
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular dynamics simulation of potentiometric sensor response: the effect of biomolecules, surface morphology and surface charge†
B. M.
Lowe
 *a,
C.-K.
Skylaris
b,
N. G.
Green
c,
Y.
Shibuta
a and
T.
Sakata
*a
*a,
C.-K.
Skylaris
b,
N. G.
Green
c,
Y.
Shibuta
a and
T.
Sakata
*a
aDepartment of Materials Engineering, School of Engineering, The University of Tokyo 7-3-1 Hongo, Bunkyo, Tokyo 113-8656, Japan. E-mail: sakata@biofet.t.u-tokyo.ac.jp
bSchool of Chemistry, University of Southampton, UK
cDepartment of Electronics and Computer Science, Nano Research Group, University of Southampton, UK. E-mail: ng2@ecs.soton.ac.uk
First published on 16th April 2018
Abstract
The silica–water interface is critical to many modern technologies in chemical engineering and biosensing. One technology used commonly in biosensors, the potentiometric sensor, operates by measuring the changes in electric potential due to changes in the interfacial electric field. Predictive modelling of this response caused by surface binding of biomolecules remains highly challenging. In this work, through the most extensive molecular dynamics simulation of the silica–water interfacial potential and electric field to date, we report a novel prediction and explanation of the effects of nano-morphology on sensor response. Amorphous silica demonstrated a larger potentiometric response than an equivalent crystalline silica model due to increased sodium adsorption, in agreement with experiments showing improved sensor response with nano-texturing. We provide proof-of-concept that molecular dynamics can be used as a complementary tool for potentiometric biosensor response prediction. Effects that are conventionally neglected, such as surface morphology, water polarisation, biomolecule dynamics and finite-size effects, are explicitly modelled.
1 Introduction
During the last century, significant progress in the study of electrified charged interfaces has enabled the development of a wide range of new technologies, such as electrochemical fuel cells1 and the ability to engineer colloids2 and materials for water purification.3,4 Despite this progress, improved understanding is required for the reliable design and modelling of these technologies. In particular, the complex interaction between surface charge and the electrodynamics of water polarisation, charged analyte molecules and electrolyte ions remains a topic of active research.An example of one such technology are potentiometric sensors. The response of potentiometric sensors originates from a change in electric potential within the system resulting from interfacial electrodynamics. A popular class of potentiometric sensors are ion-sensitive field-effect transistor (IS-FET) sensors. IS-FETs were initially popularised in the 1970s by Bergveld5,6 and can detect changes in pH due to surface charging reactions and ion-adsorption at the oxide–electrolyte interface. Starting with the work of Cui et al.7 in 2001, field-effect sensors have been functionalised with receptors specific to the biomolecular analyte for the task of sensing the biomolecular analyte, often referred to as ‘BioFET’ sensors. This functionalisation facilitates a type of biosensor which has many advantages over standard immunological detection methodologies, for example, the capability for label-free, low-cost electrostatic analyte detection.8 In the present work, field-effect sensors are focused on as an example application system; however, the interfacial physics described is relevant to all kinds of potentiometric biosensors.9,10
The silica–water interface, in particular, was investigated in the present work as it is a popular choice as oxide material for potentiometric sensors given its low-cost and simple production by thermal treatment of silicon wafers.11–13 Silica–water interfaces are among the most abundant on the planet, and therefore the relevance of understanding their interfacial electrodynamics extends beyond sensor response into understanding geochemical processes such as dissolution kinetics14–16 and prebiotic biochemistry.17
Reliable and accurate quantitative predictions of the response of field-effect sensors due to interaction of analyte molecules are currently unavailable even when the most sophisticated models are used, and qualitative predictions of the response of field-effect sensors remain challenging.8,13 The majority of field-effect sensor models incorporate a model of the electrical double layer primarily based on either the Poisson–Boltzmann equation18–28 or the linearised Poisson–Boltzmann equation,13,29–31 which were first developed in the early 20th century.32 This approach neglects effects that are expected to be important to the generation of a potentiometric signal, examples of which would include, but are not limited to, the effects of van der Waals interactions, water polarisation, electrolyte–protein ion dynamics and the finite size of biomolecules. Furthermore, difficulty in controlling the experimental conditions necessary for reproducible response, such as the analyte–receptor density at the sensor surface,8 has contributed to difficulties in validating and improving the predictive power of field-effect response models.
There are several research questions which are particularly problematic to understand via Poisson–Boltzmann-based models, for example, the experimental observation of potentiometric response due to the binding of electrically neutral non-polar organic molecules33,34 and the anomalously large signal of hexanol compared to butanol on bare silica.33 Other examples include the enhancement of potentiometric biosensor response at high ionic strength by the addition of a biomolecule-permeable polyethylene glycol surface layer35 and the changes in high frequency signal induced by biomolecule–electrolyte dynamics.8,35–37
The aim of the present work is to use molecular dynamics simulations as a novel tool for prediction of potentiometric biosensor response and for improving our understanding of the interfacial electrodynamics. The simulated surface potential is calculated as a function of surface charge for a simple experimentally well-characterised system – the silica–water interface. The results are validated against analytical models and experimental pH sensing data. Using the relationship between response and surface charge as a baseline, the magnitude of response due to the addition of a model charged biomolecule – DNA – is investigated. For biomolecular systems, a molecular dynamics-based approach has several advantages over conventional mean-field models due to its explicit treatment of biomolecule dynamics, water polarisation and finite-size effects.
Various durations of molecular dynamics simulation have been used in the literature to calculate the surface potential at the silica–water interface: ∼1 ns (ref. 38) (reactive forcefield), 3 ns (ref. 39–42), 20 ns (ref. 43) and ∼30 ns.44 Although sufficient for obtaining qualitative changes in potential, this duration is too short to reliably quantitatively distinguish the millivolt changes in surface potential expected from biomolecular signal and pH changes. For example, DNA is known to take at least 300 ns for its ionic atmosphere to equilibrate45 and at least 90 ns is required to obtain a silica–water surface potential with a mean potential stable to within several millivolts.46 In the present work, we perform 24 simulations of the silica–water interface for 320 ns and several simulations including DNA molecules for 480–680 ns, totalling an extensive total simulation duration of at least 8 μs between all simulations. Furthermore, compared to our previous work,39–42 we use a forcefield which has increased reliability for describing the silica–water-bio interface and a methodology for evaluating the electrostatics with increased accuracy.43,46,47 Combined, these considerations ensure that the present work is amongst the most rigorous molecular dynamics simulations of the silica–water interfacial potential and electric field to date. This work is of particular novelty because molecular dynamics-based oxide–water surface potential calculations remain rare, likely due to historical difficulties in obtaining accurate forcefields which integrate both organic and inorganic molecules.
The surface potential calculation methodology used has been detailed and discussed in our most recent work for a simple crystalline silica–water system.46 In the present work, we focus on the application of understanding potentiometric sensor response and nanoscale electrodynamics. We report a novel prediction and explanation of the effects of nano-morphology on surface potential changes, and provide the first proof-of-concept that molecular dynamics can be used as a complementary tool for potentiometric biosensor response prediction.
The Background section is structured as follows: section 2.1 presents commonly used models for the electrical double layer as these will be directly compared to the molecular dynamics simulations of the present work. Then, in section 2.2, the commonly-used site-binding model for the prediction of the response of potentiometric sensors to changes in pH and its limitations is introduced. In section 2.3, a brief outline of the surface chemistry of silica is provided because accurate modelling of the surface chemistry is crucial for an accurate molecular dynamics model of the interface. Finally, the literature experimental data are presented on the effects of the surface morphology on sensor response (section 2.4) and the addition of DNA on sensor response (section 2.5).
2 Background
2.1 Mean-field electrical double layer models
In this section, a brief overview of analytical mean-field electrical double layer models relevant to potentiometric sensor response modelling is presented. A more detailed background for these models can be found in ESI 1.†The simplest and most common model of field-effect sensor response treats the response due to biomolecules as charge on a parallel plate capacitor model of the surface (also called the ‘constant capacitance’ model or the Helmholtz–Perrin model48). In such models, the measured change in potential is assumed to be equal to the change in surface charge from biomolecule binding divided by a capacitance value which describes the coupling of the analyte to the FET semiconducting channel.49,50 Even though this method may be useful for estimating the density of bound molecules from an experiment, it provides no generally transferable insight into the precise relationship between the contents of the electrical double layer (i.e. biomolecule binding, electrolyte ionic strength etc.) and sensor response and is therefore not considered in the present work.
Aside from the Helmholtz–Perrin model, field-effect sensor response models almost exclusively utilise the Poisson–Boltzmann equation (or the more general Poisson–Nernst–Plank equation) to describe the electrical double layer. Such models therefore explicitly incorporate the effect of ionic strength on the response. Primary differences between field-effect biosensor models lie in the way that the biomolecular component is treated in a mean-field manner: for example, whether the biomolecular charge is treated as a ‘smeared out’ surface charge over an infinitely thin surface charge layer27,51 or as an ion-permeable membrane of finite thickness.52,53 Some recent attempts at modelling field-effect sensor response model the charges on the biomolecule discretely in a continuum solvent.13,30,54,55 Such an approach provides a good compromise between atomistic and continuum approaches, but neglects the important effects of biomolecule dynamics, surface morphology and water polarisation.
In the present work, the surface charge to surface potential relationship for the Poisson–Boltzmann equation, the Debye–Hückel theory and the modified Poisson–Boltzmann equation is presented for comparison with the molecular dynamics results; expressions will be labelled using subscripts pb, dh and mpb, respectively. The surface potential φs refers to the potential only at the surface. The equations relating to the position dependent potential are shown in ESI 1.† These models were chosen in the present work due to their simplicity and widespread usage. In addition, they require no or minimal system-specific empirical parameters, and therefore provide predictions that are transferable across oxide systems without overfitting to a particular oxide–electrolyte system.
From the Poisson–Boltzmann equation, an analytical expression for the surface charge density (σ) to surface potential (φs) relationship can be obtained, often termed the Grahame equation (ESI 1†). Rearranging the Grahame equation for surface potential provides the following expression, shown assuming a symmetric valency electrolyte:56
 | (1) |
Assuming φ is small  , the Poisson–Boltzmann equation can be linearised and rearranged resulting in eqn (2), the Debye–Huckel model57–60 surface–charge potential relationship:56
, the Poisson–Boltzmann equation can be linearised and rearranged resulting in eqn (2), the Debye–Huckel model57–60 surface–charge potential relationship:56
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 monovalent electrolyte is equal to:
:1 monovalent electrolyte is equal to: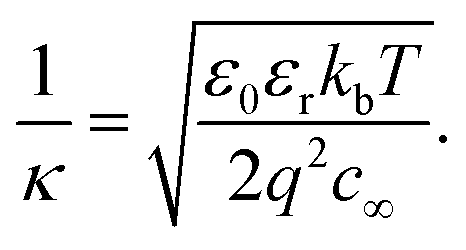 | (3) |
This approximation of assuming φ is small is termed the Debye–Hückel approximation. Despite this approximation not being strictly valid in most situations of interest in colloid science and electrochemistry,57 it is commonly used. One reason for this is its convenience, whereby electrostatic screening by electrolyte can be described in a simple parameter – the Debye length, which is inversely proportional to the square root of the ionic strength.
The Poisson–Boltzmann equation neglects finite size effects, and thus provides inaccurate results for highly concentrated systems. One way of dealing with this is the incorporation of a parallel plate-like layer at the surface which represents accumulated charge, called the ‘Stern’ layer.48,61 In reality, the region modelled by the Stern layer contains both highly polarised water molecules and accumulated electrolyte ions.61 An alternative method which does not require empirical fitting of a Stern layer capacitance is the ‘modified’ Poisson–Boltzmann equation, as shown in eqn (4) for a z:z symmetric electrolyte.62 The modified Poisson–Boltzmann equation simply constrains the maximum density of counterions permissible at the surface to an (semi-)empirical finite value based on packing constraints.62,63 The corresponding surface charge to surface potential relationship, which can be solved numerically for φs,mpb, is:
 | (4) |
An alternative which is more justifiable at high surface potentials is the Poisson–Boltzmann equation without linearisation (Grahame equation, eqn (1)). The Grahame equation has commonly been used to calculate the surface potential due to binding of charged biomolecular analyte such as DNA or, conversely, to calculate the charge due to bound biomolecules, given a measured surface potential.65,67–69 For example, the change in surface potential can be calculated before and after analyte binding, assuming that the intrinsic (i.e. silanolate, in the present work) surface charge (σsurf) is additive with the charge contribution from DNA (σDNA). In other words, the DNA is assumed to be an infinitely thin smeared layer of uniform charge density on the surface which does not change the intrinsic charge, i.e. the biomolecule does not affect the charging of silanolate groups:
| Δφ = φ(σsurf + σDNA) − φ(σsurf) | (5) |
The calculated shift in surface potential is strongly influenced by the choice of intrinsic surface charge density, and often this information is not measured but estimated, providing a large source of error in such calculations. Such calculations also often result in signals too weak compared to experimental response and thus the theoretical basis for the observed biomolecular signals remains unclear.70 This has provided motivation for the creation of improved models68 and the present work.
2.2 Site binding models
The Nikolsky–Eisenman equation and ion exchange theory is widely used to describe glass-membrane electrodes and ion-sensitive electrodes,71 however this approach is limited to estimating the selectivity of the sensor against competing ions.72 In the field of IS-FET sensing research, site-binding models are instead commonly used to describe the surface potential as a function of pH,73–75 in which the surface charge is calculated based on several chemical equations for reactions of hydroxyl groups at the surface combined with a model of the electrical double layer. Potentiometric ion sensor modelling has been extensively reviewed by Bermejo76 and Bobacka et al.71In the site-binding model,73,77 for an oxide with a ‘sensitivity factor’ of 1, the change in potential due to change in pH is the same as that predicted from the Nernst equation for a semi-permeable membrane with a change in proton activity (i.e. ∼59 mV per pH at room temperature). For many oxides, including silica, a potentiometric response weaker than 59 mV per pH is often measured and in the site-binding model; this is explained via a reduced value of the sensitivity factor. The sensitivity factor is a function of both the ability of the oxide surface to deliver or take up protons (‘surface buffer capacity’) and the differential double-layer capacitance, with this capacitance being primarily determined by the ion concentration of the bulk solution.73,77
Even though site-binding models have proved useful in IS-FET design and will likely continue to do so, they present several fundamental limitations. Firstly, they do not provide any information on the effect of surface morphology on response and, secondly, they rely upon empirical parametrisation of many properties. These properties can vary greatly even for a single material, for example, there is debate over the correct acid–base dissociation constant value for silica78 and the density of surface sites is preparation dependent,79 so there is a risk of the model being overfit, and used descriptively rather than predictively. Finally, site-binding models are not suitable for prediction of sensor response to binding of the biomolecular analyte. In the present work, we propose molecular dynamics as a complementary approach to existing models to explore the response to surface morphology and biomolecular binding.
2.3 Silica surface chemistry
Silanol groups (Si–OH) at the silica surface react with H+/OH− in water to form a charged surface layer.80 These chemical reactions are evidenced to be the primary surface charging mechanism for hydrated silica,58 with electrolyte effects having a measurable but less significant effect.81 Ionic strength can be important, for example, for ultrapure water (<0.001 M); negligible surface ionisation can occur due to the lack of availability of charge stabilising cations.82 In the present work, the ionic strength of the simulated system was 300 mM. This ionic strength is relevant to physiological conditions (e.g. high ionic strength biosensing experiments), biomimetic synthesis,83 or applications such as ocean geochemistry (salt water = ∼0.7 M (ref. 84)).The precise extent of surface ionisation depends on the pH, silanol density, the type of silanol group (e.g. isolated, vicinal or occluded in pores), the ionic strength of the solution and the type of cations and anions.85 At pH values relevant to most biosensing conditions (pH 6–9), silica is negatively charged and only gathers a positive charge under extremely acidic conditions (pH <2),86,87 as evidenced by the measured point-of-zero charge (i.e. the pH for which the net charge of the surface is zero) of pH between 2 and 4.88
2.4 Nano-morphology and potentiometric sensor response
When comparing two systems with identical hydroxyl densities and compositions, commonly-used site-binding models predict no effect on sensor response from changes in surface morphology. Nonetheless, some experiments suggest that amorphous surfaces are beneficial to pH sensor response: for example, sputtered silica was shown to have a higher pH sensitivity than thermally grown silica,89 the addition of nanowires to a planar Al2O3/SiO2 surface increased pH sensitivity by 5 mV per pH (55 mV per pH to 60 mV per pH),90 and texturing a silanised silica surface with silanised silica nanoparticles increased pH sensitivity by 11 mV per pH (43 mV per pH to 54 mV per pH).91,92 However, other experiments have shown no effect of increased surface porosity on pH sensitivity, but it did show an increase in capacitance.93,94 It is clear that further work is needed to clarify the precise relationship between surface morphology and pH sensitivity. Molecular dynamics simulations provide the unique ability to directly investigate the effect of nanoscale changes in morphology on the electrodynamics at the interface. In the present work, novel molecular dynamics simulations are used to investigate this relationship.2.5 Experimental potentiometric sensing of DNA
Experimental DNA sensing experiments often involve first the immobilisation of single stranded (‘probe’) DNA, followed by hybridisation with a complementary strand. In the present work, the absence of DNA versus the presence of DNA is simulated, which is analogous to the sum of hybridisation and immobilisation signals. Experimental field-effect biosensor DNA hybridisation signals were reviewed by Poghossian et al., ranging from no significant response to ∼120 mV response.70 Nishio et al. reviewed immobilisation signals to be 32–100 mV with hybridisation signals generally being weaker, between 11–14 mV.55 Such a large variation between experiments is due to variation in factors such as probe density, buffer composition, reference electrode setup, DNA sequence and surface chemistry. This variation makes rational design difficult and is part of the motivation for the present work which can systematically explore changes in the electric double layer due to biomolecules.At high ionic strength, the signal from DNA is commonly expected to be reduced due to screening. However, DNA has been demonstrated to produce signals with a magnitude of several millivolts even in high ionic strength solutions of 0.5 M or 1 M.95,96 This suggests that the 0.3 M ionic strength used in the present work should generate a detectable signal provided there is sufficient bound DNA. The density of the surface bound DNA molecules used in the present work (8.58 × 1012 molecules cm−2) is likely higher than typical densities but is not unreasonable given that a density of surface bound DNA molecules (‘probe density’) as high as 1 × 1013 molecules cm−2 has been found by Surface Plasmon Resonance measurements.97
3 Computational methods
3.1 Model setup
This section is divided into three parts: the setup of the silica surface models (section 3.1.1), the surface charge and electrolyte setup (section 3.1.2) and finally the DNA molecule setup (section 3.1.3). Analytical model results were calculated at 298.15 K assuming a relative permittivity of 78.5, an ionic strength of 0.3 M and a 1:1 monovalent electrolyte.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) cleavage plane of α-cristobalite, with a silanol density of 4.804 OH per nm2 in its fully protonated state. A silanol density of ∼5 OH per nm2 has been used by other authors successfully to reproduce experimental IS-FET data,98 is often used in IS-FET modelling73 and is expected to be the density of fully hydrated silica as discussed in ESI 2.†
) cleavage plane of α-cristobalite, with a silanol density of 4.804 OH per nm2 in its fully protonated state. A silanol density of ∼5 OH per nm2 has been used by other authors successfully to reproduce experimental IS-FET data,98 is often used in IS-FET modelling73 and is expected to be the density of fully hydrated silica as discussed in ESI 2.†
A different silica model was used to emulate an amorphous silica surface. The model consists of a periodic unit cell of silica with little internal ordering and a similar silanol density to the crystalline model of 5.09 OH per nm2. This model has an increased surface area compared to the crystalline surface and has silanol groups distributed over a greater distance from the surface. A side-by-side comparison of the two models can be seen in Fig. 1(a) and (b).
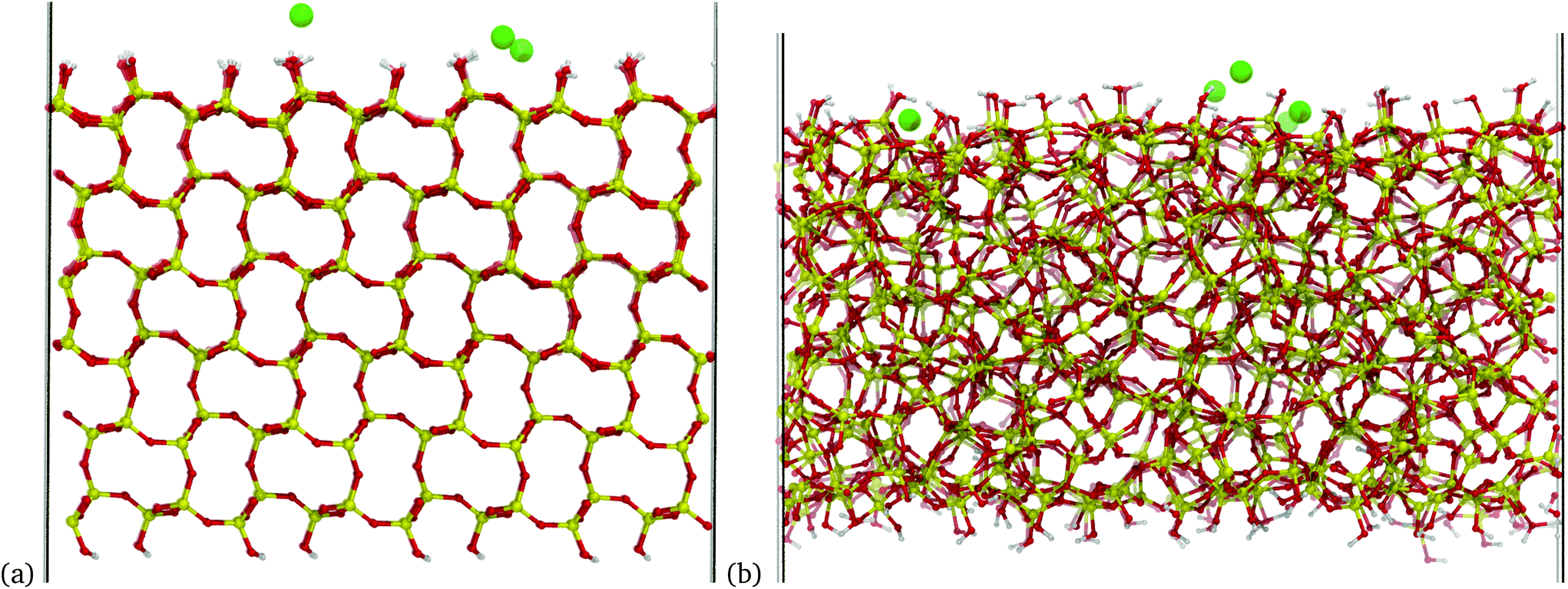 | ||
| Fig. 1 Periodic slabs used to model the silica surface in the present work. Green, red, yellow and grey atoms are sodium, oxygen, silicon and hydrogen, respectively: (a) side-on view of crystalline silica surface, (b) side-on view of amorphous silica surface. Both surfaces are analogous in that they contain approximately the same surface silanol density and surface charge density and are only terminated by Q3 silanol groups. These images were taken from a trajectory of the −0.083 C m−2 models, with the sodium ions introduced to maintain charge neutrality. For the crystalline system, the negative silanolate charges are all located randomly within the first atomic layer, whereas for the amorphous system, charges were distributed randomly between the first atomic layer and 5 Å below. | ||
Both silica models originate from the INTERFACE model database.85 Details such as cell dimensions are summarised in Table 1. In both models, atoms within ∼11 Å of the base were rigidly constrained for all simulations to emulate a silica bulk. Comprehensive details of the silica model setup can be found in ESI 3.†
| Model | ρ(pHeff) | SiO− | Cl− | Na+ | Model | ρ(pHeff) | SiO− | Cl− | Na+ |
|---|---|---|---|---|---|---|---|---|---|
| 0 (∼3.0) | 0 | 15 | 0 + 15 | 0 (∼3.0) | 0 | 22 | 0 + 22 | ||
| Crystalline | −0.041 (∼4.7) | 3 | 15 | 3 + 15 | Amorphous | −0.048 (∼5.0) | 5 | 22 | 5 + 22 |
| 4.81 OH per nm2 | −0.083 (∼6.4) | 6 | 15 | 6 + 15 | 5.09 OH per nm2 | −0.086 (∼6.6) | 9 | 22 | 9 + 22 |
| 33.5 Å × 34.9 Å | −0.12 (∼8.1) | 9 | 15 | 9 + 15 | 40.3 Å × 41.4 Å | −0.12 (∼8.2) | 13 | 22 | 13 + 22 |
| ∼300 mM NaCl | −0.17 (∼9.9) | 12 | 15 | 12 + 15 | ∼300 mM NaCl | −0.16 (∼9.8) | 17 | 22 | 17 + 22 |
| −0.39 (∼19) | 28 | 15 | 28 + 15 | −0.38 (∼19) | 40 | 22 | 40 + 22 |
Various silica models with differing extents of surface charge were prepared by deprotonating the top-surface silanol groups. For each system, neutralising Na+ counterions were added to maintain charge neutrality. This procedure is analogous to the addition of NaOH to a neutral silica surface in an experimental setup, where negatively charged silanolate groups would form at the surface due to reaction with hydroxide ions and there would be a simultaneous increase in the bulk Na+ concentration.
Once electroneutrality was attained, NaCl was added to set the bulk ionic strength. NaCl of approximately 300 mM ionic strength was used based on the initial volume of the water box, with the specific number of ions used and the cell size shown in Table 1. Details of the solvent preparation are presented in ESI 3.† In brief, two different initial configurations of counterions were considered in order to test convergence to thermodynamic equilibrium. A TIP3P solvent box with an initial height of 73 Å was used with ions placed randomly. A control system at low ionic strength was also investigated as detailed in ESI 4.†
3.2 Simulation parameters
Mean-field (analytical model) calculations in the present work were performed at 298.15 K assuming a relative permittivity for the medium of 78.5.For molecular dynamics simulations, the INTERFACE forcefield was used. The forcefield provides CHARMM forcefield compatible parameters for silica and sodium ions.101 Unlike most forcefields of silica–water interfaces, the INTERFACE forcefield has parameters to treat the charged silica–water interface and has been shown capable of accurately reproducing a broad range of experimental observables such as water contact angle, adsorption energy of peptides (at a charged surface), water adsorption isotherm, immersion energy in water and the cell parameters of quartz.85,101,102 The forcefield likely represents the state-of-the-art for accurate classical molecular dynamics simulation of the electrified silica–water(–biomolecule) interface. A discussion of the forcefield parameters and validation is found in the ESI 6.†
Details of the molecular dynamics software parameters can be found in our previous work46 and in ESI 7.† In brief, a 2 fs time step was used with NVT Langevin dynamics at 298.15 K with the SETTLE algorithm for all hydrogen atoms.103 A minimum of 320 ns of dynamics was performed for each simulation, with analysis over the last 180 ns. A simple harmonic restraint was applied to water molecules, which evaporated to return them to the bulk. The electrostatics were evaluated using the particle-mesh Ewald (PME) method using the EW3DC correction, which provides enhanced accuracy for polarised systems.43,46,47
3.3 Analysis methodology
The analysis methodology is detailed in ESI 7† and was discussed in our previous work.46 In summary, the charge density was calculated by averaging the charge density into xy slabs along the z-axis, thus reducing the system to a one-dimensional grid. The electric potential was calculated by double integration of the charge density from Poisson's equation,43,47 with integration performed using the trapezium rule and the electric potential and field set to zero at z = 0 Å.43,104 In order to calculate the surface potential the surface was defined as the z position of minimum electric potential in the region where there is a large potential drop due to silanolate charge; the surface location is indicated as a vertical solid black line in Fig. 2 with the dashed lines indicating the maximum and minimum z-positions. The surface potential was then calculated as the difference between the mean potential in bulk water (between z = 80–85 Å) and at the surface. For potential calculations the analysis was performed over the last 180 ns of the trajectory in three 60 ns parts; the standard error of the mean (ddof = 1) was calculated based on the mean of these three parts. For the plot showing electric field, the field was calculated every 100 ps over the last 180 ns (1800 frames) for both repeats of each simulation. The resulting data for the two repeat simulations were combined (3600 frames) and the mean electric field plotted, with 95% confidence intervals shown, using 1000 bootstrap intervals. For the water polarisation plot, the z-component of the water polarisation was calculated as a function of water molecule orientation multiplied by water number density.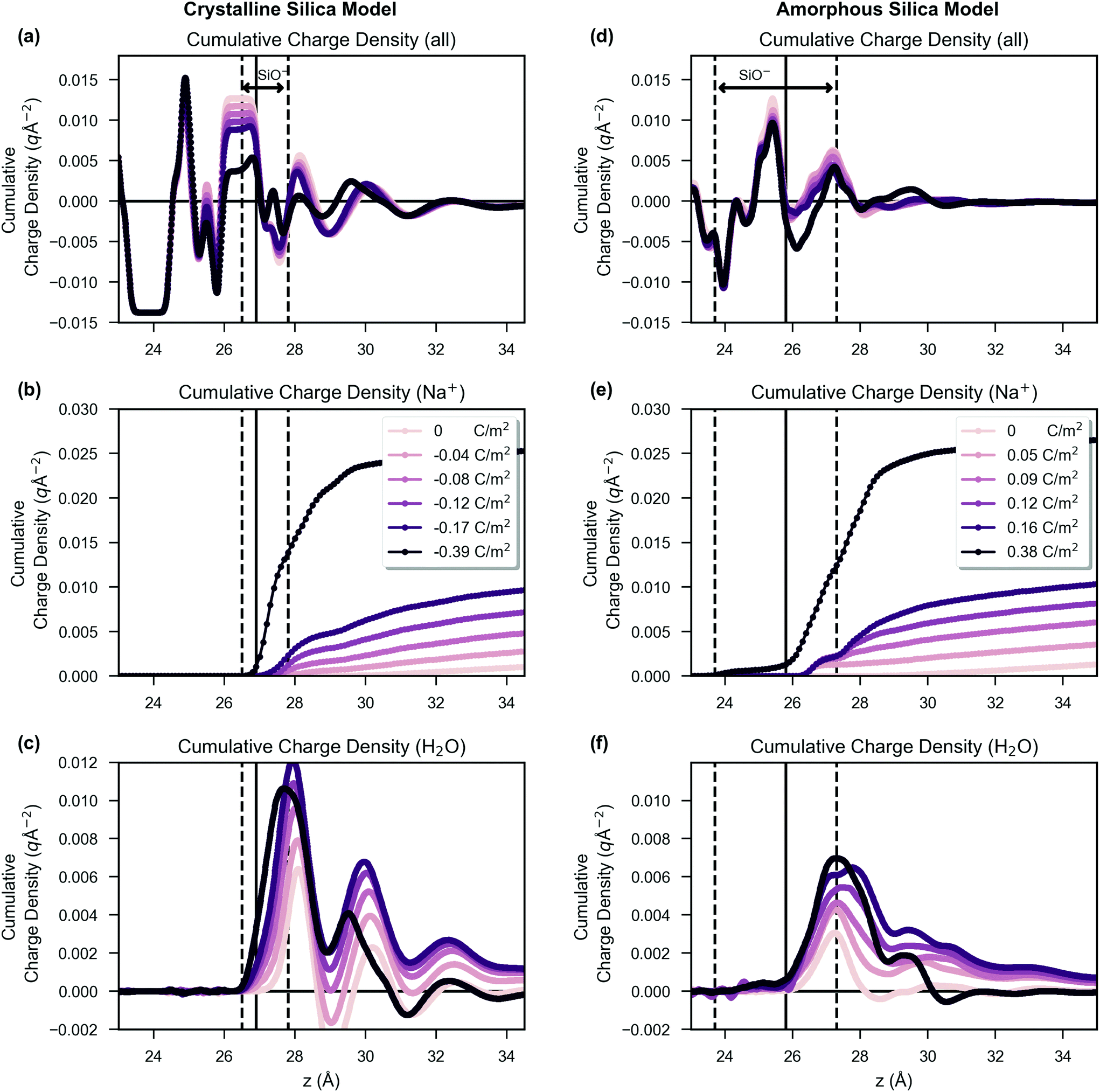 | ||
| Fig. 2 The integrated charge density of the system as a function of the z position for increasing surface charge density (from light to dark lines) with the crystalline silica model shown on the left (a, b, c) and the amorphous silica model shown on the right (d, e, f). Subfigures are shown for the combined charge density of all atoms (a, d), only the sodium ions (b, e) and only the oxygen and hydrogen atoms in water molecules (c, f). For each plot, the integration was performed from the silica (at z = 0 Å) to the bulk water. For the crystalline model, the silanol density showed two peaks, the largest at 27.0 ± 0.1 Å and a smaller peak at 27.5 ± 0.1 Å. For the amorphous system the silanol density showed silanols distributed between 22.0 ± 0.1 Å and 28.4 ± 0.1 Å with the highest density at 25.6 ± 0.1 Å. Shown on the figure by solid black vertical lines is the surface definition described in the Methods section. Dashed vertical black lines indicate the minimum and maximum z-positions at which charged silanolate groups were present. Both (b) and (e) show that with increased surface charge density there was an increase in sodium ion accumulation at the surface. Comparing (a) to (d), it can be seen that the amorphous structure resulted in a more diffuse charge distribution than the crystalline system primarily due to less structured water layers, evident on comparing (c) to (f). | ||
4 Results and discussion
The results are split into five subsections. Firstly, in section 4.1 the charge structure of the electric double layer is analysed. In the following sections, metrics directly related to the response of many potentiometric sensors are calculated and discussed. Specifically, section 4.2 presents the electric field below the silica substrate, with the aim of emulating the ‘field effect’ which controls the response of field-effect sensors. The change in surface potential due to changes in surface charge density/‘effective pH’ is presented in section 4.3, and in section 4.4 the change in surface potential due to addition of DNA is presented. Finally, in section 4.5, the effect of molecular dynamics simulation duration on the accuracy and precision of calculated electrostatic properties for biosensing applications is presented.4.1 Charge density profile
In Fig. 2, the distribution of charge as a function of z is shown. The crystalline system, shown in subfigures (a), (b) and (c), is compared side-by-side with the amorphous system, shown in subfigures (d), (e) and (f). The z position defined as the surface for surface potential calculations is indicated by a vertical black line, and the coloured curves are drawn with increasing darkness corresponding to increased surface charge density, as shown in the figure legend. The charge is shown as integrated charge density of the system from z = 0 and therefore shows the total charge density in the unit cell up to a given z position (i.e. in the bulk the value is zero as the system is net electroneutral). In subfigures (a) and (d), the total charge density is presented, but in subfigures (b) and (e) only the contribution from sodium ions is presented. In subfigures (c) and (f), only the contribution from water charges is presented (i.e. the negative oxygen and positive hydrogen atoms of the TIP3P water model).The positions of the dashed vertical black lines in Fig. 2 show that the silanols were distributed over a broader range of z positions in the amorphous structure than in the crystalline structure. An important result of this, combined with the effect of the irregular morphology of the amorphous system, is that the amorphous model charge distribution, shown in Fig. 2(d), becomes less structured compared to the charge distribution of the crystalline system, as shown in Fig. 2(a). The sodium ion distribution was similar between crystalline and amorphous systems (Fig. 2(b) versus (e)). Comparison of Fig. 2(c) and (f) reveals that the water in the crystalline model had three discrete layers, in contrast to the amorphous system in which the layers were less distinguishable, showing broader water density peaks, with less structure. The formation of three discrete layers on crystalline glass surfaces is supported by ab initio molecular dynamics studies.105
In Fig. 3(a) the density of sodium ions within the Stern-like layer increased approximately linearly with an increase of surface charge density for both amorphous and crystalline silica models. This is in agreement with the molecular dynamics simulation of Lee et al., which showed similar behaviour at high surface charge densities, upon an ideal structureless surface106 and is a result of increased electrostatic attraction between the negatively charged surface and positively charged sodium ions. The sodium ion density for the amorphous system was higher than that for the crystalline system, which is explained by the increased surface area and amorphous morphology providing cavities with improved favourability for sodium binding. The surface accessible surface area (1.4 Å solvent probe radius) was 53% larger for the amorphous model compared to the crystalline model (details in ESI 3†). Comparison of the amorphous and crystalline charge density and water polarisation profiles will be discussed later.
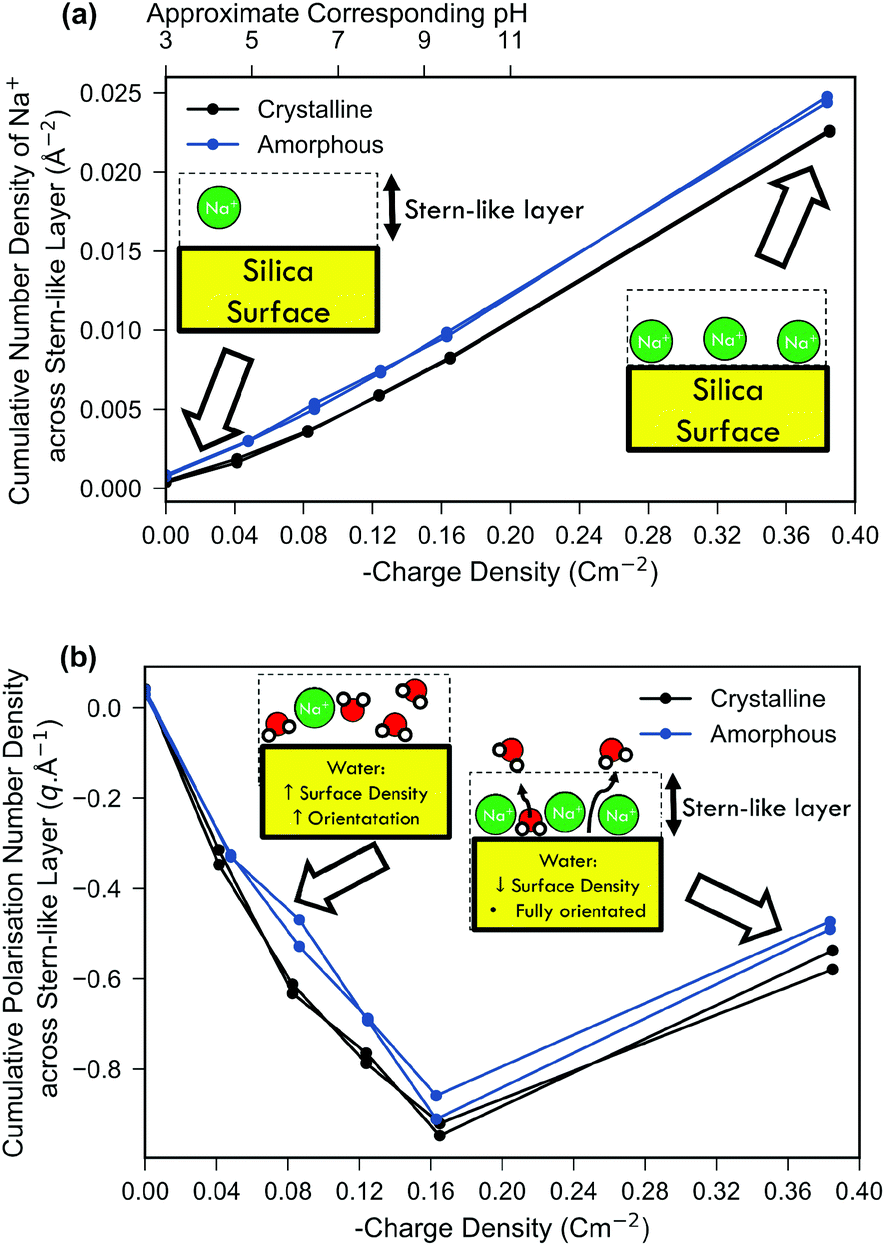 | ||
| Fig. 3 Comparison of both the cumulative water polarisation (a) and sodium ion number density (b) across the Stern-like layer near the surface as a function of the surface charge density. Schematic insets are included to show qualitative differences between low and high surface charge systems. Both the crystalline system (black) and the amorphous system (blue) are shown; for each surface charge density the result of the repeat simulation is also shown. The cumulative number density was calculated as the integral of the number density across the Stern-like layer similar to the work of Lee et al.106 The approximate ‘effective pH’ (see Methods) is shown on the top-most x-axis of subfigure (a). The cumulative polarisation density was calculated by integrating the polarisation across the Stern-like layer as described in the Methods section. | ||
In Fig. 3(b) it can be seen that net polarisation of water within the Stern-like layer increased approximately linearly with charge density up to −0.17 C m−2 for both the amorphous and crystalline model systems. The polarisation is a measure of both the net orientation of waters in the z direction and their density, and thus is proportional to the electric-field imparted by the molecules and their effect on surface potential.
Fig. 3 includes one result at very high surface charge density which is not relevant to typical experimental conditions of silica–water interfaces as a surface charge density above −0.17 C m−2 would require a highly alkaline medium (>pH 11)85,99 to form. Furthermore, experiments show that highly alkaline conditions (>pH 9) result in significant dissolution of silica14 which remains a challenging task to simulate even using ‘reactive’ molecular dynamics forcefields.38 Nonetheless, simulation of high surface charge densities is of interest both for exploring the limiting case and because it is attainable by other oxide systems. For example, δMnO2 demonstrates a surface charge of −1 C m−2 at pH 8 despite having a similar point-of-zero charge to silica.107 At a surface charge density of −0.38 C m−2 the linear trend in Fig. 3(b) ceased and there was instead a relative decrease in water polarisation. This decrease was not a result of decreased orientational ordering (as evidenced in ESI 8†), but rather due to a decrease in the density of bound water due to displacement by the particularly high density of sodium ions bound at these extreme surface charges as depicted in the schematic inset of Fig. 3(b).
4.2 Electric field and the field-effect
Field-effect sensors measure the surface potential change (or, more precisely, a threshold voltage shift) due to binding of analyte molecules or changes in pH. The underlying physical mechanism is that the long-range electric field (‘field effect’) from the analyte-bound surface extends through the oxide, and thus modulates the carrier concentration in the semiconducting layer below the analyte-binding surface, resulting in a change in threshold voltage. To emulate this property via simulation, the electric field at a distance from the oxide interface was calculated via the force on a positive test charge centred below the oxide slab.Prediction of biomolecular potentiometric response is a key motivation for the present work and thus the electric-field response due to DNA specifically was investigated. DNA sensing experiments often use an oxide surface which has been chemically functionalised with a layer of material such as (3-aminopropyl)triethoxysilane (APTES). This layer is chemically bonded to single stranded DNA to provide a highly selective template for the binding of specific single stranded DNA via a hybridisation reaction. In the present work, we are primarily interested in evaluating whether the simulation methodology can provide potentiometric-response prediction for charged biomolecules in general and so we modelled DNA on a bare silica surface to eliminate additional complexity and to provide a more general model system given that many different surface functionalisation layers are used in the literature. The CHARMM forcefield used in this work is well suited to the incorporation of organic, solvated molecules like APTES, and has been used to model APTES in the literature.108
DNA was added to the crystalline silica system with an intrinsic silanolate surface charge density of −0.083 C m−2 and constrained near the surface and the electric field response for this DNA system is shown in the blue bar of Fig. 4. A shift in the electric field due to the addition of DNA and due to increased negative surface charge density was observed. The direction of the shift is consistent with an increase in negative charge at the surface, as expected for negatively charged DNA. The graph shows a response due to changes in effective pH of 0.007 V nm−1 pH−1 (linear regression of data from green bars of Fig. 4). A shift of 0.007 V nm−1 was observed for the addition of DNA (blue bar), and given that a typical pH sensor can detect changes of at least 1 pH unit, the simulated change in electric field of 0.007 V nm−1 pH−1 due to DNA is significant with regard to the expected limit-of-detection of a sensor.
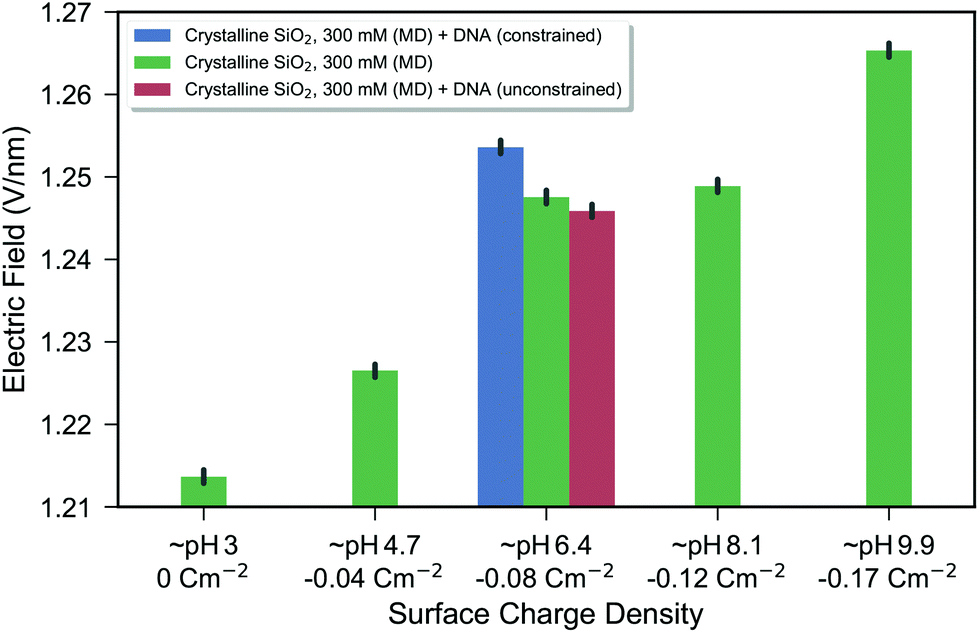 | ||
| Fig. 4 Bar chart of the electric field as a function of surface charge density for the crystalline silica model. The electric field is calculated from the force on a test charge below the silica substrate with an increasingly positive value expected from a more negatively charged surface. The green bars show the electrolyte-only system, where each bar represents the combined data from two repeat simulations. The black bars show the bootstrapped 95% confidence interval as described in the Methods. The blue and red bars show systems including DNA constrained near the surface (blue) and free to diffuse (red). From the blue bar, it can be seen that there is a change in electric field due to the introduction of DNA which was constrained near the surface (within 1 nm of the surface). After the constraints were removed, the DNA diffused slightly away from the surface (the lowest position of the DNA fluctuating between approximately 1 and 3 nm from the surface), resulting in no significant change in the electric field compared to the control. To illustrate the difference between constrained and unconstrained systems, snapshots of the DNA system are shown in ESI 5.† | ||
This response to DNA provides a proof-of-concept for this molecular dynamics simulation methodology as a novel way of investigating the field effect for biosensing applications at a molecular scale and can provide a platform for systemically exploring molecular scale effects which may be relevant for optimising FET-sensor response such as surface morphology, buffer composition, biomolecule structure and biomolecule dynamics.
:MgCl2) and a statistically significant change in electric field due to DNA addition was calculated. It was not possible to determine whether the signal was significant with regard to the resolution of detection of experimental measurements. As an improvement over the previous work, in the present work the surface charge density–electric field relationship was used to provide a model calibration method and evidence that this DNA induced change in electric field would be large enough to be experimentally measurable.
The effect of distance of DNA from the surface was also observed in the present work. In the red bar of Fig. 4, the result from the system after the DNA was permitted to freely diffuse from its initial surface position (unconstrained) is shown as a red bar. The DNA moved greater than 1 nm away from the surface and the electric field returned to that which was indistinguishable from the control shown in green. The distance-dependent reduction in electric field was a result of screening from polarised water and ions in solution. Experiments also show a strong distance-dependent reduction of DNA signal.65 At 300 mM NaCl, the Debye length is 0.55 nm and therefore based on the Debye length screening arguments (section 2.1.1), the influence on the interface is expected to be negligible after the DNA reaches 1–3 nanometres distance, which is in agreement with the present molecular dynamics simulation result.
While the electric field was used for this analysis, there was a strong correlation between the total electrostatic energy of the system (double integration of the entire charge density) and the electric field below the silica–water interface, as shown in ESI 9.† Given this correlation, it is possible that the total stored electrostatic energy may also be a viable metric of estimating the sensor response from molecular dynamics simulations.
4.3 Surface potential: effect of pH
:1 electrolyte is assumed (e.g. NaCl) with two ionic strengths shown: 300 mM (blue) corresponding to the same ionic strength as the molecular dynamics simulations performed, and a more dilute 100 mM (green) for comparison.
 | ||
| Fig. 5 Calculated surface potential as a function of surface charge from three mean-field models—the Grahame equation (eqn (1), solid line), the Debye–Hückel model (eqn (2), short dashed line), and the modified Poisson–Boltzmann model (eqn (4), circles for a = 0.189 nm and long dashed line for a = 1 nm). Two ionic strengths of a monovalent 1:1 electrolyte are shown: 300 mM (blue) and 100 mM (green). Simulated data are plotted as a function of surface charge density, with the approximate linear relationship between surface charge density and pH shown as described in the Methods section. | ||
Fig. 5 shows that the surface potential became increasingly negative with an increase in the surface charge density in all cases. A linear surface charge–surface potential relationship occurs for the Debye–Hückel model due to the constant capacitance of this model whereby the Debye length is constant as a function of surface charge.48 In contrast, both the Poisson–Boltzmann and modified Poisson–Boltzmann models have a non-linear surface charge/surface potential relationship.48,106 At low surface potentials, they provide similar predictions; however, at high surface potentials they differ because the modified Poisson–Boltzmann model prevents unphysically dense accumulation of ions at the surface, and thus has a lower differential capacitance62 or, equivalently, a steeper slope in Fig. 5.
The a parameter for the modified Poisson–Boltzmann equation represents the maximum density of ions possible at the surface, with smaller values meaning higher maximal density. a = 0.189 nm is the hydrodynamic radius of a sodium ion64 and a = 1 nm a plausible higher value considering the contributions from solvent and ion-correlation effects resulting in more disperse ions at the surface.62 The a parameter is being used here to provide a range of predictions from the modified Poisson–Boltzmann model, without resorting to empirical parametrisation. It can seen that at these charge densities, if a = 0.189 nm, then the results are equivalent to the Poisson–Boltzmann equation but that if a = 1 nm the modified Poisson–Boltzmann equation predicts a larger potential response. In all cases, the Debye–Hückel model predicts the largest response, but is technically invalid at potentials much greater than kbT/q (∼26 mV).
All three mean-field models show a strong dependence on ionic strength, which is contradicted by experimental evidence.
 | ||
| Fig. 6 Comparison of the surface potential simulated via molecular dynamics (solid lines) with experimental data (dashed lines) as a function of surface charge. The simulated surface potentials are presented relative to the system with no surface charge by subtraction of the surface potential for the zero surface-charge system. The literature data are displayed with the electrolyte used in the legend, and were measured by using methods indicated by markers: ◆ X-Ray Photoelecton Spectroscopy (XPS),109 ■ Electrolyte-on-insulator (EOS),88, ★ Impedance110 ● IS-FET.89,111–113 Experimental data are plotted as a function of measured pH, whereas simulated data are plotted as a function of surface charge density, with both the axes aligned using the approximate linear empirical relationship between surface charge density and pH described in the Methods section. The high pH sensitivity of the data of Sakata et al. was obtained using a sputtered silica sample, as opposed to conventional thermally grown or native oxide, and for these data, the point-of-zero charge was not identified, so only potential differences can be shown, with the data manually aligned to a point-of-zero charge of ∼3 in agreement with typical experiments. Molecular dynamics (MD) simulation of the crystalline silica–electrolyte system (black lines) and amorphous silica–electrolyte system (blue lines) are shown. The error bars indicate 95% confidence interval over three 60 ns repeats and quantify uncertainty in the mean due to temporal fluctuations, with two repeat simulations for each surface charge performed to quantify thermodynamic convergence. | ||
A comparison to experimental pH response data was discussed in our previous work.46 In summary, the experimental data are highly variable due to variation between silica surface preparation and electrolyte composition; however, molecular dynamics simulations and mean-field models show a qualitative agreement with some experimental data up to highly effective pH.46 Silica often shows a sub-Nernstian response114 in the 30–40 mV per pH range,8,46,112,114 but the ideal Nernst response (∼59 mV per pH) has been reported89 as shown in Fig. 6 (pink circles). This untypically high pH response was likely due to differences in silica structure as the silica was sputtered as opposed to being thermally grown or naturally formed.
The amorphous system showed a decrease in the polarisation of water compared to the crystalline system (Fig. 3(b)), due to the amorphous structure reducing the ability of water to form a highly ordered and oriented monolayer. This decrease in polarisation would be expected to cause a smaller magnitude surface potential; however, it is counteracted by increased sodium ion binding (Fig. 3(a)), resulting in the observed larger magnitude of surface potential for the amorphous system versus the crystalline system.
Comparison of Fig. 5 and 6 shows that the mean-field models at 300 mM ionic strength significantly underestimates the potential response of the two experiments shown at 1 M ionic strength110,111 and that predicted by the molecular dynamics models. The reason for this underestimation is in part due to ignoring the effect of water polarisation. The mean-field models use a constant permittivity for the liquid throughout the system, which is in contrast to the present molecular dynamics simulations which simulate the spatially-dependent polarisation of water. If a lower permittivity is used in the mean-field model, a larger potential results.
Another reason for the underestimation is due to the strong ionic-strength dependence observed in the mean-field models. Experiments show ionic strength to have a weaker effect, with pH being the dominant determinant of response.81 The strong ionic strength dependence compared to that observed experimentally is due to neglect of the acid–base chemical equilibria; for example, in ultrapure water (ionic strength less than 0.001 M), negligible surface ionisation can occur due to the lack of availability of charge stabilising cations.82 Modelling such equilibria is often performed empirically via site-binding models as it remains too computationally intensive to simulate the effects of ionic strength of acid–base equilibria from first principles.80 While site-binding models can provide accurate prediction of potentiometric pH response with sufficient parametrisation, they cannot provide molecular-scale insight such as the discussed effects of surface morphology.
4.4 Surface potential: the effect of DNA
As presented in the background (section 2.5) potentiometric sensor response to DNA binding is highly variable due to difficulties in controlling experimental conditions, and the theoretical basis for the magnitude of experimentally observed response remains an open research question. Thus in this section the effect of DNA on potentiometric response is investigated.The shift in surface potential due to addition of DNA (i.e. combined immobilisation and hybridisation signal) was calculated to be −5 ± 12 mV in one simulation and −24 ± 8 in a repeat simulation (95% confidence intervals calculated as per Fig. 6), with only the latter simulation being statistically significant (unpaired t-test p = 0.48 and 0.011, respectively). The high uncertainty of the potential calculations can be contrasted with the electric field results for the same simulations, the data for which were previously shown in Fig. 4. The electric field calculations showed strong statistical significance between the absence and presence of DNA, with p = 0.0005 and p = 0.0001, respectively. As discussed in our previous work,46 surface potential calculations via molecular dynamics are highly sensitive to changes at the position selected as the ‘surface’ layer, whereas the long-range electric field calculations performed in this work are more reliable. The electric field calculations showed a response equivalent to one effective pH unit, which is consistent in magnitude with the approximately 25 mV surface potential shift observed due to DNA in one of the two DNA simulations.
In the present work, the simulated DNA–crystalline silica model system has a DNA density of 8.58 × 1012 molecules cm−2 and 12 base pairs (24 negative charges per DNA) and thus a surface charge density of σDNA = −0.33 C m−2. The silanolate surface charge density was σsurf = −0.083 C m−2. Eqn (5) can be used with the Grahame equation (eqn (1)) to predict a shift in surface potential of 77 mV. Thus the Grahame equation predicts a shift in surface potential greater than that predicted by the molecular dynamics simulation of 5–24 mV; however, given the variation in experimental DNA sensing data, the quantitative accuracies of the molecular dynamics and mean-field models are currently difficult to evaluate.
Further complicating the issue, the predictions between different mean-field models are highly variable. For example, while response calculated using eqn (5) is often presented using the Grahame equation in the literature, it is instead possible to use eqn (5) with either the Debye–Hückel model or the modified Poisson–Boltzmann equation. In such cases, different predictions are obtained; specifically −79 mV and −756 mV for the modified Poisson–Boltzmann equation with a = 0.184 nm and 1 nm, respectively, and −264 mV for the Debye–Hückel model. ESI 1† presents visualisations of the differences in predictions of each model. As these equations have broad utility to the biosensing community, easy-to-use open-source code is also provided to facilitate members of the biosensing community to easily investigate the effects of changing surface charge (intrinsic or biomolecule), ionic strength and electrolyte composition for this set of three equations.
Future work will further validate the model by the investigation of biomolecular systems which show more reproducible experimentally measured surface potential shifts such as polystyrenesulfonate (PAS) and polyallyamine hydrochloride (PAH), which form a polyelectrolyte multilayer.67,69
4.5 Effect of simulation duration and convergence
Large fluctuations in potential and electric field occur at the 1 ps timescale; for example, as discussed, a change in pH was shown to correspond to approximately 0.007 V nm−1, which can be compared with a typical standard deviation for the electric field of 0.025 V nm−1.‡ Calculation of potentiometric (bio-)sensor response therefore requires the ability to distinguish a small change in a noisy signal; this problem is somewhat alleviated in experimental systems by measurement equipment averaging over macroscopic timescales (microseconds to seconds). Potential and electric field calculations are shown as a function of simulation duration in ESI 10.† The results show that the duration used in this work can provide precise calculations of the mean electrostatic properties and evidences thermodynamic convergence, thereby demonstrating the merits of the longer duration of simulations in the present work compared to other reported calculations of the silica–water interfacial potential.38–41,43,445. Conclusions
Understanding the electrodynamics of the silica–water interface is vital to many modern technologies such as electrochemical fuel cells,1 water filtration3 and biosensing.77 In particular, the electrostatic potential and electric field are particularly relevant for understanding the response of potentiometric biosensors such as field-effect transistor-based sensors. With a particular focus on understanding the field-effect biosensors, in the present work, molecular dynamics simulations of the electrostatic properties at the silica–electrolyte-biomolecule interface have been presented. The effect of varying surface charge density and the addition of a highly charged model biomolecule (DNA) was investigated and the results were compared against three commonly used analytical mean-field models of the electrical double layer. Both a crystalline and an amorphous silica surface model were investigated, and their differences compared and contrasted. Molecular dynamics simulations were performed for significantly longer duration than in the related literature, facilitating increased precision and accuracy of the electrostatic properties.The first few molecular layers at the surface dominate the electrostatic properties of highly charged interfaces and therefore a layer close to the surface was analysed in more detail, referred to as the ‘Stern-like’ layer within the present work. Water formed three highly ordered surface layers in the crystalline silica model, in contrast to the amorphous model which showed much less water structuring. Sodium ion density in the Stern-like layer was found to increase approximately linearly with surface charge density. Water polarisation increased approximately linearly up to a surface charge density of −0.17 C m−2, but at the very high density of −0.38 C m−2, water polarisation was found to decrease due to displacement by the high density of sodium ions present at the interface. An empirical relationship was used to relate the surface charge density to the measured pH for these systems. Using this relationship, these simulations predict that, for both amorphous and crystalline silica systems, from pH 2 to 12, both sodium ion accumulation and water polarisation do not reach a maximum. At higher pH, and therefore at higher surface charge, maximal water polarisation occurs; however, at such a high pH the effects of silica surface dissolution become significant. These effects cannot be simulated using the current methodology but attempts to describe them have been made in other ‘reactive’ forcefields.38
The surface potential properties were calculated and compared to transferable models of the electrical double layer. The amorphous surface showed a larger change in surface potential for a given change in surface charge density or effective pH than the crystalline model, despite both systems being approximately equal in hydroxyl density, charge density and electrolyte ionic strength. The greater surface potential shift of the amorphous system was explained to be as a result of increased sodium ion accumulation due to a higher surface area and an increased availability of favourable sodium ion binding sites. This novel result suggests that for pH sensor design, amorphous surfaces will have enhanced pH response compared to more structured surfaces with a lower surface area. By contrast, commonly used models for describing pH response of potentiometric sensors, such as site-binding models, cannot describe such differences due to surface morphology.
The electric field below the silica substrate was calculated as a measure of the ‘field effect’, the phenomenon which drives the response of field-effect transistor-based sensors. A shift of 0.007 V per nm per effective pH was calculated. When DNA was introduced, a similar magnitude shift of 0.007 V nm−1 was calculated, and given that typical IS-FET sensors can resolve changes of at least one pH unit, this result predicts that this DNA-system should be experimentally detectable. The response rapidly diminished with distance of the DNA from the surface, in agreement with expectations based on the Debye–Hückel model. This result provides a first proof-of-concept for this type of simulation applied to potentiometric biosensing applications. The effects of biomolecule dynamics, biomolecule–ion interactions (e.g. ion displacement by biomolecules), the finite size of the biomolecule and the surface morphology are all explicitly treated, providing a wealth of information unavailable in current potentiometric biosensor models. Thus we posit that molecular dynamics provides a novel complementary tool to existing potentiometric biosensor models.
Conventional mean-field models provide predictions that are often insufficient for the rational design of potentiometric sensors with regard to the binding of molecular analytes. For example, recent studies have shown potentiometric detection of neutral alkanes when applied in nitrogen gas33 or humid vapour,34,115 both of which are predicted to produce no signal using Poisson–Boltzmann-based models due to the lack of charge on each alkane molecule. The measured response is likely due to changes in electric field induced by water polarisation and analyte dipole orientation, both of which would be described by the present molecular dynamics model and will be investigated in future work.
A unique capability of potentiometric biosensors is detection of electrostatic properties, in contrast to conventional biosensors which often operate via mass and optical detection.116,117 As a result of this capability, properties unmeasurable using conventional biosensors can be determined such as conformational changes of the analyte.118 It is therefore anticipated that simulation of the response of potentiometric biosensors via methods which are capable of modelling the dynamics of molecules will become increasingly important as potentiometric sensing becomes more widespread in the form of point-of-care diagnostic devices and environmental sensing applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the use of both the Reedbush HPC facilities at the University of Tokyo and the IRIDIS HPC Facility, and the associated HPC and linux support services at the University of Southampton, for the completion of the present work. The NAMD software was used in this work, which was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign. This work was completed by an overseas researcher under the Postdoctoral Fellowship of the Japan Society for the Promotion of Science 2017/18. The authors acknowledge Dr In-Chul for providing code to implement the EW3DC correction used in this work and Dr Christoph Riedel for proofreading and typography corrections.References
- J. M. Andújar and F. Segura, Renewable Sustainable Energy Rev., 2009, 13, 2309–2322 CrossRef.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- M. Noked, A. Soffer and D. Aurbach, J. Solid State Electrochem., 2011, 15, 1563–1578 CrossRef CAS.
- T. Metke, A. S. Westover, R. Carter, L. Oakes, A. Douglas and C. L. Pint, Sci. Rep., 2016, 6, 24680 CrossRef CAS PubMed.
- P. Bergveld, IEEE Trans. Biomed. Eng., 1972, BME-19, 342–351 CrossRef PubMed.
- P. Bergveld, IEEE Trans. Biomed. Eng., 1970, BME-17, 70–71 CrossRef.
- Y. Cui, Q. Wei, H. Park and C. M. Lieber, Science, 2001, 293, 1289–1292 CrossRef CAS PubMed.
- B. M. Lowe, K. Sun, I. Zeimpekis, C.-K. Skylaris and N. G. Green, Analyst, 2017, 142, 4173–4200 RSC.
- K. Y. Chumbimuni-Torres, Z. Dai, N. Rubinova, Y. Xiang, E. Pretsch, J. Wang and E. Bakker, J. Am. Chem. Soc., 2006, 128, 13676–13677 CrossRef CAS PubMed.
- R. Thürer, T. Vigassy, M. Hirayama, J. Wang, E. Bakker and E. Pretsch, Anal. Chem., 2007, 79, 5107–5110 CrossRef PubMed.
- M.-Y. Shen, B.-R. Li and Y.-K. Li, Biosens. Bioelectron., 2014, 60, 101–111 CrossRef CAS PubMed.
- G. Shalev, Y. Rosenwaks and I. Levy, Biosens. Bioelectron., 2012, 31, 510–515 CrossRef CAS PubMed.
- L. De Vico, L. Iversen, M. H. Sørensen, M. Brandbyge, J. Nygård, K. L. Martinez and J. H. Jensen, Nanoscale, 2011, 3, 3635 RSC.
- P. M. Dove and S. F. Elston, Geochim. Cosmochim. Acta, 1992, 56, 4147–4156 CrossRef CAS.
- P. M. Dove and C. J. Nix, Geochim. Cosmochim. Acta, 1997, 61, 3329–3340 CrossRef CAS.
- P. M. Dove, Am. J. Sci., 1994, 294, 665–712 CrossRef CAS.
- J. V. Smith, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 3370–3375 CrossRef CAS.
- P. Nair and M. Alam, IEEE Trans. Electron Devices, 2007, 54, 3400–3408 CrossRef CAS.
- C. Heitzinger, Y. Liu, N. J. Mauser, C. Ringhofer and R. W. Dutton, J. Comput. Theor. Nanosci., 2010, 7, 2574–2580 CrossRef CAS.
- C. Heitzinger and G. Klimeck, J. Comput. Electron., 2007, 6, 387–390 CrossRef CAS.
- C. Heitzinger, R. Kennell, G. Klimeck, N. Mauser, M. McLennan and C. Ringhofer, J. Phys.: Conf. Ser., 2008, 107, 012004 CrossRef.
- C. Heitzinger, N. J. Mauser and C. Ringhofer, SIAM J. Appl. Math., 2010, 70, 1634–1654 CrossRef.
- S. Baumgartner, C. Heitzinger, A. Vacic and M. A. Reed, Nanotechnology, 2013, 24, 225503 CrossRef PubMed.
- S. Baumgartner, M. Vasicek, A. Bulyha and C. Heitzinger, Nanotechnology, 2011, 22, 425503 CrossRef CAS PubMed.
- T. Windbacher, V. Sverdlov and S. Selberherr, Computational Electronics, 2009. IWCE'09. 13th International Workshop On, 2009, pp. 1–4 Search PubMed.
- T. Windbacher, V. Sverdlov, S. Selberherr, C. Heitzinger, N. Mauser and C. Ringhofer, AIP Conf. Proc., 2010, 1199, 507 CrossRef.
- P. R. Nair and M. A. Alam, Nano Lett., 2008, 8, 1281–1285 CrossRef CAS PubMed.
- T. Windbacher, V. Sverdlov and S. Selberherr, Biomedical Engineering Systems and Technologies, Springer, New York City, 2010, pp. 85–95 Search PubMed.
- M. H. Sørensen, N. A. Mortensen and M. Brandbyge, Appl. Phys. Lett., 2007, 91, 102105 CrossRef.
- L. De Vico, M. H. Sørensen, L. Iversen, D. M. Rogers, B. S. Sørensen, M. Brandbyge, J. Nygård, K. L. Martinez and J. H. Jensen, Nanoscale, 2011, 3, 706 RSC.
- N. Lloret, R. S. Frederiksen, T. C. Møller, N. I. Rieben, S. Upadhyay, L. De Vico, J. H. Jensen, J. Nygård and K. L. Martinez, Nanotechnology, 2013, 24, 035501 CrossRef PubMed.
- R. R. Pethig and S. Smith, Introductory Bioelectronics: For Engineers and Physical Scientists, John Wiley & Sons, Hoboken, New Jersey, 2012 Search PubMed.
- N. Swaminathan, A. Henning, T. Jurca, J. Hayon, G. Shalev and Y. Rosenwaks, Sens. Actuators, B, 2017, 248, 240–246 CrossRef CAS.
- B. Wang and H. Haick, ACS Appl. Mater. Interfaces, 2013, 5, 5748–5756 CAS.
- N. Gao, W. Zhou, X. Jiang, G. Hong, T.-M. Fu and C. M. Lieber, Nano Lett., 2015, 15, 2143–2148 CrossRef CAS PubMed.
- G. Zheng, X. P. A. Gao and C. M. Lieber, Nano Lett., 2010, 10, 3179–3183 CrossRef CAS PubMed.
- I.-Y. Chung, J. Lee, M. Seo and C. H. Park, Jpn. J. Appl. Phys., 2016, 55, 127001 CrossRef.
- J. C. Fogarty, H. M. Aktulga, A. Y. Grama, A. C. T. van Duin and S. A. Pandit, J. Chem. Phys., 2010, 132, 174704 CrossRef PubMed.
- Y. Maekawa, Y. Shibuta and T. Sakata, Jpn. J. Appl. Phys., 2013, 52, 127001 CrossRef.
- Y. Maekawa, Y. Shibuta and T. Sakata, Chem. Phys. Lett., 2015, 619, 152–157 CrossRef CAS.
- Y. Maekawa, Y. Shibuta and T. Sakata, ChemElectroChem, 2014, 1, 1516–1524 CrossRef CAS.
- B. M. Lowe, Y. Maekawa, Y. Shibuta, T. Sakata, C.-K. Skylaris and N. G. Green, Phys. Chem. Chem. Phys., 2017, 19, 2687 RSC.
- I.-C. Yeh and A. Wallqvist, J. Chem. Phys., 2011, 134, 055109 CrossRef PubMed.
- C. D. Lorenz, P. S. Crozier, J. A. Anderson and A. Travesset, J. Phys. Chem. C, 2008, 112, 10222–10232 CAS.
- R. Lavery, J. H. Maddocks, M. Pasi and K. Zakrzewska, Nucleic Acids Res., 2014, 42, 8138–8149 CrossRef CAS PubMed.
- B. M. Lowe, C.-K. Skylaris, N. G. Green, Y. Shibuta and T. Sakata, Jpn. J. Appl. Phys., 2018, 57, 04FM02 CrossRef.
- I.-C. Yeh and M. L. Berkowitz, J. Chem. Phys., 1999, 111, 3155–3162 CrossRef CAS.
- J. O. Bockris, A. K. Reddy and M. E. Gamboa-Aldeco, Modern Electrochemistry 2A, Kluwer Academic Publishers, Boston, 2002, vol. 2A Search PubMed.
- X. Duan, Y. Li, N. K. Rajan, D. A. Routenberg, Y. Modis and M. A. Reed, Nat. Nanotechnol., 2012, 7, 401–407 CrossRef CAS PubMed.
- X. Wen, S. Gupta, Y. Wang, T. R. N. Iii, S. C. Lee and W. Lu, Appl. Phys. Lett., 2011, 99, 043701 CrossRef.
- B. K. Wunderlich, P. A. Neff and A. R. Bausch, Appl. Phys. Lett., 2007, 91, 083904 CrossRef.
- D. Landheer, G. Aers, W. R. McKinnon, M. J. Deen and J. C. Ranuarez, J. Appl. Phys., 2005, 98, 044701 CrossRef.
- D. Landheer, W. McKinnon, G. Aers, W. Jiang, M. Deen and M. Shinwari, IEEE Sens. J., 2007, 7, 1233–1242 CrossRef CAS.
- S. Uno, M. Iio, H. Ozawa and K. Nakazato, Jpn. J. Appl. Phys., 2010, 49, 01AG07 Search PubMed.
- Y. Nishio, S. Uno and K. Nakazato, Jpn. J. Appl. Phys., 2013, 52, 04CL01 CrossRef.
- H. Ohshima, Biophysical Chemistry of Biointerfaces, John Wiley & Sons, 2011 Search PubMed.
- R. J. Hunter, Foundations of Colloid Science, Oxford University Press, Oxford, UK, 2001 Search PubMed.
- J. Lyklema, Fundamentals of Interface and Colloid Science: Solid-Liquid Interfaces, Academic Press, Cambridge, Massachusetts, 1995 Search PubMed.
- D. Grahame, Chem. Rev., 1947, 41, 441–501 CrossRef CAS PubMed.
- J. O. Bockris and A. K. Reddy, Modern Electrochemistry 1, Kluwer Academic Publishers, Boston, 1998, vol. 1 Search PubMed.
- M. A. Brown, G. V. Bossa and S. May, Langmuir, 2015, 31, 11477–11483 CrossRef CAS PubMed.
- M. S. Kilic, M. Z. Bazant and A. Ajdari, Phys. Rev. E, 2007, 75, 021502 CrossRef PubMed.
- M. S. Kilic, M. Z. Bazant and A. Ajdari, Phys. Rev. E, 2007, 75, 021503 CrossRef PubMed.
- P. W. Atkins, Physical Chemistry, W H Freeman & Co, New York, 1994 Search PubMed.
- T. Sakata and Y. Miyahara, Angew. Chem., Int. Ed., 2006, 45, 2225–2228 CrossRef CAS PubMed.
- F. N. Ishikawa, M. Curreli, H.-K. Chang, P.-C. Chen, R. Zhang, R. J. Cote, M. E. Thompson and C. Zhou, ACS Nano, 2009, 3, 3969–3976 CrossRef CAS PubMed.
- F. Uslu, S. Ingebrandt, D. Mayer, S. Böcker-Meffert, M. Odenthal and A. Offenhäusser, Biosens. Bioelectron., 2004, 19, 1723–1731 CrossRef CAS PubMed.
- K. Jayant, K. Auluck, S. Rodriguez, Y. Cao and E. C. Kan, Phys. Rev. E, 2014, 89, 052817 CrossRef PubMed.
- J. Fritz, E. B. Cooper, S. Gaudet, P. K. Sorger and S. R. Manalis, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14142–14146 CrossRef CAS PubMed.
- A. Poghossian, A. Cherstvy, S. Ingebrandt, A. Offenhäusser and M. J. Schöning, Sens. Actuators, B, 2005, 111–112, 470–480 CrossRef CAS.
- J. Bobacka, A. Ivaska and A. Lewenstam, Chem. Rev., 2008, 108, 329–351 CrossRef CAS PubMed.
- A. Tarasov, M. Wipf, R. L. Stoop, K. Bedner, W. Fu, V. A. Guzenko, O. Knopfmacher, M. Calame and C. Schönenberger, ACS Nano, 2012, 6, 9291–9298 CrossRef CAS PubMed.
- R. E. G. van Hal, J. C. T. Eijkel and P. Bergveld, Adv. Colloid Interface Sci., 1996, 69, 31–62 CrossRef CAS.
- D. E. Yates, S. Levine and T. W. Healy, J. Chem. Soc., Faraday Trans. 1, 1974, 70, 1807–1818 RSC.
- R. E. G. van Hal, J. C. T. Eijkel and P. Bergveld, Sens. Actuators, B, 1995, 24, 201–205 CrossRef CAS.
- S. Bermejo, Chemical Sensors: Comprehensive Sensor Technologies Volume 5: Electrochemical and Optical Sensors, Momentum Press, New York, 1st edn, 2011, pp. 201–247 Search PubMed.
- P. Bergveld, Sens. Actuators, B, 2003, 88, 1–20 CrossRef CAS.
- A. Rimola, D. Costa, M. Sodupe, J.-F. Lambert and P. Ugliengo, Chem. Rev., 2013, 113, 4216–4313 CrossRef CAS PubMed.
- L. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
- B. M. Lowe, C.-K. Skylaris and N. G. Green, J. Colloid Interface Sci., 2015, 451, 231–244 CrossRef CAS PubMed.
- E. Tombácz, Chem. Eng., 2009, 53, 77–86 Search PubMed.
- G. H. Bolt, J. Phys. Chem., 1957, 61, 1166–1169 CrossRef CAS.
- V. Puchol, J. El Haskouri, J. Latorre, C. Guillem, A. Beltrán, D. Beltrán and P. Amorós, Chem. Commun., 2009, 2694 RSC.
- B. B. Owen and S. R. Brinkley, Chem. Rev., 1941, 29, 461–474 CrossRef CAS.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2014, 26, 2647–2658 CrossRef CAS.
- Y. Duval, J. A. Mielczarski, O. S. Pokrovsky, E. Mielczarski and J. J. Ehrhardt, J. Phys. Chem. B, 2002, 106, 2937–2945 CrossRef CAS.
- K. Leung, I. M. B. Nielsen and L. J. Criscenti, J. Am. Chem. Soc., 2009, 131, 18358–18365 CrossRef CAS PubMed.
- W. Siu and R. Cobbold, IEEE Trans. Electron Devices, 1979, 26, 1805–1815 CrossRef.
- T. Sakata, K. Nishimura, Y. Miyazawa, A. Saito, H. Abe and T. Kajisa, Anal. Chem., 2017, 89, 3901–3908 CrossRef CAS PubMed.
- J. Y. Oh, H.-J. Jang, W.-J. Cho and M. S. Islam, Sens. Actuators, B, 2012, 171–172, 238–243 CrossRef CAS.
- S. Dastidar, A. Agarwal, N. Kumar, V. Bal and S. Panda, IEEE Sens. J., 2015, 15, 2039–2045 CrossRef CAS.
- N. Kumar, J. Kumar and S. Panda, ECS J. Solid State Sci. Technol., 2015, 4, N18–N23 CrossRef CAS.
- M. J. Schöning, F. Ronkel, M. Crott, M. Thust, J. W. Schultze, P. Kordos and H. Lüth, Electrochim. Acta, 1997, 42, 3185–3193 CrossRef.
- M. J. Schöning, Ü. Malkoc, M. Thust, A. Steffen, P. Kordoš and H. Lüth, Sens. Actuators, B, 2000, 65, 288–290 CrossRef.
- F. K. Perkins, L. M. Tender, S. J. Fertig and M. C. Peckerar, Nanostructure Science, Metrology and Technology, Gaithersburg, MD, United States, 2002, pp. 251–265 Search PubMed.
- F. Wei, B. Sun, Y. Guo and X. S. Zhao, Biosens. Bioelectron., 2003, 18, 1157–1163 CrossRef CAS PubMed.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS PubMed.
- A. Bandiziol, P. Palestri, F. Pittino, D. Esseni and L. Selmi, IEEE Trans. Electron Devices, 2015, 62, 3379–3386 CrossRef CAS.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2016, 28, 406–407 CrossRef CAS.
- K. Hart, N. Foloppe, C. M. Baker, E. J. Denning, L. Nilsson and A. D. MacKerell, J. Chem. Theor. Comput., 2012, 8, 348–362 CrossRef CAS PubMed.
- H. Heinz, T.-J. Lin, R. Kishore Mishra and F. S. Emami, Langmuir, 2013, 29, 1754–1765 CrossRef CAS PubMed.
- F. S. Emami, V. Puddu, R. J. Berry, V. Varshney, S. V. Patwardhan, C. C. Perry and H. Heinz, Chem. Mater., 2014, 26, 5725–5734 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- A. A. Gurtovenko and I. Vattulainen, J. Chem. Phys., 2009, 130, 215107 CrossRef PubMed.
- F. Musso, P. Mignon, P. Ugliengo and M. Sodupe, Phys. Chem. Chem. Phys., 2012, 14, 10507 RSC.
- J. W. Lee, A. Mani and J. A. Templeton, Langmuir, 2015, 31, 7496–7502 CrossRef CAS PubMed.
- J. W. Murray, J. Colloid Interface Sci., 1974, 46, 357–371 CrossRef CAS.
- C. C. Bueno, A. M. Amarante, G. S. Oliveira, D. K. Deda, O. Teschke, E. d. F. Franca and F. L. Leite, IEEE Sens. J., 2014, 14, 1467–1475 CrossRef CAS.
- M. A. Brown, Z. Abbas, A. Kleibert, R. G. Green, A. Goel, S. May and T. M. Squires, Phys. Rev. X, 2016, 6, 011007 Search PubMed.
- J. Diot, J. Joseph, J. Martin and P. Clechet, J. Electroanal. Chem. Interfacial Electrochem., 1985, 193, 75–88 CrossRef CAS.
- C. Fung, P. Cheung and W. Ko, Electron Devices Meet., 1980, 689–692 Search PubMed.
- K. Sun, I. Zeimpekis, C. Hu, N. M. J. Ditshego, O. Thomas, M. R. R. de Planque, H. M. H. Chong, H. Morgan and P. Ashburn, Nanotechnology, 2016, 27, 285501 CrossRef CAS PubMed.
- L. Bousse, N. F. D. Rooij and P. Bergveld, IEEE Trans. Electron Devices, 1983, 30, 1263–1270 CrossRef.
- S. Chen, J. G. Bomer, E. T. Carlen and A. van den Berg, Nano Lett., 2011, 11, 2334–2341 CrossRef CAS PubMed.
- N. Shehada, J. C. Cancilla, J. S. Torrecilla, E. S. Pariente, G. Brönstrup, S. Christiansen, D. W. Johnson, M. Leja, M. P. A. Davies, O. Liran, N. Peled and H. Haick, ACS Nano, 2016, 10, 7047–7057 CrossRef CAS PubMed.
- H. H. Nguyen, J. Park, S. Kang and M. Kim, Sensors, 2015, 15, 10481–10510 CrossRef CAS PubMed.
- S. Sang, Y. Wang, Q. Feng, Y. Wei, J. Ji and W. Zhang, Crit. Rev. Biotechnol., 2015, 1–17 CrossRef PubMed.
- D. Cahen, R. Naaman and Z. Vager, Adv. Funct. Mater., 2005, 15, 1571–1578 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C8NR00776D |
| ‡ Calculated over 180 ns, with snapshots taken at 100 ps intervals for the −0.041 C m−2 crystalline silica model. |
| This journal is © The Royal Society of Chemistry 2018 |