
Searching for avidity by chemical ligation of combinatorially self-assembled DNA-encoded ligand libraries†
Stefan
Matysiak
*a,
Klaus
Hellmuth
a,
Afaf H.
El-Sagheer
 bc,
Arun
Shivalingam
b,
Yavuz
Ariyurek
d,
Marco
de Jong
a,
Martine J.
Hollestelle
e,
Ruud
Out
a and
Tom
Brown
*b
bc,
Arun
Shivalingam
b,
Yavuz
Ariyurek
d,
Marco
de Jong
a,
Martine J.
Hollestelle
e,
Ruud
Out
a and
Tom
Brown
*b
aPiculet-Biosciences BV, Galileiweg 8, 2333BD Leiden, The Netherlands. E-mail: S.Matysiak@gmx.net
bDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk
cChemistry Branch, Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
dLeiden Genome Technology Center, Leiden University Medical Center, Leiden, The Netherlands
eDep. Immunophathology and Blood Coagulation, Sanquin Diagnostic Services, Amsterdam, The Netherlands
First published on 7th December 2017
Abstract
DNA encoded ligands are self-assembled into bivalent complexes and chemically ligated to link their identities. To demonstrate their potential as a combinatorial screening platform for avidity interactions, the optimal bivalent aptamer design (examplar ligands) for human alpha-thrombin is determined in a single round of selection and the DNA scaffold replaced with minimal impact on the final design.
Nucleic acid aptamers are useful ligands in biotechnological, diagnostic and therapeutic application and they possess molecular recognition properties that rival those of antibodies. Aptamers can be engineered to bind to various molecular targets through repeated rounds of in vitro selection by SELEX (systematic evolution of ligands by exponential enrichment).1–3 They have several attractive features; they can be produced by chemical synthesis, they possess excellent storage properties, and can be designed to elicit little or no immunogenicity in therapeutic applications. However, they tend to bind less tightly to their substrates than antibodies.
To overcome this limitation efforts have been made to chemically modify aptamers or enhance their interaction via avidity effects. For example, many non-polymerase compatible modifications of thrombin binding aptamers have been reported,4 whilst two different thrombin exosite aptamers have been linked through carbon chains, poly(dA)/poly(dT) repeats, spacer phosphoramidites or sequence optimised spacers to enhance avidity.5–11 However, these processes generally require iterative synthesis, testing and optimisation.
A modular synthetic approach, independent of the nature of the ligand or spacer, for the discovery of high avidity bivalent binders is therefore very compelling. In this regard DNA is a versatile platform with robust and simple rules for self-assembly of two- and three-dimensional objects, some of which have been used to develop DNA-encoded12 multivalent ligands.13–20 Yet, as complexity rises, the use of hybridisation to a template to encode recovered ligand pair identify becomes more error-prone; linking the DNA-encoded ligands to give a direct read out by next generation sequencing (NGS) would be more powerful, especially if the combinatorial aspect of self-assembly can be retained.
We have incorporated the above design aspects into a DNA-based scaffolding platform that allows the generation of large libraries of homo- or hetero-bivalent ligand pairs from a relatively small number of precursor molecules (Fig. 1). Access to SABLC (Self Assembled Bivalent Ligand Complex) libraries requires the synthesis of a number “N” of individual DNA oligomers, each covalently attached to a specific potential binding sequence, e.g. a small sub-library of aptamer motifs.
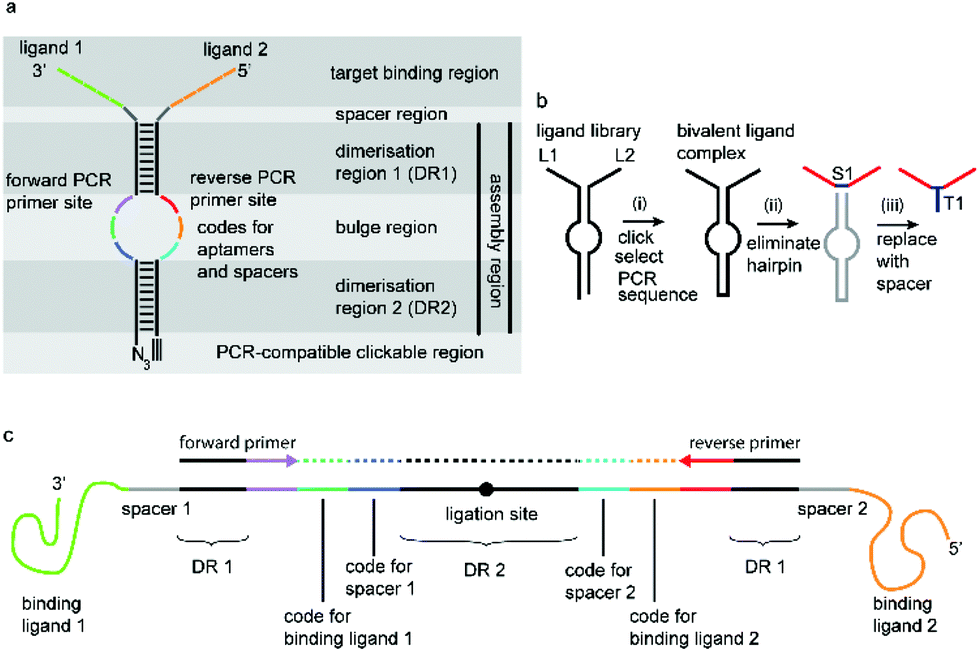 | ||
| Fig. 1 a: The general SABLC design: this comprises a binding region, an assembly region and optional spacer region in between. The assembly region is a linear mostly self-complementary DNA sequence, which forms a hairpin structure with an internal bulge. It functions first as a molecular scaffold via the dimerisation regions DR1 and DR2, and secondly serves as a rigid scaffold to stabilize the distance between the ligands and thirdly encodes all information about the binding and spacer region in the form of two separate short nucleotide sequences, opposite to each other in the bulge region. A key prerequisite is that exponential enzymatic amplification of the crosslinked assembly region must be possible. b: The general SABLC concept: (i) Firstly a library consisting of pairs of hybridized monovalent ligand–oligonucleotide conjugates is covalently crosslinked to generate a homo- or hetero-bivalent complex library. Thousands to millions of such complexes presenting individual combinations of ligands can be generated, only limited by the number of starting monovalent precursor molecules. Next, after selection, the best target-binding complexes can be amplified by PCR and sequenced to decode the ligands L1 and L2 and their spatial distance. (ii) Optionally, the DNA scaffold can subsequently be replaced by a spacer molecule S1 to simplify synthesis and to optimize the bioavailability of the complex e.g. as a therapeutic agent. (iii) The spacer can be linked to a tag T1 such as a fluorophore for diagnostic applications or an anchor group can be introduced for immobilisation on a solid support e.g. for chromatographic applications. c: SABLC from (a) in open form showing PCR primers and primer binding sites. Colour code as in (a). | ||
Mixing, self-assembly and chemical fixation of these precursor molecules (Scheme 1) in a single tube will result in N2 combinations. The benefit of the SABLC principle is not only in the relatively small number of starting molecules, but also in the fact that the ligand complexes can be derived from a diverse range of molecules which do not have to be read through by DNA polymerases during the decoding process. Therefore the platform can be designed to allow ligands such as peptides, carbohydrates and small molecules to be incorporated. Like in a classic SELEX selection step the target molecule can be immobilized on a magnetic bead and incubated with the library. Unbound library members are washed away and the remaining bound SABLCs are eluted under stringent conditions. Their embedded identifier sequence is then decoded by first amplifying the DNA scaffold by PCR followed by Next Generation Sequencing (NGS).
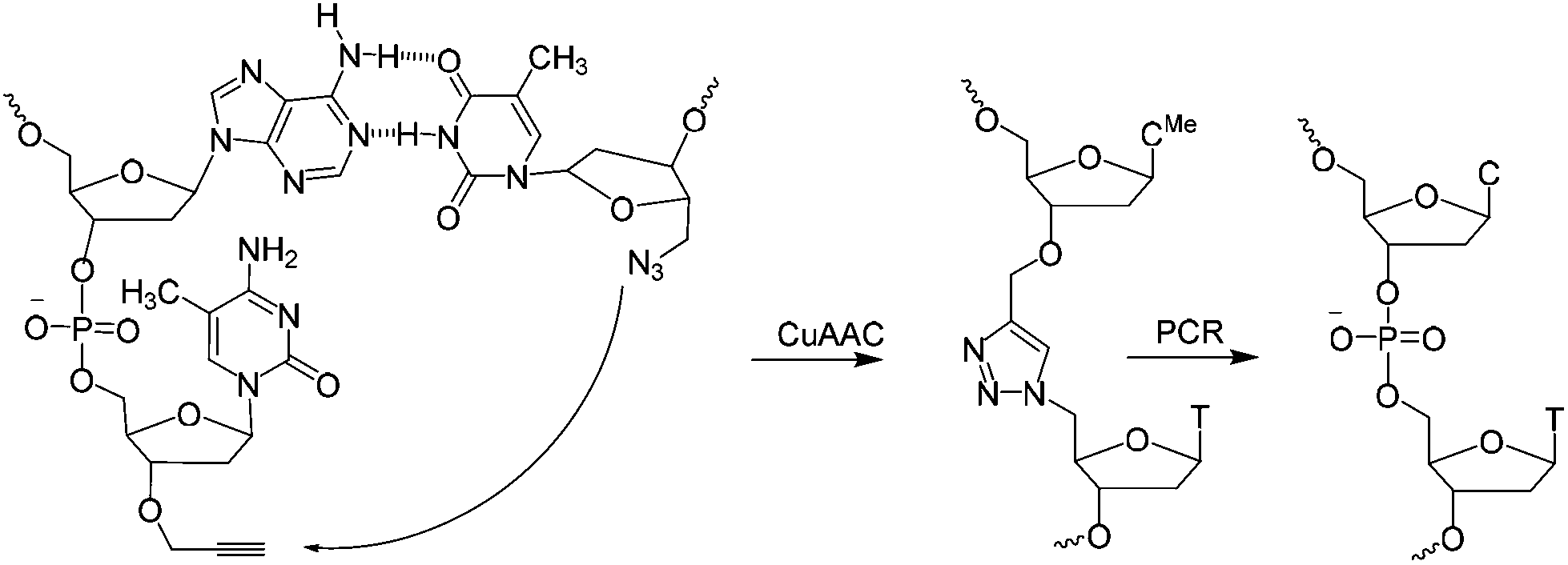 | ||
| Scheme 1 CuAAC mediated ligation between 3′-propargyl dmC and 5′-azide T across the dimerization region 2 at the end of the SABA DNA hairpin to form biocompatible triazole (ct) linkage, followed by PCR amplification of the ligated construct (see Fig. 1). | ||
After decoding the SABLC ligands and spacers, optional replacement of the scaffold can be carried out by synthesising analogues that include a short DNA hairpin loop, oligo dT or non-nucleosidic linkers to generate much smaller bivalent ligand molecules, e.g. for therapeutic applications (Fig. 1b). As DNA is simple to modify, visualization tags such as fluorophores or other labels can be readily attached. The SABL complex is then ready to be used in various applications including immuno-PCR.21
Prerequisites for a universal DNA scaffold are; (i) sufficient structural stability to maintain a fixed distance between the two attached ligands and (ii) the ability to denature during PCR to allow amplification and subsequent sequencing. A DNA hairpin of about 20 base pairs fits these criteria perfectly.
A short sequence, specific for each ligand is needed in each strand to code for the ligands and their corresponding separation. We reasoned that 7 nucleobases, which can code for 47 = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 ligand/spacer combinations, should be suitable. The scaffolding hairpin must also include a contiguous DNA sequence in each arm to function as a primer-binding site for PCR amplification and sequencing of the identifier codes for the ligands and spacers. Finally the scaffold should not be difficult or expensive to synthesize. Our chosen structure is shown in Fig. 1a. The target-binding regions of a SABL complex are a combination of two identical or different ligands. The distance between them is determined by the double-helical DNA scaffold and the two identical or different spacer molecules that are situated between the ligands and the dimerization region. This partially double stranded DNA complex, which is already quite stable under physiological conditions, is further stabilized by chemical ligation. This ensures that there is no possibility of cross-exchange between DNA strands during the subsequent selection step, so that the embedded information on the specific combination of ligands and their distance relative to each other is not lost during the target-binding, handling and decoding steps. To allow enzymatic DNA amplification for decoding, the two arms need to be covalently linked by means of a biocompatible linker so that a DNA polymerase can read through the two linked DNA strands during PCR. This is achieved via a Cu(I)-catalysed CuAAC reaction between an alkyne moiety at the 3′-terminus of each member of the first library (linked to a dmC residue for synthetic ease) and a 5′-azide group on each member of the second library (Scheme 1).22,23
384 ligand/spacer combinations, should be suitable. The scaffolding hairpin must also include a contiguous DNA sequence in each arm to function as a primer-binding site for PCR amplification and sequencing of the identifier codes for the ligands and spacers. Finally the scaffold should not be difficult or expensive to synthesize. Our chosen structure is shown in Fig. 1a. The target-binding regions of a SABL complex are a combination of two identical or different ligands. The distance between them is determined by the double-helical DNA scaffold and the two identical or different spacer molecules that are situated between the ligands and the dimerization region. This partially double stranded DNA complex, which is already quite stable under physiological conditions, is further stabilized by chemical ligation. This ensures that there is no possibility of cross-exchange between DNA strands during the subsequent selection step, so that the embedded information on the specific combination of ligands and their distance relative to each other is not lost during the target-binding, handling and decoding steps. To allow enzymatic DNA amplification for decoding, the two arms need to be covalently linked by means of a biocompatible linker so that a DNA polymerase can read through the two linked DNA strands during PCR. This is achieved via a Cu(I)-catalysed CuAAC reaction between an alkyne moiety at the 3′-terminus of each member of the first library (linked to a dmC residue for synthetic ease) and a 5′-azide group on each member of the second library (Scheme 1).22,23
As a first example we used specific nucleic acid aptamers as ligands, which are inhibitors of the blood coagulation factor thrombin. An initial test library was generated from a combination of 8 sequences; comprising ligands derived from aptamers TBA27 and G15D24,25 and mutated versions (Table 1). These motifs, which are known to bind to different exosites on thrombin, were either directly attached to the DNA scaffold or linked via a simple T6 or T12 spacer. Individual complementary oligonucleotide pairs were combined and clicked after hybridization to yield self-assembled bivalent aptamers (Table 1). Larger libraries could be made by a simultaneous chemical ligation of all pre-hybridized ligand combinations. These complexes were analysed and purified by polyacrylamide gel electrophoresis (PAGE). DNA sequencing was then performed to prove the sequence fidelity. The triazole linker did not lead to any nucleotide misincorporations, deletions or insertions (ESI Fig. S7†).
| ID no. | Name/description | Spacer | Other details |
|---|---|---|---|
| 1 | TBA27-c | T0 | 3′-Propyne dC |
| 2 | t-T0-G15D | T0 | 5′-Azido-T |
| 3 | t-T6-G15D | T6 | 5′-Azido-T |
| 4 | t-T12-G15D | T12 | 5′-Azido-T |
| 5 | TBA27s-c | T0 | 3′-Propyne dC |
| 6 | t-T12-G15Ds | T12 | 5-Azido-T |
| 7 (1 + 2) | TBA27-ct-T0-G15D | T0 | CuAAC ligated |
| 8 (1 + 3) | TBA27-ct-T6-G15D | T6 | CuAAC ligated |
| 9 (1 + 4) | TBA27-ct-T12-G15D | T12 | CuAAC ligated |
| 10 (1 + 6) | TBA27-ct-T12-G15Ds | T12 | CuAAC ligated |
| 11 (5 + 2) | TBA27s-ct-T0-G15D | T0 | CuAAC ligated |
| 12 (5 + 3) | TBA27s-ct-T6-G15D | T6 | CuAAC ligated |
| 13 (5 + 4) | TBA27s-ct-T12-G15D | T12 | CuAAC ligated |
| 14 (5 + 6) | TBA27s-ct-T12-G15Ds | T12 | CuAAC ligated |
| 15 | G15D | NA | |
| 16 | TBA27 | NA | |
| 17 | G15D-T0-TBA27 | T0 | Synthesised |
| 18 | G15D-T6-TBA27 | T6 | Synthesised |
| 19 | G15D-T12-TBA27 | T12 | Synthesised |
| 20 | TBA27-T0-G15D | T0 | Synthesised |
| 21 | TBA27-T6-G15D | T6 | Synthesised |
| 22 | TBA27-T12-G15D | T12 | Synthesised |
| 23 | Reference | NA | Reference |
A model self-assembled bivalent aptamer (SABA) library was prepared as follows: oligonucleotides 7–13 (each containing at least one thrombin aptamer) were mixed in 1:1 ratios and background DNA 14 (containing two scrambled thrombin aptamers) was added (Table 1). The final mixture comprised 99.125% background DNA (scrambled aptamers) and 0.125% of each of SABA sequences 7–13. The protein target (thrombin) was then immobilized onto magnetic beads.
Thrombin-coupled beads, negative control beads and the SABA test library were combined and the bound DNA was amplified by PCR followed by a second PCR to include the adapter sequences for the Illumina NGS instrument (details in the ESI section 4.vi., Fig. S12†). NGS is essential to provide sufficient sequence information to allow selection of the best binders from a single round of selection. SABA 8 and 9 with T6 and T12 spacers between the binding motifs were most abundant (>10-fold enrichment, Fig. 2). If no spacer was used (SABA 7) or one of the binding motifs is scrambled (SABA 10–13) the enrichment was far smaller (less than 10-fold), with active deselection of the construct when both motifs are scrambled (SABA-14). This trend is in accordance with the need for both motifs to bind the two exosites and the ∼4.5 nm separation between the exosites (Fig. 3),26,27 which is comparable to the DNA hairpin loop and T6 spacer size of SABA 8 and less than the T12 spacer size of SABA 9.
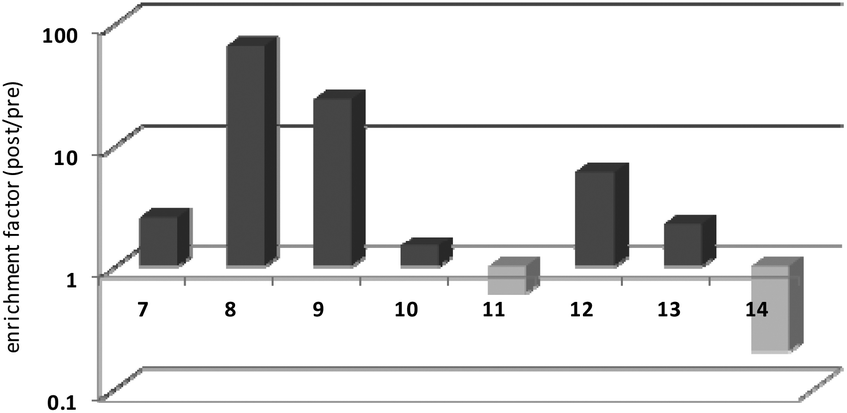 | ||
| Fig. 2 SABA capture experiment: results of Illumina sequencing of the model SABA library before and after selection (enrichment). Burrows–Wheeler alignment and mapping; NGS was carried out on an Illumina MiSeq instrument. Data are shown in logarithmic scale, normalized. | ||
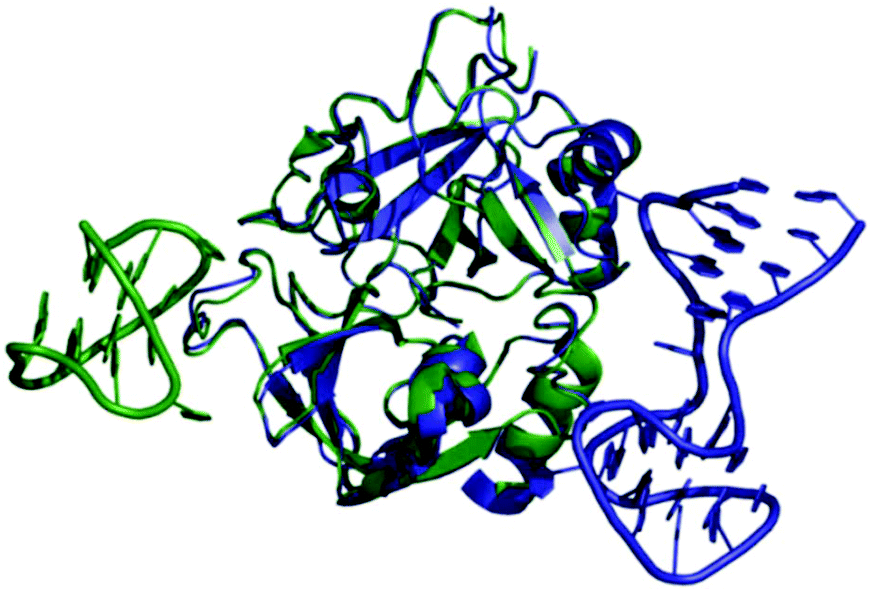 | ||
| Fig. 3 Overlay of X-ray crystal structures of human alpha-thrombin in complex with G15D aptamer bound at exosite I (green; PDB ID 4DIH); and TBA27 aptamer bound at exosite II (blue; PDB ID 4I7Y). The distance between the aptamers is approx. 4.5 nm. | ||
Next, activity was measured using aPTT values determined in the presence of various aptamer constructs using normal human plasma (a pooled plasma from a pool of more than 32 healthy adult donors derived from the Sanquin Bloodbank, Fig. 4).28 The aPTT value of the blank (buffer/H2O, below 30 s) was identical to the non-thrombin binding sequence 23 (ΔaPTT ∼0 s relative to control). Consistent with NGS results, SABA 8 and 9 (ΔaPTT > 20 s) outperformed those with a T0 spacer (SABA 7), one of the two motifs being scrambled (SABA 11 and 12) or only the monomeric motif (Table ID 15/16; all ΔaPTT ∼5 s). Interestingly, SABA 10 and 13, with only functional TBA27 and G15D motifs respectively but T12 spacers, performed better than expected (ΔaPTT ∼15 s). This is possibly due to unstructured polyanionic T12 spacer being of sufficient size to interact with thrombin (akin to heparin interactions with thrombin), an assertion supported by the greater than blank ΔaPTT of SABA 14 (∼5 s), where both aptamers are scrambled but the T12 spacer is retained.
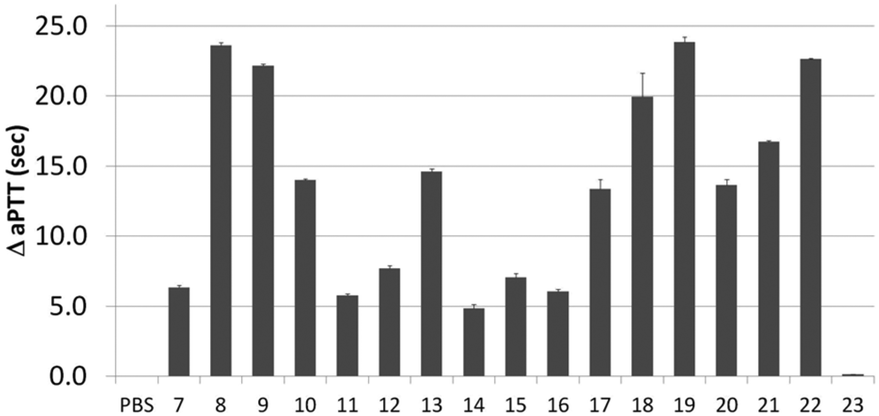 | ||
| Fig. 4 Activated Partial Thromboplastin time (aPTT) values (background subtracted) for each individual or ligand composition. | ||
To refine the bivalent aptamers, DNA scaffolds were replaced with no (T0), T6 or T12 spacers between the two aptamer motifs (ID 17, 18, 19 respectively). For ID 20–22 the order of the aptamer motifs was changed relative ID 17–19 (TBA27 at 5′-side of G15D) whilst the spacers stayed the same. In both cases similar aPTT values for the same linker lengths were observed but with some differences relative to the SABA 7, 8 and 9. Firstly, the T6 spaced aptamers (18 and 21) perform slightly worse than their SABA 8 counterpart, whilst T12 spaced aptamers are unaffected. This is possibly due to the absence of the hairpin scaffold reducing the separation of the ligands by ∼2 nm, which is compensated for by the additional length of the T12, but not T6, spacer. Secondly, T0 spaced aptamers (17 and 20) perform better than their monomeric motifs or SABA 7 counterpart suggesting the display orientation of the aptamers by the duplex in SABA 7 is important when a linker is completely omitted. In such cases, SABA refinement may require minimal duplex motifs (e.g. very stable 7 base hairpins29).
Finally, disassociation rates as a proxy for avidity were determined for monomeric (15 and 16) and bivalent aptamers (20–22) using surface plasmon resonance (Fig. 5). Unlike association rates, disassociation rates are concentration independent making their modelling, interpretation and comparison between aptamers far simpler – the slower the decay rate and the larger its fractional contribution the stronger the avidity effect. To ensure concentration independence, aptamer disassociation from the immobilised thrombin was normalised to the start point of decay and averaged over various aptamer concentration injections (1000 to 7.8 nM); the standard deviation was less than 0.04 normalised response units. All sequences required a bi-exponential decay to accurately fit the data, with one component of similar rate and fractional contribution (kd,1 ∼0.01–0.02 s−1, ∼0.12) shared by all sequences, suggesting it is a non-specific interaction with the immobilised thrombin. The second component clearly demonstrates the potency of the bivalency effect as the disassociation of monomeric motifs is at least an order of magnitude faster than bivalent complexes (kd,2, Fig. 5). Notably the trends for 20–22 are consistent with aPPT results; kd,2 of 22 (T12 spacer) is slightly slower than 21 (T6), which in turn is slower than 20 (T0) mirroring the greater activity of 22 cf. 21 and 20 in aPPT. These trends were retained upon increasing the immobilised thrombin ∼5-fold (Fig. S14†), suggesting that of the tested designs T12 spacing of G15D and TBA27 is optimal.
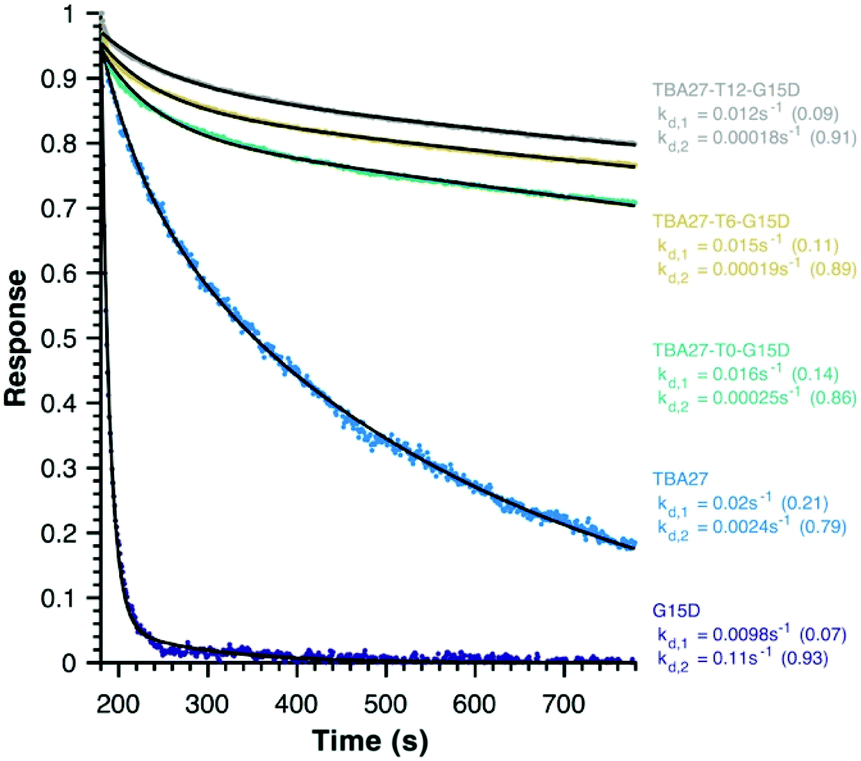 | ||
| Fig. 5 Average normalised decay curves for monomeric (ID 15 and 16) and bivalent aptamers (ID 20–22) from surface plasmon resonance. The aptamers and raw data are colour-coded, with the modelled fit to a bi-exponential decay in black. The fractional contribution of each disassociation component is in brackets. Immobilized biotinylated human alpha-thrombin = 615.5 RU. | ||
In summary, we have developed a self-assembled DNA scaffold platform that is designed to allow the generation of large libraries of bivalent ligand complexes (SABLCs) from a relatively small number of starting molecules comprised of single stranded DNA sequences tagged with potential binding motifs. As a first example of the general concept we have evaluated a small self-assembled bivalent aptamer (SABA) library based on two nucleic acid aptamer sequences that bind to different exosites of thrombin. The general SABLC concept opens up the possibility of using a wide range of ligands with great chemical diversity such as non-natural nucleotides and backbone linkages, peptides, carbohydrates or small molecules. It will also allow optimization of combinations of known monovalent ligands to improve binding affinity and enzymatic stability. From an economic standpoint it is reasonable to propose the synthesis of large SABLC libraries; oligonucleotide synthesis facilities typically generate hundreds to thousands of oligonucleotides per day. The same is true for peptides manufacturing, and other potential ligands.30 Moreover, several conjugation chemistries will allow automated large scale synthesis of monovalent ligand libraries tagged with individual nucleic acids bearing specific identifier sequences. Most importantly the universal nature of the SABLC approach and the sensitivity of the decoding process allow the re-use of aliquots of larger scale pre-synthesized libraries of monovalent DNA tagged ligands for multiple selection experiments. Their reuse in combination with other sets of complementary arms is also possible. Such libraries can be stored for future use, thus expanding the libraries and accumulating a valuable resource over time.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TB and AHE-S are grateful to the UK BBSRC for funding via the sLOLA grant BB/J001694/2: “Extending the Boundaries of Nucleic Acid Chemistry.” SM would like to thank Joop van Helvoort, Xia Teng and Jelle Oostmejier at PBS for critical comments. MH appreciated the technical support of Martin Madikrama.Notes and references
- M. Darmostuk, S. Rimpelová, H. Gbelcová and T. Ruml, Biotechnol. Adv., 2015, 15, S0734–S9750 Search PubMed.
- M. Mascini, I. Palchetti and S. Tombelli, Angew. Chem., Int. Ed., 2012, 51, 1316–1332 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- A. Anna, F. Carme, T. Maria and E. Ramon, Curr. Pharm. Des., 2012, 18, 2036–2047 CrossRef.
- K. M. Ahmad, Y. Xiao and H. T. Soh, Nucleic Acids Res., 2012, 40, 11777–11783 CrossRef CAS PubMed.
- Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664–5669 CrossRef CAS PubMed.
- Y.-H. Lao, K. Peck and L.-C. Chen, Anal. Chem., 2009, 81, 1747–1754 CrossRef CAS PubMed.
- J. Müller, B. Wulffen, B. Pötzsch and G. Mayer, ChemBioChem, 2007, 8, 2223–2226 CrossRef PubMed.
- D. Musumeci and D. Montesarchio, Pharmacol. Ther., 2012, 136, 202–215 CrossRef CAS PubMed.
- L. Tian and T. Heyduk, Biochemistry, 2009, 48, 264–275 CrossRef CAS PubMed.
- R. Wilson, C. Bourne, R. R. Chaudhuri, R. Gregory, J. Kenny and A. Cossins, PLoS One, 2014, 9, e100572 Search PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS.
- F. Abendroth, A. Bujotzek, M. Shan, R. Haag, M. Weber and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 8592–8596 CrossRef CAS PubMed.
- B. M. G. Janssen, E. H. M. Lempens, L. L. C. Olijve, I. K. Voets, J. L. J. van Dongen, T. F. A. de Greef and M. Merkx, Chem. Sci., 2013, 4, 1442–1450 RSC.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- S. Rinker, Y. Ke, Y. Liu, R. Chhabra and H. Yan, Nat. Nanotechnol., 2008, 3, 418–422 CrossRef CAS PubMed.
- K. K. Sadhu, M. Roethlingshoefer and N. Winssinger, Isr. J. Chem., 2013, 53, 75–86 CrossRef CAS.
- C. Scheibe, A. Bujotzek, J. Dernedde, M. Weber and O. Seitz, Chem. Sci., 2011, 2, 770–775 RSC.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- B. A. R. Williams, C. W. Diehnelt, P. Belcher, M. Greving, N. W. Woodbury, S. A. Johnston and J. C. Chaput, J. Am. Chem. Soc., 2009, 131, 17233–17241 CrossRef CAS PubMed.
- A. Pinto, M. C. Bermudo Redondo, V. Cengiz Ozalp and C. K. O'Sullivan, Mol. BioSyst., 2009, 5, 548–553 RSC.
- A. H. El-Sagheer, A. P. Sanzone, R. Gao, A. Tavassoli and T. Brown, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11338–11343 CrossRef CAS PubMed.
- C. N. Birts, A. P. Sanzone, A. H. El-Sagheer, J. P. Blaydes, T. Brown and A. Tavassoli, Angew. Chem., Int. Ed., 2014, 53, 2362–2365 CrossRef CAS PubMed.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- D. M. Tasset, M. F. Kubik and W. Steiner, J. Mol. Biol., 1997, 272, 688–698 CrossRef CAS PubMed.
- I. Russo Krauss, A. Pica, A. Merlino, L. Mazzarella and F. Sica, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 2403–2411 CAS.
- I. Russo Krauss, A. Merlino, A. Randazzo, E. Novellino, L. Mazzarella and F. Sica, Nucleic Acids Res., 2012, 40, 8119–8128 CrossRef CAS PubMed.
- R. D. Langdell, R. H. Wagner and K. M. Brinkhous, J. Lab. Clin. Med., 1953, 41, 637–647 CAS.
- S. Yoshizawa, G. Kawai, K. Watanabe, K.-i. Miura and I. Hirao, Biochemistry, 1997, 36, 4761–4767 CrossRef CAS PubMed.
- R. Pipkorn, C. Boenke, M. Gehrke and R. Hoffmann, J. Pept. Res., 2002, 59, 105–114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of oligonucleotide synthesis, PCR amplification, DNA sequencing, blood clotting assays. See DOI: 10.1039/c7ob02119d |
| This journal is © The Royal Society of Chemistry 2018 |