomer 4-(phenylthio)biphenyl (1.8%) was also formed, but very little of the meta-isomer (0.6%) was observed. These are the primary products of TPS photochemistry, arising from the cleavage of the S-aryl bond followed by in-cage fragment recombination that strongly favors the formation of the ortho-isomer over the para-isomer, both being electronically preferred compared to the meta-isomer (Scheme 3).
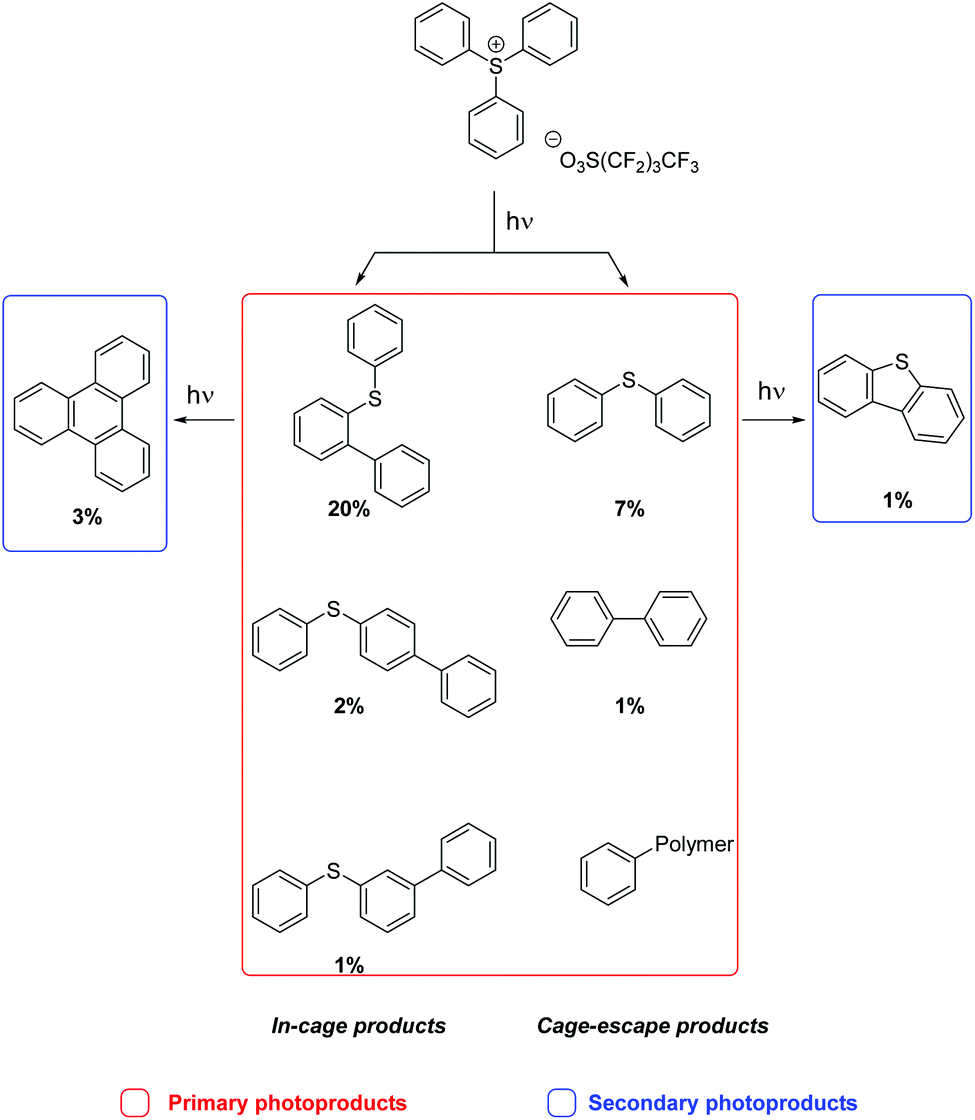 | ||
| Scheme 3 Photoproducts obtained upon 266 nm irradiation (2631 mJ cm−2) of a PMMA/TPS film. | ||
As expected, the formation of the cage-escape photoproduct, diphenylsulfide (maximum of 7.1%), was less favored compared to in-cage products. Diphenylsulfide was produced by loss of a phenyl radical (homolysis pathway) or a phenyl cation (heterolysis pathway, Scheme 1). Biphenyl can be formed by the recombination of two phenyl radicals or more likely by secondary photolysis of one or more (phenylthio)biphenyl isomers.3 A small induction period was observed in the formation of biphenyl consistent with secondary photolysis. The reaction with the polymer matrix is possible as only 42% of the TPS consumed led to molecular photoproducts. Longer exposure of the film allowed the formation of dibenzothiophene (1%) and triphenylene (3%). The long induction periods before the formation of these products (especially for dibenzothiophene) suggest that secondary photolysis was in play (Scheme 3).
The formation of the two main photoproducts, 2-(phenylthio)biphenyl and diphenyl sulfide, reached plateaus after 658 mJ cm−2 exposure, in accord with the TPS consumption curve (Fig. 8); after that, both products were slowly consumed. Indeed, upon the exposure of PMMA/diphenylsulfide and PMMA/2-BiPhSPh films to 266 nm pulsed laser irradiation, dibenzothiophene and triphenylene were obtained as photolysis products, respectively. No intermediates were observed during the irradiation of a PMMA-2-BiPhSPh film, only primarily triphenylene, in accord with an in-cage mechanism. Furthermore, the irradiation of PMMA films containing 3- or 4-(phenylthio)biphenyl did not produce any triphenylene. We conclude that the presence of an ortho phenyl substituent is essential for the formation of triphenylene.
Conclusion
The photochemistry of TPS-PMMA films was studied at different irradiation wavelengths (193, 254 and 266 nm). In all cases, two previously unreported TPS photoproducts, triphenylene and dibenzothiophene, were detected. The formation of both molecules occurred by secondary photolysis of two primary photoproducts, 2-BiPhSPh and PhSPh. Such reactivity is attributable to molecular confinement in the polymer matrix. Further mechanistic investigations are in progress in our laboratory, especially regarding triphenylene formation; this photochemical transformation is particularly interesting as it involves the loss of one sulfur and two hydrogen atoms.Most importantly, our findings highlight the role of the polymer in the microlithography process. Before our work in films, neither triphenylene nor dibenzothiophene had been observed in TPS photodecomposition, the former photoproduct resulting from the complete loss of sulfur.3,5,18 Moreover, although these photoproducts are generated via successive photolysis processes, our work demonstrates that this sequence can occur with low exposure under lithographically relevant conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr VanderVelde for his help on the DOSY-NMR experiment. This work was supported by the National Science Foundation Center for Chemical Innovation in Solar Fuels (CHE-1305124) and in part by The Dow Chemical Company through a university partnership program (Agreement # 227027AK).Notes and references
- G. S. Egerton, J. M. Gleadle and A. G. Roach, Studies on aminoanthraquinone compounds IV–Photochemistry of some simple derivatives in solution and on polymer films, J. Soc. Dyers Colour., 1966, 82, 369 CrossRef CAS; G. Olovsson, J. R. Scheffer, J. Trotter and C.-H. Wu, Novel differences between the solid state and solution phase photochemistry of 1,2-cyclodecanedione, Tetrahedron Lett., 1997, 38, 6549 CrossRef; C.-H. Tung and Y.-M. Ying, Photochemistry of phenyl phenylacetates adsorbed on pentasil andfaujasite zeolites, J. Chem. Soc., Perkin Trans. 2, 1997, 1319 RSC; T. Weisheit, D. Escudero, H. Petzold, H. Görls, L. González and W. Weigand, Photochemical behavior of (diphosphine)(η2-tolane)Pt0 complexes. Part A: Experimental considerations in solution and in the solid state, Dalton Trans., 2010, 39, 9493 RSC; A. Papagni, P. Del Buttero, C. Bertarelli, L. Miozzo, M. Moret, M. T. Pryce and S. Rizzato, Novel fluorinated amino-stilbenes and their solid-state photodimerization, New J. Chem., 2010, 34, 2612 RSC; V. Ramamurthy and S. Gupta, Supramolecular photochemistry: from molecular crystals to water-soluble capsules, Chem. Soc. Rev., 2015, 44, 119 RSC; A. Bricen and A. M. Escalona, Exploiting the use of multivalent interactions in the design of photoreactive supramolecular assemblies. From solution to crystal engineering, Photochemistry, 2016, 43, 286 Search PubMed.
- J. F. Cameron, N. Chan, K. Moore and G. Pohlers, Comparison of acid-generating efficiencies in 248 and 193-nm photoresists, Proc. SPIE, 2001, 4345, 106 CrossRef CAS.
- J. L. Dektar and N. P. Hacker, Photochemistry of triarylsulfonium salts, J. Am. Chem. Soc., 1990, 112, 6004 CrossRef CAS.
- J. W. Knapczyk and W. E. McEwen, Photolysis of triarylsulfonium salts in alcohol, J. Org. Chem., 1970, 35, 2539 CrossRef CAS; J. L. Dektar and N. P. Hacker, A new mechanism for photodecomposition and acid formation from triphenylsulphonium salts, J. Chem. Soc., Chem. Commun., 1987, 1591 RSC.
- S. Tagawa, S. Nagahara, T. Iwamoto, M. Wakita, T. Kozawa, Y. Yamamoto, D. Werst and A. D. Trifunac, Radiation and photochemistry of onium salt acid generators in chemically amplified resists, Proc. SPIE, 2000, 3999, 204 CrossRef CAS.
- For a study on the reactivity of the diphenylsulfinyl radical cation, see: Y. Matsui, H. Sugawara, S. Seki, T. Kozawa, S. Tagawa and T. Itani, Difference in reaction schemes in photolysis of triphenylsulfonium salts between 248 nm and dry/wet 193 nm resists, Appl. Phys. Express, 2008, 1, 036001 CrossRef.
- K. M. Welsh, J. L. Dektar, M. A. Garcia-Garibaya, N. P. Hacker and N. J. Turro, Photo-CIDNP and nanosecond laser flash photolysis studies on the photodecomposition of triarylsulfonium salts, J. Org. Chem., 1992, 57, 4179 CrossRef CAS.
- N. P. Hacker, D. V. Leff and J. L. Dektar, Cationic photoinitiators: Solid state photochemistry of triphenylsulfonium salts, Mol. Cryst. Liq. Cryst., 1990, 183, 505 CrossRef CAS.
- N. P. Hacker, J. L. Dektar, D. V. Leff, S. A. Macdonald and K. M. Welsh, The importance of the cage versus escape reactivity in the photochemistry of onium salts, J. Photopolym. Sci. Technol., 1991, 4, 445 CrossRef CAS.
- N. P. Hacker and K. M. Welsh, Fluorescence Spectroscopy and Photochemistry of Poly(4-oxystyrenes) with Triphenylsulfonium Salts, Insight into the Photoinitiation of Chemically Amplified Resists, Adv. Chem. Ser., 1993, 236, 557 CrossRef CAS; N. P. Hacker and K. M. Welsh, Photochemistry of triphenylsulfonium salts in poly[4-[(tert-butoxycarbonyl)oxy]styrene]: evidence for a dual photoinitiation process, Macromolecules, 1991, 24, 2137 CrossRef.
- S. Tsuji, S. Seki, T. Kozawa and S. Tagawa, Reaction mechanisms of excimer resists studied by laser flash photolysis, J. Photopolym. Sci. Technol., 2000, 13, 729 CrossRef CAS.
- V. V. Boldyrev, Topochemistry and topochemical reactions, React. Solids, 1990, 8, 231 CrossRef CAS.
- E. Kapetanakis, A. M. Douvas, P. Argitis and P. Normand, Radiation sensors based on the generation of mobile protons in organic dielectrics, ACS Appl. Mater. Interfaces, 2013, 5, 5667 Search PubMed; D. G. Georgiadou, L. C. Palilis, M. Vasilopoulou, G. Pistolis, D. Dimotikali and P. Argitis, Incorporating triphenyl sulfonium salts in polyfluorene PLEDs: an all-organic approach to improved charge injection, J. Mater. Chem., 2011, 21, 9296 RSC.
- N. Park, K. Park, M. Jang and S. Lee, One-pot synthesis of symmetrical and unsymmetrical aryl sulfides by Pd-catalyzed couplings of aryl halides and thioacetates, J. Org. Chem., 2011, 76, 4371 CrossRef CAS PubMed.
- M. D. Pelta, H. Barjat, G. A. Morris, A. L. Davis and S. Hammond, Pulse sequences for high-resolution diffusion-ordered spectroscopy (HR-DOSY), Magn. Reson. Chem., 1998, 36, 706 CrossRef CAS.
- H. Kunkely and A. Vogler, Photolysis of the ion pair triphenylsulfonium thiophenolate, Inorg. Chim. Acta, 2004, 357, 1292 CrossRef CAS.
- W. W. Simons, The Sadtler Handbook of Ultraviolet Spectra, Sadtler Research Laboratories, Philadelphia, 1979 Search PubMed.
- J. L. Dektar and N. P. Hacker, Photochemistry of diaryliodonium salts, J. Org. Chem., 1990, 55, 639 CrossRef CAS.
- F. H. Dill, W. P. Hornberger, P. S. Hauge and J. M. Shaw, Characterization of positive photoresist, IEEE Trans. Electron Devices, 1975, 445 CrossRef CAS.
- C. R. Szmanda, R. L. Brainard, J. F. Mackevich, A. Awaji, T. Tanaka, Y. Yamada, J. Bohland, S. Tedesco, B. Dal'Zotto, W. Bruenger, M. Torkler, W. Fallmann, H. Loeschner, R. Kaesmaier, P. M. Nealy and A. R. Pawloski, Measuring acid generation efficiency in chemically amplified resists with all three beams, J. Vac. Sci. Technol., B, 1999, 17, 3356 CAS.
- The periphery of the disc was not irradiated which is why TPS conversion does not reach 100%.
Footnote |
| † Electronic supplementary information (ESI) available: (i) Emission spectra of the different irradiation sources, (ii) UV-Vis spectra over time under 193 and 254 nm irradiation, (iii) UV-Vis spectra of TPS films with different polymer matrixes, (iv) UV-Vis spectra and HPLC trace of the solution and wafer experiments, (v) NMR spectra of the cross-experiment, and (vi) DOSY-NMR experiment. See DOI: 10.1039/c7pp00324b |
| This journal is © The Royal Society of Chemistry and Owner Societies 2018 |