
Rh(III)-Catalyzed ortho-C–H alkynylation of N-phenoxyacetamides with hypervalent iodine-alkyne reagents at room temperature†
Sisheng
Hu
a,
Liang
Lu
a,
Tongyang
Zhu
a,
Qian
Wu
b,
Ying
Chen
b,
Jie Jack
Li
*c and
Jing
Zhao
 *ab
*ab
aState Key Laboratory of Pharmaceutical Biotechnology, School of Chemistry and Chemical Engineering, Institute of Chemistry and BioMedical Sciences, Nanjing University, Nanjing, 210093, China. E-mail: jingzhao@nju.edu.cn
bGuangdong Key Lab of Nano-Micro Material Research, School of Chemical Biology and Biotechnology, Shenzhen Graduate School of Peking University, Shenzhen, 518055, China
cDepartment of Chemistry, University of San Francisco, San Francisco, CA 94117, USA
First published on 28th November 2017
Abstract
A Rh(III)-catalyzed C–H alkynylation of substituted N-phenoxyacetamides has been developed with the aid of hypervalent iodine-alkyne reagents. Complementary to the Sonogashira coupling reaction, this protocol provides an efficient and straightforward method to access aryl alkynes at room temperature. The multifunctional directing group is preserved which can be further employed for ortho-directed functionalizations to obtain additional new complex products.
Alkynes are one of the important functional groups in synthetic chemistry and materials science due to their diverse transformations, and existence in natural products, medicines, and advanced materials.1 The most common method for the synthesis of alkynes is the Sonogashira cross-coupling reaction.2 To circumvent the need for pre-activated starting materials (such as halides) and with the great advancement of C–H functionalization, direct alkynylation of the C–H bond using different transition metals and alkyne sources has captured much attention.3–5 Among them, alkynylated hypervalent iodines have been well studied thanks to their stability and ease of handling. Many excellent studies have been reported by Waser,5a–n Loh,5o,p Li,5q,r,u Nachtsheim,5t Cheng5v and others5 in which hypervalent iodine-alkyne reagents could efficiently react with different substrates to afford alkynylation products by the catalysis of different transition metals (Scheme 1). Our group has recently reported a number of transition metal-catalyzed C–H bond functionalizations based on a powerful directing group, –O–NHAc (oxyacetamide), which showed a preferable metal chelation ability in C–H activation reactions.6 We herein report a Rh-catalyzed C–H activation/alkynylation reaction between N-phenoxyacetamides and hypervalent iodine-alkyne reagents at room temperature.
 | ||
| Scheme 1 Metal-catalyzed alkynylation of arenes. | ||
Being cognizant of the high reactivity of hypervalent iodine reagents,5 we commenced our studies using 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one 2a (TIPS-EBX) as the coupling partner. When 2-methyl N-phenoxyacetamide 1a was treated with partner 2a using [Cp*RhCl2]2 (2.5 mol%) as the catalyst and CsOAc (0.3 equiv.) as the base in DCM at room temperature under a N2 atmosphere for 5 h (Table 1, entry 1), 3a was obtained as the major product in good yield. The structure of 3a was proved to be that of the desired ortho-alkynylated product which was different from our previous work where the O–N bond of the oxyacetamide group was preserved. In most of our previous studies, the O–NHAc group played multiple roles while the O–N bond, as an internal oxidant, was broken along with the interaction with the catalyst.6c–e In contrast, the O–N bond was also observed to be preserved in the final products under several conditions.6a,b
|
|
|||
|---|---|---|---|
| Entry | Base (equiv.) | Solvent | Yield (NMR) |
| a Conditions: 1a (0.1 mmol), 2a (0.12 mmol), [Cp*RhCl2]2 (2.5 mol%), and base in solvent (0.5 mL) at room temperature for 5 h under N2. Crude yield, as determined by 1H NMR spectroscopy using 1,4-dimethoxybenzene as an internal standard. | |||
| 1 | CsOAc (0.3) | DCM | 70% |
| 2 | CsOAc (0.3) | DCE | 69% |
| 3 | CsOAc (0.3) | CH3CN | 69% |
| 4 | CsOAc (0.3) | Toluene | 70% |
| 5 | CsOAc (0.3) | THF | 84% |
| 6 | CsOAc (0.3) | 1,4-Dioxane | 85% |
| 7 | CsOAc (0.3) | MeOH | 95% |
| 8 | CsOAc (0.3) | EtOH | 78% |
| 9 | CsOAc (0.3) | Isopropanol | 74% |
| 10 | CsOAc (0.3) | t-BuOH | 78% |
| 11 | AgOAc (0.3) | MeOH | 74% |
| 12 | NaOAc (0.3) | MeOH | 76% |
| 13 | Cs2CO3 (0.3) | MeOH | 82% |
| 14 | Ag2CO3 (0.3) | MeOH | 73% |
| 15 | Na2CO3 (0.3) | MeOH | 76% |
| 16 | K2CO3 (0.3) | MeOH | 73% |
| 17 | CsOAc (0.5) | MeOH | 90% |
| 18 | CsOAc (1) | MeOH | 89% |

Other solvents were also screened and MeOH dramatically enhanced the yield to 95% (Table 1, entries 1–10). We also examined different bases and found CsOAc to be the most effective (Table 1, entries 11–16). But increasing the equivalence of CsOAc did not significantly boost the yields (Table 1, entries 17 and 18). Finally, we set the standard reaction conditions to be [Cp*RhCl2]2 (2.5 mol%) and CsOAc (0.3 equiv.) in MeOH under nitrogen at room temperature.
With the optimized conditions in hand, we proceeded to explore the scope of N-phenoxyacetamides (Table 2). N-Phenoxyacetamides bearing electron-donating groups such as methyl (1a, 1b, 1c,), tert-butyl (1d), and dimethyl (1f) and electron-withdrawing groups such as halides (1h–1m) and ester (1n) all proceeded smoothly to afford the corresponding products in moderate to high yields ranging from 77% to 95%. When non-, meta- or para-substituted N-phenoxyacetamides reacted with 1.2 equiv. of TIPS-EBX, the dialkynylation products were obtained as the major products. Therefore, when 4 equiv. of N-phenoxyacetamides were used, the mono-alkynylation products were isolated in excellent yields. In this case, the reaction occurred at the less-hindered position when the meta-position of N-phenoxyacetamides (1h, 1j, 1l) was occupied. This regioselectivity showcased the importance of the steric effect in this reaction. In addition, we also examined the substituents on the nitrogen atom of N-phenoxyacetamides. When substrate 1o containing two competitive directing groups was used, the reaction took place regiospecifically at the position ortho to the oxyamine directing group, affording product 3o in 80% yield, which was different from what was previously reported.6b,7 However, the use of the bulky tert-butyryl group instead of the acetyl group only gave 3p in a trace amount. Meanwhile, we explored the alkynylation of various hypervalent iodine-alkyne reagents containing different aryl and alkynyl substituents (2b–2d). The corresponding products were obtained in good yields (Table 3).

|
|
|||
|---|---|---|---|
| EBX reagent | Product | EBX reagent | Product |
| a Conditions: 1 (0.1 mmol) and 2 (0.9 mmol) were used. [{Cp*RhCl2}2] (2.5 mol%), and additives in solvent (1 mL) at room temperature for 5 h under N2. Isolated yields. | |||

|

|

|

|

|

|
||

We investigated the reactivity of the obtained alkynyl N-phenoxyacetamides. Product 3f was explored as a model substrate. Treating 3f with the iridium catalyst [Cp*IrCl2]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6e led to the ortho-oxidation of phenol by O–NHAc directed second C–H activation in the internal oxidation pathway (4a). The TIPS protecting group can also be removed under basic conditions (TBAF/THF) without changing the O–NHAc group (4b) (Scheme 2).
6e led to the ortho-oxidation of phenol by O–NHAc directed second C–H activation in the internal oxidation pathway (4a). The TIPS protecting group can also be removed under basic conditions (TBAF/THF) without changing the O–NHAc group (4b) (Scheme 2).
 | ||
| Scheme 2 Derivatisation of reaction products. a3f (0.2 mmol), [Cp*IrCl2]2 (0.25 mol%), CH2(COOH)2 (0.5 mmol) in MeOH, r.t., 8 h. Isolated yields. | ||
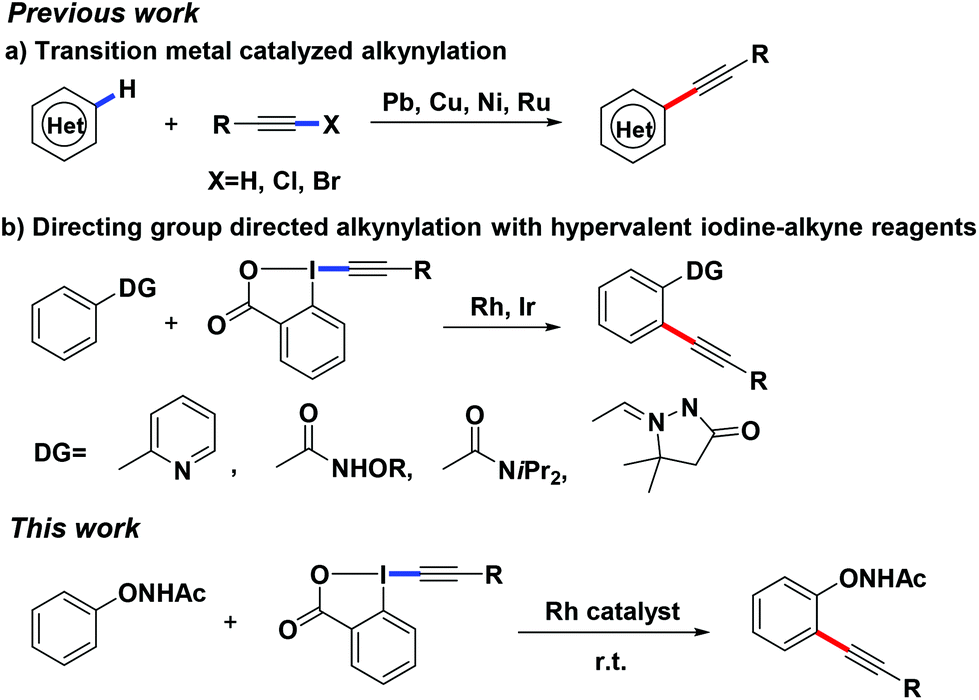
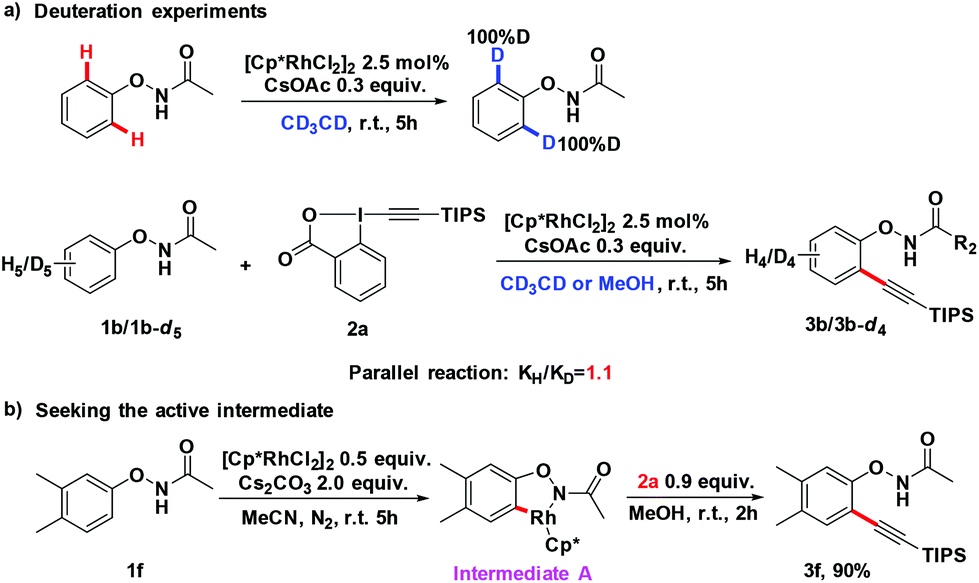
To gain a better understanding of the mechanism, we treated 1b with [Cp*RhCl2]2 (2.5 mol%) and CsOAc (0.3 equiv.) in CD3CD (Scheme 3a). After stirring at room temperature for 5 h, the reaction mixture was directly subjected to 1H NMR measurement. It was found that deuterium was incorporated exclusively at the ortho position to the directing group. This experiment indicated that the first reaction step may be a facile and reversible cyclometalation. To determine the turnover-limited step of the catalytic cycle, the kinetic isotope effect (KIE) was measured. Parallel experiments of 1b and d5-1b with TIPS-EBX gave a result as KH/KD = 1.1, indicating that the C–H bond cleavage process was unlikely involved in the rate-determining step (Scheme 3a).8 We successfully obtained the intermediate A by reacting 1f with the rhodium catalyst [Cp*RhCl2]2. Intermediate A smoothly produced the expected product 3f, suggesting that intermediate A was the active intermediate in this reaction (Scheme 3b).
 | ||
| Scheme 3 Mechanistic study. | ||
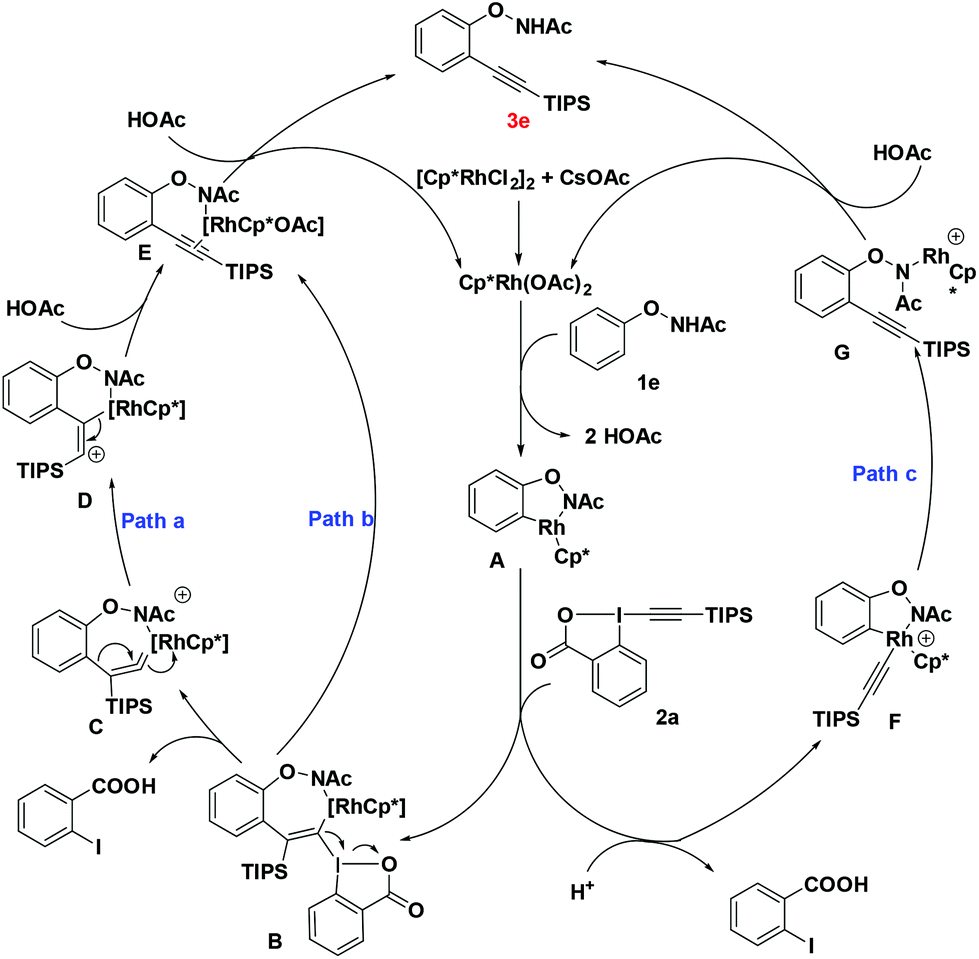
On the basis of these observations and literature precedence, we proposed the following mechanism. First, the active catalyst [Cp*RhIII(OAc)2] is generated from [Cp*RhCl2]2 and CsOAc. Intermediate A is then formed via reversible C–H activation between substrate 1e and the catalyst [Cp*RhIII(OAc)2] through a concerted metalation–deprotonation (CMD) pathway. The following regioselective carborhodation of TIPS-EBX generates intermediate B. Then α-elimination occurs to produce the rhodium vinylidene C, with the release of 2-iodobenzoic acid.9 There are two kinds of possible pathways for further process, in pathway a, the species undergoes intramolecular concerted 1,2-aryl migration and breaking of the C–Rh bond to generate the intermediate E.5a,10 In pathway b, C could follow stepwise silicium group migration and elimination viaD to produce the same intermediate E. Finally, the protonation of E with another molecule HOAc will produce the desired alkynylation product 3e, and the released active catalyst will trigger the next cycle.5p,q Another probable mechanism is also considered in pathway c. The oxidative addition of TIPS-EBX to intermediate A generates Rh(V) intermediate F which undergoes reductive elimination to obtain product 3e directly (Scheme 4).
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
It is interesting to note that the hypervalent iodine-alkynes could act as both alkynylation agents and oxidants, which might explain the fact that the O–N bond was preserved in the final products.
Conclusions
In summary, we have developed a Rh(III)-catalyzed ortho-C–H alkynylation with a broad scope of N-phenoxyacetamides with hypervalent iodine-alkyne reagents as the alkynyl source at room temperature. The reaction is complementary to the Sonogashira coupling reaction. Furthermore, in this reaction, the O–N bond of the directing group, –O–NHAc, is preserved which can be further employed for ortho-directed functionalizations to obtain additional new complex products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Science Foundation of China (21622103, 21571098 and 21671099), the Natural Science Foundation of Jiangsu Province (BK20160022), and the Shenzhen Basic Research Program (JCYJ201704131505 38897).Notes and references
- (a) P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632 CrossRef CAS; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (c) R. R. Tykwinski, Angew. Chem., Int. Ed., 2003, 42, 1566 CrossRef CAS PubMed; (d) F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material ScienceWiley-VCH, Weinheim, Germany, 2005 Search PubMed; (e) A. Fürstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC; (f) S. Toyota, Chem. Rev., 2010, 110, 5398 CrossRef CAS PubMed; (g) M. G. Finn and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1231 RSC; (h) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC; (i) I. V. Alabugin and B. Gold, J. Org. Chem., 2013, 78, 7777 CrossRef CAS PubMed; (j) R. Hu, J. W. Y. Lam and B. Z. Tang, Macromol. Chem. Phys., 2013, 214, 175 CrossRef CAS; (k) R. Chinchilla and C. Nájera, Chem. Rev., 2014, 114, 1783 CrossRef CAS PubMed.
- (a) K. Sonogashira, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 1998, p. 203 Search PubMed; (b) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS; (c) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS PubMed; (d) A. O. King and N. Yasuda, Top. Organomet. Chem., 2004, 6, 205 CrossRef CAS; (e) R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (f) H. Doucet and J.-C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 CrossRef CAS PubMed; (g) H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954 CrossRef CAS PubMed; (h) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC.
- For selected examples of C–H alkynylation with terminal alkynes, see: (a) Y. Wei, H. Zhao, J. Kan, W. Su and M. Hong, J. Am. Chem. Soc., 2010, 132, 2522 CrossRef CAS PubMed; (b) N. Matsuyama, M. Kitahara, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2358 CrossRef CAS PubMed; (c) T. Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512 CrossRef PubMed; (d) L. Yang, L. Zhao and C.-J. Li, Chem. Commun., 2010, 46, 4184 RSC; (e) S. H. Kim, J. Yoon and S. Chang, Org. Lett., 2011, 13, 1474 CrossRef CAS PubMed; (f) F. Shibahara, Y. Dohke and T. Murai, J. Org. Chem., 2012, 77, 5381 CrossRef CAS PubMed; (g) X. Jie, Y. Shang, P. Hu and W. Su, Angew. Chem., Int. Ed., 2013, 52, 3630 CrossRef CAS PubMed; (h) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (i) J. Zhou, J. Shi, Z. Qi, X. Li, H. E. Xu and W. Yi, ACS Catal., 2015, 5, 6999 CrossRef CAS.
- For selected examples of C–H alkynylation with alkynyl halides, see: (a) K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528 CrossRef CAS PubMed; (b) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742 CrossRef CAS PubMed; (c) Y. Gu and X. Wang, Tetrahedron Lett., 2009, 50, 763 CrossRef CAS; (d) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250 CrossRef CAS PubMed; (e) N. Matsuyama, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 4156 CrossRef CAS PubMed; (f) F. Besselièvre and S. Piguel, Angew. Chem., Int. Ed., 2009, 48, 9553 CrossRef PubMed; (g) S. H. Kim and S. Chang, Org. Lett., 2010, 12, 1868 CrossRef CAS PubMed; (h) T. Kawano, N. Matsuyama, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2010, 75, 1764 CrossRef CAS PubMed; (i) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096 CrossRef CAS PubMed; (j) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS PubMed; (k) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 14, 354 CrossRef CAS PubMed; (l) Y. Ano, M. Tobisu and N. Chatani, Org. Lett., 2012, 23, 2763 CAS; (m) J. He, M. Wasa, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 3387 CrossRef CAS PubMed; (n) P. Wang, G. C. Li, P. Jain, M. E. Farmer, J. He, P. X. Shen and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092 CrossRef CAS PubMed.
- For selected examples of C–H alkynylation with hypervalent iodine reagents, see: (a) J. P. Brand, J. Charpentier and J. Waser, Angew. Chem., Int. Ed., 2009, 48, 9346 CrossRef CAS PubMed; (b) J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 7304 CrossRef CAS PubMed; (c) D. Fernández González, J. P. Brand and J. Waser, Chem. – Eur. J., 2010, 16, 9457 CrossRef PubMed; (d) S. Nicolai, S. Erard, D. Fernández González and J. Waser, Org. Lett., 2010, 12, 384 CrossRef CAS PubMed; (e) J. P. Brand, C. Chevalley and J. Waser, J. Org. Chem., 2011, 7, 565 CAS; (f) J. P. Brand, D. Fernández González, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC; (g) J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. – Eur. J., 2012, 18, 5655 CrossRef CAS PubMed; (h) J. P. Brand and J. Waser, Org. Lett., 2012, 14, 744 CrossRef CAS PubMed; (i) J. P. Brand and J. Waser, Synthesis, 2012, 1155 CAS; (j) D. Fernández González, J. P. Brand, R. Mondière and J. Waser, Adv. Synth. Catal., 2013, 355, 1631 CrossRef; (k) R. Frei Waser and J. Waser, J. Am. Chem. Soc., 2013, 135, 9620 CrossRef PubMed; (l) Y. Li, J. P. Brand and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 6743 CrossRef CAS PubMed; (m) Y. Li and J. Waser, Beilstein J. Org. Chem., 2013, 9, 1763 CrossRef PubMed; (n) G. L. Tolnai, S. Ganss, J. P. Brand and J. Waser, Org. Lett., 2013, 15, 112 CrossRef CAS PubMed; (o) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722 CrossRef CAS PubMed; (p) C. Feng, D. Feng, Y. Luo and T. P. Loh, Org. Lett., 2014, 16, 5956 CrossRef CAS PubMed; (q) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed; (r) X. Zhang, Z. Qi, J. Gao and X. Li, Org. Biomol. Chem., 2014, 12, 9329 RSC; (s) K. D. Collins, F. Lied and F. Glorius, Chem. Commun., 2014, 50, 4459 RSC; (t) P. Finkbeiner, U. Kloeckner and B. J. Nachtsheim, Angew. Chem., Int. Ed., 2015, 54, 4949 CrossRef CAS PubMed; (u) Y. Li, F. Xie and X. Li, J. Org. Chem., 2016, 81, 715 CrossRef CAS PubMed; (v) R. Boobalan, P. Gandeepan and C. H. Cheng, Org. Lett., 2016, 18, 3314 CrossRef CAS PubMed.
- (a) P. Duan, Y. Yang, R. Ben, Y. Yan, L. Dai, M. Hong, Y.-D. Wu, D. Wang, X. Zhang and J. Zhao, Chem. Sci., 2014, 5, 1574 RSC; (b) P. Duan, X. Lan, Y. Chen, S.-S. Qian, J. J. Li, L. Lu, Y. Lu, B. Chen, M. Hong and J. Zhao, Chem. Commun., 2014, 50, 12135 RSC; (c) Y. Chen, D. Wang, P. Duan, R. Ben, L. Dai, X. Shao, M. Hong, J. Zhao and Y. Huang, Nat. Commun., 2014, 5, 4610 CAS; (d) Q. Wu, Y. Chen, D. Yan, M. Zhang, Y. Lu, W.-Y. Sun and J. Zhao, Chem. Sci., 2017, 8, 169 RSC; (e) Q. Wu, D. Yan, Y. Chen, T. Wang, F. Xiong, W. Wei, Y. Lu, W.-Y. Sun, J. J. Li and J. Zhao, Nat. Commun., 2017, 8, 14227 CrossRef PubMed.
- (a) S. Lu, Y. Lin, H. Zhong, K. Zhao and J. Huang, Tetrahedron Lett., 2013, 54, 2001 CrossRef CAS; (b) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed.
- (a) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS PubMed; (b) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed.
- (a) V. V. Zhdankin, C. J. Kuehl, A. P. Krasutsky, J. T. Bolz and A. J. Simonsen, J. Org. Chem., 1996, 61, 6547 CrossRef CAS PubMed; (b) R. Knorr, Chem. Rev., 2004, 104, 3795 CrossRef CAS PubMed; (c) J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. – Eur. J., 2012, 18, 5655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. See DOI: 10.1039/c7ob02438j |
| This journal is © The Royal Society of Chemistry 2018 |