Ligand-free, palladacycle-facilitated Suzuki coupling of hindered 2-arylbenzothiazole derivatives yields potent and selective COX-2 inhibitors†
Received
23rd September 2017
, Accepted 22nd November 2017
First published on 4th December 2017
Abstract
A similarity search and molecular modeling study suggested the 2′-aryl-2-arylbenzothiazole framework as a novel scaffold for the design of COX-2-selective inhibitors. Conventional Suzuki coupling conditions did not furnish the designed compounds in good yield from 2′-bromo-2-arylbenzothiazole as the starting material. A novel ligand-free Suzuki–Miyaura coupling methodology was developed for sterically hindered 2′-bromo-2-arylbenzothiazoles. The reaction depends on the coordination properties of the benzothiazole ring nitrogen, which is involved in the formation of a palladacyclic intermediate that was synthesized independently and converted to the final product. The new method provides good to excellent yields (up to 99%) with favorable functional group tolerability. Six compounds had potencies in the submicromolar range against COX-2 and higher selectivity for COX-2 vs. COX-1 compared to the currently used drug celecoxib. Molecular modeling was used to investigate the possible binding mode with COX-2.
Introduction
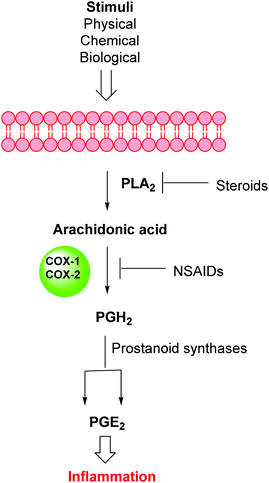
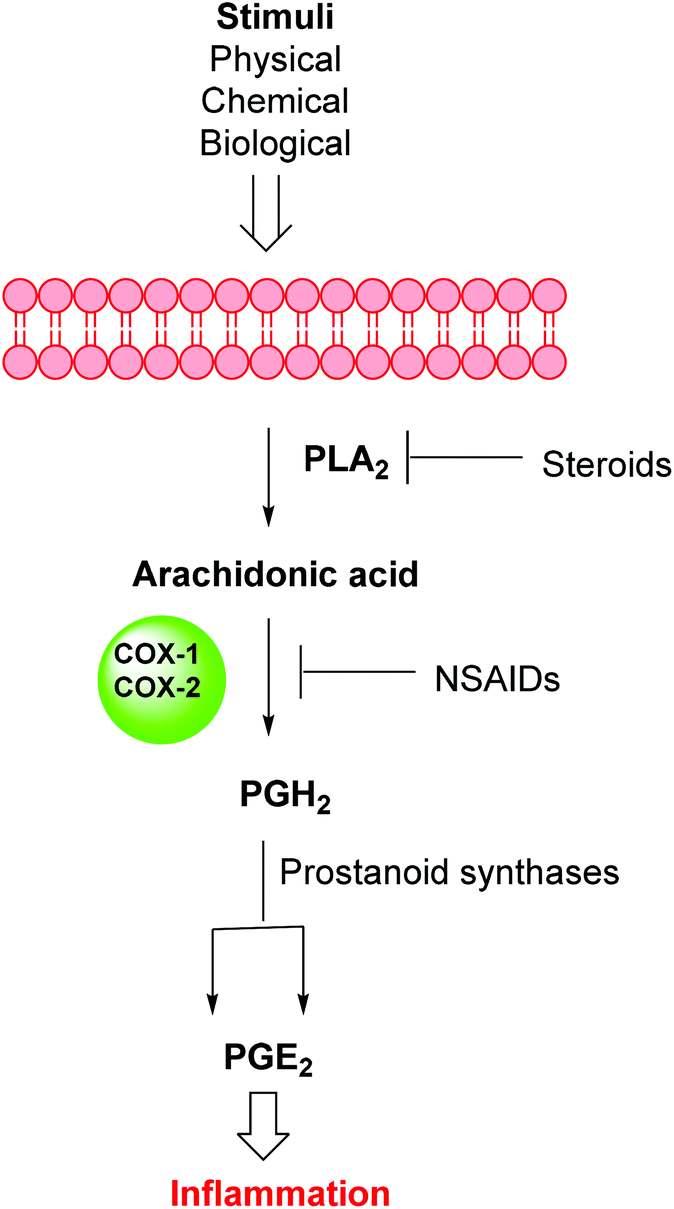
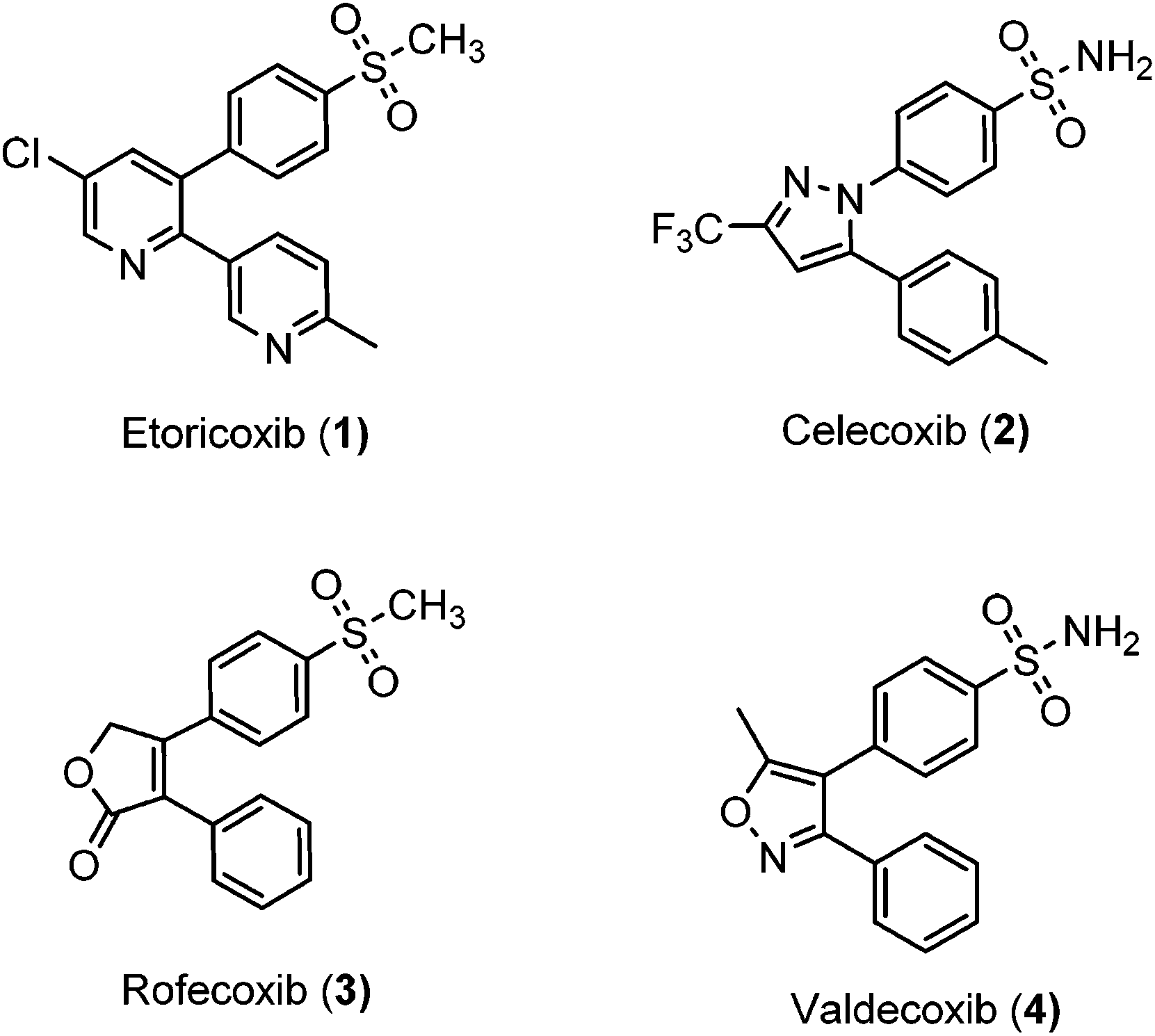
Inflammation is a complex defensive process initiated by the immune system in response to various internal or external stimuli. Although it starts as a defensive mechanism, prolonged inflammation is a major cause of tissue damage.1 Inflammation is also involved in the pathogenesis of many chronic peripheral and central nervous system disorders.2,3 Prostaglandins (PGs) are the major chemical mediators of inflammation and they are responsible for various chemical and cellular events leading to inflammation (Fig. 1).4 For many years, inflammation has been treated with steroids, that inhibit the release of arachidonic acid, and non-steroidal anti-inflammatory drugs (NSAIDs), that non-selectively inhibit cyclooxygenases 1 and 2 (COX-1 and COX-2), the key enzymes responsible for the synthesis of PGs.5 Non-selective NSAIDs suffer from many side effects which, in many cases, are not tolerable because these drugs are usually used for the treatment of chronic diseases such as rheumatoid arthritis.6,7 These side effects range from gastric ulcers to renal failure.8 The side effects associated with non-selective COX inhibitors are mainly due to the loss of the beneficial and homeostatic roles of PGs.8,9 COX-2-selective inhibitors (Fig. 2) have been introduced as safer anti-inflammatory agents with fewer side effects than are usually associated with NSAIDs.10 COX-2 inhibitors have also been investigated for the treatment of inflammation-related cancers and neurological disorders.11–16
 |
| | Fig. 1 Inflammation pathway. | |
 |
| | Fig. 2 Selective COX-2 inhibitors. | |
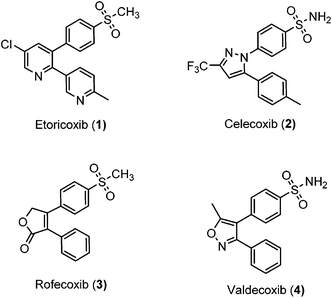
Unfortunately, two of these COX-2 selective inhibitors (rofecoxib and valdecoxib) were withdrawn from the US market due to cardiovascular side effects.17 This underscores the fact that there is still a need for the development of safe, effective and selective COX-2 inhibitors to overcome these drawbacks.
The Suzuki–Miyaura coupling reaction is an extremely useful method for C–C bond formation. Over the past few years, this reaction has been optimized for the synthesis of various compounds and hundreds of ligands have been developed to provide better yields or to increase functional group tolerability.18,19 General procedures for Suzuki–Miyaura coupling involve the use of a Pd catalyst, a ligand, and a base in a suitable solvent. Unfortunately, not all compounds are good substrates for the usual Suzuki conditions and some functional groups are not tolerated very well. It has been reported that Suzuki reactions involving ortho-heterocycle-tethered, sterically hindered substrates are challenging and need special conditions to proceed. In addition, the reported procedures either give very low yields or require complex and expensive catalysts or ligands.20–22 The present report describes a simple and efficient ligand-free Suzuki–Miyaura type coupling reaction with good to excellent yields for the synthesis of 2′-aryl-2-aryl/heteroarylbenzothiazole heterocyclic frameworks.
Results and discussion
Chemistry
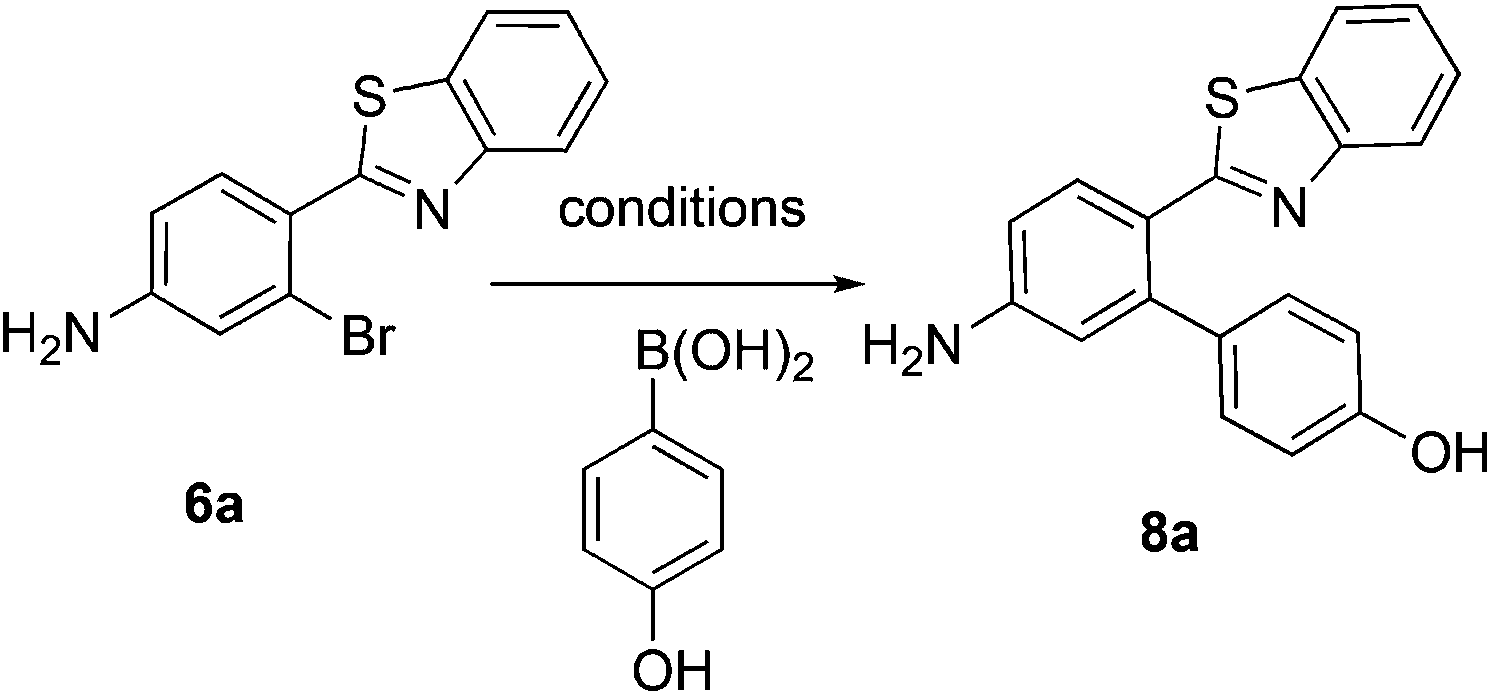
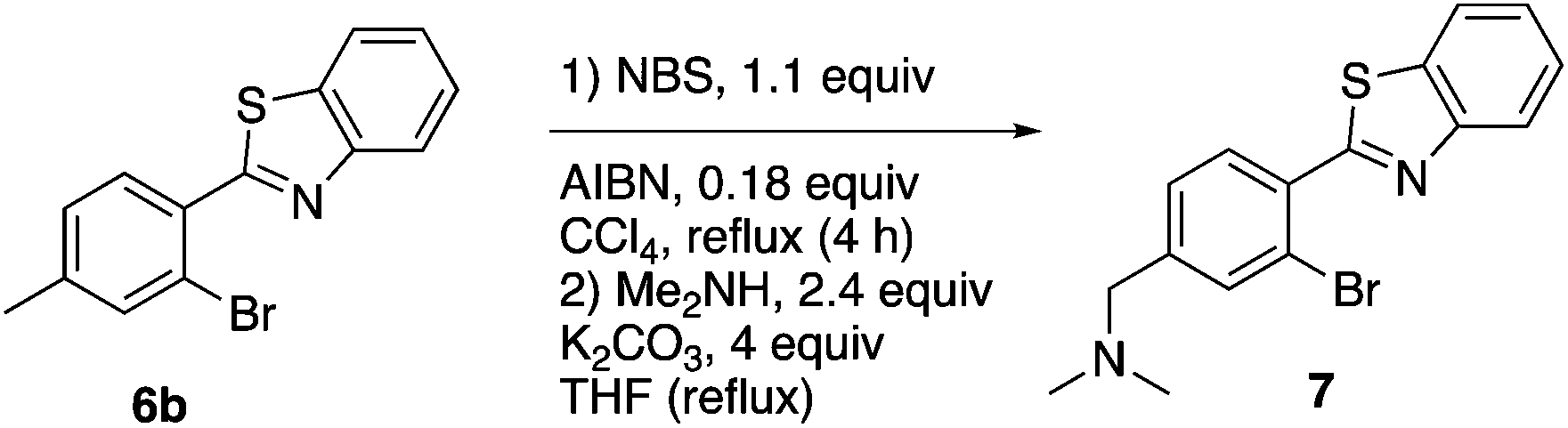
This project originated from a study of the pharmacology of 2-arylbenzothiazoles. Compound 8a (Fig. 4) was suggested as a potential COX-2 inhibitor based on a similarity search and a molecular modeling study, and therefore coupling chemistry was investigated as a possible strategy for its synthesis. First, 2-aryl-2′-bromobenzothiazoles 6a–d were prepared from the corresponding commercially available carboxylic acids 5a–d using the reported literature procedures (Scheme 1).23 Compound 7 was synthesized from 6b by benzylic bromination followed by nucleophilic substitution of the bromine atom with dimethylamine as shown in Scheme 2.
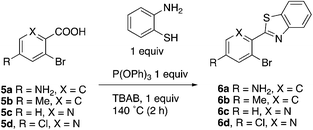 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
The reported conventional Suzuki coupling reaction conditions for the synthesis of compounds similar to 8a were investigated (Table 1).22 The reported conditions involved the use of tetrakis(triphenylphosphine)palladium(0) as the catalyst complex in a mixture of ethanol and water as the solvent. Unfortunately, the reaction of 6a proceeded with only 2% yield (entry 1, Table 1). As this coupling step was critical for the synthesis of the target compounds, an attempt was made to optimize it. Various reaction conditions, catalysts, solvents and bases were applied in order to find the optimal conditions (Table 1). Since oxidative addition is the rate-determining step in the Suzuki reaction,24 the literature was explored for similar hindered ring systems. Because the nitrogen atom in the benzothiazole ring of the reactant could possibly function as a ligand and as a directing group for the oxidative addition step, the ligand was removed from the reaction mixture and Pd(OAc)2 was applied as the catalyst to enhance the yield to 65% (entry 11, Table 1). This encouraged further optimization by changing the catalysts, base and solvent system. The use of Pd2(dba)3 slightly increased the yield to 68% (entry 14). Changing the solvent to dioxane increased the yield further (entries 15–17). The employment of Pd(0) as in Pd2(dba)3 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture of dioxane and water in the presence of sodium carbonate as the base provided the optimum conditions for this reaction, resulting in a yield of 83% (entry 16). Although the base was not absolutely necessary for this reaction, its presence produced higher yields (entries 17 and 18).
:1 solvent mixture of dioxane and water in the presence of sodium carbonate as the base provided the optimum conditions for this reaction, resulting in a yield of 83% (entry 16). Although the base was not absolutely necessary for this reaction, its presence produced higher yields (entries 17 and 18).
Table 1 Optimization of the Suzuki coupling step
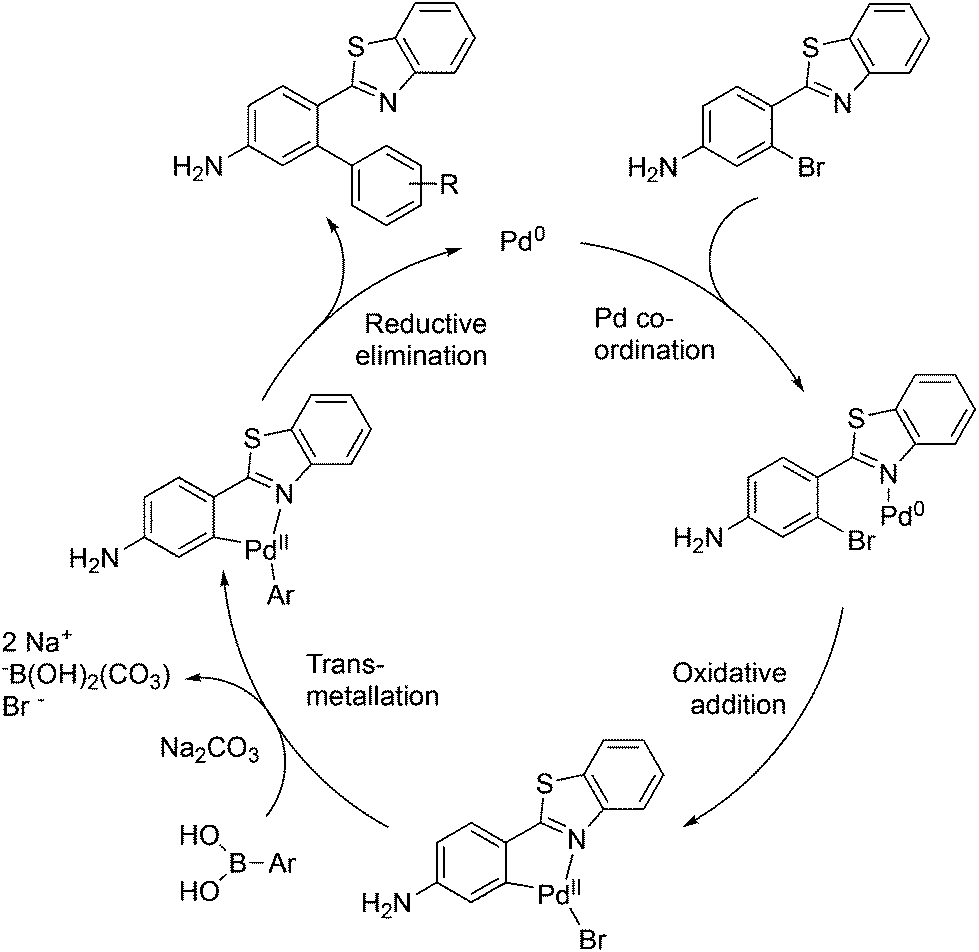
The mechanism proposed for the coupling reaction takes into consideration the coordination properties of the benzothiazole nitrogen (Fig. 3). The reaction is hypothesized to proceed via a Pd(0)/Pd(II) catalytic cycle. The nitrogen atom of the benzothiazole ring is assumed to coordinate to Pd, which will facilitate the rate-limiting oxidative addition step. The organopalladium complex will then react with the boronic acid in the presence of the base via a transmetallation process. The final step involves the coupling of the two aromatic rings and reductive elimination of the Pd metal.
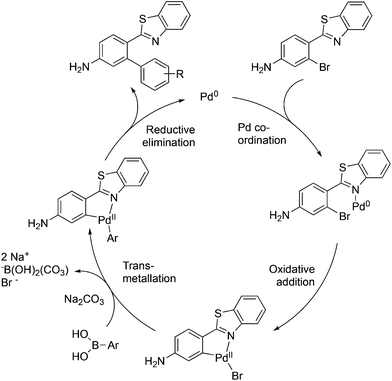 |
| | Fig. 3 Hypothetical mechanism of the Pd(0)-mediated ligand-free Suzuki coupling reaction. | |
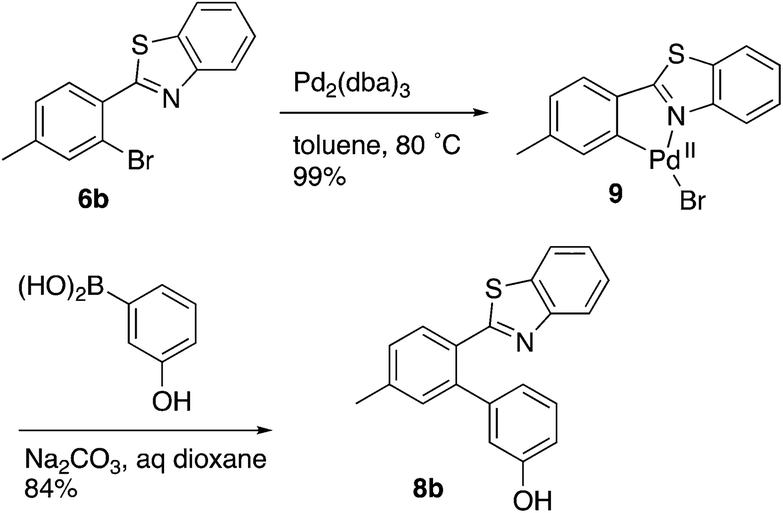
Evidence was sought in support of the proposed palladium complex in Fig. 3 (bottom). The reaction of compound 6b, having a methyl replacement for the amino group of 6a, with Pd2(dba)3 in toluene at 80 °C led to the isolation of the palladium complex 9 as a gold amorphous solid in 99% yield (Scheme 3). The MALDI mass spectrum of 9 displayed the isotope cluster with twelve peaks, expected for a substance having one palladium atom and one bromine atom, and the 1H NMR spectrum of the complex integrated for seven aromatic protons relative to the three methyl hydrogens. The complex had very limited solubility in a wide variety of different solvents and it failed to crystallize even after exhaustive experimentation. However, it could be converted to the expected product 8b by subjecting it to the conditions used in the overall Suzuki coupling reaction (Na2CO3 in aqueous dioxane) in the presence of 3-hydroxyphenylboronic acid, thus establishing the palladium complex 9 as a true reaction intermediate (Scheme 3).
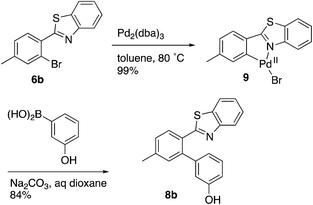 |
| | Scheme 3 | |
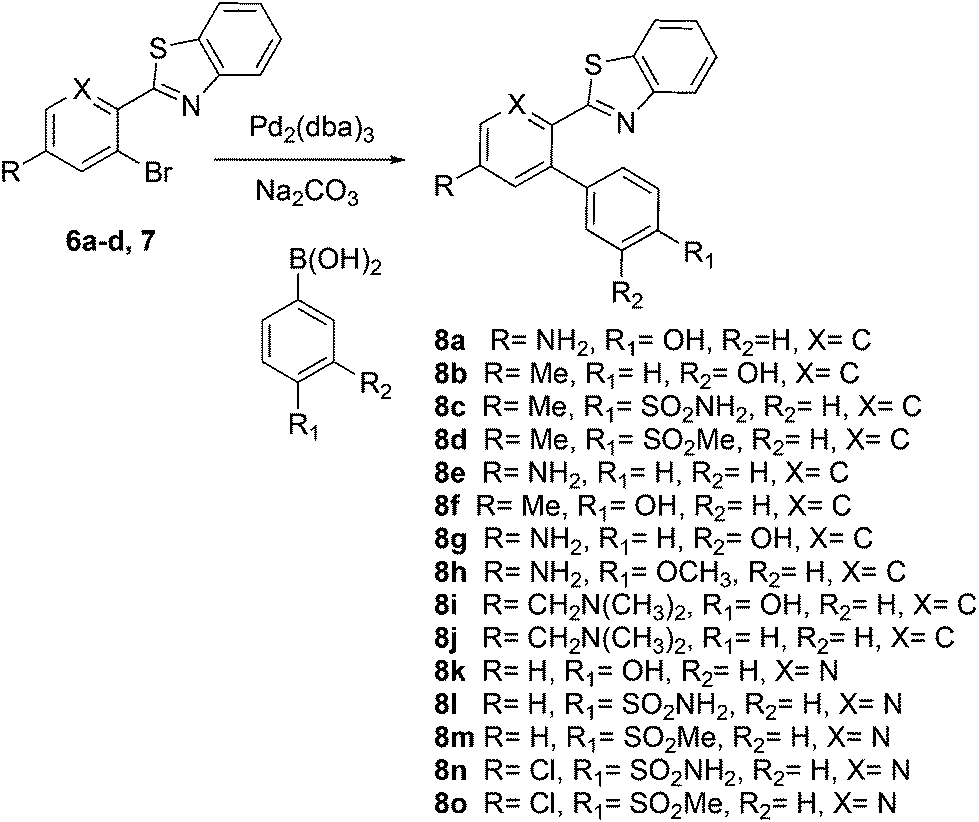
The similarity search mentioned above was conducted based on the structure of 8a using the ChEMBL database in order to find structurally similar biologically active compounds.25 Compound 8a was indicated to have high structural similarity to the selective COX-2 drug etoricoxib.26 Based on this, analogs of 8a were synthesized in order to explore their structure–activity relationships. At the same time, the scope of the reaction was defined. Scheme 4 and Fig. 4 show the scope and the reaction yields. The optimized reaction conditions provided good to excellent yields with broad functional group tolerability. Free phenols, amines, sulfonamides and sulfones reacted easily with up to 99% yield.
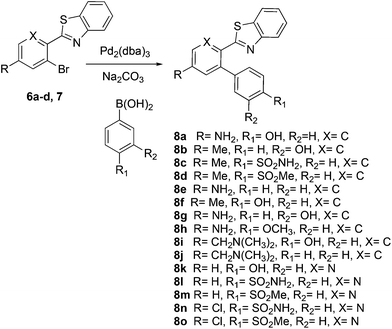 |
| | Scheme 4 | |
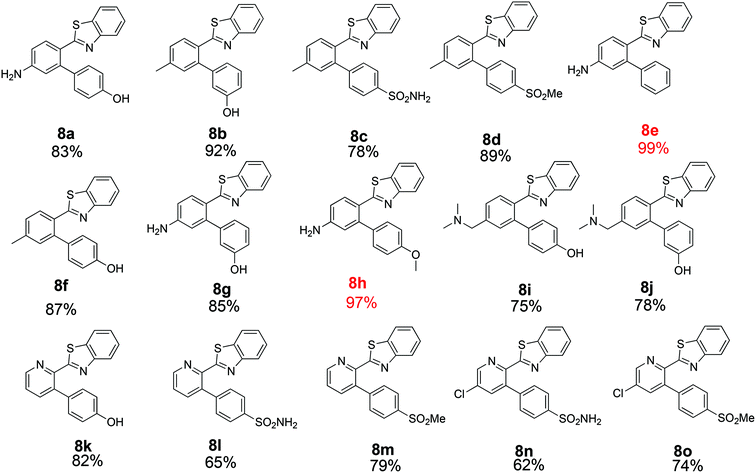 |
| | Fig. 4 Scope and yields of ligand-free Suzuki–Miyaura reaction. | |
The 2-aryl ring of the reactant can also be phenyl or pyridyl without affecting the reactivity. This demonstrates that the new coupling reaction has potential versatility for the general synthesis of this type of triaryl ring system.
In order to further explore the scope of this approach, it was applied to the 2-arylbenzoxazole 10. Use of the NiCl2(PCy3)221 catalyst and the mixed Ni2·6H2O/PdCl2 catalyst20 was considered, and a decision was made to carry out a direct comparison of the latter with Pd2(dba)3 to see if the complex palladium–nickel binary nanocluster catalyst could be avoided and still maintain a high yield. The yield reported for the mixed Ni2·6H2O/PdCl2 nanocluster catalyst was 82%, while the same authors reported 16–27% and 12–14% yields when Pd(OAc)2/biphenyl or Pd(OAc)2/PCy3 were used alone.20 The present optimized methodology using Pd2(dba)3 in aqueous dioxane provided compound 11 in 95% yield with a simple catalyst and synthetic operation as shown in Scheme 5.
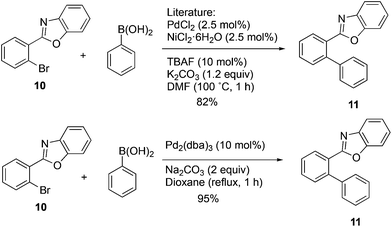 |
| | Scheme 5 | |
Biological assay
All of the products were tested for their inhibitory activities against COX-1 and COX-2. First, the percent inhibition of the enzyme activity was measured at a single concentration of 10 μM. Afterwards, the IC50 values for compounds that produced more than 50% inhibition were determined. Table 2 lists the biological activities of the compounds against COX-1 and COX-2. A total of 15 compounds were tested in the single dose assay, of which 6 compounds showed higher than 50% inhibition of COX-2 with very low inhibition of COX-1 compared to celecoxib as a positive control. Compounds 8c and 8d with a methyl group in the 2-phenyl ring and compounds 8l–o with a 2-pyridyl ring have very good potencies and selectivities. The IC50 values of these 6 compounds were determined against the two enzymes. Compound 8n with a benzene sulfonamide side chain was the most active and selective compound with weaker potency (0.21 μM) than celecoxib (0.079 μM) and slightly more favorable selectivity (>476 compared to 442). On the other hand, the IC50 values vs. COX-2 for the six active compounds reported in Table 2 compare favorably with the 5.0 μM value reported for etoricoxib.27 The IC50 values of celecoxib (positive control) against COX-2 and COX-1 were very close to the values reported in the literature (0.069 and 30 μM, respectively).28,29 The activities of the 6 compounds against COX-1 were very weak with the IC50 values higher than 100 μM. Compounds 8c, 8d and 8o were also very active and selective with the IC50 values of 0.25, 0.23 and 0.23 μM, respectively, and all showed a selectivity index of >400 for COX-2 vs. COX-1. Compounds 8a, 8b and 8e–k were not very active against COX-2 at the concentration of 10 μM with percent inhibition values from 0 to 35%, suggesting that a hydroxyphenyl side chain is not beneficial for the activity of this class of COX-2 inhibitors. Compound 8h displayed good percent inhibition against COX-1 and weak activity against COX-2, suggesting that it might be optimized as a COX-1 inhibitor. This was the only compound bearing a methoxy group, which might play a role in binding to COX-1 more strongly than COX-2. The benzenesulfonamide side chain was the best side chain for activity and selectivity as shown for compounds 8c and 8n. Moreover, the chloropyridyl ring system present in 8n and 8o is similar to that of etoricoxib and contributed very good activity.
Table 2 Enzyme inhibition activity of COX-1 and COX-2
| Compound |
COX-2% inhibition at 10 μM |
COX-2 IC50 |
COX-1% inhibition at 10 μM |
COX-1 IC50 |
Selectivity |
|
NT = not tested.
|
|
Celecoxib
|
100
|
0.079 ± 0.02
|
0
|
31 ± 2
|
442
|
|
8a
|
15.3 |
NTa |
14.2 |
NT |
1.1 |
|
8b
|
29.0 |
NT |
11.7 |
NT |
2.5 |
|
8c
|
87.3
|
0.25 ± 0.01
|
9.6
|
>100
|
>400
|
|
8d
|
72.0
|
0.23 ± 0.08
|
9.7
|
>100
|
>434
|
|
8e
|
12.4 |
NT |
59.1 |
NT |
0.21 |
|
8f
|
35.4 |
NT |
8.4 |
NT |
4.2 |
|
8g
|
33.3 |
NT |
27.7 |
NT |
1.2 |
|
8h
|
10.3 |
NT |
65.4 |
NT |
0.16 |
|
8i
|
0 |
NT |
14.2 |
NT |
0 |
|
8j
|
1.0 |
NT |
30.7 |
NT |
0.1 |
|
8k
|
8 |
NT |
12.5 |
NT |
0.6 |
|
8l
|
88.5
|
0.69 ± 0.12
|
0
|
>100
|
>144
|
|
8m
|
84.7
|
0.86 ± 0.05
|
21.5
|
>100
|
>116
|
|
8n
|
100
|
0.21 ± 0.07
|
6.6
|
>100
|
>476
|
|
8o
|
98.0
|
0.23 ± 0.01
|
0
|
>100
|
>434
|
Molecular modeling
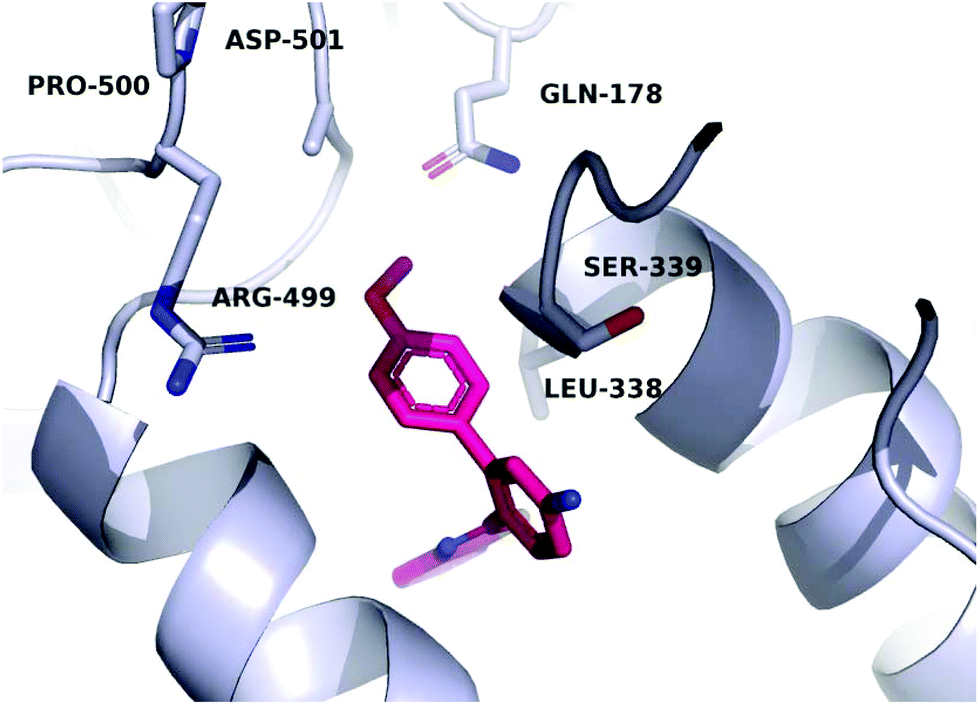
A molecular docking study was conducted in order to investigate the possible binding mode of our compounds to COX enzymes. The active compounds were docked in COX-2 using GOLD and then the selected poses were energy minimized. The docked compounds had a very similar binding mode to celecoxib. Fig. 5 illustrates the binding mode of compound 8n with COX-2. The sulfonamide group makes two hydrogen bonds with Arg499 and Gln178, while the benzothiazole ring is directed to the hydrophobic pocket deep inside the enzyme. Compared to the binding mode of 8n, celecoxib revealed that the pyridine ring of 8n is superimposed on the pyrazole ring of celecoxib while the benzothiazole moiety is aligned with the tolyl ring. At the same time the sulfonamides of the two compounds form a hydrogen bond similarly to the enzyme.
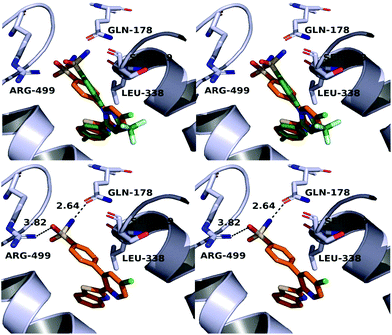 |
| | Fig. 5 Top: Alignment between celecoxib (green) and compound 8n (orange) in the active site of COX-2. Bottom: Binding mode of 8n in COX-2. The stereoview is programmed for wall-eyed (relaxed) viewing. | |
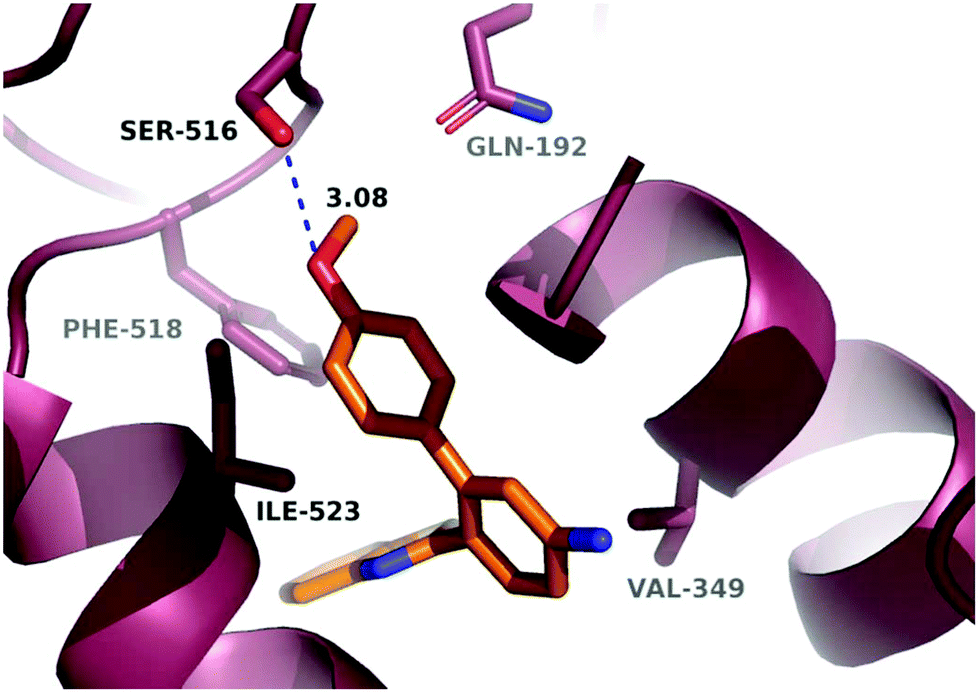
In order to investigate the relatively good inhibitory activity of compound 8h against COX-1, it was docked in the active site of the enzyme (PDB: 3KK6). The docking calculations indicated that the methoxy group is involved in a hydrogen bond interaction with the Ser516 residue, which is not present in COX-2 (Fig. 6). Compound 8h was also docked in the active site of COX-2 to compare the binding interaction and the reason behind the selectivity. Fig. 7 illustrates the docking pose obtained from the docking calculations and shows the absence of any hydrogen bonding interaction with the enzyme. The methoxy group in Fig. 7 is far from the Arg499 residue that makes the critical interaction with compound 8n (Fig. 5).
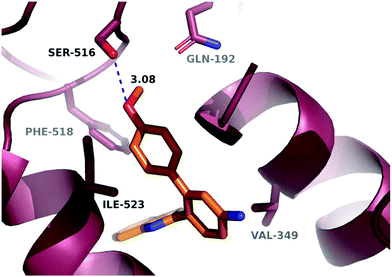 |
| | Fig. 6 Hypothetical binding mode of 8h in the active site of COX-1. | |
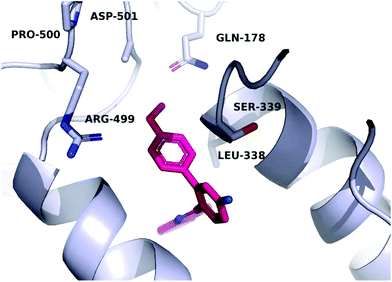 |
| | Fig. 7 Hypothetical binding mode of compound 8h in the active site of COX-2. | |
Conclusion
An efficient, simple and high-yielding ligand-free Suzuki–Miyaura coupling reaction was optimized for the synthesis of a variety of 2′-aryl-2-aryl-benzothiazoles. The hypothetical palladacyclic intermediate was independently synthesized and converted to the Suzuki coupling product, thus providing direct evidence for its role in the overall reaction. A similarity search was conducted to identify the biological target of the new compounds. Fifteen compounds were synthesized via the optimized reaction and tested for their inhibitory activities against COX-2 and COX-1. Compound 8n was found to be the most potent and selective COX-2 inhibitor of the series. Molecular docking was used to explore the possible binding mode of the new compounds with COX-2.
Experimental
General
Melting points were determined with a Mel-Temp apparatus using capillary tubes and are uncorrected. The proton nuclear magnetic resonance spectra (1H NMR) were recorded using an ARX300 300 MHz Bruker or a DRX500-2 500 MHz NMR spectrometer. IR spectra were obtained with a PerkinElmer 1600 series FTIR spectrometer. For purities estimated by HPLC, the major peak accounted for ≥95% of the combined total peak area when monitored using a UV detector at 254 nm. HPLC analyses were performed on a Waters 1525 binary HPLC pump/Waters 2487 dual λ absorbance detector system. Analytical thin-layer chromatography was conducted on Baker-flex silica gel IB2-F plates, and compounds were visualized with UV light at 254 nm. Silica gel flash chromatography was performed using 230–400 mesh silica gel.
Chemistry
4-(Benzo[d]thiazol-2-yl)-3-bromoaniline (6a).
A mixture of the acid 5a (1.96 g, 8 mmol), 2-aminothiophenol (1 g, 8 mmol), tetrabutylammonium bromide (2.58 g, 8 mmol) and triphenyl phosphite (2.47 g, 8 mmol) was heated in an oil bath at 140 °C for 2 h. After cooling the reaction mixture down to room temperature, methanol (20 mL) was added and the reaction mixture was stirred vigorously for 10 min. The orange solid was filtered off and washed thoroughly with methanol to give 6a as an orange powder (1.2 g, 44.8%): mp 212–216 °C. 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 8.1 Hz, 1 H), 7.90 (d, J = 8.5 Hz, 1 H), 7.84 (d, J = 7.8 Hz, 1 H), 7.43 (t, J = 7.7 Hz, 1 H), 7.33 (t, J = 7.6 Hz, 1 H), 7.02 (d, J = 2.3 Hz, 1 H), 6.72 (dd, J = 8.6, 2.3 Hz, 1 H); MALDIMS m/z (rel. intensity) 304/306 (MH)+; HRESIMS calcd for C13H9BrN2S (MH)+ 304.9743, found 304.9744.
2-(2-Bromo-4-methylphenyl)benzo[d]thiazole (6b).
This compound was prepared using the same procedure as that used for compound 6a but using EtOAc–hexanes (1:9) column chromatography for purification to give 6b as a white solid (1.4 g, 57%): mp 124–126 °C. 1H NMR (300 MHz, CDCl3) δ 8.13 (dd, J = 8.7, 0.5 Hz, 1 H), 7.94 (ddd, J = 8.0, 2.4, 1.8 Hz, 2 H), 7.57 (s, 1 H), 7.55–7.49 (m, 1 H), 7.46–7.39 (m, 1 H), 7.25 (q, J = 1.1 Hz, 2 H), 2.41 (s, 3 H); MALDIMS m/z (rel. intensity) 303/305 (MH)+; HRESIMS calcd for C14H10BrNS (MH)+ 303.9790, found 303.9791.
2-(3-Bromopyridin-2-yl)benzo[d]thiazole (6c).
This compound was isolated as a white solid (0.56 g, 39%): mp 123–125 °C. 1H NMR (300 MHz, CDCl3) δ 8.68 (dd, J = 4.5, 1.3 Hz, 1 H), 8.20 (dd, J = 7.6, 1.0 Hz, 1 H), 8.12 (dd, J = 8.1, 1.4 Hz, 1 H), 7.97 (dd, J = 7.1, 0.9 Hz, 1 H), 7.49 (dtd, J = 15.0, 7.3, 1.3 Hz, 2 H), 7.26 (q, J = 4.8 Hz, 2 H); MALDIMS m/z (rel. intensity) 290/292 (MH)+; HRESIMS calcd for C12H7BrN2S (MH)+ 290.9586, found 290.9587.
2-(3-Bromo-5-chloropyridin-2-yl)benzo[d]thiazole (6d).
This compound was isolated as a white powder (0.36 g, 23%): mp 135–137 °C. 1H NMR (500 MHz, CDCl3) δ 8.61 (d, J = 2.1 Hz, 1 H), 8.18 (d, J = 8.2 Hz, 1 H), 8.13 (d, J = 2.1 Hz, 1 H), 7.96 (d, J = 8.0 Hz, 1 H), 7.56–7.49 (m, 1 H), 7.46 (td, J = 7.7, 1.1 Hz, 1 H); 13C NMR (126 MHz, CDCl3) δ 166.20, 154.05, 146.77, 146.49, 142.14, 136.31, 132.36, 126.18, 124.33, 121.49, 118.00; MALDIMS m/z (rel. intensity) 324/326 (MH)+; HRESIMS calcd for C12H6BrClN2S (MH)+ 324.9196, found 324.9193.
1-(4-(Benzo[d]thiazol-2-yl)-3-bromophenyl)-N,N-dimethylmethanamine (7).
Compound 6b (0.5 g, 1.64 mmol), NBS (0.32 g, 1.80 mmol) and AIBN (50 mg, 0.3 mmol, 0.18 equiv.) were mixed in CCl4 (50 mL). The reaction mixture was heated under reflux for 4 h and then cooled down to room temperature, filtered, and the filtrate was evaporated to give the benzyl bromide derivative, which was used without further purification. The product from the previous reaction was dissolved in THF (20 mL) and then potassium carbonate (900 mg, 6.5 mmol, 4 equiv.) and excess dimethylamine (2 M solution in THF, 2 mL) were added. The reaction mixture was heated under reflux for 18 h and then THF was evaporated and the residue was dissolved in CHCl3 (30 mL). The organic layer was washed with water (1 × 20 mL) and brine (1 × 20 mL), dried with anhydrous sodium sulfate and evaporated to give a yellow residue. Compound 7 was purified using column chromatography with CHCl3–MeOH (9.5:0.5) as the solvent system to give a brownish solid (0.35 g, 63%): mp 185–187 °C. 1H NMR (500 MHz, CDCl3) δ 8.13 (d, J = 8.1 Hz, 1 H), 7.96 (dd, J = 14.6, 7.9 Hz, 2 H), 7.72 (d, J = 1.1 Hz, 1 H), 7.56–7.50 (m, 1 H), 7.46–7.37 (m, 2 H), 3.47 (s, 2 H), 2.28 (s, 6 H); 13C NMR (126 MHz, CDCl3) δ 165.49, 152.60, 142.77, 136.00, 134.23, 133.04, 131.91, 128.06, 126.18, 125.33, 123.40, 121.87, 121.33, 63.09, 45.27; MALDIMS m/z (rel. intensity) 347/349 (MH)+; HRESIMS calcd for C16H15BrN2S (MH)+ 347.0211, found 347.0212.
General procedures for ligand-free Suzuki coupling
The bromo compounds (6a–d, 7) (50 mg) were mixed with boronic acid derivatives (1.1 equivalents), Pd2(dba)3 (10 mol%) and sodium carbonate (2 equivalents) in an oven-dried round-bottom flask. Dioxane (6 mL) and water (3 mL) were then added to the mixture. Argon was bubbled through the reaction mixture for 5 min and then the mixture was heated at reflux for 4 h under argon. After cooling down to room temperature, the reaction mixture was diluted with ethyl acetate (15 mL) and then the organic layer was separated, washed with water (2 × 15 mL) and brine (1 × 15 mL), dried with anhydrous sodium sulfate and evaporated to give a residue. The residue was purified with column chromatography, using EtOAc–hexanes (2:8) or CHCl3–MeOH (9:1), to give the coupling products (8a–o).
5′-Amino-2′-(benzo[d]thiazol-2-yl)-[1,1′-biphenyl]-4-ol (8a).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (43 mg, 83%): mp 215–217 °C. IR (thin film) 3451, 3349, 1637, 1596, 1558, 1516, 1454, 1433, 1310, 1260, 1222, 1149, 1086 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.55 (s, 1 H), 7.86–7.78 (m, 3 H), 7.36 (t, J = 7.6 Hz, 1 H), 7.22 (t, J = 7.6 Hz, 1 H), 7.03 (d, J = 8.4 Hz, 2 H), 6.74 (d, J = 8.4 Hz, 2 H), 6.61 (dd, J = 8.6, 2.2 Hz, 1 H), 6.43 (d, J = 2.2 Hz, 1 H), 5.78 (s, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 168.1, 157.2, 152.52, 151.03, 143.32, 135.78, 131.55, 131.26, 131.08, 126.02, 124.44, 122.11, 121.71, 119.74, 115.66, 112.88; MALDIMS m/z (rel. intensity) 319 (MH)+; HRESIMS calcd for C19H14N2OS (MH)+ 319.0900, found 319.0900; HPLC purity, 98.31% (MeOH–H2O, 85:15).
2′-(Benzo[d]thiazol-2-yl)-5′-methyl-[1,1′-biphenyl]-3-ol (8b).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (48 mg, 92%): mp 203–205 °C. IR (thin film) 3062, 1604, 1576, 1483, 1454, 1431, 1312, 1214, 1177, 1158, 1125 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.47 (s, 1 H), 7.97 (d, J = 8.1 Hz, 1 H), 7.94–7.89 (m, 2 H), 7.44 (t, J = 7.7 Hz, 1 H), 7.33 (t, J = 7.0 Hz, 2 H), 7.20 (s, 1 H), 7.13 (t, J = 7.8 Hz, 1 H), 6.76–6.71 (m, 1 H), 6.70–6.61 (m, 2 H), 2.38 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 167.31, 157.59, 152.54, 141.59, 141.35, 140.62, 136.31, 131.57, 130.33, 129.90, 129.54, 128.85, 126.47, 125.41, 123.04, 122.19, 120.71, 116.83, 115.34, 21.15; MALDIMS m/z (rel. intensity) 318 (MH)++; HRESIMS calcd for C20H15NOS (MH)+ 318.0947, found 318.0948; HPLC purity, 95.25% (MeOH–H2O, 85:15).
Compound 8b was also prepared from the isolated palladium complex 9 as follows. The Pd complex 9 (20 mg, 0.05 mmol) was mixed with 3-hydroxyphenyl boronic acid (13 mg, 0.1 mmol) and sodium carbonate (15 mg, 0.15 mmol) in a round-bottom flask and then dioxane (3 mL) and water (1.5 mL) were added. Argon gas was bubbled into the solution for 2 min and then the flask was evacuated and refilled with argon twice. The reaction mixture was heated at reflux for 2 h and then cooled to room temperature, diluted with water (5 mL), and extracted with EtOAc (3 × 10 mL). The organic layer was dried with anhydrous sodium sulfate, evaporated, and the residue was purified with column chromatography to afford 8b as a white solid (13 mg, 84%). The spectral data were identical to compound 8b prepared by general procedures described above.
2′-(Benzo[d]thiazol-2-yl)-5′-methyl-[1,1′-biphenyl]-4-sulfonamide (8c).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (49 mg, 78%): mp 206–210 °C. IR (thin film) 3291, 3033, 1596, 1454, 1428, 1393, 1341, 1317, 1259, 1158, 1094 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 7.97–7.92 (m, 2 H), 7.88 (d, J = 7.9 Hz, 1 H), 7.76 (d, J = 8.2 Hz, 2 H), 7.49–7.43 (m, 3 H), 7.42–7.33 (m, 4 H), 7.29 (s, 1 H), 2.41 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 166.87, 152.85, 143.66, 140.98, 139.97, 136.05, 131.93, 130.87, 130.43, 129.56, 126.69, 126.04, 125.64, 123.21, 122.32, 21.16; MALDIMS m/z (rel. intensity) 381 (MH)+; HRESIMS calcd for C20H16N2O2S2 (MH)+ 381.0725, found 381.0724; HPLC purity, 98.75% (MeOH–H2O, 85:15).
2-(5-Methyl-4′-(methylsulfonyl)-[1,1′-biphenyl]-2-yl)benzo[d]thiazole (8d).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (56 mg, 89%): mp 182–184 °C. IR (thin film) 2921, 2851, 1664, 1595, 1434, 1304, 1278, 1148, 1085 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 7.99 (d, J = 7.8 Hz, 1 H), 7.96–7.86 (m, 4 H), 7.56 (d, J = 8.4 Hz, 2 H), 7.51–7.29 (m, 4 H), 3.24 (s, 3 H), 2.44 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 166.75, 152.93, 146.37, 140.60, 139.36, 139.28, 136.12, 131.32, 130.80, 130.68, 129.85, 129.41, 127.19, 126.08, 125.06, 123.14, 121.33, 44.48, 21.28; MALDIMS m/z (rel. intensity) 380 (MH)+; HRESIMS calcd for C21H17NOS2 (MH)+ 380.0773, found 380.0774; HPLC purity, 99.89% (MeOH–H2O, 85:15).
2-(Benzo[d]thiazol-2-yl)-[1,1′-biphenyl]-4-amine (8e).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as yellow powder (49 mg, 99%): mp 148–152 °C. IR (thin film) 3356, 3210, 1593, 1556, 1431, 1335, 1258, 1137 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 7.87–7.80 (m, 2 H), 7.76 (t, J = 9.6 Hz, 1 H), 7.40–7.31 (m, 4 H), 7.27–7.17 (m, 3 H), 6.67 (dd, J = 8.5, 2.3 Hz, 1 H), 6.48 (d, J = 2.3 Hz, 1 H), 5.85 (s, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 167.86, 152.61, 151.09, 143.11, 140.99, 135.67, 134.03, 131.77, 129.85, 128.74, 128.22, 127.76, 126.17, 124.56, 122.23, 121.73, 119.54, 115.52, 113.21; MALDIMS m/z (rel. intensity) 303 (MH)+; HRESIMS calcd for C19H14N2S (MH)+ 303.0950, found 303.0953; HPLC purity, 95.96% (MeOH–H2O, 85:15).
2′-(Benzo[d]thiazol-2-yl)-5′-methyl-[1,1′-biphenyl]-4-ol (8f).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (45 mg, 87%): mp 201–203 °C. IR (thin film) 2917, 1608, 1579, 1514, 1455, 1428, 1338, 1275, 1256, 1214, 1168, 1097 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.58 (s, 1 H), 7.96 (d, J = 8.1 Hz, 1 H), 7.91 (d, J = 8.0 Hz, 2 H), 7.44 (dd, J = 11.3, 4.1 Hz, 1 H), 7.35–7.27 (m, 2 H), 7.18 (s, 1 H), 7.06 (d, J = 8.4 Hz, 2 H), 6.73 (d, J = 8.5 Hz, 2 H), 2.37 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 167.53, 157.47, 152.42, 141.62, 140.51, 136.24, 131.90, 131.22, 130.41, 130.26, 129.64, 128.42, 126.43, 125.37, 122.97, 122.16, 115.73, 21.16; MALDIMS m/z (rel. intensity) 318 (MH)+; HRESIMS calcd for C20H15NOS (MH)+ 318.0947, found 318.0947; HPLC purity, 97.94% (MeOH–H2O, 85:15).
5′-Amino-2′-(benzo[d]thiazol-2-yl)-[1,1′-biphenyl]-3-ol (8g).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (44 mg, 85%): mp 198–201 °C. IR (thin film) 3460, 3374, 2918, 1625, 1594, 1449, 1429, 1316, 1258 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.46 (s, 1 H), 7.90–7.75 (m, 3 H), 7.36 (dd, J = 11.3, 4.1 Hz, 1 H), 7.23 (t, J = 7.6 Hz, 1 H), 7.15 (t, J = 7.8 Hz, 1 H), 6.75 (dd, J = 8.0, 1.8 Hz, 1 H), 6.69–6.58 (m, 3 H), 6.45 (d, J = 2.2 Hz, 1 H), 5.83 (s, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 167.91, 157.54, 152.55, 151.01, 143.22, 142.14, 135.83, 131.65, 129.84, 126.12, 124.51, 122.20, 121.74, 120.61, 119.49, 116.73, 115.24, 113.14; MALDIMS m/z (rel. intensity) 319 (MH)+; HRESIMS calcd for C19H14N2OS (MH)+ 319.0900, found 319.0901; HPLC purity, 96.24% (MeOH–H2O, 85:15).
2-(Benzo[d]thiazol-2-yl)-4′-methoxy-[1,1′-biphenyl]-4-amine (8h).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as yellow powder (53 mg, 97%): mp 175–177 °C. IR (thin film) 3357, 1598, 1558, 1505, 1452, 1431, 1336, 1256, 1241, 1172, 1027 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 7.90 (dd, J = 8.3, 4.0 Hz, 2 H), 7.84 (t, J = 15.4 Hz, 1 H), 7.42 (t, J = 7.5 Hz, 1 H), 7.28 (t, J = 7.5 Hz, 1 H), 7.23 (d, J = 8.5 Hz, 2 H), 6.98 (d, J = 8.5 Hz, 2 H), 6.71 (dd, J = 8.5, 2.2 Hz, 1 H), 6.52 (d, J = 2.1 Hz, 1 H), 5.88 (s, 2 H), 3.81 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 168.59, 159.92, 153.13, 151.63, 143.42, 136.29, 133.55, 132.25, 131.65, 126.68, 125.05, 122.73, 122.32, 120.27, 116.39, 114.76, 113.59, 55.96; MALDIMS m/z (rel. intensity) 333 (MH)+; HRESIMS calcd for C20H16N2OS (MH)+ 333.1056, found 333.1057; HPLC purity, 95.88% (MeOH–H2O, 85:15).
2′-(Benzo[d]thiazol-2-yl)-5′-((dimethylamino)methyl)-[1,1′-biphenyl]-4-ol (8i).
This compound was prepared using the general procedures described for Suzuki coupling and it was isolated as a white solid (40 mg, 75%): mp 227–229 °C. IR (thin film) 2957, 1608, 1579, 1505, 1460, 1433, 1409, 1277, 1231 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.61 (s, 1 H), 7.98 (dd, J = 8.0, 3.6 Hz, 2 H), 7.94 (d, J = 7.9 Hz, 1 H), 7.44 (dd, J = 17.2, 8.7 Hz, 2 H), 7.33 (dd, J = 16.7, 9.5 Hz, 2 H), 7.07 (d, J = 8.5 Hz, 2 H), 6.73 (d, J = 8.5 Hz, 2 H), 3.57 (s, 2 H), 2.24 (s, 6 H); 13C NMR (126 MHz, DMSO-d6) δ 167.40, 157.77, 152.51, 141.62, 136.28, 131.57, 131.26, 131.10, 130.33, 128.03, 126.49, 125.48, 123.06, 122.22, 115.77, 62.84, 45.21; MALDIMS m/z (rel. intensity) 361 (MH)+; HRESIMS calcd for C19H14N2OS (MH)+ 361.1369, found 361.1369; HPLC purity, 97.26% (MeOH–H2O, 85:15).
2′-(Benzo[d]thiazol-2-yl)-5′-((dimethylamino)methyl)-[1,1′-biphenyl]-3-ol (8j).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as a white solid (42 mg, 78%): mp 235–237 °C. IR (thin film) 2917, 1605, 1575, 1476, 1455, 1408, 1357, 1297, 1221, 1019 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.60 (s, 1 H), 7.96 (dd, J = 22.1, 7.9 Hz, 3 H), 7.47–7.38 (m, 2 H), 7.34 (t, J = 7.6 Hz, 1 H), 7.29 (s, 1 H), 7.07 (d, J = 8.4 Hz, 2 H), 6.73 (d, J = 8.4 Hz, 2 H), 3.52 (s, 2 H), 2.21 (s, 6 H); 13C NMR (126 MHz, DMSO-d6) δ 167.18, 157.62, 152.57, 141.86, 141.50, 141.30, 136.33, 131.10, 130.82, 130.36, 129.93, 128.33, 126.51, 125.50, 123.10, 122.23, 120.73, 116.81, 115.37, 62.99, 45.39; MALDIMS m/z (rel. intensity) 361 (MH)+; HRESIMS calcd for C22H20N2OS (MH)+ 361.1369, found 361.1369; HPLC purity, 96.85% (MeOH–H2O, 85:15).
4-(3-(Benzo[d]thiazol-2-yl)pyridin-2-yl)phenol (8k).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as a white solid (43 mg, 82%): mp 187–189 °C. IR (thin film) 3251, 1610, 1517, 1497, 1448, 1430, 1315, 1272, 1211 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 9.53 (s, 1 H), 8.65 (dd, J = 4.6, 1.3 Hz, 1 H), 8.10–8.03 (m, 1 H), 7.83 (dd, J = 7.8, 1.3 Hz, 1 H), 7.71–7.64 (m, 1 H), 7.57 (dd, J = 7.8, 4.6 Hz, 1 H), 7.45–7.35 (m, 2 H), 7.14 (d, J = 8.5 Hz, 2 H), 6.72 (d, J = 8.5 Hz, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 169.08, 157.43, 153.78, 148.10, 147.81, 140.34, 137.00, 135.80, 130.86, 129.34, 126.52, 126.29, 125.36, 123.66, 122.36, 115.16; MALDIMS m/z (rel. intensity) 305 (MH)+; HRESIMS calcd for C18H12N2OS (MH)+ 305.0743, found 305.0745; HPLC purity, 95.15% (MeOH–H2O, 85:15).
4-(3-(Benzo[d]thiazol-2-yl)pyridin-2-yl)benzenesulfonamide (8l).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as a white solid (41 mg, 65%): mp 213–215 °C. IR (thin film) 2920, 2851, 1729, 1556, 1507, 1456, 1423, 1336, 1155, 1094 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.77 (dd, J = 4.7, 1.4 Hz, 1 H), 8.12–8.05 (m, 1 H), 7.91 (dd, J = 7.8, 1.4 Hz, 1 H), 7.81 (d, J = 8.3 Hz, 2 H), 7.66 (dd, J = 7.8, 4.7 Hz, 1 H), 7.59–7.51 (m, 3 H), 7.43–7.37 (m, 4 H); 13C NMR (126 MHz, DMSO-d6) δ 168.74, 153.75, 149.38, 147.38, 143.26, 142.63, 140.43, 135.73, 135.38, 130.13, 126.60, 126.33, 125.54, 123.74, 122.46; MALDIMS m/z (rel. intensity) 367 (MH)+; HRESIMS calcd for C18H13N3O2S2 (MH)+ 368.0522, found 368.0520; HPLC purity, 99.53% (MeOH–H2O, 85:15).
2-(2-(4-(Methylsulfonyl)phenyl)pyridin-3-yl)benzo[d]thiazole (8m).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as a white solid (58 mg, 92%): mp 189–192 °C. IR (thin film) 3050, 3025, 2923, 1597, 1555, 1509, 1391, 1447, 1423, 1293, 1278, 1146, 1121, 1088 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.79 (d, J = 4.6 Hz, 1 H), 8.11–8.04 (m, 1 H), 7.92 (d, J = 8.2 Hz, 3 H), 7.69–7.60 (m, 3 H), 7.56–7.51 (m, 1 H), 7.43–7.33 (m, 2 H), 3.27 (s, 3 H); 13C NMR (126 MHz, DMSO-d6) δ 168.74, 153.74, 149.57, 147.45, 144.58, 140.33, 140.14, 135.62, 135.12, 130.59, 126.84, 126.67, 126.36, 125.51, 123.76, 122.45, 43.95; MALDIMS m/z (rel. intensity) 366 (MH)+; HRESIMS calcd for C19H14N2O2S2 (MH)+ 367.0569, found 367.0568; HPLC purity, 97.56% (MeOH–H2O, 85:15).
4-(3-(Benzo[d]thiazol-2-yl)-5-chloropyridin-2-yl)benzenesulfonamide (8n).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as yellowish white powder (38 mg, 62%): mp 205–207 °C. IR (thin film) 3451, 3348, 3254, 1634, 1596, 1456, 1429, 1310, 1261, 1222, 1182, 1114, 1098, 1013 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.85 (d, J = 2.2 Hz, 1 H), 8.13–8.07 (m, 2 H), 7.81 (d, J = 8.3 Hz, 2 H), 7.59–7.54 (m, 3 H), 7.44 (d, J = 6.7 Hz, 2 H), 7.43–7.39 (m, 2 H); 13C NMR (126 MHz, DMSO-d6) δ 167.57, 153.71, 147.90, 146.19, 143.90, 141.18, 139.65, 136.62, 135.63, 132.48, 130.22, 126.73, 126.51, 125.53, 123.80, 122.51; MALDIMS m/z (rel. intensity) 401/403 (MH)+; HRESIMS calcd for C18H12ClN3O2S2 (MH)+ 402.0132, found 402.0131; HPLC purity, 97.26% (MeOH–H2O, 85:15).
2-(5-Chloro-2-(4-(methylsulfonyl)phenyl)pyridin-3-yl)benzo[d]thiazole (8o).
This compound was prepared using the general procedure described for Suzuki coupling and it was isolated as yellowish white powder (46 mg, 74%): mp 168–169 °C. IR (thin film) 2919, 1712, 1598, 1512, 1425, 1299, 1146, 1117, 1087, 1030, 1016 cm−1; 1H NMR (500 MHz, DMSO-d6) δ 8.87 (d, J = 2.2 Hz, 1 H), 8.13 (d, J = 2.2 Hz, 1 H), 8.11–8.07 (m, 1 H), 7.93 (d, J = 8.3 Hz, 2 H), 7.67 (d, J = 8.3 Hz, 2 H), 7.56–7.50 (m, 1 H), 7.40 (qd, J = 7.2, 5.7 Hz, 2 H), 3.28 (s, 3 H); 13C NMR (126 MHz, CDCl3) δ 166.62, 153.65, 147.89, 146.45, 143.42, 139.56, 138.62, 135.72, 132.48, 131.75, 130.25, 127.09, 126.03, 123.89, 121.50, 44.54; MALDIMS m/z (rel. intensity) 400/402 (MH)+; HRESIMS calcd for C19H13ClN2O2S2 (MH)+ 401.0179, found 401.0179; HPLC purity, 98.68% (MeOH–H2O, 85:15).
Palladium complex of 2-(2-bromo-4-methylphenyl)benzo[d]thiazole (9).
Compound 6b (30 mg, 0.1 mmol) was mixed with Pd2(dba)3 (90 mg, 0.1 mmol) in a glass vial and the vial was capped and evacuated and refilled with Ar two times. Anhydrous toluene (3 mL) was added via a syringe to the vial and the reaction mixture was heated up to 80 °C and stirred at that temperature under Ar for 2 h. The TLC indicated the consumption of the starting materials and the appearance of a baseline yellow spot. The reaction mixture was filtered and the yellow solid obtained was washed with methylene chloride and acetone twice to remove any dba residues. The solid was then dried in vacuo to give the Pd complex of 9 as a golden coloured solid (39 mg, 99%): mp >300 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.75 (m, 1 H), 8.24 (d, J = 7.6 Hz, 1 H), 8.02 (s, 1 H), 7.65–7.50 (m, 3 H), 7.31 (s, 1 H), 2.26 (s, 3 H); MALDIMS (2,5-dihydroxybenzoic acid matrix) MH+m/z (rel. intensity) 406 (2), 408 (20), 409 (41), 410 (72), 411 (50), 412 (100), 413 (18), 414 (67), 415 (11), 416 (22), 417 (4), 418 (2).
2-([1,1′-Biphenyl]-2-yl)benzo[d]oxazole (11).
This compound was prepared from 10 according to the general procedures described for compounds 8a–o and was isolated as white powder (0.047 g, 95%). The compound showed very similar data as reported previously.20
Biology
The determination of COX-1 and COX-2 enzyme inhibitory activity was performed using the kit supplied by Cayman Chemical (Ann Arbor, MI, USA). The kit depends on assaying the peroxidase activity of the enzymes and the colorimetric monitoring of the appearance of oxidized N,N,N,N-tetramethyl-p-phenylenediamine (TMPD) at 590 nm. In a transparent 96-well plate the enzymes [COX-1 (bovine) and COX-2 (human)] were diluted using the assay buffer supplied with the kit. The enzymes were then mixed with heme and the inhibitors (dissolved in DMSO at the required concentrations) and incubated for 5 min at 25 °C. The colorimetric substrate was then added to the reaction wells and the reaction was directly initiated by the addition of arachidonic acid. After 2 min the plate was read using a BioTek Synergy 4 spectrophotometer at 590 nm. The IC50 values were determined by plotting the % inhibition of the enzyme activity against the inhibitor concentration. The data presented are the mean of three different experiments.
Molecular modeling
The X-ray crystal structures of COX-1 (PDB ID: 3KK6) and COX-2 (PDB ID: 3LN1) were obtained from the protein data bank. The protein structure was cleaned, inspected for errors and missing residues, hydrogens were added, and the water molecules were removed using Accelrys Discovery studio 2.5 software. The structures of the inhibitors were constructed using ChemBioDraw Ultra 13 and saved in the SDF file format and were corrected and optimized using Accelrys Discovery studio 2.5 software. GOLD 4.1 was used for docking with the default parameters except that the iterations were increased to 300000 and the early termination option was disabled. The centroids of the binding sites were defined by the ligands in the co-crystal structures. The top 10 docking poses per ligand were inspected visually following the docking runs. Energy minimizations were performed for selected ligand poses. The CHARMM force field was utilized within the Accelrys Discovery Studio 2.5 for energy minimization.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors gratefully acknowledge support from the Purdue University Center for Cancer Research, NIH grant P30 CA023168. Mohamed S. A. Elsayed was partially supported by the McKeehan Graduate Scholarship in Pharmacy award.
References
- L. Ferrero-Miliani, O. H. Nielsen, P. S. Andersen and S. E. Girardin, Clin. Exp. Immunol., 2007, 147, 227–235 CAS.
- D. Laveti, M. Kumar, R. Hemalatha, R. Sistla, V. G. M. Naidu, V. Talla, V. Verma, N. Kaur and R. Nagpal, Inflammation Allergy: Drug Targets, 2013, 12, 349–361 CrossRef CAS PubMed.
- S. M. Lucas, N. J. Rothwell and R. M. Gibson, Br. J. Pharmacol., 2006, 147, S232–S240 CrossRef CAS PubMed.
- E. Ricciotti and G. A. FitzGerald, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 986–1000 CrossRef CAS PubMed.
- I. Tabas and C. K. Glass, Science, 2013, 339, 166–172 CrossRef CAS PubMed.
- Z. A. Marcum and J. T. Hanlon, Ann. Longterm Care, 2010, 10, 24–27 Search PubMed.
- L. J. Crofford, Arthritis Res. Ther., 2013, 15(Suppl 3), S2 Search PubMed.
- C. K. Ong, P. Lirk, C. H. Tan and R. A. Seymour, Clin. Med. Res., 2007, 5, 19–34 CrossRef CAS PubMed.
- T. Grosser, S. Fries and G. A. FitzGerald, J. Clin. Invest., 2006, 116, 4–15 CAS.
- R. J. Flower, Nat. Rev. Drug Discovery, 2003, 2, 179–191 CrossRef CAS PubMed.
- B. Liu, L. Y. Qu and S. G. Yan, Cancer Cell Int., 2015, 15, 6 CrossRef PubMed.
- D. Mazhar, R. Ang and J. Waxman, Br. J. Cancer, 2006, 94, 346–350 CrossRef CAS PubMed.
- N. Kumar, S. Drabu and S. C. Mondal, Arab. J. Chem., 2013, 6, 1–23 CrossRef CAS.
- C. H. Trepanier and N. W. Milgram, J. Alzheimer's Dis., 2010, 21, 1089–1099 CrossRef CAS PubMed.
- A. L. Bartels and K. L. Leenders, Curr. Neuropharmacol., 2010, 8, 62–68 CrossRef CAS PubMed.
- S. Aid and F. Bosetti, Biochimie, 2011, 93, 46–51 CrossRef CAS PubMed.
- D. L. DeWitt, Mol. Pharmacol., 1999, 55, 625–631 CAS.
- I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- K. Seth, P. Purohit and A. K. Chakraborti, Org. Lett., 2014, 16, 2334–2337 CrossRef CAS PubMed.
- K. W. Quasdorf, A. Antoft-Finch, P. Liu, A. L. Silberstein, A. Komaromi, T. Blackburn, S. D. Ramgren, K. N. Houk, V. Snieckus and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 6352–6363 CrossRef CAS PubMed.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2015, 5, 29878–29884 RSC.
- S. Meghdadi, M. Amirnasr and P. C. Ford, Tetrahedron Lett., 2012, 53, 6950–6953 CrossRef CAS.
- J. K. Stille and K. S. Y. Lau, Acc. Chem. Res., 1977, 10, 434–442 CrossRef CAS.
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich, B. Al-Lazikani and J. P. Overington, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed.
- R. Clarke, S. Derry and R. A. Moore, Cochrane Database Syst. Rev., 2014 DOI:10.1002/14651858.CD004309.pub4.
- D. Riendeau, M. D. Percival, C. Brideau, S. Charleson, D. Dube, D. Ethier, J. P. Falgueyret, R. W. Friesen, R. Gordon, G. Greig, J. Guay, J. Mancini, M. Ouellet, E. Wong, L. Xu, S. Boyce, D. Visco, Y. Girard, P. Prasit, R. Zamboni, I. W. Rodger, M. Gresser, A. W. Ford-Hutchinson, R. N. Young and C. C. Chan, J. Pharmacol. Exp. Ther., 2001, 296, 558–566 CAS.
- A. Zarghi and S. Arfaei, Iran. J. Pharm. Res., 2011, 10, 655–683 CAS.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob02386c |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Siran
Chang
and
Mark
Cushman
,
Siran
Chang
and
Mark
Cushman

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
: