
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNative chemical ligation at methionine bioisostere norleucine allows for N-terminal chemical protein ligation†
Bo-Tao
Xin‡
,
Bianca D. M.
van Tol‡
 ,
Huib
Ovaa
* and
Paul P.
Geurink
*
,
Huib
Ovaa
* and
Paul P.
Geurink
*
Oncode Institute and Department of Cell and Chemical Biology, Leiden University Medical Center, Einthovenweg 20, 2333 ZC Leiden, The Netherlands. E-mail: p.p.geurink@lumc.nl; h.ovaa@lumc.nl
First published on 9th August 2018
Abstract
The development of γ-thionorleucine (ThioNle) as a handle for native chemical ligation–desulfurization is reported here. ThioNle is a new addition to the expanding thiolated amino acid toolbox and serves as a methionine substitute in NCL with the advantage that it lacks the undesirable oxidation-prone thioether moiety. Its usefulness for N-terminal ubiquitination is demonstrated by efficient preparation of fully synthetic linear diubiquitin with preserved protein folding compared to the expressed material. Interestingly, gel-based deubiquitinating assays revealed that the methionine to norleucine substitution did affect diubiquitin cleavage, which may indicate a more profound role for methionine in the interaction between ubiquitin and the deubiquitinating enzymes than has been known so far.
Introduction
The development of native chemical ligation (NCL) by Kent and co-workers1 caused a major improvement in the synthesis of peptides and proteins. This technique allows for the conjugation of two unprotected peptide segments, a C-terminal thioester-containing peptide and a peptide bearing an N-terminal cysteine. Dawson and Yan2 expanded the scope of NCL beyond the N-terminal cysteine requirement by introducing a catalytic desulfurization step, which effectively turns cysteine into alanine post-NCL (Fig. 1A). The subsequent development of a mild metal-free desulfurization procedure by Wan and Danishefsky3 opened the way to the application of other proteinogenic amino acids as cysteine surrogates by instalment of a β- or γ-thiol moiety and this has resulted in the expansion of possible ligation sites to Phe,4 Val,5 Thr,6 Leu,7 Pro,8 Glu,9 Arg,10 Asp,11 Gln12 and Trp13 over the last decade. In addition, the preparation of δ- and γ-thioLys allowed for the formation of isopeptide bonds by NCL, which was applied in chemical ubiquitination.14 Another development of NCL was the use of thioester surrogates, such as peptide hydrazides,15 which expanded the scope of thioester formation.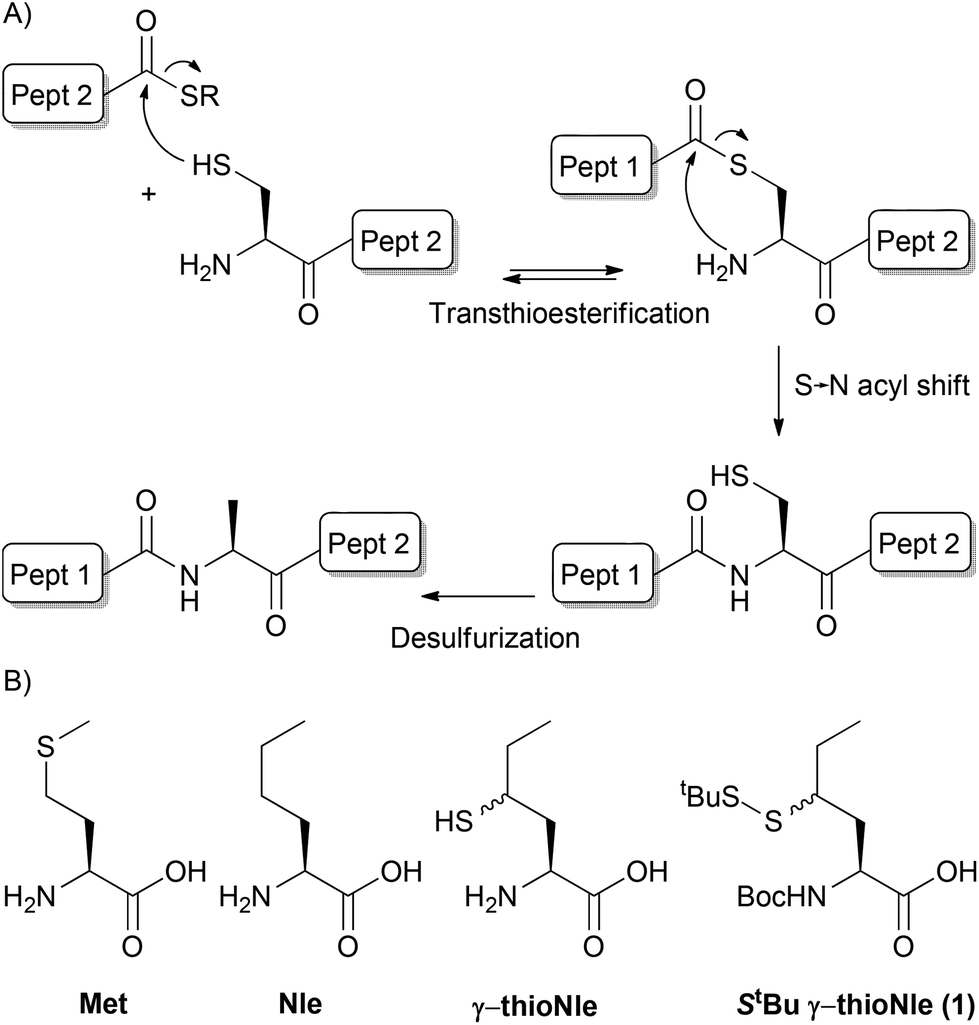 | ||
| Fig. 1 (A) Native chemical ligation–desulfurization. (B) Structures of methionine, norleucine, γ-thionorleucine and target compound 1. | ||
As methionine is encoded by the universal start codon in protein translation and, as a result, each protein is translated with an N-terminal methionine residue, NCL at these sites would allow for the N-terminal modification of proteins.16 A well-known N-terminal modification is linear ubiquitination which is an important post-translational modification.17 NCL at internal methionine sites has been performed by applying homocysteine as a thiol donor followed by S-methylation under carefully controlled conditions to prevent over- and undermethylation of homocysteine or mismethylation of other residues.18 Desulfurization of the homocysteine ligation product has also been reported which effectively leads to the mutation of methionine into 2-aminobutyric acid in the final peptide.2 Thiomethionine is currently missing from the thiolated amino acid collection; yet it would serve as an attractive handle for the N-terminal modification of proteins by means of NCL. On the other hand, the thioether moiety in methionine is susceptible to oxidation into a sulfoxide or even a sulfone, and this occurs rapidly under aerobic conditions, which often results in a significant loss of bioactivity of the synthesized protein.19 In addition, the different oxidation states of methionine often lead to a mixture of different molecular weights for a single protein, which complicates the analysis by mass spectrometry. This is especially detrimental to the desulfurization reaction, typically monitored by mass spectrometry, since double oxidation or single oxidation of two methionine residues results in a net mass increase of 32 Da, which is exactly the mass decrease upon effective removal of sulfur during desulfurization. Hence, an overall change in mass is not observed although desulfurization is completed. In order to overcome these limitations, methionine is typically substituted by its closer isostere norleucine (Nle) (Fig. 1B), without substantially affecting the peptide or protein structure and function as described in the literature.19 We here present the synthesis of γ-thionorleucine (Fig. 1B) and its application in NCL for N-terminal ubiquitination.
Results and discussion
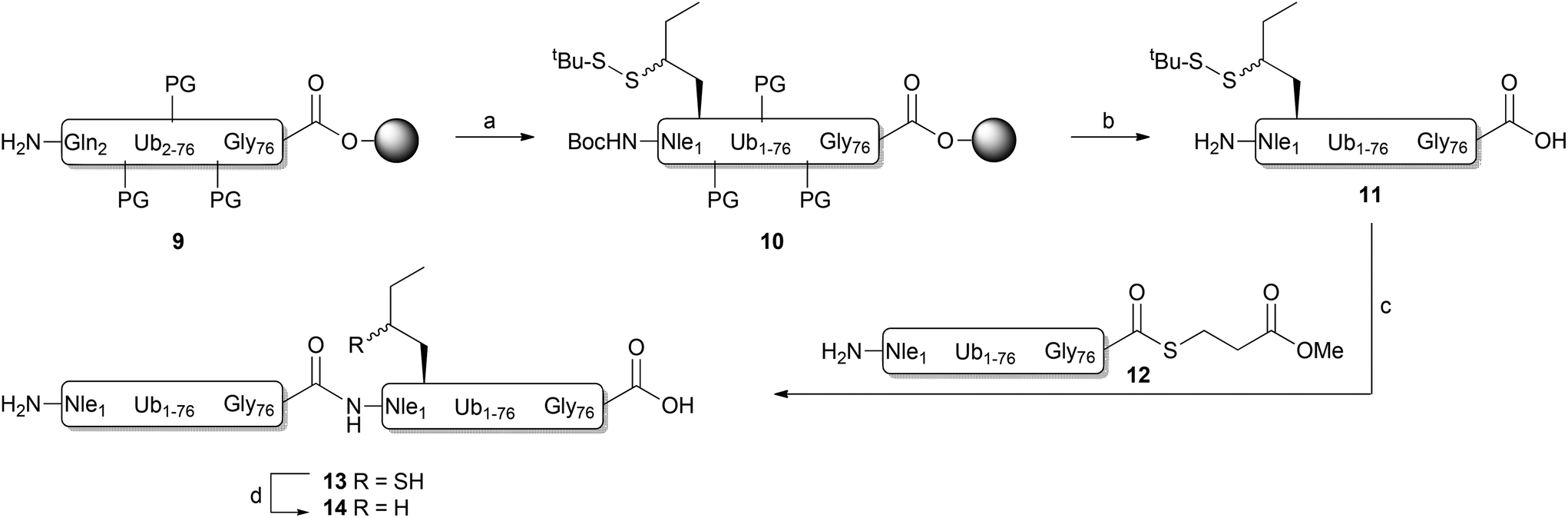
As γ-thionorleucine is installed onto peptides by means of solid-phase peptide synthesis (SPPS) we prepared appropriately protected N-Boc, S-tert-butylsulfide γ-thionorleucine 1 (Fig. 1B), as depicted in Scheme 1. The synthesis commenced with the preparation of tert-butyl (S)-2,2-dimethyl-4-(2-oxoethyl)oxazolidine-3-carboxylate 2 according to literature procedures.20 Addition of ethylmagnesium bromide to aldehyde 2 yielded compound 3 as a mixture of two diastereomers and the synthesis was continued with this mixture. The free hydroxyl was protected as benzyl ether (4), which was subsequently treated with Jones reagent to hydrolyse the acetonide and concomitantly oxidize the resulting alcohol to a carboxylic acid. This was converted into the corresponding tert-butyl ester 5 upon treatment with O-tert-butyl N,N′-diisopropylisourea.21 Palladium-catalysed hydrogenation and subsequent mesylation of the alcohol intermediate resulted in methanesulfonate 6, which was transformed to acetylated thiol 7 upon treatment with potassium thioacetate. tert-Butyl disulfide 8 was obtained after treating compound 7 with S-tert-butyl methane thiosulfonate, hydroxylamine and Et3N.22 TFA treatment and subsequent instalment of a Boc protecting group resulted in target compound 1. | ||
| Scheme 1 Synthesis of γ-thionorleucine. Reagents and conditions: (a) EtMgBr, Et2O, 93%; (b) BnBr, NaH, n-Bu4NI, DMF, 0 °C, 44%; (c) Jones reagent, acetone; (d) O-tert-butyl N,N′-diisopropylisourea, THF, 60 °C, 70%; (e) Pd/C (10% wt), H2 (4 bar), MeOH, 48 h; (f) MsCl, Et3N, DCM, 63%; (g) KSAc, 65 °C, 18 h, 60%; (h) S-tert-butyl methane thiosulfonate, HONH2·HCl, Et3N, MeOH, 63%; (i) TFA; (j) Boc2O, K2CO3, THF, H2O, 50%. | ||
The ability of the thioNle building block to function as a new native chemical ligation handle was assessed by N-terminal protein ubiquitination. We chose to ubiquitinate ubiquitin (Ub), which effectively results in a linear diubiquitin (diUb) species. Ubiquitination is a post-translational protein modification that plays an important role in virtually all biological processes.23 Poly-ubiquitination involves the instalment of multiple successively linked Ubs to a protein and the amino acid residue involved in the linkage between two Ubs (any of its seven lysine residues or the N-terminal Met) determines the eventual biological signal. For example, the canonical polyUb linkage Lys-48 targets the tagged protein for proteasomal degradation. Linear Ub chains (e.g. coupled via Met-1) on the other hand play a key role in the regulation of NF-κB signalling and cell death.24 We and others have developed chemical synthesis methods for the generation of all seven isopeptide-linked (e.g. via a Lys side chain) Ub linkages using NCL which have led to many new biological insights.14a,25 However, a method to synthesize the linear Ub linkage has been lacking so far.
Linear diUb was constructed by NCL between a Ub-thioester and γ-thioNle-containing Ub (Scheme 2). The individual Ub proteins were synthesized by Fmoc-based linear SPPS.14a Compound 1 was coupled to Ub(2-76) 9 on resin under standard coupling conditions, followed by global deprotection under strong acidic conditions and RP-HPLC purification, which resulted in Ub(1-76, ThioNle1) 11 in multi-milligram amounts. LC-MS analysis of compound 11 resulted in two similar peaks at different retention times but of identical mass (Fig. 2A). We believe that these represent the two diastereomers of compound 11, having the racemic γ-carbon atom in thioNle as indicated in Scheme 2. Ub(1-75) was prepared by SPPS on a hyper-acid-labile trityl resin and protected with a Boc group at the N-terminus. Subsequently, the protein was cleaved from the resin under mild acidic conditions (20 vol% HFIP in DCM) which liberated only the C-terminal carboxylic acid without affecting the other protecting groups. The C-terminus was activated after which methyl-3-(glycylthio)-propionate was coupled, followed by global deprotection and RP-HPLC purification to result in Ub(1-76)-thioester 12. A first attempt for the NCL between Ub-thioester 12 and γ-thioNle-Ub 11 under previously reported conditions (e.g. 50 mg mL−1 in 6 M Gnd·HCl/0.15 M sodium phosphate buffer, pH 7.6, 250 mM MPAA)26 resulted in only trace amounts of the desired dimer 13, which according to LC-MS analysis was caused by the slow reduction of the tert-butyldisulfide moiety in γ-thioNle. Apparently, compared to the reported correspondingly thio-protected γ-thioLys, which is readily reduced by MPAA, the tert-butyldisulfide moiety in γ-thioNle is much more stable. A preincubation of 11 with 100 mM TCEP for 90 min at 37 °C readily resulted in the fully liberated thiol moiety as evidenced from LC-MS analysis shown in Fig. 2B. Efficient NCL was achieved using a 40 mg mL−1 final Ub concentration and 250 mM MPAA for 2 hours at 37 °C according to LC-MS analysis (Fig. 2C and D), which indicated nearly full consumption of the thioNle-containing Ub and hydrolysis of remaining Ub-thioester excess. Intermediate 13 was obtained after RP-HPLC purification. The desulfurization under standard radical conditions proceeded smoothly and a subsequent purification by RP-HPLC and gel filtration yielded the target linear diUb 14 in a good overall yield (2.5 mg, 25% after NCL-deS) and purity, as confirmed by LC-MS (Fig. 2E and ESI†) and SDS-PAGE analysis (Fig. S1 in the ESI†).
 | ||
| Scheme 2 Synthesis of linear diUb. Reagents and conditions: (a) compound 1, PyBOP, DiPEA, NMP; (b) TFA, H2O, TIS, phenol, 38%; (c) TCEP, MPAA, 6 M Gnd·HCl, 0.15 M sodium phosphate, pH 7.5, 37 °C, 27%; (d) VA-044, TCEP, GSH, 6 M Gnd·HCl, 0.15 M sodium phosphate, pH 7.0, 37 °C, 93%. | ||
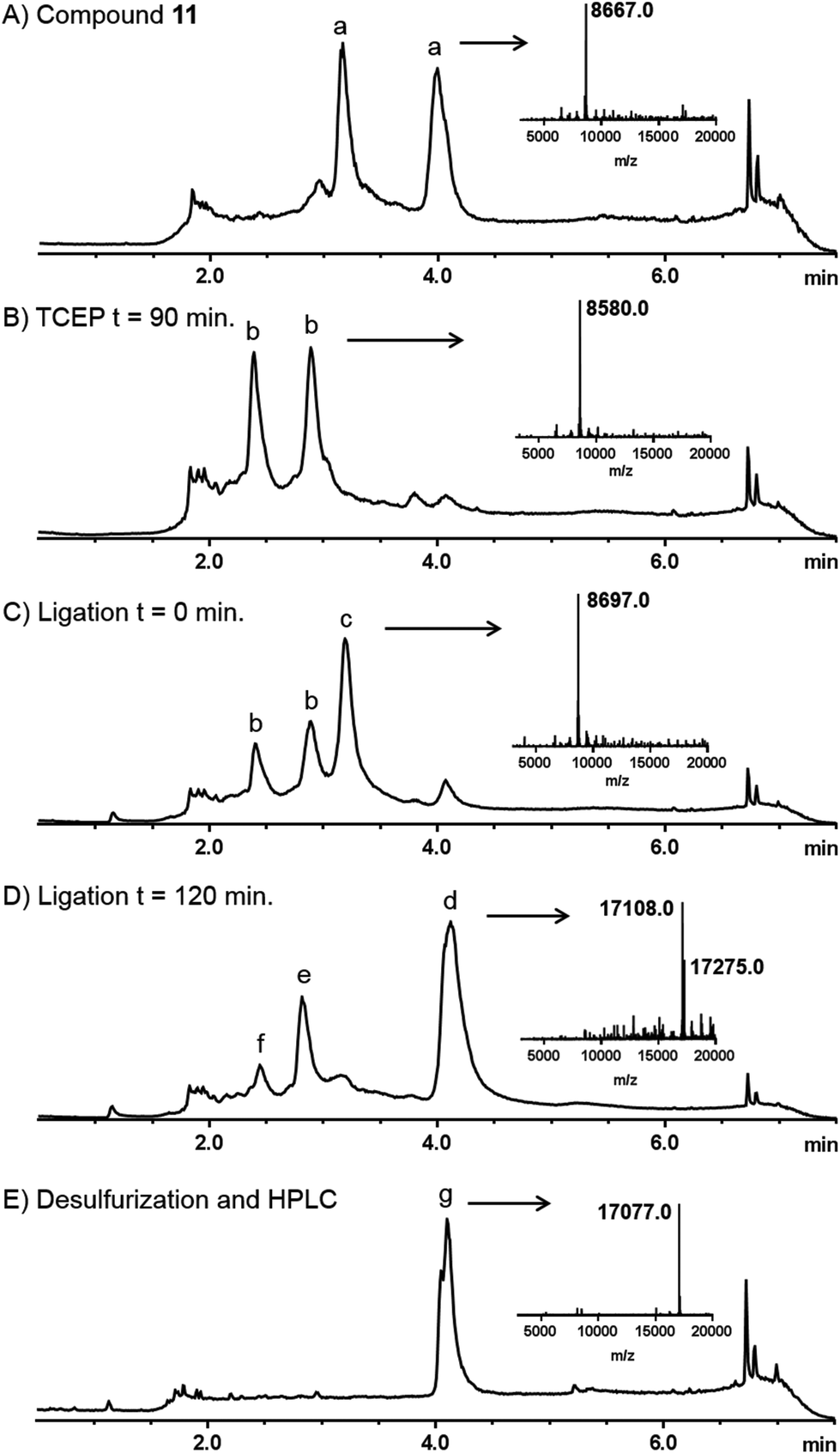 | ||
| Fig. 2 Analytical LC-MS analysis of the NCL-deS reactions. Mass traces of (A) purified compound 11, (B) preincubation of 11 with TCEP, (C) ligation reaction between 11 and 12 at t = 0 min, (D) ligation reaction between 11 and 12 at t = 120 min, and (E) product after desulfurization and purification. The insets in each trace represent the deconvoluted mass spectra of the indicated peaks. a = compound 11, b = reduced disulfide of 11 (free thiol), c = MPAA ester of 12, d = ligation product (mixture of free thiol and MPAA disulfide), e = hydrolysed Ub-thioester (8547 Da), f = assumed Ub-Gnd. Adduct (8588 Da), g = compound 14 (the small shoulder has a mass identical to the main peak and might indicate a different conformation; see also the ESI†). | ||
Correct folding of the purified synthetic linear diUb was verified by circular dichroism (CD) spectroscopy (Fig. 3A). The spectra of synthetic and purified expressed recombinant linear diUb were recorded and compared. Similar curves were obtained for both constructs, which indicates correct protein folding of synthetic linear diUb. To verify the biochemical function we compared synthetic and expressed linear diUb by enzymatic cleavage experiments. Deubiquitinase (DUB)-mediated cleavage of synthetic and expressed linear diUb was assessed using OTULIN, USP16, and USP21, three well-studied DUBs from the two largest DUB families, which are known to cleave the linear Ub linkage.25 Synthetic and expressed diUb were incubated with the three DUBs at 37 °C and the reaction samples were taken and immediately denatured at different time points. All proteins were resolved by SDS-PAGE and visualized by InstantBlue staining. The cleavage of diUb into monoUb is revealed by the disappearance of the diUb protein band and appearance of the monoUb protein band (Fig. 3B). Indeed, synthetic linear diUb is recognized and appropriately processed by all three DUBs, demonstrating proper protein folding and biochemical function.
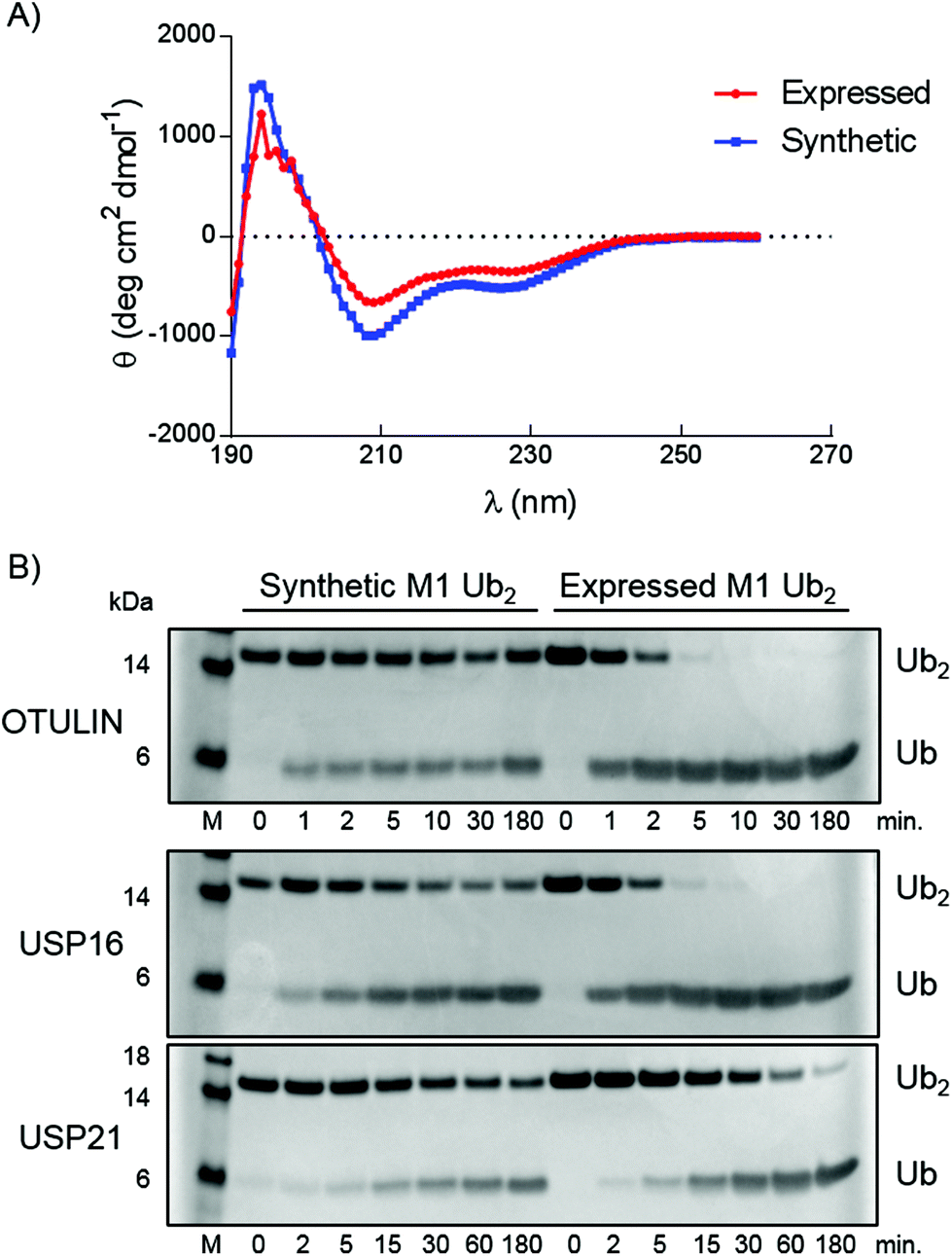 | ||
| Fig. 3 Characterization of synthetic linear diUb (14). (A) CD measurements of synthetic and expressed linear diUb. (B) SDS-PAGE analysis of linear diUb cleavage by OTULIN, USP16 and USP21. | ||
Interestingly, there appears to be a difference in the cleaving efficiency by the DUBs of synthetic diUb compared to expressed linear diUb, although for USP21 this difference is very small. As proper folding of the synthetic construct was confirmed by CD measurements (Fig. 3A) the observed difference in hydrolysis rates could likely be attributed to the methionine to norleucine substitution. OTULIN is specific for linear Ub chains, and the positioning of the Ub–Ub linkage in the active site was assigned with atomic resolution.27 From the crystal structures (PDB: 3ZNZ and 5OE7) it becomes apparent that the Ub methionine side chain points outwards from the active site and is probably not directly involved in the binding between enzyme and substrate. Our observation that the methionine to norleucine substitution affects the hydrolysis rate may therefore indicate that the thioether moiety in methionine is important for the interaction between Ub and OTULIN. Except for the finding that Met-1 sulfur can form a hydrogen bond with the Lys-63 backbone amine and that oxidation of this sulfur or Met-1 deletion affects the Ub folding below pH 4,28 little is known about the contribution of methionine to the biochemical function of Ub. As no structural data on the linear Ub–Ub linkage within the USP16 and USP21 active sites are available, the importance of the methionine residue for these DUBs remains to be investigated.
The development of γ-thionorleucine proved valuable as the NCL-deS construction of linear diUb proceeded efficiently and concomitantly omitted all mass spectrometry disadvantages associated with methionine oxidation. Unexpectedly, the methionine to norleucine substitution did affect the DUB mediated diUb cleavage, which may indicate a more profound role for methionine in the interaction between Ub and DUB than has been known so far.
Conclusions
In summary, we presented thioNle as a new handle for NCL and showed its feasibility for the N-terminal modification of proteins by preparing linear diubiquitin in a fully synthetic way for the first time. ThioNle is a new addition to the expanding thiolated amino acid toolbox and serves as a suitable methionine substitute in NCL with the advantage that it lacks the undesirable oxidation-prone thioether moiety. In addition, the fully synthetic preparation of linear diUb opens the way for the creation of linear diUb-based constructs, such as activity-based probes and assay reagents, which will benefit the field of Ub research.27bExperimental section
General
General reagents were obtained from Sigma Aldrich, Biosolve, Fluka and Acros, and used as received. Solvents were purchased from Biosolve or Sigma Aldrich. Dry THF and DCM were obtained using an Innovative Technology PureSolv Micro Solvent Purification System. Peptide synthesis reagents were purchased from NovaBiochem and Rapp Polymers. Analytical thin layer chromatography (TLC) was performed on Merck aluminium sheets (pre-coated with silica gel 60 F254). Compounds were visualized by UV adsorption (254 nm) and/or by using a solution of KMnO4 (7.5 g L−1) and K2CO3 (50 g L−1) in water and charring. Column chromatography was carried out on silica gel (40–63 u, 60 Å, Fluorochem). NMR spectra (1H, 13C) were recorded on a Bruker Ultrashield 300 Spectrometer (1H: 300.17 MHz, 13C: 75.47 MHz) at 298 K. Peak shapes in NMR spectra are indicated with symbols ‘d’ (doublet), ‘s’ (singlet), ‘t’ (triplet) and ‘m’ (multiplet). Chemical shifts (δ) are given in ppm relative to CDCl3 as an internal standard.LC-MS
LC-MS measurements were performed on an LC-MS system equipped with a Waters 2795 Separation Module (Alliance HT), a Waters 2996 Photodiode Array Detector (190–750 nm), a Waters Xbridge C18 column (2.1 × 30 mm, 3.5 μm) or a Waters Xbridge C18 column (2.1 × 100 mm, 3.5 μm) and a LCT ESI-Orthogonal Acceleration Time of Flight Mass Spectrometer. Samples were run using 2 mobile phases: A = 1% CH3CN and 0.1% formic acid in water and B = 1% water and 0.1% formic acid in CH3CN. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1 (deconvolution with Maxent1 function).Program 1: Waters Xbridge C18 column (2.1 × 30 mm, 3.5 μm); flow rate = 0.8 mL min−1, runtime = 6.2 min, column T = 40 °C, mass detection: 300–2000 Da. Gradient: 0–0.2 min: 5% B; 0.2–3.2 min: 5% → 95% B; 3.2–4.2 min: 95% B; 4.2–4.4 min: 95% → 5% B; 4.4–6.2 min: 5% B.
Program 2: Waters Xbridge C18 column (2.1 × 100 mm, 3.5 μm); flow rate = 0.4 mL min−1, runtime = 13 min, column T = 40 °C, mass detection: 300–2000 Da. Gradient: 0–0.4 min: 5% B; 0.4–9.0 min: 5% → 95% B; 9.0–11.2 min: 95% B; 11.2–11.3 min: 95% → 5% B; 11.3–13.00 min: 5% B.
LC-MS analysis of diUb as well as the TCEP reduction, NCL and desulfurization reactions (as shown in Fig. 2) were recorded on a Waters XEVO-G2 XS Q-TOF mass spectrometer equipped with an electrospray ion source in positive mode (capillary voltage: 1.2 kV, desolvation gas flow: 900 L h−1, temperature: 60 °C) with a resolution R = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Samples were run using 2 mobile phases: A = 0.1% formic acid in water and B = 0.1% formic acid in CH3CN on a Waters Acquity UPLC Protein BEH C4 column, 300 Å, 1.7 μm (2.1 × 50 mm); flow rate = 0.6 mL min−1, runtime = 10.00 min, column T = 60 °C, mass detection: 50–1500 Da. Gradient: 0–0.80 min: 2% B; 0.80–1.00 min: 2% → 23% B; 1.00–1.50: 23% B; 1.50–3.00 min: 23% → 25.5% B; 3.00–3.30 min: 25.5% B; 3.30–3.50 min: 25.5% → 29% B; 3.50–4.50: 29% → 32% B; 4.50–6.50 min: 32% → 100% B; 6.50–8.00 min: 100% B; 8.00–8.10 min: 100% → 2% B; 8.10–10.00 min: 2% B. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1.
000. Samples were run using 2 mobile phases: A = 0.1% formic acid in water and B = 0.1% formic acid in CH3CN on a Waters Acquity UPLC Protein BEH C4 column, 300 Å, 1.7 μm (2.1 × 50 mm); flow rate = 0.6 mL min−1, runtime = 10.00 min, column T = 60 °C, mass detection: 50–1500 Da. Gradient: 0–0.80 min: 2% B; 0.80–1.00 min: 2% → 23% B; 1.00–1.50: 23% B; 1.50–3.00 min: 23% → 25.5% B; 3.00–3.30 min: 25.5% B; 3.30–3.50 min: 25.5% → 29% B; 3.50–4.50: 29% → 32% B; 4.50–6.50 min: 32% → 100% B; 6.50–8.00 min: 100% B; 8.00–8.10 min: 100% → 2% B; 8.10–10.00 min: 2% B. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1.
HRMS measurements
High resolution mass spectra were recorded on a Waters XEVO-G2 XS Q-TOF mass spectrometer equipped with an electrospray ion source in positive mode (capillary voltage: 3.0 kV, desolvation gas flow: 900 L h−1, temperature: 60 °C) with a resolution R = 22000 and 200 pg μL−1 Leu-Enk (m/z = 556.2771) as a “lock mass”. Samples were run using 2 mobile phases: A = 0.1% formic acid in water and B = 0.1% formic acid in CH3CN on a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm); flow rate = 0.6 mL min−1, runtime = 3.00 min, column T = 60 °C, mass detection: 50–1500 Da. Gradient: 0–0.15 min: 2% B; 0.15–1.85 min: 2% → 100% B; 1.85–2.05: 100% B; 2.05–2.10 min: 100% → 2% B; 2.10–3.00 min: 100% B. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1.
HPLC purification
HPLC purifications were performed on a Waters HPLC equipped with a Waters 2489 UV/Vis detector, Waters fraction collector III and Waters XBridge prep C18 OBD (30 × 150 mm, 5 μm). Flowrate = 37.5 mL min−1. Mobile phase: A = H2O, B = CH3CN and C = 1% TFA in H2O. Gradient: 0–5 min: 90% A, 5% B, 5% C; 5–7 min: 5 → 20% B, 5% C; 7–18 min: 20 → 45% B, 5% C; 18–18.5 min: 45% → 95% B, 5% C; 18.5–21.6 min: 95% B, 5% C; 21.6–25 min: 95% → 5% B, 5% C.Alternatively, a Shimadzu LC-20AT equipped with a Shimadzu SPD-20A UV/Vis detector and a Shimadzu FRC-10A fraction collector and a Waters XBridge C18-Prep column (10 × 150 mm, 5 μm) was used. Flowrate = 4.00 or 6.50 mL min−1. Mobile phase: A = 0.05% TFA in H2O and B = 0.05% TFA in CH3CN. T = 40 °C. Gradient: 0–8.20 min: 5% B (4.00 mL min−1) and 0–1 min: 5% B; 1 → 2 min 5% → 10% B; 2–17 min: 10% → 70% B; 17–17.10 min: 70% → 95% B; 17.10–19.10 min: 95% B; 19.10–22.10: 5% B.
Synthesis of γ-thionorleucine
Synthesis of linear diubiquitin
The polypeptide was deprotected and detached from the resin by treatment with TFA/H2O/phenol/iPr3SiH (90.5/5/2.5/2; v/v/v/v; 2 mL) for 3 h. The reaction mixture was filtered directly into ice-cold Et2O/n-pentane (3/1; v/v; 15 mL) and the resin was spooled with TFA (2 × 2 mL). The solution was centrifuged and Et2O/n-pentane (supernatant) was removed. The pellet was washed with Et2O (3 × 15 mL), the solution was vortexed, the suspension was centrifuged and Et2O was removed. The wash step was repeated twice. The pellet was dissolved in H2O/CH3CN/formic acid (65/25/10; v/v/v; 5 mL) and lyophilized. The protein was subsequently purified using RP-HPLC.
Yields:
Ub(1-76, Nle1)-S(CH2)2CO2Me (12) = 229.19 mg, 26.44 μmol, 52.88%. LC-MS: Rt: 4.74 min: ESI MS+ (amu) calcd: 8649.0 [M], found 8650.0 (deconv.).
Ub(1-76, ThioNle1) (11) = 32.81 mg, 3.79 μmol, 37.9%. LC-MS: Rt: 4.92 min: ESI MS+ (amu) calcd: 8667.1 [M], found 8668.0 (deconv.).
The product was purified by gel filtration using a Bio-Rad NGC Chromatography system on a size exclusion S75 16/600 superdex PG-GE healthcare column with a volume bed of 120 mL and 3–70 kDa separation range using a filtered aqueous buffer containing 50 mM TRIS·HCl and 100 mM NaCl at pH 7.55 at a flow rate of 1 mL min−1. The sample was prepared by dissolving the product in DMSO (250 μL), and dropwise addition of this solution to MilliQ (2450 μL) and dropwise addition of 10× TRIS buffer (300 μL). The mixture was centrifuged for 5 min @3500 rpm. The fractions were analysed by SDS-PAGE analysis and pure fractions were pooled.
The product was obtained as a colourless solution containing 50 mM TRIS·HCl and 100 mM NaCl buffer at pH 7.55. LC-MS analysis (Program 2 and XEVO) was done to check the purity.
The protein concentration (and synthesis yield) was determined by SDS-PAGE analysis and quantification of band intensities after InstantBlue™ (Expedeon) staining using a GE Healthcare Amersham Imager 600 with ImageQuant TL 8.1 GE Healthcare Life Sciences software. Different amounts mono-ubiquitin (0.5 μg, 1 μg, 2 μg, and 4 μg) were included on the same gel to calculate the concentration of the final compound. Although quantification of bands from SDS-PAGE analysis is not the most accurate way of quantification, it is the most reliable one due to the relatively low concentration of the solution. Yield: linear diUb (14) = 2.54 mg, 0.15 μmol, 93.4%. LC-MS (Program 2): Rt: 5.18 min: ESI MS+ (amu) calcd: 17075.7 [M], found 17075.00 (deconv.). LC-MS (XEVO): Rt: 4.10 min: ESI MS+ (amu) calcd: 17075.7 [M], found 17077.00 (deconv.)
Expression of linear diubiquitin
Linear diubiquitin was expressed using a pET17b vector by inducing with 250 μM IPTG in BL21 (DE3) cells at an OD of 0.6. Purification was done as described for the yeast ubiquitin proprotein by Larsen et al., 1998.30 The concentration was determined using a NanoDrop spectrophotometer and estimated as 20.21 mg mL−1 (1.18 mM).Characterization of synthetic and expressed linear diubiquitin
Purity check and concentration normalization were performed using SDS-PAGE gel analysis. Synthetic (14) and expressed linear diubiquitin were diluted to ∼5.85 μM, ∼11.7 μM and ∼17.55 μM (∼1, ∼2 and ∼3 μg linear diUb per lane). 10 μL of each sample was diluted with 5 μL sample buffer (3×), containing NUPAGE® LDS sample buffer (4×, Invitrogen) (900 μL), β-mercaptoethanol (90 μL) and water (210 μL), heated at 95 °C for 5 minutes and loaded on 12% NUPAGE® Novex® Bis-Tris Mini Gels (Invitrogen) using MES-SDS running buffer. SeeBlue Pre-stained Plus2 Standard (Invitrogen, LC5925) was used as a marker. InstantBlue™ (Expedeon) stains were scanned using a GE Healthcare Amersham Imager 600. InstantBlue band intensities were determined using ImageQuant TL 8.1 (GE Healthcare Life Sciences).Circular dichroism (CD) analysis
The CD measurements were carried out on a JASCO J-815 CD spectrometer fitted with a Peltier temperature controller set to 25 °C. Samples were measured in a quartz cuvette with a 1 mm path length. Spectra were recorded from 260 to 190 nm at 1 nm intervals with a 1 nm bandwidth. The scan speed was 100 nm min−1 and the response time was 1 s. Data were obtained by averaging 5 scans. Data were converted to the mean residue molar ellipticity θ (deg cm2 dmol−1) according to the equation:31
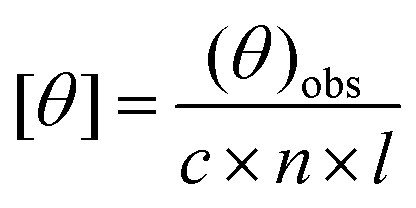
DUB cleavage assays
USP16 (human, full length (1-823), produced in-house as previously described25b), USP21 (human, cat. domain (196-565), Ubiquigent 64-0037-050) and OTULIN (human, full length (1-352), Ubiquigent 64-0048-050) were diluted to 2× final concentration (150 nM, 740 nM and 13 nM, respectively) in a buffer containing 50 mM Tris·HCl, 100 mM NaCl, pH 7.6, 5 mM DTT and 1 mg mL−1 3-[(3-cholamidopropyl) dimethylammonio] propanesulfonic acid (CHAPS). Subsequently, 40 μL of enzyme was mixed with 40 μL of 2× final concentration of synthetic or recombinant diubiquitin in the same buffer (30.8 μM or 27.3 μM respectively). The samples were incubated at 37 °C for 1, 2, 5, 10, 30 and 180 minutes followed by quenching using a sample buffer containing β-mercaptoethanol and subsequent SDS gel electrophoresis.A sample of the reaction mixture was diluted with a sample buffer (3×), containing NUPAGE® LDS sample buffer (4×, Invitrogen) (900 μL), β-mercaptoethanol (90 μL) and water (210 μL), heated at 95 °C for 5 minutes and loaded on 12% NUPAGE® Novex® Bis-Tris Mini Gels (Invitrogen) using MES-SDS running buffer. A SeeBlue Pre-stained Standard (Invitrogen, LC5925) was used as a marker. InstantBlue™ (Expedeon) stains were scanned using a GE Healthcare Amersham Imager 600.
Conflicts of interest
HO is a shareholder of UbiQ Bio BV.Acknowledgements
We thank Gabriëlle van Tilburg and Angela Elhebieshy for linear diubiquitin expression and purification, Dris El Atmioui and Cami Talavera Ormeño for SPPS and Bjorn van Doodewaerd for LC-MS assistance. This work was supported by NWO (VICI grant 724.013.002 to HO).References
- P. E. Dawson, T. W. Muir, I. Clarklewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef PubMed.
- L. Z. Yan and P. E. Dawson, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef PubMed.
- D. Crich and A. Banerjee, J. Am. Chem. Soc., 2007, 129, 10064–100065 CrossRef PubMed.
- (a) J. Chen, Q. Wan, Y. Yuan, J. L. Zhu and S. J. Danishefsky, Angew. Chem., Int. Ed., 2008, 47, 8521–8524 CrossRef PubMed; (b) C. Haase, H. Rohde and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef PubMed.
- J. Chen, P. Wang, J. L. Zhu, Q. Wan and S. J. Danishefsky, Tetrahedron, 2010, 66, 2277–2283 CrossRef PubMed.
- Z. Harpaz, P. Siman, K. S. A. Kumar and A. Brik, ChemBioChem, 2010, 11, 1232–1235 CrossRef PubMed.
- S. Y. Shang, Z. P. Tan, S. W. Dong and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 10784–10786 CrossRef PubMed.
- K. M. Cergol, R. E. Thompson, L. R. Malins, P. Turner and R. J. Payne, Org. Lett., 2014, 16, 290–293 CrossRef PubMed.
- L. R. Malins, K. M. Cergol and R. J. Payne, ChemBioChem, 2013, 14, 559–563 CrossRef PubMed.
- (a) X. Y. Guan, M. R. Drake and Z. P. Tan, Org. Lett., 2013, 15, 6128–6131 CrossRef PubMed; (b) R. E. Thompson, B. Chan, L. Radom, K. A. Jolliffe and R. J. Payne, Angew. Chem., Int. Ed., 2013, 52, 9723–9727 CrossRef PubMed.
- P. Siman, S. V. Karthikeyan and A. Brik, Org. Lett., 2012, 14, 1520–1523 CrossRef PubMed.
- L. R. Malins, K. M. Cergol and R. J. Payne, Chem. Sci., 2014, 5, 260–266 RSC.
- (a) F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef PubMed; (b) K. S. A. Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef PubMed; (c) K. K. Pasunooti, R. L. Yang, S. Vedachalam, B. K. Gorityala, C. F. Liu and X. W. Liu, Bioorg. Med. Chem. Lett., 2009, 19, 6268–6271 CrossRef PubMed; (d) R. Merkx, G. de Bruin, A. Kruithof, T. van den Bergh, E. Snip, M. Lutz, F. El Oualid and H. Ovaa, Chem. Sci., 2013, 4, 4494–4498 RSC.
- G. M. Fang, Y. M. Li, F. Shen, Y. C. Huang, J. B. Li, Y. Lin, H. K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef PubMed.
- S. Varland, C. Osberg and T. Arnesen, Proteomics, 2015, 15, 2385–2401 CrossRef PubMed.
- (a) A. Hershko, H. Heller, E. Eytan, G. Kaklij and I. A. Rose, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 7021–7025 CrossRef PubMed; (b) A. Ciechanover and R. Ben-Saadon, Trends Cell Biol., 2004, 14, 103–106 CrossRef PubMed.
- (a) J. P. Tam and Q. T. Yu, Biopolymers, 1998, 46, 319–327 CrossRef PubMed; (b) P. Van de Vijver, L. Scheer, J. van Beijnum, A. Griffioen and T. M. Hackeng, Chem. Commun., 2012, 48, 9403–9405 RSC.
- L. Moroder, J. Pept. Sci., 2005, 11, 187–214 CrossRef PubMed.
- G. M. Ksander, R. deJesus, A. Yuan, R. D. Ghai, A. Trapani, C. McMartin and R. Bohacek, J. Med. Chem., 1997, 40, 495–505 CrossRef PubMed.
- E. Huerta, B. van Genabeek, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Macromol. Rapid Commun., 2014, 35, 1320–1325 CrossRef PubMed.
- G. J. van der Heden van Noort, R. Kooij, P. R. Elliott, D. Komander and H. Ovaa, Org. Lett., 2017, 19, 6490–6493 CrossRef PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef PubMed.
- (a) H. Walczak, K. Iwai and I. Dikic, BMC Biol., 2012, 10, 23 CrossRef PubMed; (b) K. Iwai, H. Fujita and Y. Sasaki, Nat. Rev. Mol. Cell Biol., 2014, 15, 503–508 CrossRef PubMed.
- (a) T. E. T. Mevissen, M. K. Hospenthal, P. P. Geurink, P. R. Elliott, M. Akutsu, N. Arnaudo, R. Ekkebus, Y. Kulathu, T. Wauer, F. El Oualid, S. M. V. Freund, H. Ovaa and D. Komander, Cell, 2013, 154, 169–184 CrossRef PubMed; (b) A. C. Faesen, M. P. A. Luna-Vargas, P. P. Geurink, M. Clerici, R. Merkx, W. J. van Dijk, D. S. Hameed, F. El Oualid, H. Ovaa and T. K. Sixma, Chem. Biol., 2011, 18, 1550–1561 CrossRef PubMed.
- P. P. Geurink, B. D. M. van Tol, D. van Dalen, P. J. G. Brundel, T. E. T. Mevissen, J. N. Pruneda, P. R. Elliott, G. B. A. van Tilburg, D. Komander and H. Ovaa, ChemBioChem, 2016, 17, 816–820 CrossRef PubMed.
- (a) K. Keusekotten, P. R. Elliott, L. Glockner, B. K. Fiil, R. B. Damgaard, Y. Kulathu, T. Wauer, M. K. Hospenthal, M. Gyrd-Hansen, D. Krappmann, K. Hofmann and D. Komander, Cell, 2013, 153, 1312–1326 CrossRef PubMed; (b) A. Weber, P. R. Elliott, A. Pinto-Fernandez, S. Bonham, B. M. Kessler, D. Komander, F. El Oualid and D. Krappmann, Cell Chem. Biol., 2017, 24, 1299–1313 CrossRef PubMed.
- S. Bamezai, M. A. T. Banez and E. Breslow, Biochemistry, 1990, 29, 5389–5396 CrossRef PubMed.
- G. Galley, A. Goergler, Z. K. Groebke Zbinden and R. Norcross, Patent US2011/0144333A1, 2010 Search PubMed.
- C. N. Larsen, B. A. Krantz and K. D. Wilkinson, Biochemistry, 1998, 37, 3358–3368 CrossRef PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2527–2535 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR and LC-MS spectra and full gel images. See DOI: 10.1039/c8ob01627e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |