
Self-discriminating fluorescent hybrid nanocrystals: efficient and accurate tracking of translocation via oral delivery†
Chengying
Shen‡
a,
Yinqian
Yang‡
b,
Baode
Shen‡
ac,
Yike
Xie
b,
Jianping
Qi
b,
Xiaochun
Dong
b,
Weili
Zhao
b,
Weifeng
Zhu
c,
Wei
Wu
 b,
Hailong
Yuan
*a and
Yi
Lu
*b
b,
Hailong
Yuan
*a and
Yi
Lu
*b
aDepartment of Pharmacy, Air Force General Hospital of PLA, Beijing 100142, China. E-mail: yhlpharm@126.com
bKey Laboratory of Smart Drug Delivery of Ministry of Education and PLA, School of Pharmacy, Fudan University, Shanghai 201203, China. E-mail: fd_luyi@fudan.edu.cn
cKey Lab of Modern Preparation of Traditional Chinese Medicine (TCM), Ministry of Education, Jiangxi University of TCM, Nanchang 330004, China
First published on 11th December 2017
Abstract
The in vivo fate of nanocrystals is a controversial topic, i.e. dissolving versus integral absorption through the intestinal membrane. This is due to the lack of functional strategies to identify integral nanocrystals. In this study, the in vivo fate of quercetin hybrid nanocrystals (QT-HNCs) via the oral route is explored by physically embedding an environment-responsive probe in the crystal lattices of quercetin. The specific property of the probe is the water-initiated aggregation-caused quenching (ACQ) ability, by which integral QT-HNCs can be self-discriminated. Instead of dissolving instantly, QT-HNCs can be retained in the gastrointestinal tract for 12–16 h, and can then be absorbed and distributed into various organs with the liver as the primary terminal. The ileum provides better absorption than the jejunum. Cellular studies prove that both trans-epithelial and M cell-mediated routes are involved in the absorption of integral QT-HNCs, which may be impeded by the mucous layer. Moreover, the particle size affects the in vivo behavior and the ex vivo cellular interaction of QT-HNCs, with moderate size, such as 550 nm, being preferred. The results not only validate the idea of using ACQ fluorophores for bioimaging of integral nanocrystals but also support the intestinal absorption of nanocrystals.
Introduction
The delivery of poorly water-soluble drugs is currently one of the top challenges in the pharmaceutical industry.1 Nanocrystals have long been established as one of the functional approaches to overcome the formulation challenges of poorly water-soluble drugs.2 By definition, nanocrystals, also known as nanosuspensions, are crystalline nanoparticles of pure drugs with mean particle size less than 1 μm, typically between 200 and 500 nm.3 The high specific surface area of nanocrystals owing to the nanometer particle sizes leads to significant improvements in apparent solubility and dissolution, which consequently facilitates absorption of poorly water-soluble drugs via diverse administrative routes, such as oral, ocular, pulmonary, transdermal and intravenous routes.4–8 Generally speaking, nanocrystals are more advantageous than polymeric carrier-based nanoparticles due to high drug loading (theoretically 100%), high stability, easy scale-up and less toxicity.3 In spite of the success of tens of nanocrystal products, both commercially and clinically,9 the underlying mechanisms of enhanced performance have yet to be explored. None the less, relevant mechanistic investigations are very difficult to perform because of the inability to “see” nanocrystals while they are busy working in the body. Elucidation of the in vivo fate of nanocrystals themselves presents as a prerequisite for the full understanding of the performance of nanocrystals.However, it is a great challenge to study the in vivo fate of nanocrystals because of the difficulties in discriminating integral nanocrystals from the biologic environment. Currently, the in vivo behaviors of nanocrystals are mainly obtained from the pharmacokinetic and biodistribution data obtained by monitoring drug levels.10,11 Since the measured results encompass both dissolved and non-dissolved drugs, the virtual in vivo fate of nanocrystals per se (non-dissolved) might be different from that of the drugs. Several strategies have been adopted to visualize the in vivo fate of nanocrystals, including direct observation of nanocrystal morphology in biological samples using transmission electron microscopy (TEM),12 tracking nanocrystals with autofluorescence (e.g. curcumin and coumarin 6)13,14 and tracking fluorescently hybridized nanocrystals (HNCs).15–17 However, these strategies are not without defects.18 For TEM, possible recrystallization of either the drug or other reagents used during sample preparation might mess up the images. The autofluorescence strategy is merely applicable to a limited number of drugs that emit fluorescence. Obviously, each kind of nanocrystals exhibits specific in vitro and in vivo behaviors, so the results acquired from a definite model of nanocrystals are not applicable universally. In order to probe nanocrystals of different drugs, a universal tool is highly demanded. By embedding a trace amount of fluorophores into the lattice of nanocrystals, HNCs are invented as such a valuable and universal tool for bioimaging of nanocrystals.15,16 Albeit innovative, HNCs failed to discriminate themselves from the bulk background because released probes are still fluorescent and thus bring about significant interference. Along with the dissolution of nanocrystals, the fraction of free probes increases gradually, and the bulk fluorescence becomes more and more disqualified to represent integral nanocrystals. Increasing discrepancy in biodistribution between the fluorescence signals, which comes from a dynamic mix of both nanocrystals and free probes, and the drug signals makes the interpretation of the in vivo fate erroneous.17
In our previous studies, a class of near-infrared (NIR) fluorescent probes sharing a BODIPY or aza-BODIPY parent structure was developed and successfully used to monitor the in vivo fate of a series of nanocarriers.19–24 One of the distinct properties of these probes is the aggregation-caused quenching (ACQ) effect due to intermolecular π–π stacking upon contact with water.25 The fluorescent probes are able to emit fluorescence whenever they are dispersed in a molecular state, such as in an organic solvent, but form aggregates and quench when they are exposed to an unfavourable solvent, such as water. The same holds true when the probes are immobilized and dispersed molecularly in the matrix of the nanoparticles. The nanoparticles are illuminated by encapsulating the probes, whereas the released probes, for instance, due to degradation of the matrix, quench immediately and absolutely in the aqueous media. Thus, the free probes would not cause much interference, making the nanoparticles self-discriminative. Inspired by the successful application in various polymeric carrier-based nanoparticles, we managed to adapt this class of probes to visualize the in vivo fate of nanocrystals by fashioning HNCs.
In this proof-of-concept study, the feasibility of probing the in vivo fate of nanocrystals through hybridizing with ACQ probes is systematically studied using quercetin (QT) as a model drug. As illustrated in Fig. 1, when physically embedded inside the lattice of nanocrystals, the probes emit strong fluorescence, which however quenches immediately following the dissolution of nanocrystals. A correlation can be established between the fluorescence and the particulate mass. Based on the measurement of the fluorescence, the kinetics of the nanocrystals can be investigated, at least semi-quantitatively. It is reasonable to deduce that the same protocol also works well for crystals with larger sizes, such as drug crystals that present universally in oral solid dosage forms. In this study, the in vivo fate of nanocrystals is investigated by tracking the translocation of integral nanocrystals after oral administration. This self-discriminative HNCs-based platform technology might be adaptable to study drug crystals with a wide span of particle sizes and via diverse administrative routes.
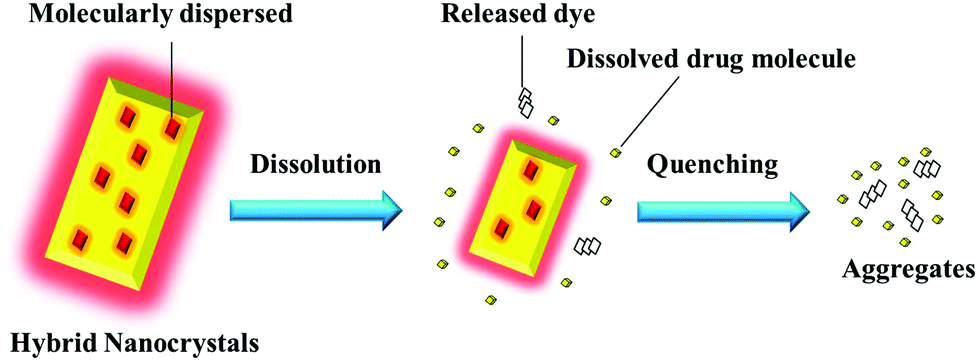 | ||
| Fig. 1 Schematic presentation of the rationale for monitoring integral nanocrystals by hybridization with ACQ fluorescent probes. | ||
Results and discussion
Preparation and characterization of QT-HNCs
Hybridized QT nanocrystals (QT-HNCs) were prepared by an anti-solvent crystallization method using ACQ dyes of P2 and P4 for live imaging and confocal laser scanning microscopy (CLSM) observation, respectively. The physicochemical properties of the HNCs formulations are shown in ESI Table S1.† The mean particle sizes of the HNCs, encoded as QT-HNCs-280, QT-HNCs-550 and QT-HNCs-1100, are around 280, 550 and 1100 nm, respectively (Fig. 2b). The polydispersity index (PDI) is less than 0.25 for all formulations, indicating a narrow size distribution. Scanning electron microscopy (SEM) of the QT-HNCs reveals well-defined, short rod-like morphology with the estimated average sizes of 100–400 nm, 400–800 nm and 700–1400 nm in length, respectively (Fig. 2c), which are close to the results obtained from a particle sizer. The zeta potentials of all formulations are found to be around −20 mV, which is attributed to the dissociation of the phenolic hydroxyl group of QT molecule (Fig. 2a).26 The electric charge is believed to be advantageous for stabilizing the QT-HNCs due to electrostatic repulsion in collaboration with the stabilizer poloxamer 188 (P188), which provides steric hindrance simultaneously.27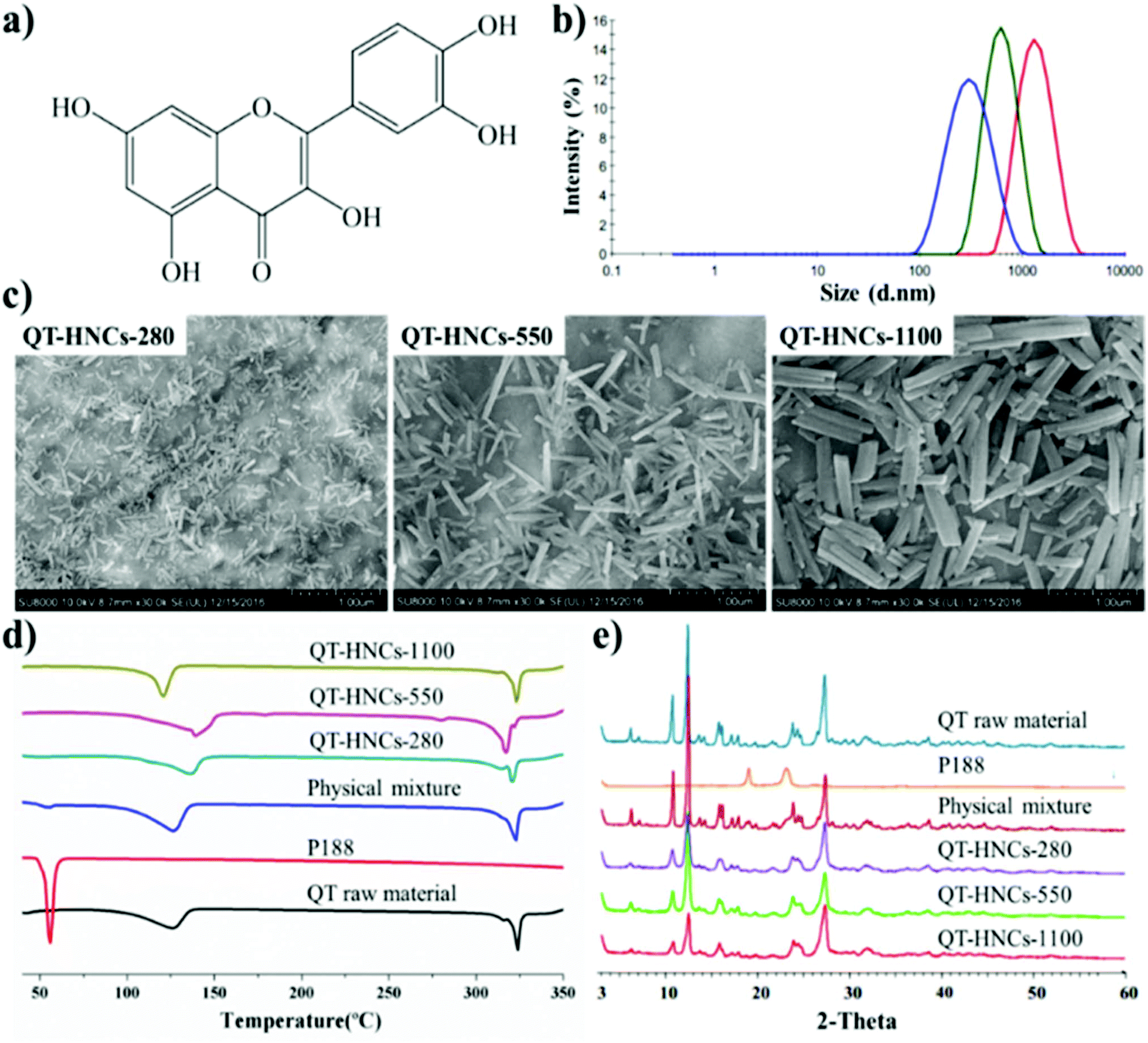 | ||
| Fig. 2 Physicochemical properties of QT-HNCs. (a) Chemical structure of QT; (b) size distribution of QT-HNCs formulations; (c) SEM photographs of QT-HNCs formulations; (d) DSC thermograms; and (e) powder X-ray diffractograms. | ||
The QT-HNCs show fluorescence emission (ESI Fig. S1c–e†) as observed under an IVIS Spectrum Live Imaging System (PerkinElmer, Waltham, MA, USA). Owing to the loss in preparation, the fluorescence intensity of the probe solution in ethanol is stronger than that of the QT-HNCs (ESI Fig. S1a†). On the contrary, the probe solution in water and P188 solution (0.016%, w/v) gives no fluorescence due to the ACQ effect (ESI Fig. S1b and f†). Besides, no fluorescence is found for the QT nanocrystals (QT-NCs) without probes (ESI Fig. S1g, h and i†). These results confirm that the fluorescence stems from probes embedded inside the QT-HNCs. P2 was entrapped in the crystalline lattices of QT with an entrapment efficiency around 60% and a loading of 0.13‰ (w/w), while P4 had a higher entrapment efficiency (>95%) in the nanocrystal lattices and a loading of about 0.22‰ (w/w).19 Albeit at relatively low levels of P2 loading, the QT-HNCs emit strong fluorescence exceeding 5 × 109 μW cm−2 owing to the high quantum yield of the probes embedded, sufficient for live imaging. With even higher fluorescence intensity, P4/QT-HNCs were diluted to equivalent fluorescence levels to P2/QT-HNCs in studies of in situ perfusion and cellular interaction.
The differential scanning calorimetry (DSC) thermograms and powder X-ray diffractometry (PXRD) patterns of crude QT crystals, P188, physical mixture, and QT-HNCs are shown in Fig. 2d and e, respectively. Two endothermic peaks are found in the DSC curves of both QT and QT-HNCs. The broad endothermic peaks between 100 °C and 140 °C are ascribed to the loss of bound water, while the sharp peak at around 318 °C belongs to the melting point of QT. These results are consistent with reports in the literature.28–30 It should be noted that the water content in the dried QT-HNCs samples for DSC detection was not strictly controlled. Thus, the endothermic peaks corresponding to water evaporation varied among the QT-HNCs samples. In addition, characteristic PXRD peaks observed from crude QT crystals are found in QT-HNCs as well, which confirms that hybridization with a trace amount of probes does not change the crystallinity of QT.
Validation of the self-discriminativity of QT-HNCs
Albeit theoretically workable, the self-discriminativity of HNCs depends on several assumptions. First of all, the possible fluorescence quenching due to water infiltration or probe leakage should be excluded. Otherwise, the premature release and subsequent quenching would bring about interference to the measurement of fluorescence. Secondly, the fluorescence of the HNCs should be instantly quenched upon dissolution; if not, the fluorescence measured would be bigger than the virtual value, causing a false impression of reduced dissolution. Finally, the fading of the fluorescence of the particulate mass should be synchronous to the dissolution rate to guarantee accurate monitoring of the dissolution kinetics. The following is the validation of these assumptions step by step by simulating a virtual environment that would be encountered in the in vivo fate study.In order to prevent fluorescence quenching from HNCs dissolving, we purposely created a situation with as little dissolution of the nanocrystals as possible to test the robustness of the nanocrystal mass per se. This was easily done by testing in a small volume of pure water and different buffers with limited dissolution capacity. As shown in Fig. 3a–c, the fluorescence intensity of QT-HNCs, irrespective of the particle size, was quite stable for up to 48 h in water and a series of buffers. The overall fluorescence fluctuates between 90 and 100% relative to original fluorescence, which is more or less due to measurement deviation. These results suffice to prove that the fluorescence of HNCs stays stable within an observation duration of 48 h and any attenuation due to water infiltration or probe leakage can be excluded. For comparison, the fluorescence stability of QT-HNCs in different simulated media (ESI Table S2† for their compositions) was also studied. The fluorescence intensity of the QT-HNCs is also quite stable for up to 48 h in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) with minor fluctuations between 90 and 105%. The results indicate that enzymes such as pepsin and trypsin do not damage the structure of the HNCs. However, a decrease of 10–60% in fluorescence intensity is observed under the same conditions in both fasted-state simulated small intestine fluid (FaSSIF) and fed-state simulated small intestine fluid (FeSSIF) for all size groups (Fig. 3d–f). Especially for QT-HNCs-280 with the smallest particle size, the fluorescence intensity is reduced to as low as 38% at 2 h in FeSSIF. The decrease of fluorescence is attributed to the solubilizing effects of fortified surfactants (phospholipids and bile salts) in FaSSIF and FeSSIF, which facilitate dissolution of HNCs and lead to fluorescence quenching. Since FeSSIF has higher levels of surfactants than FaSSIF, the fraction of dissolution is higher as well. Consequently, the fluorescence intensity is reduced more in FeSSIF than in FaSSIF. Moreover, an apparent negative correlation exists between particle size and fluorescence attenuation, which can be explained by the correlation of faster dissolution due to substantially increased specific surface areas (Fig. 3d–f).
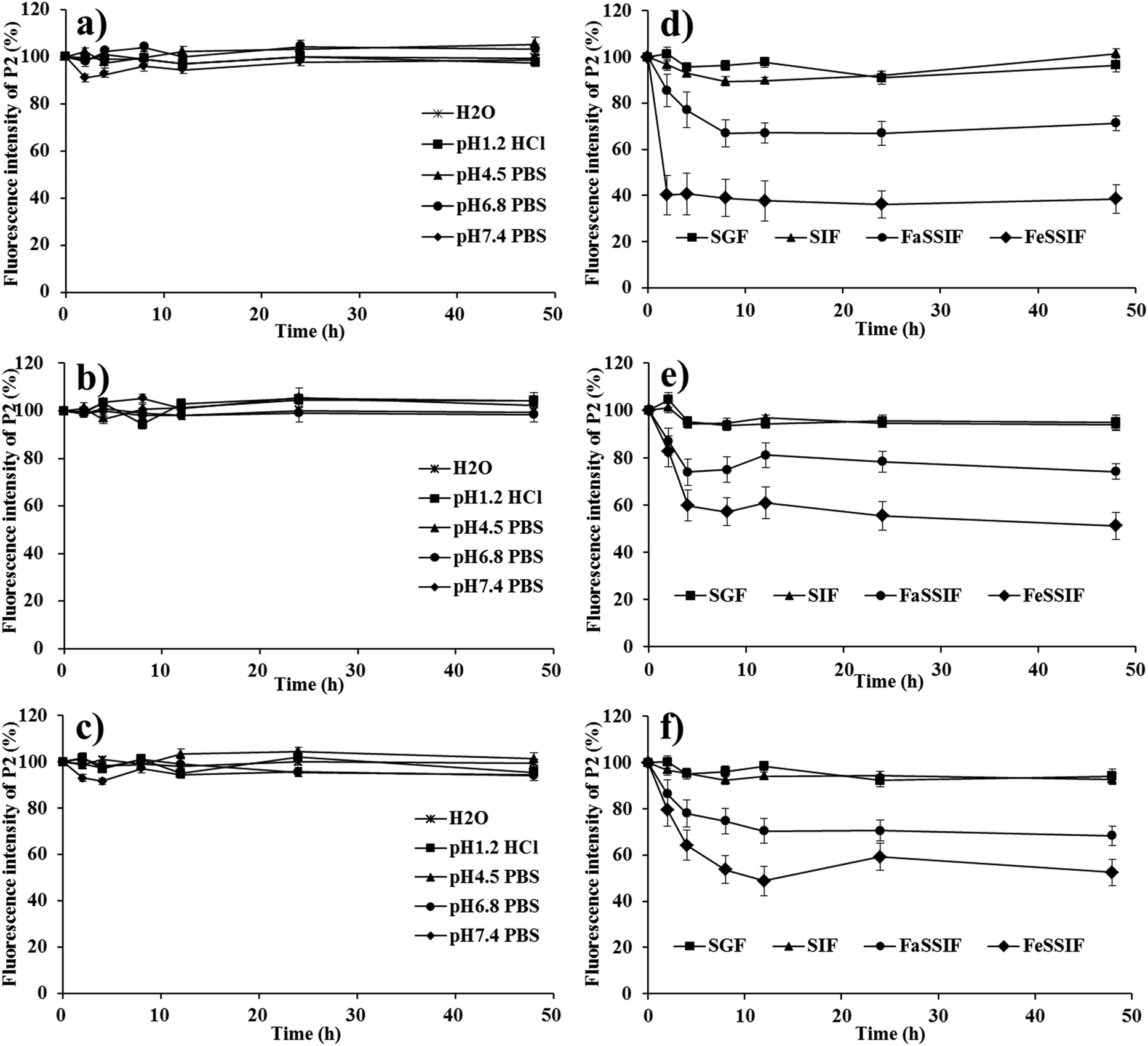 | ||
| Fig. 3 (a), (b), (c) In vitro fluorescence stability of different QT-HNCs formulations in different pH or water and (d), (e), (f) different simulated gastric or intestinal fluids. (a) and (d): QT-HNCs-280; (b) and (e): QT-HNCs-550; (c) and (f): QT-HNCs-1100. | ||
In order to test the water-quenching sensitivity, we simulate a fast dissolution situation by using aqueous alcohol with incremental fractions of water. If immediate fluorescence quenching is observed, excellent water sensitivity would be ensured. It is found that for all particle groups the fluorescence disappears abruptly in media with water content exceeding 80% (v/v) (Fig. 4), which is similar to the probe molecules themselves.25 Mainly owing to the fast dissolution of HNCs, probe molecules are released into the aqueous alcohol solution and exhibit the ACQ effects. Due to the lipophilicity, solutions with higher ethanol content have a better capacity to dissolve the probe molecules, leading to retention of the fluorescence. Thus, the fluorescence declines with increasing water content in the media. Following this observation, it is reasonable to deduce that under an absolute aqueous environment the dissolved fractions would show immediate quenching as well. Therefore, high water sensitivity of the fluorescence can be concluded upon dissolution of the nanocrystals.
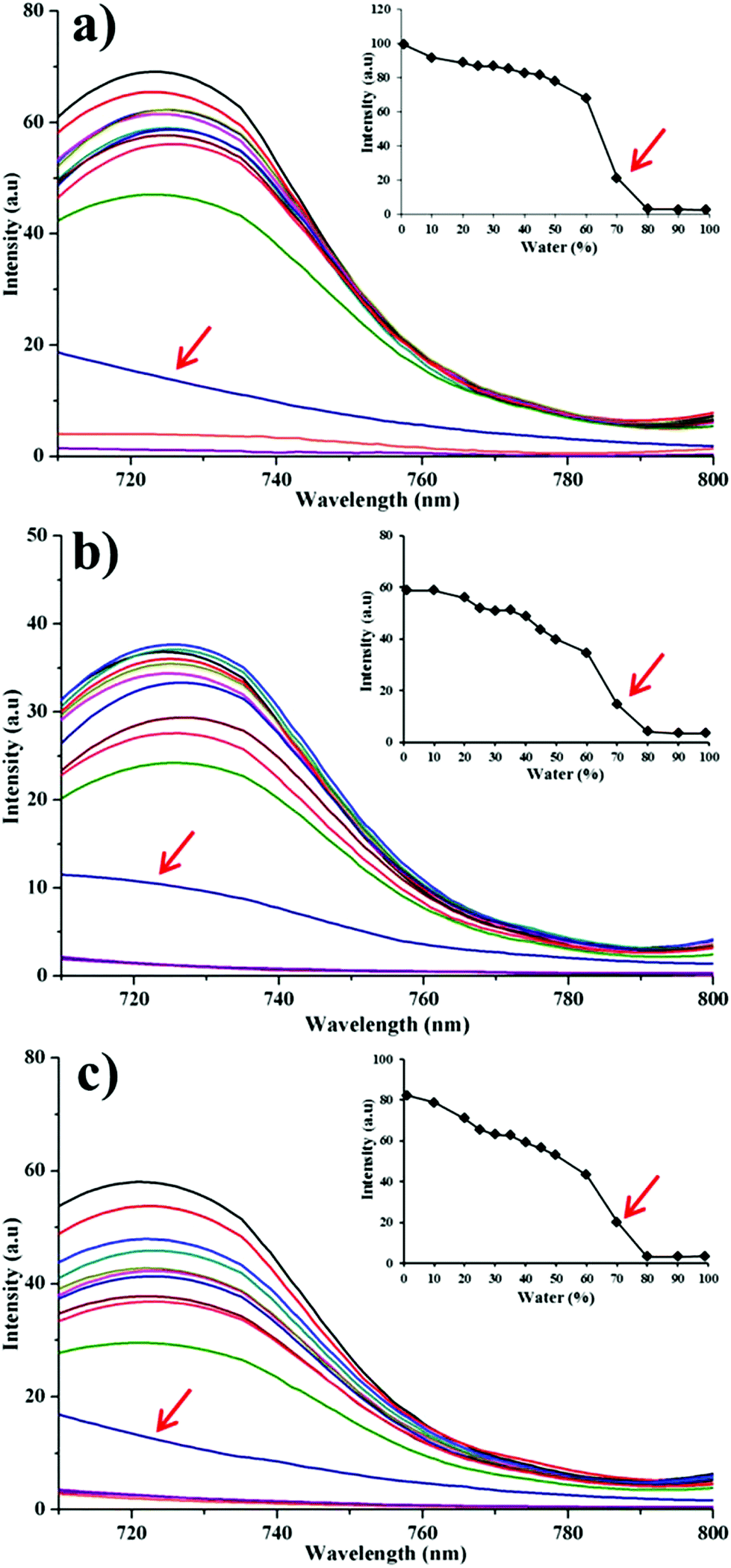 | ||
| Fig. 4 Fluorescence emission of formulations in ethanol–water binary solution at different ratios. (a): QT-HNCs-280; (b): QT-HNCs-550; (c): QT-HNCs-1100. The inset shows the peak value of fluorescence intensity changes upon dilution by water. | ||
In order to track the real-time in vivo translocation of QT-HNCs, it is essential to build up the synchronicity between the dissolution of HNCs and fluorescence quenching. Therefore, the correlation between the dissolution of QT-HNCs and fluorescence quenching was explored. To expedite the dissolution, aqueous ethanol was used as the dissolution media, and a concentration of 20% (v/v) was chosen because this is the highest safe level of ethanol that shows absolute and immediate fluorescence quenching (Fig. 4), above which fluorescence would not quench spontaneously. The dissolution kinetics of HNCs of different sizes, presented as the residual percentage of non-dissolved HNCs, and fluorescence quenching kinetics are shown in Fig. 5. Owing to the particle size effects, the smaller HNCs present a faster dissolution rate than the other two groups, especially at the initial stages. For all groups, the dissolution profiles correlate well with the fluorescence quenching profiles with correlation coefficients exceeding 0.9 (Fig. 5). It is obvious that dissolution slightly outpaces fluorescence quenching in the first hour. However, it should not be attributed to the lack of sensitivity of the probes to water upon dissolution. The deviation of the fluorescence quenching profiles from the dissolution profiles can be explained by a similar reason as observed in previous studies that attenuation of the particulate suspension helps to strengthen the fluorescence temporarily.19 Therefore, it is concluded that very good synchronicity can be established between dissolution and fluorescence quenching of HNCs.
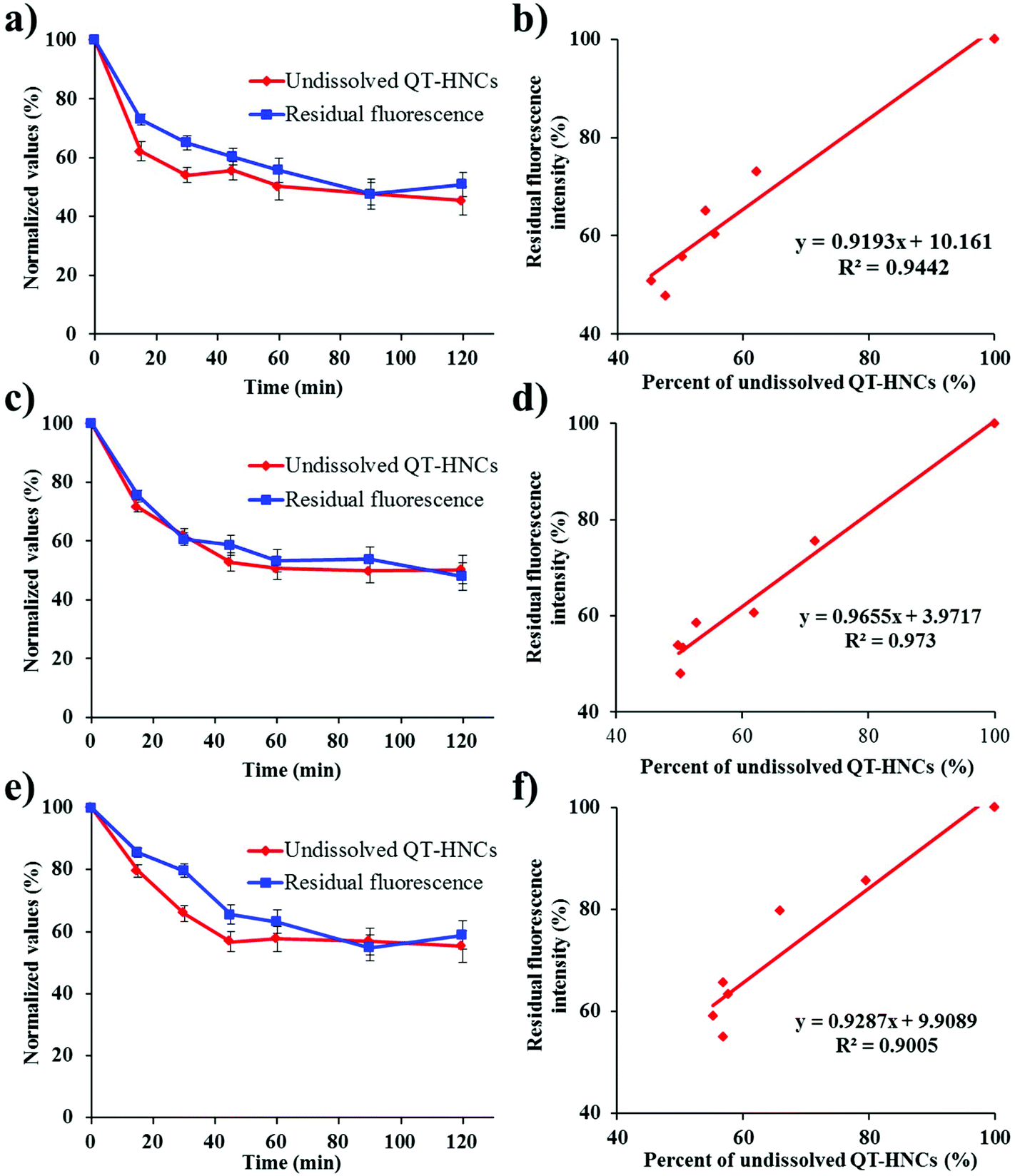 | ||
| Fig. 5 (a), (c) and (e) Normalized value of non-dissolved QT-HNCs and residual fluorescence intensity percentage versus time during dissolution. (b), (d) and (f) Correlation between dissolution and fluorescence quenching. (a) and (b): QT-HNCs-280; (c) and (d): QT-HNCs-550; (e) and (f): QT-HNCs-1100. | ||
Altogether, the feasibility of using ACQ probes to track HNCs has been validated under secure simulated conditions.
Live imaging
Fig. 6a shows the live images after oral administration of QT-HNCs with a quenched P2 solution as a reference. Similar to our previous results, oral administration of the quenched P2 solution only produces negligible fluorescence.19 This eliminates concerns over the interference from the reillumination of the quenched probes. For all groups, fluorescence signals are only observable in the abdomen region. Theoretically, the fluorescence of live imaging might follow a profile of gradual attenuation along with dissolution. Nonetheless, the fluorescence intensifies gradually, peaks at around 2 h, and drops afterwards (Fig. 6b). The same reason as proposed in our previous study with solid lipid nanoparticles can be employed to interpret this phenomenon as well.19 It is postulated that QT-HNCs accumulate within the confined space of the stomach for a short time after oral gavage, resulting in fluorescence quenching to a certain degree. Along with transportation, the QT-HNCs are dispersed throughout the whole gastrointestinal tract (GIT), leading to a resurgence in fluorescence intensity. Notably, the gradual fading out of fluorescence in the later stage reflects the dynamics of dissolution as well as the kinetics of absorption.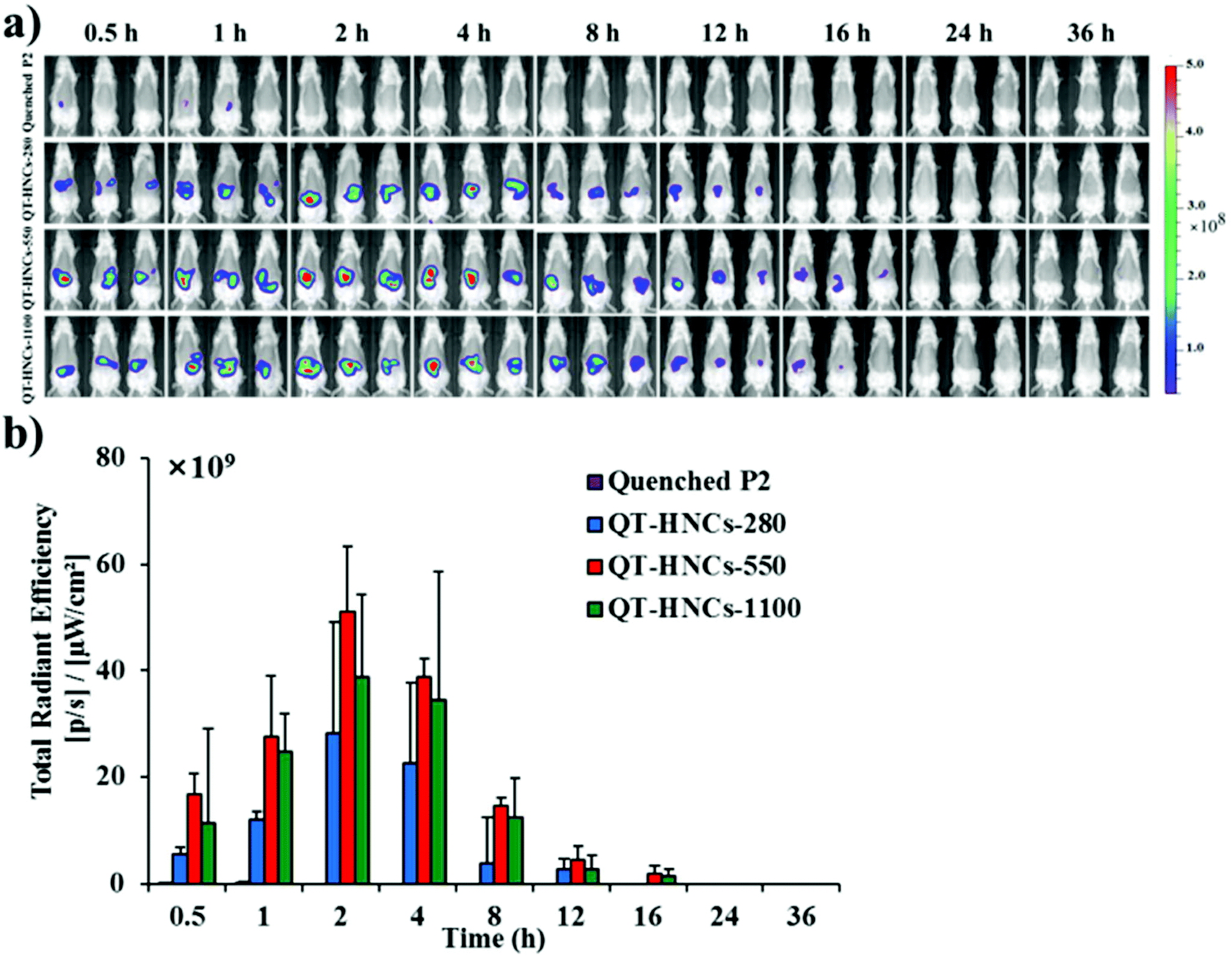 | ||
| Fig. 6 (a) In vivo live imaging and (b) quantification of fluorescence for SD rats after gavage administration of quenched P2 solution and QT-HNCs. | ||
The fluorescence signals can be observed for as long as 12–16 h in the GIT, highlighting an unexpectedly long biological life of the HNCs in the GIT. These findings provide solid evidence to rebut the general perception that nanocrystals dissolve quickly following oral administration, which is more or less due to speculation from in vitro dissolution. As a result, those non-dissolved HNCs surviving the GIT environment have good chances of interacting with the enteric epithelia. Particle size affects the retention of QT-HNCs in the GIT. QT-HNCs-280 is able to be retained in the GIT for 12 h, while for the other two groups it is 16 h (Fig. 6a). Furthermore, quantification of fluorescence gives total radiation efficiency (TRE) values in an order of QT-HNCs-550 > QT-HNCs-1100 > QT-HNCs-280 (Fig. 6b). It is well accepted that the dissolution rate of nanocrystals increases with decreasing particle size.31,32 As a result, QT-HNCs-280, with a much smaller particle size, dissolves faster than the other two groups, leading to less in vivo retention time and lower TRE values. However, the TRE value of QT-HNCs-550 is higher than that of QT-HNCs-1100, which might be due to higher bioadhesion associated with smaller particles and thus enhanced retention in spite of faster dissolution.33,34
In order to further explore the in vivo translocation of QT-HNCs, the whole GIT and various vital organs are dissected and observed under IVIS (Fig. 7a and 8a). Similarly, the water-quenched probe control shows only negligible signals. The overall residence time of QT-HNCs in the GIT is in line with the results of live imaging. The fluorescence in the stomach remains steady within the first 1 h post administration and starts to drop afterwards. At 4 h, stomach-borne fluorescence almost disappears due to gastric emptying and dissolution of the QT-HNCs. Intestinal fluorescence peaks at around 1–2 h post administration for all groups, coinciding with gastric emptying. Interestingly, the fluorescence intensity in both the stomach and the intestine follows the following sequence: QT-HNCs-550 > QT-HNCs-1100 > QT-HNCs-280. The reason is just the same as for live imaging. The overall intestinal fluorescence for QT-HNCs-550 and QT-HNCs-1100 is about 3–5-fold higher than that in the stomach, whereas it is twofold higher for QT-HNCs-280. This is mainly due to the faster dissolution of the smaller QT-HNCs in the upper GIT. Furthermore, strong fluorescence can be observed in the intestine for up to 12–16 h post administration, while only faint signals can be observed in the cecocolon at 2, 4 and 8 h, highlighting the intestine as the main site for dissolution and absorption of QT-HNCs.
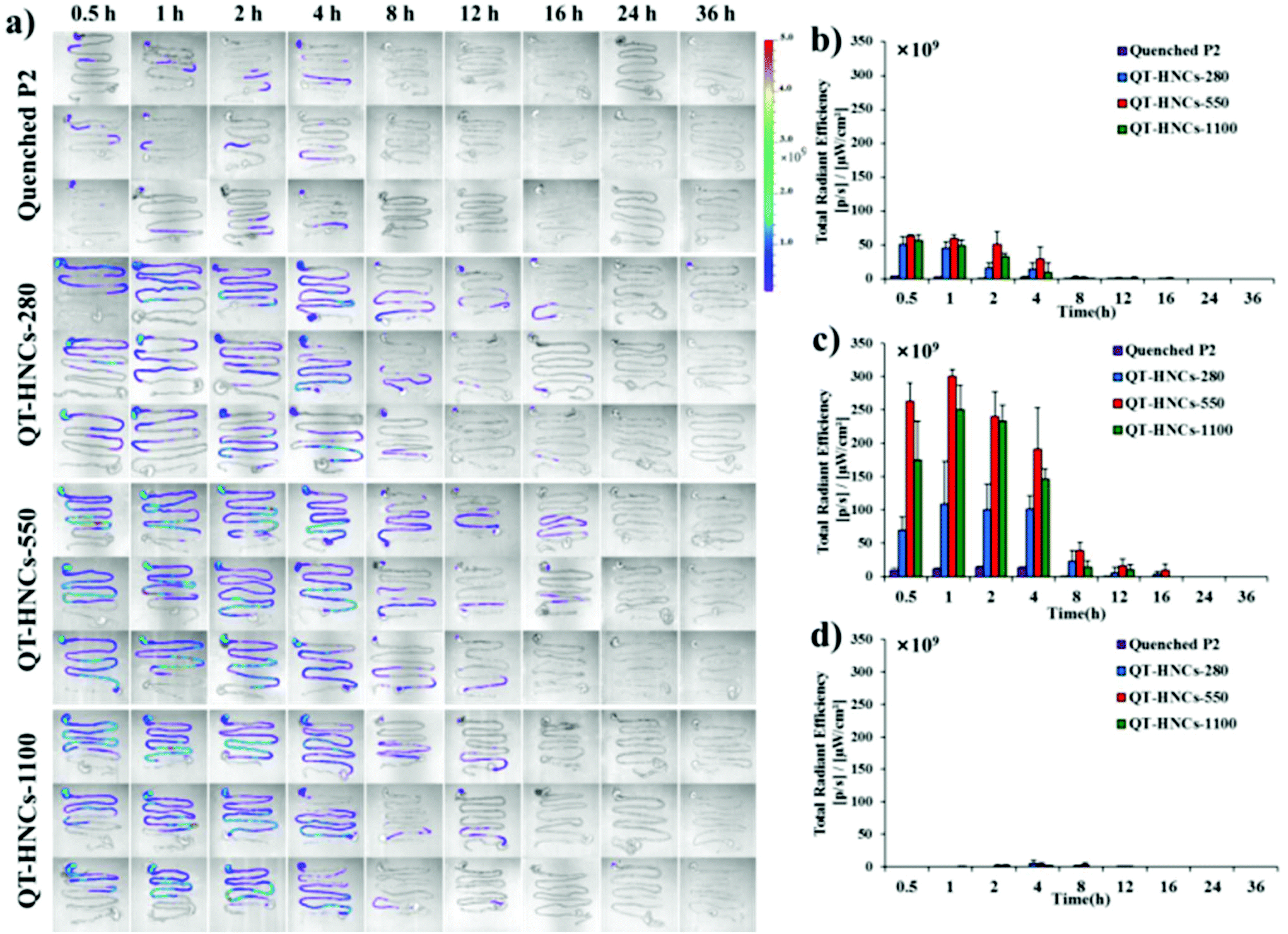 | ||
| Fig. 7 (a) Ex vivo live imaging of the whole isolated GIT after gavage administration of QT-HNCs and quenched P2 solution. Quantification of total fluorescence in (b) stomach, (c) small intestine and (d) colon. | ||
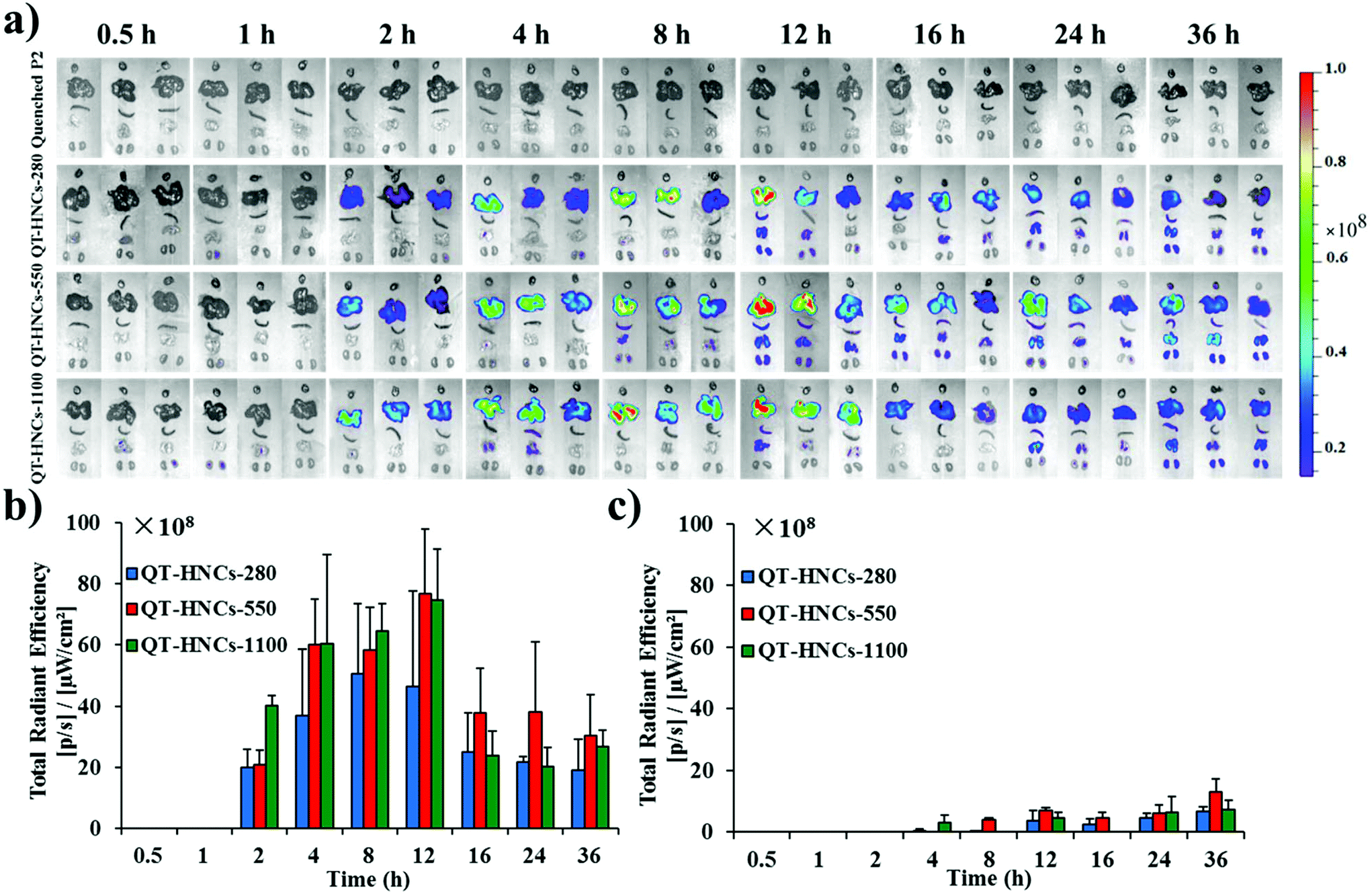 | ||
| Fig. 8 (a) Ex vivo live imaging of main organs after gavage administration of QT-HNCs and quenched P2 solution. Quantification of total fluorescence in (b) liver and (c) lung. The organs in each panel are placed from top to bottom in the order of heart, liver, spleen, lung and kidney. | ||
It is surprising to obtain fluorescence in several organs, such as the liver, lung, spleen and kidney, for all HNC groups after oral administration (Fig. 8). We regard these images as concrete evidence of oral absorption of integral HNCs. Although it is arguable that the fluorescence might be due to absorption and subsequent accumulation of free probes, the absence of fluorescence in related organs after gavage administration of the quenched probe solution as a control helps relieve this concern. The liver is the main terminal and shows the densest fluorescence with fluorescence signals appearing as early as 2 h post administration. For other organs, fluorescence signals, however, start to appear from 4 h on with the lung showing the second densest accumulation. It is easy to understand that particulates entering the circulation are doomed to be opsonized by the complementary system and captured by macrophages residing in various vital organs. A relevant example is with paclitaxel hybrid nanocrystals; approximately 40% of intravenously injected nanocrystals are captured by the liver within 11 min post administration.17 Hepatic accumulation follows a pattern of gradual increment from 2 h to 12 h for all size groups but an abrupt drop to half of the 12 h level afterwards, which is maintained to as long as 36 h. Moreover, the particle size affects the exposure rate and extent of QT-HNCs. The overall fluorescence intensity from QT-HNCs-280 is lower than that of the other two groups. The smaller nanoparticles may achieve a faster absorption rate across the intestinal epithelium.35,36 As for nanocrystals, the smaller ones dissolve faster due to the increased specific surface area and apparent solubility. Therefore, a lower amount of QT-HNCs-280 remains integral in the GIT than the other two groups (Fig. 7), leading to lower liver exposure and a shorter peak time. QT-HNCs-550 and QT-HNCs-1100 show similar liver exposure at the first 12 h post administration. Although the peak time is a little bit lagged in comparison with QT-HNCs-280, the hepatic fluorescence is higher for QT-HNCs-550 and QT-HNCs-1100.
Moreover, although QT-HNCs, represented by fluorescence, were found trapped in the liver for a prolonged time, it does not mean that QT is mainly absorbed in nanocrystal form, trapped in the liver and unable to enter the systemic circulation. In fact, the observation of fluorescence only reflects the fate of the QT “nanocrystals” but not free QT molecules. The majority of QT is released in the GIT, absorbed in its molecular form and reaches the systemic circulation to elicit a pharmacological effect. However, since it is difficult to differentiate QT-HNCs from free QT quantitatively, the percentage of absorbed QT-HNCs cannot be accurately measured at present. Theoretically, free QT can be separated from QT-HNCs by using centrifugation or a Sephadex column but it does not work on biosamples because of the fact that the handling process of biosamples might accelerate the dissolution of QT-HNCs and thus bring about systemic errors. In practice, we did try to measure the total amount of QT in biosamples through extraction and the crystalline QT amount through P2 fluorescence, respectively, hoping that the differentiation of these two values gave the amount of free QT. The key point is the accurate measurement of the crystalline QT amount in biosamples. We tried to establish standard curves for the measurement of crystalline QT by mixing different QT-HNCs in plasma or tissue homogenates but failed due to the dissolution of QT-HNCs, as mentioned above. To the best of our knowledge, there are no workable approaches available to solve this problem. However, roughly estimated from the fluorescence intensity in the small intestine and the liver, approximately 3% QT-HNCs was absorbed. The majority of QT-HNCs may be dissolved in the GIT and absorbed in the molecular form. Apparently, the fraction of QT-HNCs trapped in the liver might not exert a substantial influence on overall bioavailability. The physiological and therapeutic significance of hepatic QT-HNCs are still awaiting appraisal.
Mucosal retention of QT-HNCs
The fact that QT-HNCs can reside in the GIT for a certain time raises the chance of bioadhesion of nanocrystals to the intestinal wall. Therefore, we employed an in situ perfusion model to investigate the bioadhesion of QT-HNCs in the intestinal tract and try to find evidence of internalization of integral QT-HNCs by intestinal epithelia. The control group of water-quenched P4 solution shows negligible fluorescence, excluding the contribution of fluorescence rekindling to overall fluorescence measurement (Fig. 9). Fig. 9a shows the fluorescent images of ex vivo jejunum and ileum segments after in situ perfusion. QT-HNCs-550 and QT-HNCs-1100 show stronger fluorescence than QT-HNCs-280 in both intestinal segments. By measuring the average radiant efficiency (ARE), the retention of various formulations is quantified and shown in Fig. 9b. Although without significance, the ARE values of all size groups in the ileum are higher than those in the jejunum, indicating good bioadhesion of QT-HNCs in the ileum. The ARE values show an order of QT-HNCs-1100 > QT-HNCs-550 > QT-HNCs-280 in the jejunum and an order of QT-HNCs-550 > QT-HNCs-1100 > QT-HNCs-280 in the ileum. The lower ARE values of QT-HNCs-280 might be due to their faster dissolution in comparison with larger ones. No significance is observed in ARE values between QT-HNCs-550 and QT-HNCs-1100, indicating comparable bioadhesion. The QT-HNCs-550 shows significantly higher ARE values than QT-HNCs-280 in the ileum, suggesting easier retention in the ileum.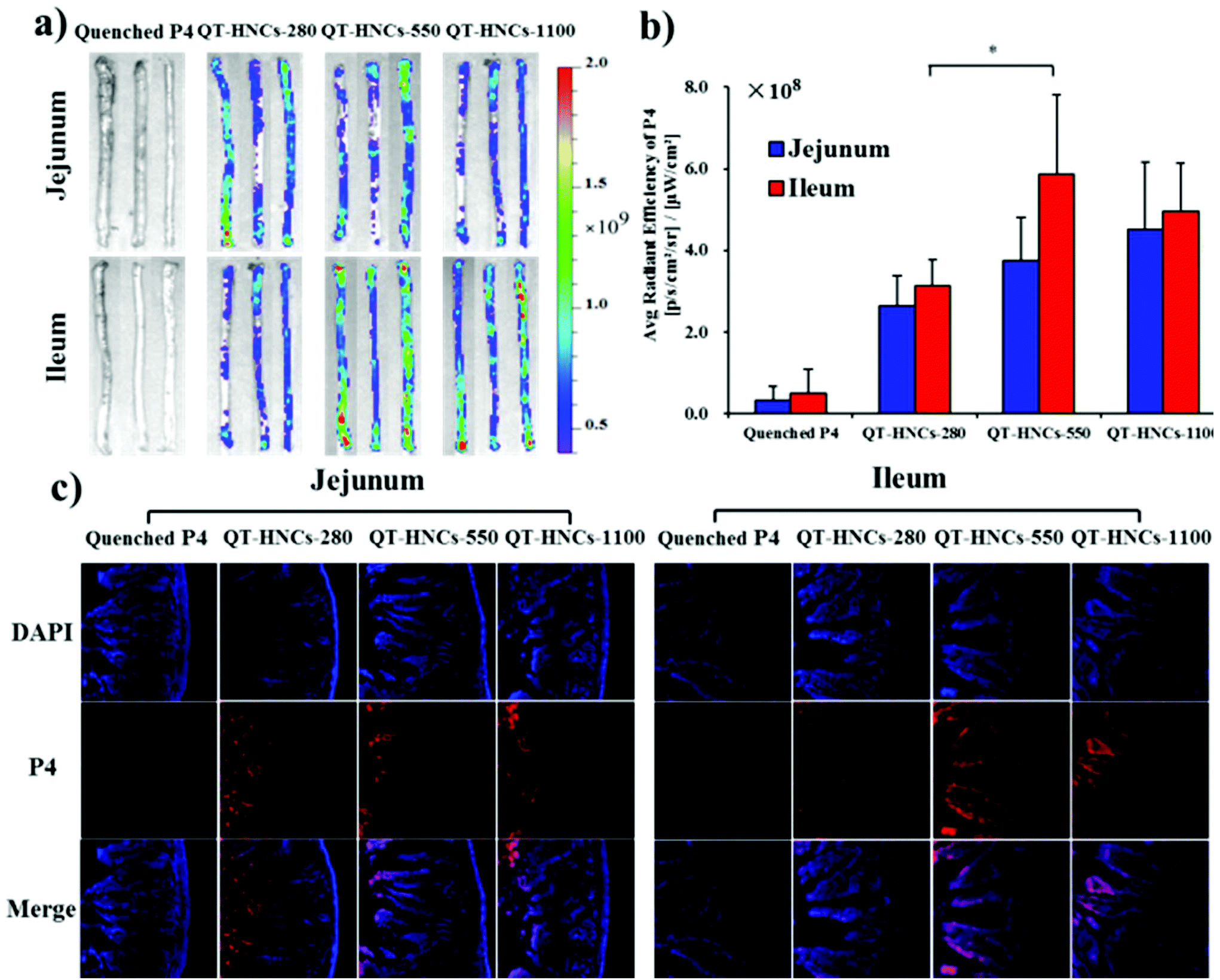 | ||
| Fig. 9 (a) Live images of the ex vivo jejunum and ileum segments after in situ perfusion with different formulations and (b) the corresponding ARE values by measuring P4 fluorescence; (c) the CLSM photos of the frozen section of the rat jejunum and ileum. *p < 0.05, **p < 0.01. | ||
After live imaging, the intestine segments are freeze-sectioned and observed by CLSM to identify traces of fluorescence that represent internalization of the particles (Fig. 9c). The P4 signals can be observed in the basolateral tissues in the jejunum and ileum for all three groups, which serves as strong evidence to confirm the internalization of integral QT-HNCs by enteric epithelia. However, the fluorescence signals in the ileum, especially for QT-HNCs-550 and QT-HNCs-1100, are stronger than those in the jejunum and the signals can be seen throughout the whole population of villi. The results indicate that the bigger particle size may facilitate absorption of integral nanocrystals through the jejunum. The primary differences between the jejunum and the ileum are the existence of special structure such as Peyer's patches (PPs) in the ileum.37 The follicular-associated epithelial cells (FAE) are enriched in the PPs, where many M cells reside in the FAE region. M cells have been extensively reported to recognize and transport foreign particulates from the lumen to basolateral lymphoid tissues.38–40 Therefore, the presence of M cells may favor the absorption and transport of nanocrystals as integral particles. Furthermore, QT-HNCs-550 shows stronger fluorescence than the other two groups, indicating that the moderate size favors absorption.
Cellular uptake and trans-monolayer transport
To find more evidence to support our hypothesis that QT-HNCs can be internalized as integral particles, a series of Caco-2 cell models was used to study the interaction of QT-HNCs with cells. The Caco-2/HT29-MTX cell co-cultures form cell monolayers that mimic a layer of mucous on the surface, while the Caco-2/HT29-MTX/Raji co-cultures mimic the function of M cells. Fig. 10a and b show the live imaging of cellular uptake of QT-HNCs, as well as TRE by measuring P4 fluorescence. The cellular uptake of integral QT-HNCs in Caco-2 cell lines is higher than in Caco-2/HT29-MTX co-cultures, suggesting that the presence of a mucous layer due to secretion by HT29-MTX cells creates a barrier for the interaction between cells and QT-HNCs. In addition, the cellular uptake of QT-HNCs-550 was higher than that of QT-HNCs-280 and QT-HNCs-1100, indicating that QT-HNCs-550 favor cellular uptake. Although this is consistent with the results of live imaging of ex vivo organs, the exact mechanisms are yet to be explored. In addition, it should be noted that the observed fluorescence comprises both the internalized nanocrystals and the ones adhering to the surfaces of the cells. Then multiple CLSM scanning modes are used to confirm the cellular uptake of QT-HNCs (Fig. 10c). P4 signals, which stand for integral nanocrystals, can be found in the same horizontal plane as the cell nucleus, indicating the cellular uptake of QT-HNCs as integral particles. The fluorescence signals of QT-HNCs in the Caco-2 cell models are generally stronger than those in the Caco-2/HT29-MTX cell models due to the mucous barrier. Besides, in both cell models, the overall fluorescence intensity is in a descending sequence of QT-HNCs-550, QT-HNCs-1100 and QT-HNCs-280 as well. Moreover, the presence of P4 signals in the basolateral side from z-axis scanning indicates penetration of integral QT-HNCs across the cell monolayers.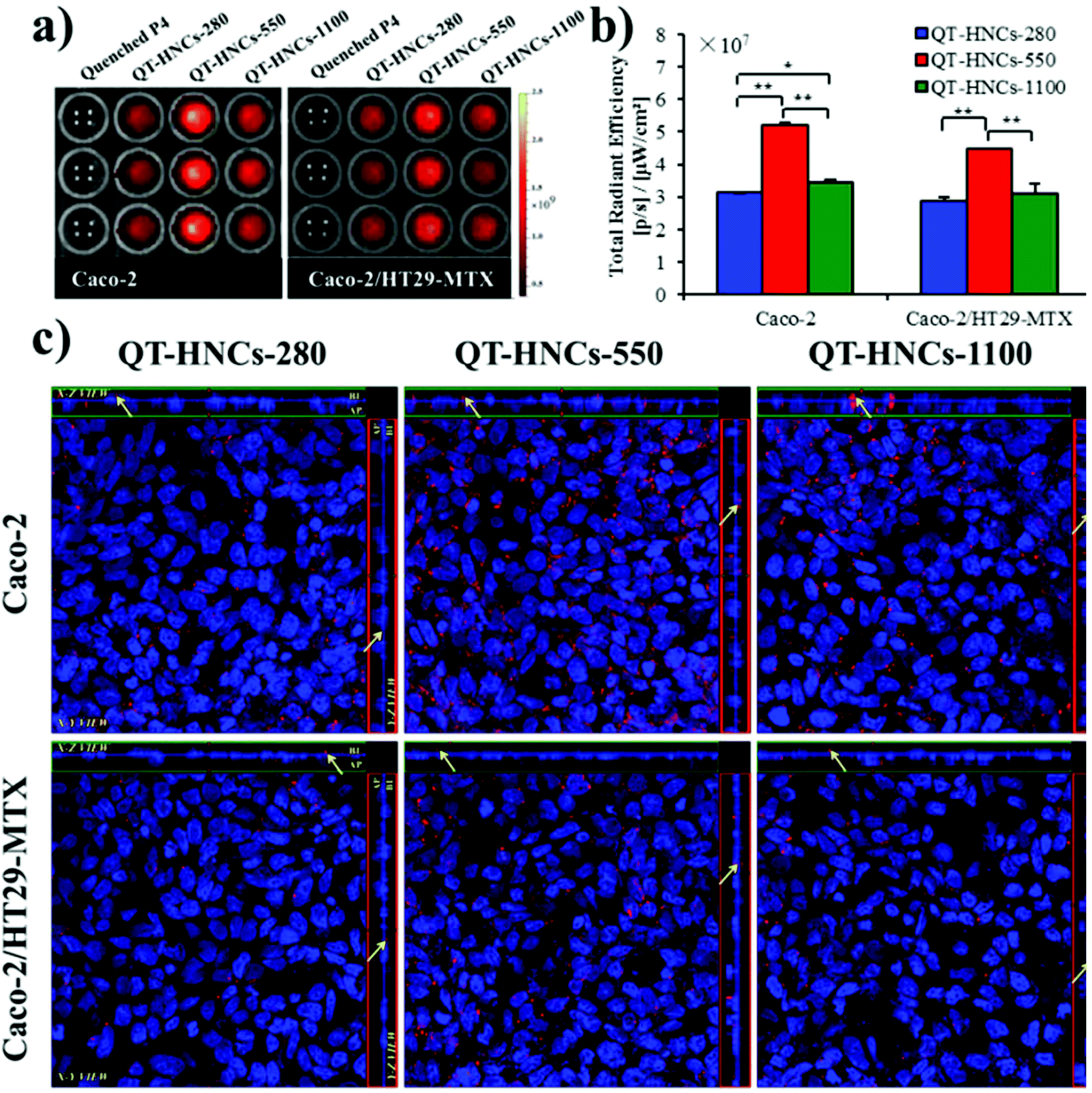 | ||
| Fig. 10 (a) Live images and (b) TRE quantification of cellular uptake of QT-HNCs from Caco-2 and Caco-2/HT29-MTX cell models. (c) Confocal microscopic images of Caco-2 and Caco-2/HT29-MTX monolayers incubated with different QT-HNCs. *p < 0.05, **p < 0.01. | ||
In comparison with cellular uptake, the study of trans-monolayer transportation is more efficient at testing the transiting behavior of integral QT-HNCs by measuring fluorescence in the receiving chamber. As shown in Fig. 11a and b, P4 fluorescence can be detected in the receptor chamber for both Caco-2 and Caco-2/HT29-MTX/Raji cell monolayers, which confirms the trans-monolayer transportation of integral QT-HNCs. However, no fluorescence signals could be observed in the receiving chamber of Caco-2/HT29-MTX cell monolayers, although fluorescence signals are found in the basolateral side by CLSM (Fig. 10c). Owing to the presence of the mucous barriers, only a limited amount of QT-HNCs is able to reach the cell surfaces and even less can be transported across the monolayers. The fluorescence levels in the receptor chamber might be lower than the detection limit of the IVIS system; in addition, nanocrystals transported to the receptor chamber might dissolve and thus lead to fluorescence quenching. P4 fluorescence in the receptor chamber in the Caco-2/HT29-MTX/Raji model is stronger than that in the Caco-2 models. Since Raji cells are generally employed to mimic the function of M cell-associated phagocytosis, the findings above imply that trans-epithelial permeation of integral vehicles, if any, is prone to the M cell pathway. The P4 fluorescence of QT-HNCs-280 and QT-HNCs-550 in the receiving chamber of the Caco-2 cell monolayers is stronger than that of QT-HNCs-1100, while the P4 fluorescence of QT-HNCs-280 in the receiving chamber of the Caco-2/HT29-MTX/Raji cell monolayers is weaker than that of QT-HNCs-550 and QT-HNCs-1100. In general, QT-HNCs-550 showed high transport for both Caco-2 and Caco-2/HT29-MTX/Raji cell monolayers.
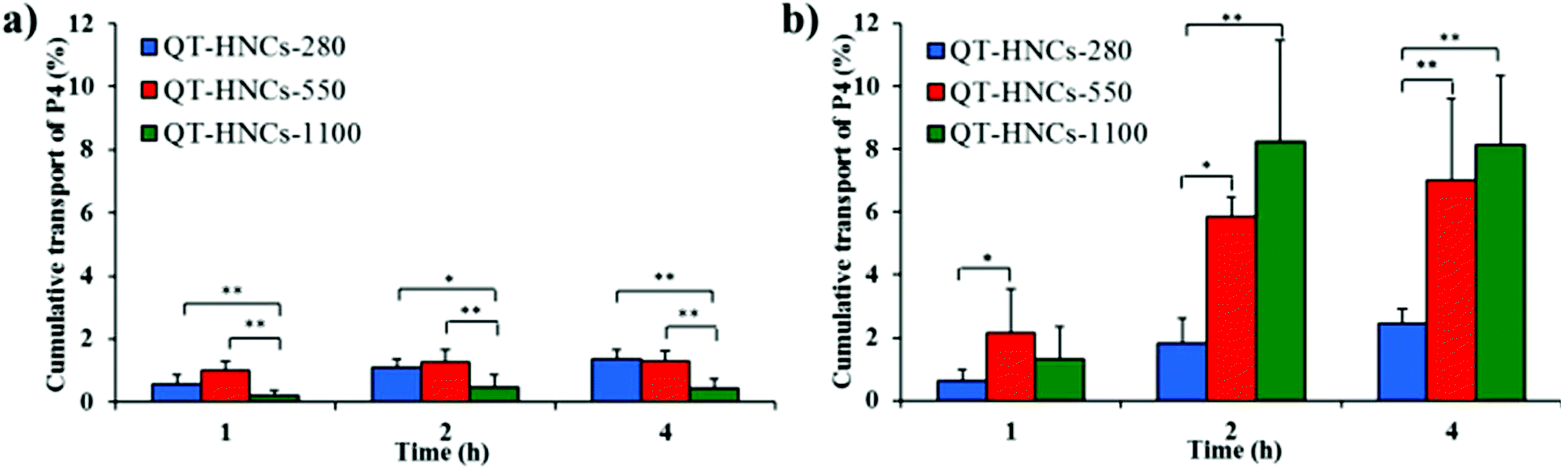 | ||
| Fig. 11 Cumulative transport of QT-HNCs across (a) Caco-2 and (b) Caco-2/HT29-MTX/Raji co-culture monolayers. *p < 0.05, **p < 0.01. | ||
Experimental
Materials
QT was purchased from Shaanxi Jintai Biological Engineering Co. Ltd (Taiyuan, China). P188 was obtained from Sigma-Aldrich (St Louis, USA). ACQ probes, P2 (λabs/λem = 708 nm/732 nm) and P4 (λabs/λem = 651 nm/662 nm) were synthesized according to previous procedures.22 Powders for reconstitution of simulated gastrointestinal fluids were obtained from Biorelevant.Com Ltd (Croydon, U.K.). Isoflurane was obtained from Shandong Keyuan Pharmaceutical Co., Ltd (Jinan, China). 4% Paraformaldehyde was from Fortune bio-tech Co., Ltd (Shanghai, China). 4′,6-Diamidino-2-phenylindole (DAPI) was from Yeasen Bio-tech Co., Ltd (Shanghai, China). Caco-2 cells and Raji cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HT29-MTX was purchased from the China Center for Type Culture Collection (Wuhan, China). Hank's balanced salt solution (HBSS) was purchased from Gibco Invitrogen Co., Ltd (Carlsbad, USA). Other reagents were of at least analytical grade and purchased from local distributors.Preparation and characterization of QT-HNCs
QT-HNCs were prepared using an anti-solvent crystallization method with modification.17 Briefly, QT (100 mg) and ACQ probes (20 μg), either P2 or P4, were dissolved in 6 mL of ethanol, which was rapidly instilled into 120 mL of P188 aqueous solution (0.016%, w/v). For the preparation of QT-HNCs-280, simultaneous high shearing at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and ultrasound (250 W, 40 kHz) for 5 min was adopted to facilitate crystallization during the mixing of the solutions, while the resulting dispersions were sonicated for another 10 min in a water bath at 4 ± 0.5 °C. For the preparation of QT-HNCs-550 and QT-HNCs-1100, magnetic stirring at 1000 and 600 rpm, respectively, was applied during the mixing step and lasted for 20 min. The retentate was collected by filtration through a 50 nm polycarbonate membrane (Sigma-Aldrich, St Louis, MO, USA). The filter cake was rinsed with 10 mL of water to remove free probes that may adsorb on the surface of the HNCs. Finally, the filter cake was re-dispersed in 5 mL of P188 (0.016%, w/v) aqueous solution under ultrasound (250 W, 40 kHz) for 15 min.
000 rpm and ultrasound (250 W, 40 kHz) for 5 min was adopted to facilitate crystallization during the mixing of the solutions, while the resulting dispersions were sonicated for another 10 min in a water bath at 4 ± 0.5 °C. For the preparation of QT-HNCs-550 and QT-HNCs-1100, magnetic stirring at 1000 and 600 rpm, respectively, was applied during the mixing step and lasted for 20 min. The retentate was collected by filtration through a 50 nm polycarbonate membrane (Sigma-Aldrich, St Louis, MO, USA). The filter cake was rinsed with 10 mL of water to remove free probes that may adsorb on the surface of the HNCs. Finally, the filter cake was re-dispersed in 5 mL of P188 (0.016%, w/v) aqueous solution under ultrasound (250 W, 40 kHz) for 15 min.
Water-quenched P2 or P4 solutions were prepared as controls to evaluate possible interference due to the rekindling of fluorescence. Briefly, 1 mL of acetonitrile containing 100 μg of P2 or P4 was mixed with 25 mL of water, followed by evaporation with an R-3 rotatory evaporator (BUCHI, Basel, Switzerland) to remove acetonitrile. Then the sample was diluted with water to a probe concentration equal to that of the QT-HNCs.
The particle size, size distribution, and zeta potential were determined with a Malvern Nano-ZS Zetasizer (Worcestershire, U.K.). QT-HNCs (200 μL) were put into 96-well plates for measurement of fluorescence intensity using IVIS by a region of interest quantification method. After appropriate dilution, the QT-HNCs formulations were dropped onto the tin-foil paper and then dried for SEM (SU8000, Hitachi, Ltd, Tokyo, Japan) observation.
The DSC of the QT raw materials, P188, physical mixture, QT-HNCs-280, QT-HNCs-550 and QT-HNCs-1100 was performed using a 204A/G thermal analyzer (Netzsch, Bühl, Germany). The QT-HNCs formulations were filtrated through a 50 nm polycarbonate membrane and then dried at 50 °C to obtain a solid powder. The samples (5 mg) were weighed into an aluminium pan and heated at a speed of 10 °C min−1 from 40 °C to 350 °C under nitrogen atmosphere. The PXRD spectra of the same samples were recorded by an X-ray diffractometer (D/MAX-2500/PC, Rigaku, Tokyo, Japan) with a Cu source of radiation over the 2θ range from 3° to 60° at a rate of 2° min−1.
QT-HNCs were diluted using ethanol 250-fold for QT concentration (CQ) measurement by a UV-visible spectrophotometer (Agilent, Santa Clara, CA, USA) at the wavelength of 375 nm while QT-HNCs were diluted using ethanol by 20-fold for the measurement of the fluorophore concentration (CF) by a Cary Eclipse fluorospectrophotometer (Agilent, Santa Clara, CA, USA). The entrapment efficiency (EE, %) and probe loading (LD, %) were calculated using eqn (1) and (2).
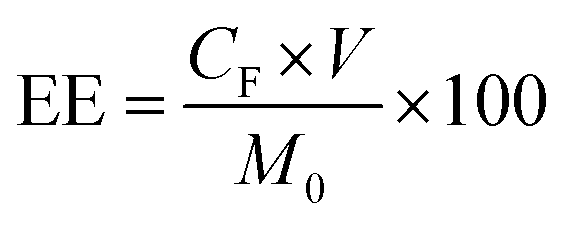 | (1) |
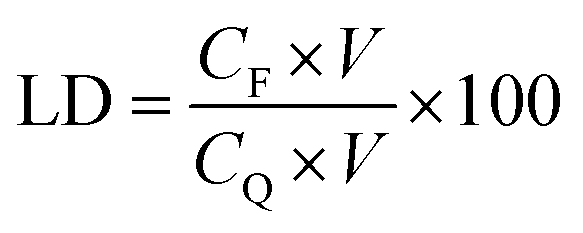 | (2) |
Fluorescence stability of QT-HNCs
The fluorescence stability of QT-HNCs in buffers of different pHs, water and simulated physiological media was assessed. The compositions of SGF, SIF, FaSSIF and FeSSIF are shown in ESI Table S2.† In brief, 2 mL of QT-HNCs was added to 8 mL of the above medium and incubated at 37 °C. At selected time points, 0.4 mL samples were withdrawn for fluorescence measurement.ACQ effects of QT-HNCs in water
Briefly, 7.5 μL of QT-HNCs was mixed with 5 mL of aqueous alcohol with incremental volumes of water for fluorescence scanning using a Cary Eclipse fluorescence spectrophotometer.Correlation between dissolution and fluorescence quenching
The dissolution of QT-HNCs was investigated with a dissolution tester (ZRS-8G, Tianda Tianfa Co., Ltd, Tianjin, China) through Chinese Pharmacopeia Method II (paddle method). QT-HNCs (1 mL) were added to 900 mL of an ethanol/water (80:20, v/v) solution stirred at 50 rpm and thermostated at 37 ± 0.5 °C. Samples of 5 mL were collected at time intervals with concurrent replacement with an equal volume of dissolution medium. One millilitre of the sample was directly measured for fluorescence intensity (Ft). The residual 4 mL was filtered through a 50 nm membrane for measurement of the dissolved QT amount (Wt).
QT-HNCs (1 mL) were dispersed in 900 mL of dissolution medium and measured instantly for the initial fluorescence intensity (F0). The percentage of residual fluorescence intensity (Y1) was calculated using eqn (3):
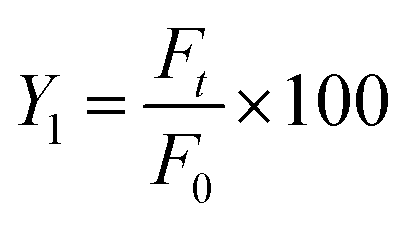 | (3) |
The percent of residual QT-HNCs (Y2) was calculated using eqn (4):
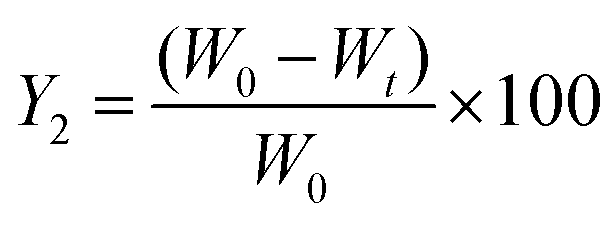 | (4) |
Either residual QT-HNCs or residual fluorescence was plotted versus time. Then Y1 was plotted with Y2 to evaluate the correlation between in vitro dissolution and fluorescence quenching.
In vivo live imaging
Sprague-Dawley (SD) rats (male, 200–220 g) were raised in the Experimental Animal Center of the School of Pharmacy, Fudan University. Guidelines on animal welfare during experiments issued by the Institutional Animal Care and Use Committee at the School of Pharmacy, Fudan University were followed. Before the experiment, the animals were placed in individual cages and fasted overnight for 12 h but allowed free access to water. The animals were randomly divided into four groups, i.e. QT-HNCs-280, QT-HNCs-550, QT-HNCs-1100 and quenched P2 solution. The formulations, 1 mL each, were gavage administered to the rats. The in vivo fate was monitored using the IVIS. During the imaging process, animals were anesthetized by isoflurane through Matrix VIP 3000 Isoflurane Vaporizer (Midmark Corporation, Versailles, USA).Ex vivo imaging
The grouping and administration are similar to the in vivo live imaging but rats were sacrificed at time intervals. The whole of the GIT and various organs were dissected and observed using the IVIS. The fluorescence intensity in each sample was quantified and compared.In situ bioadhesion and ex vivo translocation study
SD rats were anesthetized using phenobarbital (40 mg kg−1) intraperitoneally and fixed in a supine position, the abdomen of the animals was cut open with a midline incision, and a 10 cm segment of either the jejunum or ileum was measured, isolated, cannulated with polyethylene tubing and ligated using silk sutures. The dissected intestinal segments were gently rinsed with pre-warmed saline (37 ± 1 °C) and then attached to the perfusion assembly comprising a peristaltic pump (BT 100-2J/DG-2, Longer Pump Co., Ltd, Baoding, China). The entire surgical area was covered with a piece of sterile absorbent gauze wetted with saline (37 ± 1 °C). At the beginning of the experiment, the blank K-R solution was perfused at a flow rate of 0.5 mL min−1 for 30 min to ensure steady-state conditions. Then, QT-HNCs, diluted sixfold using K-R solution beforehand, were perfused at a flow rate of 0.5 mL min−1 for 90 min. After perfusion, the residues in the intestinal lumen were forced out by air, and the intestinal segments were cut off immediately and observed using the IVIS.The jejunum and ileum segments were sliced into rings and fixed in 4% paraformaldehyde for 12 h, then dehydrated in 30% sucrose solution. Frozen sections of the specimen were collected after being embedded in the OCT (optimum cutting temperature) compound, stained using DAPI to mark the nucleus, and observed by CLSM (LSM510, Carl Zeiss, Jena, Germany).
Cellular uptake study
The uptake of QT-HNCs by Caco-2 cells and Caco-2/HT29-MTX co-culture cells was assessed at 37 °C. The two cellular uptake models were established according to our pre-established procedures.19 The cells were seeded into black cell-culture plates for live imaging and glass cell-culture dishes for CLSM. QT-HNCs were diluted 10-fold using HBSS and 0.1 mL of this suspension was added to the culture for incubation for 2 h. After that, the test samples were discarded and each cell-culture well was washed with D-Hank's solution three times. After addition of 200 μL of D-Hank's solution, the fluorescence of each well was scanned and quantified by IVIS or observed by CLSM.Trans-monolayer transport study
The Caco-2, Caco-2/HT29-MTX monolayers and Caco-2/HT29-MTX/Raji co-culture models were used to assess the transport of QT-HNCs. The models were established and validated according to our previous study.19 QT-HNCs (0.4 mL) were added to the apical sides, and 0.6 mL of HBSS was added to the basolateral sides. At various time intervals, 0.2 mL of the sample was withdrawn from the basolateral side and the same volume of fresh HBSS was simultaneously added. The fluorescence intensity of QT-HNCs in the basolateral side was measured by IVIS.Conclusions
QT-HNCs with three different particle sizes (280 nm, 550 nm and 1100 nm) were successfully prepared by an anti-solvent crystallization method. ACQ probes can be embedded in the QT-HNCs without affecting the crystallinity of QT. The feasibility of the tracking strategies was validated by evaluation of stability, water sensitivity and dissolution synchronicity under secure simulated conditions. Instead of dissolving instantly post oral administration, a fraction of QT-HNCs survives the gastrointestinal environment for as long as 12–16 h. Integral QT-HNCs can be absorbed from the GIT with the liver as the primary terminal. Owing to the prolonged GIT retention, QT-HNCs with bigger particle sizes achieve higher liver exposure, whereas smaller QT-HNCs show a faster absorption rate. In addition, the ileum provides better absorption for integral QT-HNCs than the jejunum. Integral QT-HNCs can be internalized by the Caco-2 cells, and impeded by the presence of the mucous layer. Both trans-epithelial and M cell-mediated routes are involved in the trans-cellular transport of QT-HNCs. The particle size plays an important role in the in vivo behavior as well as the ex vivo cellular interaction of QT-HNCs. The moderate size (550 nm) is preferable for the in vivo retention and absorption of integral QT-HNCs. The results not only validate the idea of using ACQ fluorophores for bioimaging of integral nanocrystals but also support the intestinal absorption of nanocrystals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81573363, 81573697, 81690263); the Military Logistics Comprehensive Fund (BKJ16J025, BKJ16J011) and the Natural Science Foundation of Shanghai (16ZR1403500).Notes and references
- V. K. Pawar, Y. Singh, J. G. Meher, S. Gupta and M. K. Chourasia, J. Controlled Release, 2014, 183, 51–66 CrossRef CAS PubMed.
- L. Gao, G. Liu, J. Ma, X. Wang, L. Zhou, X. Li and F. Wang, Pharm. Res., 2013, 30, 307–324 CrossRef CAS PubMed.
- R. H. Müller, S. Gohla and C. M. Keck, Eur. J. Pharm. Biopharm., 2011, 78, 1–9 CrossRef PubMed.
- B. Shen, N. Wu, C. Shen, F. Zhang, Y. Wu, P. Xu, L. Zhang, W. Wu, Y. Lu, J. Han, Y. Wang and H. Yuan, Drug Dev. Ind. Pharm., 2016, 42, 1772–1781 CrossRef CAS PubMed.
- G. B. Romero, C. M. Keck, R. H. Müller and N. A. Bou-Chacra, Eur. J. Pharm. Biopharm., 2016, 107, 215–222 CrossRef CAS PubMed.
- M. Malamatari, S. Somavarapu, K. M. Taylor and G. Buckton, Expert Opin. Drug Delivery, 2016, 13, 435–450 CrossRef CAS PubMed.
- C. Y. Shen, P. H. Xu, B. D. Shen, H. Y. Min, X. R. Li, J. Han and H. L. Yuan, Drug Delivery, 2016, 23, 610–618 CrossRef CAS PubMed.
- K. Sigfridsson, P. Skantze, U. Skantze, L. Svensson, L. Löfgren, P. Nordell, E. Michaëlsson, B. Smedsrød, B. Fuglesteg, K. Elvevold and L. Lindfors, Int. J. Pharm., 2017, 524, 248–256 CrossRef CAS PubMed.
- R. Shegokar and R. H. Müller, Int. J. Pharm., 2010, 399, 129–139 CrossRef CAS PubMed.
- D. Dong, X. Wang, H. Wang, X. Zhang, Y. Wang and B. Wu, Int. J. Nanomed., 2015, 10, 2521–2535 CAS.
- L. Gao, G. Liu, J. Ma, X. Wang, L. Zhou and X. Li, J. Controlled Release, 2012, 160, 418–430 CrossRef CAS PubMed.
- Q. Fu, J. Sun, X. Ai, P. Zhang, M. Li, Y. Wang, X. Liu, Y. Sun, X. Sui, L. Sun, X. Han, M. Zhu, Y. Zhang, S. Wang and Z. He, Int. J. Pharm., 2013, 448, 290–297 CrossRef CAS PubMed.
- L. Vidlářová, G. B. Romero, J. Hanuš, F. Štěpánek and R. H. Müller, Eur. J. Pharm. Biopharm., 2016, 104, 216–225 CrossRef PubMed.
- X. Miao, Y. Li, X. Wang, S. M. Lee and Y. Zheng, ACS Appl. Mater. Interfaces, 2016, 8, 12620–12630 CAS.
- C. P. Hollis, H. L. Weiss, M. Leggas, B. M. Evers, R. A. Gemeinhart and T. Li, J. Controlled Release, 2013, 172, 12–21 CrossRef CAS PubMed.
- R. Zhao, C. P. Hollis, H. Zhang, L. Sun, R. A. Gemeinhart and T. Li, Mol. Pharm., 2011, 8, 1985–1991 CrossRef CAS PubMed.
- C. P. Hollis, H. L. Weiss, B. M. Evers, R. A. Gemeinhart and T. Li, Pharm. Res., 2014, 31, 1450–1459 CrossRef CAS PubMed.
- Y. Lu, J. Qi, X. Dong, W. Zhao and W. Wu, Drug Discovery Today, 2017, 22, 744–750 CrossRef CAS PubMed.
- X. Hu, W. Fan, Z. Yu, Y. Lu, J. Qi, J. Zhang, X. Dong, W. Zhao and W. Wu, Nanoscale, 2016, 8, 7024–7035 RSC.
- H. He, J. Zhang, Y. Xie, Y. Lu, J. Qi, E. Ahmad, X. Dong, W. Zhao and W. Wu, Mol. Pharm., 2016, 13, 4013–4019 CrossRef CAS PubMed.
- E. Ahmad, Y. Feng, J. Qi, W. Fan, Y. Ma, H. He, F. Xia, X. Dong, W. Zhao, Y. Lu and W. Wu, Nanoscale, 2017, 9, 1174–1183 RSC.
- Y. Xie, X. Hu, H. He, F. Xia, Y. Ma, J. Qi, X. Dong, W. Zhao, Y. Lu and W. Wu, J. Mater. Chem. B, 2016, 4, 2864–2873 RSC.
- F. Xia, W. Fan, S. Jiang, Y. Ma, Y. Lu, J. Qi, E. Ahmad, X. Dong, W. Zhao and W. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 21660–21672 CAS.
- R. Su, W. Fan, Q. Yu, X. Dong, J. Qi, Q. Zhu, W. Zhao, W. Wu, Z. Chen, Y. Li and Y. Lu, Oncotarget, 2017, 8, 38214–38226 Search PubMed.
- X. Hu, J. Zhang, Z. Yu, Y. Xie, H. He, J. Qi, X. Dong, Y. Lu, W. Zhao and W. Wu, Nanomedicine, 2015, 11, 1939–1948 CrossRef CAS PubMed.
- J. Varshosaz, A. Jafarian, G. Salehi and B. Zolfaghari, J. Liposome Res., 2014, 24, 191–203 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- L. Gao, G. Liu, X. Wang, F. Liu, Y. Xu and J. Ma, Int. J. Pharm., 2011, 404, 231–237 CrossRef CAS PubMed.
- G. S. Borghetti, I. S. Lula, R. D. Sinisterra and V. L. Bassani, AAPS PharmSciTech, 2009, 10, 235–242 CrossRef CAS PubMed.
- Y. Zheng and A. H. L. Chow, Drug Dev. Ind. Pharm., 2009, 35, 727–734 CrossRef CAS PubMed.
- C. Bi, X. Q. Miao, S. F. Chow, W. J. Wu, R. Yan, Y. H. Liao, A. H. Chow and Y. Zheng, Nanomedicine, 2017, 13, 943–953 CrossRef CAS PubMed.
- D. Liu, H. Pan, F. He, X. Wang, J. Li, X. Yang and W. Pan, Int. J. Nanomed., 2015, 10, 6425–6434 CrossRef CAS PubMed.
- A. Lamprecht, U. Schäfer and C. M. Lehr, Pharm. Res., 2001, 18, 788–793 CrossRef CAS.
- J. Youshia and A. Lamprecht, Expert Opin. Drug Delivery, 2016, 13, 281–294 CrossRef CAS PubMed.
- M. Yao, L. He, D. J. McClements and H. Xiao, J. Agric. Food Chem., 2015, 63, 8044–8049 CrossRef CAS PubMed.
- K. M. Williams, K. Gokulan, C. E. Cerniglia and S. Khare, J. Nanobiotechnol., 2016, 14, 62–74 CrossRef PubMed.
- E. M. Kuhn and F. J. Kaup, Anat., Histol., Embryol., 1996, 25, 65–69 CrossRef CAS.
- R. L. Owen, Semin. Immunol., 1999, 11, 157–163 CrossRef CAS PubMed.
- T. E. Rajapaksa, M. Stover-Hamer, X. Fernandez, H. A. Eckelhoefer and D. D. Lo, J. Controlled Release, 2010, 142, 196–205 CrossRef CAS PubMed.
- T. Jiang, B. Singh, H. S. Li, Y. K. Kim, S. K. Kang, J. W. Nah, Y. J. Choi and C. S. Cho, Biomaterials, 2014, 35, 2365–2373 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06052a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |