
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface transmetalation of metalloporphyrins†
Diana
Hötger
a,
Paula
Abufager
b,
Claudius
Morchutt
ac,
Patrick
Alexa
a,
Doris
Grumelli
 d,
Jan
Dreiser
e,
Sebastian
Stepanow
f,
Pietro
Gambardella
f,
H. Fabio
Busnengo
b,
Markus
Etzkorn‡
a,
Rico
Gutzler
*a and
Klaus
Kern
ac
d,
Jan
Dreiser
e,
Sebastian
Stepanow
f,
Pietro
Gambardella
f,
H. Fabio
Busnengo
b,
Markus
Etzkorn‡
a,
Rico
Gutzler
*a and
Klaus
Kern
ac
aMax Planck Institute for Solid State Research, 70569 Stuttgart, Germany. E-mail: r.gutzler@fkf.mpg.de
bInstituto de Física Rosario and Universidad Nacional de Rosario, CONICET-UNR, S2000EZO Rosario, Argentina
cInstitute de Physique, Ecole Polytechnique Fédérale de Lausanne, 1015 Lausanne, Switzerland
dInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas (INIFTA), Facultad de Ciencias Exactas, Universidad Nacional de La Plata, CONICET, 1900 La Plata, Argentina
ePaul Scherrer Institute, Swiss Light Source, 5232 Villigen PSI, Switzerland
fDepartment of Materials, ETH Zürich, 8093 Zürich, Switzerland
First published on 26th October 2018
Abstract
Increasing the complexity of 2D metal–organic networks has led to the fabrication of structures with interesting magnetic and catalytic properties. However, increasing complexity by providing different coordination environments for different metal types imposes limitations on their synthesis if the controlled placement of one metal type into one coordination environment is desired. Whereas metal insertion into free-base porphyrins at the vacuum/solid interface has been thoroughly studied, providing detailed insight into the mechanisms at play, the chemical interaction of a metal atom with a metallated porphyrin is rarely investigated. Herein, the breadth of metalation reactions is augmented towards the metal exchange of a metalloporphyrin through the deliberate addition of atomic metal centers. The cation of Fe(II)-tetraphenylporphyrins can be replaced by Co in a redox transmetalation-like reaction on a Au(111) surface. Likewise, Cu can be replaced by Co. The reverse reaction does not occur, i.e. Fe does not replace Co in the porphyrin. This non-reversible exchange is investigated in detail by X-ray absorption spectroscopy complemented by scanning tunneling microscopy. Density functional theory illuminates possible reaction pathways and leads to the conclusion that the transmetalation proceeds through the adsorption of initially metallic (neutral) Co onto the porphyrin and the expulsion of Fe towards the surface accompanied by Co insertion. Our findings have important implications for the fabrication of porphyrin layers on surfaces when subject to the additional deposition of metals. Mixed-metal porphyrin layers can be fabricated by design in a solvent-free process, but conversely care must be taken that the transmetalation does not proceed as an undesired side reaction.
Introduction
Porphyrins are ubiquitous molecules that offer a tremendous broadness of functionality in diverse domains ranging from biology to technologically relevant devices. In their metalated state, porphyrin derivatives operate as carrier molecules for respiratory gases,1 are key to photosynthesis,2 find application as (electro)catalysts,3,4 or in molecular (opto)electronics.5 Their seemingly unlimited functional properties have promoted research on porphyrins and related molecules in thin films, as monolayers, and at the single-molecule level.6,7The metal cation embedded in the tetrapyrrole macrocycle is of particular interest as it governs many chemical and physical properties of the molecule. At well-defined interfaces, for example, the gas-fixating properties of the metal centers,8,9 their magnetic properties,10,11 or catalytic activity12–15 were investigated. The almost planar shape of the molecular macrocycle ensures that the coordinatively unsaturated cation is accessible for reactants, which in part explains their high reactivity towards small molecules. At the same time the flat geometry also results in a strong influence of the surface to the central metal atom.16 A standard procedure to metalate free-base porphyrins is through the uptake of metal atoms at the vacuum/solid interface and the accompanied release of H2.17 The metalation can occur through the incorporation of readily available metal atoms from the metallic support,18 or alternatively through the allocation of a different metal by thermal evaporation.19–21 The metalation can thus be achieved with different metal types and is routinely used to synthesize metalloporphyrins on surfaces on demand.22,23 The mechanism by which the two pyrrolic hydrogens are expelled and replaced by a metal atom is well understood.17,24
However, the metal exchange, in which an existing metal cation is replaced by a second metal, is rarely investigated in surface-supported porphyrins. Two studies describe the replacement of Ni and Co by Cu, which is supplied through the adatom gas on the Cu(111) surface in ultra-high vacuum.25,26 A third study describes the exchange of Zn by Cu at the solid/liquid interface.27 No clear trends can be extracted from the existing literature: on the one hand Franke et al.27 report Zn substitution by Cu in a tetraphenylporphyrin network at the solid–liquid interface. On the other hand Shi et al.28 report no such substitution of Zn-tetrapyridylporphyrin and co-deposited Cu in vacuum. Recently, the hierarchical exchange of Cu by additionally supplied Ni and Fe in organic macrocycles was reported at elevated temperatures.29 Whereas these exchange reactions can be controlled regarding the exchanging metal centres, the reaction yield appears less controllable. Furthermore, so far systems with only one coordination environment, the porphyrin macrocycle, were investigated, limiting the exchange to this one site. Certainly, further studies are required for a complete understanding of the exchange mechanism and to achieve a more controlled reaction with possibly quantitative yield. In particular, it is interesting to study systems which offer a second coordination environment that might function as a trap for additionally supplied metal centres, thus inhibiting the exchange process. This is particularly important in the light of the recent trend of increasing the complexity of heterometallic metal–organic networks with two or more different coordination environments.15,28,30 In this context of ever more elaborate metal–organic networks fabricated on surfaces with synthesis procedures of increasing complexity, it becomes of fundamental importance to investigate the competitive interactions between different building units such as organic molecules and metal centres. Where the cation in the molecular environment defines electronic and physicochemical properties – molecular magnetism, advanced (bio)materials, sensors, to name a few6 – it must be ensured that synthesis procedures yield the desired structure. As will be shown in the following, the sublimation of a metal onto a metalloporphyrin monolayer in vacuum can be used to fabricate complex mixed-metal porphyrin networks.
Here, we describe the transmetalation of Fe- and Cu-porphyrins in ultra-high vacuum (UHV). Deposition of Co spontaneously substitutes Fe/Cu and occupies its place in the tetrapyrrole ring on a Au(111) surface at room temperature. The metal exchange is investigated using X-ray absorption spectroscopy (XAS) supported by scanning tunnelling microscopy (STM). Porphyrins with different embedded cations and different peripheral functional groups were studied, Fe-tetraphenylporphyrin (FeTPP) was used alongside Fe-tetrapyridylporphyrin (FeTPyP) and Cu-tetrapyridylporphyrin (CuTPyP), see Fig. 1a. In all three molecules the partial replacement of the metal centre by additionally sublimed Co is observed, whereas the replacement of Co by Fe is not observed. The pyridyl groups can coordinate metal centres, but do not inhibit the replacement in the macrocycle. Density functional theory (DFT) calculations reveal the exchange mechanism and barrier height of the transmetalation reaction.
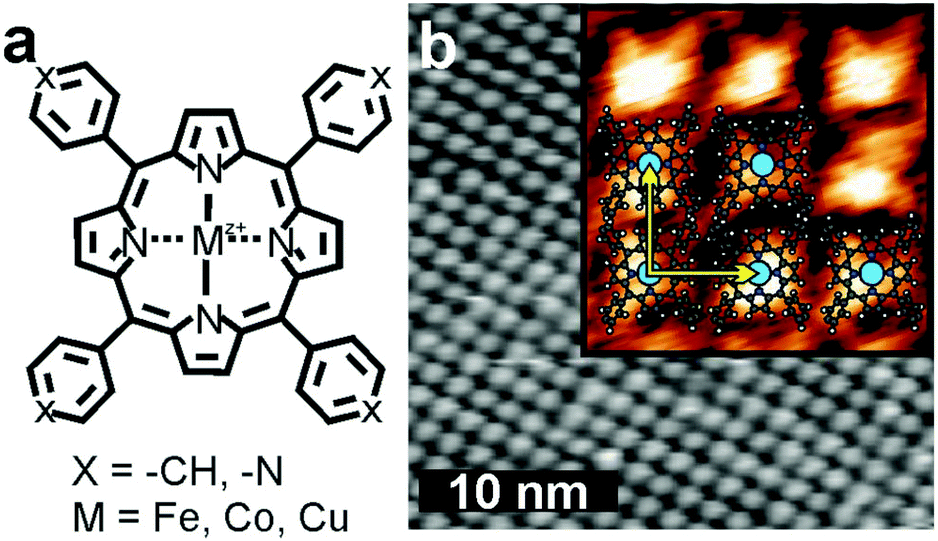 | ||
| Fig. 1 (a) Structural formula of metalloporphyrin with M = Fe, Co or Cu and different peripheric functional groups: phenyl (X = −CH) and pyridyl (X = N). (b) STM image of a FeTPP monolayer (X = –CH, Itunnel = 1.0 nA; VBias = 1.0 V); inset FeTPyP monolayer (X = N, Itunnel = 0.2 nA; VBias = −0.7 V) the lattice vectors are indicated by yellow arrows and the porphyrin molecule is superimposed. | ||
Methods
The Au(111) substrates (single crystals and gold on mica) were cleaned by repeating Ar+ ion sputtering and thermal annealing cycles at 823 K. Molecules (purchased from Frontier Scientific) were sublimed using a molecular beam evaporator to form self-assembled networks. FeTPP was evaporated at 620 K, FeTPyP at 744 K, CoTPyP at 683 K, and CuTPyP at 673 K with the substrate held at room temperature. Deposition times were adjusted for full monolayer or submonolayer coverage. Co and Fe were sublimed from an electron beam evaporator, again with the sample held at room temperature. The molecular network's topography was investigated in two different UHV-STM setups (base pressure <5 × 10−10 mbar). Images were analysed using WsXM.31 X-ray absorption spectra were recorded at 300 K with linear horizontal (σh) or linear vertical (σv) polarization at the X-Treme beamline32 of the Swiss Light Source. The experimental geometry was grazing incidence (θ = 60°), and the spectra were recorded in total electron yield. A negligibly small magnetic field of 50 mT was applied, helping the electrons to leave the surface and thereby reducing the noise on the measured drain current. For comparison the spectra were normalized to equivalent integral.DFT calculations were performed with the VASP code33 within the slab-supercell approach and using the projector augmented-wave (PAW) method.34 Wave functions were expanded using a plane wave basis set with an energy cut-off of 450 eV. The spin-polarized generalized gradient approximation (GGA-PBE) was used. In order to improve the description of dispersion forces, the D3 van der Waals correction method proposed by Grimme35 was employed. The unreconstructed Au(111) surface was modelled using a slab geometry with three Au layers and a vacuum region of 17 Å. All calculations were carried out for one M1TPP molecule and a M2 atom in the same 7 × 7 unit cell (M1, M2 = Co, Fe) and for a single k-point (Γ). Charge analysis was performed using Bader's method.36
Results and discussion
The iron-containing porphyrins (Fig. 1a) self-assemble spontaneously into densely packed monolayers on Au(111) (Fig. 1b). The distance between molecules on the square lattice measures approximately 1.5 nm indicated by yellow vectors. The additional sublimation of Co to the iron porphyrin assembly at room temperature does not alter the appearance of the molecules in STM. Replacing Fe by Co or Cu, or replacing phenyl by pyridyl groups has no effect on the molecular arrangement.To address potential chemical changes that come along with the availability of the second metal type, we used XAS at the Fe and Co L2,3-edges (Fig. 2). The Fe L3 absorption edge of approximately one monolayer FeTPP exhibits a multiplet structure with maxima located at 706.5 eV and 707.6 eV (Fig. 2a, grey) expected for coordinated Fe.37 The spectra σv (yellow) and σh (red) exhibit distinct multiplet splitting – a consequence of the ligand field created by the four pyrrole nitrogen atoms37 – leading to an appreciable linear dichroism (Fig. 2a, lower panel). The multiplet structure is diminished after deposition of Co slightly exceeding the stoichiometric amount of Fe (Co/Fe ≈ 1.2), and the absorption spectrum suggests a metallic state38,39 (Fig. 2b, blue). The loss in multiplet structure is likewise visible in σv and σh, which become more isotropic, leading to a reduced linear dichroism (Fig. 2b, lower panel). The change upon Co deposition is particularly appreciable in the σh spectrum, in which the second peak at higher photon energy is reduced to a faint shoulder. In the (σv + σh) of FeTPP + Co XAS the maximum of L3 sits at 706.5 eV in accordance with the first peak of pristine FeTPP. A shoulder located at 707.6 eV reminiscent to the second peak of (σv + σh) of pristine FeTPP indicates a reduced influence of pristine FeTPP to the XAS signal. The spectrum of FeTPP + Co can therefore be understood as a superposition of molecular FeTPP and metallic Fe. The X-ray linear dichroism (XLD) of FeTPP (Fig. 2a, lower panel) is larger than that of FeTPP + Co (Fig. 2b, lower panel) although the shape remains similar except for the high relative weight of the L3 shoulder at ∼708 eV. The Co edge exhibits for FeTPP + Co a structured L3 peak (Fig. 2c), which is accompanied by a linear dichroism (Fig. 2c, lower panel). The two L3 maxima in the (σv + σh) spectrum are located at 776.8 eV and 778.5 eV and result from discrete peaks in the respective σv and σh spectra.
 | ||
| Fig. 2 Characterization of FeTPP and FeTPP + Co at Fe and Co L2,3-edges. (a) Fe L2,3-edge of FeTPP (upper panel) and XLD (lower panel); (b) Fe L2,3-edge of FeTPP + Co (upper panel) and XLD (lower panel); (c) Co L2,3-edge of FeTPP + Co (upper panel) and XLD (lower panel); (d) Co L2,3-edge of CoTPyP (upper panel) and XLD (lower panel). | ||
The clear multiplet structure and linear dichroism of the Co L2,3-edge are a result of well separated 3d orbital states, which cannot be expected for Co cluster,39,40 but likely result from the ligand field created by the four pyrrole nitrogen atoms of the porphyrin ring.11 For Co to be able to interact strongly with the nitrogen atoms, Fe has to be reductively expulsed from the porphyrin. Indeed, the Co spectra shown in Fig. 2c can be well described by a superposition of CoTPP and metallic Co (Fig. SI1†). For comparison, the Co L2,3-edge of Co-tetrapyridylporphyrin (CoTPyP) is shown in Fig. 2d. In the XAS the multiplet structure is difficult to compare directly because of the contributions of metallic Co in FeTPP + Co. In the dichroism, in which metallic Co does contribute only weakly, the strong similarities between FeTPP + Co and CoTPyP become obvious. This clearly confirms the presence of Co within the molecule. This also explains the transition from multiplet to featureless structure of the Fe L3-edge after Co deposition, i.e. Fe is expelled from its coordination environment in the porphyrin. Both Fe and Co absorption spectra consistently demonstrate a substitution of Fe by Co. For a quantitative evaluation of the amount of the metal exchanged, the FeTPP + Co XAS signal was decomposed into the sum of the XAS of FeTPP and metallic Fe in separately grown clusters, measured independently, by varying the ratio between these two spectra (Fig. SI1†). The sum of FeTPP and metallic Fe signal at the Fe edge reproduce well the shape of the XAS signal for FeTPP + Co and gives a roughly 50% exchange yield, even in excess of Co (Co/Fe ≈ 1.2). Consistently, the spectral shape of the equivalent Co L2,3-edge can be decomposed into a mixture of signals from pure CoTPP and Co clusters (Fig. SI1†). Note that although the signal from Fe cluster is used for the decomposition, the expelled Fe atoms likely remain underneath the porphyrin molecule in a metallic state as will be discussed in the following section.
DFT calculations provide further support to the proposed metal exchange in the porphyrin, which is thermodynamically driven and accompanied by a small reaction barrier. A Co atom impinging from the metal beam can spontaneously replace the Fe centre of the molecule through a billiard-like direct mechanism. This process in which the Fe atom is pushed down to the surface by the arriving Co atom is non-activated and highly exothermic (ΔE = −5.6 eV). In Fig. 3a, we schematically show the corresponding minimum energy pathway. Along this path there is only a shallow potential well associated with the Co atom adsorbed above the Fe centre of the molecule (II), from which the system can evolve to the final state (III) with a minor activation energy barrier of ∼0.2 eV (defined with respect to the energy of state II). Since the exothermicity of the non-activated initial Co adsorption is 2.7 eV (see Fig. 3a) and thus very large compared to the reaction barrier, the Fe → Co replacement process can take place in a direct billiard-like mechanism before settling in state II, which would demand the dissipation of a large amount of energy. On the other hand, if such a thermalization process should take place, the Fe → Co replacement could still proceed easily along this pathway at room temperature due to thermal fluctuations.
 | ||
| Fig. 3 DFT calculations. (a) Calculated reaction barriers of (I) initial, (II) intermediate, and (III) final state of the transmetalation. (b) Projected density of states onto Fe and Co d-atomic orbitals in the final state. | ||
As schematically shown in Fig. 3a, in the final state III the Fe atom is adsorbed on the surface and acts as an extra link between the CoTPP molecule and the gold surface (see Fig. SI2† for an atomistic representation). Thus, the surface plays a key role in the stabilization of the final state as well as for the reduction of the energy barrier for the exchange (see Fig. SI3† for a comparison with the energetics of the replacement reaction in absence of a substrate). It is important to mention that state III is the one with the lowest energy among the many investigated structures with a M1 atom and a M2TPP molecule near the Au(111) surface (M1, M2 = Co, Fe) and thus constitutes the thermodynamically most stable state. The full list of investigated structures can be found in Table SI1 in the ESI.† The energy cost that accompanies the diffusion of the expulsed Fe atom from underneath the newly formed CoTPP molecule is 1.9 eV (see Table SI1G†), which hinders Fe segregation and formation of iron clusters on the surface.
It might be argued that the Fe → Co replacement can also proceed through an indirect multiple-step mechanism involving (i) Co adsorption on a clean patch of Au(111), (ii) Co diffusion underneath the FeTPP molecule, and (iii) Fe → Co replacement. However, such a mechanism is much less likely to take place than the billiard-like one because during arrival and diffusion of the Co atom on the surface most of its adsorption and kinetic energy will dissipate and will not be available anymore to drive the reaction. In addition, step iii involves a larger activation energy barrier of ∼0.9 eV (see Table SI1D and E†), which, although accessible at room temperature, would yield much lower reaction rates than the billiard-like mechanism.
The exchange M1(II)TPP + M2(0) → M2(II)TPP + M1(0) occurs in the trans configuration and is substantially different to the common transmetalation of porphyrins in solution,41–45 for which an SE2-mechanism involving an atop complex is usually proposed. Bader charge analysis shows how charges redistribute during the transmetalation. Fe in FeTPP/Au(111) has a charge of 6.9e and is slightly reduced with respect to the FeTPP molecule far from the surface (charge on Fe 6.8e). In the initial state with Co sitting slightly off centre on top of the molecule, Fe is further reduced to 7.0e, while Co is oxidized compared to metallic Co (8.4e vs. 8.9e, respectively). The interaction of Co with the pyrrolic nitrogen and the Fe cation reorganizes the charge distribution such that Co is oxidized and Fe reduced, which can be viewed as the first step in the redox transmetalation. In the final state, the Fe atom sandwiched between the CoTPP molecule and the substrate is further reduced to 7.2e, whereas Co is oxidized to 8.2e, completing the redox process of charge transfer from one metal to the other. Though charge assignment to atoms in an organometallic molecule is difficult in general,46 the Bader analysis should provide a qualitatively correct picture of the charge transfer processes taking place during transmetalation. From the calculated projected density of states (PDOS) for the final state, i.e., Co in the porphyrin and Fe underneath, it becomes clear that the metallic character of the Fe XAS signal results from a hybridization of the unoccupied Fe d-levels with the underlying gold substrate and the interaction of Fe with CoTPP, leading to a broad and finite PDOS for energies near the Fermi level (Fig. 3b, red curve). In a similar analysis, the PDOS of a Co centre underneath a FeTPP molecule (structure D in Table SI1†) shows that in this case Co is in a metallic state and Fe shows discrete d-levels as expected for coordinated metal centres (Fig. SI4†). This rules out the possibility of such a structure being compatible with the multiplet splitting observed in the Co XAS in Fig. 2.
Within the picture offered here, sublimed Co impinging onto a porphyrin during the deposition process spontaneously replaces Fe in the porphyrin, whereas Co that lands first on the surface has a larger transmetalation barrier and possibly also a larger kinetic barrier if diffusion underneath the molecules is hindered. This part of deposited Co thus cannot contribute to the exchange process. The replaced Fe atom most likely remains underneath the molecule at room temperature as its diffusion from underneath the molecule is accompanied by a large energetic barrier of >1.9 eV.
This model together with further calculations also helps to rationalize the 50% exchange yield. The analysis of the interaction of Co with FeTPP shows that the molecular macrocycle attracts Co atoms favouring the encounter with Fe centres. For instance, at 2.15 Å above the molecular plane (∼5.45 Å above the outermost Au layer), the potential energy over the macrocycle is ∼−2 eV (with respect to Co far from the molecule in vacuum) and ∼1 eV smaller than on an uncovered surface patch between two molecules. Since the area of the molecular macrocycle represents ∼50% of the total area of FeTPP in the densely packed monolayer, this provides a possible explanation for the ∼50% exchange yield observed experimentally. Roughly half of the sublimed Co impinges onto a macrocycle during the deposition process and spontaneously replace Fe in the porphyrin. The rest of Co atoms land on the metal surface and encounter a significantly larger transmetalation barrier. This reasoning is further corroborated by the observed clustering of Fe on top of CoTPP at low temperature, possibly due to the funnelling of impinging atoms onto the macrocycle to a stable adsorption site close to the embedded metal centre.11
Identical observations on the transmetalation were also made for FeTPyP + Co, where peripheral pyridyl groups offer additional coordination sites and might capture Co and thus suppress the exchange. However, transmetalation occurs in FeTPyP + Co (Fig. SI5†) as well as in CuTPyP + Co (Fig. SI6†). The backreaction, as can be expected from thermodynamic considerations offered by DFT for M1 + M2TPP/Au(111), is experimentally not observed when Fe is deposited in approximately stoichiometric amounts onto a CoTPyP monolayer (Fig. SI7†). We note that local tunnelling spectroscopy was inconclusive to demonstrate the exchange process at the molecular scale as spectra of FeTPyP, CoTPyP, and FeTPyP + Co show very similar occupied and unoccupied states (see Fig. SI8†) with no appreciable energy shifts as a function of metal ion in the porphyrin or the coordination of a second metal centre to its pyridyl groups.
The partial transmetalation observed here stands in contrast to the metalation of free-base porphyrins at surfaces, which can be routinely employed to achieve complete synthesis of metalloporphyrins with high yield.17,19,20 However, optimizing deposition coverage and deposition times should in principle lead to higher yields also for transmetalation. The very low barrier that can be surmounted also at low temperature separates our approach from previous studies, in which Cu adatoms were used to replace cations in porphyrins.25,26 The high annealing temperatures needed for these reactions are presumably due to the different starting configuration, in which the replacing metal centre starts from underneath the molecule, which is, as shown above, accompanied by a larger barrier.
Conclusions
The presented experimental data provide evidence for the transmetalation of metalloporphyrin monolayers at the solid/vacuum interface. Co replaces Fe and Cu in porphyrins, driven by favourable thermodynamics of stronger binding energy of Co in the porphyrin. In contrast to the common SE2-mechanism of porphyrin transmetalation in solution, a redox transmetalation is proposed for the molecules bound to a surface in vacuum. A very low reaction barrier of 0.2 eV accompanies the exchange and the expelled metal centre resides underneath the molecule in the final state. The availability of a second coordination environment in the form of pyridyl groups, which is freely accessible to the newly introduced metal centres, does not inhibit transmetalation. This is possibly a result of the particular exchange process presented here, in which the sublimed atoms arriving onto the macrocycle are funnelled towards the embedded metal centre.Our results are of particular interest for the fabrication of well-defined metallo-molecular structures on surfaces for applications such as molecular spintronics47 or catalysis,15 and the implications are far reaching everywhere the cation defines chemical or physical properties of the structure. This might open the way for the synthesis of novel metalloporphyrins that are not accessible via solution chemistry. Since the metal centre in the porphyrin substantially contributes to chemical, electronic, and magnetic properties of the molecules, uncontrolled transmetalation may prove diametrical to the tailored synthesis of porphyrin structures with desired properties, and may hinder or inhibit interpretation of experimental data. On the other hand, once kinetics and thermodynamics of the transmetalation can be controlled it will offer a novel route towards complex multimetallic-organic nanostructures. In particular, the reaction yield should be improved from the fair 50% yield to a more quantitative yield. For this goal to be achieved, further studies are required that address the interplay between the two metal centre to be exchanged, the exact function of the substrate, and possibly also the functionalization of porphyrin/phthalocyanine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by ANPCyT project PICT 2016-2750. We acknowledge computer time provided by the CCT-Rosario Computational Center and the Computational Simulation Center (CSC) for Technological Applications, members of the High Performance Computing National System (SNCAD, MincyT-Argentina). Open Access funding provided by the Max Planck Society.References
- C. C. W. Hsia, N. Engl. J. Med., 1998, 338, 239–248 CrossRef CAS PubMed.
- L. O. Björn, G. C. Papageorgiou, R. E. Blankenship and Govindjee, Photosynth. Res., 2009, 99, 85–98 CrossRef PubMed.
- S. Chunnian and F. C. Anson, Inorg. Chem., 1990, 29, 4298–4305 CrossRef.
- C. Costentin, H. Dridi and J. M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535–13544 CrossRef CAS PubMed.
- M. Jurow, A. E. Schuckman, J. D. Batteas and C. M. Drain, Coord. Chem. Rev., 2010, 254, 2297–2310 CrossRef CAS PubMed.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- D. den Boer, M. Li, T. Habets, P. Iavicoli, A. E. Rowan, R. J. M. Nolte, S. Speller, D. B. Amabilino, S. De Feyter and J. A. A. W. Elemans, Nat. Chem., 2013, 5, 621–627 CrossRef CAS PubMed.
- K. Seufert, W. Auwärter and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 18141–18146 CrossRef CAS PubMed.
- M. Bernien, J. Miguel, C. Weis, M. E. Ali, J. Kurde, B. Krumme, P. M. Panchmatia, B. Sanyal, M. Piantek, P. Srivastava, K. Baberschke, P. M. Oppeneer, O. Eriksson, W. Kuch and H. Wende, Phys. Rev. Lett., 2009, 102, 047202 CrossRef CAS PubMed.
- S. Vijayaraghavan, W. Auwärter, D. Ecija, K. Seufert, S. Rusponi, T. Houwaart, P. Sautet, M. L. Bocquet, P. Thakur, S. Stepanow, U. Schlickum, M. Etzkorn, H. Brune and J. V. Barth, ACS Nano, 2015, 9, 3605–3616 CrossRef CAS PubMed.
- K. Suto, S. Yoshimoto and K. Itaya, Langmuir, 2006, 22, 10766–10776 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Rodríguez, M. L. Pegis and J. M. Mayer, J. Am. Chem. Soc., 2014, 136, 12544–12547 CrossRef CAS PubMed.
- P. Vasudevan, Santosh, N. Mann and S. Tyagi, Transition Met. Chem., 1990, 15, 81–90 CrossRef CAS.
- B. Wurster, D. Grumelli, D. Hötger, R. Gutzler and K. Kern, J. Am. Chem. Soc., 2016, 138, 3623–3626 CrossRef CAS PubMed.
- W. Hieringer, K. Flechtner, A. Kretschmann, K. Seufert, W. Auwärter, J. V. Barth, A. Görling, H.-P. Steinrück and J. M. Gottfried, J. Am. Chem. Soc., 2011, 133, 6206–6222 CrossRef CAS PubMed.
- T. E. Shubina, H. Marbach, K. Flechtner, A. Kretschmann, N. Jux, F. Buchner, H. P. Steinrück, T. Clark and J. M. Gottfried, J. Am. Chem. Soc., 2007, 129, 9476–9483 CrossRef CAS PubMed.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. V. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- F. Buchner, V. Schwald, K. Comanici, H. P. Steinrück and H. Marbach, ChemPhysChem, 2007, 8, 241–243 CrossRef CAS PubMed.
- W. Auwärter, A. Weber-Bargioni, S. Brink, A. Riemann, A. Schiffrin, M. Ruben and J. V. Barth, ChemPhysChem, 2007, 8, 250–254 CrossRef PubMed.
- J. M. Gottfried, K. Flechtner, A. Kretschmann, T. Lukasczyk and H. P. Steinrück, J. Am. Chem. Soc., 2006, 128, 5644–5645 CrossRef CAS PubMed.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS PubMed.
- K. Diller, A. C. Papageorgiou, F. Klappenberger, F. Allegretti, J. V. Barth and W. Auwärter, Chem. Soc. Rev., 2016, 45, 1629–1656 RSC.
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H. P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 CrossRef CAS PubMed.
- C. M. Doyle, J. P. Cunniffe, S. a. Krasnikov, A. B. Preobrajenski, Z. Li, N. N. Sergeeva, M. O. Senge and A. a. Cafolla, Chem. Commun., 2014, 50, 3447–3449 RSC.
- K. Shen, B. Narsu, G. Ji, H. Sun, J. Hu, Z. Liang, X. Gao, H. Li, Z. Li, B. Song, Z. Jiang, H. Huang, J. W. Wells and F. Song, RSC Adv., 2017, 7, 13827–13835 RSC.
- M. Franke, F. Marchini, N. Jux, H. P. Steinrück, O. Lytken and F. J. Williams, Chem. – Eur. J., 2016, 22, 8520–8524 CrossRef CAS PubMed.
- Z. Shi and N. Lin, ChemPhysChem, 2010, 11, 97–100 CrossRef CAS PubMed.
- A. Rieger, S. Schnidrig, B. Probst, K. H. Ernst and C. Wäckerlin, J. Phys. Chem. Lett., 2017, 8, 6193–6198 CrossRef CAS PubMed.
- D. Heim, D. Écija, K. Seufert, W. Auwärter, C. Aurisicchio, C. Fabbro, D. Bonifazi and J. V. Barth, J. Am. Chem. Soc., 2010, 132, 6783–6790 CrossRef CAS PubMed.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- C. Piamonteze, U. Flechsig, S. Rusponi, J. Dreiser, J. Heidler, M. Schmidt, R. Wetter, M. Calvi, T. Schmidt, H. Pruchova, J. Krempasky, C. Quitmann, H. Brune and F. Nolting, J. Synchrotron Radiat., 2012, 19, 661–674 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- J. Miguel, C. F. Hermanns, M. Bernien, A. Krüger and W. Kuch, J. Phys. Chem. Lett., 2011, 2, 1455–1459 CrossRef CAS.
- A. Scherz, E. K. U. Gross, H. Appel, C. Sorg, K. Baberschke, H. Wende and K. Burke, Phys. Rev. Lett., 2005, 95, 3–6 CrossRef PubMed.
- C. T. Chen, Y. U. Idzerda, H.-J. Lin, N. V. Smith, G. Meigs, E. Chaban, G. H. Ho, E. Pellegrin and F. Sette, Phys. Rev. Lett., 1995, 75, 152–155 CrossRef CAS PubMed.
- T. Regan, H. Ohldag, C. Stamm, F. Nolting, J. Lüning, J. Stöhr and R. White, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 1–11 CrossRef.
- H. Baker, P. Hambright, L. Wagner and L. Ross, Inorg. Chem., 1973, 12, 2200–2202 CrossRef CAS.
- J. Reid and P. Hambright, Inorg. Chim. Acta, 1979, 33, L135–L136 CrossRef CAS.
- A. Shamim and P. Hambright, J. Inorg. Nucl. Chem., 1980, 42, 1645–1647 CrossRef CAS.
- C. Grant and P. Hambright, J. Am. Chem. Soc., 1969, 91, 4195–4198 CrossRef CAS.
- P. Hambright and P. B. Chock, J. Am. Chem. Soc., 1974, 96, 3123–3131 CrossRef CAS PubMed.
- P. Karen, P. Mcardle and J. Takats, Pure Appl. Chem., 2014, 86, 1017–1081 CAS.
- N. Ballav, C. Wäckerlin, D. Siewert, P. M. Oppeneer and T. A. Jung, J. Phys. Chem. Lett., 2013, 4, 2303–2311 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional XAS, STM/STS, and DFT data. See DOI: 10.1039/c8nr04786c |
| ‡ Present Address: Institut für Angewandte Physik, TU Braunschweig, 38106 Braunschweig, Germany. |
| This journal is © The Royal Society of Chemistry 2018 |