
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-invasive 19F NMR analysis of a protein-templated N-acylhydrazone dynamic combinatorial library†
Alexander G.
Ekström
,
Jue Theresa
Wang
,
Juraj
Bella
and
Dominic J.
Campopiano
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: Dominic.Campopiano@ed.ac.uk
First published on 4th October 2018
Abstract
Dynamic combinatorial chemistry (DCC) is a powerful tool to identify ligands for biological targets. We used 19F NMR as an in situ, non-invasive technique for measuring the composition of a dynamic combinatorial library (DCL) of N-acylhydrazones (NAHs). An NAH DCL, constructed from a fluoro-aromatic aldehyde and a small set of hydrazides, was targetted at ecFabH, an essential enzyme in bacterial fatty acid biosynthesis. Our NMR analysis identified a tert-butyl NAH as the best binder which was confirmed by enzymatic assay.
Introduction
Dynamic combinatorial chemistry (DCC) is a technique derived from fragment-based screening that exploits chemically-reversible reactions to generate a thermodynamically equilibrated library from a pool of building blocks.1 The reversibility of the dynamic combinatorial library (DCL) allows perturbation of the equilibrium through the addition of a template. Since its inception, DCC has been applied to a variety of fields including self-assembly systems, dynamic polymers and host–guest chemistry, however an exciting application is in the field of drug discovery, whereby a protein is used to self-select a ligand through amplification of the best binding species at the expense of other combinations (see Fig. 1). | ||
| Fig. 1 Formation of a DCL and analysis of the blank and protein-templated DCL product distribution by 19F NMR. | ||
The constantly expanding toolbox of DCC-compatible reversible reactions2,3 has helped facilitate the wide range of applications of the technique as a whole, however the number of bio-compatible reactions is limited by the requirement for the reaction to proceed on a reasonable timescale under physiological conditions (temperature, solvent, pH). From this panel of suitable reactions, the chemistry of N-acylhydrazone (NAH) exchange has been well documented and successfully used to identify ligands for a number of different protein targets.4–8 The reaction between an aldehyde and a hydrazide occurs rapidly at pH 4 with an equilibrium constant and stability that strongly favours product formation.9 In order to allow the reaction to proceed at a physiologically relevant pH, Bhat et al. applied early work from Jencks on oxime exchange catalysis.10 By using aniline as a nucleophilic catalyst they showed that their 10-member N-acylhydrazone library equilibrated within a few hours at pH 6.2.5,10,11
A DCC experiment typically involves 3 main steps: firstly, the DCL is equilibrated under conditions promoting a thermodynamic equilibrium. Secondly, the protein template is added and the DCL is allowed to re-equilibrate establish a new product distribution. Thirdly, the final and arguably most vital step, is the analysis of the DCL in the presence and absence of the template. Various techniques to deconvolute the library mixture have been demonstrated,12 including HPLC,5,13–15 mass spectrometry8,16–181H NMR and Saturation Transfer Difference (STD) NMR,7,19,20 dynamic deconvolution21 and X-ray crystallography.22 Recent reviews of this area have highlighted both experimental and theoretical approaches used to analyse DCLs.12,23
The main difficulty lies in not perturbing the DCL during the analysis. This has previously been achieved by either chemically or kinetically freezing the exchange reaction. For example, NAH exchange is effectively stopped by increasing the pH to 9. The second hurdle is ensuring that the complete product population is analysed. This problem is exemplified in HPLC analysis of protein-templated DCL, where the protein must be removed from the mixture prior to analysis to prevent column fouling.5 In the absence of denaturation agents which may perturb the finely balanced protein-templated DCL equilibrium, target binders may remain bound to the protein and are excluded from the DCL analysis. This technique of “ligand fishing” has in fact been demonstrated in the DCC context to find competitive inhibitors of lysozyme.24
Analysis by non-denaturing electrospray ionisation mass spectrometry (ESI-MS) has been demonstrated in a number of elegant DCC experiments,8,20,25 however the technique relies on the protein ionising efficiently under non-denaturing conditions and is therefore not universal. STD-NMR has also been used to identify binders from a DCL, however the technique requires upwards of a 20-fold molar excess of ligand. At such stoichiometry, STD experiments do not leverage the dynamic self-selection qualities central to the DCC concept. Instead, they exploit NAH formation as a facile, one-pot synthetic route to a library of compounds for binding assay. With the exception of STD-NMR, the aforementioned techniques are all destructive in a sense that a portion of the DCL is consumed in the analysis.
Our preliminary HPLC analyses of a protein-templated NAH DCL delivered striking differences in the product distribution depending on the method of protein removal (see Fig. S1†). To resolve this discrepancy, we set out to employ a non-destructive technique that would allow the DCL composition to be monitored in pseudo real-time.
Fluorine has become a central part of drug discovery, with approximately 25% of all marketed drugs containing at least one fluorine atom.26 As a bioisostere of hydrogen, its inclusion can help medicinal chemists to modulate the pharmacokinetic, pharmacodynamic and physical binding properties of a compound.26 Subsequently 19F NMR has developed into a popular technique for screening fluorine-based fragment libraries, with the ability to screen up to 20 fragments in one experiment. The high gyromagnetic ratio and chemical shift anisotropy of the 19F nucleus allow for well-resolved signals that relay information on protein binding through signal broadening or chemical shift perturbation. The scarce biological abundance of 19F allows for background-free spectra, unaffected by protonated buffers and solvents, to be recorded in minutes on a low-field NMR spectrometer. The caveat of 19F screening is the requirement of individual well-resolved signals. This can be achieved though intelligent library design facilitated by increasingly accurate chemical shift prediction software (e.g. MNova, TopSpin). More complex NMR experiments have been used to resolve overlapping signals, both by 2D homonuclear correlation-ordered experiments (1H–19F COSY) and pseudo-2D diffusion-ordered (19F DOSY) experiments, where the signals are separated on second axis by diffusion coefficient.26–28 The elegance of 19F NMR analysis has already been demonstrated in a number of abiotic DCC examples.29–31 This method was also used to monitor binding of a 4-component imine library to a domain of a human β-catenin target.32 Herein, we describe our design and analysis of a protein-templated NAH DCL to further demonstrate the additional advantages of 19F NMR in DCC for drug discovery.
Results and discussion
We chose to design a library targeting β-ketoacyl-ACP synthase III (FabH) from E. coli, the initial condensing enzyme in bacterial fatty acid biosynthesis (FAS II). Although the active site residues and primary sequence remain highly conserved across both Gram-positive and Gram-negative bacteria, small differences in the binding pocket architecture determine the substrate specificity across bacterial species. This makes FabH a plausible target for novel narrow-spectrum antibiotics.33 There are currently few known FabH inhibitors, and none that report good in vivo efficacy.34–36 Zhang et al. have recently published a number of reports on the discovery of NAH inhibitors targeting FabH where each compound was prepared individually by organic synthesis. Since each molecule was composed of A–B ring systems joined by an NAH linker it provided the basis around which our dynamic library was designed.37–41The 5-membered proof-of-concept library was based around commercially available fluoro-aromatic aldehyde A as the central core along with five commercially available aromatic hydrazides 1–5 with differing chemical and physical properties (Fig. 2). The high degree of conjugation through the system allowed us to observe sufficiently resolved 19F chemical shifts of individual NAHs despite the differences in chemistry being up to 13 bonds away42 (see Fig. S2†).
 | ||
| Fig. 2 Rationale for the DCL and composition of the 5-membered proof-of-concept 19F-labelled DCL. To note, hydrazide 1 (isoniazid) is a front-line TB therapy with another FAS II target. | ||
To minimise the time required to reach equilibrium, the use of aniline as a nucleophilic catalyst for NAH exchange was introduced by Dawson,11 and has since been successfully used in many protein-templated DCC experiments.5,6 From preliminary 1H-STD-NMR experiments we observed that aniline was binding to FabH and would potentially interfere with the DCL. Issues with aniline were noted by Blanden et al. in their attempts to optimise hydrazone ligation for biomolecular labelling. They identified 4-amino-L-phenylalanine (4-APA) as a suitable replacement for aniline so we investigated if 4-APA could be substituted for aniline in a DCL.43 We validated that 4-APA has comparable catalytic efficiency to aniline with our NAH DCL, both in the forward reaction (Fig. 3 and Fig. S4†) and the reverse reaction (Fig. S5†). When a library is prepared from product A5 and hydrazides 3 and 4 (see Fig. 2), the same product distribution is observed as when a library is prepared from hydrazides 3, 4 and 5 and aldehyde A (Fig. S4 and S5†). Binding of 4-APA to FabH was not observed by 1H-STD-NMR.
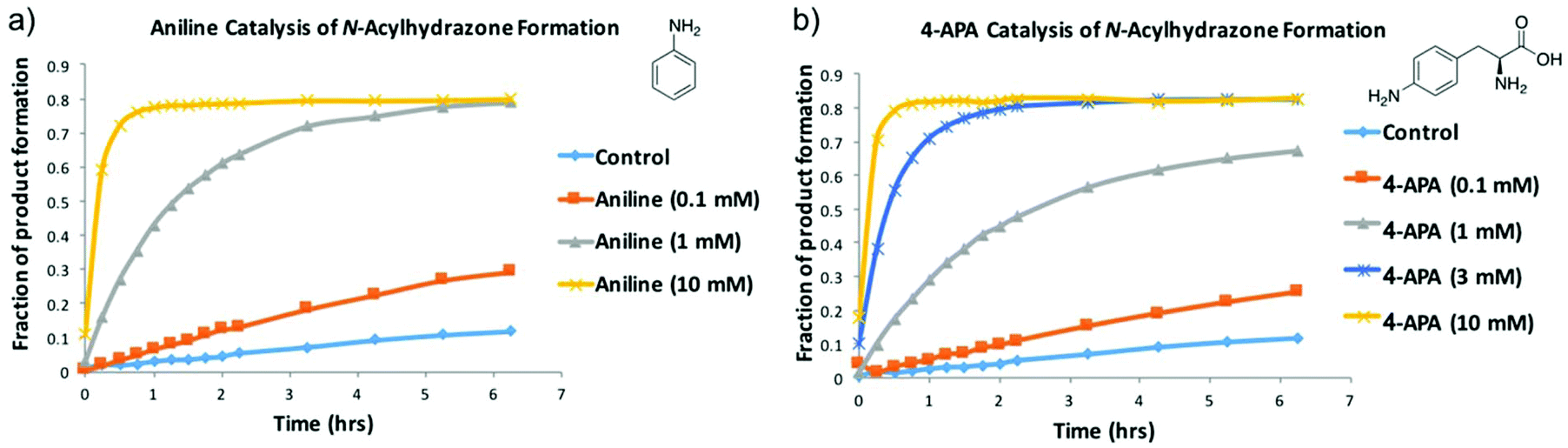 | ||
| Fig. 3 Validation of 4-APA as an alternative to aniline. (a) shows the formation of NAH A4 from aldehyde A and hydrazide 4 over time in the presence of different concentrations of aniline. (b) shows the same experiment, catalysed by different concentrations of 4-APA. The data shown is a compilation of the NAH A4 HPLC peaks recorded at given time points (see Fig. S3†). | ||
The DCL was assembled with each of the hydrazides 1–5 and aldehyde A at a final concentration of 200 μM, buffered at pH 6.2 using 50 mM sodium phosphate buffer with 10% DMSO and 3 mM 4-APA. The DCL cocktail was spiked with 200 μM 5-fluorouracil as an internal chemical shift reference and either 200 μM ecFabH or an equivalent volume of buffer. Each library was transferred to an NMR tube and a 19F spectrum was acquired every two hours over a twelve-hour period. In agreement with the 4-APA characterisation experiment (Fig. 3), the DCL reached equilibrium within 2 hours. Fig. 4a shows the blank library 8 hours after mixing. Notably not all products are at an equal concentration, suggesting the electronic substituents of the ring have an effect on the intrinsic thermodynamic stability of each NAH. Fig. 4b shows the 19F spectrum of the library templated by the target, ecFabH. Slight signal broadening is observable for all DCL members, suggesting a slow exchange process is occurring between bound and unbound ligand states. Most impressively, the signal broadening and upfield chemical shift perturbation of compound A4 is indicative of the compound being present in its bound state at a relatively higher proportion to the other library members.27
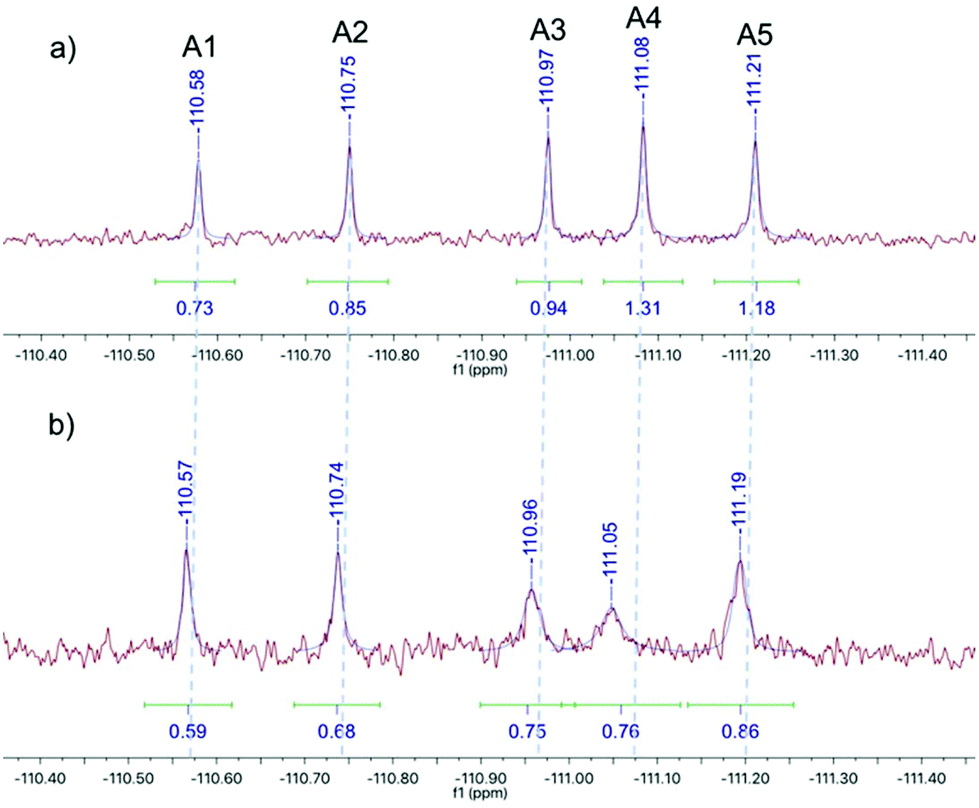 | ||
| Fig. 4 19F NMR spectra of the 19F-labelled DCL: (a) the DCL equilibrated in the absence of the protein target, (b) the protein-templated DCL equilibrated in the presence of FabH. | ||
It must be noted that there are two opposing forces at play when considering signal integral: on one hand Le Chatelier's principle will strive toward a product distribution proportional to the depth of the energy well of each species, although this is not always the case.2 Therefore, if product A4 is selected by the protein its relative concentration and corresponding signal integral should increase. Concomitantly, the signal broadening resulting from exchange between chemical shifts corresponding to bound and unbound states of the ligand may present itself as an apparent integral decrease of the bound ligand. Due to the breadth and low intensity of the bound state signal we have as yet been unable to determine the chemical shift of the bound state by 19F COSY-NMR. This method is therefore not strictly quantitative, but acts as a qualitative indicator of potential binders whose properties can be further characterised by quantitative techniques.
Using individually synthesised DCL members an in vitro assay validated the results from the 19F NMR DCC experiment. All 5 NAHs showed inhibitory activity at concentrations of 3 mM. Gratifyingly, compound A4 showed the strongest inhibition, causing a 50% decrease in activity at 3 mM compared to the DMSO vehicle control and the FabH inhibitor HR45 as a negative control44,45 (Fig. S6†). That such a weak inhibitor could have been picked out of the NMR analysis is encouraging. It also leads to the question of what effect would a tight nM binder have? Broadening of the signal such that it disappears altogether into the baseline may also be useful in identifying hits by comparison with protein-free controls. We do not seek to claim that the NAH molecules described here may be of therapeutic value, rather we present this as a proof of concept that 19F NMR analysis can be used to interrogate an NAH DCL derived from appropriately 19F-labelled building blocks.
Conclusions
In summary, we have demonstrated a non-invasive analysis of a protein-templated DCL by 19F NMR, using 4-APA as a biologically benign alternative to aniline-catalysed NAH exchange. The results from the DCL agreed well with preliminary inhibition data from an in vitro FabH assay. Screening of much larger compound libraries will no doubt require multiple methods to identify, then validate, hit molecules. A combination of 19F fragment screening and MS-based methods are complementary analytical tools suitable for protein-templated DCC. Development of such rapid, cost-effective, and universal methods should help DCC become more widely used in hit discovery.Experimental
Nuclear magnetic resonance (NMR) spectra were recorded at 298 K on Bruker PRO500, AVA400 or AVA500 spectrometers running at 500 MHz (1H spectra), 126 MHz (12C spectra) or 94 MHz (19F spectra). Chemical shift values (δ) are reported in parts per million (ppm) relative to tetramethylsilane (δTMS = 0 ppm) and are referenced to the residual solvent peak, or to the signal of internal standard 5-fluorouracil (δ5FU = −169.19 ppm) in the case of the 19F-labelled DCL. 1H NMR data are reported in the format: chemical shift, relative intensity, multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constant (J value, Hz), and assignment. 13C NMR data are reported in the format: chemical shift and assignment.Expression and purification of ecFabH
The ecFabH/pET-28a construct (4 μL) was transformed into an aliquot (50 μL) of BL21(DE3) cells and set on ice for 25 minutes. The cells were heat shocked at 42 °C for 40 seconds and set back on ice for a further 2 minutes. SOC media (100 μL) was added and the mixture was agitated at 37 °C for 1 hour. The mixture was spread on LB agar (30 μg mL−1 kanamycin) and incubated overnight at 37 °C. A single transformant was used to inoculate two seed cultures of sterile LB broth (2 × 250 mL, 30 μg mL−1 kanamycin) and agitated overnight at 37 °C. One of the overnight seed cultures was used to sub-culture sterile LB broth (5 × 500 mL, 30 μg mL−1 kanamycin) to an OD600 of 0.1. The cultures were agitated at 37 °C until the OD600 reached 0.6, at which point expression was induced by addition of IPTG (final conc. 0.1 mM). Cells were harvested by centrifugation after a further 3 hours at 30 °C and subsequently stored at −20 °C.N-Terminal histidine-tagged ecFabH was purified at 4 °C by Ni-affinity chromatography followed by size exclusion chromatography. The BL21 (DE3) cell pellet expressing ecFabH was resuspended in lysis buffer (30 mL, 20 mM Tris-HCl pH 7.6, 300 mM NaCl, 5 mM imidazole) and lysed for 15 minutes with rounds of 30 seconds of sonication followed by 30 seconds of rest. The cell lysate was clarified by centrifugation (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 30 minutes, 4 °C) and the cell-free extract was injected onto a HisTrap 5 mL (GE Healthcare) Ni2+-affinity chromatography column pre-equilibrated in lysis buffer. The column was washed with lysis buffer (5 CV) before the histidine-tagged protein was eluted using a gradient (0–100%) of lysis buffer to elution buffer (20 mM Tris-HCl pH 7.6, 300 mM NaCl, 400 mM imidazole) over 20 CV. Each elution fraction was analysed by SDS-PAGE, and the fractions containing His-tagged ecFabH were pooled, and concentrated to a volume less than 5 mL. ecFabH was further purified by size exclusion chromatography (HiLoad Superdex 200 16/60, GE Healthcare) with an isocratic elution of mobile phase buffer (20 mM Tris-HCl pH 7.6, 100 mM NaCl, 10% glycerol) at 1 mL min−1 over 120 minutes. ecFabH eluted at approximately 70 minutes and the most concentrated fractions were pooled and flash frozen and stored at −80 °C.
000g, 30 minutes, 4 °C) and the cell-free extract was injected onto a HisTrap 5 mL (GE Healthcare) Ni2+-affinity chromatography column pre-equilibrated in lysis buffer. The column was washed with lysis buffer (5 CV) before the histidine-tagged protein was eluted using a gradient (0–100%) of lysis buffer to elution buffer (20 mM Tris-HCl pH 7.6, 300 mM NaCl, 400 mM imidazole) over 20 CV. Each elution fraction was analysed by SDS-PAGE, and the fractions containing His-tagged ecFabH were pooled, and concentrated to a volume less than 5 mL. ecFabH was further purified by size exclusion chromatography (HiLoad Superdex 200 16/60, GE Healthcare) with an isocratic elution of mobile phase buffer (20 mM Tris-HCl pH 7.6, 100 mM NaCl, 10% glycerol) at 1 mL min−1 over 120 minutes. ecFabH eluted at approximately 70 minutes and the most concentrated fractions were pooled and flash frozen and stored at −80 °C.
DCL conditions
The library was assembled with each of the hydrazides 1–5 and aldehyde A at a final concentration of 200 μM, buffered at pH 6.2 using 50 mM sodium phosphate with 50% D2O, 10% DMSO, 50 mM NaCl and 3 mM 4-APA. The library cocktail also contained 200 μM 5-fluorouracil as a non-competing internal reference and either 200 μM ecFabH or an equivalent volume of enzyme purification buffer. Each library was transferred to an NMR tube and a 19F spectrum (94 MHz, 512 scans, T1 = 1 second) was acquired every two hours over a twelve-hour period.Length of relaxation time in 19F NMR pulse sequence
A three-member library was assembled from N-acylhydrazones A1, A3 and A5 (200 μM each) in sodium phosphate buffer (50 mM, D2O, pH 6.2), 5-fluorouracil (internal standard, 200 μM) and a total of 10% DMSO. A series of 19F NMR experiments were conducted with 512 scans and relaxation times of 1, 2, 3 or 4 seconds. The integral of the signals corresponding each N-acylhydrazone relative to the internal standard were compared at different relaxation times to determine the required relaxation time for the DCL 19F NMR experiment. No difference in relative signal integral was apparent between all T1 intervals, therefore a T1 of 1 second was used in the DCL experiments.FabH assay
The activity of FabH was quantified by 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB) assay with the following procedure. An assay cocktail was prepared from DTNB (1 mL, 4 mM in 50 mM NH4OAc), HEPES buffer (1 mL, 1 M, pH 8.0) and ddH2O (8 mL). The assay was run in 96-well plate format with the following protocol (the following bracketed figures refer to final concentrations): 100 μL DTNB assay cocktail, 25 μL ecFabH (0.63 μM), 25 μL Acetyl-CoA (375 μM), 25 μL Malonyl-CoA (375 μM), 5 μL ddH2O, 20 μL NAH A1–A5 (3 mM) or HR45 (200 μM) in DMSO or DMSO control. Following 20 min incubation at 37 °C, the reaction was initiated by the addition of malonyl-CoA and followed by monitoring absorbance change at 412 nm over 20 min at 37 °C. The DMSO concentration was maintained at 10% to aid the solubility of the NAH compounds.General procedure for the synthesis of N-acylhydrazones A1–A5
2-Fluoro-5-formylbenzoic acid (50 mg, 0.30 mmol, 1.0 eq.) and the hydrazide (0.33 mmol, 1.1 eq.) were dissolved in ethanol (2 mL). A few drops of glacial acetic acid were added and the mixture was stirred overnight at room temperature. The solid formed was collected by vacuum filtration and washed in diethyl ether (5 mL) and water (5 mL) to yield the target N-acylhydrazone as a white solid.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank the Scottish Universities Life Sciences Alliance (SULSA) for funding a PhD studentship (AGE).Notes and references
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26–36 CrossRef PubMed.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- S. R. Beeren and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3804–3807 CrossRef CAS PubMed.
- V. T. Bhat, A. M. Caniard, T. Luksch, R. Brenk, D. J. Campopiano and M. F. Greaney, Nat. Chem., 2010, 2, 490–497 CrossRef CAS PubMed.
- A. J. Clipson, V. T. Bhat, I. McNae, A. M. Caniard, D. J. Campopiano and M. F. Greaney, Chem. – Eur. J., 2012, 18, 10562–10570 CrossRef CAS PubMed.
- M. Mondal, N. Radeva, H. Köster, A. Park, C. Potamitis, M. Zervou, G. Klebe and A. K. H. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 3259–3263 CrossRef CAS PubMed.
- S.-A. Poulsen, J. Am. Soc. Mass Spectrom., 2006, 17, 1074–1080 CrossRef CAS PubMed.
- J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523–7526 CrossRef CAS PubMed.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826–831 CrossRef CAS.
- A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602–15603 CrossRef CAS PubMed.
- R. Huang and I. Leung, Molecules, 2016, 21, 1–18 Search PubMed.
- M. Hochgürtel, H. Kroth, D. Piecha, M. W. Hofmann, C. Nicolau, S. Krause, O. Schaaf, G. Sonnenmoser and A. V. Eliseev, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3382–3387 CrossRef PubMed.
- I. Huc and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- O. Ramström and J. M. Lehn, ChemBioChem, 2000, 1, 41–48 CrossRef.
- T. Hotchkiss, H. B. Kramer, K. J. Doores, D. P. Gamblin, N. J. Oldham and B. G. Davis, Chem. Commun., 2005, 4264–4266 RSC.
- R. F. Ludlow and S. Otto, Components, 2008, 3, 12218–12219 Search PubMed.
- B. M. R. Liénard, N. Selevsek, N. J. Oldham and C. J. Schofield, ChemMedChem, 2007, 2, 175–179 CrossRef PubMed.
- R. Caraballo, H. Dong, J. P. Ribeiro, J. Jiménez-Barbero and O. Ramström, Angew. Chem., Int. Ed., 2010, 49, 589–593 CrossRef CAS PubMed.
- M. Demetriades, I. K. H. Leung, R. Chowdhury, M. C. Chan, M. A. McDonough, K. K. Yeoh, Y. M. Tian, T. D. W. Claridge, P. J. Ratcliffe, E. C. Y. Woon and C. J. Schofield, Angew. Chem., Int. Ed., 2012, 51, 6672–6675 CrossRef CAS PubMed.
- T. Bunyapaiboonsri, O. Ramström, S. Lohmann, J. M. Lehn, L. Peng and M. Goeldner, ChemBioChem, 2001, 2, 438–444 CrossRef CAS PubMed.
- M. S. Congreve, D. J. Davis, L. Devine, C. Granata, M. O′Reilly, P. G. Wyatt and H. Jhoti, Angew. Chem., Int. Ed., 2003, 42, 4479–4482 CrossRef CAS PubMed.
- M. Misuraca and E. Moulin, New J. Chem., 2014, 38, 3336–3349 RSC.
- Z. Fang, W. He, X. Li, Z. Li, B. Chen, P. Ouyang and K. Guo, Bioorg. Med. Chem. Lett., 2013, 23, 5174–5177 CrossRef CAS PubMed.
- E. C. Y. Woon, M. Demetriades, E. A. L. Bagg, W. Aik, S. M. Krylova, J. H. Y. Ma, M. Chan, L. J. Walport, D. W. Wegman, K. N. Dack, M. A. McDonough, S. N. Krylov and C. J. Schofield, J. Med. Chem., 2012, 55, 2173–2184 CrossRef CAS PubMed.
- A. Vulpetti and C. Dalvit, ChemMedChem, 2013, 8, 2057–2069 CrossRef CAS PubMed.
- C. Dalvit and A. Vulpetti, Magn. Reson. Chem., 2012, 50, 592–597 CrossRef CAS PubMed.
- C. Dalvit, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 243–271 CrossRef CAS.
- J. W. Sadownik and D. Philp, Org. Biomol. Chem., 2015, 13, 10392–10401 RSC.
- T. Kosikova, H. MacKenzie and D. Philp, Chem. – Eur. J., 2016, 22, 1831–1839 CrossRef CAS PubMed.
- J. W. Sadownik and D. Philp, Angew. Chem., Int. Ed., 2008, 47, 9965–9970 CrossRef CAS PubMed.
- M. A. Henen, L. Abou-Zeid, A. El-Din Barghash and H. Eisa, Curr. Top. Pept. Protein Res., 2014, 15, 35–39 Search PubMed.
- Z. Nie, C. Perretta, J. Lu, Y. Su, S. Margosiak, K. S. Gajiwala, J. Cortez, V. Nikulin, K. M. Yager, K. Appelt and S. Chu, J. Med. Chem., 2005, 48, 1596–1609 CrossRef CAS PubMed.
- J. W. Campbell and J. E. Cronan Jr., Annu. Rev. Microbiol., 2001, 55, 305–332 CrossRef CAS PubMed.
- J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, A. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully and S. B. Singh, Nature, 2006, 441, 358–361 CrossRef CAS PubMed.
- J. Wang, S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. L. Colletti, K. Herath, R. Cummings, O. Salazar, I. González, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. F. Cully and S. B. Singh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7612–7616 CrossRef CAS PubMed.
- F. Zhang, Q. Wen, S.-F. Wang, B. Shahla Karim, Y.-S. Yang, J.-J. Liu, W.-M. Zhang and H.-L. Zhu, Bioorg. Med. Chem. Lett., 2014, 24, 90–95 CrossRef CAS PubMed.
- Y. Li, C. P. Zhao, H. P. Ma, M. Y. Zhao, Y. R. Xue, X. M. Wang and H. L. Zhu, Bioorg. Med. Chem., 2013, 21, 3120–3126 CrossRef CAS PubMed.
- L. Shi, R.-Q. Fang, Z.-W. Zhu, Y. Yang, K. Cheng, W.-Q. Zhong and H.-L. Zhu, Eur. J. Med. Chem., 2010, 45, 4358–4364 CrossRef CAS PubMed.
- X. L. Wang, Y. Bin Zhang, J. F. Tang, Y. S. Yang, R. Q. Chen, F. Zhang and H. L. Zhu, Eur. J. Med. Chem., 2012, 57, 373–382 CrossRef CAS PubMed.
- X. Song, Y. Yang, J. Zhao and Y. Chen, Chem. Pharm. Bull., 2014, 62, 1110–1118 CrossRef CAS PubMed.
- J. T. Gerig, Methods Enzymol., 1989, 177, 3–23 CAS.
- A. R. Blanden, K. Mukherjee, O. Dilek, M. Loew and S. Bane, Bioconjugate Chem., 2011, 22, 1954–1961 CrossRef CAS PubMed.
- X. He, A. M. Reeve, U. R. Desai, E. Glen, K. A. Reynolds and G. E. Kellogg, Antimicrob. Agents Chemother., 2004, 48, 3093–3102 CrossRef CAS PubMed.
- A. G. Ekström, V. Kelly, J. Marles-Wright, S. L. Cockroft and D. J. Campopiano, Org. Biomol. Chem., 2017, 15, 6310–6313 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01918e |
| This journal is © The Royal Society of Chemistry 2018 |