
Self-assembled N-graphene nanohollows enabling ultrahigh energy density cathode for Li–S batteries†
Hanting
Tang
,
Jinlong
Yang
,
Guangxing
Zhang
,
Chaokun
Liu
,
Han
Wang
,
Qinghe
Zhao
,
Jiangtao
Hu
,
Yandong
Duan
and
Feng
Pan
 *
*
School of Advanced Materials, Peking University, Shenzhen Graduate School, Shenzhen 518055, People's Republic of China. E-mail: panfeng@pkusz.edu.cn
First published on 27th November 2017
Abstract
Functional porous carbon materials are widely used to solve the low conductivity and shuttle effect of Li–S batteries; however, the common carbon/sulfur composite electrodes based on traditional technology (with conducting agents and binders) make it difficult for a battery to work stably at an ultra-high sulfur loading of 10 mg cm−2. Herein, an appropriate content of sulfur was injected into a pomegranate-like structure self-assembled with nanohollows (PSSN) of N-graphene. The Li-PSSN/S battery based on traditional technology displays a large-capacity, high-rate and long-life at an ultra-high areal-sulfur loading of 10.1 mg cm−2. The excellent performance with ultra-high areal-sulfur loading can be attributed to the hierarchal nanohollows with graphene-shells being in close contact to build a 3D-electronic conduction network and promoting electrolyte adsorption into the entire electrode to maintain rapid Li-ion transport, while stopping the shuttle-effect via the strong interaction of polysulfide with the doped N elements on graphene-shells. In addition, the exact sulfur content can provide just enough space to maintain the huge volume change and constant thickness of the S-electrodes during the charge–discharge process to enhance the cycling stability.
Introduction
Rechargeable lithium-ion batteries, which play a key role in propelling electric vehicles and storing residual electricity, have attracted increasing attention during the past decade.1 Li–S batteries are considered to be a promising substitution for the next generation of batteries because of their high theoretical specific capacity (1675 mA h g−1) and energy density (2600 W h kg−1).2,3 However, the low electronic conductivity of sulfur, “shuttle effect” of polysulfide, huge volume expansion and lithium dendrites observed during the charge–discharge process leads to the rapid capacity decay of Li–S batteries and constrain their use in practical applications.4,5 In order to solve the abovementioned problems, a series of studies including the development of advanced sulphur cathodes,6 electrolytes7 and protected anodes8 were proposed to improve the overall performance of lithium–sulfur batteries. Among them, the carbon materials with electronic conduction, high specific surface area and suitable pore structures used in cathodes can remarkably improve the electrochemical performance of the battery and have aroused a great deal of interest from researchers.9The carbon materials applied in Li–S batteries can be segmented into the following species: net structure carbons,10 hollow carbon nanospheres,11,12 carbon nanofibers/nanotubes13,14 and graphene,15 which can provide a large pore volume for the storage of sulfur and greatly improve the electric conductivity of the sulfur cathode. However, most of these porous carbon materials have two main problems: first is that the interaction of the porous carbon and the polysulfide is so weak that it cannot effectively inhibit the dissolution of the polysulfide into the electrolyte and therefore cannot solve the “shuttle effect” of polysulfide. The second problem is that the volume expansion of sulfur is too large in the charge–discharge process, which leads to the destruction of porous carbon structure when the content and position of loading sulfur are not controllable during the process. To improve the interaction between porous carbon and sulfur, an effective method that can be used is chemical modification via elemental doping. Many studies16,17 have shown that the addition of N, O and other elements into the porous carbon structure can greatly increase the interaction between sulfur and the shells of porous carbon. In addition, the doped elements can form chemical bonds with sulfur and the polysulfide compounds to fix the sulfur on the shells of porous carbon and inhibit the transmission of the polysulfide compounds. In order to solve the volume expansion during charging and discharging, the usual method is to provide sufficient space for the volume change.18 Cui et al. developed a Sulphur-TiO2 yolk–shell nanoarchitecture with internal void space for long-cycle Li–S batteries19 and a pomegranate-inspired nanoscale design for large-volume-change lithium battery electrodes.20
The conventional sulfur loading is the proportion of sulfur in the cathode material, while the areal sulfur loading is the mass of sulfur per unit electrode area. Although many approaches have achieved the trapping of sulfur in the carbon matrix and have achieved a sulfur content up to 90% and even higher, it should be noted that the areal sulfur loadings on the final cathode are usually lower than 2 mg cm−2. It has been proven difficult to reach an areal capacity of 4 mA h cm−2, which is required for the present day practical applications of Li-ion batteries.21 Increasing the areal sulfur loading would require increasing the electrode thickness, which greatly limits the conduction of Li-ions and electrons.22 Prior to this, there have been some reports, in which high areal sulfur loadings have been achieved. For example, Song et al.23 reported highly crumpled nitrogen-doped graphene sheets, exhibiting a high sulfur loading of 5 mg cm−2. Zhou et al.24 reported 3D N,S-doped graphene sponge electrodes, showing a high sulfur loading of 8.5 mg cm−2. Yuan et al.25 reported hierarchical CNT-S paper electrodes with 3D interconnected conductive frameworks, showing an ultra-high sulfur loading of 17.3 mg cm−2. However, there still exist a series of problems: (1) when using the traditional electrode preparation method with conducting carbon black and PVDF/PTFE binders,26 it is difficult to make the sulfur loading increase to 10 mg cm−2 or more; (2) when the sulfur loading increases to 10 mg cm−2 or more, the batteries mostly require a special or freeing conductive agent/binder27 rather than traditional carbon black or PVDF/PTFE; (3) at high sulfur loadings, the Li–S batteries usually charge and discharge at only 0.1 C or even lower current density, and show poor cycle stability at high rates due to polarization.28 Therefore, to overcome the above difficulties, the following factors in regard to the electrodes need to be considered: (1) a 3D conductive framework structure is indispensable to achieve electron conduction and (2) the electrolyte should quickly infiltrate the entire electrode to achieve lithium-ion conduction.
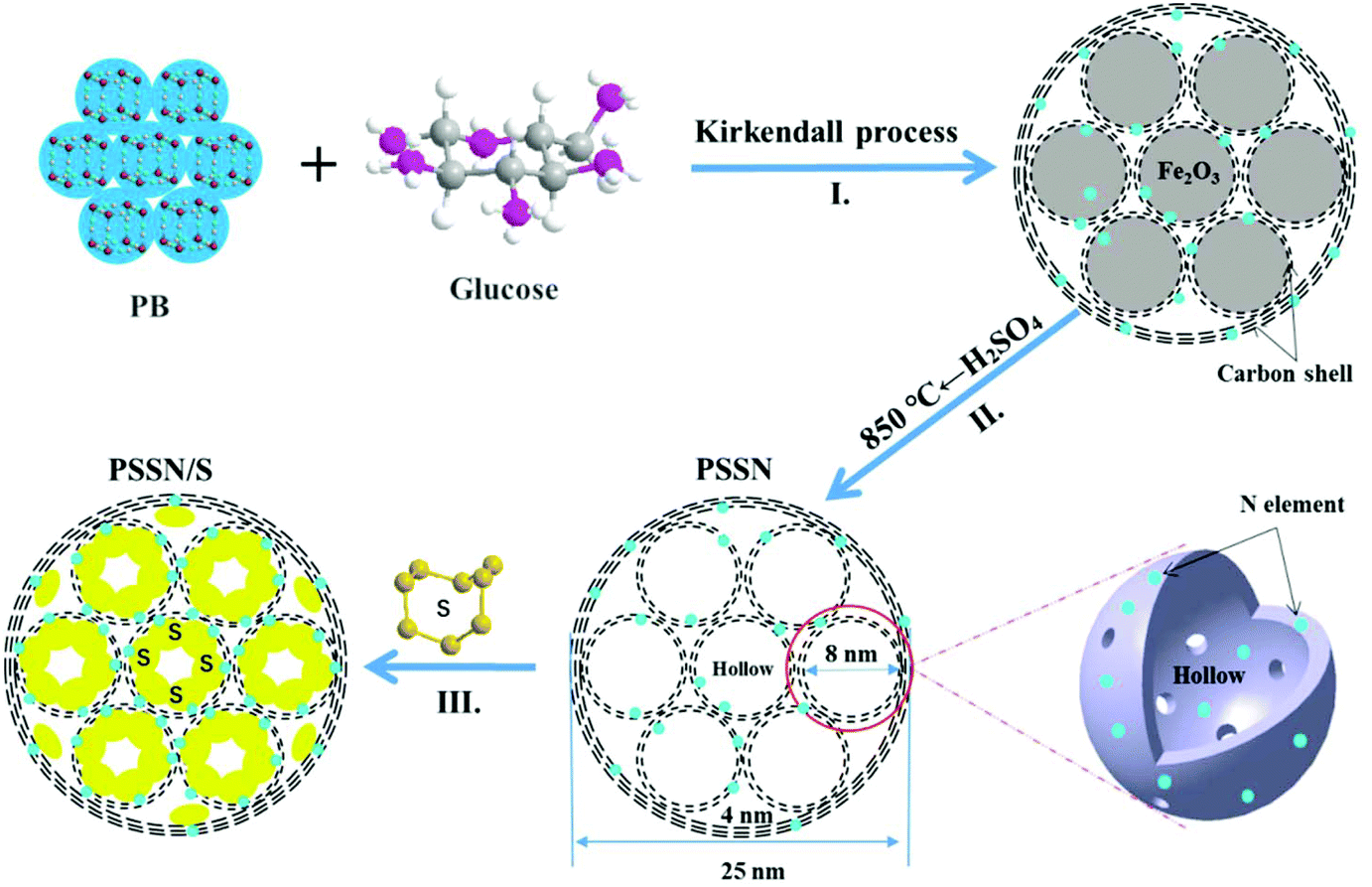
In this study, a ultra-high areal-sulfur loading (10.1 mg cm−2) in Li–S batteries was enabled by a pomegranate-like structure self-assembled with nanohollows (PSSN). The novel structure is composed of smaller hollows with diameters of about 8 nm and thinner N-doped graphene shells, which were fully embedded in the larger hollows with diameters of about 25 nm and thicker N-graphene-shells. It was easily synthesized by employing Prussian blue and glucose as the precursors. PSSN was applied in Li–S batteries with an ultra-high areal-sulfur loading of 10.1 mg cm−2, large-rate-capacity performance and long-life. After impregnating sulfur into the PSSN, the sulfur was stored in the smaller nanohollows encapsulated with N-doped graphene, which were further self-assembled to come in close contact to build a 3D-electronic conduction network. The sulfur content was controllably loaded at 60 wt% in the special network due to the interaction of sulfur with the doped N-elements at an optimized concentration. Because of this, PSSN with an exact sulfur content by interaction with the N-elements on the shells of the nanohollows can provide just enough space to maintain the huge volume change in the lithium polysulfide (Li2Sn) observed during the charge–discharge process so as to enhance the cycling stability. In addition, the graphene layers, interconnected inside and outside, construct a 3D electron conductive framework and the pores in the 3D structure promote electrolyte adsorption into the entire electrode to maintain the rapid Li-ion transport in the cathode with ultra-high areal-sulfur loading, which further ensures its large-capacity and high-rate as well as long-life.
Results and discussion
The synthesis route for the preparation of PSSN/S is illustrated in Fig. 1. First, a core–shell self-assembled nanohollows γ-Fe2O3@graphene hybrid was synthesized by employing Prussian blue (PB) and glucose (mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3) as the precursors (step I in Fig. 1). In this typical route, for the minimization of the total energy of the system, the small primary PB nanoparticles aggregated together to form homogeneous microspheres. Calcination of the PB microspheres and glucose under an argon atmosphere gave rise to the decomposition of the cyano groups and the formation of Fe nanocrystals. Thus, pomegranate-like structures with Fe nanocrystals were self-assembled, in which the small primary nanoparticles resembled the pomegranate fruit and the microspheres resembled the pomegranate peel.
:0.3) as the precursors (step I in Fig. 1). In this typical route, for the minimization of the total energy of the system, the small primary PB nanoparticles aggregated together to form homogeneous microspheres. Calcination of the PB microspheres and glucose under an argon atmosphere gave rise to the decomposition of the cyano groups and the formation of Fe nanocrystals. Thus, pomegranate-like structures with Fe nanocrystals were self-assembled, in which the small primary nanoparticles resembled the pomegranate fruit and the microspheres resembled the pomegranate peel.
 | ||
| Fig. 1 Synthesis route for preparation of the PSSN/S. | ||
When exposed to air, the Fe nanocrystals were oxidized and a core–shell self-assembled nanohollows γ-Fe2O3@graphene hybrid was obtained via the Kirkendall process.29 Subsequently, the γ-Fe2O3 core was etched using concentrated sulfuric acid and the initial PSSN shell was collected. In view of our previous study,30 the PSSN was reheated to 850 °C under an argon atmosphere to improve its conductivity without losing much nitrogen (step II in Fig. 1). Third, sulfur was impregnated into the PSSN using a melt-diffusion method31 in an open system and the sulfur content was perfectly controlled by interacting with the doped N element at an optimized concentration to obtain a certain amount of space and allow for the volume expansion observed during lithium intercalation (step III in Fig. 1). It should be noted that the partial amount of sulfur in the nanohollows, which does not interact with the doped N elements on the graphene-shells, could be sublimed during the melt-diffusion process at a temperature of about 155 °C, which is important for controlling the volume expansion and eliminating the “shuttle effect” of polysulfide by subliming the non-bonded sulfur on N-graphene.
The morphologies and structural features of PSSN and PSSN/S were confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 2a, PSSN is composed of numerous nanoparticles wound and linked by few thin mesh-like structures. The size of the nanoparticles is similar to that of PB (Fig. S1a†), indicating that the carbon framework of PB was retained after high temperature pyrolysis. The thin mesh-like carbon originates from the pyrolyzed glucose, which plays a key role in building the 3D conductive framework. The TEM image (Fig. 2b) further proves that the nanoparticles in PSSN were hollow. Fig. 2c shows that the shell of the nanoparticles is made up of 5–10 layers of graphene and inside, the less layers (1–4 layers) of graphene revolve around few small holes (less than 10 nm), which are the pomegranate-like structures self-assembled by nanohollows with graphene-shells, which is consistent with the pore structure probed using the N2 adsorption/desorption isotherms. The SEM images (Fig. 2d and S1 b, c†) show that after sulfur injection, the graphene shells with thin mesh-like structure in PSSN/S are the same as that in PSSN, indicating that the graphene structure was very strong and hard to crack during the sulfur injection process. The TEM images (Fig. 2e and f) show the microstructures of PSSN/S. It is clearly observed that the sulfur is filled into the pores, which are encapsulated by the thin layer graphene shells. Energy dispersive X-ray spectroscopy (EDX) (Fig. 2g) clearly confirms the uniform distribution of the C, N, O, and S elements in PSSN/S. The C, N, and O elements are contributed by the PSSN (EDX in Fig. S1d†) and the small-size sulfur particles are stored in the nanohollows surrounded by the N,O-doped graphene. The sulfur in the holes can interact with the doped N and O elements to reduce the shuttle effect of Li2Sn during the charge/discharge process.32,33
 | ||
| Fig. 2 SEM and TEM images of (a–c) PSSN and (d–f) PSSN/S, and (g) EDX mapping of C, S, N, O elements collected from a selected area in PSSN/S. | ||
X-ray diffraction (XRD) was carried out to trace the phase composition of the materials. Fig. S2† shows that the phase composition of Prussian blue (PB) and the glucose precursors transformed into γ-Fe2O3@graphene after the Kirkendall process. The XRD patterns of PSSN, elemental S and PSSN/S are shown in Fig. 3a. In the XRD pattern of PSSN, it can be observed that the Fe2O3 peaks are absent, indicating that iron was completely removed from PSSN by the acid treatment step. The only diffraction peak at 26.3° is ascribed to the characteristic peak of graphite (JPCDS No. 41-1487). Sharp diffraction peaks are observed in the spectra of PSSN/S, which correspond to the rhombic structure of S (JPCDS No. 08-0247). The S content in the PSSN/S composite was further measured using thermogravimetric analysis (TGA), which showed a sulfur loading ratio of 60 wt% (Fig. S3†). Raman spectroscopy was used to investigate the structural features of sulfur and carbon in PSSN/S as shown in Fig. S4.† Two main characteristic peaks for carbon were observed at 1359 cm−1 and 1590 cm−1 corresponding to the D band and G band of carbon, respectively. The G band is usually assigned to sp2-graphitic carbon, while the D band is assigned to defects or disorder in the carbon matrix.34 The density ratio of IG/ID in PSSN/S was approximately 0.95, which is almost the same as that of PSSN (0.97), indicating that the sulfur in nanohollows does not affect the conductivity of the graphene framework in PSSN/S. In addition, the main characteristic peaks (at 470, 217 and 150 cm−1)35 of elemental S are not visible in the Raman spectrum of PSSN/S, indicating that sulfur was encapsulated completely inside the PSSN. The N2 adsorption/desorption isotherms of PSSN and PSSN/S in Fig. 3b show type IV isotherms with type H3 hysteresis loops, indicating that mesopores account for majority of the structure. The Brunauer–Emmett–Teller (BET) surface area of PSSN was calculated to be 422 m3 g−1. After impregnating with sulfur, the specific surface area of PSSN/S was 42 m3 g−1. The pore sizes were derived using the BJH model and calculated from the desorption branch, which revealed that the pores in PSSN were primarily mesopores with sizes centered at 4 nm and 8 nm, and a small quantities of micropores with an average size of 1.8 nm. Typically, the pore volume of PSSN and PSSN/S was 2.035 and 0.328 cm3 g−1, respectively, which can be calculated by the fact that the volume of sulfur accounted for 59.7% of the volume of the carbon pores. The theoretical results19 showed that the volume of sulfur expands ∼80% during the charge–discharge process. According to the above data, the carbon pores have just enough space for the sulfur expansion (the volume of sulfur expands to 105% of the volume of carbon pores when discharging).
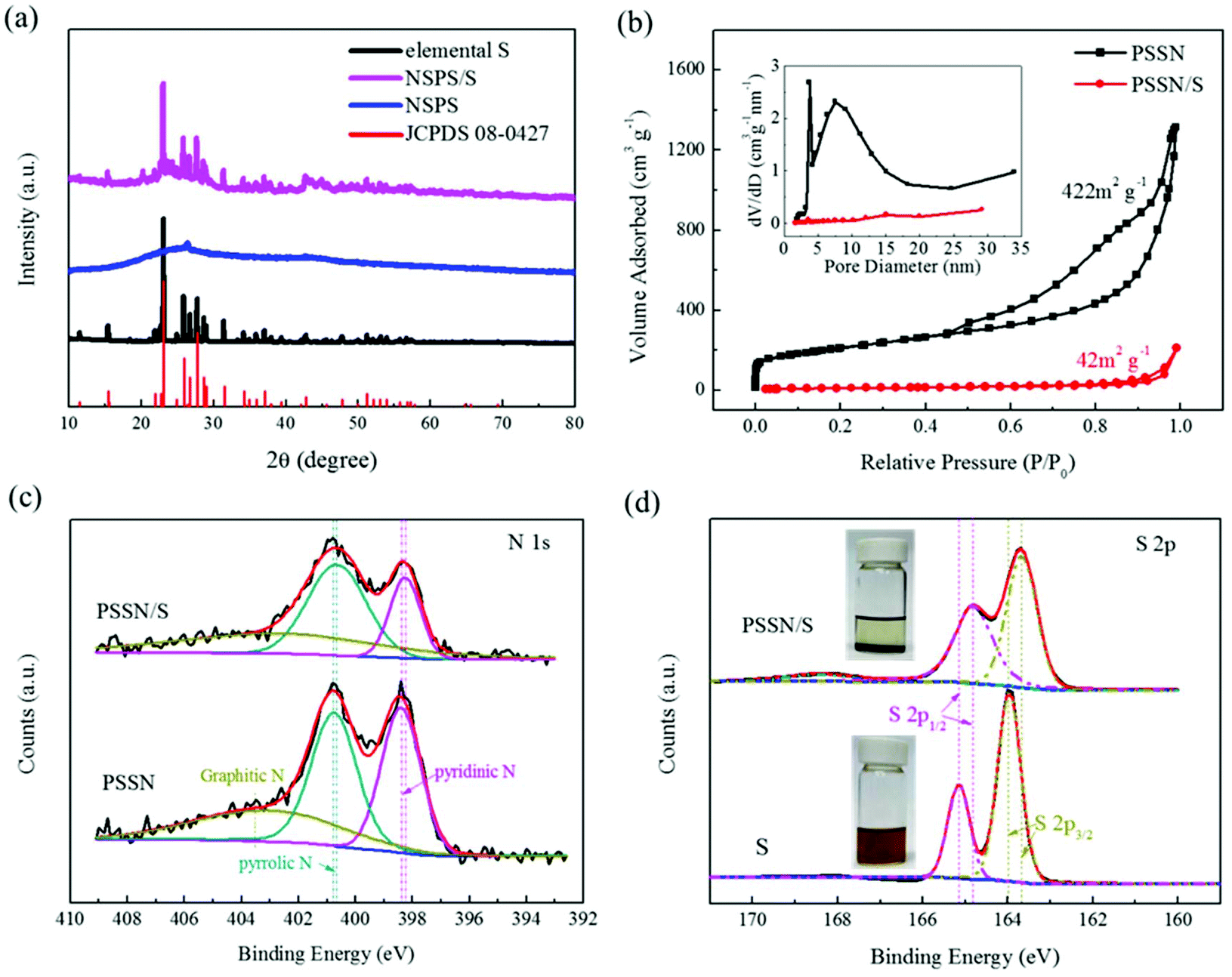 | ||
| Fig. 3 (a) XRD patterns of PSSN, elemental S and PSSN/S. (d) Nitrogen adsorption–desorption isotherms of PSSN and PSSN/S, inset of (b) is the corresponding pore-size distribution. (c) High-resolution N 1s spectra of PSSN and PSSN/S. (d) High-resolution S 2p XPS spectra of pure S and PSSN/S, inset of (d) is polysulfide adsorption experiments of PSSN (up) and contrast (down). | ||
X-ray photoelectron spectroscopy (XPS) was conducted to confirm the interactions between sulfur and PSSN. The C, N, O, and S elements were observed in the overall spectra of PSSN/S (Fig. S5a†). The N 1s spectra (Fig. 3c) of PSSN can be deconvoluted into three peaks:30 the pyridinic N (398.4 eV), pyrrolic N (400.8 eV), and graphitic N (402.6 eV). The peaks of pyridinic N (398.3 eV) and pyrrolic N (400.7 eV) in PSSN/S shift to low energy by 0.1 eV, which confirms the interaction of N and S. The N content (Table S1†) in PSSN measured using XPS was 7.4%, which is a large number for N-element doping, indicating that the N–S interaction could play an important role in trapping sulfur and polysulfide generated in the cycling of the batteries. Moreover, as the PSSN changed to PSSN/S, it can be observed that the content of pyridinic N decreases, suggesting that part of the carbon–nitrogen bonds change from a double bond to a single bond with the participation of sulfur. In addition, the weakening of the carbon–nitrogen bonds indicates that there can be a strong bonding between sulfur and nitrogen, which could be one of the important reasons why the sulfur can be stabilized in the carbon shell during S-loading. The C 1s spectra (Fig. S5b†) of PSSN can be deconvoluted into four peaks:30 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.6 eV), C–C (285.1 eV), C–N/O (286.5 eV) and O/N–CN/O (289.4 eV). The peaks of CC, C–C, C–N/O in PSSN/S shift to lower energy by 0.1 eV when compared to PSSN, thus once again proving the interaction of C and S elements in PSSN/S. The S content (Table S1†) of PSSN/S measured by XPS was only 18.3%, which is much lower than that measured by TGA analysis, indicating that most of the sulfur was stored in the carbon shell rather than on the surface. The high-resolution XPS spectrum of PSSN/S in the S 2p region shows two main peaks at 164.9 eV and 163.7 eV (Fig. 3d), which can be assigned to S 2p1/2 and S 2p3/2, respectively.36 It is well-known that the peaks of S8 molecules assigned to S 2p1/2 and S 2p3/2 are located at 165.1 eV and 163.9 eV, respectively. The 0.2 eV gap between S and PSSN/S could be caused by the interaction of S and PSSN. A broad peak at 168.4 eV is ascribed to sulfate species, demonstrating the presence of S–O bonding. To confirm the existence of the interaction between sulfur and PSSN, polysulfide adsorption experiments were carried out taking Li2S6 as a polysulfide representative, which was prepared using a previously reported method.37 As shown in Fig. 3d, the solution containing PSSN turned completely colorless when compared to that without PSSN. Carbon black and PTFE, used in the PSSN cathode, were also tested (Fig. S6†), which exhibited almost no change in color. These results proved that PSSN had a strong interaction with polysulfide and had a superb polysulfide trapping capability in the electrode, while other materials in the electrode could not be replaced.
C (284.6 eV), C–C (285.1 eV), C–N/O (286.5 eV) and O/N–CN/O (289.4 eV). The peaks of CC, C–C, C–N/O in PSSN/S shift to lower energy by 0.1 eV when compared to PSSN, thus once again proving the interaction of C and S elements in PSSN/S. The S content (Table S1†) of PSSN/S measured by XPS was only 18.3%, which is much lower than that measured by TGA analysis, indicating that most of the sulfur was stored in the carbon shell rather than on the surface. The high-resolution XPS spectrum of PSSN/S in the S 2p region shows two main peaks at 164.9 eV and 163.7 eV (Fig. 3d), which can be assigned to S 2p1/2 and S 2p3/2, respectively.36 It is well-known that the peaks of S8 molecules assigned to S 2p1/2 and S 2p3/2 are located at 165.1 eV and 163.9 eV, respectively. The 0.2 eV gap between S and PSSN/S could be caused by the interaction of S and PSSN. A broad peak at 168.4 eV is ascribed to sulfate species, demonstrating the presence of S–O bonding. To confirm the existence of the interaction between sulfur and PSSN, polysulfide adsorption experiments were carried out taking Li2S6 as a polysulfide representative, which was prepared using a previously reported method.37 As shown in Fig. 3d, the solution containing PSSN turned completely colorless when compared to that without PSSN. Carbon black and PTFE, used in the PSSN cathode, were also tested (Fig. S6†), which exhibited almost no change in color. These results proved that PSSN had a strong interaction with polysulfide and had a superb polysulfide trapping capability in the electrode, while other materials in the electrode could not be replaced.
The electrochemical performance of PSSN/S as the cathode was evaluated in Li–S batteries. The cyclic voltammetry curves are shown in Fig. 4a. In the cathodic scan, two well-fined peaks are observed at 2.04 V and 2.30 V. The discharge plateau of 2.30 V is associated to the reduction of elemental sulfur to lithium polysulfide (Li2Sn, 4 < n < 8) and the discharge plateau of 2.04 V is associated to the further reduction of lithium polysulfide to solid Li2S.38 The same phenomenon appears in the anodic scan, in which the curve exhibits two close peaks at 2.30 V and 2.38 V. The oxidation peak at 2.30 V corresponds to the oxidation of Li2S and Li2S2 to lithium polysulfide and the peak at 2.38 V corresponds to the oxidation of lithium polysulfide to S8.39 We can draw a conclusion from the cyclic voltammetry that PSSN/S cathode has a reversible two-step process during the charge and discharge process, which is a significant premise for Li–S batteries to obtain a high capacity. The cyclic voltammetry curves quickly stabilized in the subsequent cycles, indicating its superb cycling stability.
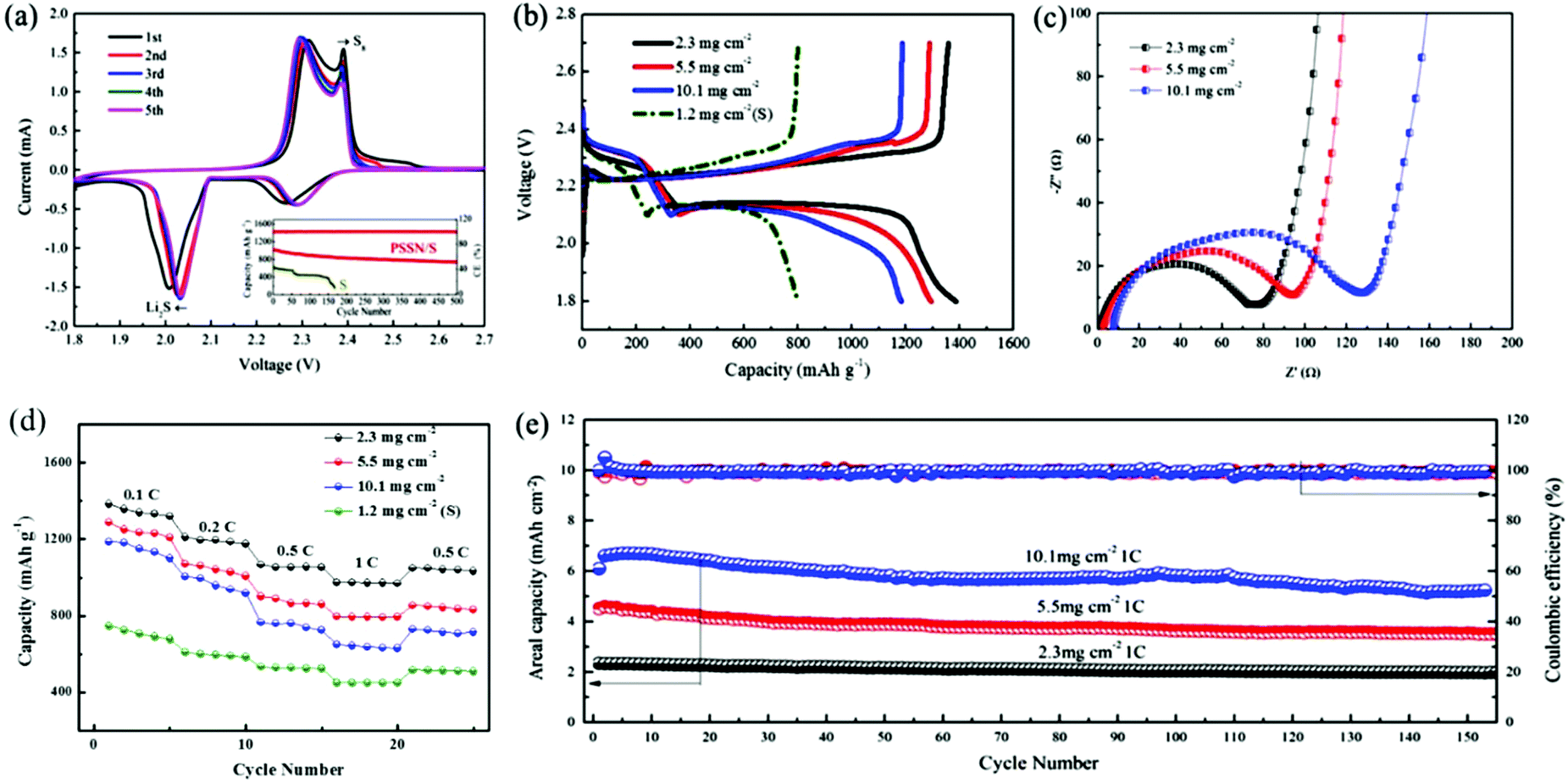 | ||
| Fig. 4 (a) Cyclic voltammetry curves of PSSN/S (scan rate: 0.1 mV s−1); inset of (a) is the long-term cycling stability of PSSN/S and S electrodes at 1.0 C over 500 cycles. (b) Galvanostatic charge/discharge curves obtained at 0.1 C, (c) electrochemical impedance spectra, (d) rate capabilities and (e) areal capacities of PSSN/S cathodes with sulfur loading of 2.3, 5.5 and 10.1 mg cm−2 and S cathode with a sulfur loading of 1.2 mg cm−2. | ||
The galvanostatic charge/discharge profiles of the PSSN/S cathode (Fig. S7a†) are consistent with the cyclic voltammetry curves. The discharge capacity of the first cycle reached ∼1700 mA h g−1, but quickly decayed to 1400 mA h g−1 in the second cycle and stabilized in the later cycles. The ultra-high capacity in the first cycle should be attributed to the solid-electrolyte interlayer (SEI) formation involving LiNO3 in the electrolyte.40 The typical galvanostatic charge/discharge profiles of PSSN/S at different current rates are shown in Fig. S7b.† It can be observed that the gap between the platforms of the charge and discharge increases as the current rates increase. Nevertheless, the platforms are still clear and show two charging/discharging plateaus, indicating that the charge/discharge process maintains the two-step process even at high current density. The long-term cycling stabilities of the PSSN/S and S cathodes are shown in the inset of Fig. 4a. It can be observed that the capacity of the S cathode rapidly decayed to 0 mA h g−1 within 170 cycles. For PSSN/S, the initial charge and discharge capacities were 1016 and 1013 mA h g−1. After 500 cycles, the specific discharge capacity is well-maintained at 735 mA h g−1 and the coulombic efficiency stabilized at 99.6%, which corresponds to a low capacity delay of only 0.055% per cycle, implying that most of the polysulfide intermediates were successfully immobilized within the nanohollow graphene during the electrochemical processes.
To maximize the actual energy density of Li–S batteries, it is critical to investigate the electrochemical behavior of the cathodes with higher areal sulfur loadings. Herein, we increased the areal sulfur loading from 2.3 to 10.1 mg cm−2, which is extremely high when compared to previous studies33,41,42 based on the traditional electrode technology with conducting agents and binders. The galvanostatic charge/discharge profiles obtained at 0.1 C for the cathodes with different sulfur loadings are shown in Fig. 4b. The discharge capacities of the PSSN/S cathode with a sulfur loading of 2.0, 5.5 and 10.1 mg cm−2 were 1400, 1310 and 1190 mA h g−1, respectively, which are far higher than that obtained for the S cathode. As the sulfur loading increased, the capacity decreased slightly and the gap between the charge and discharge platforms increased a slightly. This can be attributed to the increase in polarization caused by the longer electronic and Li-ionic transportation pathways as the electrodes become thicker. The changes in the capacity and gap between the platforms remain at a low level, which indicates that the ability for electronic transfer and Li-ions transportation changes slightly and the cathode comprised of PSSN/S has good electrolyte accessibility and can still prevent the sulfur from taking part in the shuttle reaction at a high sulfur loading. Electrochemical impedance spectroscopy (EIS) was used to characterize the internal resistance and charge-transfer process of the electrodes with different sulfur loadings. Fig. 4c shows that the plots obtained for the three electrodes before cycling are composed of a depressed semicircle in the high-to-medium frequency region and an inclined line at low frequencies, which can be ascribed to the charge-transfer resistance (Rct) and a mass-transfer process, respectively.43 The value of Rct rises as the sulfur loading increases. However, when the sulfur loading is as high as 10.1 mg cm−3, the charge-transfer resistance was still only 130 Ω, which is a smaller value when compared with those reported in previous studies.44,45 This could be attributed to the fact that our electrode design of a 3D porous contacts with graphene shells self-assembled into pomegranate-like structures can significantly decrease the electrochemical reaction resistance and facilitate charge transfer. The EIS spectra recorded after five cycles are shown in Fig. S8.† Unlike the spectra before cycling, the spectra after cycling were composed of two overlapped semicircles at the high-to-medium frequency region and an inclined line at low frequencies. The semicircle from high to medium frequency corresponds to the solid-electrolyte interface (SEI) layer resistance and the second semicircle at medium frequency was related to the charge-transfer resistance.46 All these three electrodes show drastically low charge-transfer resistance values, indicating good electrolyte infiltration and fast charge transport due to the 3D porous contacts with graphene-shell structure.
The rate capacities of the PSSN/S cathodes with different sulfur loadings are shown in Fig. 4d. For PSSN/S with a sulfur loading of 2.3 mg cm−2 at a low current density of 0.1 C, a discharge capacity of 1400 mA h g−1 was observed. When the current rates increased to 0.2 and 0.5 C, a capacity of 1200 and 1067 mA h g−1 are obtained, respectively. Specifically, it can be clearly observed that the capacity at 1 C was still up to 973 mA h g−1, which is much higher than that obtained for the S cathode, which should be attributed to the ultra-fast electron and lithium-ion transport with depolarization effect in the PSSN/S cathode. For the cathode with a sulfur loading of 10.1 mg cm−2 at a low current density of 0.1 C, the discharge capacity was still 1180 mA h g−1. When the current rates increased to 0.2, 0.5 and 1 C, the cathode showed a capacity of 996, 763, 648 mA h g−1, respectively. The capacity recovered as the current rate was reduced back to 0.5 C, indicating the good stability and structural invariability of PSSN/S with a high sulfur loading. The plot in Fig. S9† shows that the electrodes with high sulfur loading cycled at 1 C still have good capacity retention. After 150 cycles, the capacity of the cathodes with a sulfur loading of 5.5 and 10.1 mg cm−2 is 660 and 576 mA h g−1, respectively. As shown in Fig. 4e, the cathodes with a sulfur loading of 2.3, 5.5 and 10.1 mg cm−2 correspond to areal capacities of 2.4, 4.6 and 6.8 mA h cm−2, respectively. The Coulombic efficiency was almost 100% during the cycling process, indicating that the electrodes remain stable in the batteries. In addition, the areal capacities and cycling stability are much better than the most of the previously reported data (Table S2†)8,24,33,41,42,44,47–51 using the same sulfur loading cathodes.
The mechanism of the excellent electrochemical performance of the PSSN/S cathode was further investigated. Beaker cell tests were carried out to understand the excellent capability of PSSN/S for trapping soluble polysulfide. The electrolyte color was recorded at different times during the first discharge process at 0.1 C (Fig. 5a). The electrolyte in the cell with the S cathode quickly turned to a yellow color in 2 h and the color gradually changed to deep yellow as the time progressed to 10 h, indicating the dissolution and leakage of lithium polysulfide into the electrolyte. Unlike the S cathode, the electrolyte in the PSSN/S cell remained colorless throughout the first charge process, suggesting the remarkable polysulfide trapping capability of the PSSN/S cathode in Li–S batteries. The cycled cells were disassembled and examined after 50 cycles. As shown in Fig. S11,† for cells using the S cathodes, polysulfide in the separator and a damaged Li anode surface are clearly noted. In contrast, for the cells using PSSN/S cathodes, a clean separator and smooth Li anode were observed, which also proved the superb ability of PSSN/S to stabilize polysulfide. This could be attributed to the sublimation of sulfur when heating; the sulfur left in the PSSN/S cathode was tightly trapped around the nitrogen atoms in PSSN. Thus, the shuttle effect can effectively avoided during the charge–discharge process. To prove the importance of the carbon pores to provide a sufficient expansion volume for sulfur, the performance of the PSSN/S electrode with a sulfur loading of 80% (PSSN/S-80%) and areal sulfur loading of 2.3 mg cm−2 was tested and the results are shown in Fig. S12.† It can be observed that the capacity of PSSN/S-80% rapidly decays to 600 mA h g−1 after 200 cycles, indicating that a certain content of sulfur overflows from the carbon shell during the charge/discharge process. However, PSSN/S-60% (Fig. 4a) has much more excellent cycle stability. As we mentioned earlier, PSSN/S-60% has just enough space for the sulfur expansion; thus, we can draw a conclusion that the lack of volume leads to a significant decrease in performance. 3D confocal microscopy was first used to measure the thickness of the cathodes with different sulfur loadings before and after cycling. As shown in Fig. 5b and S10a–c,† the 113, 230, and 465 μm thickness corresponding to the cathodes with 2.0, 5.5, and 10.1 mg cm−2 of sulfur loading, respectively, showing the direct proportion to the sulfur content. After 100 cycles (Fig. 5c), the cathode thickness with 10.1 mg cm−2 sulfur loading was 470 nm, almost maintaining its original value, indicating that the expansion of sulfur did not cause the expansion of the cathode. A comparison of the electrode thickness before and after cycling was undoubtedly further powerful evidence to account for sufficient stress and space allowing for sulfur expansion. Fig. 5d and e shows the working principle of these thick electrodes, including the electronic transfer and Li-ion transport. The electrode was composed of two main types of materials, PSSN/S and electronic conductive carbon (acetylene black (AB)). The electrons transfer through the carbon layer of acetylene black and PSSN/S, while the lithium ions transport through the electrolyte absorbed in the pores on the carbon layer and the holes between the edge of PSSN/S and acetylene black. Both paths are continuous and efficient, ensuring regular operation of the electrodes with a high sulfur loading.
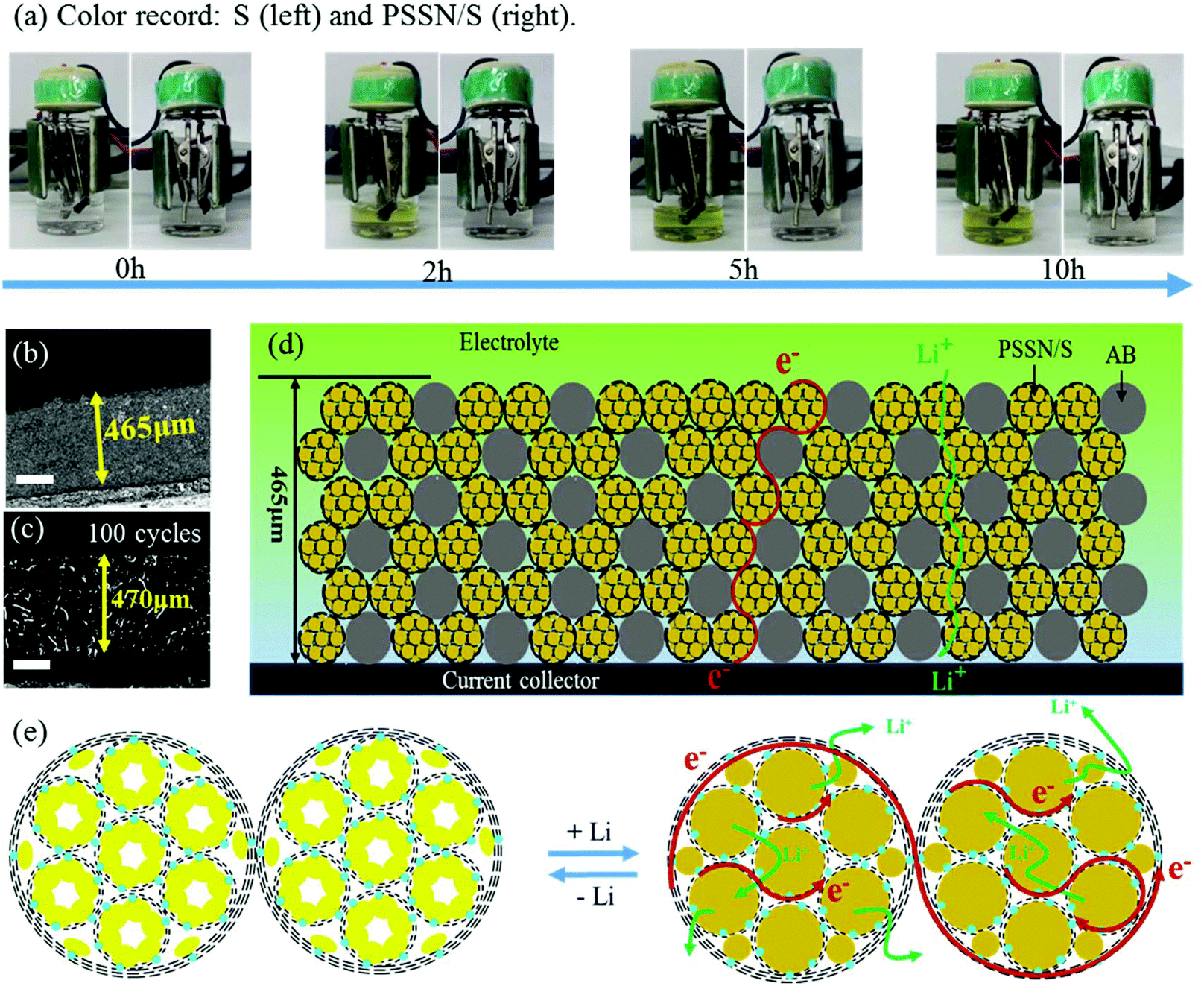 | ||
| Fig. 5 (a) A comparison of the electrolyte color recorded at different times in a beaker cell with the S and PSSN/S cathodes. 3D confocal microscope images of the PSSN/S cathodes with a sulfur loading of 10.1 mg cm−2 before (b) and after (c) cycling (the length of the blockage represents 100 nm). (d) Structural schematic and (e) electron and Li-ion transport mechanism of the thick cathode with a high sulfur loading. | ||
Experimental
Materials synthesis
:0.3 using ethanol as the solvent. The mixture was then dried at 80 °C for 4 h. The dried material was placed in a tube furnace and heated to 650 °C (2 °C min−1) under an argon atmosphere for 6 h. Following this, the high valence Fe in the PB was reduced to Fe nanometals by the action of glucose, which was bound to the middle by the nitrogen-doped carbon graphene. After cooling to room temperature, the nano-iron spontaneously burned to produce γ-Fe2O3 when the material was removed from the tube furnace and exposed to air. Agitation was necessary to heat the material evenly. The core–shell nanohollows γ-Fe2O3@graphene hybrid was finally obtained when the Fe nano-metals were completely oxidized by O2.
:3. The two reactants were thoroughly ground in a mortar until they completely turned black. The mixture was then placed into an open crucible and heated to 155 °C (1 °C min−1) under an argon atmosphere for 12 h and cooled to room temperature.
Materials characterization and electrochemical measurements
X-ray powder diffraction patterns were obtained using a Bruker D8 Advance diffractometer with Cu Kα (λ = 0.15418 nm). The diffraction data were recorded in the 2θ range of 10–120° with a step of 0.02° and a count time of 1 s. The morphologies of the materials were observed by scanning electron microscopy (FE-SEM, ZEISS Supra 55) and transmission electron microscopy (HRTEM, Tecnai G2 F20 S-TWIN, 200KV). Thermogravimetric analysis (TGA) data was collected on a TGA/DSC system at a heating rate of 10 °C min−1 under nitrogen flow. The surface area and the pore size distribution were determined using Brunauer–Emmett–Teller nitrogen adsorption/desorption analyses (BET, Micromeritics ASAP 2020 HD88). XPS analyses were performed using an ESCALAB 250XL. Raman spectra were recorded on a iHR 320. The thickness of the working electrode was measured using a Keyence VK-x200 3D confocal microscope.A coin cell type CR2032 was used for the electrochemical tests in a non-aqueous system. The PSSN/S composite electrode was prepared by mixing the PSSN/S composite, acetylene black and polytetrafluoroethylene (PTFE) at a weight ratio of 7:2:1. The mixture was dissolved in isopropanol to form the electrode slurry. The slurry was then ground in a mortar to form the electrode film. The films with different sulfur loadings were obtained by compressing at different pressures on a tablet press. Following this, the film was cut into round pieces for use as the working electrode and dried at 60 °C for 24 h in the vacuum oven. Lithium metal was used as the counter electrode. The S electrode was prepared using the same method by mixing sulfur, acetylene black and PTFE at a weight ratio of 2.5:3:2. The electrolyte was consisted of 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in 1,3-dioxolane/1,2-dimethoxyethane (DOL/DME) (1:1, v/v) containing 0.3 mol L−1 of LiNO3. All the cells were assembled in a glove box with a water/oxygen content lower than 0.1 ppm and tested at room temperature. The charge/discharge performance was tested between 1.8 and 2.7 V at different C-rates on a NEWARE battery cycler. The electrochemical impedance spectra were recorded on a CHI 660E electrochemical workstation from 100000 Hz to 0.01 Hz. The cyclic voltammetry curves were recorded on a CHI 660E electrochemical workstation between 1.8 and 2.7 V at a scan rate of 0.0001 V s−1.
Conclusions
In summary, we demonstrated PSSN with N-graphene-shells using a facile thermal treatment process. The temperature was used to control the nitrogen content, while the nitrogen content and melt-diffusion method in an open system were further used to control the sulfur content at 60 wt%. The special feature of PSSN was that the large (∼25 nm)-small (∼8 nm) self-assembled pomegranate-like structure with N-graphene shells not only has enough stress and space for sulfur expansion but can also trap polysulfide to stop the shuttle-effect from occurring during the charge/discharge process via the strong interaction of polysulfide with the doped N-elements. In addition, the less-layered graphene, interconnected inside and outside, constructed a 3D conductive framework and the pores in the 3D structure promote electrolyte adsorption into the entire electrode to maintain the rapid Li-ion transport. Therefore, the above advantages of the PSSN/S cathode ensure its large-capacity, high-rate and long-life at a ultra-high areal-sulfur loading for Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the National Science Foundation of China (No. 51602009), Postdoctoral Science Foundation (2016M600008), National Materials Genome Project (2016YFB0700600), Shenzhen Science and Technology Research Grant (No. JCYJ20150729111733470 and JCYJ20151015162256516) and Natural Science Foundation of Guangdong Province (2015A030310138).Notes and references
- J. Lu, Z. H. Chen, Z. F. Ma, F. Pan, L. A. Curtiss and K. Amine, Nat. Nanotechnol., 2016, 11, 1031–1038 CrossRef CAS PubMed.
- N. R. Van, Nature, 2013, 498, 416–417 CrossRef PubMed.
- W. Lv, Z. J. Li, Y. Q. Deng, Q. H. Yang and F. Y. Kang, Energy Storage Mater., 2016, 2, 107–138 CrossRef.
- B. Dominic, P. Stefano and S. Bruno, Chem. Commun., 2013, 49, 10545–10562 RSC.
- A. Manthiram, Y. Z. Fu and S. H. Chung, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Z. Lin, Z. Liu, W. Fu, N. J. Dudney and C. Liang, Angew. Chem., Int. Ed., 2013, 125, 7608–7611 CrossRef.
- C. Huang, J. Xiao, Y. Y. Shao, J. M. Zheng, W. D. Bennett, D. P. Lu, L. V. Saraf, M. Engelhard, L. W. Ji, J. G. Zhang, X. L. Li, G. L. Graff and J. Liu, Nat. Commun., 2015, 5, 3015–3021 Search PubMed.
- Z. W. Seh, Y. M. Sun, Q. F. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Z. Li, Y. Jiang, L. X. Yuan, Z. Q. Yi, C. Wu, Y. Liu, P. Strasser and Y. H. Huang, ACS Nano, 2014, 8, 9295–9303 CrossRef CAS PubMed.
- G. He, S. Evers, X. Liang, M. Cuisinier, A. Garsuch and L. F. Nazar, ACS Nano, 2013, 7, 10920–10930 CrossRef CAS PubMed.
- C. F. Zhang, H. B. Wu, C. Z. Yuan, Z. P. Guo and X. W. Lou, Angew. Chem., Int. Ed., 2012, 124, 9730–9733 CrossRef.
- S. T. Lu, Y. W. Cheng, X. H. Wu and J. Liu, Nano Lett., 2013, 13, 2485–2489 CrossRef CAS PubMed.
- L. Sun, M. Y. Li, Y. Jiang, W. B. Kong, K. L. Jiang, J. P. Wang and S. S. Fan, Nano Lett., 2014, 14, 4044–4049 CrossRef CAS PubMed.
- G. M. Zhou, S. F. Pei, L. Li, D. W. Wang, S. G. Wang, K. Huang, L. C. Yin, F. Li and H. M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef CAS PubMed.
- Y. C. Li, R. Mi, S. M. Li, X. C. Liu, W. Ren, H. Liu, J. Mei and W. M. Lau, Int. J. Hydrogen Energy, 2014, 39, 16073–16080 CrossRef CAS.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- B. Li, S. M. Li, J. H. Liu, B. Wang and S. B. Yang, Nano Lett., 2015, 15, 3073–3079 CrossRef CAS PubMed.
- Z. W. Seh, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. Mcdowell, P. C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331–1336 CrossRef PubMed.
- N. Liu, Z. Lu, J. Zhao, M. T. Mcdowell, H. W. Lee, W. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- L. X. Miao, W. K. Wang, K. G. Yuan, Y. S. Yang and A. B. Wang, Chem. Commun., 2014, 50, 13231–13233 RSC.
- D. Lv, J. Zheng, Q. Li, X. Xie, S. Ferrara, Z. Nie, L. B. Mehdi, N. D. Browning, J. G. Zhang, G. L. Graff, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1402290 CrossRef.
- J. Song, Z. Yu, M. L. Gordin and D. Wang, Nano Lett., 2016, 16, 864–870 CrossRef CAS PubMed.
- G. M. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760–7765 CrossRef CAS PubMed.
- Z. Yuan, H. J. Peng, J. Q. Huang, X. Y. Liu, D. W. Wang, X. B. Cheng and Q. Zhang, Adv. Funct. Mater., 2015, 24, 6244–6251 CrossRef.
- O. Ogoke, G. Wu, X. Wang, A. Casimir, L. Ma, T. Wu and J. Lu, J. Mater. Chem. A, 2017, 5, 448–452 CAS.
- H. J. Peng, J. Q. Huang, X. B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 1700260 CrossRef.
- J. Zheng, J. Lu, K. Amine and F. Pan, Nano Energy, 2017, 33, 497–507 CrossRef CAS.
- J. Hu, J. Zheng, L. Tian, Y. Duan, L. Lin, S. Cui, H. Peng, T. Liu, H. Guo, X. Wang and F. Pan, Chem. Commun., 2015, 51, 7855–7858 RSC.
- J. Hu, J. Yang, Y. Duan, C. K. Liu, H. T. Tang, L. P. Lin, Y. Lin, H. B. Chen and F. Pan, Chem. Commun., 2017, 53, 857–860 RSC.
- J. Jiang, J. H. Zhu, W. Ai, X. L. Wang, Y. L. Wang, C. J. Zou, W. Huang and T. Yu, Nat. Commun., 2015, 6, 8622–8629 CrossRef CAS PubMed.
- Y. J. Li, J. M. Fan, M. S. Zheng and Q. F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 CAS.
- C. Tang, Q. Zhang, M. Q. Zhao, J. Q. Huang, X. B. Cheng and G. L. Tian, Adv. Mater., 2014, 26, 6100–6105 CrossRef CAS PubMed.
- F. Wu, J. Qian, R. J. Chen, T. Zhao, R. Xu, Y. S. Ye, W. H. Li, L. Li, J. Lu and K. Amine, Nano Energy, 2015, 12, 742–749 CrossRef CAS.
- T. Chen, B. R. Cheng, R. Y. Zhu, R. P. Chen, Y. Hu, L. B. Ma, H. L. Lv, Y. R. Wang, J. Liang, Z. X. Tie, Z. Jin and J. Liu, Nano Lett., 2017, 17, 437–444 CrossRef CAS PubMed.
- J. H. Yan, B. Y. Li and X. B. Liu, Nano Energy, 2015, 18, 245–252 CrossRef CAS.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759–4766 CrossRef CAS PubMed.
- J. T. Yoo, S. J. Cho, G. Y. Jung, H. K. Su, K. H. Choi, J. H. Kim, C. K. Lee, K. K. Song and S. Y. Lee, Nano Lett., 2016, 16, 3292–3300 CrossRef CAS PubMed.
- Z. B. Xiao, Z. Yang, L. Wang, H. G. Nie, M. Zhong, Q. Q. Lai, X. G. Xu, L. G. Zhang and S. M. Huang, Adv. Mater., 2015, 27, 2891–2898 CrossRef CAS PubMed.
- S. Y. Wei, L. Ma, K. E. Hendrickson, Z. Y. Tu and J. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143–12152 CrossRef CAS PubMed.
- L. F. Nazar and Q. Pang, ACS Nano, 2016, 10, 4111–4118 CrossRef PubMed.
- W. Zhou, B. Guo, H. Gao and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502059 CrossRef.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A920–A923 CrossRef CAS.
- R. P. Fang, S. Y. Zhao, P. X. Hou, M. Cheng, S. G. Wang, H. M. Cheng, C. Liu and F. Li, Adv. Mater., 2016, 2, 3374–3382 CrossRef PubMed.
- G. M. Zhou, S. F. Pei, L. Li, D. W. Wang, S. G. Wang, K. Huang, L. C. Yin, F. Li and H. M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef CAS PubMed.
- J. H. Yan, X. B. Liu, M. Yao, X. F. Wang, T. K. Wafle and B. Y. Li, Chem. Mater., 2015, 27, 5080–5087 CrossRef CAS.
- Z. Li, J. T. Zhang, Y. M. Chen, J. Li and X. W. Lou, Nat. Commun., 2015, 6, 8850–8859 CrossRef CAS PubMed.
- G. J. Hu, C. Xu, Z. H. Sun, S. G. Wang, H. M. Cheng, F. Li and W. C. Ren, Adv. Mater., 2016, 28, 1603–1609 CrossRef CAS PubMed.
- L. Qie, C. X. Zu and A. Manthiram, Adv. Energy Mater., 2016, 6, 1502459 CrossRef.
- Q. Pang, Q. Liang, C. Y. Kwok, J. Kulisch and L. F. Nazar, Adv. Energy Mater., 2017, 7, 1601630 CrossRef.
- N. Osada, C. B. Bucur, H. Aso and J. Muldoon, Energy Environ. Sci., 2016, 9, 1668–1673 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C7NR06731C |
| This journal is © The Royal Society of Chemistry 2018 |