
Synthesis of polysubstituted pyridines from oxime acetates using NH4I as a dual-function promoter†
Yujia
Xia‡
,
Jinhui
Cai‡
,
Huawen
Huang
 * and
Guo-Jun
Deng
*
* and
Guo-Jun
Deng
*
Key Laboratory for Green Organic Synthesis and Application of Hunan Province, Key Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, China. E-mail: hwhuang@xtu.edu.cn; gjdeng@xtu.edu.cn
First published on 27th November 2017
Abstract
Pyridine formation with oxime acetates as the building blocks under metal-free conditions is described. Ammonium iodide has proved to be a highly efficient promoter for oxime N–O bond reduction and subsequent condensation reactions, whereby it played a dual-function role in the transformation. While the three-component reaction of oxime acetates, benzaldehydes, and 1,3-dicarbonyls proceeded well with the assistance of a stoichiometric amount of ammonium iodide, the condensation of oximes and acroleins was enabled by using a catalytic initiator to afford substituted pyridines. By this protocol, substituted pyridine products were generated in moderate to excellent yields with tolerance towards a broad range of functional groups.
Introduction
Pyridine-containing compounds are found in natural products and spices, and have wide applications in the fields of pharmaceuticals and functionalized organic materials. Consequently, beyond the classic Chichibabin and Hantzsch pyridine synthesis, the development of methodologies for pyridine formation remains a topic of considerable interest in modern synthetic chemistry.1,2Oxime derivatives provide structurally advantaged and versatile building blocks for pyridine synthesis under transition-metal catalysis.3 For example, vinyl ketoximes undergo formal [4 + 2] cycloaddition4 with alkenes or alkynes to afford highly substituted pyridines (Scheme 1a), in which copper-catalyzed electrophilic amination4a or rhodium-catalyzed directed C–H activation4b–d occurs as the initial step. Furthermore, oxime acetates bearing α-protons have also been widely used as C2N1 units for pyridine construction. The activation of oximes by metal salts followed by the condensation of two molecules of oxime acetates with a C1 source such as aldehydes, dimethylformamide (DMF), or dimethylaniline proceeds through formal [3 + 2 + 1] annulation to give structurally symmetrical pyridines or 2,4-diarylpyridines (Scheme 1b).5 Furthermore, [3 + 3] pyridine formation has been disclosed6 independently by Yoshikai,6a,e Cui,6b and Jiang6c,d groups, wherein copper-catalyzed oxime activation initiates subsequent condensation with acroleins, α,β-unsaturated ketimines, or α,β-unsaturated nitriles and ketones in situ generated by three-component assembly (Scheme 1c).
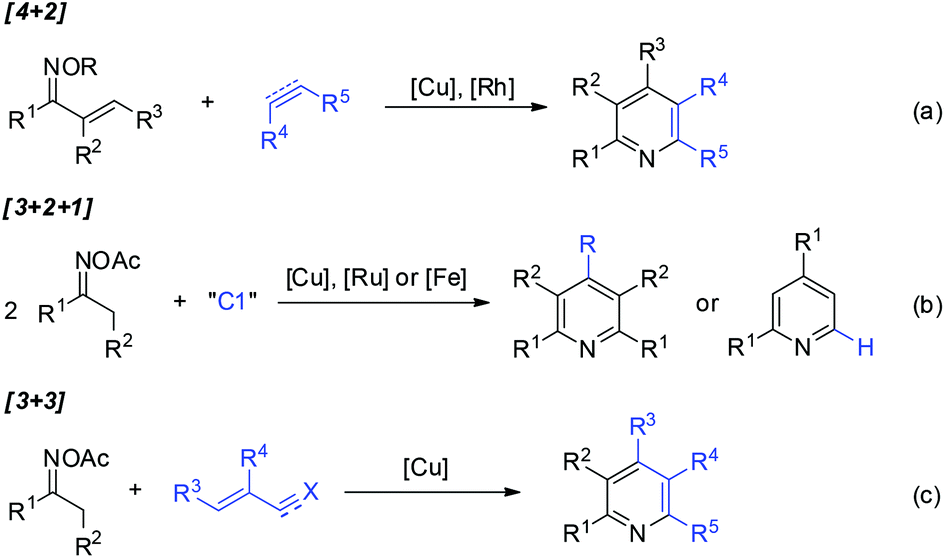 | ||
| Scheme 1 Synthesis of pyridines from oximes under metal catalysis. | ||
The activation of the oxime N–O bond could also proceed with a non-metal initiator, which was originally applied for intramolecular cyclization and N-heterocycle synthesis.7 In our own study on sustainable metal-free synthesis,8 recently, we developed an efficient I2/Et3N-mediated pyridine synthesis from ketoximes and acroleins (Scheme 2a).8a Thereafter, the construction of trifluoromethyl pyridines was enabled by a NH4I-based system combined with Na2S2O4 as the reducing agent (Scheme 2b).8b The experimental results of these studies encouraged us to explore the broader generality of N-heterocycle synthesis from oximes under metal-free systems. Herein, we disclose the NH4I-initiated effective formation of polysubstituted pyridines through [3 + 3] annulation of oxime acetates and α,β-unsaturated carbonyls (Scheme 2c). The present work provides an alternative entry to oxime-based pyridine synthesis in a reaction complementary to previous copper-catalyzed methods.6a,c Moreover, the dual-function iodide catalysis would inspire other cases of iodine-based synthetic chemistry.9
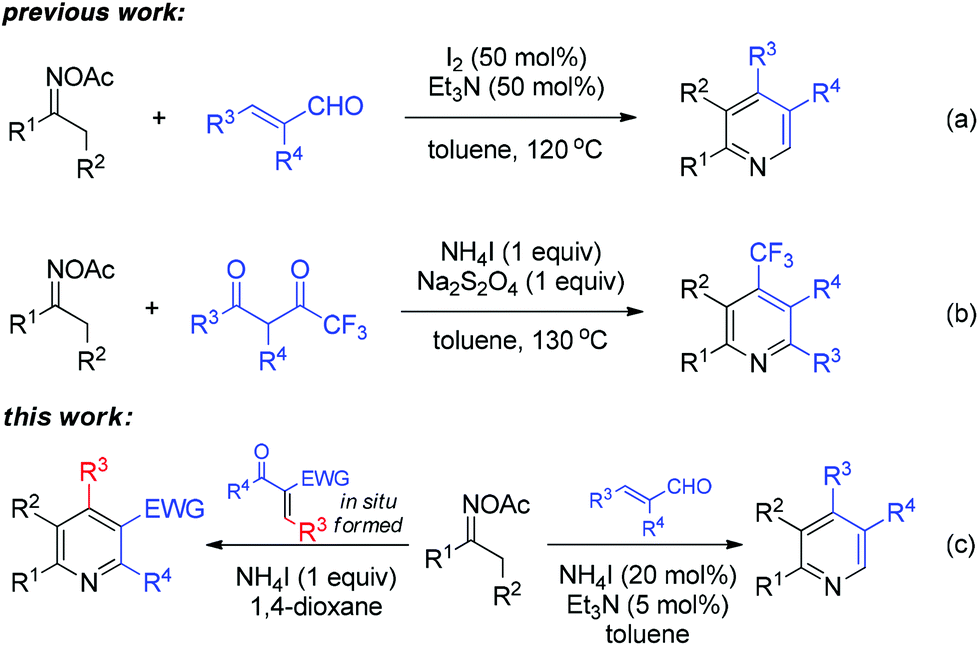 | ||
| Scheme 2 Synthesis of pyridines from oximes under metal-free conditions. | ||
Results and discussion
In a reaction complementary to our previous synthesis of fluorinated pyridines, the three-component reaction of ketoximes, benzaldehydes, and trifluoromethyldiketones had been preliminarily investigated, and moderate yields were generally obtained in the NH4I/Et3N/toluene system.8b We suspected that a more efficient reaction system could be explored for the three-component assembly of pyridines. Thereby ketoxime acetate 1a, benzaldehyde 2a, and acetylacetone 3a were chosen as the model system to screen the reaction conditions for the pyridine formation (Table 1). When a catalytic amount of NH4I was used, solvent screening indicated that 1,4-dioxane was superior to the previously used toluene and others (entries 1–4). Other ammonium halides such as NH4Cl and NH4Br featured no catalytic activity for this reaction (entries 5 and 6), as did other iodides such as KI, NaI and Bu4NI (entries 7–9). These results revealed that NH4I might play a dual-function role, as previously proposed, serving as an electron-donor to enable the oxime N–O bond cleavage and as an acid to promote the condensation process. Notably, elemental iodine and NIS gave a lower yield of 4a compared with NH4I (entries 10 and 11). To our delight, in the 1,4-dioxane medium, the reaction yield increased with the increased amount of NH4I (entries 12 and 13), and excellent yield of 4a (87%) was obtained with 1 equiv. of initiator. Finally, the reaction performed at 100 °C or 130 °C gave inferior results (entries 14 and 15).|
|
||||
|---|---|---|---|---|
| Entry | Add. (mol%) | Solvent | Temp. (°C) | Yieldb (%) |
| a The reactions were performed with 0.2 mmol of 1a, 0.3 mmol of 2a, and 0.4 mmol of 3a in 1 mL of solvent, 12 h. b Yields of isolated product 4a are given. | ||||
| 1 | NH4I (20) | Toluene | 120 | 6 |
| 2 | NH4I (20) | PhCl | 120 | 0 |
| 3 | NH4I (20) | DMSO | 120 | 0 |
| 4 | NH4I (20) | 1,4-Dioxane | 120 | 17 |
| 5 | NH4Cl (20) | 1,4-Dioxane | 120 | 0 |
| 6 | NH4Br (20) | 1,4-Dioxane | 120 | 0 |
| 7 | KI (20) | 1,4-Dioxane | 120 | 0 |
| 8 | NaI (20) | 1,4-Dioxane | 120 | 0 |
| 9 | Bu4NI (20) | 1,4-Dioxane | 120 | 0 |
| 10 | I2 (20) | 1,4-Dioxane | 120 | 10 |
| 11 | NIS (20) | 1,4-Dioxane | 120 | 12 |
| 12 | NH4I (50) | 1,4-Dioxane | 120 | 53 |
| 13 | NH4I (100) | 1,4-Dioxane | 120 | 87 |
| 14 | NH4I (100) | 1,4-Dioxane | 100 | 51 |
| 15 | NH4I (100) | 1,4-Dioxane | 130 | 78 |
With the optimized reaction conditions in hand, we explored the generality of the three-component pyridine formation by employment of a broad range of substrates (Scheme 3). First, acetophenone oxime acetates bearing various functionalities at the benzene ring were subjected to this system and proved to be effective, affording the corresponding pyridine products in moderate to good yields (4a–4k). Among them, alkyl, methoxyl, chloro, and bromo functional groups were well tolerated. Naphthyl and thienyl ketoximes were also compatible with this reaction (4l and 4m, respectively). Then, a series of substituted benzaldehydes attached with alkyl, methoxyl, halo (F, Cl, Br), nitro, and amino substituents worked smoothly to give the respective tetrasubstituted pyridines in good yields (5a–5o). Thereafter, we studied the scope of viable 1,3-dicarbonyls 3. Thereby, among others 1,3-diketone was successfully coupled with oxime 1a and benzaldehyde 2a to afford the target pyridines 6a in moderate yields. Then various β-ketone esters bearing functional groups (6b–6e) including allyl ester (6d) were all accommodated with the present NH4I system. Moreover, β-ketoamide furnished the desired nicotinamide product 6f in modest yield.
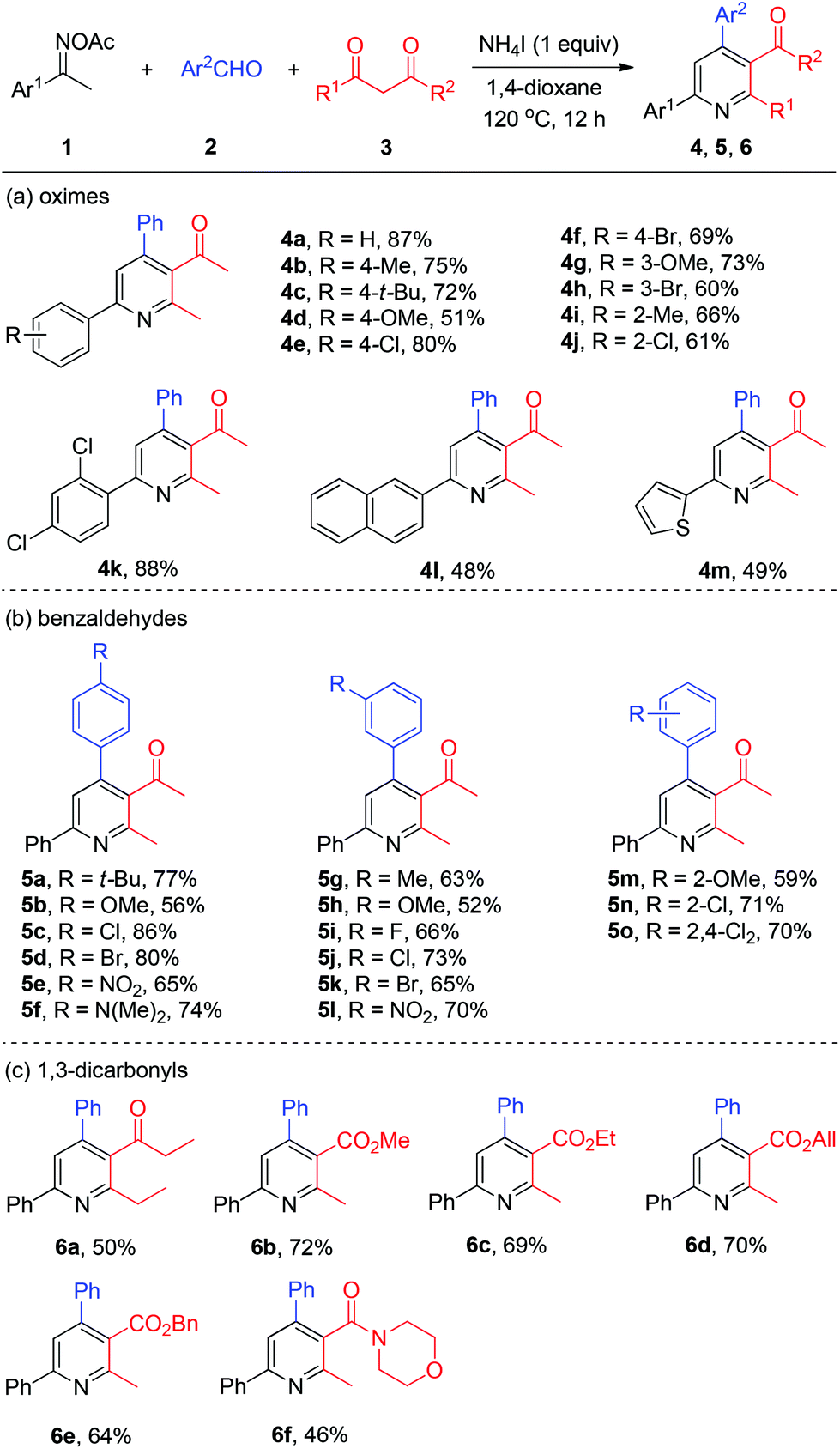 | ||
| Scheme 3 Three-component pyridine synthesis by the NH4I initiator. | ||
With the three-component pyridine formation established by the NH4I system, we speculate that this viable dual-function initiator could be applicable to other cases of oxime-based condensation reactions, whereby we reexamined the pyridine formation from ketoxime 1a and cinnamic aldehyde 7a (Table 2). Initially, we subjected them to the NH4I-mediated system for the three-component reaction, and obtained the 2,6-diphenyl pyridine 8a in 22% yield (entry 1). A very slightly reduced yield of 8a was observed when using a catalytic amount of NH4I (entry 2). Likewise, other ammonium salts and iodides also gave no pyridine product and elemental iodine as well as NIS was inferior (entries 3–8). Then we found that the addition of triethylamine in a catalytic amount dramatically improved the transformation (entries 9 and 10), furnishing the product in good yield. In combination of NH4I (20 mol%) with Et3N (25 mol%), the examination of solvent (entries 11–13) revealed that toluene was the best medium for this pyridine formation, giving 8a in 85% yield.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (mol%) | Add. (mol%) | Solvent | Yieldb (%) |
| a The reactions were performed with 0.2 mmol of 1a, 0.3 mmol of 7a in 1 mL of solvent at 120 °C for 12 h. b Yields of isolated product 8a are given. | ||||
| 1 | NH4I (100) | — | 1,4-Dioxane | 22 |
| 2 | NH4I (20) | — | 1,4-Dioxane | 19 |
| 3 | NH4Br (20) | — | 1,4-Dioxane | 0 |
| 4 | KI (20) | — | 1,4-Dioxane | 0 |
| 5 | NaI (20) | — | 1,4-Dioxane | 0 |
| 6 | I2 (20) | — | 1,4-Dioxane | 9 |
| 7 | Bu4NI (20) | — | 1,4-Dioxane | 0 |
| 8 | NIS (20) | — | 1,4-Dioxane | 7 |
| 9 | NH4I (20) | Et3N (50) | 1,4-Dioxane | 68 |
| 10 | NH4I (20) | Et3N (25) | 1,4-Dioxane | 70 |
| 11 | NH4I (20) | Et3N (25) | Toluene | 85 |
| 12 | NH4I (20) | Et3N (25) | PhCl | 26 |
| 13 | NH4I (20) | Et3N (25) | DMSO | 35 |
Comparably, while elemental iodine could be used as a soft acid and electron-donor for oxime N–O bond reduction in the previous work,8a,10 the NH4I/Et3N system featured obviously higher dual-function catalytic activity in the current observation. Furthermore, the catalytic NH4I system made the reaction handling either in the pre- or late-stage much easier.
Thereafter, we probed the scope of ketoximes and acroleins for the NH4I/Et3N-catalyzed pyridine generation (Scheme 4). In general, moderate to excellent yields were observed and the electronic and steric hindrance effects of the substrates on the yield were proved not to have a strong impact on the catalytic reactivity. This catalytic system allowed a broad range of oxime acetates derived from acetoarenes to couple with cinnamic aldehyde 7a, with functional groups such as alkyl (8b and 8c), methoxyl (8e and 8j), and nitro (8i, 8l, and 8n) being tolerated well. To our delight, halo-functionalities such as F, Cl, Br, and I were all tolerated well (8f–8h, 8k, and 8m). And heteroaromatic ketoximes bearing furan (8q), thiophene (8r), and pyridine (8s) were all accommodated. Then, 2,3,4-trisubstituted pyridines 8t and 8u were generated in good yield by the use of oxime acetates derived from propiophenone and 1,2-diphenylethanone. The investigation of cinnamic aldehydes indicated that both electron-donating methoxy and electron-deficient nitro attached to the benzene ring were compatible with this reaction, affording the products 9a and 9b in 74% and 66% yield, respectively. Finally, we found that (E)-4-phenylbut-3-en-2-one reacted smoothly to give 2-methyl-4,6-diphenylpyridine 9d in modest yield.
 | ||
| Scheme 4 Pyridine synthesis catalyzed by NH4I. | ||
Next, a number of control experiments were performed to gain the mechanistic information of the ammonium iodide initiated oxime reduction and annulation. The addition of a scavenger such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and butylated hydroxytoluene (BHT) did not dramatically affect the yields of both reaction systems (Scheme 5, reactions (a) and (b)). Compared with the iodine-promoted oxime reduction probably involving the radical pathway,8a ammonium iodide may directly serve as an electron- and proton-donor to reduce the N–O bond of oxime acetate to give imine and acetic acid, in which the radical intermediate would not be formed, while elemental iodine would be generated (Scheme 5d, 1a to B). The viable reductive reactivity of ammonium iodide was further demonstrated by the experimental result of oxime acetate 1d, which furnished the expected ketone 10 with excellent yield in the absence of any carbonyls (Scheme 5, reaction (c)). Concerning the broad application of oximes as linkage groups in biochemistry,11 the present simple and mild reductive system could be applied in a broader field of chemistry. Thereafter, the imine intermediate B undergoes tautomerization to form enamine B′, which proceeds through a Michael addition to α,β-unsaturated carbonyls with the assistance of iodine12in situ generated (Scheme 5d, B′ to C). Then, the intramolecular condensation/annulation of intermediate C occurs to deliver dihydropyridine D. Finally, the desired pyridine products are obtained through the I2-mediated oxidative aromatization.
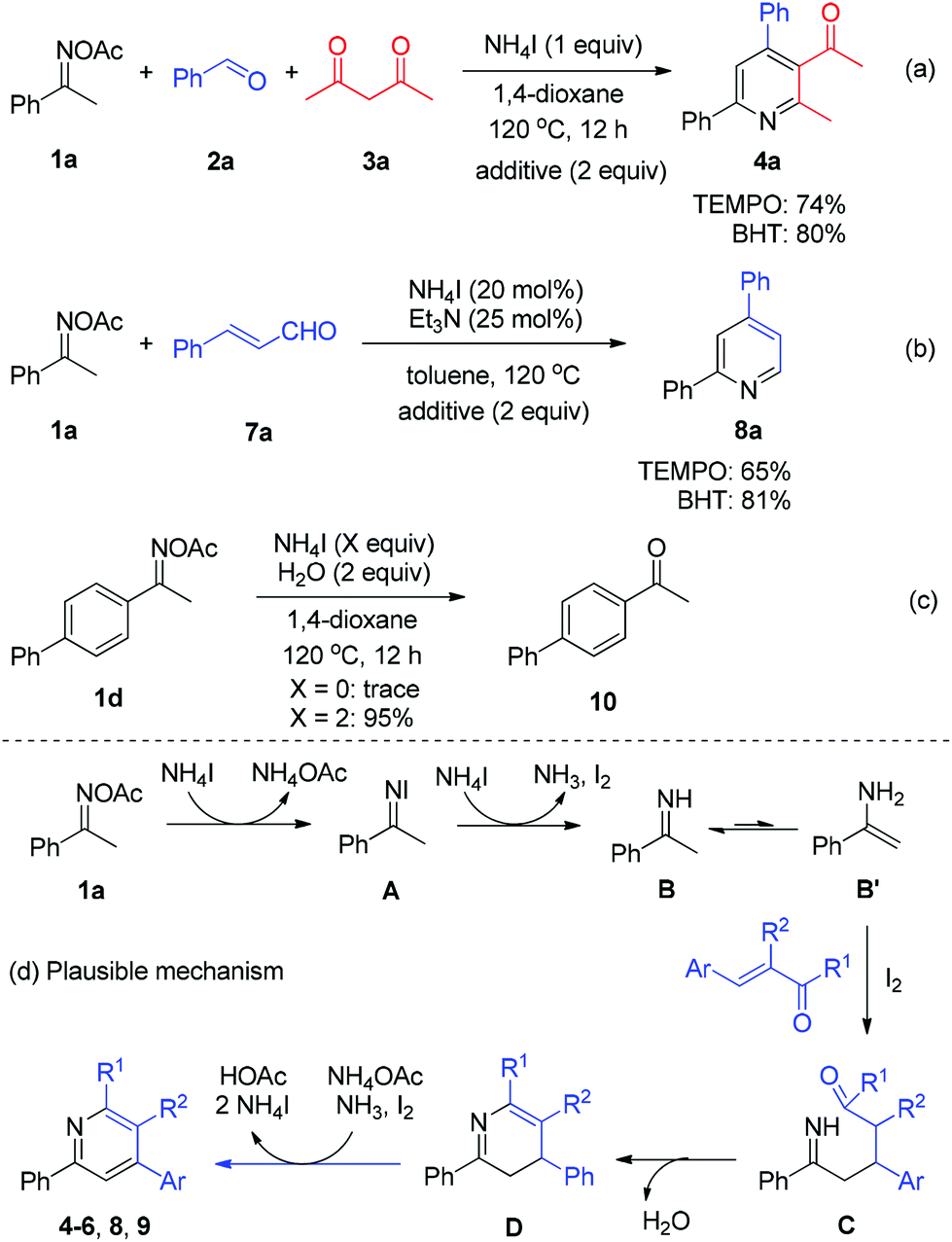 | ||
| Scheme 5 Mechanistic studies and proposal of pyridine synthesis catalyzed by NH4I. | ||
Conclusions
In summary, we developed the efficient ammonium iodide-assisted N–O bond reduction of oxime acetates, which was applied to the condensation reactions with α,β-unsaturated carbonyls to provide alternative methods for pyridine synthesis. In complementary with previous copper- and iodine-promoted reactions, this protocol allows a modular generation of multisubstituted pyridines from oxime acetates and α,β-unsaturated carbonyls. More importantly, the present metal-free oxime reduction by the simple and mild system with ammonium iodide may be applied to the cleavage of oxime linkage groups in the field of biochemistry.Experimental
General information
All reactions were carried out under the standard conditions unless otherwise noted. Column chromatography was performed using silica gel (200–300 mesh). 1H NMR and 13C NMR spectra were recorded on a 400 MHz NMR spectrometer, and the chemical shifts are referenced to signals at 7.26 and 77.0 ppm, respectively. Generally, chloroform was used as the solvent with TMS as the internal standard. MS analyses were performed on an Agilent 5975 GC-MS instrument (EI). HRMS was carried out on a high-resolution mass spectrometer (ESI, LCMS-IT-TOF). The structure of known compounds was further corroborated by comparing their 1H NMR, 13C NMR data and MS data with those in the literature. Melting points were measured with a BÜCHI B-545 melting point instrument without correction. All reagents were obtained from commercial suppliers and used without further purification.General procedure for pyridine synthesis by three-component reaction (4, 5 and 6)
A 10 mL reaction vessel was charged with NH4I (0.2 mmol, 1.0 equiv.), oxime acetate (1, 0.2 mmol, 1.0 equiv.), aldehyde (2, 0.3 mmol, 1.5 equiv.), and 1,3-dicarbonyl (3, 0.4 mmol, 2.0 equiv.). The sealed reaction vessel was purged with argon three times. 1,4-Dioxane (0.5 mL) was added to the sealed reaction vessel by using a syringe. The resulting solution was stirred at 120 °C for 12 h. The mixture was then allowed to cool down to room temperature and flushed through a short column of silica gel with ethyl acetate. After rotary evaporation, the residue was purified by column chromatography (silica gel, petroleum ether/EtOAc = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 50:1) to obtain 4, 5, and 6.
:1 to 50:1) to obtain 4, 5, and 6.
General procedure for the synthesis of pyridines 8 and 9
A 10 mL reaction vessel was charged with NH4I (0.04 mmol, 20 mol%), Et3N (0.05 mmol, 25 mol%), oxime acetate (1, 0.2 mmol, 1.0 equiv.), and aldehyde (7, 0.3 mmol, 1.5 equiv.). The sealed reaction vessel was purged with argon three times. Toluene (0.5 mL) was added to the sealed reaction vessel by using a syringe. The resulting solution was stirred at 120 °C for 12 h. The mixture was then allowed to cool down to room temperature and flushed through a short column of silica gel with ethyl acetate. After rotary evaporation, the residue was purified by column chromatography (silica gel, petroleum ether/EtOAc = 20:1 to 50:1) to obtain 8 and 9.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21602187, 21572194), the Collaborative Innovation Center of New Chemical Technologies for Environmental Benignity and Efficient Resource Utilization, the Hunan Provincial Innovative Foundation for Postgraduate (CX2017B293), and the Hunan Provincial Natural Science Foundation of China (2017JJ3299).Notes and references
- For reviews, see: (a) G. D. Henry, Tetrahedron, 2004, 60, 6043–6061 CrossRef CAS; (b) M. D. Hill, Chem. – Eur. J., 2010, 16, 12052–12062 CrossRef CAS PubMed; (c) C. Allais, J. M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829–10868 CrossRef CAS PubMed.
- For recent representative examples of pyridine synthesis, see: (a) P. Kumar, S. Prescher and J. Louie, Angew. Chem., Int. Ed., 2011, 50, 10694–10698 CrossRef CAS PubMed; (b) M. Ohashi, I. Takeda, M. Ikawa and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 18018–18021 CrossRef CAS PubMed; (c) M. Z. Chen and G. C. Micalizio, J. Am. Chem. Soc., 2012, 134, 1352–1356 CrossRef CAS PubMed; (d) G. Onodera, Y. Shimizu, J. N. Kimura, J. Kobayashi, Y. Ebihara, K. Kondo, K. Sakata and R. Takeuchi, J. Am. Chem. Soc., 2012, 134, 10515–10531 CrossRef CAS PubMed; (e) N. S. Loy, A. Singh, X. Xu and C. M. Park, Angew. Chem., Int. Ed., 2013, 52, 2212–2216 CrossRef CAS PubMed; (f) S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326–6329 CrossRef CAS PubMed; (g) Z. Shi and T. P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584–8587 CrossRef CAS PubMed; (h) J. Wu, W. Xu, Z. X. Yu and J. Wang, J. Am. Chem. Soc., 2015, 137, 9489–9496 CrossRef CAS PubMed; (i) Y. Wang, L. J. Song, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 9704–9708 CrossRef CAS PubMed; (j) L. G. Xie, S. Shaaban, X. Chen and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 12864–12867 CrossRef CAS PubMed; (k) T. Hille, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 371–374 CrossRef CAS PubMed.
- For reviews, see: (a) M. Kitamura and K. Narasaka, Chem. Rec., 2002, 2, 268–277 CrossRef CAS PubMed; (b) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505–4519 CrossRef CAS; (c) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (d) H. Huang, J. Cai and G. J. Deng, Org. Biomol. Chem., 2016, 14, 1519–1530 RSC , and references cited therein.
- (a) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918–6919 CrossRef CAS PubMed; (b) K. Parthasarathy, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2008, 10, 325–328 CrossRef CAS PubMed; (c) I. Nakamura, D. Zhang and M. Terada, J. Am. Chem. Soc., 2010, 132, 7884–7886 CrossRef CAS PubMed; (d) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846–11848 RSC; (e) R. M. Martin, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2012, 77, 2501–2507 CrossRef CAS PubMed; (f) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66–69 CrossRef CAS PubMed; (g) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2014, 136, 2735–2738 CrossRef CAS PubMed.
- (a) Z.-H. Ren, Z.-Y. Zhang, B.-Q. Yang, Y.-Y. Wang and Z.-H. Guan, Org. Lett., 2011, 13, 5394–5397 CrossRef CAS PubMed; (b) M. N. Zhao, R. R. Hui, Z. H. Ren, Y. Y. Wang and Z. H. Guan, Org. Lett., 2014, 16, 3082–3085 CrossRef CAS PubMed; (c) M. N. Zhao, Z. H. Ren, L. Yu, Y. Y. Wang and Z. H. Guan, Org. Lett., 2016, 18, 1194–1197 CrossRef CAS PubMed.
- (a) Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756–3759 CrossRef CAS PubMed; (b) Q. Wu, Y. Zhang and S. Cui, Org. Lett., 2014, 16, 1350–1353 CrossRef CAS PubMed; (c) H. Jiang, J. Yang, X. Tang, J. Li and W. Wu, J. Org. Chem., 2015, 80, 8763–8771 CrossRef CAS PubMed; (d) M. Zheng, P. Chen, W. Wu and H. Jiang, Chem. Commun., 2016, 52, 84–87 RSC; (e) W. W. Tan, Y. J. Ong and N. Yoshikai, Angew. Chem., Int. Ed., 2017, 56, 8240–8244 CrossRef CAS PubMed.
- (a) T. Mikami and K. Narasaka, Chem. Lett., 2000, 338–339 CrossRef CAS; (b) M. Kitamura, Y. Mori and K. Narasaka, Tetrahedron Lett., 2005, 46, 2373–2376 CrossRef CAS; (c) M. Yoshida, M. Kitamura and K. Narasaka, Chem. Lett., 2002, 144–145 CrossRef CAS; (d) M. Yoshida, M. Kitamura and K. Narasaka, Bull. Chem. Soc. Jpn., 2003, 76, 2003–2008 CrossRef CAS.
- (a) H. Huang, J. Cai, L. Tang, Z. Wang, F. Li and G.-J. Deng, J. Org. Chem., 2016, 81, 1499–1505 CrossRef CAS PubMed; (b) H. Huang, J. Cai, H. Xie, J. Tan, F. Li and G.-J. Deng, Org. Lett., 2017, 19, 3743–3746 CrossRef CAS PubMed; (c) Y. Xie, X. Chen, Z. Wang, H. Huang, B. Yi and G.-J. Deng, Green Chem., 2017, 19, 4294–4298 RSC; (d) X. Che, J. Jiang, F. Xiao, H. Huang and G.-J. Deng, Org. Lett., 2017, 19, 4576–4579 CrossRef CAS PubMed; (e) H. Huang, F. Li, Z. Xu, J. Cai, X. Ji and G.-J. Deng, Adv. Synth. Catal., 2017, 359, 3102–3107 CrossRef CAS.
- For selective iodine-based synthesis, see: (a) S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496–4499 RSC; (b) A. Verma, S. Patel, Meenakshi, A. Kumar, A. Yadav, S. Kumar, S. Jana, S. Sharma, C. D. Prasad and S. Kumar, Chem. Commun., 2015, 51, 1371–1374 RSC; (c) A. Verma and S. Kumar, Org. Lett., 2016, 18, 4388–4391 CrossRef CAS PubMed.
- (a) B. K. Banik, S. Samajdar and I. Banik, J. Org. Chem., 2004, 69, 213–216 CrossRef CAS PubMed; (b) H. Togo and S. Iida, Synlett, 2006, 2159–2175 CrossRef CAS.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- For iodine-catalyzed Michael additions, see: (a) K. J. Borah, M. Phukan and R. Borah, Synth. Commun., 2010, 40, 2830–2836 CrossRef CAS; (b) M. Breugst, E. Detmar and D. von der Heiden, ACS Catal., 2016, 6, 3203–3212 CrossRef CAS; (c) D. von der Heiden, S. Bozkus, M. Klussmann and M. Breugst, J. Org. Chem., 2017, 82, 4037–4043 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob02471a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |