
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare-earth-doped fluoride nanoparticles with engineered long luminescence lifetime for time-gated in vivo optical imaging in the second biological window†
Meiling
Tan‡
a,
Blanca
del Rosal‡
 b,
Yuqi
Zhang
a,
Emma
Martín Rodríguez
*dg,
Jie
Hu
cd,
Zhigang
Zhou
e,
Rongwei
Fan
e,
Dirk H.
Ortgies
cd,
Nuria
Fernández
f,
Irene
Chaves-Coira
h,
Ángel
Núñez
h,
Daniel
Jaque
cd and
Guanying
Chen
*a
b,
Yuqi
Zhang
a,
Emma
Martín Rodríguez
*dg,
Jie
Hu
cd,
Zhigang
Zhou
e,
Rongwei
Fan
e,
Dirk H.
Ortgies
cd,
Nuria
Fernández
f,
Irene
Chaves-Coira
h,
Ángel
Núñez
h,
Daniel
Jaque
cd and
Guanying
Chen
*a
aMIIT Key Laboratory of Critical Materials Technology for New Energy Conversion and Storage, School of Chemistry and Chemical Engineering & Key Laboratory of Micro-systems and Micro-structures, Ministry of Education, Harbin Institute of Technology, 150001 Harbin, People's Republic of China. E-mail: chenguanying@hit.edu.cn
bCentre for Micro-Photonics, Faculty of Science, Engineering and Technology, Swinburne University of Technology, PO Box 218, Hawthorn, VIC 3122, Australia
cDepartamento de Física de Materiales, Universidad Autónoma de Madrid, Madrid 28049, Spain. E-mail: emma.martin@uam.es
dInstituto Ramón y Cajal de Investigación Sanitaria, IRYCIS, Ctra. Colmenar km. 9.100, Madrid 28034, Spain
eNational Key Laboratory of Tunable Lasers, Institute of Optical-Electronics, Harbin Institute of Technology, 150001 Harbin, People's Republic of China
fDepartamento de Fisiología, Facultad de Medicina, Avda. Arzobispo Morcillo 2, Universidad Autónoma de Madrid, 28029 Madrid, Spain
gDepartamento de Física Aplicada, Universidad Autónoma de Madrid, Madrid 28049, Spain. E-mail: emma.martin@uam.es
hDepartamento de Anatomía, Histología y Neurociencia, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
First published on 23rd August 2018
Abstract
Biomedicine is continuously demanding new luminescent materials to be used as optical probes for the acquisition of high resolution, high contrast and high penetration in vivo images. These materials, in combination with advanced techniques, could constitute the first step towards new diagnosis and therapy tools. In this work, we report on the synthesis of long lifetime rare-earth-doped fluoride nanoparticles by adopting different strategies: core/shell and dopant engineering. The here developed nanoparticles show intense infrared emission in the second biological window with a long luminescence lifetime close to 1 millisecond. These two properties make the here presented nanoparticles excellent candidates for time-gated infrared optical bioimaging. Indeed, their potential application as optical imaging contrast agents for autofluorescence-free in vivo small animal imaging has been demonstrated, allowing high contrast real-time tracking of gastrointestinal absorption of nanoparticles and transcranial imaging of intracerebrally injected nanoparticles in the murine brain.
1. Introduction
Fluorescence-based optical imaging utilizes color-encoded emissions (typically in the visible range) from endogenous or exogenous fluorophores to acquire detailed images of organs and tissues as well as subcellular structures to unravel biological complexities.1 This technique is non-invasive and non-ionizing, and allows video-rate imaging with high spatial resolutions not only at the optical microscopy level (sub-micrometre, ca. 250 nm), but also at the optical diffraction-limited level (optical super-resolution, ca. 20 nm).1–5 As a result, fluorescence imaging is applied in a plethora of fields ranging from pre-clinical testing and clinical diagnosis, to precise image-guided surgical excision of tumors, and to molecular-level monitoring of disease progression.6–12 However, fluorescence imaging has been limited by the low penetration depth of light into tissues (<1 mm for wavelengths in the visible range of 400–650 nm) and by the autofluorescence of biological tissues.13,14 These two problems result in the loss of spatial resolution and the decrease of the signal to noise ratio, and thus in a serious deterioration of the acquired in vivo images. The problem of penetration has been alleviated in the past years, as it has been shown that centimeter-scale imaging depths can be accomplished when shifting both fluorescence and excitation wavelengths from the visible to the near infrared (NIR) optical biological windows, where the absorption of tissues reaches a minimum. The NIR region of 650–950 nm is known as the first biological window (NIR I), while the NIR region of 1000–1350 nm is known as the second biological window (NIR II) (Fig. 1a). Recent experimental results have demonstrated that using fluorescent labels emitting in NIR II instead of NIR I result in an improved contrast and imaging depth due to the reduction of light scattering,15,16 which scales as λ−α, where λ is the wavelength and α has a value from 0.2 to 4 depending on the composition of the tissue.17 Therefore, a range of fluorescent materials with excitation in NIR I and emission in NIR II have been developed and tested for small animal imaging. These materials span from small organic NIR fluorescent dyes to inorganic nanoparticles (NPs), such as carbon nanotubes,18 rare earth doped nanocrystals,19 and NIR quantum dots20–22 (among them, Ag2S quantum dots can be highlighted),23,24 enabling through-skull fluorescence imaging of the murine brain with sub-10 μm resolution.16 Yet, even in NIR II, infrared-excited autofluorescence from tissues remains substantial, as some biological components show non-negligible emissions beyond 1000 nm, resulting in a dramatic reduction of the image contrast.13 Optical probes with longer emission wavelengths (>1300 nm) can help improve the contrast by minimizing spectral overlap with autofluorescence; however, such probes remained rare.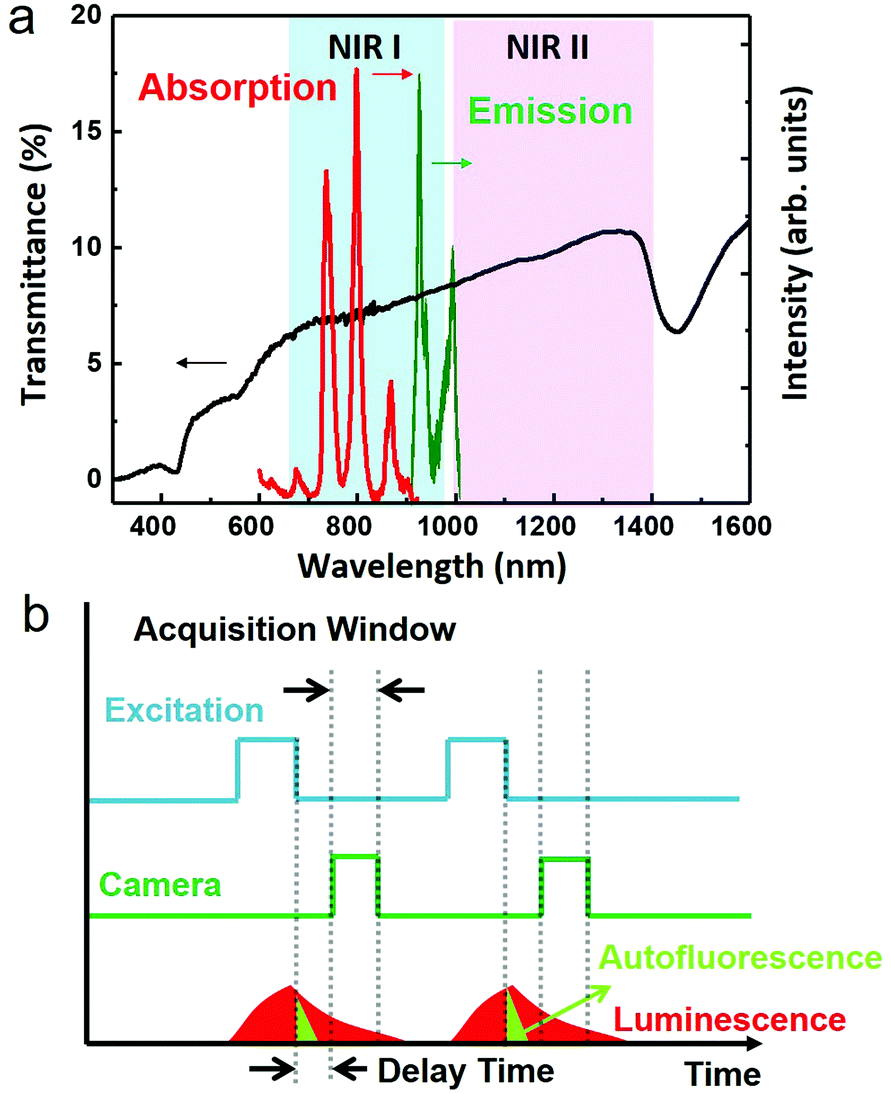 | ||
| Fig. 1 a. Schematic illustration of the first (NIR I) and second (NIR II) biological windows. The black curve represents the transmission of light through a nude mouse skin (thickness of 1 mm). The absorption and emission spectra of our synthesized NaYF4:Yb3+,Nd3+@CaF2 NPs are also included in this figure for reference. b. A schematic depiction of the general principle of time-gated optical imaging. Periodic excitation is usually triggered by high repetition pulsed lasers. Image acquisition triggered a determined delay time after the end of each laser pulse. | ||
Time-gated optical imaging is an established stroboscopic technique, which can eliminate the nuisance of autofluorescence by exploiting the long lifetime of emissions from optical probes (∼μs–ms) against the short lifetime (∼ns) of tissue autofluorescence.25–28 Despite their spectral overlap, the difference of emission profiles in the time domain allows a straightforward removal of the autofluorescence background by capturing images in a time window that excludes the unwanted natural emissions from tissue endogenous luminophores (Fig. 1b).28,29 This technique employs periodic laser pulses to perform light excitation, while the excitation train of pulses is synchronized, but with a precisely defined time delay (longer than the lifetime of the autofluorescence), with the activation of the camera. It has been shown that time-gated imaging, with long lifetime lanthanide complexes (typically containing europium or terbium), can allow high contrast fluorescence microscopy imaging of cells in the visible range.30 Moreover, in combination with the long emission lifetime (5–13 μs) of porous silicon nanoparticles, time-gated optical imaging enables a >20-fold improvement of the signal to background contrast ratio in vivo when imaging with photoluminescence (600–900 nm) in the NIR I window.29,31 Despite these advantages, the use of time-gated techniques for in vivo optical imaging in the NIR II window remains elusive, because most NIR II luminophores have emission lifetimes commensurate with that of autofluorescence, significantly limiting their usefulness in this regard.
Lanthanide-based luminescent nanomaterials constitute an emerging class of promising biolabels for in vivo time-gated optical imaging in NIR II. This is because lanthanide ions have not only abundant energy levels in the infrared range, but also typically long emission lifetimes (on the order of 10−4–10−3 s).32,33 Moreover, their advantages of characteristic narrow excitation and emission bands, absence of photobleaching, and no known significant toxic effects neither in in vivo nor in in vitro, make them perfect as biolabels for optical bioimaging in the time domain.28 Though particles doped with infrared-emitting lanthanide ions (such as neodymium, Nd3+, or erbium, Er3+) have been reported for steady-state in vivo imaging in the NIR II window, the advantages of NIR II lanthanide-doped particles with a tailored long lifetime emission for time domain imaging have not yet been revealed.34–41 Recently, we demonstrated in a proof-of-concept that NaGdF4:Nd3+ particles with a size of 600–800 nm could enable time-gated optical imaging of mice at 1050 nm, entailing a remarkable improvement in the contrast of fluorescence images due to the removal of autofluorescence.37 However, to reach sufficiently long lifetimes (200 μs) and efficient luminescence, micrometer-scale particle size had to be employed, which is too large for many bioapplications. It is, therefore, important to develop small-sized lanthanide-doped nanocrystals with tailored long lifetime luminescence for time-gated optical imaging in the NIR II window.
By examining the energy level structures of lanthanide ions, we found that ytterbium (Yb3+) ions possess a unique structure and have only one single excited state (2F5/2), which produces an emission centered at 1000 nm (the 2F7/2 → 2F5/2 transition) lying right across the NIR I and NIR II windows (Fig. 1a).42,43 Moreover, the incorporation of Yb3+ ions in crystallized materials typically results in emission lifetimes of hundreds of microseconds, several orders of magnitudes above the autofluorescence lifetime.44 Both features make Yb3+ ion-containing nanocrystals attractive for NIR II time-gated optical imaging. Note that Yb3+ ions are commonly used as sensitizers for many activators (Er3+, Ho3+, Tm3+, etc.) in doped NPs to produce intense photon upconversion.42,45 In fact, they do have excellent radiative properties to be utilized as emitters, as many types of commercial lasers have employed Yb3+ ions to realize a tunable wavelength lasing output at ∼1000 nm.46,47 However, to excite Yb3+ ion emitters, a laser output with a wavelength at ∼980 nm has to be used, which, unfortunately, overlaps largely with the absorption peak of water molecules that are dominant in biological samples. Overexposure of biological species to 980 nm light would cause overheating issues, resulting in significant cell death and tissue damage.43,48–51 This problem can be addressed by co-doping NPs with neodymium (Nd3+) ions, which act as sensitizers for Yb3+ ions (Fig. 2a) and have an intense absorption band at around 800 nm where water molecules and biological tissues have an about 10 times lower absorption coefficient. As a result, we reason that small-sized Nd3+/Yb3+ co-doped NPs with engineered bright and long lifetime luminescence at 1000 nm hold promise for employment as fluorescent biolabels for in vivo time-gated optical imaging in the NIR II window.
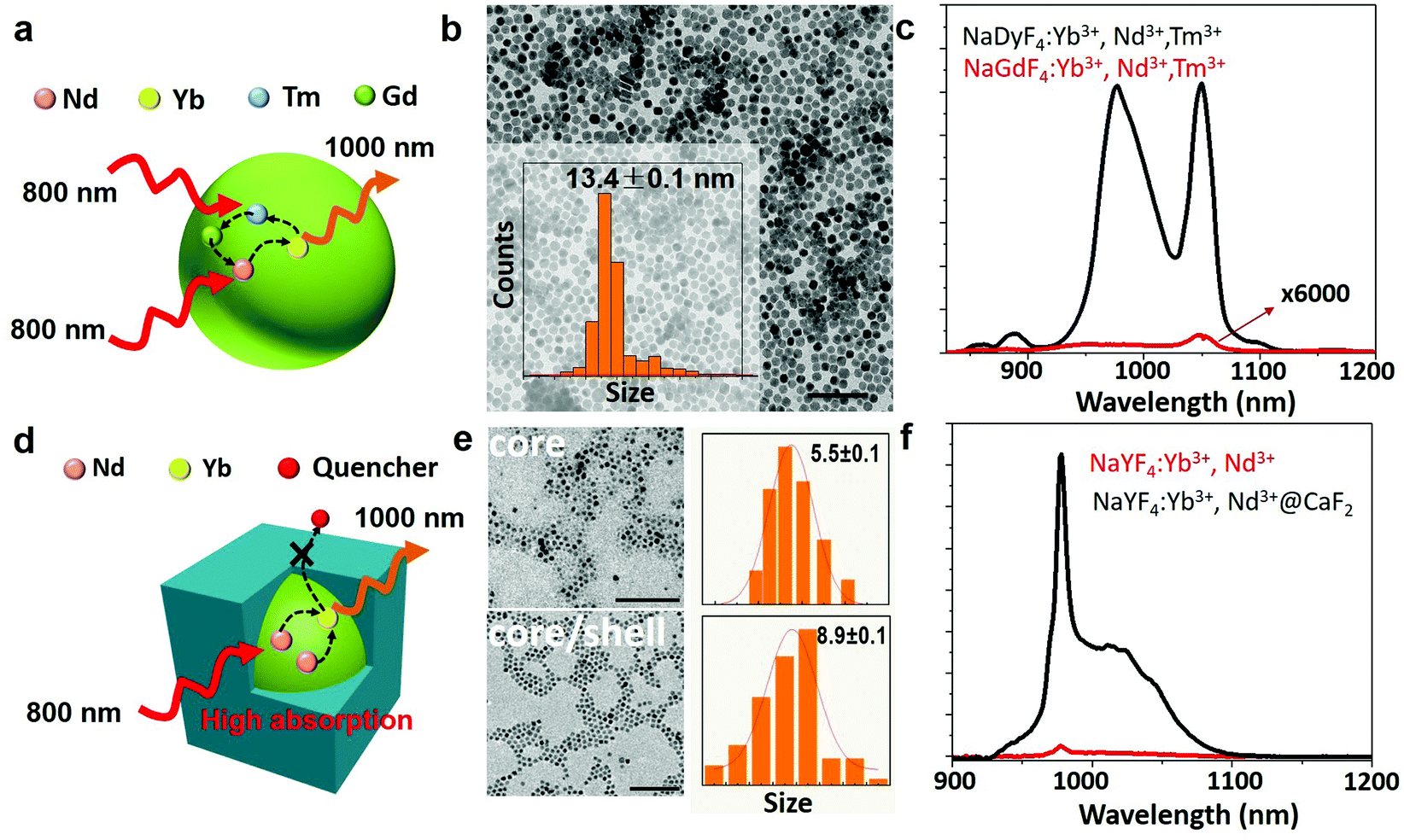 | ||
| Fig. 2 a. Schematic illustration of energy transfer in the NaGdF4:2% Yb,3% Nd,0.2% Tm NPs; b. TEM image of the NaGdF4:2% Yb,3% Nd,0.2% Tm NPs (scale bar, 100 nm), with their size distribution shown in the inset; c. emission spectra from the NaGdF4:2% Yb,3% Nd,0.2% Tm NPs and the NaDyF4:2% Yb,3% Nd,0.2% Tm NPs in hexane under excitation at 790 nm; d. schematic illustration of the core/shell structure designed to suppress the surface quenching effect; e. TEM image of the core NaYF4:10% Yb3+,30% Nd3+ and core/shell NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs (scale bar, 100 nm), with their size distribution shown (right); and f. emission spectra from the core and the core/shell NPs dispersed in hexane under excitation at 800 nm. | ||
In this work, we report on two approaches to produce a class of sub-15 nm Nd/Yb-codoped luminescent NPs with enhanced emission efficiency and long lifetime, allowing high contrast time-gated in vivo imaging in the NIR II window. The first one is to incorporate thulium (Tm3+) dopants in Yb3+/Nd3+ codoped fluoride NPs (∼13 nm) devoid of shell protection, while the second one is to utilize a core/shell structure to produce enhanced and long lifetime emissions from NaYF4:Yb3+,Nd3+@CaF2 core/shell NPs (∼9 nm). For the first case, we compared the effect of Tm3+ doping in two fluoride hosts (NaGdF4 and NaDyF4) and observed that the presence of Gd3+ in the host crystal gives NPs with a lifetime longer than 1 millisecond. In the second case, we performed a systematic optimization of doping concentrations of both Nd3+ and Yb3+ ions in the NaYF4 core, which enabled enhancing the absorption of excitation light at 800 nm while shortening the distance between Nd3+ and Yb3+ for an improved energy transfer efficiency. Coating with an inert biocompatible CaF2 shell served the purpose of reducing surface-related quenching, thereby increasing the luminescence intensity at 1000 nm by about 45 times and lifetime from about 50 to 830 μs. The suitability of the NPs developed in both approaches for in vivo time-gated optical imaging in the NIR II window has been demonstrated, showing superior performance to that of commercially available Ag2S NPs.
2. Experimental
All synthesis, characterization, and imaging details are described in detail in the ESI.† All in vivo experiments carried out in this work were approved by the Ethics Committee from Universidad Autónoma of Madrid (CEIT) in the frame of the project MAT2010-21270-C04-01 supported by the Spanish Ministerio de Economía y Competitividad.3. Results and discussion
We first explored the possibility of obtaining long luminescence-lifetime NPs in single core NPs. We selected NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs, due to their good luminescence properties already reported.52 The synthesis procedure of NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs is described in the ESI.† The host materials and dopants that enhance the energy transfer process can lead to an intense and long lifetime Yb3+ emission. The mechanism is represented schematically in Fig. 2a and in detail in Fig. S1.† The co-doped Nd3+ and Tm3+ ions, which increase the absorption at 800 nm, and Gd3+ acted as the energy bridge for the energy transfer from Tm3+ to Nd3+, enhancing the emission of Yb3+ under 800 nm excitation. The size of NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs was found to be approximately 13 nm, according to the transmission electron microscopy (TEM) image and size distribution shown in Fig. 2b. For comparison, NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs were also synthesized, to study the role of Gd3+ in the Nd3+ → Yb3+ energy transfer process. Fig. 2c shows the emission spectra of NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ and NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs under 790 nm excitation under the same experimental conditions. The NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs show two intense emission bands in the region from 850 nm to 1100 nm, the first one at 980 nm, corresponding to the 2F7/2 → 2F5/2 transition of Yb3+; and the second one at 1060 nm corresponding to the 4F3/2 → 4I11/2 transition of Nd3+. In comparison, the NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ sample shows an emission intensity about 6 × 104 times weaker. It is also remarkable that the spectrum is dominated by the 1060 nm band from Nd3+, demonstrating the importance of Gd3+ in the energy transfer processes that leads to the Yb3+ emission. Dispersibility in water was achieved by coating the NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ and NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs with a PEGylated lipid (DSPE-PEG-amine). But due to the existence of surface-related quenching effects, the single core NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs cannot support the high doping concentration of lanthanides to further enhance the absorption of excitation light at 800 nm and increase the number of emitters for higher luminescence intensity. Recent results show that a core/shell structure is able to prevent the migration process of excited energy from lanthanide dopants to surface quenching sites, which mediates in part the lanthanide cross relaxation induced concentration quenching effect.53 As a result, a core/shell structure is crucial for high lanthanide doped NPs, along with the suppression of surface-related quenching.In the case of the core/shell NPs, NaYF4:Yb3+,Nd3+ NPs were used as seeds for the epitaxial growth of an inert calcium fluoride (CaF2) shell (Fig. 2d). CaF2 was chosen as the shell layer because it has a low lattice mismatch with the core, broad spectral range of optical transparency and high stability in aqueous environments; additionally, because its constituents (calcium and fluoride ions) are common components of biological tissues, which can enhance the biocompatibility of the resulting core/shell NPs.54 Both the NaYF4:Yb3+,Nd3+ core and the NaYF4:Yb3+,Nd3+@CaF2 core/shell NPs doped with varied concentrations of Yb3+ and Nd3+ were synthesized via the thermal decomposition of metallic trifluoroacetates at high temperatures (see the ESI†). The TEM image of the synthesized core NaYF4:Yb3+,Nd3+ NPs reveals a spherical shape with an average diameter of about 5.5 nm (Fig. 2e). The obtained core/shell NPs are monodisperse with a uniform, cubic shape, having an average size close to 9 nm. The X-ray diffraction (XRD) patterns indicate that both the core and the core/shell structures are of the cubic crystallographic phase (Fig. S2†). All the XRD peaks are consistent with the standard patterns of α-NaYF4 (JCPDS No. 77-2042) and CaF2 (JCPDS No. 77-2096). Energy-dispersive X-ray (EDX) spectra confirm the presence of Ca in the core/shell structure, suggesting the successful preparation of the designated NaYF4:Yb3+,Nd3+@CaF2 core/shell NPs (Fig. S3†).
The absorption spectrum of the core/shell NPs (Fig. S4†) displays the characteristic absorption bands of Nd3+ ions (at 890, 800 and 750 nm) and the absorption band of Yb3+ ions (centered at around 980 nm). The photoluminescence spectra of the core and core/shell NPs, obtained under optical excitation at 800 nm, are included in Fig. 2f. The emission spectra show two emission peaks (ca. 1000 nm), centered at 980 and 1011 nm, both of which arise from radiative transitions between Stark energy sublevels of the excited (2F5/2) and ground states (2F7/2) of Yb3+ ions. No luminescence from Nd3+ ions was observed in those NPs doped with a high lanthanide concentration. Note that the excitation spectrum of the core/shell NPs presents three emission peaks centered at 750, 800 and 860 nm, which matches well with the absorption peaks of Nd3+ ions (Fig. S4†). This spectral match unequivocally demonstrates the possibility of Nd3+ → Yb3+ energy transfer processes, thus enabling the excitation of core/shell NPs through Nd3+ absorption in the NIR I window (Fig. 2f).55,56Fig. 2f also includes the emission spectrum from the corresponding core NPs. It is evidenced that the addition of an inert CaF2 shell results in a 45 times increase in the emission intensity. This can be attributed to the reduction of surface lattice defects of the core nanocrystals that act as luminescence quenchers, as well as to the decrease of nonradiative interactions between surface lanthanide ions and luminescence quenchers from the surrounding environment (solvents, ligands, etc.), created by the spatial isolation of the epitaxial shell (Fig. 2d).57,58
To probe the impact of the absorption-enhanced fluorescence and to optimize the energy transfer between Nd3+ and Yb3+ ions, we investigated the concentration effect of sensitizer Nd3+ ions on the overall emission intensity of the core/shell structure. For this purpose, we prepared a set of α-NaYF4:10% Yb3+,x% Nd3+ core NPs doped with different Nd3+ contents (x = 10, 20, 40, 60), and then utilized the same amount of the CaF2 shell precursor for coating. All synthesized core and core/shell NPs present virtually identical sizes, as shown by the TEM images and size distribution (Fig. S5†). Increasing the Nd3+ doping concentration from 10% to 30% results in a relevant increase in the fluorescence emission intensity. However, a decrease in the emission intensity is observed if the Nd3+ doping level is further increased (50% and 80%) (Fig. S6†). No significant changes in the shape of the emission spectrum were observed, revealing a minimum distortion of the crystalline field of the host lattice (NaYF4) even at high dopant concentrations of Nd3+ ions. Whereas the absorption of the excitation light can be enhanced by increasing the Nd3+ doping concentration, the deleterious cross-relaxation processes between Nd3+ ions are simultaneously activated, which results in a reduced energy transfer to Yb3+ ions. The final emitted intensity generated by Yb3+ ions would result from the balance between these two effects, indicating an optimized Nd3+ concentration of 30%.
We next verified the role of the concentration of the activator Yb3+ ions in the overall energy transfer process by evaluating the emission intensity as a function of Yb3+ concentration in the range from 10% to 60%. As occurred when changing the concentration of Nd3+ ions, no effects on the morphology and size of the NPs were observed (Fig. S7†). Increasing the Yb3+ concentration resulted in a progressive reduction in the emission intensity (Fig. S8†). This result indicates that a long Yb3+–Yb3+ interionic distance is essential for efficient energy transfer from Nd3+ to Yb3+ ions. This has been explained in the past in terms of the activation of an Yb3+ → Nd3+ back energy transfer process that could occur for high Yb3+ concentrations (note that in a back transfer process, the Yb3+ ion acts as a sensitizer).59 Additionally, the reduction of ytterbium emission at high doping levels could also be due to the activation of concentration quenching effects as described in the literature.60 The optimal doping levels for Yb3+ and Nd3+ in the NaYF4:Yb3+,Nd3+@CaF2 core/shell NPs were determined to be 10% and 30%, respectively.
Finally, to verify that the co-doping approach (simultaneous incorporation of both Nd3+ and Yb3+ in the core) was the best choice for optimizing the energy transfer rate from Nd3+ to Yb3+, we evaluated the energy transfer process when the sensitizer and activator ions are spatially separated in the core/shell structure. In particular, we prepared the NaYF4:10% Yb3+@CaF2:30% Nd3+ core/shell structure, and compared the NIR II emission at 1000 nm with that from single-core co-doped NaYF4:10% Yb3+,30% Nd3+@CaF2 core/shell NPs. We observed that the separation of Nd3+ and Yb3+ ions in the core/shell structure lets the emission peak at 860 nm from Nd3+ ions dominate over the emission peak at 1000 nm from Yb3+ ions, as opposed to the result achieved with the co-doping approach. Moreover, the interesting NIR II emission at 1000 nm is about 10 times weaker than that of the co-doped NaYF4:10% Yb3+@CaF2:30% Nd3+ core/shell NPs (Fig. S9†). This result confirms the importance of positioning both Nd3+ and Yb3+ ions in the core of a core/shell structure to produce a strong NIR emission at 1000 nm.
Prior to their application in time-gated in vivo imaging experiments, the optimal core/shell NPs (NaYF4:10% Yb3+,30% Nd3+@CaF2) were provided with dispersibility in aqueous media by replacing the oleate molecules present on the surface of the NPs with poly(acrylic acid) (PAA, MW = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). The procedure is described in detail in the ESI.† The Fourier transform infrared (FTIR) spectra of the as-synthesized and modified NPs (Fig. S10†) showed an increased intensity of the band at 1724 cm−1 (that can be assigned to –C
000). The procedure is described in detail in the ESI.† The Fourier transform infrared (FTIR) spectra of the as-synthesized and modified NPs (Fig. S10†) showed an increased intensity of the band at 1724 cm−1 (that can be assigned to –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups) demonstrating that a large amount of COOH groups had been added to the core/shell NP surface. This is further supported by the increased intensity of the band at 3454 cm−1 (3100–3600 cm−1, OH groups), and the decreased intensity of the band at 2927 cm−1 (CH3 groups). This indicates a successful replacement of the original oleic acid ligand by PAA. In addition, the hydrodynamic size of the PAA-coated core/shell NPs was evaluated to be about 37 nm using dynamic light scattering (DLS) (Fig. S11†), much larger than the size shown in the TEM (∼9 nm) image. This larger size can be attributed to the presence of PAA in contact with the aqueous environment, evidencing the successful ligand transfer, and demonstrating that, even after the ligand transfer, the NPs are small enough for biological applications. The emission intensity from the water-dispersible core/shell NPs was about two times lower than that from the hexane-dispersed form (Fig. S12†), which could be attributed to the imperfectness of the shell and the existence of a large amount of –OH groups (a high phonon energy of ∼3500 cm−1) in water, resulting in the non-radiative depopulation of the 2F5/2 state of Yb3+ ions.61 The cytotoxicity of the NPs was evaluated via a methyl thiazolyl tetrazolium (MTT) assay on HeLa cells, which indicated no significant effects of the NPs on cell viability (Fig. S13†).
O groups) demonstrating that a large amount of COOH groups had been added to the core/shell NP surface. This is further supported by the increased intensity of the band at 3454 cm−1 (3100–3600 cm−1, OH groups), and the decreased intensity of the band at 2927 cm−1 (CH3 groups). This indicates a successful replacement of the original oleic acid ligand by PAA. In addition, the hydrodynamic size of the PAA-coated core/shell NPs was evaluated to be about 37 nm using dynamic light scattering (DLS) (Fig. S11†), much larger than the size shown in the TEM (∼9 nm) image. This larger size can be attributed to the presence of PAA in contact with the aqueous environment, evidencing the successful ligand transfer, and demonstrating that, even after the ligand transfer, the NPs are small enough for biological applications. The emission intensity from the water-dispersible core/shell NPs was about two times lower than that from the hexane-dispersed form (Fig. S12†), which could be attributed to the imperfectness of the shell and the existence of a large amount of –OH groups (a high phonon energy of ∼3500 cm−1) in water, resulting in the non-radiative depopulation of the 2F5/2 state of Yb3+ ions.61 The cytotoxicity of the NPs was evaluated via a methyl thiazolyl tetrazolium (MTT) assay on HeLa cells, which indicated no significant effects of the NPs on cell viability (Fig. S13†).
To evaluate the suitability of the NPs for time-gated imaging in the second biological window, the decay curve of the NIR II emission at 1000 nm (corresponding to the 2F5/2 → 2F7/2 transition of Yb3+ ions) from both types of NPs was acquired (Fig. 3). The decay curve could be fitted to a single exponential giving a value of τ = (1350 ± 50) μs for the NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs (Fig. 3a). This extremely long lifetime makes these particles an ideal fluorophore for time-gated imaging, as the detection can be delayed without observing a decrease in the emitted intensity from the NPs (for a delay of 1 μs the remaining signal will be 99.93% of the original signal). In good agreement with the observed emitted intensity, the lifetime of the NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs was τ = (55 ± 5) μs (Fig. 3a), supporting once more the importance of Gd3+ in the emission mechanism of Yb3+. For the PAA-coated NaYF4:10% Yb3+,30% Nd3+ and NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs, whereas only core NPs presented a lifetime of 51 μs for Yb3+ ions, the lifetime of the core/shell NPs in aqueous dispersion was determined to be as long as 833 μs, indicating their suitability for time-gated imaging (Fig. 3b). Note that the significant difference of the lifetime for the NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ and NaYF4:10% Yb3+,30% Nd3+ core NPs is due to both particles having a distinct concentration of lanthanide dopants. We also compared the emission intensity of the core/shell NPs developed in this work with that of commercial Ag2S NPs, whose emission band lies at around 1230 nm (Fig. S14†), and which constitute one of the few probes available for high contrast imaging in the second biological window.62,63 For an equivalent concentration of NPs in the aqueous dispersion (10 mg mL−1), our optimized core/shell NPs present a much brighter emission signal than Ag2S NPs. In addition, the short lifetime of Ag2S NPs (tens of nanoseconds) does not allow their application in time-gated imaging, as is demonstrated in Fig. S15,† in which a delay time of 10 μs is enough to eliminate the fluorescence signal generated by Ag2S NPs. The absolute quantum yield of our core/shell NPs was estimated to be ∼11 ± 1% using an integrating sphere method, which is much higher than the emission quantum yield of Ag2S of 0.15–4.7%.64,65 This is in agreement with the observed comparison of emission brightness.
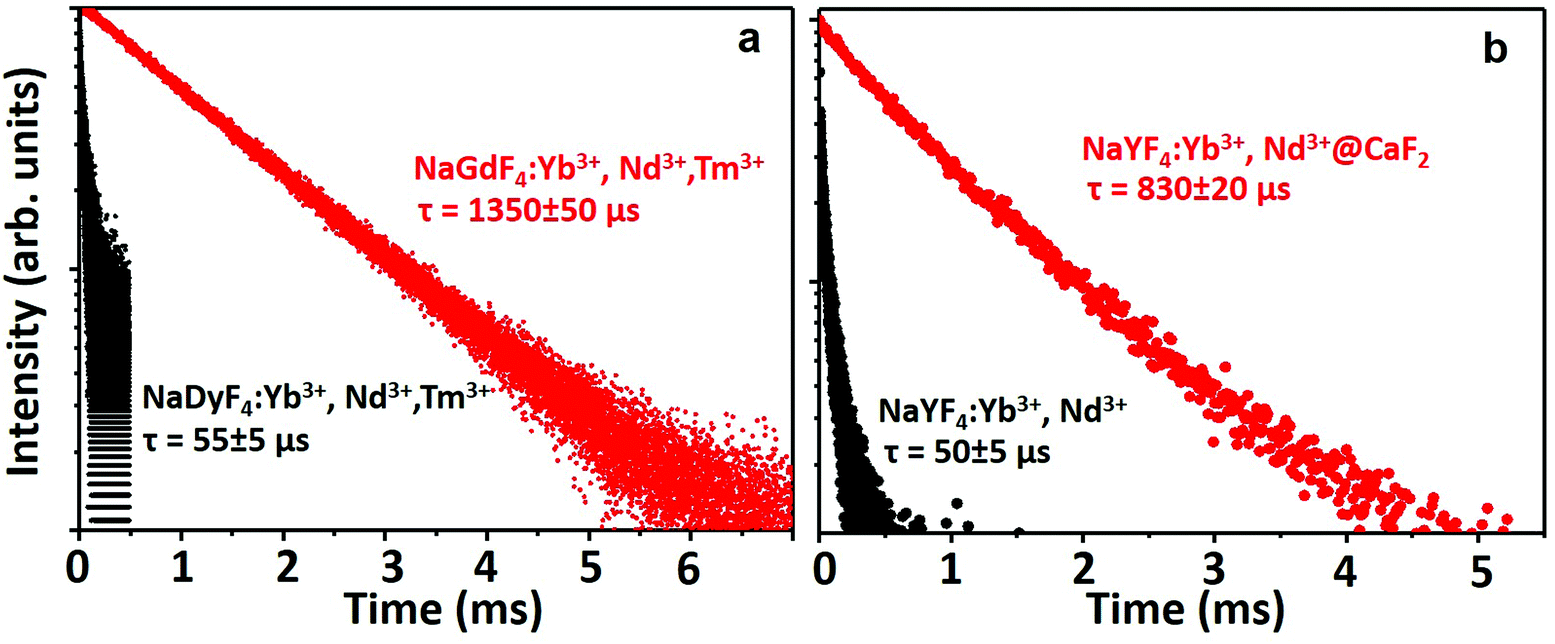 | ||
| Fig. 3 Fluorescence decay curves of the emission observed at 1000 nm corresponding to: a. NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs and NaDyF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs. b. PAA-coated NaYF4:10% Yb3+,30% Nd3+@CaF2 core/shell NPs. | ||
Their excellent optical properties led us to test both types of NPs for autofluorescence-free time-gated imaging. In particular, NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs were tested for real-time tracking of the distribution of NPs after an oral administration of 200 μL of a 4 mg mL−1 dispersion of NPs in water. To test the NPs under the most adverse conditions, C57BL/6 mice were used, as they present a very intense autofluorescence due to the skin pigmentation.13 This can be seen in Fig. 4a, which shows the ventral fluorescence image of a mouse immediately after NP administration and without applying any delay. The strong autofluorescence caused by the skin pigmentation of the animal is clearly observed, together with a strong signal coming from the abdomen, which can be attributed to the autofluorescence of the animal's diet and the liver. Once a delay of 1 μs is applied (Fig. 4b), the signal generated by the NPs can be easily localized and isolated from the autofluorescence signal. To obtain these images, only one accumulation with an integration time of 30 ms was required, which makes this system compatible with real-time NP tracking. In particular, we monitored the transit of the administered NPs from the stomach to the intestine, as shown in Fig. 4c and d. Two combined effects can be observed over time: first, as the NPs are deposited at the bottom of the stomach, the fluorescence signal displaces up and to the left, in the direction of the duodenum; and second, once the NPs have reached the exit of the stomach, the intensity decreases. This decrease in the intensity indicates the progressive transit of the NPs to the intestine. In the particular case shown in Fig. 4c, it took around 75 minutes for the majority of the NPs to exit the stomach. The lack of signals from the intestine indicates that the NPs are rapidly absorbed at the beginning of the tube. Fig. 4d shows a color-coded time-lapse image composed with the images shown in Fig. 4c. The images have been generated by overlapping the position of the maxima of fluorescence in a different color for each time. The figure indicates that the fluorescence signal is initially (reddish color) located at the bottom of the stomach, whereas later (greenish color) the NPs are concentrated in an upper-left position, which matches the position of the valve that connects the stomach to the duodenum. The ex vivo study of the organs confirmed that most of the NPs had abandoned the stomach, as only a weak emission was registered in that organ, and that the NPs had been absorbed in the intestine, as no signal from the NPs could be observed (Fig. S16†). The importance of this study stems from the fact that the transit time of substances from the stomach to the intestine, and subsequently, to the bloodstream, depends on many factors that cannot be externally controlled (e.g. the amount of food present in the stomach or the level of activity of the animal can affect the speed of the process).66,67 The transit rate, in turn, has a strong influence on the absorption of orally administered drugs as, among other reasons, it determines the concentration of the drug in the plasma and its bioavailability.68 Our approach provides a powerful alternative to the models and simulations that are usually employed to study gastrointestinal transit,67,69 because it allows real time tracking of the NP distribution thanks to their fluorescence.
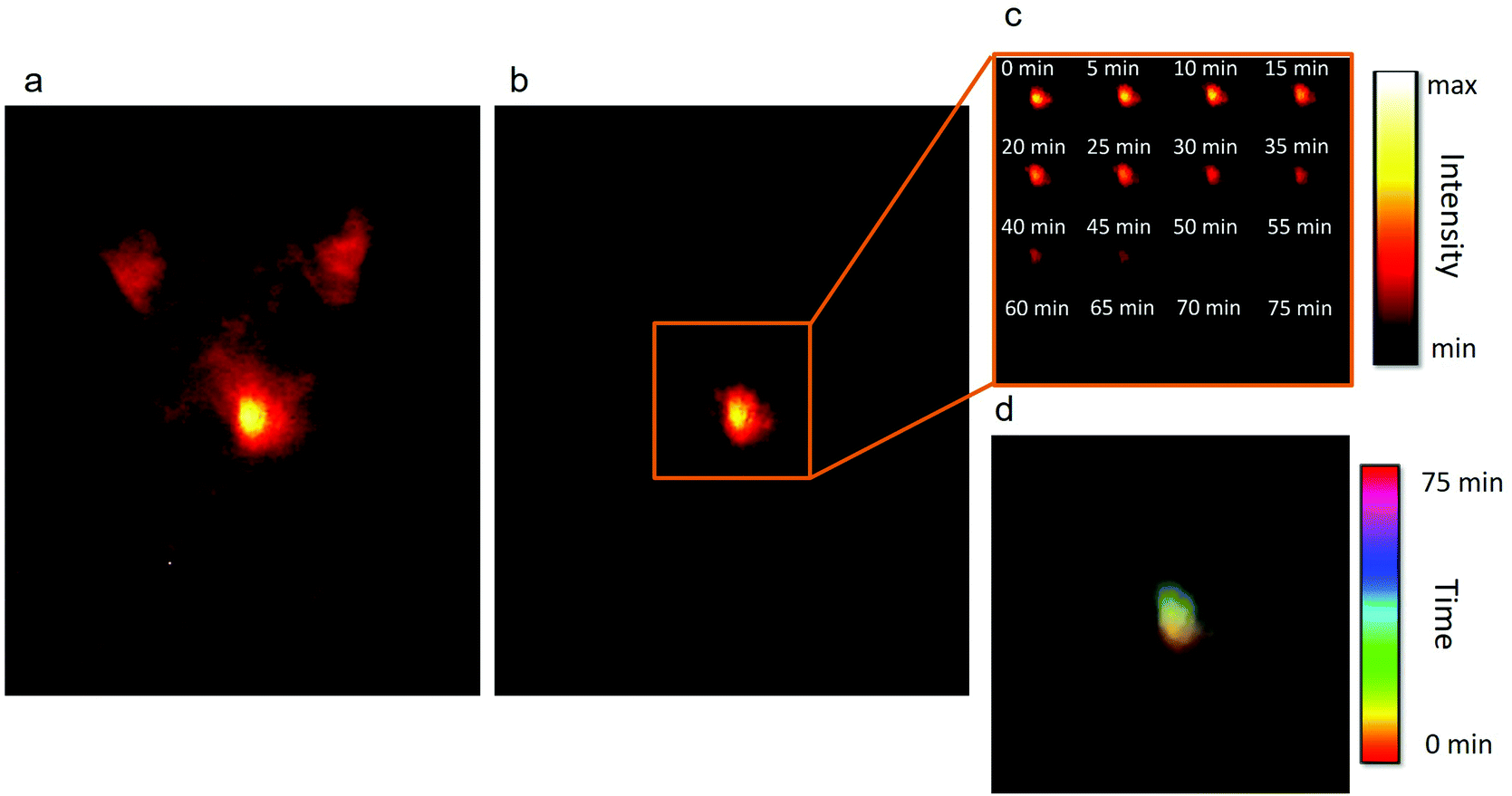 | ||
| Fig. 4 a. Fluorescence ventral image of a black CD1 mouse after oral administration of a dispersion of NaGdF4:Yb3+,Nd3+,Tm3+ NPs in water. The image was taken without time delay so the autofluorescence of the mouse is clearly seen, especially from the region of the stomach. b. Image of the same animal after applying a delay time of 1 μs. Only the signal coming from the NPs can be observed. c. Intensity-based real-time tracking of the distribution of NPs in the stomach. The signal displaces towards the duodenum and the intensity decreases as the NPs get absorbed in the intestine. d. Time-lapse color-coded composition of the images shown in c, showing the position of the NPs in one color for each time. It can be seen that the NPs are initially located at the bottom of the stomach (reddish color) but in the final moments the NPs are mainly located close to the duodenum (greenish color). | ||
A different set of experiments, also designed for their potential of autofluorescence-free in vivo imaging, was performed with the NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs. In this case, the core/shell NPs were first used for in vivo autofluorescence-free time-gated imaging. In our experiment, we compared the NIR fluorescence images obtained for a C57BL/6 mouse subcutaneously injected with our core/shell NPs and a control mouse (to which no NPs were administered) when no delay between laser pulse and image collection was applied and when a 1 μs delay time was applied (Fig. 5a). In the absence of any time delay, a strong background of autofluorescence is evidenced in both control and injected mice. Indeed, the autofluorescence-related background is so strong that it is difficult to elucidate the position of the injection, whereas an introduction of a time delay leads to a complete removal of the autofluorescence background, making it possible to trace clearly the location of the subcutaneous NP injection in the mouse. These images unmistakably indicate the capability of time-gated imaging for complete elimination of the autofluorescence background in the NIR. This is useful not only to avoid skin autofluorescence, but also to avoid undesirable organ autofluorescence, which can be affected by animal diet. In Fig. 5b, it is shown that the NIR emission signal generated by a food pellet can be successfully removed using time-gated imaging. When no decay is established, the foodstuff displays a weak but non-negligible emission signal, which disappears when time-gated imaging is applied.
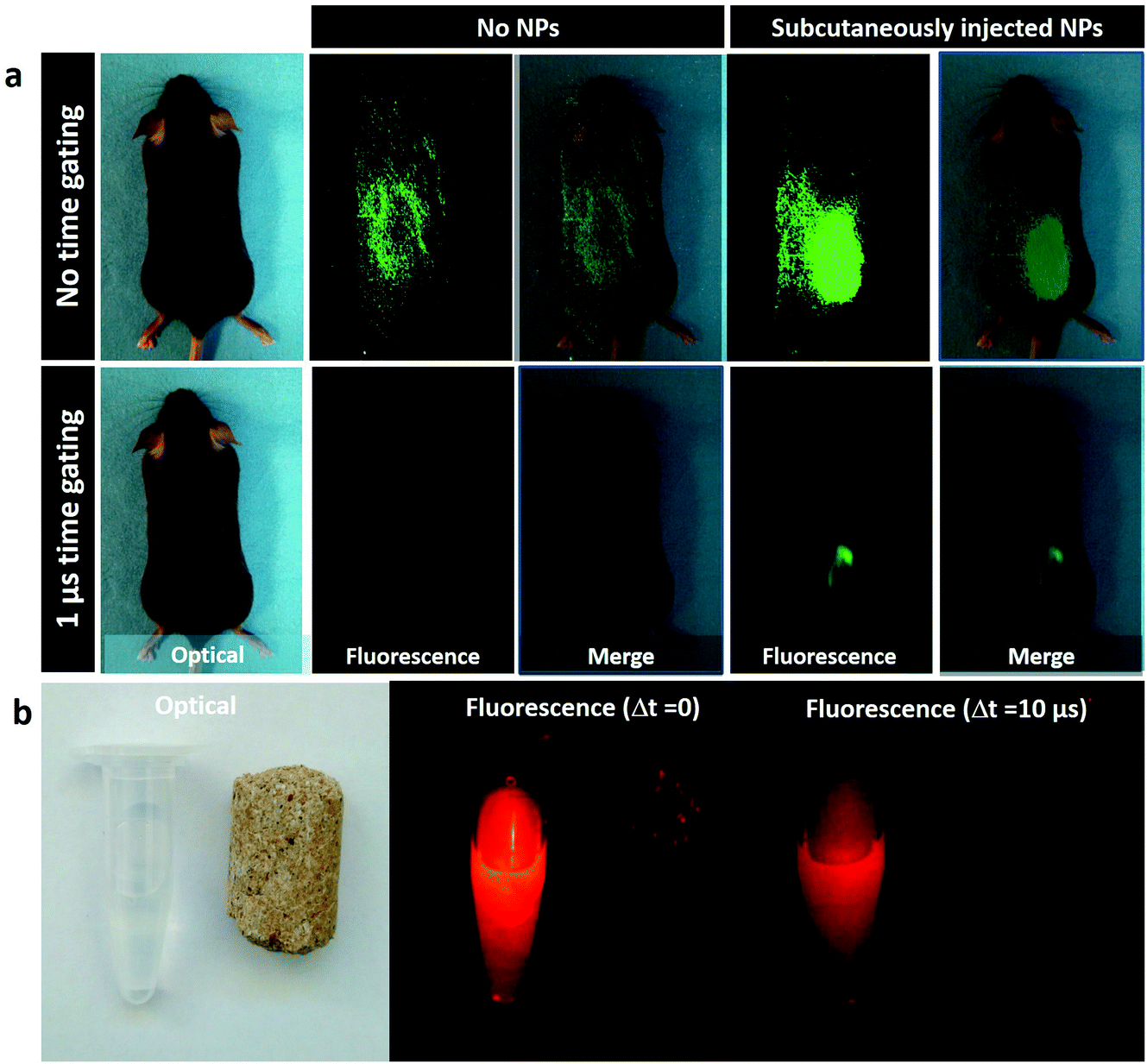 | ||
| Fig. 5 a. Optical and NIR (900–1700 nm) images of C57/Bl6 mice, one of them injected with 50 μl NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs and the other acting as a control. Fluorescence images were recorded for no delay and for a 1 μs delay time. b. NIR images of NaYF4:10% Yb3+,30% Nd3+@CaF2 dispersion and feedstuff detected with zero and 10 μs time delay. | ||
Then, the core/shell NPs were used for transcranial fluorescence imaging. To do so, the core/shell NPs were directly injected into a CD1 mouse brain, as indicated in Fig. 6a. The mouse was injected intracerebrally with 100 nL of a dispersion of NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs. The extremely low volume of NPs that can be injected into the brain to avoid adverse effects makes it of paramount importance to increase the contrast of the image by eliminating any nonspecific background such as that generated by autofluorescence of the eyes. In particular, when dealing with head and brain imaging, the NIR fluorescence signal is hindered by the autofluorescence emission of the eyes in both NIR I and NIR II windows, which can be attributed to the presence of melanin. This is evidenced in Fig. 6b, where the NIR image (obtained under continuous wave 808 nm excitation) of a mouse with an intracranial injection of our infrared emitting core/shell NPs is shown. As shown in Fig. 6c, the autofluorescence emission at the eye can be completely eliminated by performing time-gated imaging with a time delay of 1 μs, allowing for a straightforward localization of the NPs inside the brain. As was the case for orally administrated NPs, the image shown in Fig. 6b corresponds to a single accumulation with a 30 ms integration time. This indicates the adequacy of our strategy for performing autofluorescence-free real time monitoring of brain structures and processes even at very low NP amounts. The results included in Fig. 6 open the way for the use of rare-earth-doped NPs for brain imaging as the basis of advanced cerebral studies.
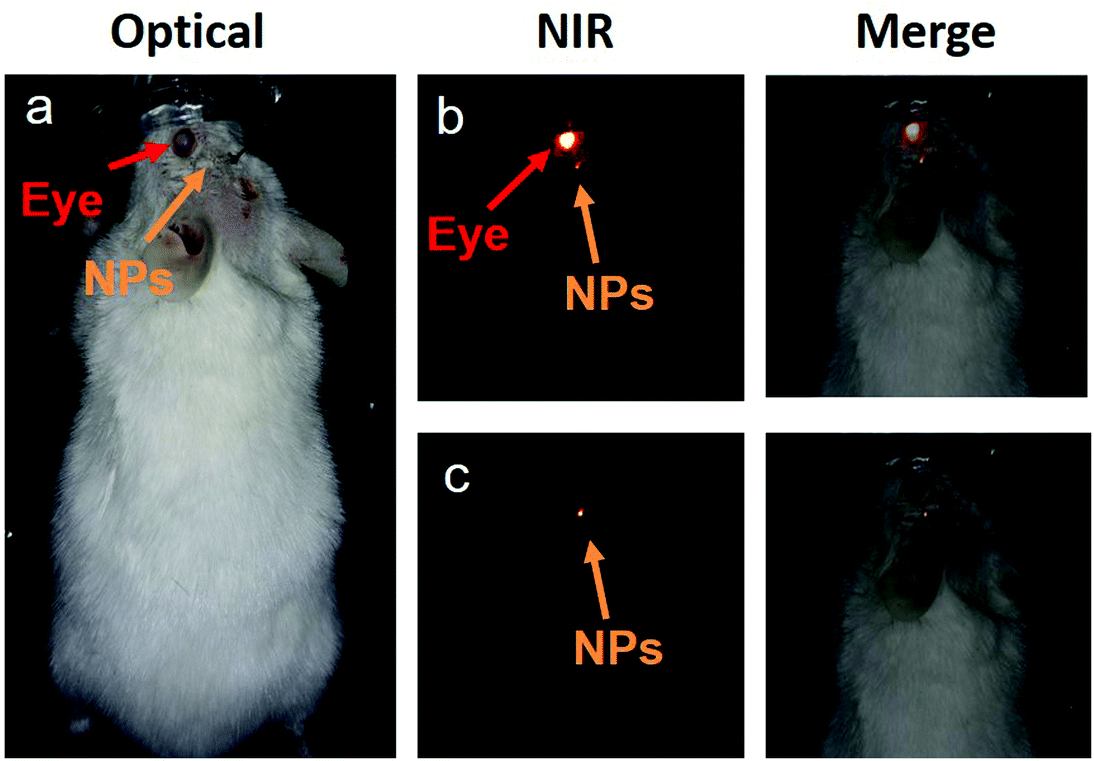 | ||
| Fig. 6 a. Optical image of a CD1 mouse after intracerebral injection of NaYF4: 10% Yb3+,30% Nd3+@CaF2 NPs. b. NIR fluorescence image of the mouse in (a) as obtained with no time delay between excitation pulse and image collection. The merge between optical and fluorescence images is shown to indicate that the brightest fluorescence emission corresponds to eye autofluorescence. c. NIR time-gated fluorescence image, as obtained for the mouse in (a) when a 1 μs time delay is introduced between excitation and image collection. The merged images show that the autofluorescence generated by the eye was successfully removed. | ||
4. Conclusion
In conclusion, we have developed two different approaches for obtaining NPs with intense luminescence and long fluorescence lifetime in NIR II. First, the effect of the selection of dopants to optimize the energy transfer processes and the obtained NaGdF4:2% Yb3+,3% Nd3+,0.2% Tm3+ NPs (∼13 nm) with a bright emission at 980 nm and a lifetime of 1.3 ms was explored. Second, we synthesized sub-10 nm monodisperse core/shell NaYF4:10% Yb3+,30% Nd3+@CaF2 NPs with high dopant concentrations, which show a strong NIR II emission at 1000 nm with a long lifetime close to 1 millisecond. The advantages of core/shell engineering and selective doping were systematically investigated, allowing for a fine optimization of the fluorescence properties. The possibility of efficient optical excitation in NIR I and bright emission in NIR II with fluorescence decay times around 1 millisecond (orders of magnitude longer than that of tissue autofluorescence) makes our NPs superior candidates for high penetration, autofluorescence-free in vivo NIR imaging using a time-gated approach. This was experimentally demonstrated through the performance of simple but conclusive in vivo experiments in murine models: real-time tracking of gastrointestinal absorption of orally administered NPs and transcranial autofluorescence-free imaging of intracerebrally injected NPs. This fact, coupled with their outstanding brightness, which surpasses that of commonly used NIR-emitting Ag2S nanoparticles, makes our NPs great candidates as contrast agents for high-contrast deep tissue bioimaging in the time domain.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the grants from the Fundamental Research Funds for the Central Universities, China (HIT. BRETIV.201503 and AUGA5710052614) and the National Natural Science Foundation of China (51672061). We thank Dr Lina Wu at the Fourth Hospital of Harbin Medical University for her kind help with the MTT assay, and Dr Tymish Y. Ohulchanskyy at Shenzhen University for his kind help with the fluorescence lifetime measurement. The work was also supported by the Ministerio de Economia y Competitividad of Spain (grant MAT2016-75362-C3-1-R). Jie Hu acknowledges the scholarship from the China Scholarship Council (No. 201506650003). Dirk H. Ortgies is grateful to the Spanish Ministry of Economy and Competitiveness for a Juan de la Cierva scholarship (No. FJCI-2014-21101) and the Spanish Institute of Health (ISCIII) for a Sara Borell Fellowship (No. CD17/00210).References
- F. Leblond, S. C. Davis, P. A. Valdés and B. W. Pogue, J. Photochem. Photobiol., B, 2010, 98, 77–94 CrossRef PubMed.
- C.-H. Quek and K. W. Leong, Nanomaterials, 2012, 2, 92–112 CrossRef PubMed.
- K.-T. Yong, Y. Wang, I. Roy, H. Rui, M. T. Swihart, W.-C. Law, S. K. Kwak, L. Ye, J. Liu and S. D. Mahajan, Theranostics, 2012, 2, 681 CrossRef PubMed.
- M. Sauer and M. Heilemann, Chem. Rev., 2017, 117, 7478–7509 CrossRef PubMed.
- B. Huang, W. Q. Wang, M. Bates and X. W. Zhuang, Science, 2008, 319, 810–813 CrossRef PubMed.
- S. Andersson-Engels, C. af Klinteberg, K. Svanberg and S. Svanberg, Phys. Med. Biol., 1997, 42, 815 CrossRef PubMed.
- Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317–4322 CrossRef PubMed.
- S. Gioux, H. S. Choi and J. V. Frangioni, Mol. Imaging, 2010, 9, 237 CrossRef PubMed.
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef PubMed.
- J. V. Frangioni, Curr. Opin. Chem. Biol., 2003, 7, 626–634 CrossRef PubMed.
- E. A. Cowles, J. L. Kovar, E. T. Curtis, H. Xu and S. F. Othman, BioRes. Open Access, 2013, 2, 186–191 CrossRef PubMed.
- A. P. Patterson, S. A. Booth and R. Saba, BioMed Res. Int., 2014, 2014, 1–14 CrossRef PubMed.
- B. del Rosal, I. Villa, D. Jaque and F. Sanz-Rodríguez, J. Biophotonics, 2016, 9, 1059–1067 CrossRef PubMed.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710–711 CrossRef PubMed.
- G. S. Hong, S. Diao, J. L. Chang, A. L. Antaris, C. X. Chen, B. Zhang, S. Zhao, D. N. Atochin, P. L. Huang, K. I. Andreasson, C. J. Kuo and H. J. Dai, Nat. Photonics, 2014, 8, 723–730 CrossRef PubMed.
- G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris, Q. Wang and H. Dai, Angew. Chem., 2012, 51, 9818–9821 CrossRef PubMed.
- A. N. Bashkatov, E. A. Genina, V. I. Kochubey and V. V. Tuchin, J. Phys. D: Appl. Phys., 2005, 38, 2543 CrossRef.
- J. T. Robinson, G. Hong, Y. Liang, B. Zhang, O. K. Yaghi and H. Dai, J. Am. Chem. Soc., 2012, 134, 10664–10669 CrossRef PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 2199–2199 CrossRef PubMed.
- C. Caltagirone, A. Bettoschi, A. Garau and R. Montis, Chem. Soc. Rev., 2015, 44, 4645–4671 RSC.
- G. Hong, J. T. Robinson, Y. Zhang, S. Diao, A. L. Antaris, Q. Wang and H. Dai, Angew. Chem., Int. Ed., 2012, 51, 9818–9821 CrossRef PubMed.
- A. Benayas, F. Ren, E. Carrasco, V. Marzal, B. del Rosal, B. A. Gonfa, Á. Juarranz, F. Sanz-Rodríguez, D. Jaque and J. García-Solé, Adv. Funct. Mater., 2015, 25, 6650–6659 CrossRef.
- C. Li, Y. Zhang, M. Wang, Y. Zhang, G. Chen, L. Li, D. Wu and Q. Wang, Biomaterials, 2014, 35, 393–400 CrossRef PubMed.
- Y. Zhang, G. Hong, Y. Zhang, G. Chen, F. Li, H. Dai and Q. Wang, ACS Nano, 2012, 6, 3695–3702 CrossRef PubMed.
- L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326 CrossRef PubMed.
- H. Osaki, C. M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito and A. Fukazawa, Angew. Chem., Int. Ed., 2016, 128, 7247–7251 CrossRef.
- D. Jin, J. A. Piper and A. Chem, Anal. Chem., 2011, 83, 2294–2300 CrossRef PubMed.
- X. Zheng, X. Zhu, Y. Lu, J. Zhao, W. Feng, G. Jia, F. Wang, F. Li and D. Jin, Anal. Chem., 2016, 88, 3449–3454 CrossRef PubMed.
- L. Gu, D. J. Hall, Z. Qin, E. Anglin, J. Joo, D. J. Mooney, S. B. Howell and M. J. Sailor, Nat. Commun., 2013, 4, 2326–2326 CrossRef PubMed.
- M. Rajendran and L. W. Miller, Biophys. J., 2015, 109, 240–248 CrossRef PubMed.
- J. Joo, X. Liu, V. R. Kotamraju, E. Ruoslahti, Y. Nam and M. J. Sailor, ACS Nano, 2015, 9, 6233 CrossRef PubMed.
- W. T. Carnall, P. R. Fields and K. Rajnak, J. Chem. Phys., 1968, 49, 4424–4442 CrossRef.
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- R. Wang, X. Li, L. Zhou and F. Zhang, Angew. Chem., Int. Ed., 2014, 53, 12086–12090 CrossRef PubMed.
- E. S. Levy, C. A. Tajon, T. S. Bischof, J. Iafrati, A. Fernandezbravo, D. J. Garfield, M. Chamanzar, M. M. Maharbiz, V. S. Sohal and P. J. Schuck, ACS Nano, 2016, 10, 8423–8433 CrossRef PubMed.
- L. Ma, Y. Liu, L. Liu, A. Jiang, F. Mao, D. Liu, L. Wang and J. Zhou, Adv. Funct. Mater., 2017, 1705057 Search PubMed.
- B. del Rosal, D. H. Ortgies, N. Fernández, F. Sanz-Rodríguez, D. Jaque and E. M. Rodríguez, Adv. Mater., 2016, 28, 10188–10193 CrossRef PubMed.
- D. J. Naczynski, M. C. Tan, M. Zevon, B. Wall, J. Kohl, A. Kulesa, S. Chen, C. M. Roth, R. E. Riman and P. V. Moghe, Nat. Commun., 2013, 4, 1345–1346 CrossRef PubMed.
- Y. Zhong, Z. Ma, S. Zhu, J. Yue, M. Zhang, A. L. Antaris, J. Yuan, R. Cui, H. Wan and Y. Zhou, Nat. Commun., 2017, 8, 737 CrossRef PubMed.
- W. Shao, G. Chen, A. Kuzmin, H. L. Kutscher, A. Pliss, T. Y. Ohulchanskyy and P. N. Prasad, J. Am. Chem. Soc., 2016, 138, 16192 CrossRef PubMed.
- M. Kamimura, N. Kanayama, K. Tokuzen, K. Soga and Y. Nagasaki, Nanoscale, 2011, 3, 3705–3713 RSC.
- H. Dong, L. D. Sun and C. H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- Y. F. Wang, G. Y. Liu, L. D. Sun, J. W. Xiao, J. C. Zhou and C. H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef PubMed.
- A. Brenier, J. Opt. Soc. Am. B, 2006, 23, 2209–2216 CrossRef.
- L. Tu, X. Liu, F. Wu and H. Zhang, Chem. Soc. Rev., 2015, 44, 1331–1345 RSC.
- Y. Jeong, J. Sahu, D. Payne and J. Nilsson, Opt. Express, 2004, 12, 6088–6092 CrossRef PubMed.
- W. F. Krupke, IEEE J. Sel. Top. Quantum Electron., 2002, 6, 1287–1296 Search PubMed.
- J. Shen, G. Chen, A. M. Vu, W. Fan, O. S. Bilsel, C. C. Chang and G. Han, Adv. Opt. Mater., 2014, 1, 644–650 CrossRef.
- Y. T. Zhong, G. Tian, Z. Gu, Y. J. Yang, L. Gu, Y. L. Zhao, Y. Ma and J. N. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef PubMed.
- H. Wen, H. Zhu, X. Chen, T. F. Hung, B. Wang, G. Zhu, S. F. Yu and F. Wang, Angew. Chem., 2013, 52, 13419–13423 CrossRef PubMed.
- X. Xie, N. Gao, R. Deng, S. Qiang, Q. H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef PubMed.
- X. Zhang, Z. Zhao, X. Zhang, D. B. Cordes, B. Weeks, B. Qiu, K. Madanan, D. Sardar and J. Chaudhuri, Nano Res., 2015, 8, 636–648 CrossRef.
- N. J. Johnson, S. He, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139, 3275–3282 CrossRef PubMed.
- G. Y. Chen, J. Shen, T. Y. Ohulchanskyy, N. J. Patel, A. Kutikov, Z. P. Li, J. Song, R. K. Pandey, H. Agren, P. N. Prasad and G. Han, ACS Nano, 2012, 6, 8280–8287 CrossRef PubMed.
- K. Prorok, M. Pawlyta, W. Stręk and A. Bednarkiewicz, Chem. Mater., 2016, 28, 2295–2300 CrossRef.
- L. Wang, H. Dong, Y. Li, R. Liu, Y. F. Wang, H. K. Bisoyi, L. D. Sun, C. H. Yan and Q. Li, Adv. Mater., 2015, 27, 2065 CrossRef PubMed.
- Y. F. Wang, L. D. Sun, J. W. Xiao, W. Feng, J. C. Zhou, J. Shen and C. H. Yan, Chemistry, 2012, 18, 5558 CrossRef PubMed.
- N. J. J. Johnson, H. Sha, S. Diao, E. M. Chan, H. Dai and A. Almutairi, J. Am. Chem. Soc., 2017, 139, 3275 CrossRef PubMed.
- D. Jaque, M. O. Ramirez, L. E. Bausá, J. G. Solé, E. Cavalli, A. Speghini and M. Bettinelli, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 035118 CrossRef.
- A. C. Berends, F. T. Rabouw, F. C. M. Spoor, E. Bladt, F. C. Grozema, A. J. Houtepen, L. D. A. Siebbeles and C. de Mello Donegá, J. Phys. Chem. Lett., 2016, 7, 3503–3509 CrossRef PubMed.
- R. Arppe, I. Hyppanen, N. Perala, R. Peltomaa, M. Kaiser, C. Wurth, S. Christ, U. Resch-Genger, M. Schaferling and T. Soukka, Nanoscale, 2015, 7, 11746–11757 RSC.
- X. Zhang, Y. Gu and H. Chen, J. Innovative Opt. Health Sci., 2014, 7, 1350059 CrossRef.
- X. Jia, D. Li, J. Li and E. Wang, RSC Adv., 2015, 5, 80929–80932 RSC.
- G. Jingwen, W. Chuanli, D. Dan, W. Ping and C. Chenxin, Adv. Healthcare Mater., 2016, 5, 2437–2449 CrossRef PubMed.
- D. H. Zhao, J. Yang, R. X. Xia, M. H. Yao, R. M. Jin, Y. D. Zhao and B. Liu, Chem. Commun., 2018, 54, 527 RSC.
- M. Rowland, C. Peck and G. Tucker, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 45–73 CrossRef PubMed.
- A. Y. Abuhelwa, D. B. Williams, R. N. Upton and D. J. R. Foster, Eur. J. Pharm. Biopharm., 2017, 112, 234–248 CrossRef PubMed.
- T. Kimura and K. Higaki, Biol. Pharm. Bull., 2002, 25, 149–164 CrossRef.
- W. Huang, S. L. Lee and L. X. Yu, AAPS J., 2009, 11, 217–224 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr02382d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |