
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinoline–galactose hybrids bind selectively with high affinity to a galectin-8 N-terminal domain†
Kumar Bhaskar
Pal‡
a,
Mukul
Mahanti‡
a,
Xiaoli
Huang
b,
Stella
Persson
a,
Anders P.
Sundin
a,
Fredrik R.
Zetterberg
c,
Stina
Oredsson
b,
Hakon
Leffler
d and
Ulf J.
Nilsson
 *a
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, SE-221 00, Lund, Sweden. E-mail: ulf.nilsson@chem.lu
bDepartment of Biology, Lund University, Sölvegatan, 35/37, SE-223 62, Lund, Sweden
cGalecto Biotech AB, Sahlgrenska Science Park, Medicinaregatan 8 A, SE-413 46 Gothenburg, Sweden
dSection MIG, Department of Laboratory Medicine, Lund University, BMC-C1228b, Klinikgatan 28, SE-221 84, Lund, Sweden
First published on 3rd August 2018
Abstract
Quinolines, indolizines, and coumarins are well known structural elements in many biologically active molecules. In this report, we have developed straightforward methods to incorporate quinoline, indolizine, and coumarin structures into galactoside derivatives under robust reaction conditions for the discovery of glycomimetic inhibitors of the galectin family of proteins that are involved in immunological and tumor-promoting biological processes. Evaluation of the quinoline, indolizine and coumarin-derivatised galactosides as inhibitors of the human galectin-1, 2, 3, 4N (N-terminal domain), 4C (C-terminal domain), 7, 8N, 8C, 9N, and 9C revealed quinoline derivatives that selectively bound galectin-8N, a galectin with key roles in lymphangiogenesis, tumor progression, and autophagy, with up to nearly 60-fold affinity improvements relative to methyl β-D-galactopyranoside. Molecular dynamics simulations proposed an interaction mode in which Arg59 had moved 2.5 Å and in which an inhibitor carboxylate and quinoline nitrogen formed structure-stabilizing water-mediated hydrogen bonds. The compounds were demonstrated to be non-toxic in an MTT assay with several breast cancer cell lines and one normal cell line. The improved affinity, selectivity, and low cytotoxicity suggest that the quinoline–galactoside derivatives provide an attractive starting point for the development of galectin-8N inhibitors potentially interfering with pathological lymphangiogenesis, autophagy, and tumor progression.
1. Introduction
Galectins are an ancient family of glycan binding proteins found in most organisms, including 15 mammalian members.1 Galectins are illustrated as a developmental preserved sub-class of endogenous lectins based on the structures of their carbohydrate recognition domain (CRD) and binding to β-D-galactoside-containing ligands.2 Galectins contribute to a large diversity of biologically important functions including cell–cell and cell–matrix interactions, immune and inflammatory responses, induction of apoptosis for T-cells, anti-apoptotic functions, modulation of cell adhesion and migration.3–5 The dysfunction of such galectin-related biological mechanisms has been demonstrated to influence cancer biology, such as tumor cell survival, neoplastic metamorphosis, angiogenesis, and tumour metastasis. Thus, there is a pressing need for molecules towards the discovery of galectin-blocking drug leads.6–9Quinolines, indolizines, and coumarins and substituted derivatives thereof are common structures in medicinal and synthetic organic chemistry and frequently display distinct biological activities.10–13 As galectins specifically bind galactose-containing glycoconjugates and have been demonstrated to be inhibited by synthetic galactosides derivatized with aromatic structures at C-3,14 we hypothesized that combining quinolines, indolizines, and coumarins with a β-D-galactoside core structure could lead to the discovery of new compounds with enhanced galectin affinities and selectivities. Here we report the design and synthesis of a series of quinoline-, indolizine-, and coumarin-carrying galactoside derivatives and their evaluation as inhibitors of galectin-1, 2, 3, 4N (N-terminal domain), 4C (C-terminal domain), 7, 8N, 8C, 9N, and 9C, as well as of their cytotoxic properties.
2. Results and discussion
2.1 Chemistry
Quinoline bromides 7a–7c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15–17 are known in the literature, while bromides 7d–7i were synthesized (Scheme 1). 2-Methyl quinolines 6e–f were synthesized from crotonaldehyde and anilines 4 and 5. Radical mono-bromination of the 2-methyl group of quinolines 6d–6i gave quinolinylmethyl bromides 7d–7i that together with the known bromides 7a–7c were used in stannylidene-acetal-mediated regioselective alkylation of methyl β-D-galactopyranoside 8 to afford compounds 1a–1i in 71–81% yield (Scheme 1). Compounds 1j and 1k were obtained in 63–66% yield by the hydrolysis of the corresponding methyl esters 1d and 1e under basic conditions.
15–17 are known in the literature, while bromides 7d–7i were synthesized (Scheme 1). 2-Methyl quinolines 6e–f were synthesized from crotonaldehyde and anilines 4 and 5. Radical mono-bromination of the 2-methyl group of quinolines 6d–6i gave quinolinylmethyl bromides 7d–7i that together with the known bromides 7a–7c were used in stannylidene-acetal-mediated regioselective alkylation of methyl β-D-galactopyranoside 8 to afford compounds 1a–1i in 71–81% yield (Scheme 1). Compounds 1j and 1k were obtained in 63–66% yield by the hydrolysis of the corresponding methyl esters 1d and 1e under basic conditions.
 | ||
| Scheme 1 Synthesis of quinoline derived methyl β-D-galactosides 1a–1k. | ||
Recently, Bonte and co-workers reported an efficient way towards the formation of substituted indolizines via the cycloaddition of pyridinium ylides with propiolic esters.18 Inspired by this report, we treated known methyl 2,4,6-tri-O-benzyl-β-D-galactopyranoside 1119 with propiolic acid under DIC-promotion to give the 3-O-propiolate 12 as an alkyne source for the synthesis of indolizine-derivatized galactosides 2a–2e (Scheme 2). Pyridinium salts 10d–10e were prepared by the alkylation of the corresponding pyridines 9d–9e. The treatment of 10d–10e and the known pyridinium salts 10a–10c18 in methanol and with K2CO3 as a base generated intermediate pyridinium ylides that were reacted with the propiolate 12 at room temperature for 12 h. The crude products were subjected to hydrogenolysis to remove benzyl ether protecting groups to give the indolizines 2a–2e in 25–34% yield over two steps.
 | ||
| Scheme 2 Synthesis of indolizidine-derivatised thiogalactosides 2a–2e. | ||
Coumarins can be easily equipped with an alkyne functionality, which would open up for use in cycloaddition reactions with the 3-azido galactoside 16. The unprotected 3-azido derivative 16 was synthesized via Zemplén de-O-acetylation20 of the known p-tolyl 2,4,6-tri-O-acetyl-3-azido-3-deoxy-1-thio-β-D-galactopyranoside 1521 (Scheme 3). Copper-(I)-catalysed cycloadditions22–24 with 6-alkoxy coumarins 14a–14d and 7-alkoxycoumarin 14e gave the triazoles 3a–3e in 59–85% yields.
 | ||
| Scheme 3 Synthesis of triazole and coumarin derivatised thiogalactosides 3a–3e. | ||
2.2 Galectin binding affinities
The quinoline- (1a–1i), indolizine- (2a–2e), and coumarin- (3a–3e) derived galactosides were evaluated as inhibitors against the human galectin-1, -2, -3. -4N- and C-terminal domains, -7, 8N-and C-terminal domains, and -9N- and C-terminal domains in a competitive protein-binding assay based on fluorescence anisotropy.25–27 Methyl β-D-galactopyranoside 8 was included as a reference27,28 (Table 1). Overall, the indolizines (2a–2e) display better inhibition potency for galectin-3 and two–five-fold selectivity over the other galectins evaluated. Compounds 2a, 2b, and 2e showed similar inhibition with dissociation constants ∼300 μM, thus resulting in 53-fold affinity enhancement over the reference compound 8. The coumarin-functionalized galactose derivatives 3a–3e bound the tested galectins with apparently no selectivity and moderate affinity enhancements.| Compounds | Galectin | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4Nb | 4Cc | 7 | 8Nb | 8Cc | 9Nb | 9Cc | |
| a The data are averaged and standard deviation of 4–8 single-double point measurements. b N-Terminal domain. c C-Terminal domain. d Non-binding. e Not available. | ||||||||||
| 1a | 1700 ± 100 | 2600 ± 340 | 620 ± 50 | 1000 ± 38 | 2700 ± 280 | NBd | 700 ± 41 | 2900 ± 38 | 470 ± 71 | 2200 ± 110 |
| 1b | 1300 ± 130 | 1900 ± 240 | 580 ± 22 | 1700 ± 52 | 2100 ± 520 | NBd | 700 ± 50 | 3100 ± 340 | 510 ± 88 | NBd |
| 1c | 1900 ± 70 | 2200 ± 100 | 510 ± 83 | 1600 ± 280 | 4000 ± 70 | NBd | 440 ± 46 | 2800 ± 21 | 550 ± 110 | 2400 ± 770 |
| 1d | 1200 ± 20 | 2000 ± 40 | 710 ± 170 | 2300 ± 210 | 4600 ± 230 | NBd | 520 ± 38 | 4600 ± 760 | 1300 ± 270 | NBd |
| 1e | 1700 ± 20 | 1600 ± 40 | 610 ± 2 | 1600 ± 130 | NBd | NBd | 630 ± 9 | 3100 ± 670 | 550 ± 53 | 1900 ± 340 |
| 1f | 1200 ± 15 | 1900 ± 600 | 410 ± 53 | 1400 ± 50 | 2800 ± 10 | NBd | 1400 ± 60 | 2700 ± 14 | 580 ± 86 | NBd |
| 1g | NBd | 2000 ± 890 | 820 ± 33 | 2100 ± 510 | NBd | NBd | 1000 ± 25 | 3500 ± 440 | 1100 ± 10 | 1300 ± 13 |
| 1h | 1300 ± 90 | 2700 ± 61 | NAd | 880 ± 160 | 2300 ± 280 | NBd | 3500 ± 650 | NBd | 750 ± 7 | NBd |
| 1i | 1400 ± 130 | 1200 ± 19 | 450 ± 84 | 470 ± 80 | 1700 ± 270 | NBd | 640 ± 80 | 2400 ± 590 | 390 ± 29 | 1700 ± 78 |
| 1j | 690 ± 8 | 1700 ± 260 | 250 ± 11 | NBd | 3800 ± 480 | NBd | 250 ± 10 | 3100 ± 41 | 1500 ± 100 | 930 ± 8 |
| 1k | 880 ± 25 | 2800 ± 370 | 380 ± 11 | NBd | 2600 ± 270 | NBd | 110 ± 6 | 3200 ± 1210 | 300 ± 16 | 3900 ± 150 |
| 2a | NBd | 1500 ± 140 | 390 ± 34 | 1400 ± 370 | 1700 ± 470 | NBd | 1000 ± 94 | NBd | NBd | 1800 ± 130 |
| 2b | 1600 ± 220 | 1200 ± 80 | 360 ± 42 | 1200 ± 10 | 2200 ± 250 | NBd | 840 ± 22 | 3200 ± 165 | NBd | 1700 ± 76 |
| 2c | 1100 ± 74 | 1300 ± 4 | 740 ± 29 | 1100 ± 120 | 3600 ± 1230 | NBd | NBd | NBd | NBd | 2600 ± 54 |
| 2d | NBd | 500 ± 30 | 960 ± 182 | 830 ± 85 | 870 ± 80 | 2270 ± 230 | 1300 ± 76 | 1700 ± 56 | NBd | NBd |
| 2e | 480 ± 34 | 750 ± 54 | 300 ± 29 | 1600 ± 260 | 1700 ± 80 | >1370 | 740 ± 39 | 1600 ± 76 | NAe | NBd |
| 3a | 500 ± 8 | 1400 ± 47 | 490 ± 67 | 650 ± 120 | NAe | NBd | 540 ± 60 | 990 ± 2 | 1600 ± 130 | 640 ± 32 |
| 3b | 820 ± 100 | 1100 ± 130 | 1370 ± 120 | 710 ± 150 | 1300 ± 26 | NBd | 480 ± 4 | 1000 ± 94 | 2600 ± 100 | 1100 ± 370 |
| 3c | 150 ± 26 | NBd | 260 ± 41 | 1300 ± 80 | 890 ± 50 | 2560 ± 560 | NBd | 260 ± 84 | NBd | NBd |
| 3d | 500 ± 8 | 620 ± 120 | 830 ± 49 | 1300 ± 270 | 2000 ± 240 | NBd | 520 ± 26 | 980 ± 130 | NBd | NBd |
| 3e | 460 ± 18 | 720 ± 76 | 480 ± 44 | 840 ± 83 | 610 ± 110 | NBd | 360 ± 9 | 950 ± 28 | NBd | 510 ± 13 |
|
827,28 |
>10000 |
13000 |
4400 | 6600 | 10000 |
4800 | 6300 | >30000 |
3300 | 8600 ± 730 |
| 17 | 670 ± 125 | NAe | NAe | NAe | NAe | NAe | 150 ± 12 | NAe | NAe | NAe |
| 18 | >4000 | >6000 | 730 ± 80 | 3100 ± 500 | 2000 ± 40 | NAe | >4000 | >4000 | 1700 ± 110 | >3500 |
The quinoline-derivatized methyl β-D-galactosides 1a–1k revealed a more varying structure–activity relationship and particularly efficient inhibition of galectin-8N with down to ≈100 μM affinities reflecting a near 60-fold affinity enhancement over the reference compound 8. In general, 1a–1k displayed somewhat higher affinity for galectin-8N than the reference compound 8. Substituents on the quinoline (1b–1i) either had no effects or slightly decreased the affinity for galectin-8N when compared to the unsubstituted 1a. In contrast, the two carboxylic acid substituted quinolines 1j and 1k proved to show promising affinities for galectin-8N with 1j and 1k having dissociation constants of 250 and 110 μM, respectively. Hence, moving the carboxylate from quinoline position 6 (1j) to position 7 (1k) doubled the affinity, suggesting the presence of a specific interaction formed by the 7-carboxylate. Compound 1k is the best β-D-galactopyranoside-based monosaccharide inhibitor for human galectin-8N reported hitherto and better than the methyl glycoside of the corresponding natural disaccharide fragment sialyl-α-(2-3)-galactoside 1729,30 (Fig. 1). Rajput et al.31 have reported coumarin α-D-galactopyranoside-based derivatives with affinities down to ≈200 μM for galectin-8N, but these compounds differed from 1k in that they were even better inhibitors of galectin-3 and thus displayed high galectin-3 selectivity. In order to investigate the role of the quinoline nitrogen in interactions enhancing the affinities for galectin-8N, we synthesized the corresponding naphthalene derivative 18via stannylidene acetal-mediated alkylation of 8 in the same manner as described for 1a–1i. The naphthalene-derivatized analog 18 was proved to be more than 36 times worse than 1k as a galectin-8N inhibitor, which indicates an important role of the quinoline nitrogen in binding to galectin-8N.
 | ||
| Fig. 1 Reference compound methyl sialyl-α-(2-3)-galactopyranoside 1729,30 and methyl 3-O-naphth-2-yl-β-D-methylgalactoside 18. | ||
2.3 Affinity enhancement through combination of 1j and 1k quinolines with an α-thiophenyl aglycon fragment
In an attempt to further enhance the affinity of the quinoline-derived ligands 1j and 1k, we hypothesised that combining the carboxy-quinolines with α-thiophenyl aglycons, recently reported to greatly enhance galectin-3 affinities,32 could be a viable strategy. Per-acetylated galactose 19 was converted to 3,4-dichlorophenyl α-thiogalactoside 20, followed by deacetylation and stannylidene-mediated regioselective 3-O-quinolylmethylation to give 22a and 22b. Alkaline hydrolysis of 22a and 22b gave the corresponding carboxy-quinoline thiogalactosides 23a and 23b (Scheme 4). | ||
| Scheme 4 Synthesis of quinoline-derivatized α-thiogalactosides 23a and 23b. | ||
Compounds 23a and 23b showed greatly increased affinity for galectin-8N as compared to 1j and 1k (128-fold and 73-fold, respectively). This observation corroborates the recent finding that m-halo-substituted α-thiophenyl aglycons greatly enhance the affinities of galactosides towards galectins.32 However, an even larger influence of the α-thiophenyl aglycon was observed for galectin-3 and -9N, as 23a and 23b bound these two galectins similarly to galectin-8N. Hence, although the α-thiophenyl aglycon as hypothesized increases the affinity for galectin-8N, the selectivities observed for 1j and 1k are lost due to the even larger influence of the α-thiophenyl aglycons on galectin-3 and -9N binding. Nevertheless, compound 23b is the best synthetic monosaccharide-based galetin-8N inhibitor reported to date (Table 2).
| Compounds | Galectin | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4Nb | 4Cc | 7 | 8Nb | 8Cc | 9Nb | 9Cc | |
| a The data are averaged and standard deviation of 5–12 single to double point measurements. b N-Terminal domain. c C-Terminal domain. d Not available. | ||||||||||
| 23a | 100 ± 5 | 110 ± 12 | 1.2 ± 0.02 | 110 ± 14 | 43 ± 8 | 32 ± 5 | 1.9 ± 0.1 | 330 ± 48 | 8.8 ± 0.5 | 27 ± 7 |
| 23b | 48 ± 4.4 | 59 ± 4 | 1.27 ± 0.07 | 43 ± 7.1 | 43 ± 5.7 | NAd | 1.5 ± 0.08 | 240 ± 15 | 2.06 ± 0.09 | 14 ± 1.3 |
2.4 Molecular modelling
In order to gain understanding of the binding of the quinoline derivatives for galectin-8N, molecular dynamics simulations were performed. Low energy conformations of 1k in pbd id 3VKO, with the galactose unit placed in the galectin core galactose binding site in the same pose as natural galactose-containing ligands, were generated by rotating the three bonds between the galactose C-3 atom and the quinoline, followed by energy minimization with the OPLS3 force field and the GB/SA solvation method for water. Initial MD simulations were performed with these low energy structures (see ESI Fig. S109†), which all drifted towards the same general structure with the quinoline group oriented in the galactose C3–O3 direction close to Arg45. Subsequently, a 1000 ns molecular dynamics simulation was performed starting from a ligand posed close to Arg45 and this converged after 300 ns to a protein–ligand 1k geometry where Gly142 was buried in the protein with hydrogen bonds from Arg59, Asp49, His65, Gln47 and a buried water molecule and simultaneously Arg59 had both hydrophobic planar sides removed from the solvent (Fig. 2). Furthermore, this interaction mode led to an altered conformation of the side chain of Arg59 (the guanidinium group moved about 2.5 Å) compared to the apo structure (2YV8) or the corresponding lactose complexes (2YXS, 3AP4, 3VKL, and 3VKM). The 1k quinoline moiety was positioned in a subsite adjacent to O3 of the bound galactopyranose with a replacement of poorly coordinated water molecules, while at the same time positioning the 7-carboxylate for multiple water-mediated hydrogen bonds with Arg45, Gln47, and Gly142 that would be less favorable for the corresponding 6-carboxylate 1j. Furthermore, in a majority of sampled complex conformations the quinoline nitrogen formed a water-mediated hydrogen bond to the galactose HO2, which led to a conformation with simultaneous ideal steric fit of both the quinoline and galactose moieties of 1k.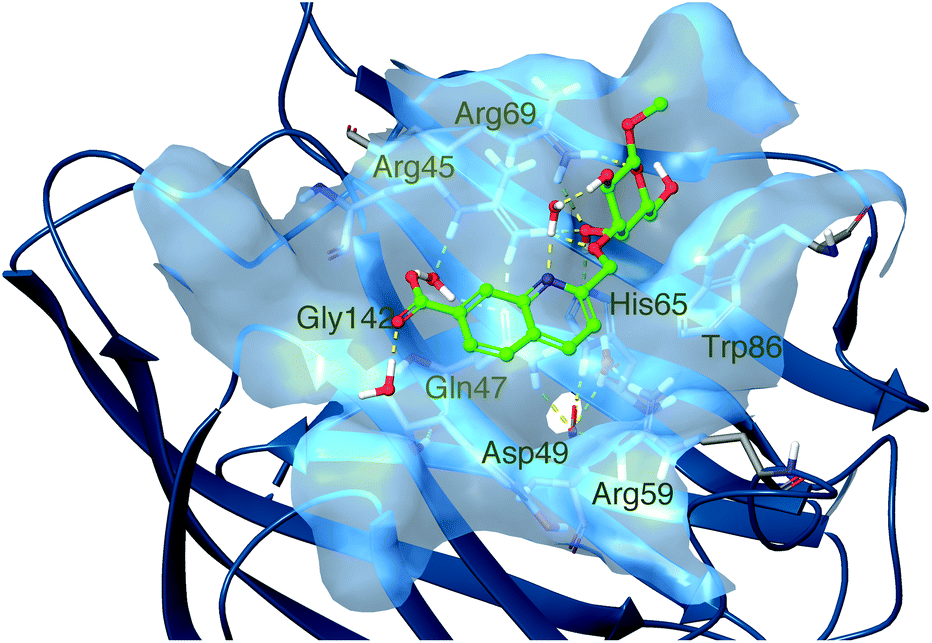 | ||
| Fig. 2 Molecular dynamics simulation snapshot of a representative low-energy conformation of 1k in complex with galectin-8N. | ||
2.5 Cytotoxicity evaluation
In addition to high galectin-8 affinity and selectivity, a criterion for compounds to be valuable as inhibitors in biological experiments is lack of or low toxicity. The cytotoxicities of all the quinoline-, indolizine-, and coumarin-derived galectin inhibitors 1a–1k, 2a–2e, 3a–3e, 23a, and 23b were investigated in JIMT-1 and MCF-7 human breast cancer cell lines, as well as in the human normal-like MCF-10A cell line using an MTT assay.33–35 The MTT assay is a colorimetric assay that is based on the reduction of the water-soluble tetrazolium salt MTT to an insoluble purple formazan in the mitochondria of viable cells. Thus, the MTT reduction is used to reflect the viable cell number. None of the inhibitors affect the cell viability at the concentrations ranging from 0.05 μM to 50 μM in either of the three cell lines except for 3d, which gave the IC50 values 33, 39, and 27 μM in JIMT-1, MCF-7, and MCF-10A cells, respectively (Fig. S103–108†). The results indicate a low cytotoxicity of the inhibitors.3. Conclusions
In conclusion, we have developed simple, efficient, and economically viable methods to synthesize quinoline, indolizine and coumarin derivatised galactosides. 3-O-(Carboxyquinoline)-derivatized methyl galactosides displayed particularly good affinity and selectivity against galectin-8N, while the 3-O-indolizinyl and coumaryl derivatives showed moderate affinity enhancements over the reference methyl β-D-galactopyranoside. Combining the 3-O-carboxyquinoline moiety with an α-3,4-dichlorophenylthio aglycon greatly enhanced affinity for galectin-8N down to 1.5 μM, but also reduced selectivity over galectin-3 and -9N. Molecular dynamics simulation of 1k in complex with galectin-8N suggested an ideal fit of the quinoline with a subsite near the galactose site, that allowed Gly142 to be buried in the protein and directly stabilized through water-mediated ligand carboxylate–protein hydrogen bonds. Furthermore, a water-mediated quinoline nitrogen hydrogen bond to galactose HO2 provided optimal steric and electronic complementarity between 1k and galectin-8N, which may explain the selectivity-induction and affinity-enhancement of the quinoline moiety.Finally, the compounds show low cytotoxicity in both tumor and normal cell lines, which further supports the hypothesis that 3-O-carboxyquinoline-derivatised galactosides may constitute a promising class of non-toxic compounds for the discovery of selective galectin-8 inhibitors. Furthermore, the inhibitory activities of the quinolines against one of the domains of galectin-8, N-terminal is probably enough to have a pharmacological activity against galectin-8, and it has been shown earlier that the inhibition of one of the galectin-8 domains can block the function of galectin-8.36 This is particularly important in light of the reported key roles of galectin-8 in autophagy,37 lymphangiogenesis,38 and cancer.39
4. Experimental
4.1 General
All reactions were carried out in oven-dried glassware. All solvents and reagents were purchased from commercial sources or synthesized via literature protocols and used without further purification. TLC analysis was performed on pre-coated Merck silica gel 60 F254 plates using UV light and charring solution (10 mL conc. H2SO4/90 mL EtOH). Flash column chromatography was performed on SiO2 purchased from Aldrich (technical grade, 60 Å pore size, 230–400 mesh, 40–63 μm). Preparative HPLC was performed on an Agilent 1260 infinity system, column SymmetryPrep-C18, and a 17 mL min−1 H2O–MeCN gradient 10–100% 15 min with 0.1% formic acid. All NMR spectra were recorded with a Bruker DRX 400 MHz spectrometer (400 MHz for 1H, 100 MHz for 13C, 376 MHz for 19F) at ambient temperature using CDCl3, CD3OD or (CD3)2SO as solvents. Chemical shifts are given in ppm relative to the residual solvent peak (1H NMR: CDCl3δ 7.26; CD3OD δ 3.31; (CD3)2SO δ 2.50; 13C NMR: CDCl3δ 77.16; CD3OD δ 49.00; (CD3)2SO δ 39.52) with multiplicity (b = broad, s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, hept = heptet, m = multiplet, app = apparent), coupling constants (in Hz) and integration. High-resolution mass analyses were performed using a Micromass Q-TOF mass spectrometer (ESI). Purities of final compounds were determined by UPLC (Waters Acquity UPLC system, column Waters Acquity CSH C18, 0.5 mL min−1 H2O–MeCN gradient 5–95% 10 min with 0.1% formic acid). Analytical data are given if the compound is novel or not fully characterized in the literature.4.2. Methyl 2-methylquinoline-7-carboxylate 6e
To 3-aminobenzoic acid 4 (2.5 g, 18.24 mmol), 6 N HCl (37 mL) was added and the mixture was refluxed for 2 h. Crotonaldehyde (1.8 mL, 21.9 mmol) was added dropwise over 45 min. After 4 h, the reaction mixture was cooled to 0 °C and the pH was adjusted to 3–5 with aqueous ammonia solution. The solid suspended in the aqueous layer was dissolved by adding DCM. The organic layer was collected, dried over Na2SO4, filtered and dried under vacuum. Recrystallization from EtOH and washing (EtOH, 7 × 50 mL) afforded 2-methylquinoline-7-carboxylic acid in 46% yield (1.56 g, 8.34 mmol) as a white solid, which was dissolved in methanol (15 mL). Sulfuric acid (1.5 mL) was added dropwise at 0 °C and then stirred at 65 °C for 12 h. The reaction mixture was concentrated and to the residue dichloromethane and aqueous sodium carbonate solutions were added. The organic layer was collected, dried with Na2SO4 and concentrated to afford 6e (1.0 g, 4.97 mmol, 59.6%) as a white solid. 1H NMR (CDCl3, 400 MHz): 9.09 (d, 1H, J 8.8 Hz, ArH), 8.09 (dd, 2H, J 2.8 Hz, J 6.8 Hz, ArH), 7.57 (t, 1H, J 8.0 Hz, ArH), 7.26 (d, 1H, J 8.8 Hz, ArH), 3.98 (s, 3H, CO2CH3), 2.64 (s, 3H, C11H8NO2CH3). 13C NMR (CDCl3, 100 MHz): 166.9 (CO2CH3), 159.2, 147.9, 134.2, 134.1, 129.8, 127.8, 126.4, 125.2, 123.4, (CH2Ar), 52.1 (CO2CH3), 34.0 (C11H8NO2CH3). HRMS calcd for C12H11NO2 + H+ (M + H)+: 202.0868, found: 202.0867.4.3. 2-Methyl-6,7-difluoroquinoline 6f
To a refluxing solution of 3,4-difluoroaniline 5 (1 mL, 10 mmol), tetrachloro-1,4-benzoquinone (2.5 g, 10 mmol), and concentrated hydrochloric acid (2.6 mL) in 2-butanol (20 mL) was added crotonaldehyde (1 mL, 12 mmol). After 2.5 h, the reaction mixture was concentrated and the resulting residue was stirred in warm (50 °C) THF (15 mL). The mixture was cooled to 0 °C and the solid was collected by filtration and washed with cold THF (3 × 15 mL). The solid was stirred in distilled water (80 mL), the resulting solution made basic with K2CO3 and extracted with EtOAc (3 × 40 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated to give 2-methyl-6,7-difluoroquinoline 6f as a black solid in 64% yield (1.15 g, 6.4 mmol) without any further purification. 1H NMR (CDCl3, 400 MHz): 7.96 (d, 1H, J 8.4 Hz, ArH), 7.74 (dd, 1H, J 7.6 Hz, J 11.6 Hz, ArH), 7.57 (dd, 1H, J 8.8 Hz, J 10.4 Hz, ArH), 7.27 (d, 1H, J 8.4 Hz, ArH), 2.71 (s, 3H, C10H7NF2CH3). 13C NMR (CDCl3, 100 MHz): 159.6 (d, J 2.8 Hz), 153.6 (d, J 15.8 Hz), 151.1 (dd, J 14.0 Hz, J 15.8 Hz), 148.5 (d, J 15.7 Hz), 145.0 (d, J 9.7 Hz), 135.5 (d, J 3.3 Hz), 123.3 (d, J 8.4 Hz), 122.3 (d, J 2.5 Hz), 115.1 (d, J 16.1 Hz), 112.8 (dd, J 1.4 Hz, J 17.4 Hz), 25.3 (s, 3H, C10H7NF2CH3). 19F NMR (CDCl3, 376 MHz): −131.9 (d, J 20.8 Hz), −137.0 (d, J 20.8 Hz). HRMS calcd for C10H7F2N + H+ (M + H)+: 180.0625, found: 180.0623.4.4. General method for the preparation of substituted-2-(bromomethyl)benzoquinolines 7d–7i
A mixture of substituted 2-methylquinolines 7d–7i (1.0 eq.), NBS (1 eq.) and AIBN (0.2 eq.) in CCl4 (5 mL for 1 mmol of 7d–7i) was stirred at 80 °C for 6 h. H2O (20 mL for 1 mmol of 7d–7i) and DCM (20 mL for 1 mmol of 7d–7i) were added to the mixture and the layers were separated. The aqueous layer was extracted with DCM (20 mL × 2). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The organic layers were concentrated and purified by column chromatography to afford substituted 2-(bromomethyl)benzoquinolines 7d–7i. See the ESI† for physical data.4.5. General method for the preparation of quinoline derived galactosides 1a–1i and 23a–23b
Methyl β-D-galactopyranoside, 8 (1 eq.), was dissolved in dry MeOH (1 mL per 15 mg) and Bu2SnO (1.1 eq.) was added to the solution. The mixture was stirred under reflux conditions for 3 hours under an N2 atmosphere. The reaction mixture became transparent after 1 h. After 3 h, the solvent was evaporated under reduced pressure and also the crude was co-evaporated with toluene to remove any remaining MeOH. The residue was dried under vacuum to give the intermediate as an amorphous white solid. The dry crude was dissolved in 1,4-dioxane and Bu4NBr and the corresponding bromides 7a–7i (1.5 eq.) were added. The reaction mixture was stirred under an N2 atmosphere overnight at 85 °C. TLC showed that no starting material remained. The solvent was removed in vacuo and the crude material was purified by flash chromatography to give quinolyl galactosides as a white amorphous solid. Compounds were further purified with preparative HPLC prior to galectin binding and cell assays. All tested compounds were >95% pure according to analytical HPLC analysis. See the ESI† for physical data.4.6. General method for the hydrolysis of methyl carboxylates 1d, 1e, 22a, and 22b
To a suspension of methyl carboxylates (1 eq.) in EtOH–H2O (3:1, 1 mL per 10 mg of sugar derivative), KOH (2 eq.) was added and it was stirred at 60 °C for 6 hours. Volatiles were evaporated under reduced pressure and the crude material was purified by preparative HPLC to obtain pure carboxylic acids 1j, 1k, 23a, and 23b as sole isolated products in yields of 66%, 63%, 69% and 65% respectively. All tested compounds were >95% pure according to analytical HPLC analysis. See the ESI† for physical data.
4.7. General method for the synthesis of pyridinium salts 10d–10e
Pyridine derivatives 9d–9e (1 eq.) and the alkylating reagent (1.5 eq.) were dissolved in dry acetone (2 mL for 1 mmol of pyridine derivative). The reaction mixture was stirred in an ultra-sound bath for 12 h. The temperature of the bath was kept under 50 °C by adding ice occasionally. Et2O (5 mL for 1 mmol of pyridine derivative) was added and the quaternary salts 10d–10e precipitated, were filtered off, and washed with Et2O. See the ESI† for physical data.4.8. Methyl (2,4,6-tri-O-benzyl-3-O-propynoyl)-β-D-galactopyranoside 12
Compound 11 (4.0 g, 8.62 mmol) was dissolved in 20 mL of dry THF. Propiolic acid (0.8 mL, 12.9 mmol) and DMAP (5 mg, 0.043 mmol) were added and the temperature lowered to 0 °C. DIC (2 mL, 12.925 mmol) was slowly added to the cold reaction mixture. The mixture was slowly allowed to warm to room temperature and stirred overnight. The reaction mixture was filtered through a fritted funnel to remove the urea precipitate. The THF mixture was exposed to reduced pressure to remove the solvent and excess propiolic acid and DIC. Compound 12 was purified by flash chromatography (heptane/EtOAc, 8:1 to 4:1) in a yield of 67% (2.98 g, 5.77 mmol) containing small amounts of DIC and DIU impurities. [α]25D +12.4 (c 1.4, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.36–7.26 (m, 15H, ArH), 5.02 (dd, 1H, J2,3 10.0 Hz, J3,4 2.8 Hz, H-3), 4.85 (d, 1H, J 11.2 Hz, CH2Ph), 4.71 (d, 1H, J 11.2 Hz, CH2Ph), 4.66 (d, 1H, J 11.2 Hz, CH2Ph), 4.50 (d, 1H, J 11.2 Hz, CH2Ph), 4.49 (d, 1H, J 11.6 Hz, CH2Ph), 4.43 (d, 1H, J 11.6 Hz, CH2Ph), 4.33 (d, 1H, J1,2 7.6 Hz, H-1), 3.98 (d, 1H, J3,4 2.8 Hz, H-4), 3.79 (dd, 1H, J1,2 7.6 Hz, J2,3 10.0 Hz, H-2), 3.67 (m, 1H, H-5), 3.64–3.60 (m, 2H, H-6a, H-6b), 3.55 (s, 3H, OCH3), 2.99 (s, 1H, COCCH). 13C NMR (CD3OD, 100 MHz): 152.2 (COCCH), 138.3, 137.81, 137.79, 128.5, 128.4, 128.0, 127.9, 127.8, 127.7 (ArC), 104.8 (C-1), 76.9, 76.7, 76.0, 75.0, 74.8, 73.7, 73.5, 73.0, 57.1 (OCH3). HRMS calcd for C31H32O7 + NH4+ (M + NH4)+: 534.2492, found: 534.2496.
4.9. General method for the preparation of indolizine derived galactosides 2a–2e
The reaction was performed with 1 eq. of quaternary salts (10a–10e), 1 eq. of the propiolate galactoside derivative, 12 and 1 eq. of K2CO3 in methanol (4 mL for 0.1 mmol of 12). The reaction mixture was stirred for 4 h at room temperature under an air atmosphere. After that the reaction mixture was quenched with water and collected in EtOAc. The EtOAc layer was washed successively with brine solution and water. The organic layer was collected, dried over Na2SO4, and evaporated under reduced pressure. The crude material was obtained, and subjected to hydrogenolysis with Pd(OH)2–C (10 wt%, 1 mg for 4 mg of crude), without any further purification. The crude was dissolved in EtOAc (3 mL for 0.1 mmol of crude) and isopropanol (9 mL per 0.1 mmol of crude) and the solution was stirred under a hydrogen atmosphere at room temperature for 12 h. After the completion of the reaction (as indicated by TLC), the reaction mixture was filtered through a Celite bed and washed with methanol. The filtrate was concentrated under reduced pressure and purified through flash column chromatography to obtain the desired compounds 2a–2e in yields of 25–34% over two steps. Finally, the compounds were purified by preparative HPLC before galectin binding and cell assays. All tested compounds were >95% pure according to analytical HPLC analysis. See the ESI† for physical data.4.10. General method for the preparation of substituted propargylated coumarins 14a–14e
To a solution of hydroxyl coumarins 13a–13e (1 eq.) in dry acetone (20 mL per 1 mmol of hydroxyl coumarin) were added K2CO3 (2 eq.) and propargyl bromide (80 wt% in toluene) (1.5 eq.). The resulting mixture was stirred at 60 °C overnight. The mixture was cooled and the solvent was removed under reduced pressure. The residue was treated with water (10 mL per 1 mmol of hydroxyl coumarin) and extracted with ethyl acetate. The combined organic phase was washed with water, dried over Na2SO4 and evaporated under vacuum, and the residue was purified through flash column chromatography to afford 14a–14e. See the ESI† for physical data.4.11. p-Tolyl 3-azido-3-deoxy-1-thio-β-D-galactopyranoside 16
p-Tolyl 2,4,6-tri-O-acetyl-3-azido-3-deoxy-1-thio-β-D-galactopyranoside 15 (700 mg, 1.6 mmol) was dissolved in MeOH (7.5 mL). NaOMe (2 mL, 0.5 M in MeOH) was added and the solution was stirred at room temperature overnight. The solution was neutralized with DOWEX 50 W H+ resin, filtered and the solvents were evaporated under reduced pressure and the crude 16 was used without any further purification. The crude product was obtained as a white amorphous solid (458 mg, 92%). [α]25D +8.9 (c 1.6, CH3OH). 1H NMR (CD3OD, 400 MHz): 7.44 (dd, 2H, J 1.6 Hz, J 6.4 Hz, ArH), 7.10 (d, 1H, J 7.6 Hz, ArH), 4.58 (d, 1H, J1,2 9.6 Hz, H-1), 3.95 (d, 1H, J3,4 2.8 Hz, H-4), 3.80 (t, 1H, J1,2, J2,3 9.6 Hz, H-2), 3.74 (dd, 1H, J5,6a 6.8 Hz, J6a,6b 11.2 Hz, H-6a), 3.68 (dd, 1H, J5,6b 5.2 Hz, J6a,6b 11.6 Hz, H-6b), 3.55 (m, 1H, H-5), 3.36 (dd, 1H, J2,3 9.6 Hz, J3,4 2.8 Hz, H-3), 2.28 (s, 3H, SC6H4CH3). 13C NMR (CD3OD, 100 MHz): 138.4, 132.8, 131.5, 130.5 (ArC), 90.9 (C-1), 80.6, 69.3, 69.2, 68.3, 62.3, 21.1(SC6H4CH3). HRMS calcd for C13H17N3O4S–H+ (M − H)+: 310.0862, found: 310.0858.4.12. General method for the preparation of coumarin derived thiogalactosides 3a–3e
To a solution of azide 16 (1 eq.) in dichloromethane (1 mL for 10 mg of sugar azide), propargylated coumarins 14a–14e (1.5 eq.), CuI (10 mol% with respect to sugar azide) and DIPEA (1.5 eq.) were added and the mixture was stirred at room temperature for 24 h, until TLC showed no trace of the starting sugar azide. The solvent was removed under reduced pressure, and the residue was dissolved in EtOAc and the solution was washed with brine, dried over Na2SO4 and concentrated in vacuo. The product was purified by flash column chromatography (DCM/MeOH, 25:1–10:1) to give the corresponding triazoles as a white amorphous solid. The compounds were further purified with preparative HPLC prior to galectin binding and cell assays. All tested compounds were >95% pure according to analytical HPLC analysis. See the ESI† for physical data.
4.13. Methyl 3-O-(napth-2-ylmethyl)-β-D-galactopyranoside 17
The reaction was performed with 8 (103 mg, 0.53 mmol) and 2-bromomethylnaphthalene (176 mg, 0.795 mmol) following general method 4.5. Methyl 3-O-(naphthalen-2-ylmethylene)-β-D-galactopyranoside 17 was obtained in 88% yield (166 mg, 0.467 mmol) as a colourless oil. [α]25D +42.7 (c 1.2, CH3OH). 1H NMR (CD3OD, 400 MHz): 8.33 (s, 1H, J 8.4 Hz, ArH), 7.85–7.80 (m, 3H, J 8.4 Hz, ArH), 7.58 (dd, 1H, J 1.6 Hz, J 8.4 Hz, ArH), 7.48–7.42 (m, 2H, ArH), 4.91 (d, 1H, J 12.0 Hz, CH2C10H7), 4.81 (d, 1H, J 12.0 Hz, CH2C10H7), 4.15 (d, 1H, J1,2 7.6 Hz, H-1), 4.07 (dd, 1H, J3,4 3.2 Hz, J4,5 0.4 Hz, H-4), 3.80–3.68 (m, 3H, H-2, H-6a, H-6b), 3.53 (s, 3H, OCH3), 3.44 (m, 1H, H-5), 3.41 (dd, 1H, J2,3 9.6 Hz, J3,4 3.2 Hz, H-3). 13C NMR (CD3OD, 100 MHz): 137.3, 134.7, 134.5, 129.0, 128.9, 128.6, 127.6, 127.1, 127.0, 126.9 (ArC), 105.9 (C-1), 82.4, 76.5, 72.6, 71.8, 67.1, 62.4, 57.2 (OCH3). HRMS calcd for C18H22O6 + H+ (M + H)+: 335.1495, found: 335.1495.4.14. 3,4-Dichlorophenyl 2,3,4,6-tetra-O-acetyl-1-thio-α-D-galactopyranoside 20
To a stirred suspension of 19 (2.0 g, 5.13 mmol) and PCl5 (1.17 g, 5.64 mmol) in dry DCM (20 mL), BF3·Et2O (32 μL, 0.26 mmol) was added. After stirring for 30 min TLC analysis (heptane:EtOAc, 1:1) showed the complete consumption of the starting material. The reaction mixture was diluted with DCM (100 mL) and then washed with ice-cold water (50 mL), saturated ice-cold NaHCO3 solution (2 × 50 mL), and again ice-cold water (2 × 30 mL). The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was co-evaporated with toluene colorless oil, which solidified slowly and was used without further purification. NaH (221 mg, 9.23 mmol) was added into dry DMF (15 mL) in a separate flask. 3,4-Dichlorobenzenethiol (1.83 g, 10.25 mmol) was added into the reaction mixture and the mixture was stirred at room temperature for 30 min. Then crude 2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl chloride was dissolved in 15 mL DMF and added to the reaction mixture. The mixture was heated to 50 °C for 12 h. When TLC showed that all the starting material was consumed, the mixture was diluted with DCM (100 mL) and water (50 mL). The organic phase was washed with water (30 mL × 3) and concentrated. The residue was purified by column chromatography (heptane:EtOAc, 6:1 to 5:2) to give 20 (1.77 g, 68%) as a white solid over two steps. [α]25D +28.6 (c 1.2, CHCl3). 1H NMR (CDCl3, 400 MHz): 7.48 (d, 1H, J 2.0 Hz, ArH), 7.27 (dd, 1H, J 8.4 Hz, ArH), 7.20 (dd, 1H, J 2.0 Hz, J 8.4 Hz, ArH), 5.90 (d, 1H, J1,2 5.6 Hz, H-1), 5.25 (dd, 1H, J1,2 5.6 Hz, J2,3 10.8 Hz, H-2), 5.15 (dd, 1H, J2,3 10.8 Hz, J3,4 3.2 Hz, H-3), 4.58 (m, 1H, H-5), 4.02 (dd, 1H, J5,6a 5.6 Hz, J6a,6b 11.6 Hz, H-6a), 3.98 (dd, 1H, J5,6b 7.2 Hz, J6a,6b 11.6 Hz, H-6b), 2.06 (s, 3H, COCH3), 2.02 (s, 3H, COCH3), 1.92 (s, 3H, COCH3), 1.89 (s, 3H, COCH3). 13C NMR (CDCl3, 100 MHz): 170.1, 169.84, 169.79, 169.6, 133.1, 132.7, 131.9, 131.0, 130.5 (ArC), 85.2 (C-1), 67.8, 67.7, 67.4, 61.8, 20.6 (COCH3), 20.40 (2× COCH3), 20.39 (COCH3). HRMS calcd for C20H22Cl2O9S + NH4+ (M + NH4)+: 526.0705, found: 526.0705.
4.15. 3,4-Dichlorophenyl 1-thio-β-D-galactopyranoside 21
Compound 20 (1.2 g, 2.36 mmol) thus obtained was dissolved in MeOH (15 mL). Then NaOMe (5 mL, 0.5 M in MeOH) was added and the solution was stirred at room temperature overnight. The solution was neutralized with DOWEX 50 W H+ resin, filtered and the solvents were evaporated under reduced pressure and the crude was used without any further purification. Compound 21 was obtained as a colourless oil (755 mg, 94%). [α]25D +3.4 (c 1.3, CH3OH). 1H NMR (CD3OD, 400 MHz): 7.70 (d, 1H, J 2.0 Hz, ArH), 7.45 (dd, 1H, J 2.0 Hz, J 8.4 Hz, ArH), 7.40 (d, 1H, J 8.4 Hz, ArH), 5.66 (d, 1H, J1,2 5.6 Hz, H-1), 4.27 (m, 1H, H-5), 4.19 (dd, 1H, J1,2 5.6 Hz, J2,3 10.4 Hz, H-2), 3.80 (dd, 1H, J3,4 3.2 Hz, J4,5 1.2 Hz, H-4), 3.74 (dd, 1H, J5,6a 5.2 Hz, J6a,6b 11.2 Hz, H-6a), 3.69 (dd, 1H, J5,6b 6.8 Hz, J6a,6b 11.2 Hz, H-6b), 3.65 (dd, 1H, J2,3 10.4 Hz, J3,4 3.2 Hz, H-3). 13C NMR (CD3OD, 100 MHz): 137.0, 134.2, 133.3, 132.5, 131.9, 131.5 (ArC), 91.1 (C-1), 73.5, 72.1, 70.6, 69.7, 62.3. HRMS calcd for C12H14Cl2O5S + Na+ (M + Na)+: 362.9837, found: 362.9835.4.16. 3,4-Dichlorophenyl 3-O-(6-methoxycarbonyl-quinolin-2-yl-methyl)-1-thio-α-D-galactopyranoside 22a
The reaction was performed with 21 (64 mg, 0.19 mmol) following the general method 4.5. Compound 22a was obtained in 73% yield (74 mg, 0.14 mmol) as a white amorphous solid. [α]25D +58.8 (c 0.7, CH3OH). 1H NMR ((CD3)2SO, 400 MHz): 8.71 (d, 1H, J 2.0 Hz, ArH), 8.61 (d, 1H, J 8.8 Hz, ArH), 8.22 (dd, 1H, J 2.0 Hz, J 8.8 Hz, ArH), 8.05 (d, 1H, J 8.8 Hz, ArH), 7.92 (d, 1H, J 8.4 Hz, ArH), 7.74 (d, 1H, J 2.0 Hz, ArH), 7.53 (d, 1H, J 8.4 Hz, ArH), 7.44 (dd, 1H, J 2.0 Hz, J 8.4 Hz, ArH), 5.91 (d, 1H, J 4.4 Hz, OH), 5.76 (d, 1H, J1,2 5.2 Hz, H-1), 5.06 (d, 1H, J 6.0 Hz, OH), 5.01 (d, 1H, J 14.8 Hz, CH2C11H8NO2), 4.92 (d, 1H, J 14.8 Hz, CH2C11H8NO2), 4.64 (t, 1H, J 5.6 Hz, OH), 4.30 (m, 1H, H-2), 4.15 (t, 1H, J4,OH 4.4 Hz, H-4), 4.00 (t, 1H, J5,6a, J5,6b 6.4 Hz, H-5), 3.93 (s, 3H, CO2CH3), 3.59 (m, 1H, H-6a), 3.52 (dd, 1H, J2,3 10.0 Hz, J3,OH 6.0 Hz, H-3), 3.41 (dd, 1H, J5,6a 6.0 Hz, J6a,6b 10.8 Hz, H-6b). 13C NMR ((CD3)2SO, 100 MHz): 166.5 (CO2CH3), 160.3, 146.7, 136.3, 134.1, 133.7, 131.9, 131.2, 130.8, 130.5, 130.1, 129.0, 128.7, 126.8, 125.2, 121.3 (ArC), 88.7 (C-1), 79.7, 72.7, 71.0, 66.9, 64.8, 60.1, 52.5 (CO2CH3). HRMS calcd for C24H23Cl2NO7S + H+ (M + H)+: 540.0651, found: 540.0648.4.17. 3,4-Dichlorophenyl 3-O-(7-methoxycarbonyl-quinolin-2-yl-methyl)-1-thio-α-D-galactopyranoside 22b
The reaction was performed with 21 (55 mg, 0.162 mmol) following the general method 4.5. Compound 22a was obtained in 67% yield (58 mg, 0.108 mmol) as a white amorphous solid. [α]25D +59.1 (c 0.6, CH3OH). 1H NMR ((CD3)2SO, 400 MHz): 9.18 (d, 1H, J 9.2 Hz, ArH), 8.24–8.21 (m, 2H, J 8.4 Hz, ArH), 7.95 (d, 1H, J 9.2 Hz, ArH), 7.87 (dd, 1H, J 7.2 Hz, J 8.4 Hz, ArH), 7.74 (d, 1H, J 2.0 Hz, ArH), 7.54 (d, 1H, J 8.4 Hz, ArH), 7.44 (dd, 1H, J 2.0 Hz, J 8.4 Hz, ArH), 5.88 (bs, 1H, OH), 5.76 (d, 1H, J1,2 5.6 Hz, H-1), 5.05 (bs, 1H, OH), 5.00 (d, 1H, J 14.4 Hz, CH2C11H8NO2), 4.91 (d, 1H, J 14.4 Hz, CH2C11H8NO2), 4.62 (bs, 1H, OH), 4.30 (dd, 1H, J1,2 5.6 Hz, J2,3 10.0 Hz, H-2), 4.14 (bs, 1H, H-4), 4.00–3.95 (m, 4H, H-5, CO2CH3), 4.58 (dd, 1H, J5,6a 6.0 Hz, J6a,6b 10.8 Hz, H-6a), 3.51 (dd, 1H, J2,3 10.0 Hz, J3,4 3.2 Hz, H-3), 3.41 (m, 1H, H-6b). 13C NMR ((CD3)2SO, 100 MHz): 165.9 (CO2CH3), 162.7, 148.5, 138.0, 136.3, 132.0, 131.3, 130.8, 130.5, 129.1, 128.8, 128.7, 127.1, 126.4, 120.7 (ArC), 88.8 (C-1), 79.8, 72.7, 71.2, 66.9, 64.8, 60.2, 52.4 (CO2CH3). HRMS calcd for C24H23Cl2NO7S + H+ (M + H)+: 540.0651, found: 540.0653.4.18. Molecular dynamics simulations
Molecular dynamics simulations were performed with the OPLS3 force field in Desmond implemented in Schrödinger Release 2017-3 using default settings except for the length of the simulation and the use of light harmonic constraints (1 kcal mol−1 Å−2) on all stranded backbone atoms and on the galactose O4 atom. A series of energy-minimized starting conformations of 1k with different dihedral angles of the three rotatable bonds linking galactose C3 to the quinoline ring system of 1k was placed in the published structure of galectin-8N in complex with sialyl-α-(2-3)-lacNAc (pdb id 3VKO) and subjected to molecular dynamics simulations. All simulations drifted towards a complex geometry where the quinoline of 1k was close to Arg45. Subsequently, a starting conformation with the 1k quinoline close to Arg45 was subjected to a 1000 ns molecular dynamics simulation.4.19. Cell lines and cell culture
The human breast cancer cell line JIMT-1 (ACC589) was purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ) and was routinely maintained in Dulbecco's modified Eagle's medium/nutrient mixture Ham's F12 medium (VWR, Lund, Sweden). The human breast cancer cell line MCF-7 (HTB-22) and human normal-like breast epithelial cell line MCF-10A (CRL-10317) were obtained from American Type Culture Collection (Manassas, VA, USA) and were cultured in RPMI1640 medium (VWR). The JIMT-1 and MCF-7 cell lines were cultured with the addition of 10% fetal calf serum (FCS) (VWR), nonessential amino acids (1 mM) (VWR), insulin (10 μg mL−1) (Sigma-Aldrich), penicillin (100 U mL−1) (VWR), and streptomycin (100 g mL−1) (VWR). The MCF-10A cells were cultured with the addition of 10% heat-inactivated FCS, nonessential amino acids (1 mM), insulin (10 μg mL−1), penicillin (100 U mL−1), streptomycin (100 g mL−1), epithermal growth factor (20 ng mL−1) (Sigma-Aldrich), cholera toxin (50 ng mL−1) (Sigma-Aldrich), and hydrocortisol (250 ng mL−1) (Sigma-Aldrich). All cell lines were maintained at 37 °C in a humidified incubator with 5% CO2.4.20. MTT assay
An MTT assay was used to evaluate the cytotoxicity of all the inhibitors as previously described.40 Briefly, the inhibitors were dissolved in DMSO and then serially diluted in PBS and used at final concentrations from 0.05 μM to 50 μM. The final DMSO concentration in the assays was 0.1% for all concentrations used. Accordingly, the control was treated with 0.1% DMSO in PBS. The cells were seeded in 96-well plates (5000 cells for JIMT-1 and MCF-7 cell lines and 3000 cells for MCF-10A cell line per well in 180 μl medium) and the plates were incubated for 24 h before the addition of compound. After 72 h of treatment, MTT solution (20 μl of 5 mg mL−1 in PBS) was added to each well and the plate was incubated for 1 h. Thereafter, the medium was removed and the purple formazan product was dissolved by the addition of 100 μl of 100% DMSO per well. The plates were swirled gently for 10 min to dissolve the precipitate and the cells. Absorbance was monitored at 540 nm using a Labsystems iEMS Reader MF (Labsystems Oy, Helsinki, Finland) and the software DeltaSoft II v.4.14 (Biometallics Inc., Princeton, NJ, USA). The software program GraphPad Prism was used to analyze the data and plot dose response curves.Conflicts of interest
F.R.Z. is an employee of and H.L. and U.J.N. are shareholders in Galecto Biotech AB, a company developing galectin inhibitors. The other authors have no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (Grant No. 621-2012-2978), the Royal Physiographic Society, Lund, a project grant awarded by the Knut and Alice Wallenberg Foundation (KAW 2013.0022), and by Galecto Biotech AB, Sweden.Notes and references
- R.-Y. Yang, G. A. Rabinovich and F. T. Liu, Expert Rev. Mol. Med., 2008, 10, e17 CrossRef PubMed.
- S. Di Lella, V. Sundblad, J. P. Cerliani, C. M. Guardia, D. A. Estrin, G. R. Vasta and G. A. Rabinovich, Biochemistry, 2011, 50, 7842 CrossRef PubMed.
- L. Johannes, R. Jacob and H. Leffler, J. Cell Sci., 2018, 131, 1 CrossRef PubMed.
- G. A. Rabinovich and D. O. Croci, Immunity, 2012, 36, 322 CrossRef PubMed.
- G. A. Rabinovich and M. A. Toscano, Nat. Rev. Immunol., 2009, 9, 338 CrossRef PubMed.
- L. Ingrassia, I. Camby, F. Lefranc, V. Mathieu, P. Nshimyumukiza, F. Darro and R. Kiss, Curr. Med. Chem., 2006, 13, 3513 CrossRef PubMed.
- H. Leffler and U. J. Nilsson, in ACS Symp. Ser, ed. A. A. Klyosov and P. G. Traber, 2013, vol. 1115, pp. 47–59 Search PubMed.
- C. Oberg, H. Leffler and U. J. Nilsson, Chimia, 2011, 65, 18 CrossRef PubMed.
- R. J. Pieters, ChemBioChem, 2006, 7, 721 CrossRef PubMed.
- S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 9, 1648 CrossRef PubMed.
- G. S. Singh and E. E. Mmatli, Eur. J. Med. Chem., 2011, 46, 5237 CrossRef PubMed.
- J. Dandriyal, R. Singla, M. Kumar and V. Jaitak, Eur. J. Med. Chem., 2016, 119, 141 CrossRef PubMed.
- K. K. Kumar, S. P. Seenivasan, V. Kumar and T. M. Das, Carbohydr. Res., 2011, 346, 2084 CrossRef PubMed.
- A. Mackinnon, W.-S. Chen, H. Leffler, N. Panjwani, H. Schambye, T. Sethi and U. J. Nilsson, in Topics in Medicinal Chemistry: Carbohydrates as Drugs, ed. C. Rademacher and P. H. Seeberger, Springer-Verlag GmbH & Co. KG, Berlin/Heidelberg, 2014, pp. 95–121 Search PubMed.
- Y. Xie and L. Li, Tetrahedron Lett., 2014, 55, 3892 CrossRef.
- N. S. Stock, G. Bain, J. Zunic, Y. Li, J. Ziff, J. Roppe, A. Santini, J. Darlington, P. Prodanovich, C. D. King, C. Baccei, C. Lee, H. Rong, C. Chapman, A. Broadhead, D. Lorrain, L. Correa, J. H. Hutchinson, J. F. Evans and P. Prasit, J. Med. Chem., 2011, 54, 8013 CrossRef PubMed.
- L. Wang, X. Hou, H. Fu, X. Pan, W. Xu, W. Tang and H. Fang, Bioorg. Med. Chem., 2015, 23, 4364 CrossRef PubMed.
- S. Bonte, I. O. Ghinea, R. Dinica, I. Baussanne and M. Demeunynck, Molecules, 2016, 21, 332 CrossRef PubMed.
- H. M. Flowers, Carbohydr. Res., 1975, 39, 245 CrossRef.
- G. Zemplen, Ber. Dtsch. Chem. Ges., 1926, 59, 1254 CrossRef.
- K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Håkansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2017, 61, 1164 CrossRef PubMed.
- C. M. Tornøe and M. Meldal, in Peptides: The Wave of the Future, Proceedings of the Second International and the Seventeenth American Peptide Symposium, Springer, San Diego, CA, United States, 2001, pp. 263–264 Search PubMed.
- C. M. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef PubMed.
- P. Sörme, B. Kahl-Knutson, U. Wellmar, U. J. Nilsson and H. Leffler, Methods Enzymol., 2003, 362, 504 Search PubMed.
- P. Sörme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36 CrossRef PubMed.
- I. Cumpstey, S. Carlsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2005, 3, 1922 RSC.
- S. Mandal, V. K. Rajput, H. Leffler, B. Mukhopadhyay and U. J. Nilsson, Can. J. Chem., 2016, 94, 936 CrossRef.
- D. Schmidt, B. Sauerbrei and J. Thiem, J. Org. Chem., 2000, 65, 8518 CrossRef PubMed.
- B. Dhakal, S. Buda and D. Crich, J. Org. Chem., 2016, 81, 10617 CrossRef PubMed.
- V. K. Rajput, H. Leffler, U. J. Nilsson and B. Mukhopadhyay, Bioorg. Med. Chem. Lett., 2014, 24, 3516 CrossRef PubMed.
- F. R. Zetterberg, K. Peterson, R. E. Johnsson, T. Brimert, M. Håkansson, D. T. Logan, H. Leffler and U. J. Nilsson, ChemMedChem, 2018, 27, 133 CrossRef PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef PubMed.
- D. Gerlier and N. Thomasset, J. Immunol. Methods, 1986, 94, 57 CrossRef PubMed.
- E. Vega-Avila and M. K. Pugsley, Proc. West. Pharmacol. Soc., 2011, 54, 10 Search PubMed.
- S. Carlsson, C. T. Öberg, M. C. Carlsson, A. Sundin, U. J. Nilsson, D. Smith, R. D. Cummings, J. Almkvist, A. Karlsson and H. Leffler, Glycobiology, 2007, 17, 663 CrossRef PubMed.
- T. L. M. Thurston, M. P. Wandel, N. V. Muhlinen, Á. Foeglein and F. Randow, Nature, 2012, 482, 414 CrossRef PubMed.
- W.-S. Chen, Z. Cao, S. Sugaya, N. Laver, H. Leffler, U. J. Nilsson, J. Fu, J. Song, L. Xia and N. Panjwani, Nat. Commun., 2016, 7, 11302 CrossRef PubMed.
- M. Friedel, S. André, H. Goldschmidt, H.-J. Gabius and R. Schwartz-Albiez, Glycobiology, 2016, 26, 1048–1058 CrossRef PubMed.
- X. Huang, B. Borgström, L. Månsson, L. Persson, S. Oredsson, C. Hegardt and D. Strand, ACS Chem. Biol., 2014, 9, 1587 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01354c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |