
Natural product syntheses via carbonylative cyclizations
Kaiqing
Ma†
 ab,
Brandon S.
Martin†
a,
Xianglin
Yin†
a and
Mingji
Dai
*a
ab,
Brandon S.
Martin†
a,
Xianglin
Yin†
a and
Mingji
Dai
*a
aDepartment of Chemistry, Center for Cancer Research, Institute for Drug Discovery, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: mjdai@purdue.edu; Fax: +1-765-496-2592; Tel: +1-765-496-7898
bModern Research Center for Traditional Chinese Medicine of Shanxi University, No. 92, Wucheng Road, Taiyuan 03006, Shanxi, China
First published on 20th June 2018
Abstract
Covering: 2000–2018
In this review, we highlight recent examples of natural product total syntheses employing transition metal-mediated/catalyzed carbonylative cyclization strategies to build key ring systems. It mainly covers carbonylative cyclizations for the construction of O-heterocycles, N-heterocycles and carbocycles including cyclic ketones and phenols. The reaction types include carbonylation of epoxide to β-lactones, carbonylative (macro)lactonization/lactamization, the Semmelhack reaction, tandem hydroformylation–cyclization, the Pauson–Khand reaction, carbonylative C–H activation cyclization, the Stille/Suzuki carbonylation, [n + m + 1] carbonylative cycloaddition, the Dötz annulation, and others.
 Kaiqing Ma | Kaiqing Ma obtained his B.S. in Pharmacy in 2005 from Shanxi Medical University under the supervision of Professor Shuang Hu. Subsequently, he started his graduate study at Fudan University and received his Ph.D. in Medicinal Chemistry in 2011 under the supervision of Professor Deyong Ye. He then took a postdoctoral position in the laboratory of Professor Xiaoguang Lei at National Institute of Biological Sciences, Beijing, China. In February 2014, he joined the Modern Research Center for Traditional Chinese Medicine in Shanxi University as a lecturer and was promoted to associate professor in 2017. Currently, he is a visiting scholar with Professor Mingji Dai at Purdue University, focusing on the total synthesis of complex alkaloids. |
 Brandon S. Martin | Brandon S. Martin received a B.S. in chemistry from Eastern Illinois University in 2014. He began graduate study under the supervision of Professor Mingji Dai at Purdue University in 2016, focusing on the total synthesis of natural products. |
 Xianglin Yin | Xianglin Yin obtained his B.E. degree in 2013 in Polymer Science and Technology from Beijing University of Chemical Technology under the supervision of Professor Liqun Zhang, and M.S. degree in Polymer Science in 2014 from The University of Akron under the supervision of Professor Coleen Pugh. He is currently a graduate student with Professor Mingji Dai at Purdue University, focusing on developing new palladium-catalyzed tandem carbonylation chemistry and their applications in total synthesis of natural products. |
 Mingji Dai | Mingji Dai grew up in Sichuan, China, and received his B.S. from Peking University in 2002. After two years of research with Professors Zhen Yang and Jiahua Chen at the same university, he went to Columbia University in the City of New York in 2004 to pursue graduate study under the guidance of Professor Samuel J. Danishefsky and earned his Ph.D. degree in 2009. He then took a postdoctoral position in the laboratory of Professor Stuart L. Schreiber at Harvard University and the Broad Institute. In August 2012, he began his independent career as an assistant professor in the Chemistry Department of Purdue University. His lab currently focuses on developing new strategies and methodologies for the synthesis of complex natural products and other medicinally and biologically important molecules. |
1. Introduction
Transition metal catalyzed reactions such as asymmetric hydrogenations, palladium-catalyzed cross coupling reactions, and olefin metathesis reactions have transformed total synthesis of natural products, discovery and development of lifesaving therapeutics, and many other fields. In this review article, we focus on the application of a series of transition metal-catalyzed/mediated carbonylative cyclization reactions in facilitating total synthesis of complex bioactive natural products. The highlighted carbonylation processes use carbon monoxide as a one-carbon linchpin to rapidly build different ring systems including O-heterocycles, N-heterocycles, cyclic ketones, and aromatic ring systems. Carbon monoxide is an abundant, cheap and readily available one-carbon chemical stock. Due to its carbene nature, it can coordinate with various transition metals to form carbonyl complexes. It can also undergo migratory insertion into metal–carbon or metal–heteroatom bond to produce acyl-metallo species, which are usually highly reactive and can be intercepted by different nucleophiles to form carbon–carbon or carbon–heteroatom bonds and provide carbonyl-containing products such as ketones, esters, amide, aldehydes, and their derivatives.1–8 In most cases, carbon monoxide gas is used directly. A variety of user-friendly carbon monoxide surrogates have been developed to avoid the direct use of carbon monoxide gas.9–11Transition metal-catalyzed carbonylation reactions offer new avenues or complementary ways to access carbonyl containing products. The carbonylative cyclization processes covered here include carbonylation of epoxide to β-lactones, carbonylative (macro)lactonization/lactamization, the Semmelhack reaction, tandem hydroformylation–cyclization, the Pauson–Khand reaction, carbonylative C–H activation cyclization, the Stille/Suzuki carbonylation, [n + m + 1] carbonylative cycloaddition, the Dötz annulation, and others. In general, these transformations feature high efficiency in building complex structures, good atom economy, and excellent chemo-, regio- and stereo-selectivity. The carbonyl group derived from carbon monoxide either locates in the ring (endo-cyclic carbonylation) or outside of the ring (exo-cyclic carbonylation, Fig. 1).
 | ||
| Fig. 1 Endo/exo carbonylative cyclization. | ||
2. Carbonylative O-heterocycle synthesis
2.1. Epoxide carbonylation to β-lactones and related products
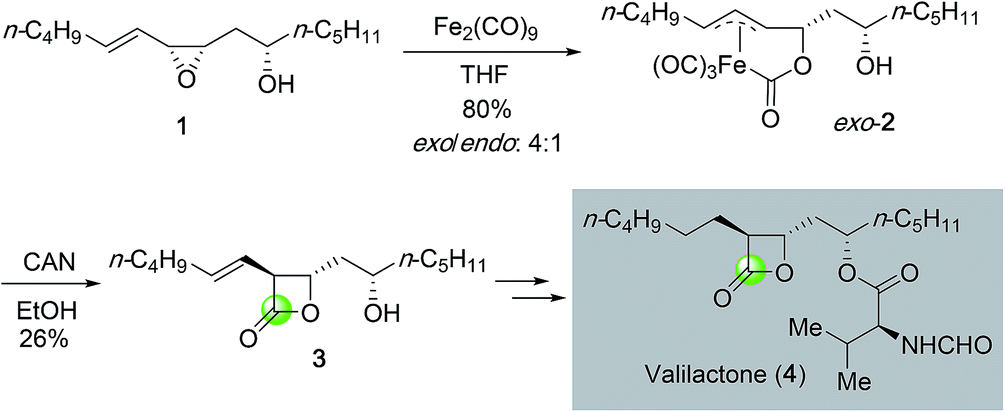
β-Lactones are important structural motifs that prevalently exist in many bioactive natural products. Conventionally, β-lactone syntheses mainly rely on the lactonization of β-hydroxy acid promoted by various activating reagents. Later ketene–aldehyde/ketone [2 + 2] cycloaddition emerged as a popular method to prepare β-lactones12 and the related enantioselective [2 + 2] cycloadditions have been developed as well to access β-lactones in optically active form.13 Due to the intrinsic ring strain, epoxides have demonstrated high reactivity and can undergo various transformations. Many epoxidation methods especially enantioselective ones such as the Sharpless epoxidation, Jacobsen–Katsuki epoxidation, Shi epoxidation, and the recent organocatalytic epoxidation have been developed to synthesize epoxides.14 Structurally, epoxides are one carbonyl group away from β-lactones. Direct carbon monoxide insertion to epoxide has become an important strategy to access β-lactones.A pioneering example of epoxide carbonylation in β-lactone total synthesis was reported by Ley and co-workers in 1991 (Fig. 2).15 Towards their synthesis of valilactone (4), they developed a method to convert epoxide 1 derived from directed epoxidation of a homoallylic alcohol to β-lactone 3 with diiron nonacarbonyl complex (Fe2(CO)9) followed by oxidative lactonization. The initial carbonylation products were isolated as a mixture of exo and endo complexes in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Oxidation of the major exo complex 2 with ceric ammonium nitrate (CAN) gave the desired β-lactone in 26% yield. The oxidation yield is higher when the secondary alcohol was protected as an acetate (50% yield). β-Lactone 3 was then advanced to valilactone in two steps.
:1 ratio. Oxidation of the major exo complex 2 with ceric ammonium nitrate (CAN) gave the desired β-lactone in 26% yield. The oxidation yield is higher when the secondary alcohol was protected as an acetate (50% yield). β-Lactone 3 was then advanced to valilactone in two steps.
 | ||
| Fig. 2 The Ley synthesis of valilactone (1991). | ||
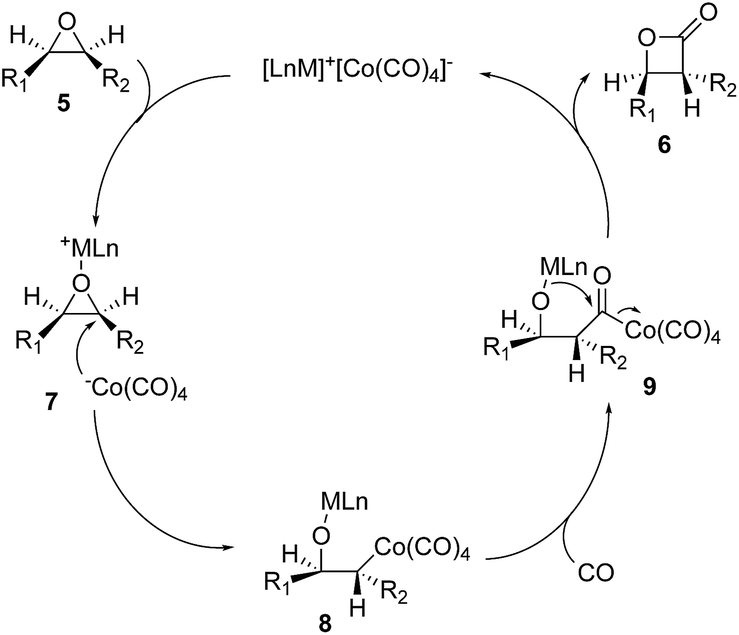
There are a few drawbacks associated with the diiron nonacarbonyl-promoted epoxide carbonylation. It requires stoichiometric amount of the diiron nonacarbonyl complex and an additional low yielding oxidation step. Coates and co-workers came up with a remarkable solution to these problems by using bimetallic [Lewis acid]+[Co(CO)4]− catalysts (Fig. 3, 5 → 6).16,17 The Lewis-acidic cation of the ion pair would coordinate and activate the epoxide to facilitate a backside attack of the activated epoxide by [Co(CO)4]−. The regioselectivity of the epoxide ring opening can be controlled by the steric and electronic properties of the epoxide. The resulting alkylcobalt intermediate (8) would undergo carbon monoxide insertion to generate acyl-species 9. Lactonization of 9 would complete the catalytic cycle to provide β-lactone 6 and regenerate the catalyst for the next catalytic cycle. This catalytic epoxide carbonylation reaction provides a direct route to β-lactones from epoxides and has been broadly used in polymer synthesis and natural product total synthesis.
 | ||
| Fig. 3 Bimetallic [Lewis acid]+[Co(CO)4]−-catalyzed carbonylative β-lactone synthesis from epoxide. | ||
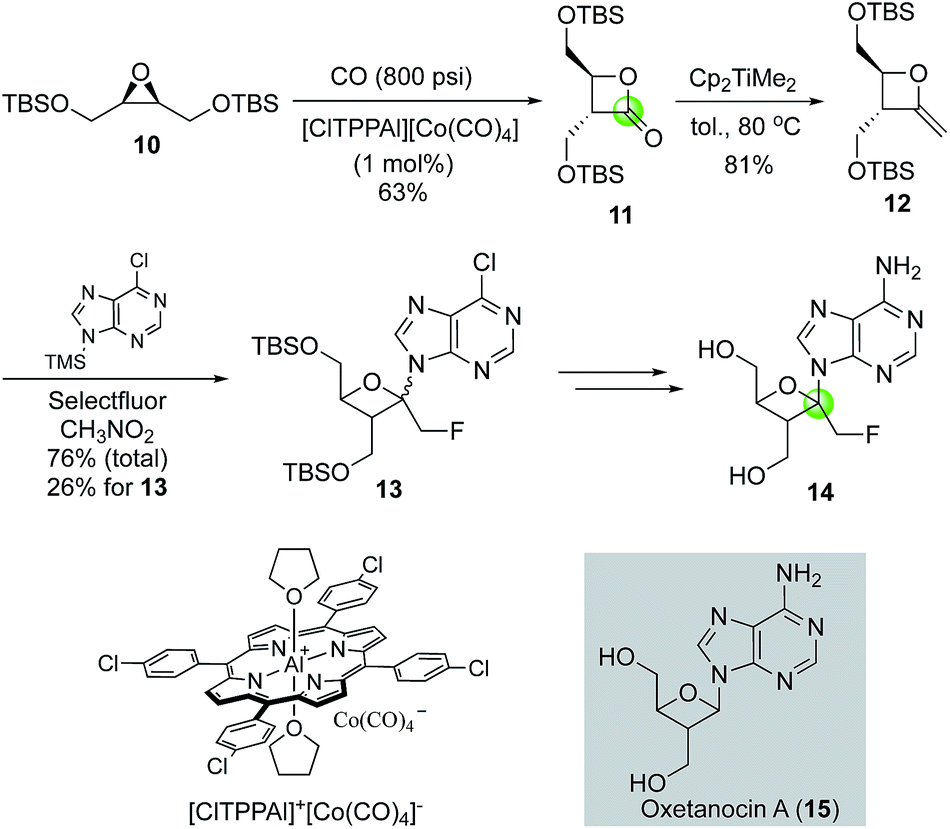
Oxetanocin A (OXT-A, 15, Fig. 4) is a natural product with an unusual nucleoside-substituted oxetanosyl sugar and has shown potent anti HSV-1, HSV-2, HCMV, and HIV-1 activity. In searching for analogs with improved function, Howell and co-workers reported the first synthesis of psico-oxetanocin analog 1′-fluoromethyl-OXT-A, 14.18 Their synthesis features the cobalt-catalyzed carbonylation of symmetrical epoxide 10 to β-lactone 11 in 63% yield by using 1 mol% of the aluminium–cobalt ion pair [ClTPPAl][Co(CO)4]. The β-lactone product was then converted to methyleneoxetane 12via a Petasis olefination. Selectfluor-promoted nucleobase installation gave a mixture of regio- and stereo-isomers with a total yield of 76% and 26% yield of 13, which was then converted to 14.
 | ||
| Fig. 4 The Howell synthesis of psico-nucleosides (2011). | ||
The same cobalt-catalyzed carbonylation of epoxide was applied by O'Doherty and co-workers to synthesize fridamycin E19 and tetrahydrolipstatin20 (Fig. 5). For the synthesis of fridamycin E, epoxide 16 derived from a sequence of Sharpless dihydroxylation and epoxide formation was treated with [ClTPPAl]+[Co(CO)4]− catalyst developed in the Coates group under 900 psi of carbon monoxide in THF at 40 °C to afford β-lactone 17 in 70% yield (Fig. 5A). Interestingly, when the reaction temperature reached to 60 °C, a significant amount of alkene byproduct was observed via a deoxygenation process. After cleavage of the β-lactone ring and removal of the benzyl protecting group, fridamycin E (18) was formed. For the tetrahydrolipstatin synthesis (Fig. 5B), O'Doherty and co-workers evaluated both an early-stage and a late-stage (last step, 19 → 20) installation of the required β-lactone. Both cases worked smoothly and demonstrated the functional group compatibility and reliability of the carbonylative β-lactone synthesis in complex settings. In the end, tetrahydrolipstatin was prepared in an impressive ten steps and 31% overall yield.
 | ||
| Fig. 5 The O'Doherty synthesis of fridamycin E (2011) and tetrahydrolipstatin (2014). | ||
In a related study, Fürstner and co-workers used a Co2(CO)8-catalyzed carbonylative epoxide ring opening developed by the Jacobsen group21 to synthesize amide 22 (Fig. 6), which was one of the key building blocks for diyne intermediate 23 for an alkyne ring closing metathesis to close the macrocycle (23 → 24). The carbon from carbon monoxide eventually served as C21 of the putative structure of anticancer mandelalide A (25).22 Notably, this synthesis also led to the structural revision of mandelalide A.
 | ||
| Fig. 6 The Fürstner synthesis of the putative structure of mandelalide A (2015). | ||
2.2. 5 or 6-membered lactone formation
 | ||
| Fig. 7 The Marshall synthesis of aristolactone (1987). | ||
The pumiliotoxin alkaloids were isolated from the skin extracts of the Panamanian poison frogs and have attracted plenty of synthetic attention.34 In their efforts toward the pumiliotoxin A (37, Fig. 8) and related analogs, Kibayashi and co-workers employed a similar palladium-catalyzed carbonylative lactonization to convert vinyliodide 35 to lactone 36, which was later rearranged to pumiliotoxin A with a 5,6-fused ring system.35 Vinyliodide 35 was prepared in three steps from proline derivative 32. The first step involves a HfCl4-catalyzed propargylation of 32 with allenylsilane 33 followed by Boc-protection. A combination of radical stannation with Ph3SnH and Et3B and iodination with NIS was able to convert alkyne 34 to vinyliodide 35.
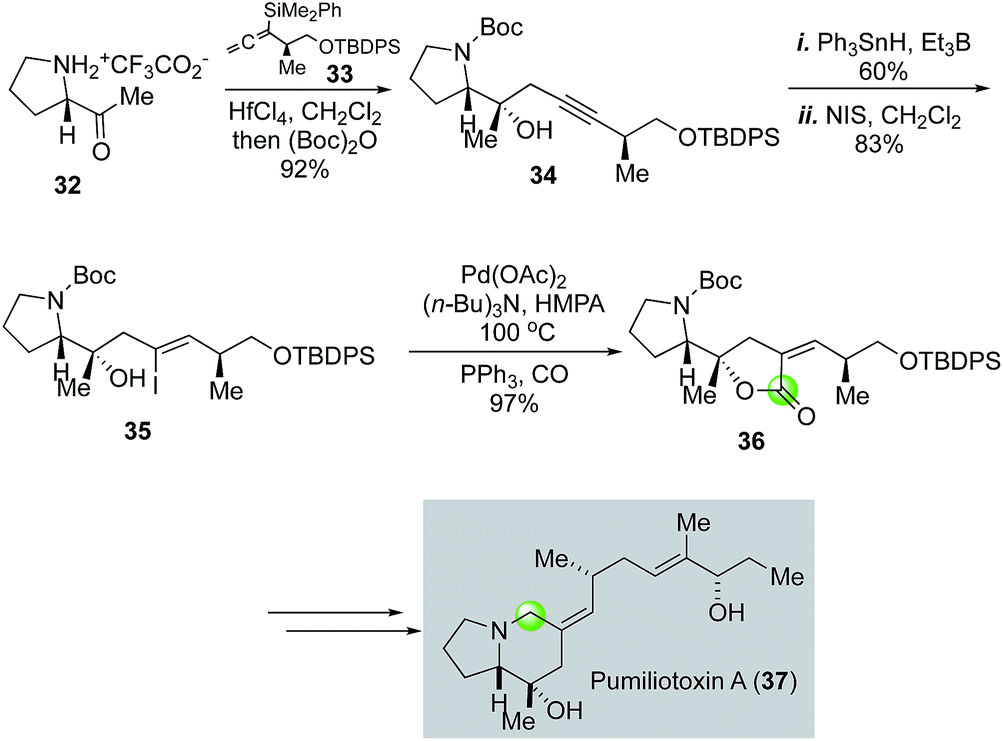 | ||
| Fig. 8 The Kibayashi synthesis of pumiliotoxin A (2002). | ||
Since their isolation, the phomoidride molecules, due to their promising cholesterol-lowering and anticancer activity and unique structural scaffold, have attracted a significant amount of synthetic attention and inspired numerous synthetic methods and strategies toward their syntheses.36,37 In their synthetic efforts toward the phomoidride B (CP-263, 114, Fig. 9, 41), Leighton and co-workers orchestrated an impressive tandem sequence of ketal formation, palladium-catalyzed carbonylative lactone formation, and [3,3]-sigmatropic rearrangement to convert vinyltriflate 38 to 40 containing the core structure of phomoidride B via spirolactone intermediate 39.38
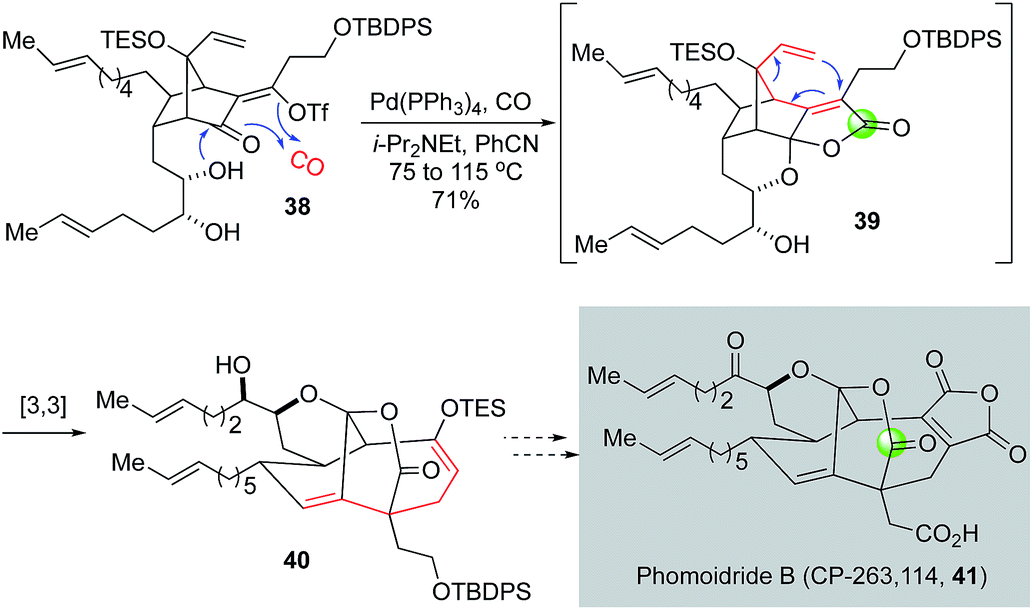 | ||
| Fig. 9 The Leighton synthesis toward phomoidride B (2003). | ||
In Larock's plicadin synthesis (Fig. 10),39 an iodine-promoted benzofuran synthesis was used to produce 3-iodobenzofuran 43 from 42 for a palladium-catalyzed carbonylation, which afforded plicadin (44) in 57% yield after removal of the tosyl group with TBAF. This strategy also led to the total synthesis of coumestan and coumestrol, and their analogs.
 | ||
| Fig. 10 The Larock synthesis of plicadin (2005). | ||
Hypoestoxide (49, Fig. 11), isolated from the tropical shrub Hypoestes rosea, has demonstrated a broad range of biological profiles including anticancer activity by inhibiting angiogenesis, antimalarial, and anti-inflammatory activity. Structurally, it contains a challenging rigid “inside–outside” bicyclo[9.3.1] ring system. In their efforts toward the total synthesis of hypoestoxide, Njardarson and co-workers used a palladium-catalyzed carbonylative lactonization to convert vinyltriflate 45 to bicyclic lactone 46.40 After DIBAL-H reduction of lactone 46 to a diol, they used a titanium-templated relay ring closing metathesis to building the “inside–outside” bicyclo[9.3.1]pentadecane ring system and eventually reached a diene intermediate (48) which is one step away from the atropisomer (49a) of hypoestoxide. However, selectively oxidative cleavage of the extra methylene group of 48 was unsuccessful. In this case, the carbon atom from carbon monoxide could potentially serve as the required exo-methylene carbon of 49 or 49a.
 | ||
| Fig. 11 The Njardarson synthesis toward hypoestoxide (2008). | ||
In 2009, Willis and co-workers reported a palladium-catalyzed intramolecular carbonylation reaction of aryl halides and enolate nucleophiles to build isocoumarins (Fig. 12).41 Unlike the previous cases which used alcohol or phenol nucleophiles to trap the acylpalladium species, their work employed an in situ generated enolate. A 2-step synthesis of thunberginol A (51) was achieved from ketone 50, which was prepared by a palladium-catalyzed α-arylation.
 | ||
| Fig. 12 The Willis synthesis of thunberginol A (2009). | ||
Architecturally unique and complex natural product salvileucalin B (57) was isolated by Takeya and co-workers.42 This molecule features an unusually stable norcaradiene embedded in a polycyclic cage-like skeleton and two five-membered lactone rings. Salvileucalin B also demonstrated promising anti cancer cell proliferation activity. Synthetically, Reisman and co-workers used a palladium-catalyzed carbonylative lactonization to convert vinyltriflate 55 to lactone 56 (Fig. 13).43 The iso-dihydrofuran ring of 56 was then oxidized to the second required lactone by chromium trioxide-3,5-dimethylpyrazole complex to complete the first total synthesis of salvileucalin B. A significant amount of byproduct 58 was also obtained under the oxidation conditions. Vinyltriflate 55 was efficiently synthesized from triyne intermediate 52via steps including a ruthenium-catalyzed [2 + 2 + 2] cycloisomerization to build the aromatic ring and a copper-catalyzed intramolecular [2 + 1] cycloaddition of α-diazo-β-ketonitrile 53 to provide the norcaradiene moiety. Additionally, in their synthetic efforts toward salvileucalin B, Taber and co-worker used a similar carbonylative lactonization to build the corresponding lactone.44
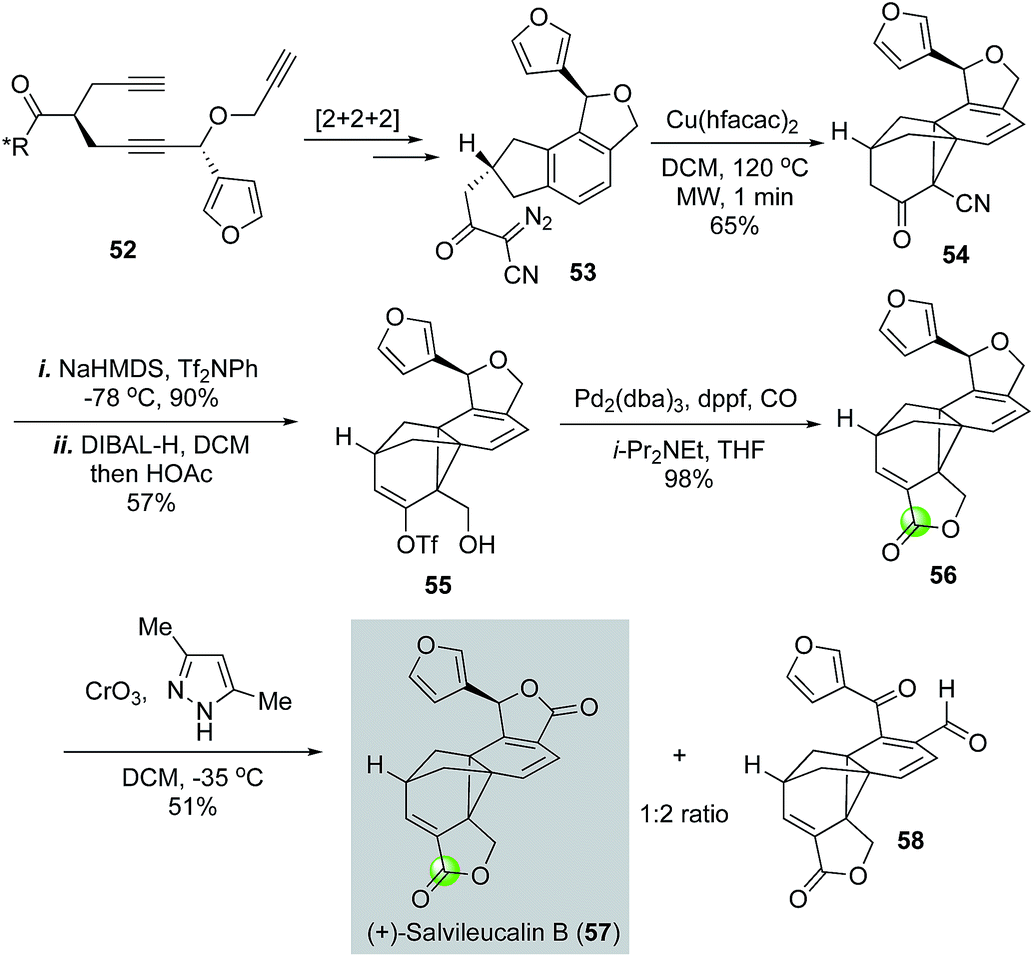 | ||
| Fig. 13 The Reisman synthesis of salvileucalin B (2010). | ||
In their total synthesis of two diterpenoids amphilectolide (62) and sandresolide B (63) and two norditerpenoids caribenols A and B (64, 65, Fig. 14),45,46 Trauner and co-workers used a palladium-catalyzed carbonylative lactonization to convert vinyltriflate 59 to bicyclic lactone 60. Notably, in this case, the combination of H2SO4 and HCO2H can be used as effective carbon monoxide gas surrogate. They then transformed the lactone moiety of 60 into furan of 61via a partial reduction of the lactone to lactol and dehydration. By taking advantage of the reactions of the electron-rich furan such as Friedel–Crafts alkylation/acylation and chiral amine-catalyzed radical cation cyclization, Trauner and co-workers were able to complete the total synthesis of the aforementioned natural molecules.
 | ||
| Fig. 14 The Trauner syntheses of amphilectolide, sandresolide B, caribenol A and B (2014 & 2017). | ||
Leucosceptroids and norleucosceptroids are complex natural products featuring a 5-6-5 tricyclic skeleton (Fig. 15). Magauer and Hugelshofer developed an elegant approach towards these natural products47,48 by employing a palladium-catalyzed carbonylative lactonization followed by a Hauser–Kraus annulation to rapidly build intermediate 69 with the 5-6-5 tricyclic core from vinyltriflate 66. Key intermediate 69 was then advanced to a few (nor)leucosceptroid natural products. Again, in this case, the combination of H2SO4 and HCO2H was used as effective carbon monoxide gas surrogate.
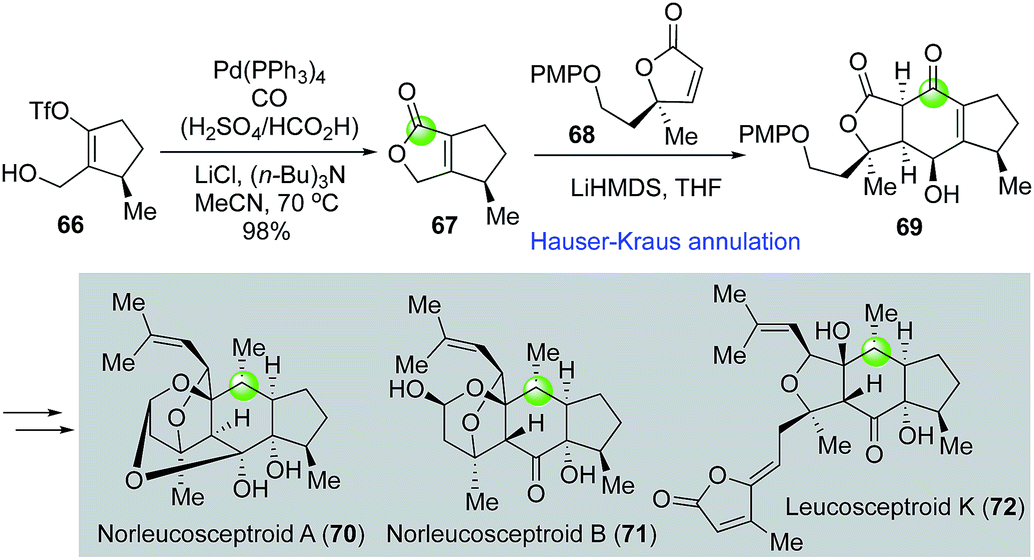 | ||
| Fig. 15 The Magauer synthesis of (nor)leucosceptroids (2014). | ||
In addition to the above highlighted cases, there are a significant amount of other natural product total syntheses involving the use of palladium-catalyzed carbonylative lactonization of the corresponding aryl/vinyl triflates or halides to build the key ring systems. Some of these natural products are listed in Fig. 16.49–53
 | ||
| Fig. 16 Selected natural products synthesized via palladium-catalyzed carbonylative lactonizations. | ||
While the palladium-catalyzed carbonylative lactonizations of aryl/vinyl triflates or halides with intramolecularly tethered alcohols are common ways to build 5- or 6-membered lactones, several other palladium-catalyzed carbonylations can be used as well. For example, Davies and co-workers used an interesting palladium-catalyzed decarboxylation/carbonylation of 1,3-dioxan-2-one 73 to synthesize γ-butyrolactone 74, which was then advanced to pilocarpine (75) and isopilocarpine (76) after PtO2-catalyzed hydrogenation (Fig. 17).54 Podlech and co-workers coupled a palladium-catalyzed carbonylation of propargylic mesylate 77 with a following iodolactonization of the resulting acid 78 to synthesize iodolactone 79, a key intermediate for their altenuic acid III synthesis (Fig. 18).55
 | ||
| Fig. 17 The Davies synthesis of pilocarpine (2009). | ||
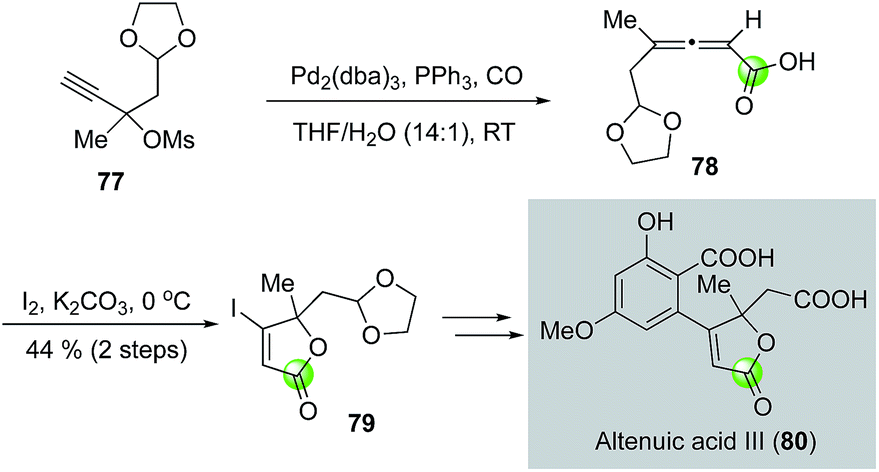 | ||
| Fig. 18 The Podlech synthesis of altenuic acid III (2013). | ||
Recently carbonylative C–H functionalizations have emerged as powerful methods for synthesizing carbonyl products.56–61 Direct C–H functionalization eliminates synthetic steps involved in pre-installation of the corresponding triflates, halides, or their equivalents. Carbonylative C–H functionalizations offer efficient ways to synthesize γ-butyrolactones and δ-valerolactones, but have not been frequently employed in natural product total synthesis. Hong and co-workers demonstrated the efficiency of such strategy in their total synthesis of frutinone A (Fig. 19), a potent inhibitor of the CYP1A2 enzyme. In their synthesis, an intermolecular oxidative C–H activation/Suzuki cross coupling was used to synthesize 83 from 81 and 82. After removal of the methyl group with BBr3, a phenol-directed C–H activation followed by carbonylative lactonization gave frutinone A (84) in only three steps.62
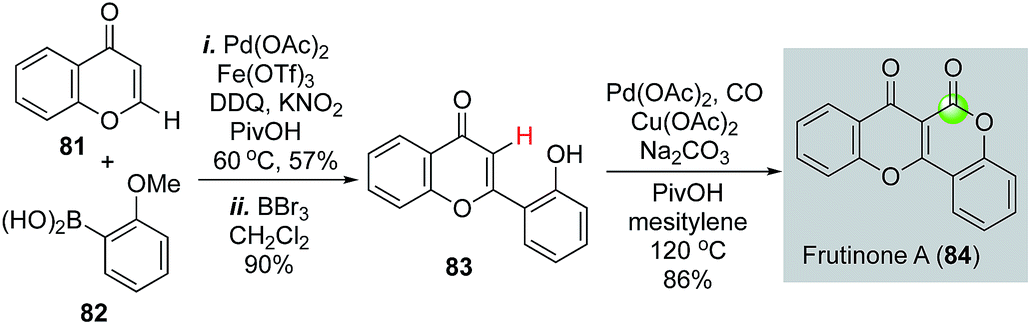 | ||
| Fig. 19 The Hong synthesis of frutinone A (2015). | ||
In addition to monocyclic lactones, palladium-catalyzed carbonylative lactonization can be used to synthesize spirocyclic ring systems. Dai, Waymouth and co-workers recently developed a tandem oxidative catalytic carbonylation reaction to convert hydroxycyclopropanols (cf.86, Fig. 20),63 which are readily available from the corresponding lactones via the Kulinkovich protocol, to oxaspirolactones (cf.89, 90) in one step. The monocationic dimeric [Pd(neoc)(OAc)]2(OTf)2 complex developed in the Waymouth group was approved to be an optimal catalyst for this transformation. The catalytic cycle was proposed to go through a palladium(II)-catalyzed β-carbon elimination to cleave the less substituted Walsh bond of cyclopropanol 86 and form palladium-homoenolate 87. Spiroketal formation followed by carbonylative lactonization gave oxaspirolactones 89 and 90 as a mixture of diastereomers and a palladium(0) catalyst. Oxidation of the latter with benzoquinone regenerated the palladium(II) catalyst and completed the catalytic cycle. The effectiveness of this method was demonstrated in their short total syntheses of α-levantanolide (89) and α-levantenolide (91).
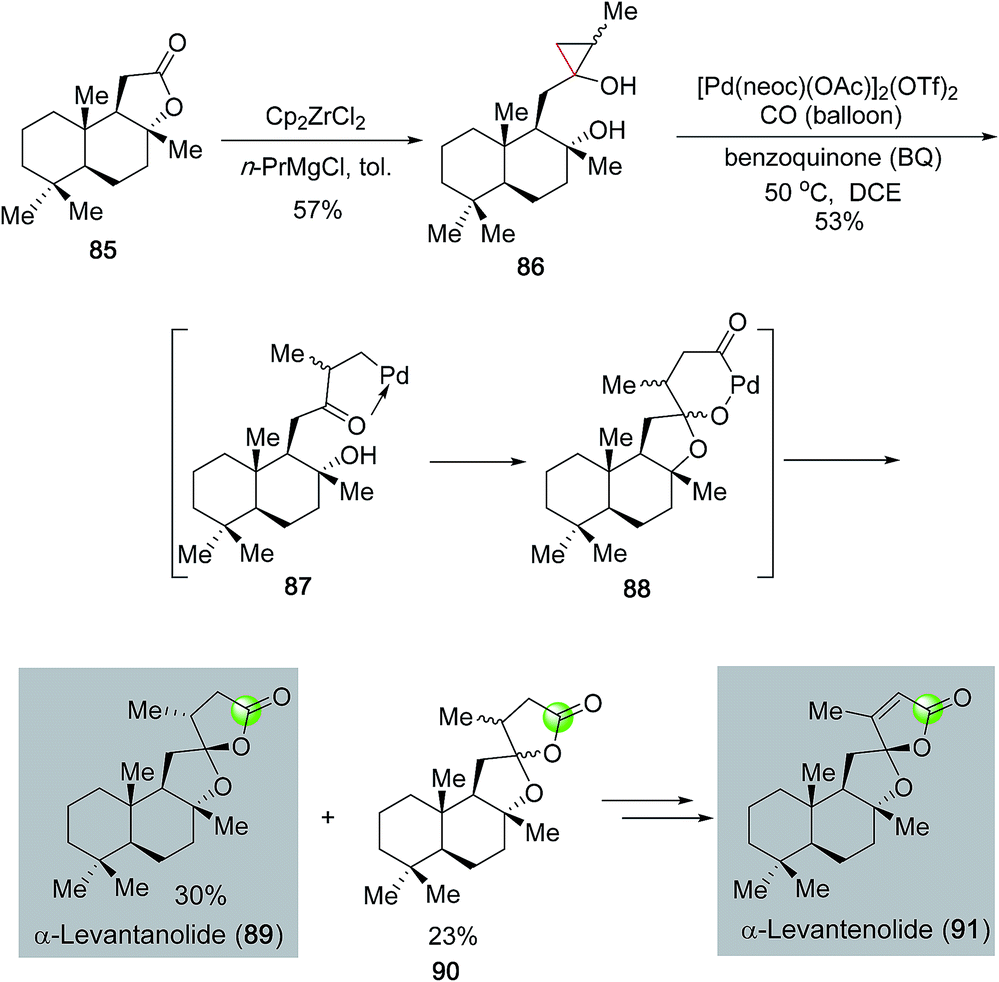 | ||
| Fig. 20 The Dai–Waymouth synthesis of levanta(e)nolide (2016). | ||
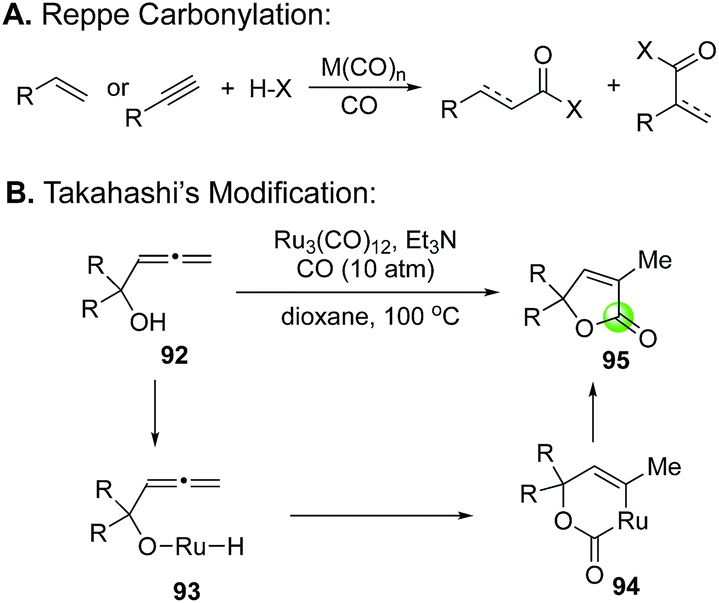 | ||
| Fig. 21 Reppe carbonylation and Takahashi's modification. | ||
The Takahashi's modified Reppe carbonylation provides a rapid approach to prepare γ-butenolides from allenyl alcohols and has been used in a few total syntheses of bioactive natural products (Fig. 22). For example, Bates et al. utilized it to synthesize bicyclic butenolide natural product mintlactone (98, Fig. 22A).66 In their case, the Feringa asymmetric conjugate addition was used to introduce the chiral center bearing the methyl group. This stereocenter then oriented the formation of the secondary alcohol center via a tin(II) chloride-mediated intramolecular 1,2-addition to provide a cis-cyclohexyl allene product 97, which was further advanced to mintlactone with the Takahashi's modified Reppe carbonylation. Hong and co-workers used the Takahashi's modified Reppe carbonylation to build the γ-butyrolactone of stemoamide (103, Fig. 22B).67 Their synthesis features an iron chloride-promoted Speckamp-type cyclization to provide a 3:1 mixture of allenes (101a and 101b) in 86% yield from starting material 99via acyliminium ion intermediate 100. The Speckamp-type cyclization favors product 101a with undesired stereochemistry at the carbon bearing the TBS ether. Interestingly, after releasing the secondary alcohol with tetrabutylammonium fluoride (TBAF), the Takahashi's modified Reppe carbonylation converted both stereoisomers to the same tricyclic γ-butenolide 102 in 81% yield. Apparently, a dynamic kinetic carbonylative lactonization process occurred under the reaction conditions. The undesired secondary alcohol derived from 101a was epimerized to the desired one via a ketone intermediate and Ru–H complex. Notably, the presence of carbon monoxide was critical for this epimerization as no epimerization was observed when carbon monoxide was excluded from the reaction system. With the γ-butenolide installed, a stereoselective nickel-hydride reduction completed the total synthesis of stemoamide 103. The third example is from the Jia group, who used the Takahashi's modified Reppe carbonylation to build the γ-butenolide moiety of rugulovasine A by converting allenyl alcohol 104 to 105 (Fig. 22C). A sequential removal of the two Boc-protecting groups completed their total synthesis of rugulovasine A.68
 | ||
| Fig. 22 The Takahashi modified Reppe carbonylation in total synthesis. | ||
 | ||
| Fig. 23 The Zhai syntheses of mintlactone, merrilactone A, and aplykurodinone-1. | ||
These hetero-Pauson–Khand reactions are highly effective in building γ-butyrolactones or γ-butenolides and have been applied in total synthesis. For example, Zhai and co-workers utilized the conditions developed by Adrio and Carretero to convert yne-aldehyde 109 derived from (−)-citronellol to mintlactone (ent-98) in just one step (Fig. 23A).74 A couple of years later, the same group utilized this hetero-Pauson–Khand strategy to complete a total synthesis of merrilactone A (Fig. 23B).75 In this case, they were able to convert yne-aldehyde 110 to tricyclic γ-butenolide in 69% by using the same Mo(CO)3(DMF)3 catalyst. A following vinylogous Mukaiyama–Michael addition to methylvinylketone gave rise to ketone 112, which then underwent SmI2-mediated reductive carbonyl-alkene cyclization to provide tetracyclic intermediate 113. From 113, merrilactone A total synthesis was achieved in five steps featuring a homo-Payne rearrangement to build the oxetane ring. In 2017, the hetero-Pauson–Khand strategy was utilized again by Zhai and co-workers for their total synthesis of aplykurodinone-1 (Fig. 23C).76 A Mo(CO)6-catalyzed hetero-Pauson–Khand reaction enabled a rapid construction of tricyclic γ-butenolide 116 from yne-aldehyde 115. The former served as a key intermediate in their aplykurodinone-1 total synthesis.
In 2015, Shenvi and co-workers reported a remarkable eight-step gram-scale synthesis of jiadifenolide (Fig. 24).77 Their synthesis features a hetero-Pauson–Khand reaction to convert yne-aldehyde 118 to bicyclic γ-butenolide 119 in the presence of Mo(CO)6 and tetra-n-butylammonium bromide. Aldehyde 118 was derived from (+)-citronellal in two steps. They then used a highly convergent and efficient tandem intermolecular Michael addition and intramolecular Michael addition to unite 119 and 120 and form ketolactone 121, which contains the entire carbon skeleton of jiadifenolide, in a single step in 70% yield with 20:1 diastereoselectivity. After an oxidative α-hydroxylation with m-CPBA and stereoselective ketone reduction, advanced intermediate 122 was obtained, which was then converted to jiadifenolide via a sequence of α-bromolactone formation with LDA/CBr4 and α-hydroxylation with the Davis' oxaziridine. Later in 2017, Shenvi and co-workers successfully converted ketolactone 121 to 11-O-debenzoyltashironin (126, Fig. 24).78 This synthesis involves a sequence of α-methylation of ketolactone, hydrolysis of the lactone, and acid-promoted decarboxylation to remove the extra carboxylate and convert 121 to 124. The latter was then advanced to 125 with a cyclopentene ring. Mukaiyama hydration via a hydrogen atom transfer process afforded an intermediate containing a trans-hydrindane ring system with an angular hydroxyl group, which was then rearranged to 11-O-debenzoyltashironin with the addition of p-toluenesulfonic acid.
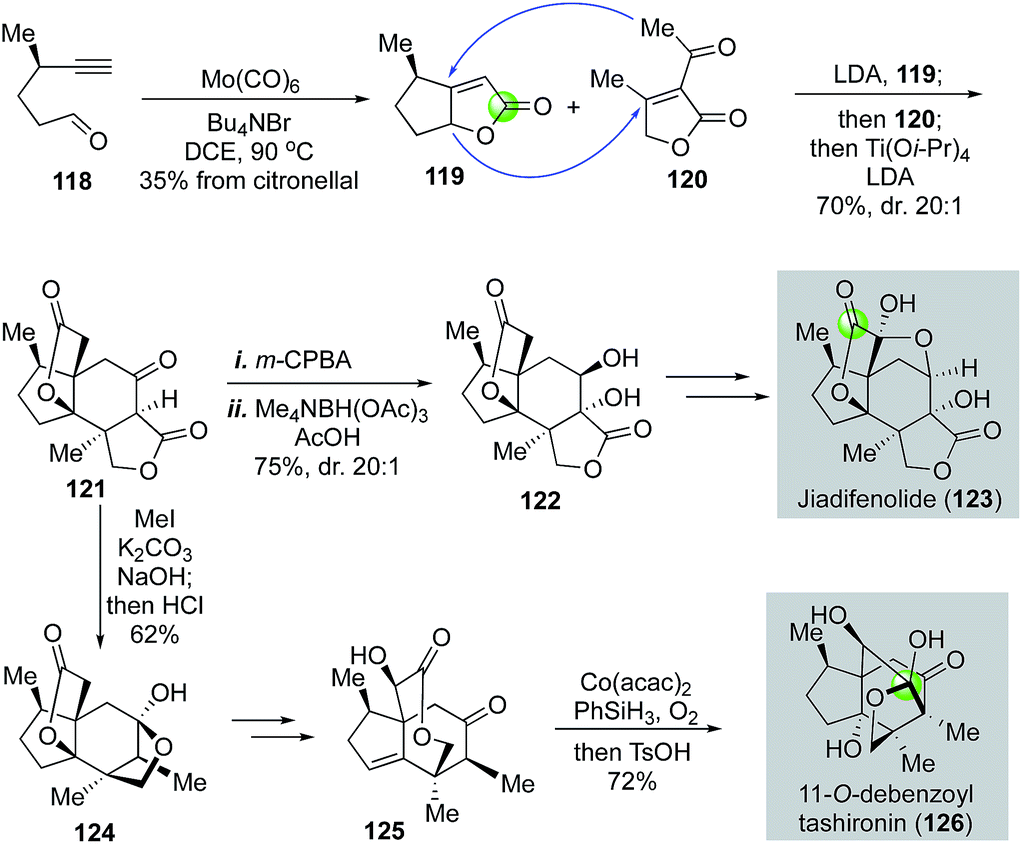 | ||
| Fig. 24 The Shenvi syntheses of Jiadifenolide (2015) and 11-O-debenzoyltashironin (2017). | ||
2.3. THP and THF ring formation
 | ||
| Fig. 25 The Semmelhack reaction. | ||
In 2000, Leighton and co-workers developed an elegant total synthesis of leucascandrolide A,83 a potent anticancer and antifungal natural product isolated from the sponge Leucascandra caveolata by Pietra et al. Their synthesis features three carbonylation reactions including one Semmelhack reaction to build the THP ring. As highlighted in Fig. 26, the Leighton synthesis commenced with known alcohol 132, which underwent Yb(OTf)3-catalyzed oxymercuration with HgClOAc to provide organomercury chloride 133. Then a rhodium-catalyzed formylation was utilized to convert 133 to aldehyde 134, which was further advanced to olefin 135 by a Brown asymmetric crotylation. Hydroformylation catalyzed by Rh(acac)(CO)2 followed by hemiacetal formation, the second carbonylation reaction, elaborated 135 to 136 in 89% as a 1:1 mixture of diastereomers. The latter was advanced to 137 in five steps including two allylation reactions to set up the stage for the Semmelhack reaction, which successfully converted 137 to THP ester 138, a key intermediate for the Leighton total synthesis of leucascandrolide A. Notably, a similar rhodium-catalyzed hydroformylation/hemiacetal formation strategy was employed by Floreancig and co-workers to build the same THP ring in their leucascandrolide A macrolactone synthesis.84
 | ||
| Fig. 26 The Leighton synthesis of leucascandrolide A (2000). | ||
In their synthetic studies toward phorboxazole A, White and co-workers utilized two Semmelhack reactions to install two of the four THP rings of phorboxazole A (Fig. 27).85 The first Semmelhack reaction converted 140 to 141 in 86% yield as a single stereoisomer. THP ester 141 was then advanced to 142 for the second Semmelhack reaction to obtain 143, the C9–C32 segment of phorboxazole A (144).
 | ||
| Fig. 27 The White synthetic study toward phorboxazole A (2001). | ||
In 2008, MacMillan and co-workers reported a total synthesis of callipeltoside C, which also led to its structural revision (Fig. 28).86 Similar to leucascandrolide A and phorboxazole A, callipeltoside C and related analogs feature a THP-bridged macrolactone ring system. However, unlike the other two, callipeltoside C contains a hemiacetal moiety in the THP ring. The MacMillan synthesis initiated with a proline-catalyzed double diastereo-differentiating aldol reaction between aldehydes 145 and 146 to provide aldehyde 147 in high stereoselectivity (12:1 anti/syn and >19:1 Felkin/anti-Felkin). Subsequent propargyl zinc addition via a Felkin-selective chelation model converted 147 to 148. They then used the Marshall modified Semmelhack protocol with a terminal alkyne87 to form THP ester 150 equipped with an acetal in one step in 75% yield. The use of an alkyne substrate offers a direct solution for the required acetal moiety and the process was proposed to go through intermediate 149. After 150 was advanced to 151, a chelation-controlled vinyl Grignard addition using 152 gave 153 in 96% yield with 16:1 diastereoselectivity. To close the macrolactone ring, MacMillan and co-workers utilized the Yamaguchi macrolactonization. The aglycone product was then converted to callipeltoside C using the glycosylation procedure developed by Tietze and co-workers.88
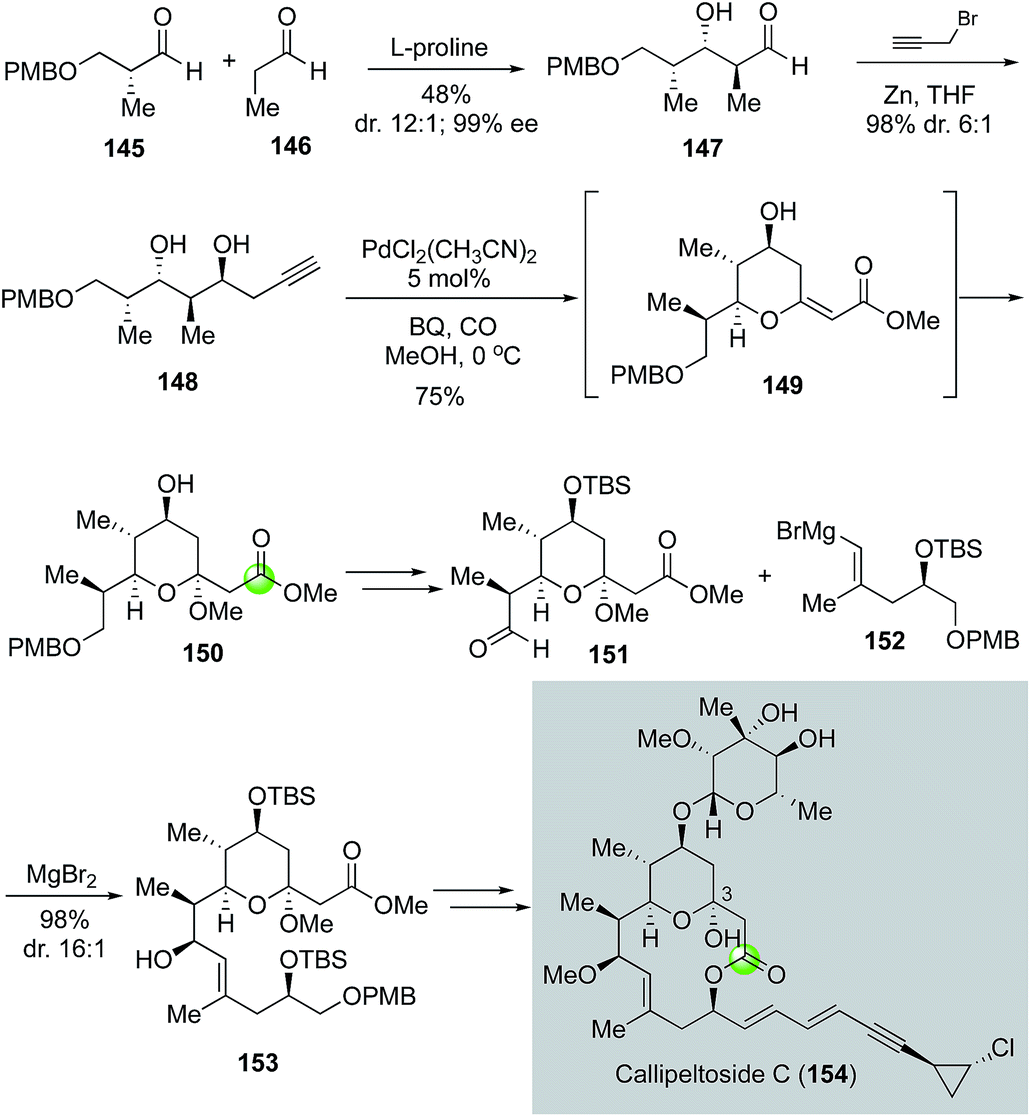 | ||
| Fig. 28 The MacMillan synthesis of callipeltoside C (2008). | ||
Another application of the Semmelhack reaction in making THP-containing bridged macrolactone came from She and co-workers (Fig. 29).89 In their total synthesis of potent anticancer natural product neopeltolide, the Semmelhack reaction was used to convert C2-symmetric diol 156 to THP ester 157 in 83% yield. Diol 156 was synthesized from 1,3-propanediol using the Krische's iridium-catalyzed asymmetric carbonyl allylation method.90,91 After 157 was converted to diene 158, ring closing metathesis with the Hoveyda–Grubbs 2nd generation catalyst was employed to form the macrocycle followed by a macrocyclic stereocontrolled hydrogenation to introduce the C9 stereocenter and removal of the benzyl group. The resulting alcohol 159 was then converted to neopeltolide (160) by using a Mitsunobu reaction to install the side chain.
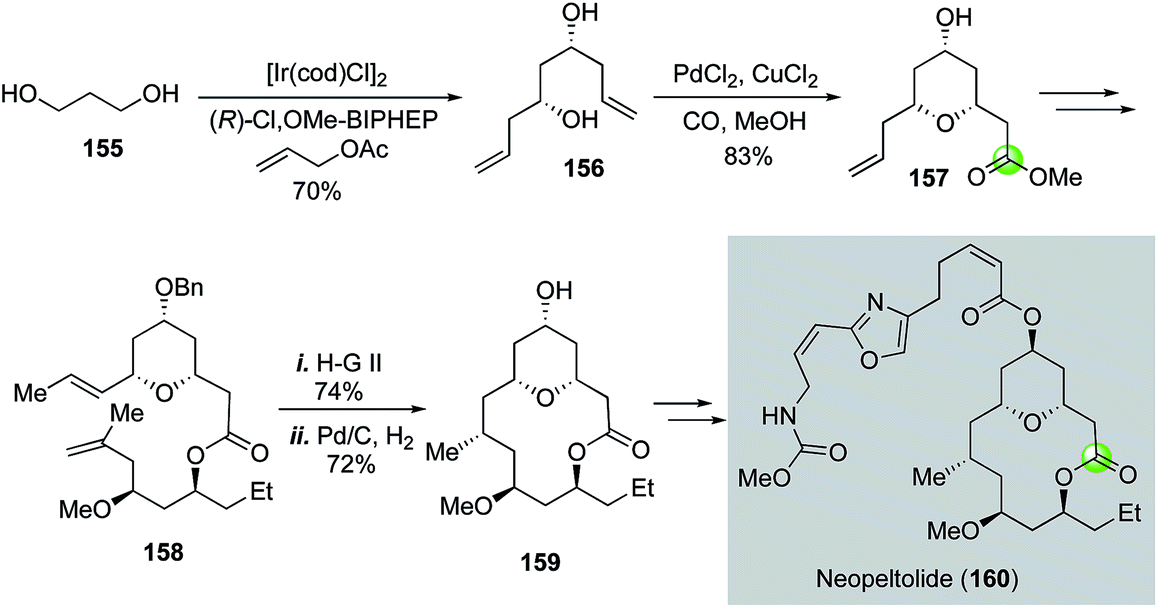 | ||
| Fig. 29 The She synthesis of neopeltolide (2011). | ||
In addition to alcohols, phenols can be used as nucleophiles for the alkoxypalladation/carbonylation process to synthesize chromane structures. Tietze and co-workers utilized such a strategy toward the total synthesis of 4-dehydroxydiversonol (163, Fig. 30).92 In this case, they have developed an enantioselective version of the Semmelhack reaction by using a combination of Pd(tfa)2 and chiral (S,S)-Bn-BOXAX ligand 164 to convert phenol 161 to chromane 162 in 80% yield and 96% ee. Compound 162 was then advanced to 4-dehydroxydiversonol in eight steps.
 | ||
| Fig. 30 The Tietze synthesis of 4-dehydroxydiversonol (2008). | ||
The Semmelhack annulation (Fig. 25, 127 → 131) is an efficient process to build fused bicyclic lactones, which are frequently found in many bioactive natural products. Synthetic chemists have been harnessing its power in making these natural products. For example, Kitching and co-workers have utilized the Semmelhack annulation to install the 5,5-cis-fused bicyclic lactone moiety of plakortone D (Fig. 31).93 In their case, a 63:37 ratio of 167 and 166 were obtained from a 60:40 mixture of diastereomers of 165 by using the standard PdCl2/CuCl2 catalyst system under carbon monoxide atmosphere in AcOH with NaOAc as base to neutralize the in situ generated HCl.
 | ||
| Fig. 31 The Kitching synthesis of plakortone D (2002). | ||
In 2008, Yang and co-workers modified the Semmelhack annulation conditions by introducing tetramethylthiourea (TMTU) as ligand, which was proposed to facilitate carbon monoxide migratory insertion.94 In addition, propylene oxide was used to quench the in situ generated HCl. Under this modified reaction conditions, they were able to convert diol 169 to bicyclic lactone 170 in 88% yield (Fig. 32).95 The latter was then advanced to crisamicin A (171) via a palladium–thiourea pincer complex-catalyzed homodimerization of the corresponding aryl boronic ester.
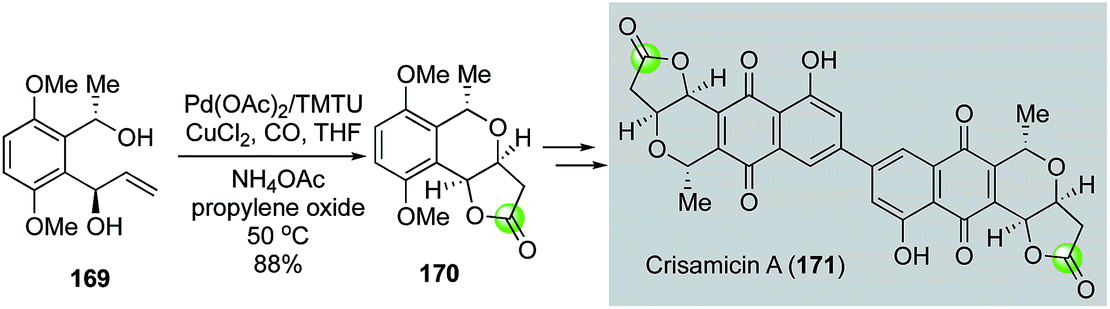 | ||
| Fig. 32 The Yang synthesis of crisamicin A (2008). | ||
Later in 2011, Tang and co-workers used the Semmelhack annulation as a key step to synthesize kumausallene (175), a nonisoprenoid sesquiterpene containing a chiral bromoallene moiety and dioxabicyclo[3.3.0]octane core (Fig. 33).96 In their case, C2-symmetric diol 172, available from acetylacetone in three steps, was successfully converted to bicyclic lactone 173 using the standard Semmelhack carbonylative annulation conditions. After advancing 173 to enyne 174, they used a biomimetic 1,4-syn-bromoetherification promoted by a combination of N-bromosuccinimide (NBS) and DMF to form the bromoallene and complete the total synthesis of kumausallene. The use of DMF is critical for the success of the bromoetherification. Notably, McErlean et al. also used a Semmelhack annulation strategy to complete a formal synthesis of kumausallene.97
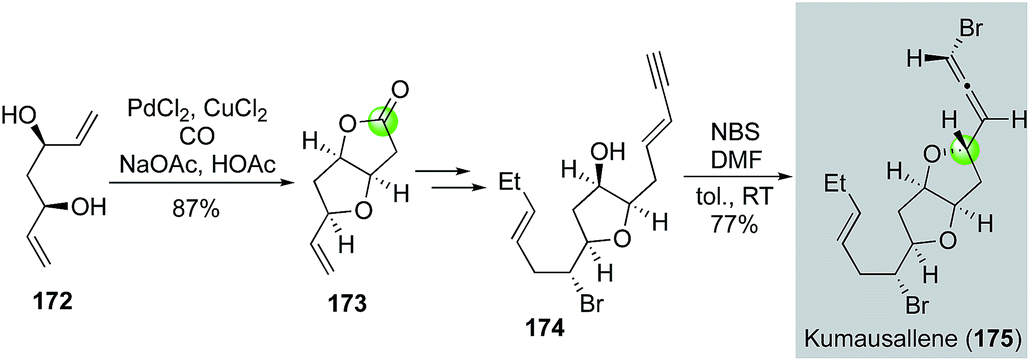 | ||
| Fig. 33 The Tang synthesis of kumausallene (2011). | ||
In another case from the Yang group, the Semmelhack carbonylative annulation was used at a late stage on a complex polycyclic intermediate to enable the first total synthesis of nortriterpenoid schindilactone A isolated by Sun and co-workers (Fig. 34).98 Their synthesis features a ring closing metathesis of diene 176 with the Grubbs 2nd generation catalyst to form the 8-membered carbocycle of 177 and a Co2(CO)8/TMTU-promoted intramolecular Pauson–Khand reaction to convert 178 to cyclopentenone 179. The use of MgBr2 is important for the ring closing metathesis because it enabled an in situ epimerization of the hemiketal carbon and led to the formation of 177 as a single diastereomer from a mixture of 176. The Pauson–Khand product 179 was then transformed to 180, the precursor for the Semmelhack annulation. In this case, Yang et al. developed a unique C2-symmetric thiourea ligand 182 to facilitate the key carbonylative annulation process99 and desired product 181 was obtained in 78% yield. The bulky C2-symmetric thiourea ligand was proved to be more effective than TMTU for promoting the carbonylation process. The complexity of the substrate and the product highlights the mildness and functional group tolerability of their modified reaction conditions. Compound 181 was then advanced to schindilactone A in six steps.
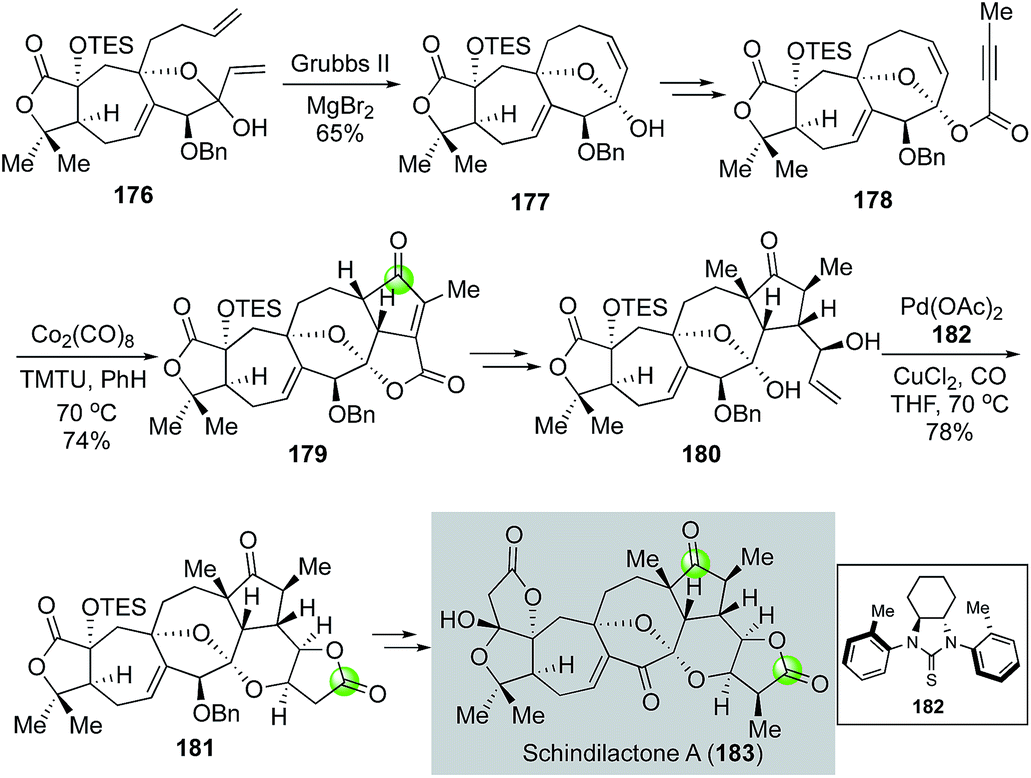 | ||
| Fig. 34 The Yang synthesis of schindilactone (2011). | ||
Yang's modified alkoxycarbonylative annulation reaction conditions were also utilized in the total syntheses of diterpenoid pallambins by both the Wong group (Fig. 35)100 and the Carreira group (Fig. 36).101 The Wong's synthesis of pallambins C and D started with the chiral pool molecule Wieland–Miescher ketone 184. After converting 184 to 185, they developed an impressive tandem Grob fragmentation-intramolecular aldol cyclization to obtain bicyclo[3.2.1] enone 187 in 61% yield via intermediate 186. Enone 187 was then elaborated to 188 for the Semmelhack carbonylative annulation. With the Yang's modified conditions, 188 was transformed to 189 in 78% yield. The total syntheses of pallambins C and D were completed in three more steps: olefination, removal of TBS group, and oxidation of the resulting allylic alcohol.
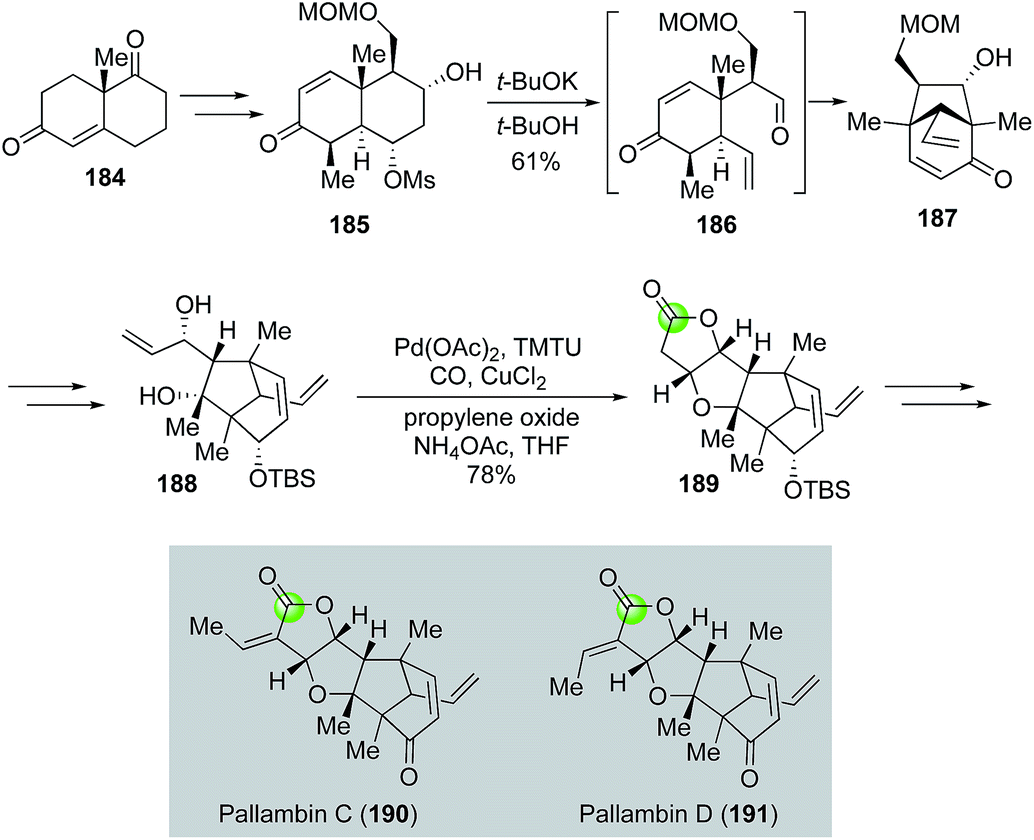 | ||
| Fig. 35 The Wong synthesis of pallambins C and D (2012). | ||
 | ||
| Fig. 36 The Carreira synthesis of pallambins A and B (2015). | ||
Structurally, in addition to the same 5-5-5-fused tricyclic ring system of pallambins C and D, pallambins A and B contain a highly congested tetracyclo[4.4.03,5.02,8]decane core (Fig. 36). The Carreira synthesis of pallambins A and B utilized a Diels–Alder reaction between methylacrylate and pentafulvene 193 (generated in situ from 192) to form 194 and a Simmons–Smith cyclopropanation to install the challenging cyclopropane motif. Product 195 was obtained in 85% yield with greater than 20:1 diastereoselectivity. After 195 was elaborated to methylketone 196, a sequence of α-diazoketone formation and rhodium-catalyzed intramolecular C–H activation provided 197 in 76% yield, which was then transformed to 198via vinyltriflate formation followed by Negishi cross coupling with Zn(Me)2 to introduce the methyl group. The resulting trisubstituted olefin then underwent [3 + 2] cycloaddition with bromonitrile oxide 1,3-dipole derived from the treatment of dibromoformaldoxime with KHCO3 to afford isoxazoline 199 in 91% yield. After converting isoxazoline 199 to aldehyde 200 then to allylic alcohol 201, Carreira and co-workers used the Yang's modified alkoxycarbonylative annulation procedure to form lactone 202, which was then converted to pallambins A and B via a sequential aldol reaction and elimination.
 | ||
| Fig. 37 The Aggarwal synthesis of avenaciolide (2004). | ||
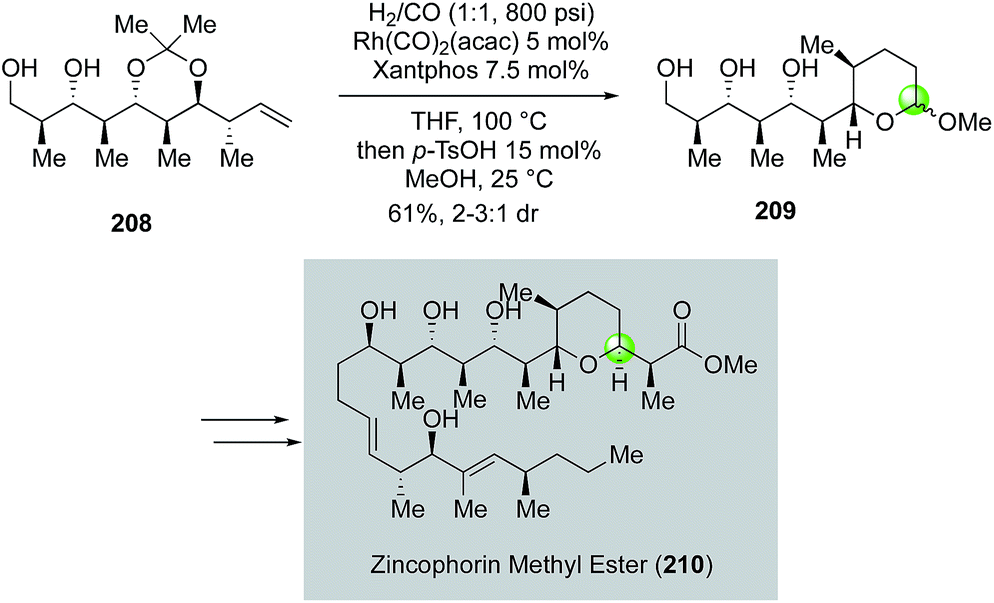 | ||
| Fig. 38 The Krische synthesis of zincophorin methyl ester (2015). | ||
2.4. Macrolactonization
Over the years, most of the macrolide syntheses have relied on various macrolactonizations of the corresponding seco acids promoted by more than stoichiometric amounts of activating reagents. Catalytic macrolactonization methods have been significantly underexplored.109 Recently, palladium-catalyzed carbonylative macrolactonization reactions have emerged as an efficient strategy for building macrolactones. This strategy offers the advantage of not pre-installing the carboxylate group. A highly reactive acylpalladium species is generated in situ; therefore, the use of activating reagents can be avoided. A few examples are highlighted below to showcase the synthetic efficiency of palladium-catalyzed carbonylative macrolactonization in total synthesis of complex natural products.In 2003, Takahashi and co-workers reported a combinatorial synthesis of a macrosphelide library.110 Their strategy features two palladium-catalyzed carbonylations on solid polymer support to build the two α,β-unsaturated carboxylates embedded in the 16-membered macrocycle (Fig. 39). They first used a three-component intermolecular carbonylative Heck reaction to synthesize ester 213 from vinyliodide 211, carbon monoxide, and alcohol 212 bearing a vinylbromide moiety. They then used an intramolecular carbonylative macrolactonization reaction to convert vinylbromide 214 with a remotely tethered secondary alcohol to macrosphelide after acid-cleavage of the polymer support. This strategy enabled the production of a 122-member macrosphelide library for focused anti-cancer study.
 | ||
| Fig. 39 The Takahashi synthesis of macrosphelide (2003). | ||
In 2015, Menche and Schmalzbauer reported a concise synthesis of the tricyclic core of salimabromide (Fig. 40).111 Salimabromide has shown antibiotic activity, but its natural scarcity has hampered further biological study. Menche and Schmalzbauer proposed to synthesize salimabromide from tricyclic intermediate 217, presumably via a sequence of transannular conjugate addition followed by an elimination. While this transformation hasn't been realized so far, Menche and Schmalzbauer have developed an efficient strategy to synthesize 217 and their synthesis features a palladium-catalyzed chemoselective carbonylative lactonization of 216 to build the desired 8-membered lactone of 217 in 31% yield.
 | ||
| Fig. 40 The Menche synthesis of the salimabromide tricyclic core (2015). | ||
Inspired by the original Semmelhack reaction, Dai and co-workers developed a palladium-catalyzed tandem alkoxycarbonylative macrolactonization to build THP/THF-containing bridged macrolactones in one step from relatively simple alkene diols (Fig. 41). In their case, the in situ generated acylpalladium species was trapped by a remotely tethered alcohol to form a macrolactone. They further demonstrated the application of this efficient method in their synthesis of 9-demethylneopeltolide.112 As shown in Fig. 41, alkene diol 219 smoothly underwent the palladium-catalyzed tandem alkoxycarbonylative macrolactonization under the mild reaction conditions to form bridged macrolactone 222 in 58% yield as a single isomer. Notably, the ketal group is critical for the observed high diastereoselectivity. Compound 222 was then advanced to 9-demethylneopeltolide 223 in three steps, namely, removal of the ketal protecting group, reduction of the resulting ketone, and Mitsunobu reaction to introduce the side chain.
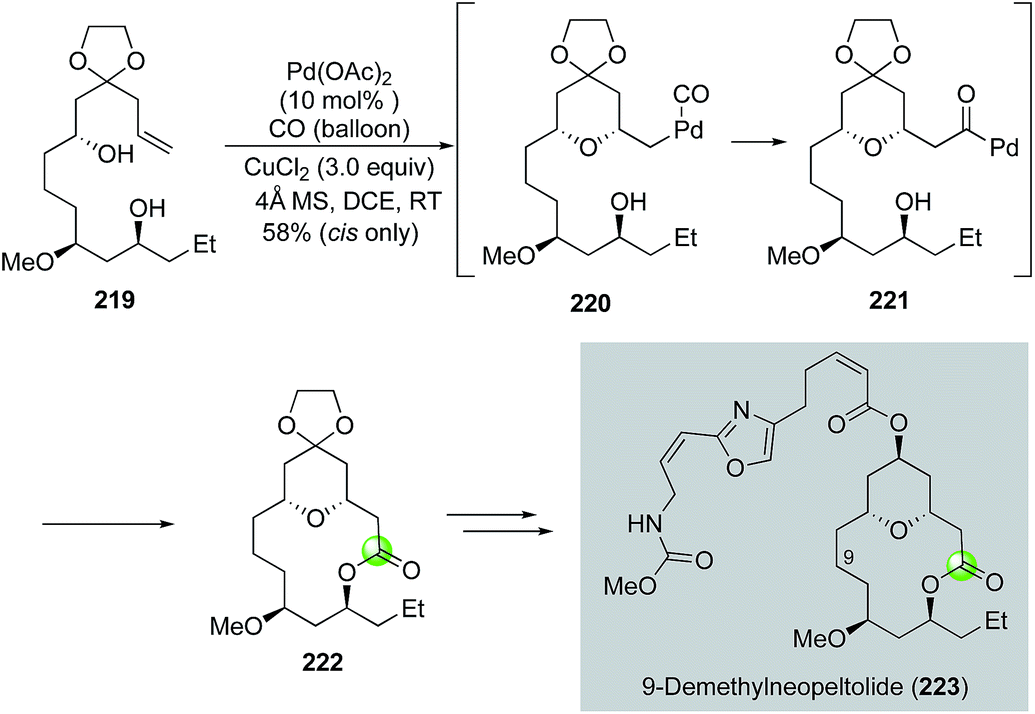 | ||
| Fig. 41 The Dai synthesis of 9-demethylneopeltolide (2014). | ||
In 2018, Harran and co-workers employed Dai's cascade variant of the Semmelhack reaction to build the key macrolactone of anticancer natural product callyspongiolide.113 As shown in Fig. 42, readily available building blocks 224 and 225 were joined together by an iridium-catalyzed diastereoselective fragment coupling developed by Krische's group114,115 to provide homoallylic alcohol 227 as a single diastereomer. Compound 227 was then advanced to ketal 228 in five steps. Harran et al. then used an oxidative fragmentation developed by Schreiber and co-workers116 to convert 228 to 230. This process initiated with perhemiketal 229 derived from treating 228 with acid and hydrogen peroxide (H2O2). Perhemiketal 229 then underwent a smooth fragmentation in MeOH upon the treatment with Cu(OAc)2 and FeSO4 to afford 230 for the palladium-catalyzed tandem alkoxycarbonylative macrolactonization, which occurred successfully at gram scale to deliver bridged macrolactone 231 as a mixture of diastereomers. The newly formed stereocenter was inconsequential because it was eliminated in the next step to form macrolactone 232, which was then converted to callyspongiolide uneventfully via key steps involving a photochemical double bond isomerization, Wittig reaction and Sonogashira coupling to append the ene-yne-ene moiety.
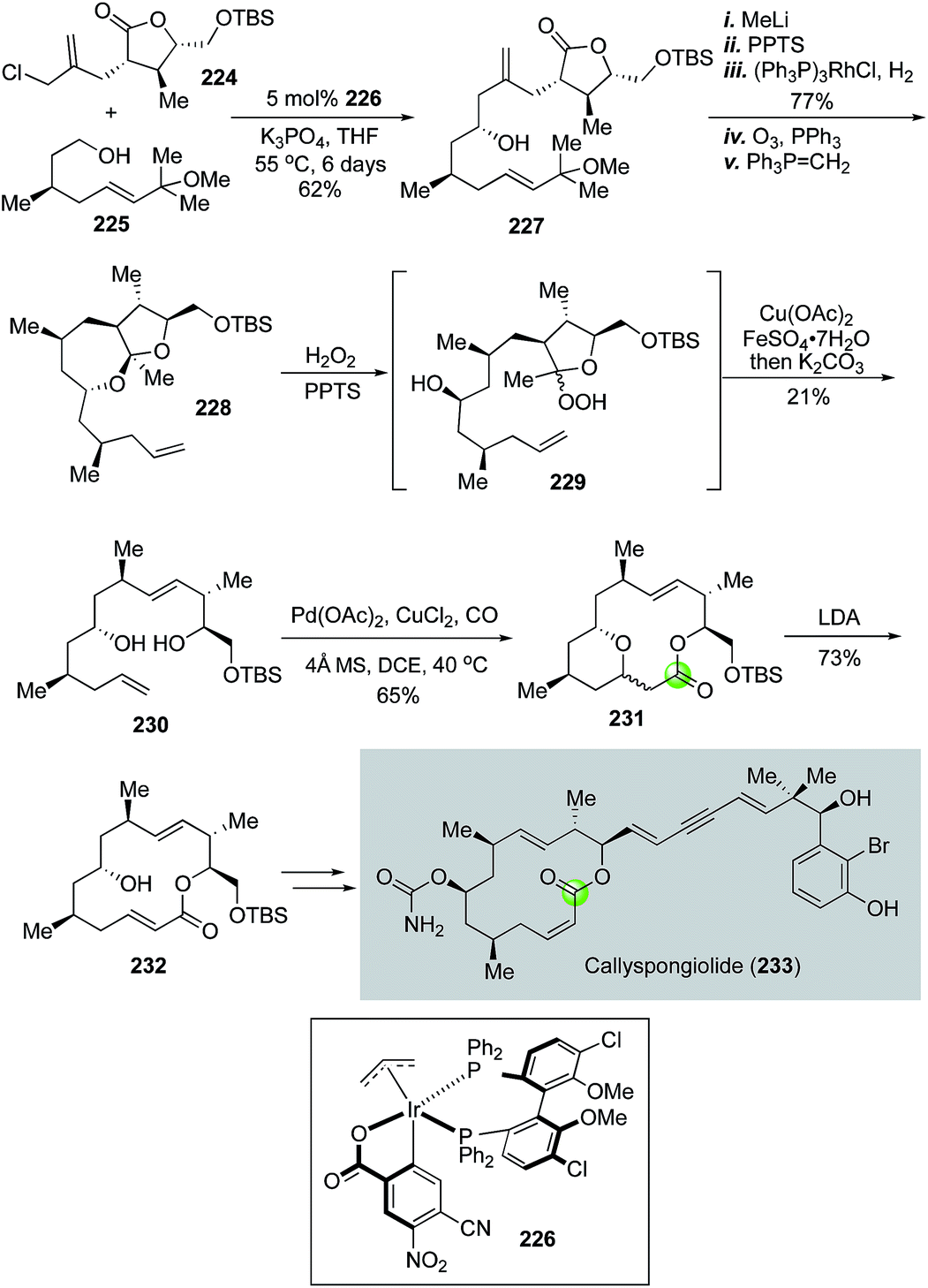 | ||
| Fig. 42 The Harran synthesis of callyspongiolide (2018). | ||
In their continued interest in developing tandem carbonylation reactions for building spirocycles and macrocycles by using carbon monoxide as a one-carbon linchpin, Dai and co-workers completed a total synthesis of spinosyn A with a palladium-catalyzed carbonylative Heck macrolactonization to form the 5,12-fused ring system embedded in its tetracyclic core.117 As highlighted in Fig. 43, their spinosyn A total synthesis features a gold-catalyzed propargylic acetate rearrangement to convert 234, derived from a Corey–Fuchs 1,2-addition of the corresponding readily available dibromoalkene and trans-5,6-fused aldehyde, to α-iodoenone 235 in 58% yield in high stereoselectivity. Additionally, NIS-mediated selenide oxidative elimination and TBS-deprotection took place under the same reaction conditions to release the required terminal olefin and secondary alcohol for the carbonylative Heck macrolactonization, which occurred smoothly with the combination of Pd(OAc)2 and P(2-furyl)3 under 3 atm of carbon monoxide in toluene. Desired tetracyclic product 238 was obtained in 43% yield presumably via alkylpalladium intermediate 236 and acylpalladium intermediate 237. The α-tri-O-methyl-L-rhamnose moiety was then installed using the Schmidt glycosylation and the challenging β-D-forosamine moiety was installed using the Yu glycosylation protocol.118 For the latter, the diastereoselectivity was about 1:1 and needed to be improved probably by developing new glycosylation chemistries.
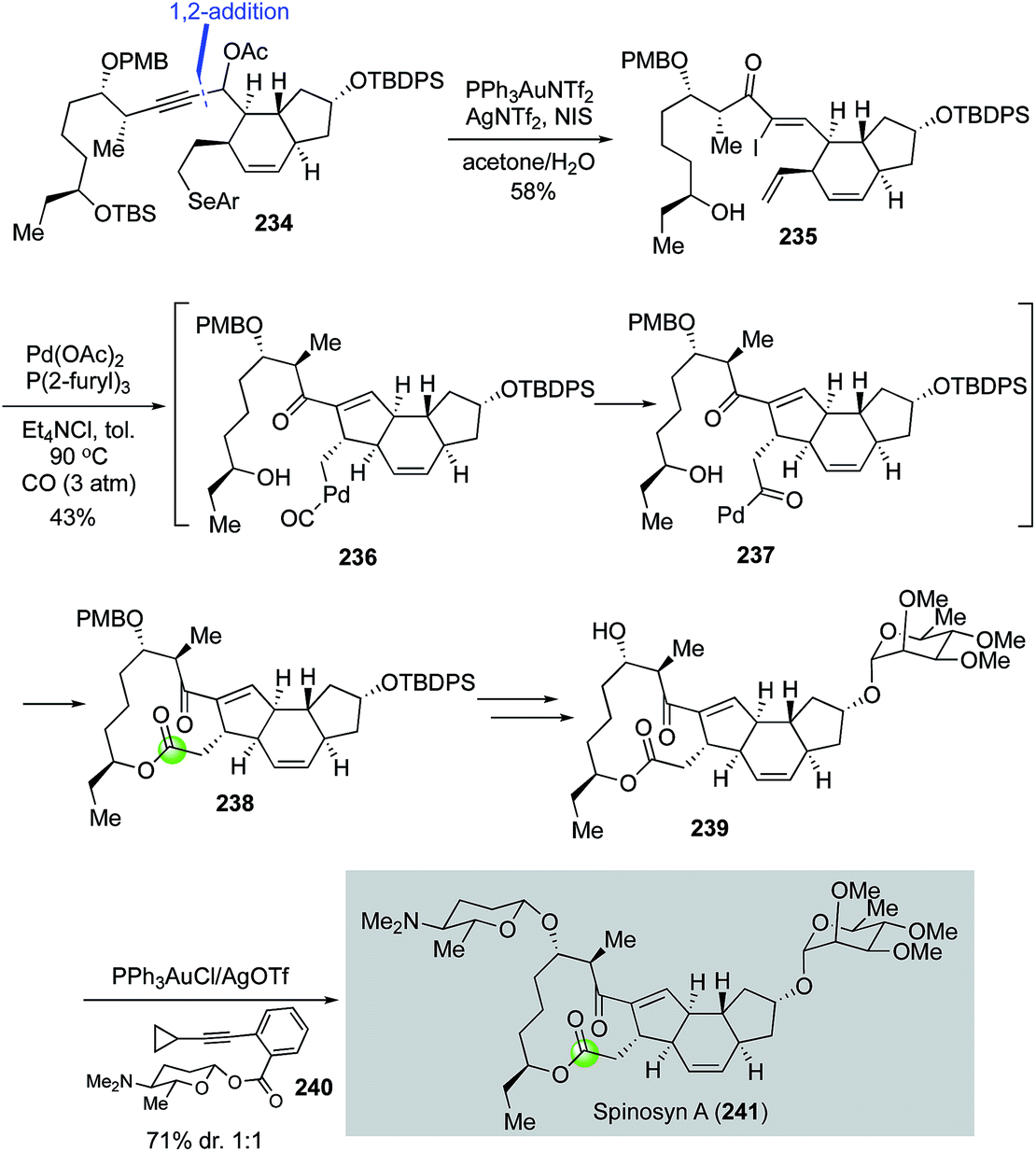 | ||
| Fig. 43 The Dai synthesis of spinosyn A (2016). | ||
In 2012, Burke and co-workers coupled an asymmetric hydroformylation and intramolecular Wittig olefination to synthesize the macrolactone ring of patulolide C (Fig. 44).119 Their synthesis initiated with (2R)-8-nonyn-2-ol 242 which underwent a hydroacetoxylation with [RhCl(COD)]2 as catalyst and 243 as ligand to yield Z-enol acetate 244 in 82% via a remarkable anti-Markovnikov process. Under the conditions of Rh(acac)(CO)2 and (SSS)-BDP ligand at 50 °C under 150 psi of CO/H2, enol acetate 244 was transformed to aldehyde 245 with 100% conversion and high diastereoselectivity and regiocontrol. Aldehyde 245, without further purification, was treated with the Bestmann ylide 246 in refluxing toluene. A remarkable tandem ester formation and intramolecular Wittig olefination took place to afford 12-membered lactone 247 in 62%. After an enzymatic acetate hydrolysis with Pseudomonas florescens lipase PFL under neutral buffer conditions, patulolide C was produced in 99% yield and 96.6:3.4 diastereoselectivity.
 | ||
| Fig. 44 The Burke synthesis of patulolide C (2012). | ||
3. Carbonylative N-heterocycle synthesis
Many carbonylative N-heterocycle synthesis strategies resemble the ones of the carbonylative O-heterocycle syntheses, but amine nucleophiles are used to trap the acyl-metallo species instead of the corresponding alcohols. The following examples highlight different N-heterocyclic natural product synthesis via various carbonylation reactions.3.1. Lactam formation
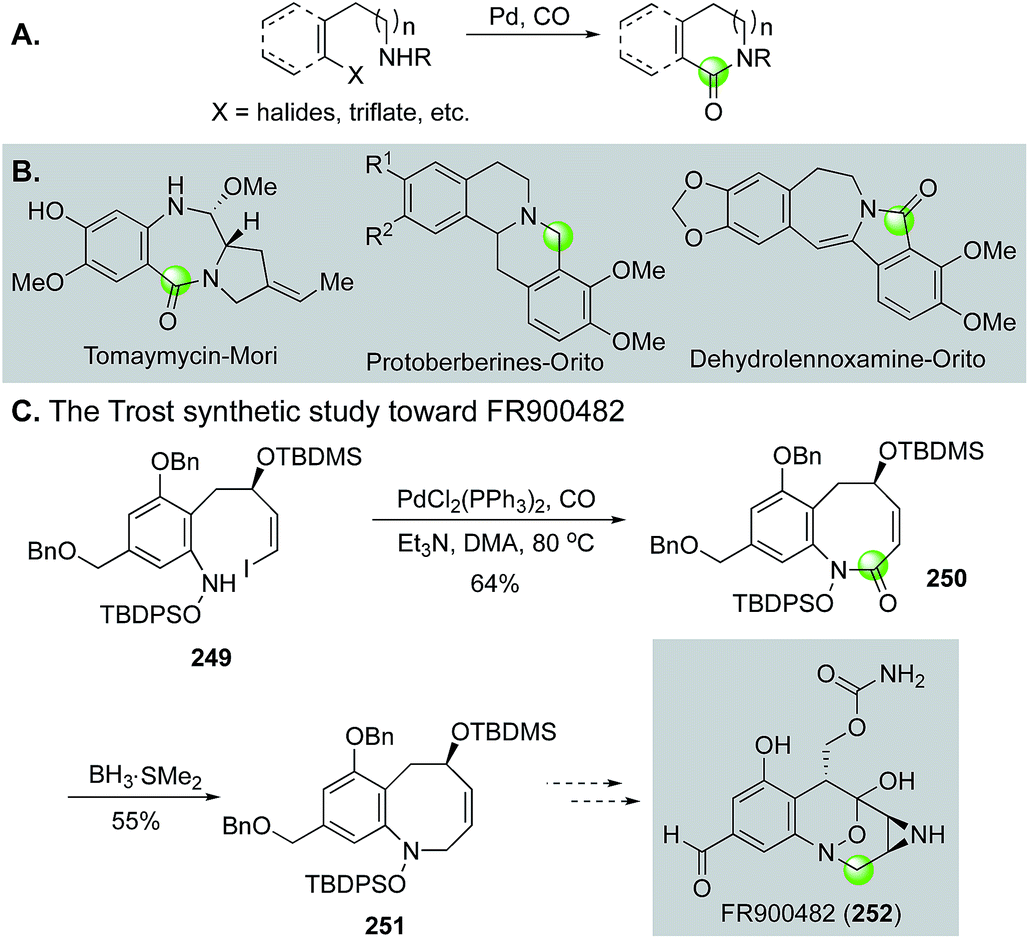 | ||
| Fig. 45 Carbonylative lactam formation in total synthesis. | ||
Additionally, in an effort to circumvent the entropic and enthalpic hurdles associated with medium-sized ring formation, Trost and Ameriks employed a Pd-catalyzed carbonylation strategy in the formation of the benzazocine core of FR900482 (252, Fig. 45C).123 Specifically, using a carbonylation strategy benefits from the absence of transannular interactions due to the increased unsaturation in the forming ring. After optimization, they found that conducting the carbonylative lactamization at 80 °C in N,N-dimethylacetamide gave 250 in 64% yield from tethered vinyl iodide-hydroxylamine intermediate 249. Reduction of the lactam gave 251 in 55% yield.
α-Cyclopiazonic acid (α-CPA, 259) is a prenylated indole alkaloid with Ca2+-dependent ATPase inhibiting activity. Inspired by the biosynthetic pathway of α-CPA, Aggarwal and co-workers developed an efficient and convergent approach to synthesize α-CPA and its analog iso-α-CPA (260) in an enantioselective manner (Fig. 46).124 The Aggarwal synthesis features an aziridination of imine 253 with chiral ylide 254 to afford aziridine 255 in 61% yield with 9:1 trans/cis selectivity and 98:2 enantioselectivity for the trans isomer. The crude aziridination product was then treated with TfOH to trigger an intramolecular [3 + 2] cycloaddition and pyrrolidine 256 was produced after removal of the nosyl protecting group. A palladium-catalyzed carbonylative lactam formation was used to convert pyrrolidine 256 to lactam 257. Due to the severe angle strain of the newly formed 5,5-fused bicyclic ring system, an in situ N–O bond reductive cleavage occurred under the carbonylation reaction conditions to open the isoxazole ring and release product 258 in 80% yield. Compound 258 was then converted to α-CPA and iso-α-CPA with basic conditions in MeOH-THF-H2O to remove the tosyl group and hydrolyze the vinylogous amide.
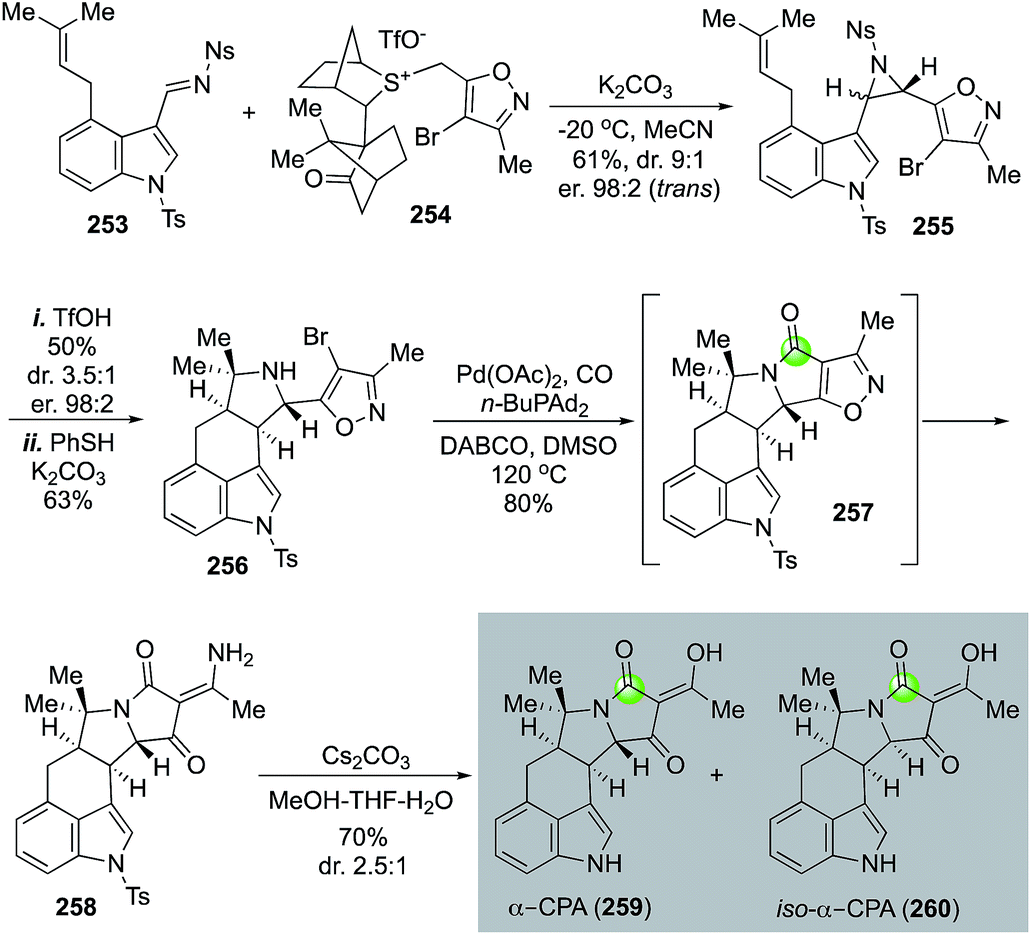 | ||
| Fig. 46 The Aggarwal synthesis of cyclopiazonic acid (CPA, 2018). | ||
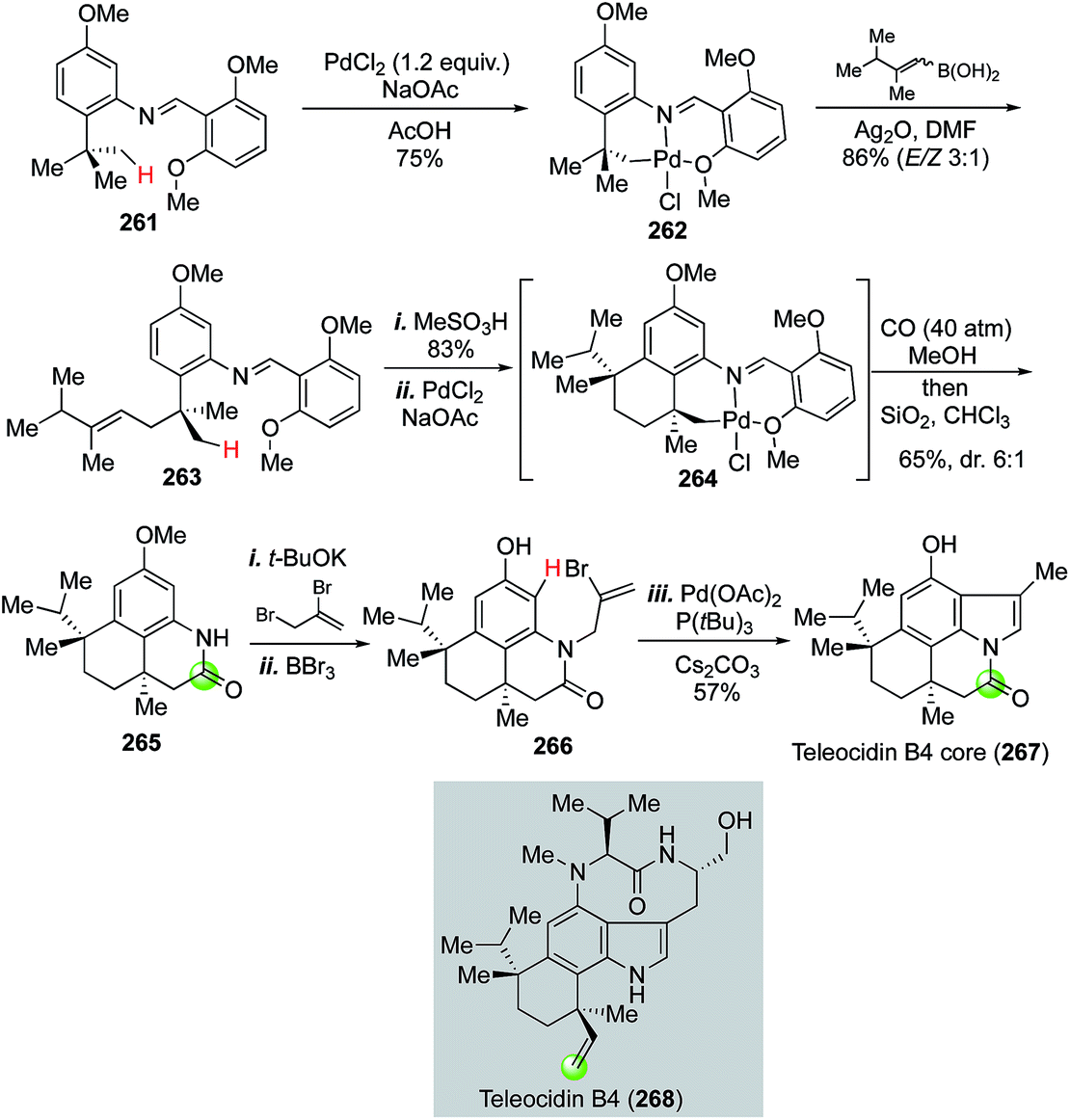 | ||
| Fig. 47 The Sames synthesis of teleocidin B4 core (2002). | ||
In 2014, Takemoto and co-workers reported their synthesis of pyrrolophenanthridine alkaloids based on two C–H functionalization reactions (Fig. 48).132 They first used a Catellani reaction133 followed by oxidation to produce 270 in 66% yield. Once carbamoyl chloride 271 was synthesized from 270, a Pd(OAc)2-catalyzed benzylic C–H functionalization between the carbamoyl chloride and the methyl group of 271 occurred to provide lactam 272 in 30% yield with 60% yield of 270. The formation of 270 indicated a decarbonylation process after oxidative addition. Therefore, carbon monoxide was utilized to help suppress this byproduct formation process, which renders this transformation a partial carbonylative cyclization. Lactam 272 was then reduced to assoanine (273), one of the pyrrolophenanthridine alkaloids.
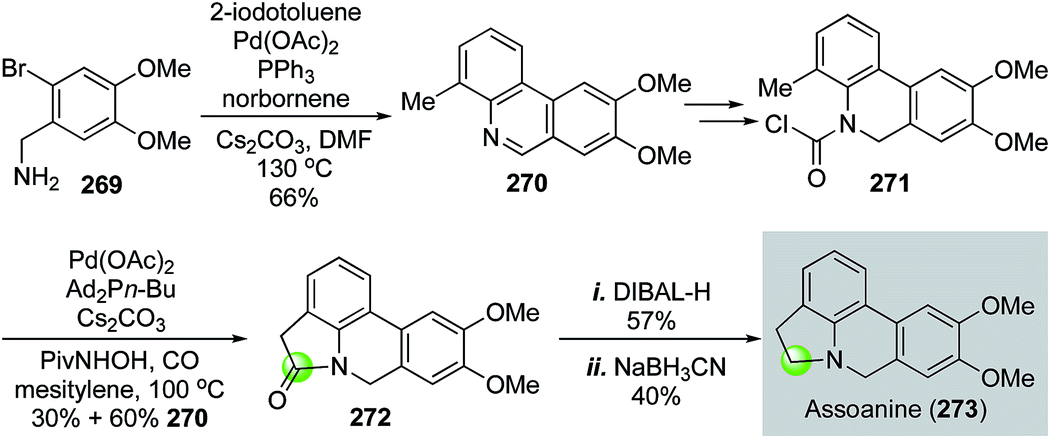 | ||
| Fig. 48 The Takemoto synthesis of assoanine (2014). | ||
Palladium-catalyzed directed C(sp3)–H carbonylative cyclization is an attractive strategy to access lactams. Recently, Wang and co-workers used a picolinamide directed C(sp3)–H carbonylative lactam formation to convert 274 to 275 (Fig. 49).134 The latter was hydrolyzed to pregabalin by removing the directing group and opening the lactam ring. Pregabalin is a clinically used drug to treat epilepsy and other neurological diseases.
 | ||
| Fig. 49 The Wang synthesis of pregabalin (2015). | ||
In 2016, Gaunt et al. disclosed an impressive synthesis of K-252c (staurosporinone) by employing a sequential C–H functionalization of arenes including three copper-catalyzed C(sp2)–H functionalizations to construct two C–C bonds and one C–N bond, one palladium-catalyzed carbonylative C–H activation to form the desired 5-membered lactam, and a Cadogan nitrene cyclization to build another key C–N bond.135 As shown in Fig. 50, they first used a copper-catalyzed C–H functionalization to unite diphenyliodonium triflate and dibenzyl p-toluidine 277 to form biaryl product 278, which was further transformed to 279 for the second copper-catalyzed C–H arylation with diphenyliodonium triflate to afford 280. After a nitration to convert 280 to 281, a copper-catalyzed C–H amination developed by Chang and co-workers was used to form a C–N bond and afford carbazole 282 in 88% yield with [bis(trifluoroacetoxy)iodo]benzene (PhI(TFA)2) as oxidant. Carbazole 282 was subjected to a radical bisbromination at the benzylic position followed by DMF and K2CO3 treatment to produce the corresponding aldehyde, which underwent reductive amination to furnish 2,6-dimethylbenzylamine 283. The use of 2,6-dimethylbenzylamine is critical for the next palladium-catalyzed carbonylative C–H lactam formation because the two methyl groups can block the potential competitive C–H activation pathways and desired lactam was produced in 96% yield with Pd(OAc)2 as catalyst and Cu(OAc)2 and air as oxidants. Finally, a Cadogan nitrene cyclization upon the treatment of nitro compound 284 with P(OEt)3 followed by removal of the 2,6-dimethylbenzyl group with BCl3 and TBAI under heating conditions completed the total synthesis of K-252c.
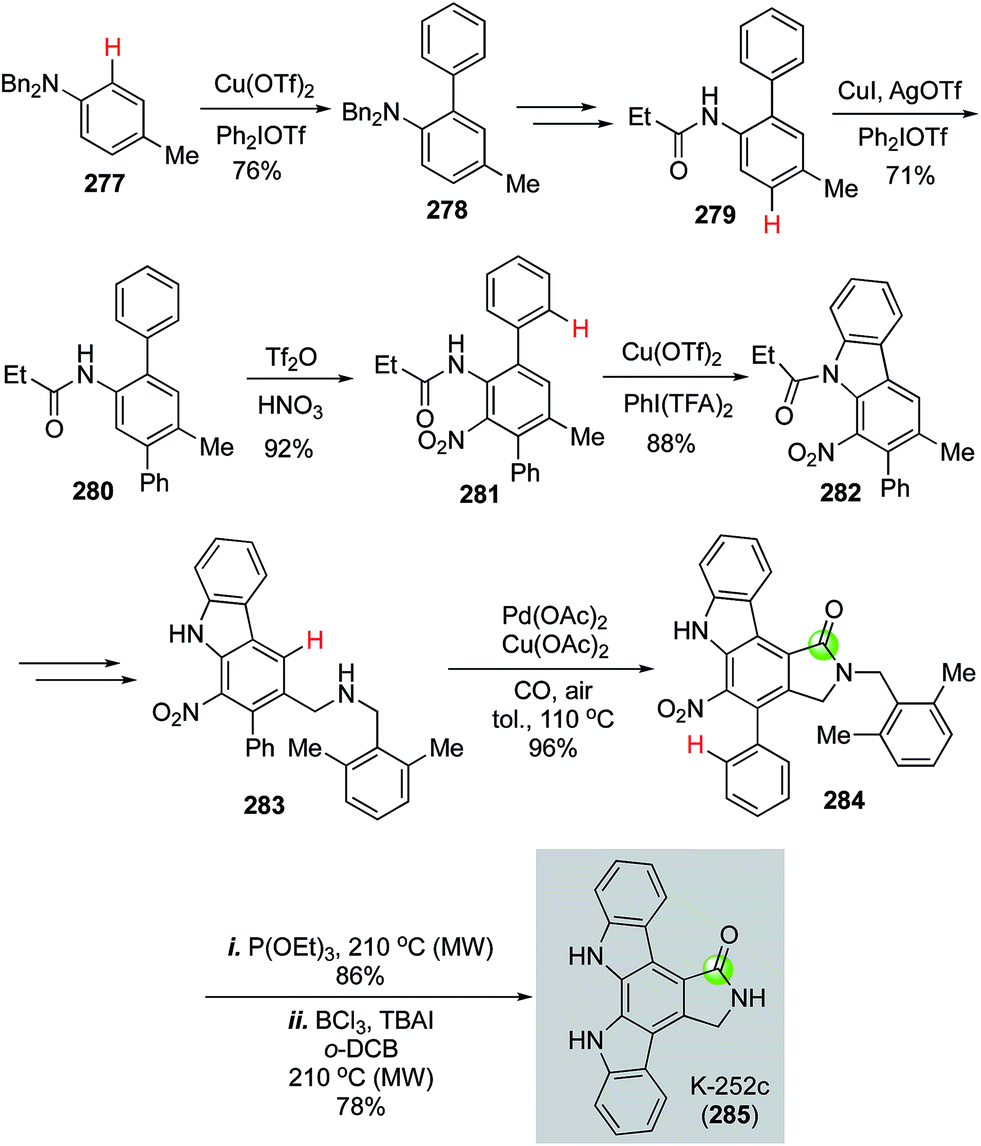 | ||
| Fig. 50 The Gaunt synthesis of K-252c (2016). | ||
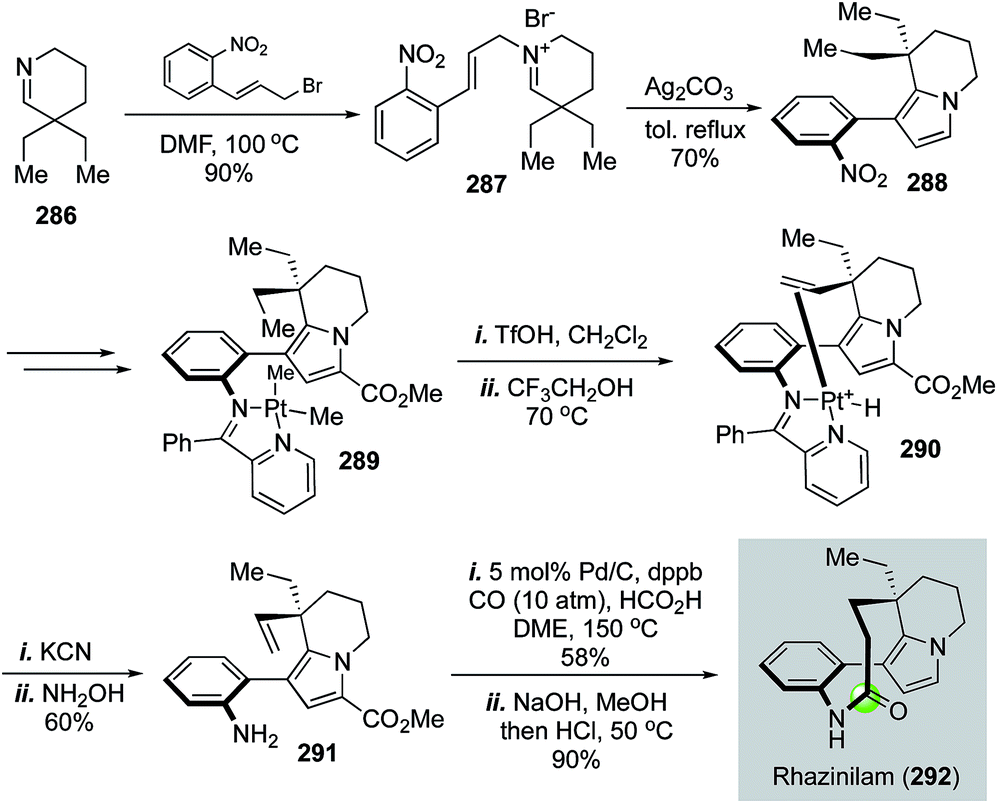 | ||
| Fig. 51 The Sames synthesis of rhazinilam (2002). | ||
 | ||
| Fig. 52 The Mukai synthesis of physostigmine (2006). | ||
The substituted maleinimide natural products, of which himanimide A and B are members, and their synthetic derivatives are known to possess various biological activities including antimicrobial activities, cytotoxicity, and kinase inhibiting properties. Beller and co-workers reported an efficient approach to these maleinimide natural products in 2010 (Fig. 53).138 In their case, internal alkyne substrate 298, prepared by Sonogashira coupling of the corresponding terminal alkyne and aryl bromide, was subjected to Fe3(CO)12-catalyzed carbonylation under 20 atm of carbon monoxide and excess NH3 in THF via a formal [2 + 1 + 1 + 1] annulation to construct the maleinimide ring of 299 in 78% yield. Oxidative dehydrogenation then gave himanimide A (300) in 48% overall yield. Conversion of himanimide A to himanimide B in 90% yield and 60% ee was realized by Sharpless asymmetric dihydroxylation.
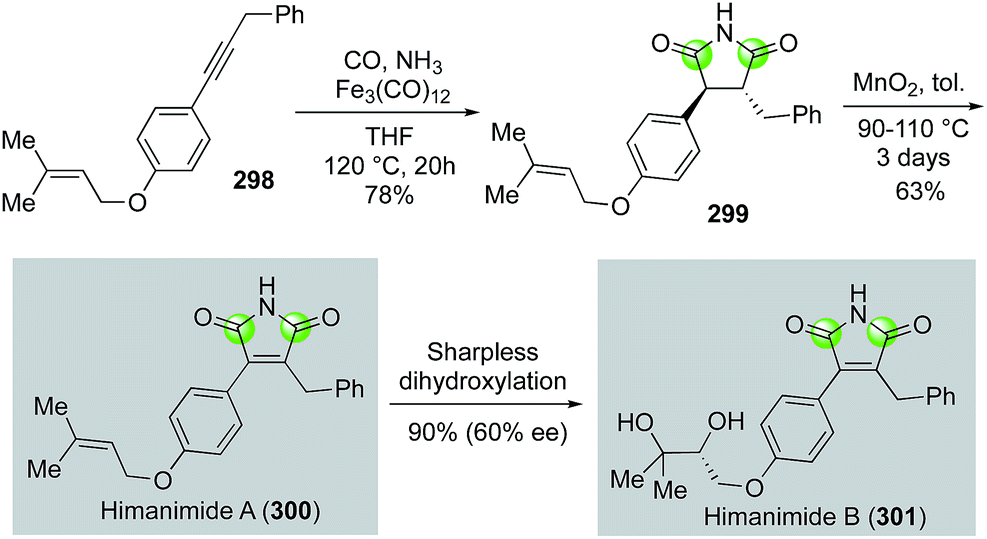 | ||
| Fig. 53 The Beller synthesis of himanimide A and B (2010). | ||
In 2017, Sarlah and co-workers reported an elegant total synthesis of pancratistatin and 7-deoxypancratistatin from benzene (Fig. 54).139 The pancratistatins belong to the Amaryllidaceae alkaloid family with substantial in vitro and in vivo anticancer activity. The Sarlah approach presents a rapid synthetically simplified entry into the pancratistatin core, which could be potentially used for supplying material for further biomedical development and future structural editing to identify analogs with improved functions. Their synthesis started with a visible-light-promoted para-cycloaddition of N–N arenophile MTAD to benzene to form a benzene–MTAD [4 + 2] cycloadduct, which without purification was then subjected to an enantioselective nickel-catalyzed aryl Grignard addition to form the trans-1,4-carboamination product 303 in 65% yield and 98:2 er at over 10 g scale with (R,Rp)-i-Pr-Phosferrox as ligand. Chemo- and diastereo-selective epoxidation and hydrolysis followed by Upjohn dihydroxylation installed the four contiguous hydroxy substituents of 305. After releasing of the free amine and bromination, they utilized a photochemical carbonylation with NaCo(CO)4 under UV light and carbon monoxide atmosphere to give (+)-7-deoxypancratistatin in 72% yield in one pot. A final C–H hydroxylation via cupration with (TMP)2Cu(CN)Li2 followed by in situ oxidation completed the total synthesis of pancratistatin in seven steps and 12% overall yield.
 | ||
| Fig. 54 The Sarlah synthesis of pancratistatins (2017). | ||
3.2. Other N-heterocycle formation
Rhodium-catalyzed hydroformylation–aminal cyclization has been useful in building N-heterocycles such as piperidine (Fig. 55) or pyrrolidine (Fig. 56), which are commonly found in bioactive natural products and lifesaving drug molecules. For the piperidine case, Ojima and co-workers used a Rh(acac)(CO)2-catalyzed tandem hydroformylation-intramolecular enamine formation sequence to convert 311 to 312, then to homokainoid 313 for the identification of kainic acid analogs as kainite receptor agonists for excitatory transmission in the central nervous system.140 Intermediate 311 can be readily accessed from (R)-serine (309). Wulff and Ren used a similar hydroformylation–intramolecular enamine formation strategy to convert homoallylic amine 310 to 311141 with a catalyst system developed by Morimoto and co-workers ([RhCl(cod)]2 as catalyst and BIPHEP and NiXantphos as ligands).142 In this case, paraformaldehyde was used as both the hydrogen and carbonyl sources. The generation of a cooperative dual catalyst system is the key for the success of this transformation because two catalytic processes are involved. One is the decarbonylation of formaldehyde and the other one is the hydroformylation of the terminal olefin of 318. To synthesize homoallylic amine 318, Wulff and Ren used a boroxinate (316)-catalyzed asymmetric aza-Cope rearrangement and hydrolysis sequence developed in the Wulff lab to realize the union of 314 and 315 and provide 317 in 61% yield, which was subsequently converted to 318 with a simple TBS protection. In 2013, Bates and co-worker used a double hydroformylation process to convert 321 to 322 after one of the two newly formed aldehydes cyclized with the sulfonamide to form an enamine (Fig. 55C).143 Enamine 322 was then advanced to azimic acid in a few steps. | ||
| Fig. 55 Hydroformylation–aminal cyclization toward piperidine-containing natural products. | ||
 | ||
| Fig. 56 Hydroformylation–aminal cyclization toward pyrrolidine-containing natural products. | ||
When allylic amines are used in the rhodium-catalyzed hydroformylation–aminal cyclization process, pyrrolidine derivatives can be obtained. Helmchen and co-workers achieved a rapid synthesis of (S)-nicotine with this strategy (Fig. 56A).144 In their case, the allylic amine substrate 329 was obtained by an iridium-catalyzed asymmetric allylic amination of allylic carbonate 327 with chiral ligand 328. Then, a Rh(acac)(CO)2/Biphephos-catalyzed tandem hydroformylation–intramolecular aminal formation followed by further hydrogenation gave (S)-nicotine 330 in 61% yield and 99% ee. In 2013, Chiou and co-workers developed an interesting synthesis of alkaloid physostigmine by employing a rhodium-catalyzed hydroformylation–double cyclization as the key step to afford pyrrolidinoindoline 332 in one step from 331 (Fig. 56B).145 Radical benzylic bromination of 332 with the treatment of 1,3-dibromo-5,5-dimethylhydantoin and AIBN afforded 334, which then underwent methylation with ZnMe2 smoothly in refluxing toluene to yield 335 for their formal synthesis of physostigmine. Additionally, the same strategy was used for their formal synthesis of physovenine.
In general, for these rhodium-catalyzed hydroformylation–aminal cyclization processes (Fig. 55 and 56), the substituents on the nitrogen and the product ring sizes affect the identity of the final cyclized products. For the case of converting 329 to 330, the use of alkyl groups such as benzyl and methyl groups are critical for the successful one-pot reduction to form pyrrolidine product directly. When tosyl or Boc groups were used, the reactions stopped at hemiaminal stage for pyrrolidines but enamine stage for piperidines.
In addition to the above endo-carbonylative cyclization pathways (carbon monoxide ends in the ring), exo-carbonylative cyclizations (carbon monoxide ends outside of the ring) are useful for preparing N-heterocycles. In 2003, Weinreb and co-workers reported an intramolecular carbonylative cyclization to yield oxindole product 339 from 338 (Fig. 57).146 The reaction initiated with a 5-exo-trig cyclization via a Heck-type olefin migratory insertion to form an all-carbon quaternary center and an alkylpalladium species. Since β-hydride elimination was blocked in this case, a carbon monoxide migratory insertion took place to generate an acylpalladium species, which was trapped by methanol to form ester 339. The carbonylation product 339 was then advanced to 340 with the two adjacent all-carbon quaternary centers which are required for synthesizing perophoramidine (341).
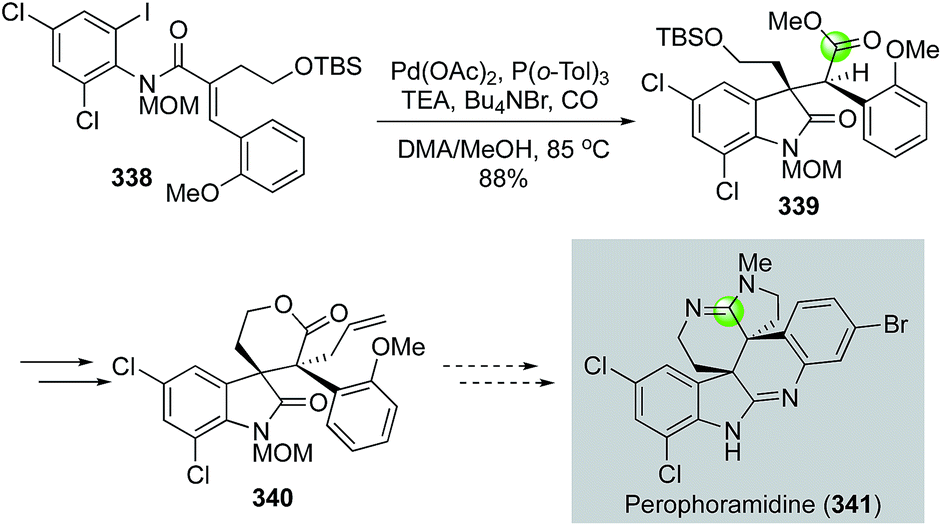 | ||
| Fig. 57 The Weinreb synthesis of perophoramidine core (2003). | ||
In 2009, Denmark and co-workers disclosed their total synthesis of isodomoic acids G and H via an innovative carbonylative silylcarbocyclization to build the key 3-carboxymethylproline moiety (Fig. 58).147 Isodomoic acids G and H differ at the configuration of the tetrasubstituted double bond and both belong to the family of kainoid amino acids. Fig. 58 highlights their total synthesis of isodomoic acid H. Treatment of enyne 342 and HSiMe2Ph with Rh(acac)(CO)2 catalyst under 500 psi of carbon monoxide at 120 °C delivered an inseparable mixture of diastereomers in an 8:1 ratio favouring the trans isomer 344. After oxidizing the newly formed aldehyde to ester 345, an ICl-promoted iododesilylation proceeded smoothly to give Z-alkenyl iodide 347 in 86% yield. A complete inversion of the tetrasubstituted double bond configuration occurred in the iododesilylation process, which was rationalized by the neighboring group participation of the ester carbonyl via intermediate 346. A Hiyama cross coupling between 347 and 348 was effective with a combination of Pd2(dba)3 and TBAF. The resulting diene 349 was elaborated to isodomoic acid H after hydrolyzing all three methyl esters with LiOH and removing the tosyl group using sodium amalgam. By using a similar approach, but a configuration retentive iododesilylation, they also completed the total synthesis of isodomoic acid G.
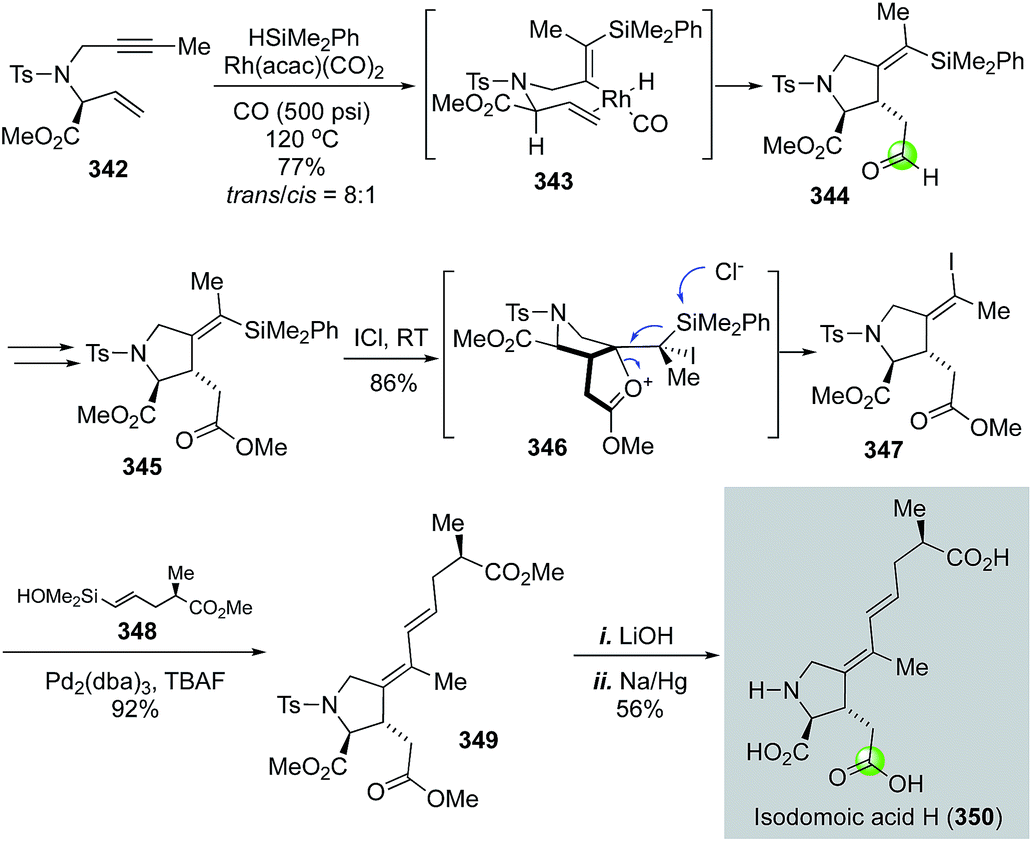 | ||
| Fig. 58 The Denmark synthesis of isodomoic acid H (2009). | ||
4. Carbonylative ketone synthesis
In addition to O- and N-heterocycles, carbonylative cyclization strategy is very effective in building carbocycles particularly cyclic ketones and aromatic products. In this review, we mainly focus on the recent use of the Stille carbonylation–Nazarov cyclization, the Pauson–Khand reaction, Rh-catalyzed [n + m + 1 (CO)] cycloaddition, and the Dötz annulation in natural product total synthesis. Notably, in the 1990s Oppolzer and co-workers developed a series of cascade palladium-catalyzed ene cyclization–carbonylation to synthesize polycyclic ketone products148 including 3-isorauniticine149 and hirsutene,150 which have been reviewed previously1 and are not discussed in detail here.4.1. Palladium-catalyzed carbonylative ketone synthesis
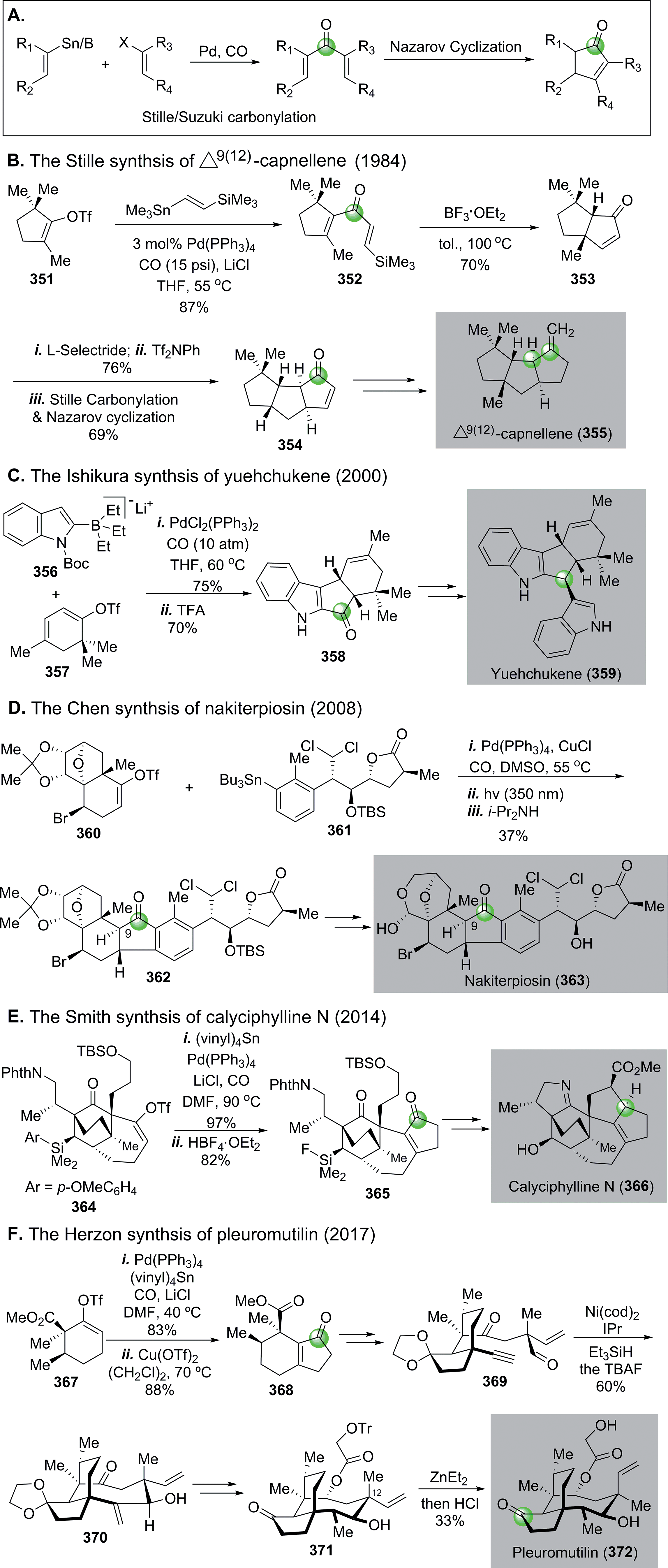 | ||
| Fig. 59 The Stille/Suzuki carbonylation–Nazarov cyclization in total synthesis. | ||
In 1984, Stille and co-workers achieved an elegant total synthesis of Δ9(12)-capnellene by orchestrating two consecutive Stille carbonylation–Nazarov cyclizations (Fig. 59B).154 The first one commenced with vinyltriflate 351 and (trimethylsilyl)vinylstannane, which were linked together with a molecule of carbon monoxide to dienone 352. A silicon-directed Lewis acid-promoted Nazarov cyclization occurred next to yield 353, which was advanced to 354, then to Δ9(12)-capnellene 355via another Stille carbonylation–Nazarov cyclization cascade to build the last cyclopentane ring.
In 2000, Ishikura and co-workers employed a combination of Suzuki carbonylation–Nazarov cyclization to their total synthesis of bis-indole alkaloid natural product yuehchukene (Fig. 59C).155 In this case, a lithiated “ate” complex 356 underwent Suzuki carbonylation smoothly with triflate 357. The newly generated indole-vinyl ketone was converted to 358via a TFA-promoted Nazarov cyclization. After ketone reduction and indole alkylation, a total synthesis of yuehchukene was reached.
Nakiterpiosin (363, Fig. 59D) is a scarce anticancer C-nor-D-homosteroid isolated from marine sponge Terpios hoshinota. To solve the material supply problem and understand its mode of actions, Chen and co-workers developed a highly convergent total synthesis of nakiterpiosin.156 Their synthesis features an effective Stille carbonylation to couple two acid- and base-sensitive building blocks 360 and 361 together. After a photochemical Nazarov cyclization and epimerization at C9, complex cyclopentanone product 362 was obtained in 37% overall yield. They completed the total synthesis of nakiterpoisin from 362 in four more steps.
In 2014, Smith and Shvartsbart completed a total synthesis of architecturally complex Daphniphyllum alkaloid calyciphylline N,157,158 isolated by Kobayashi and co-workers in 2008.159 The Smith synthesis utilized the Stille carbonylation–Nazarov cyclization sequence to convert vinyltriflate 364 to cyclopentenone 365, which was then advanced to calyciphylline N (Fig. 59E).
Very recently, the Stille carbonylation–Nazarov cyclization sequence was utilized by Herzon and co-workers in their modular and enantioselective synthesis of the pleuromutilin antibiotics (Fig. 59F).160 Pleuromutilin is a diterpene fungal metabolite with growth inhibiting activity against Gram-negative pathogens by binding to the highly conserved peptidyl transferase center of the bacterial ribosome. The Herzon synthesis used the Stille carbonylation–Nazarov cyclization sequence to provide bicyclic enone 368 from vinyltriflate 367, derived from cyclohexenone in three steps including one copper-catalyzed asymmetric conjugate addition-C-acylation to introduce the first chiral center of their synthesis. After 368 was furnished to alkynyl aldehyde 369, a nickel-catalyzed reductive cyclization built the strained macrocycle and afforded 370 in 60% yield. Compound 370 was further transformed to 371via a few functional group manipulations, but 371 contains opposite stereochemistry at the C12 all-carbon quaternary center in comparison to pleuromutilin. The final epimerization was achieved by treating 371 with Et2Zn via a retroallylation–allylation process. A following in situ removal of the trityl group afforded pleuromutilin in 33% yield along with 12-epi-pleuromutilin in 56% yield.
:1). The latter was transformed into a known intermediate for the total synthesis of neovibsanin B.
 | ||
| Fig. 60 The Fukuyama–Esumi formal synthesis of neovibsanin B (2010). | ||
Very recently, Zhao and co-workers utilized a palladium-catalyzed cascade carbonylative cyclization to rapidly build the 6-5-6 tricyclic ring system of cephanolides, which culminated in the first total synthesis of cephanolides B and C (Fig. 61).162 Their synthesis commenced with 5-bromo-2-methylanisole, which was advanced to lactone 380 in seven steps. Zhao and co-workers then developed a palladium-catalyzed carbonylative C–H annulation to convert 380 to polycyclic ketone products 381 and 382. While the palladium-catalyzed carbonylative C–H annulation was effective to build the desired cyclopentanone imbedded in the 6-5-6 tricyclic ring system, product 382 with undesired stereochemistry at the cis 5,6-fused ring junction was produced as the major stereoisomer because the lactone moiety is less sterically hindered than the methyl substituted ethylene moiety of the bicyclic [2.2.2] ring system. To circumvent this undesired stereochemical outcome, lactone 380 was then converted to acetal 383 with the thought that the acetal would be more sterically hindered than the lactone. Indeed, with this subtle change, carbonylative annulation product 384 was produced in high yield with desired the stereochemistry at the cis-fused ring junction. After oxidizing the acetal back to the corresponding lactone with m-CPBA, a formal deoxygenation with the treatment of TfOH/Et3SiH afforded 385 in 70% yield. A subsequent removal of the methyl group completed the total synthesis of cephanolide B. To convert 385 to cephanolide C, two benzylic C–H oxidation steps were employed. The first DDQ oxidation introduced the tertiary hydroxyl group and the second PCC oxidation installed the ketone functionality and delivered 387, which was then advanced to cephanolide C in three steps to remove the extra methoxy group.
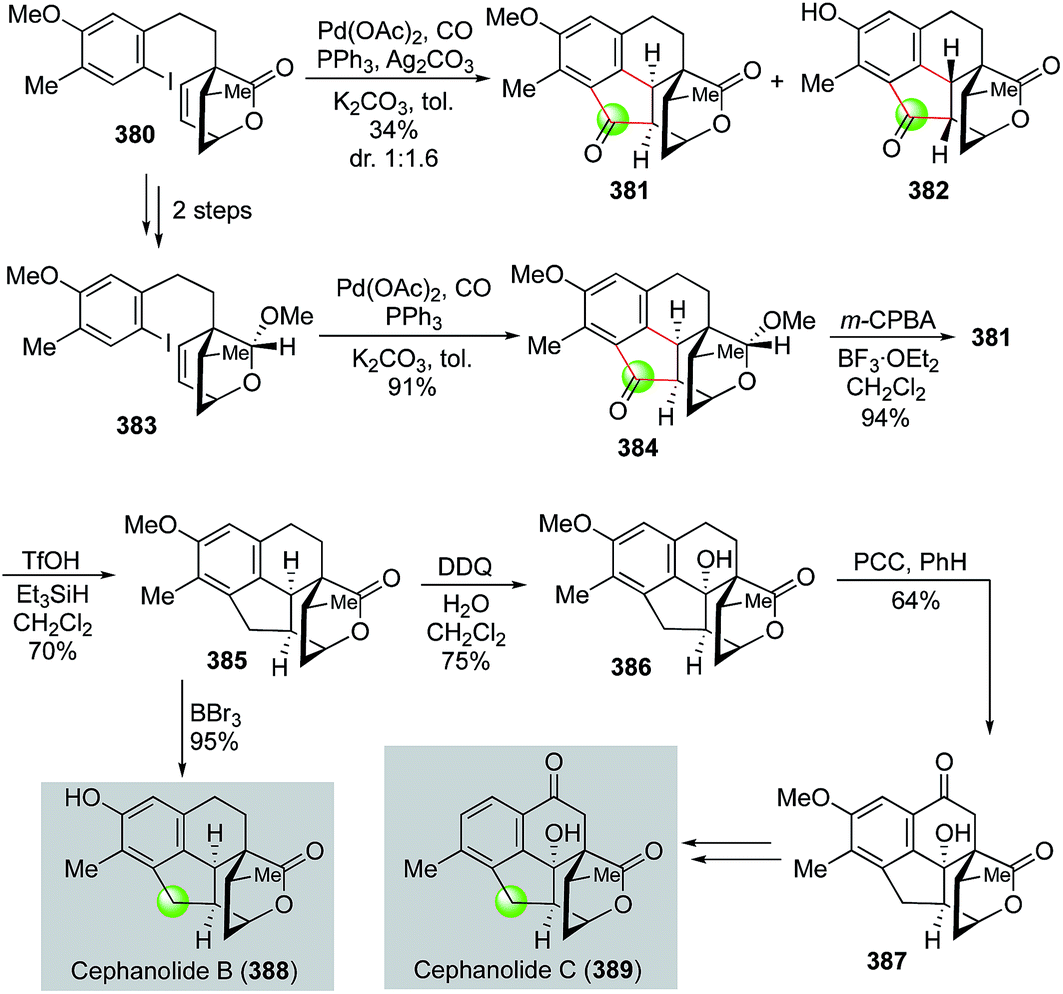 | ||
| Fig. 61 The Zhao synthesis of cephanolides B and C (2018). | ||
4.2. The Pauson–Khand reaction
The Pauson–Khand reaction, first developed in 1973, is arguably the most efficient process to construct cyclopentenones. It formally goes through a [2 + 2 + 1] cycloaddition involving an alkyne, an alkene and a carbon monoxide with a transition metal complex. Both intermolecular and intramolecular versions of the Pauson–Khand reactions have been developed. When the alkene and alkyne are tethered in the same molecule, a bicyclic ring system can be formed in one step. Since its discovery, the Pauson–Khand reaction has been widely used in natural product total synthesis. This topic has been frequently reviewed before.163–167 In this part, we highlight some recent applications of the Pauson–Khand reaction in making complex natural products.:1 diastereoselectivity, but favoring the undesired stereochemistry at C12. The remaining installation of the required strained trans-5,5-fused ring system was nontrivial and took them a detour to achieve this goal and complete the total synthesis of epoxydictymene.
 | ||
| Fig. 62 The Schreiber synthesis of epoxydictymene (1997). | ||
Serratine is a member of the Lycopodium alkaloids with a 5-6-5-6 fused tetracyclic skeleton. Zard and co-workers reported a total synthesis of 13-deoxyserratine in 2002 (Fig. 63).169 Their synthesis utilized a Pauson–Khand reaction to convert acyclic enyne 395 to bicyclic product 396 in 89% yield with 93:7 diastereoselectivity. Compound 396 was then elaborated to O-benzoyl-N-(2-chloroallyl)hydroxamine 397 for a tandem radical cyclization to install the other two ring systems. Under the conditions of slow addition of Bu3SnH and 1,1′-azobis(cyclohexanecarbonitrile) (ACCN) to a refluxing solution of 397 in trifluorotoluene, a sequential 5-exo-trig and 6-endo-trig radical cyclization took place to give tetracyclic intermediate 399 in 52% yield. Notably, the use of N-(2-chloroallyl)hydroxamine is critical for the desired 6-endo-trig cyclization process. Without the chloride, a 5-exo-trig cyclization would occur to give a pyrrolidine ring instead of the required piperidine ring. Compound 399 was then transformed to 13-deoxyserratine uneventfully. In this case, the Pauson–Khand reaction and the tandem radical cyclization enabled a rapid construction of the tetracyclic structure of 13-deoxyserratine.
 | ||
| Fig. 63 The Zard synthesis of 13-deoxyserratine (2002). | ||
Magellanine and related natural products are also family members of the Lycopodium alkaloids, but they share a common 6-5-5-6 tetracyclic framework with multiple stereogenic centers. Both Mukai's group170 and Ishizaki's group171 have employed the Pauson–Khand reaction as the key strategy to build these molecules, but their corresponding bond disconnections are quite different (Fig. 64). Ishizaki and co-workers used the Pauson–Khand reaction to build the B,C-ring system by converting 401 to 402, then to magellanine (403). Mukai and co-workers first used the Pauson–Khand reaction to form the A,B-ring system (404 → 405). They then employed a Stork radical cyclization strategy172 to elaborate 405 to 407. The latter was converted to enyne 408 for the second Pauson–Khand reaction, which built the C,D-ring system and afforded tetracyclic intermediate 409. After oxidative fragmentation of the newly formed cyclopentenone ring followed by constructing the piperidine ring, they were able to finish several magellanine family members including magellanine itself and magellaninone. Notably, in 2013, Mukai and co-workers employed a similar Pauson–Khand/Stork radical cyclization strategy to accomplish the total synthesis of several other Lycopodium alkaloids including fawcettimine, fawcettidine, lycoflexine, and lycoposerramine Q.173
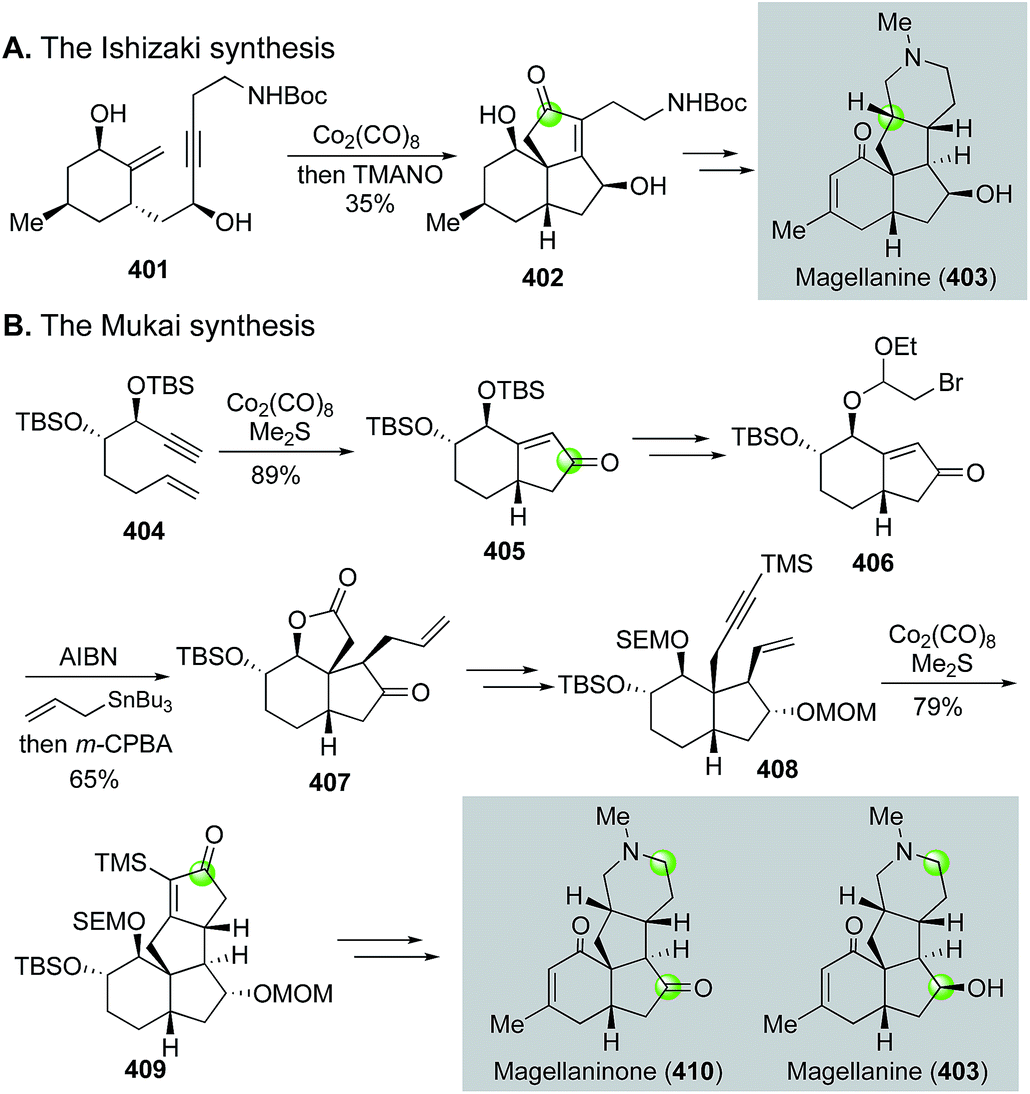 | ||
| Fig. 64 The Mukai (2007) and Ishizaki (2003) synthesis of magellanine. | ||
In 2007, Fox and co-worker reported a Pauson–Khand strategy to an enantioselective total synthesis of pentalenene, an angular triquinane natural product (Fig. 65).174 In particular, the Pauson–Khand reaction involving chiral cyclopropene 413 provided tricyclic product 414 in 80% yield and 4:1 diastereoselectivity. It also built the angular all-carbon quaternary center. Notably, the TMS group of substrate 413 is essential for the success of the Pauson–Khand reaction. Substrate 412 failed to undergo the desired Pauson–Khand reaction under various reaction conditions. The chiral cyclopropene substrate was prepared in 80% yield and 91% ee via an enantioselective and chemoselective cyclopropenation of diyne 411 using Corey's Rh2(OAc)(R,R-DPTI)3 catalyst. After removal of the silyl groups of the Pauson–Khand reaction product with TBAF, Fox et al. used a Pd/C-catalyzed hydrogenation to reduce the double and cleave the cyclopropane ring. Bicyclic product 415 was obtained and further advanced to pentalenene.
 | ||
| Fig. 65 The Fox synthesis of pentalenene (2007). | ||
Alstonerine features an azabicyclo[3.3.1] substructure that is fused with an indole ring. It has shown cytotoxic activity against a few human cancer cell lines. In 2007, Martin and Miller disclosed a concise total synthesis of alstonerine involving a remarkable Pauson–Khand reaction to convert enyne 417 to 418 and build the azabicyclo[3.3.1] substructure in 94% yield (Fig. 66).175 An oxidative cyclopentenone cleavage strategy was next developed to provide lactone 421 in high yield via a sequence of enone reduction with Karstedt's catalyst 419, Lemieux–Johnson silyl enol ether oxidative cleavage, aldehyde reduction and acid-promoted lactonization. Lactone 421 was then advance to alstonerine in six steps.
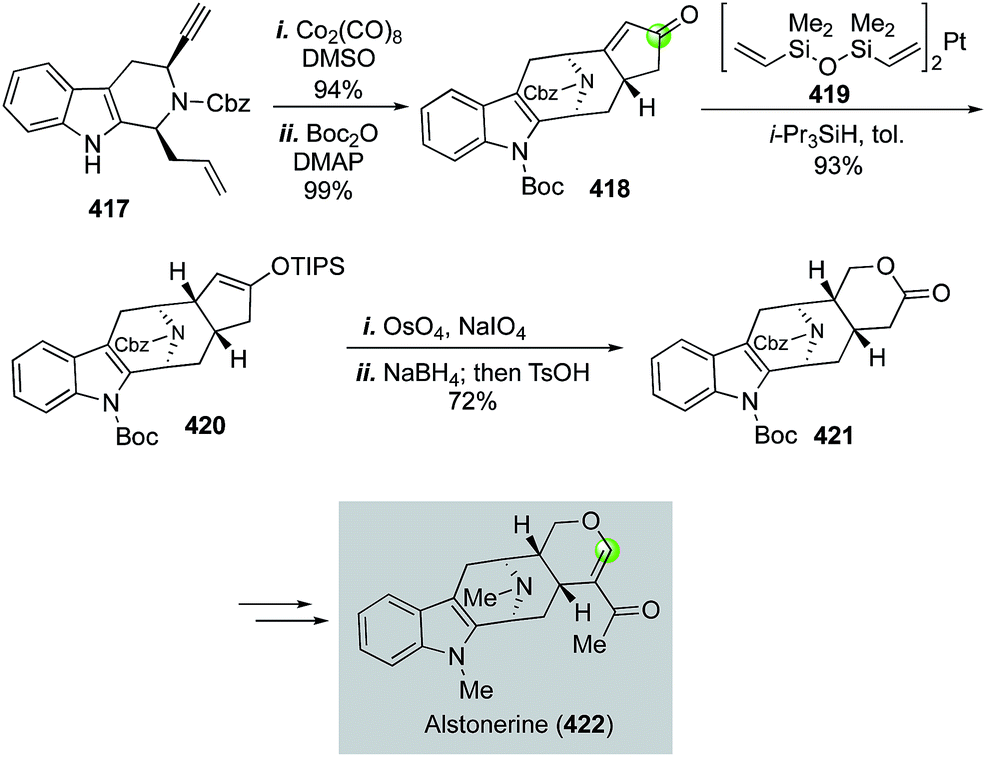 | ||
| Fig. 66 The Martin synthesis of alstonerine (2007). | ||
In the same year, Danishefsky and Min reported the first total synthesis of paecilomycine A with potent neurotrophic activity in fostering neurite outgrowth of PC12 cells (Fig. 67).176 Their synthesis commenced with a Diels–Alder reaction between diene 423 and alkyne-activated dienophile 424, which occurred smoothly even at 0 °C to form cyclohexene product 425 in 58% yield and high endo-selectivity. Cycloadduct 425 was then converted to the Pauson–Khand reaction precursor 426 in four steps. Under the conditions of Co2(CO)8 in toluene at 100 °C, tricyclic product 427 was produced in 37% yield. After Luche reduction of the ketone, a directed cyclopropanation followed by reductive ring opening strategy was used to install the required methyl group and build the all-carbon quaternary center. The Furukawa reagent was effective for the cyclopropanation. After the Ley oxidation and dissolving metal reduction, ketone 429 was obtained, then advanced to paecilomycine A. Notably, in 2012, Mehta and co-workers used a similar strategy to synthesize paecilomycine A.177
 | ||
| Fig. 67 The Danishefsky synthesis of paecilomycine A (2007). | ||
Since its isolation by Kobayashi and co-workers, the complex marine anticancer alkaloid nakadomarin A has attracted a significant amount of synthetic attention. Among these, the Mukai group178 and the Clark group179 have used a Pauson–Khand strategy to build the key 5,6-fused bicycle (Fig. 68). Mukai and co-workers used a Co2(CO)8-mediated Pauson–Khand reaction to convert 431 to 432, then to a formal synthesis of nakadomarin A. Clark and Xu used an efficient cobalt-catalyzed asymmetric Pauson–Khand reaction developed by Hiroi and co-workers180 to set the first chiral center of their entire total synthesis and produce 434 in 72% yield and 97% ee from simple enyne 433. Notably, a similar Pauson–Khand strategy was also employed by Magnus and co-workers in their synthetic efforts toward nakadomarin A and the manzamines.181
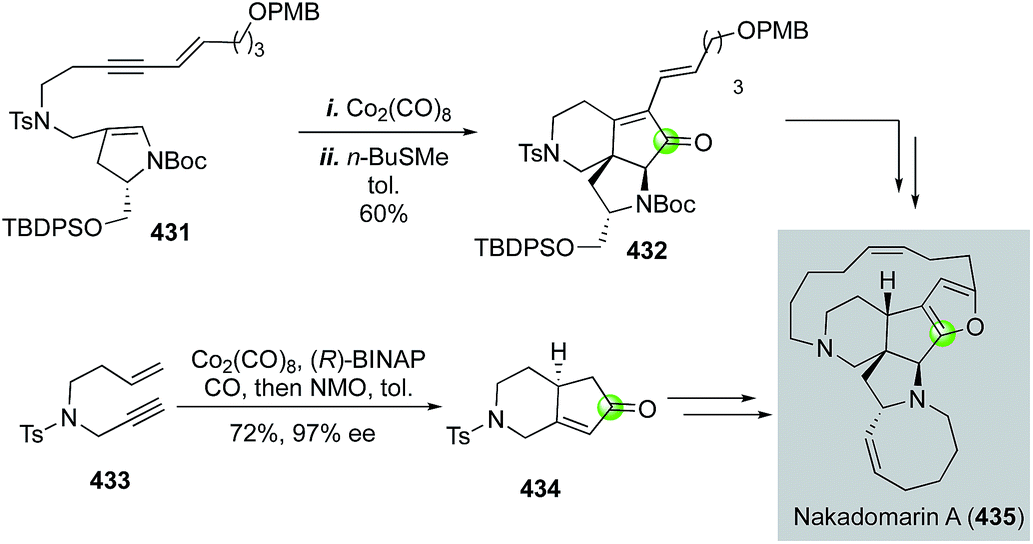 | ||
| Fig. 68 The Mukai (2010) and Clark (2016) synthesis of nakadomarin A. | ||
Axinellamines A and B (447, 448, Fig. 69) represent the most complex members of the bioactive pyrrole-imidazole marine alkaloid family. They present a daunting challenge to synthetic chemists. In 2011, Baran and co-workers revealed a remarkable synthetic approach which can be used to prepare gram scale of both axinellamines A and B.182,183 In their synthesis, a Pauson–Khand reaction between symmetric alkene 436 and terminal alkyne 437 under the conditions of Co2(CO)8, NMO and ethylene glycol in CH2Cl2 delivered cyclopentenone 438 in 46–58% yield and formed the cyclopentane core of the axinellamines. Notably, the combination of ethylene glycol and NMO is critical for the success of the Pauson–Khand reaction. It was hypothesized that they both were involved in the formation of a more reactive cobalt complex. The Pauson–Khand product was then transformed to trichloride 439via Luche reduction and trichlorination with NCS/PPh3. An unprecedented Zn/In-mediated Barbier reaction was developed for the union of trichloride 439 and aldehyde 440, producing homoallylic alcohol 441 in 61% overall yield from 438. After intermediate 441 was advanced to aminoimidazole 442 in five steps, DMDO oxidation with a subsequent TFA-promoted cyclization afforded 443 as a mixture of isomers. Baran and co-workers then employed a C–H oxidation mediated by silver(II) picolinate 444 to install the required hydroxyl group. A one-pot azide reduction with PtO2/H2 and acylation with 2,3-dibromo-5-trichloroacetylpyrrole led to the total syntheses of axinellamines A and B.
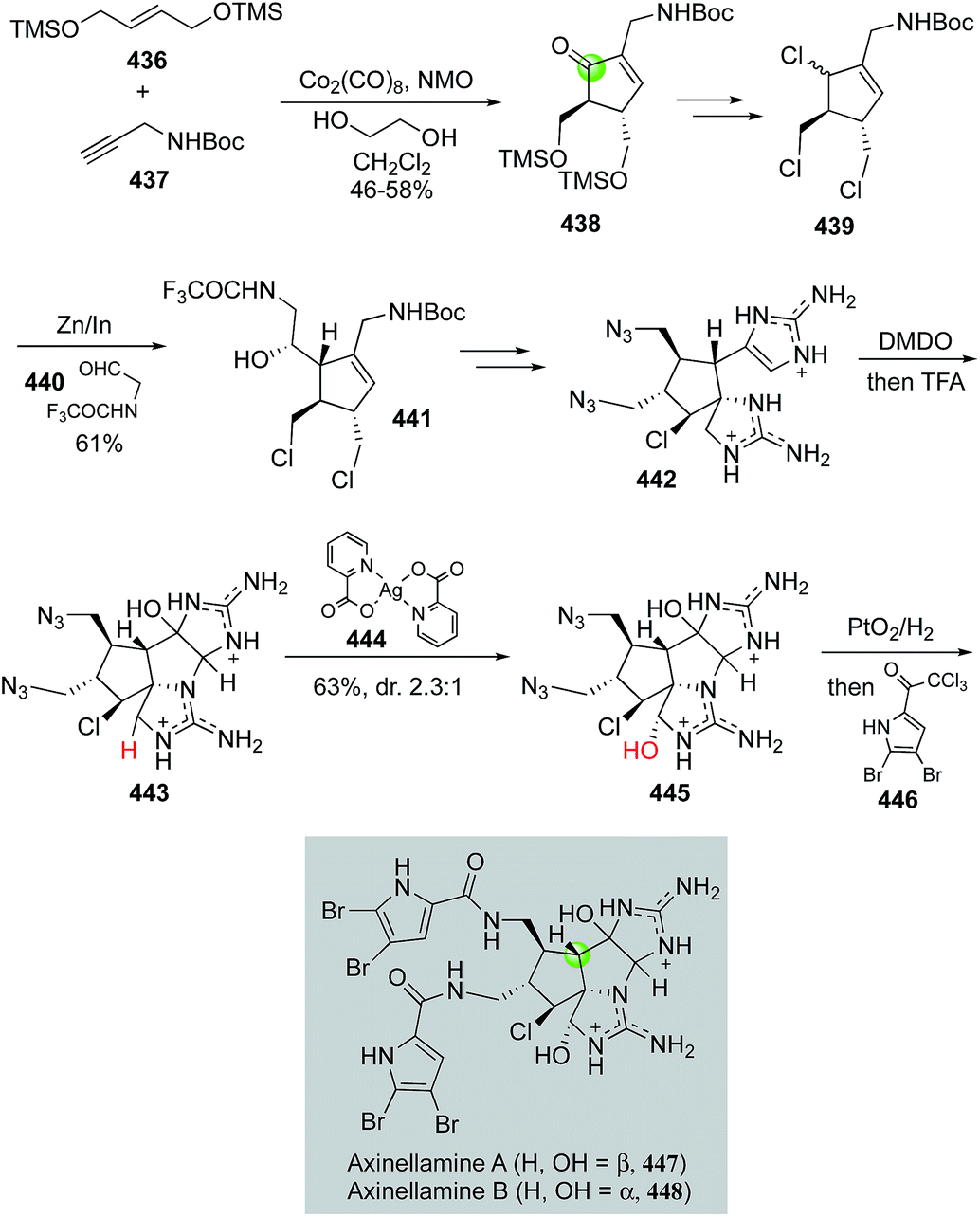 | ||
| Fig. 69 The Baran synthesis of axinellamines-Baran (2011). | ||
Jiadifenin, isolated by Fukuyama and co-workers from the pericarps of Illicium jiadifengpi, exhibited impressive neurotrophic activity in promoting neurite outgrowth of fetal rat cortical neuronal cells and has been a popular target for total synthesis. In 2012, Zhai and workers reported their total synthesis of jiadifenin, which features key steps such as the Ireland–Claisen rearrangement, the Pauson–Khand reaction, [2 + 2]-cycloaddition, and cyclobutanone fragmentation (Fig. 70).184 In details, an Ireland–Claisen rearrangement was used to convert 449 to 450 in 54% yield with 7:1 diastereoselectivity. After protecting group manipulations, a Co2(CO)8/Bu3PS-mediated Pauson–Khand reaction led to the conversion of enyne 451 to tricyclic intermediate 452, which then underwent photochemical [2 + 2] cycloaddition with an allene to afford 453 in 87% yield (dr. 4.8:1). A fragmentation process involving ozonolysis of the newly formed exo-methylene and NaOMe-promoted C–C bond cleavage gave 454 in 89% yield, which was eventually advanced to jiadifenin in five steps.
 | ||
| Fig. 70 The Zhai synthesis of jiadifenin (2012). | ||
Since 2011, Yang and co-workers disclosed a series of impressive applications of the Pauson–Khand reaction in total synthesis of polycyclic bioactive natural products (Fig. 34 and 71).185 In addition to their schindilactone A synthesis discussed in Fig. 34, their efforts toward pentalenolactone A methyl ester,186 fusarisetin A,187 propindilactone G,188 retigeranic acids,189 presilphiperfolanes,190 and lancifodilactone G191 are chronologically summarized in Fig. 71.
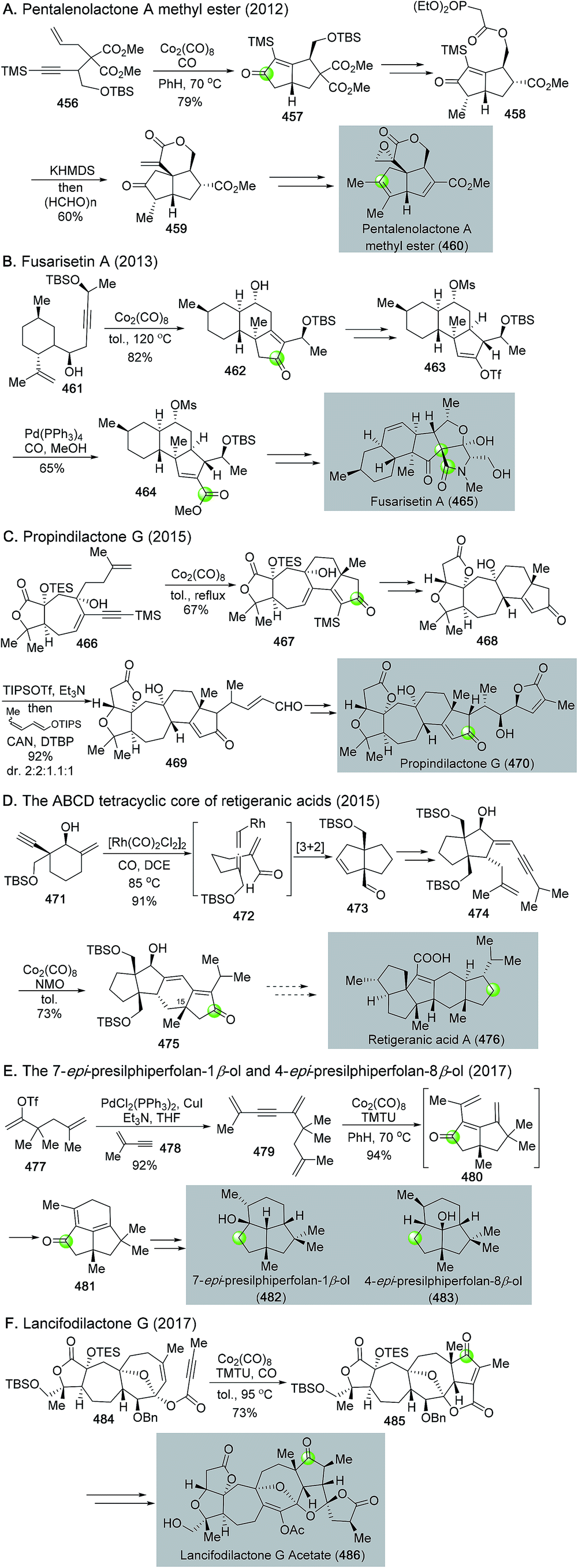 | ||
| Fig. 71 Total synthesis via Pauson–Khand reaction from Yang and co-workers. | ||
Pentalenolactones (460, Fig. 71A) are complex angular tricyclic natural products produced by prokaryotic organisms and have demonstrated a broad range of antimicrobial activities. Yang and co-workers utilized an intramolecular Pauson–Khand reaction of 456 upon the treatment of Co2(CO)8 under carbon monoxide atmosphere to furnish bicyclic enone 457 in 79% yield, which was subsequently converted to phosphonate 458. An impressive tandem conjugate addition and Horner–Wadsworth–Emmons exo-methylenation converted 458 to angular tricyclic lactone 459 in 60% yield, which was then advanced to pentalenolactone A methyl ester via a few functional group manipulations.
Fusarisetin A (465, Fig. 71B) was reported to inhibit cancer cell migration and invasion without showing any significant cytotoxicity. Its intriguing effect on cancer metastasis coupled with its novel pentacyclic structure have rendered it an appealing target for total synthesis. Yang and co-workers used a Co2(CO)8-mediated Pauson–Khand reaction to convert enyne 461 to cyclopentenone 462 containing the fused 6-6-5 tricyclic carbon core. After 462 was transformed to vinytriflate 463, a Heck carbonylation introduced the required carboxylate and resulted in 464 in 65% yield, from which fusarisetin A can be reached in seven steps.
Propindilactone G (470, Fig. 71C) belongs to a novel group of nortriterpenoid isolated by Sun and co-workers. In their continuing interest in synthesizing nortriterpenoids, Yang and co-workers accomplished an asymmetric synthesis of propindilactone G in 2015. Their synthesis features a Pauson–Khand reaction to build the fused 5-7-6-5 tetracyclic carbon skeleton by converting 466 to 467. After the latter was advanced to 468, an oxidative enolether heterodimerization initiated by ceric(IV) ammonium nitrate (CAN) oxidation was developed to append the side chain and produce 469 in high yield but as a mixture of four diastereomers. A sequence of Horner–Wadsworth–Emmons olefination, OsO4-catalyzed dihydroxylation and concurrent lactonization was able to deliver propindilactone G in good yield.
In their synthetic studies toward the retigeranic acids (cf.476, Fig. 71D), Yang and co-workers used a Co2(CO)8/TMTU-mediated Pauson–Khand reaction to deliver 475 equipped with the desired tetracyclic core skeleton. Notably, they developed a novel rhodium-catalyzed [3 + 2] cycloaddition to convert 471 to 473 in 91% yield. This highly effective transformation was proposed to go through intermediate 472, derived from a rhodium-catalyzed C–C bond cleavage. A formal [3 + 2] cycloaddition occurred next to form 473 with two challenging adjacent all-carbon quaternary centers.
In 2017, Yang and co-workers developed a tandem sequence of the Pauson–Khand reaction and 6π-electrocyclization to rapidly build the strained tricyclic core of presilphiperfolanols. As shown in Fig. 71E, a Sonogashira coupling between vinyltriflate 477 and enyne 478 led to the formation of 479 in 92% yield, which underwent a Co2(CO)8/TMTU-mediated Pauson–Khand reaction smoothly to produce triene 480. Under the same reaction conditions, a 6π-electrocyclization took place to yield 481 in 94% yield from linear precursor 479. Two presilphiperfolane analogs 482 and 483 were prepared from 481.
In the same year, they also reported an elegant total synthesis of lancifodilactone G acetate (486, Fig. 71F). Similar to their schindilactone A synthesis, a Co2(CO)8/TMTU-mediated Pauson–Khand reaction was used to build the 5,5-fused ring system and convert 484 to 485, then to lancifodilactone G acetate 486.
The complex diterpene natural product ryanodol has been a long-standing synthetic challenge. Biologically, ryanodol and related analogs modulate intracellular calcium-ion release at the ryanodine receptors. In 2016, Reisman, Chuang, and Xu disclosed a remarkable 15-step total synthesis of ryanodol (Fig. 72).192 Starting from chiral pool molecule (S)-pulegone (487), Reisman et al. quickly accessed intermediate 488, which was subjected to Grignard addition with ethoxyethynylmagnesium bromide followed by a silver(I)-catalyzed cyclization and elimination cascade to afford α,β-unsaturated lactone 489 in good yield. A subsequent conjugate addition with vinylcuprate derived from vinylMgBr and CuI led to the formation of an all-carbon quaternary center and produced enyne 490 for the following Pauson–Khand reaction. While cobalt or molybdenum carbonyl complexes are effective for the Pauson–Khand reaction, [RhCl(CO)2]2 was optimal for catalyzing the desired [2 + 2 + 1] cycloaddition under 1 atm of carbon monoxide at 110 °C. Enone 491 was produced in 5.7 g at a single pass. They then discovered an amazingly effective triple oxygenation to convert 491 to 492 by simply heating 491 in presence of SeO2. With 492 in hand, the 2-propenyl group was introduced via a sequential vinyl triflate formation using the Comins' reagent and Stille cross coupling with 2-propenylstannane. Product 493 was produced in good yield then advanced to epoxide 494 in three steps. Subjection of 494 to Li/NH3 triggered a reductive cyclization to afford ryanodol in 38% yield.
 | ||
| Fig. 72 The Reisman synthesis of ryanodol (2016). | ||
In 2016 and 2017, Trauner and co-workers reported their total synthesis of lycopalhine A193 and sinoracutine194 respectively. Both syntheses used the Pauson–Khand strategy to construct the key cyclopentenone intermediates. In particular, Co2(CO)8-mediated [2 + 2 + 1] cycloaddition converted enyne 496 to cyclopentenone 497. A subsequent conjugate addition led to the formation of 498, which was then elaborated to lycopalhine A (499) with steps involving Mannich cyclization and intramolecular aldol reaction to build the additional ring systems (Fig. 73A). In their sinoracutine synthesis, Pauson–Khand reaction of 500 under oxidative reaction conditions resulted in the formation of tricyclic intermediate 501, which was then reduced to the corresponding allylic alcohol. After the failures of using Eschenmoser–Claisen or Johnson–Claisen strategy to convert 501 to 502 with an all-carbon quaternary benzylic stereocenter, Trauner and co-workers used the Mandai two-step protocol to realize this transformation. Particularly, oxa-Michael addition to vinyl sulfoxide followed by elimination of sulfenic acid and subsequent [3,3] sigmatropic rearrangement delivered aldehyde 502 in 57% yield. A combination of reductive amination, iodoamination and Kornblum oxidation was able to convert 502 to 503. Sinoracutine was reached from 503via a sequence of Mukaiyama oxidation, elimination of the silyl alcohol and debenzylation.
 | ||
| Fig. 73 The Trauner synthesis of lycopalhine A and sinoracutine. | ||
Astellatol contains a unique bicyclo[4.1.1]octane moiety and a strained trans-hydrindane, both of which are tempting and challenging for synthetic chemists. In 2018, Xu and co-workers offered a solution to these problems and completed the first total synthesis of astellatol (Fig. 74).195 Their synthesis used an intramolecular Pauson–Khand reaction to convert enyne 505 to polycyclic compound 506, which was further advanced to 507via a sequential base-promoted elimination, ethyl ester formation, DIBAL-H reduction, and the Ley–Griffith TPAP oxidation. Compound 507 was next treated with SmI2 to trigger a reductive 1,6-radical addition to build the bicyclo[4.1.1]octane moiety and a mixture of 508 and 509 was obtained in 64% total yield. Both 508 and 509 were then converted to 510via hydrogenation to reduce the corresponding dienes and TPAP oxidation of the secondary alcohol. Compound 510 was eventually transformed to astellatol in seven steps including one hydroxy group-directed hydrogenation using Crabtree's catalyst to build the trans-hydrindane ring system.
 | ||
| Fig. 74 The Xu synthesis of astellatol (2018). | ||
The Daphniphyllum alkaloids are a large group of natural products containing fascinating polycyclic architectures that have attracted many synthetic efforts. In 2012, Dixon and co-workers reported an expedient synthesis of the 7-5-5 tricyclic core of the Daphniphyllum alkaloids daphnilongeranin B and related analogs using a key intramolecular Pauson–Khand reaction to construct the 5,5-fused ring system.196 The Pauson–Khand strategy was also employed by Li and co-workers to build the same 5,5-fused ring system, but at a much late strategy, which eventually led to the total synthesis of daphnilongeranin B, hybridaphniphylline B, and two other natural analogs (Fig. 75).197 In particular, compound 512 underwent the desired [2 + 2 + 1] cycloaddition with Co2(CO)8 to afford 513 as a mixture of two diastereomers, both of which were then converged to 514via a K2CO3-promoted isomerization process and 514 was obtained in 63% yield from 512. Reduction of the thiolactam with RANEY® Ni led to the formation of daphnilongeranin B (515) at gram scale. On the other hand, Luche reduction of 514 yielded alcohol 516 in 99% yield. When a mixture of 516 and 517 (derived from natural product genipin) was heated at 160 °C in presence of MgSO4, a tandem dehydration and intermolecular Diels–Alder reaction took place to afford a mixture of four isomers in total 61% yield. The major isomer was isolated as 518 in 24% yield, which was advanced to hybridaphniphylline B via RANEY® Ni reduction of the thiolactam and removal of all the acetyl groups.
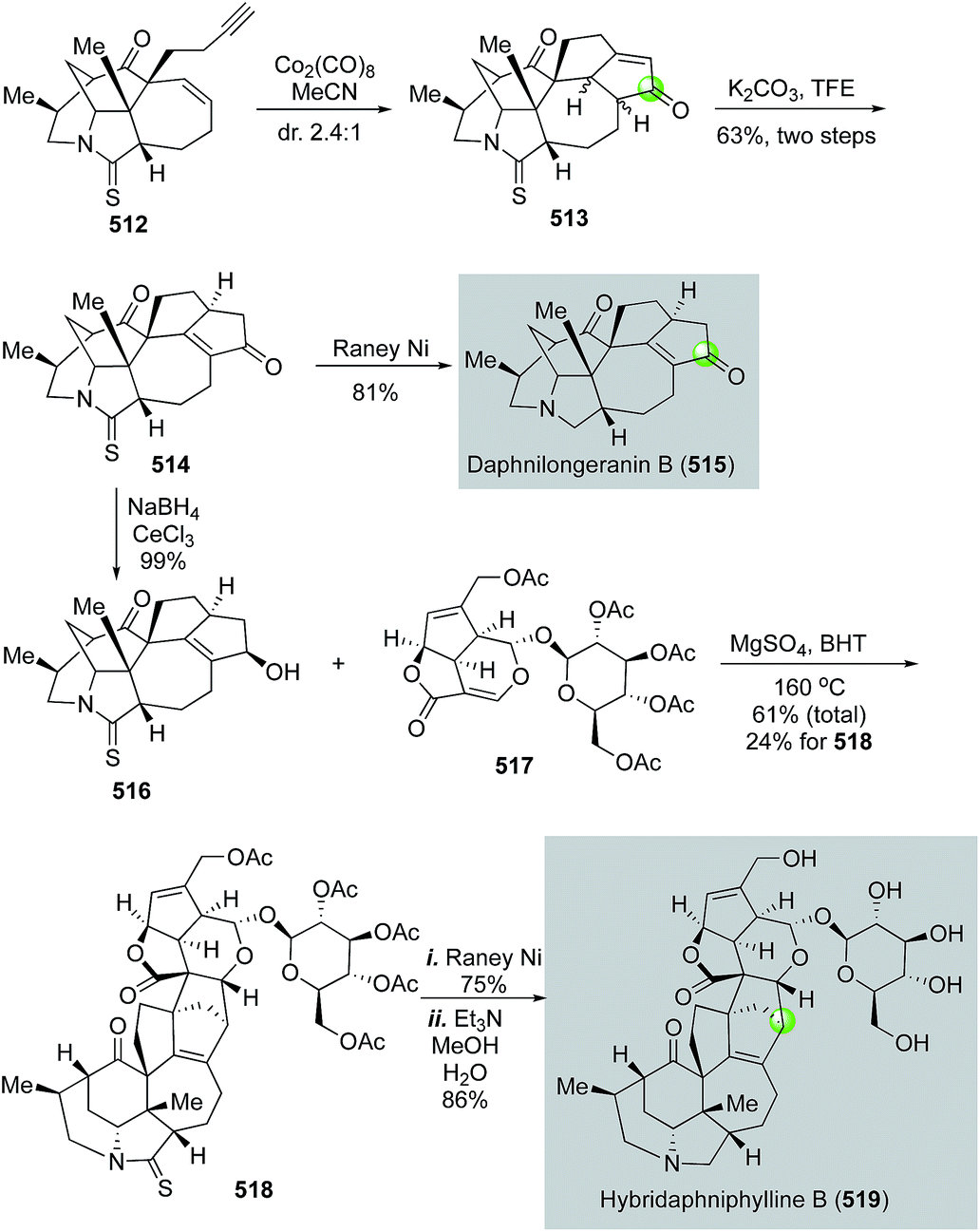 | ||
| Fig. 75 The Li synthesis of daphnilongeranin B and hybridaphniphylline B (2018). | ||
In addition to the above discussed cases, alkyne–alkene Pauson–Khand reaction has been widely used in many other natural product total syntheses as well as synthetic studies.198–204 A collection of natural products completed via an alkyne–alkene Pauson–Khand reaction as a key step is listed in Fig. 76.205–223
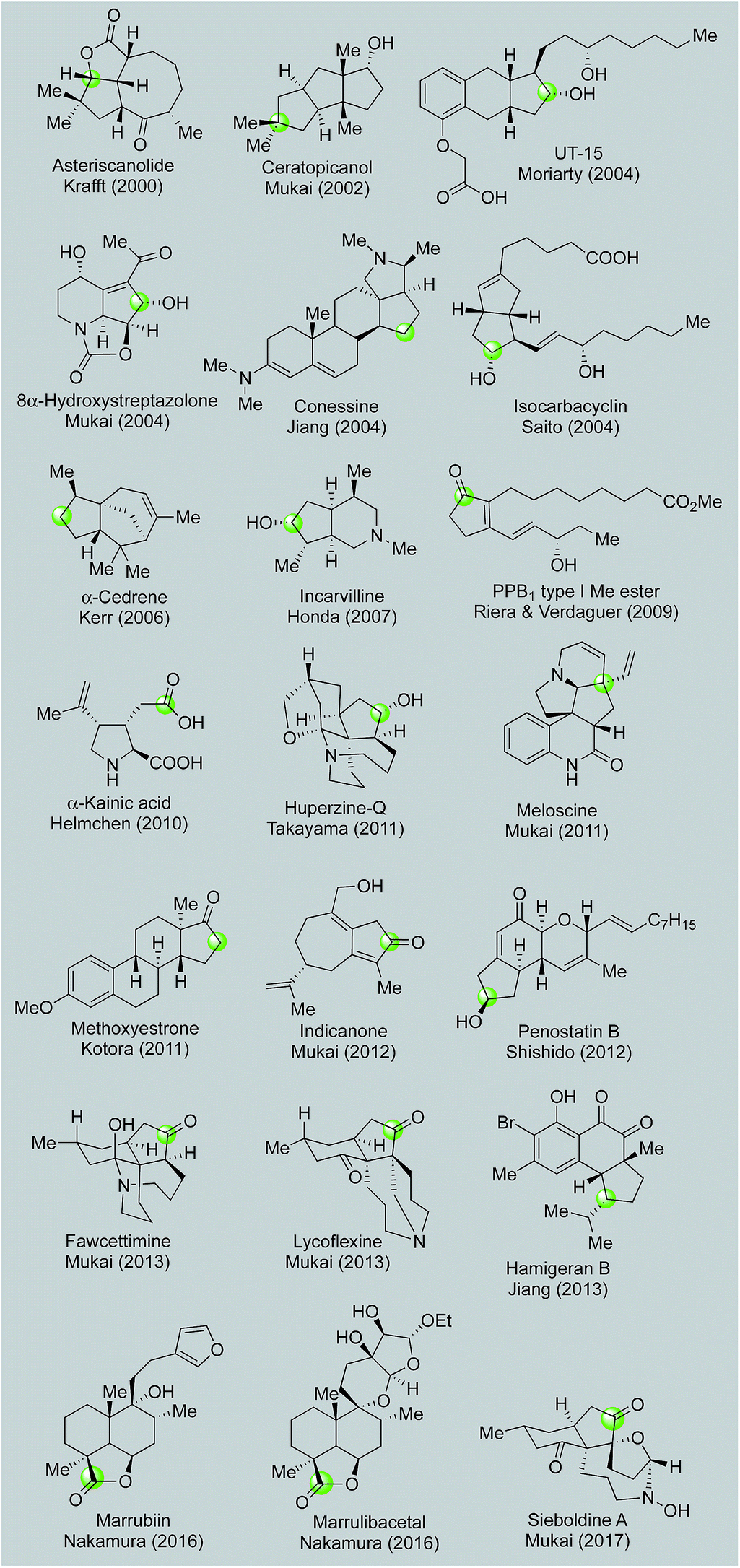 | ||
| Fig. 76 Selected natural products synthesized after 2000 via a Pauson–Khand reaction. | ||
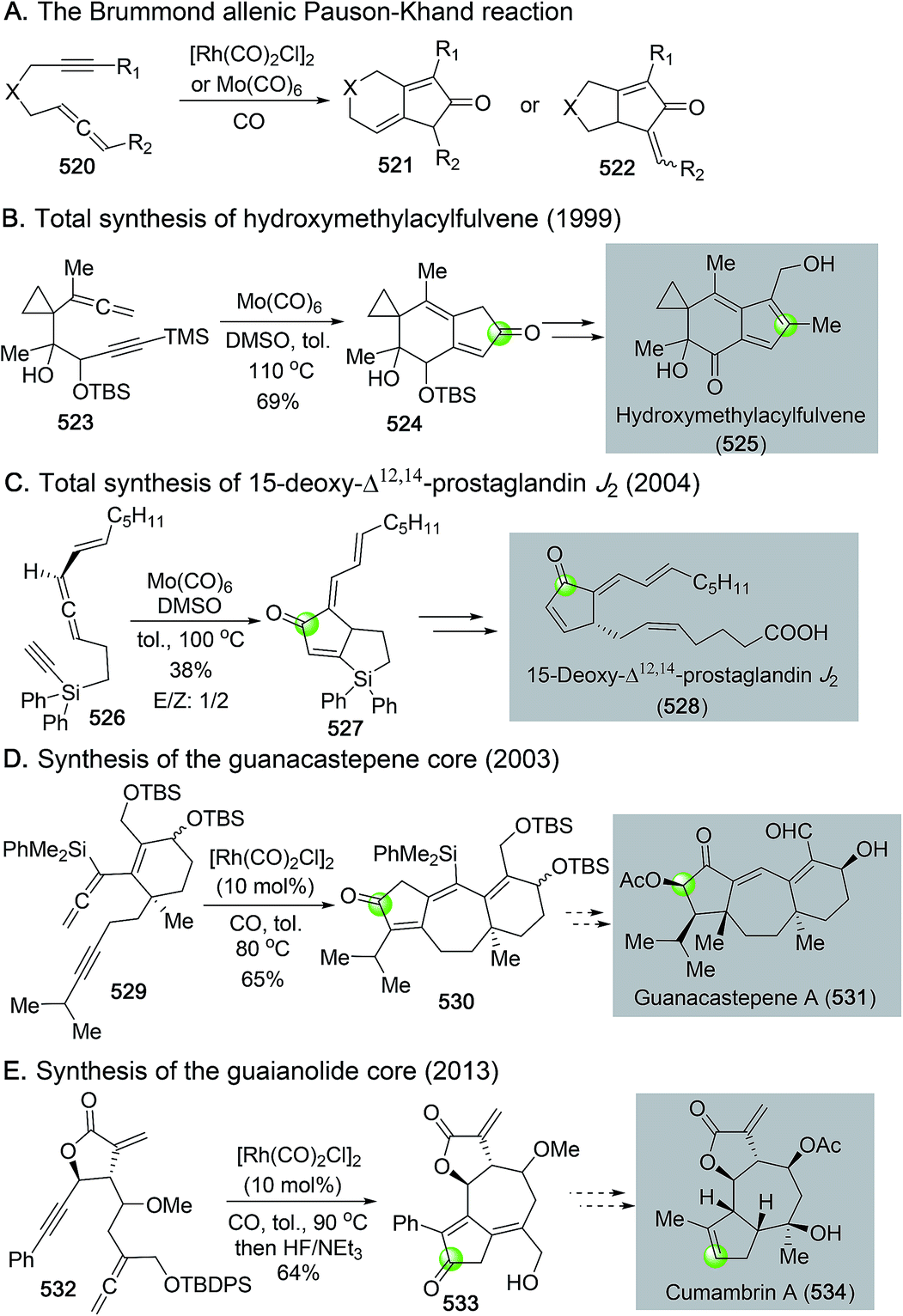 | ||
| Fig. 77 The Brummond allenic Pauson–Khand reaction. | ||
In 2013, Baran and co-workers reported a remarkable 14-step synthesis of ingenol by starting from (+)-3-carene (Fig. 78).232,233 One of their key steps involves a Brummond allenic [2 + 2 + 1] cycloaddition catalyzed by [Rh(CO)2Cl]2 to convert 535 to 536 with a fused 5-7-6-3 tetracyclic carbon skeleton. The latter was then advanced to 537 for a vinylogous pinacol rearrangement to deliver the in,out-[4.4.1] carbon skeleton of ingenol. The rearrangement was ultimately realized by treating 537 with BF3 etherate in CH2Cl2 at low reaction temperatures followed by quenching with NEt3 and MeOH and 538 was obtained in 80% yield. With 538 in hand, two SeO2-mediated allylic oxidations, one Martin's sulfurane dehydration, and removal of the carbonate delivered the final product ingenol 539 in a total fourteen steps.
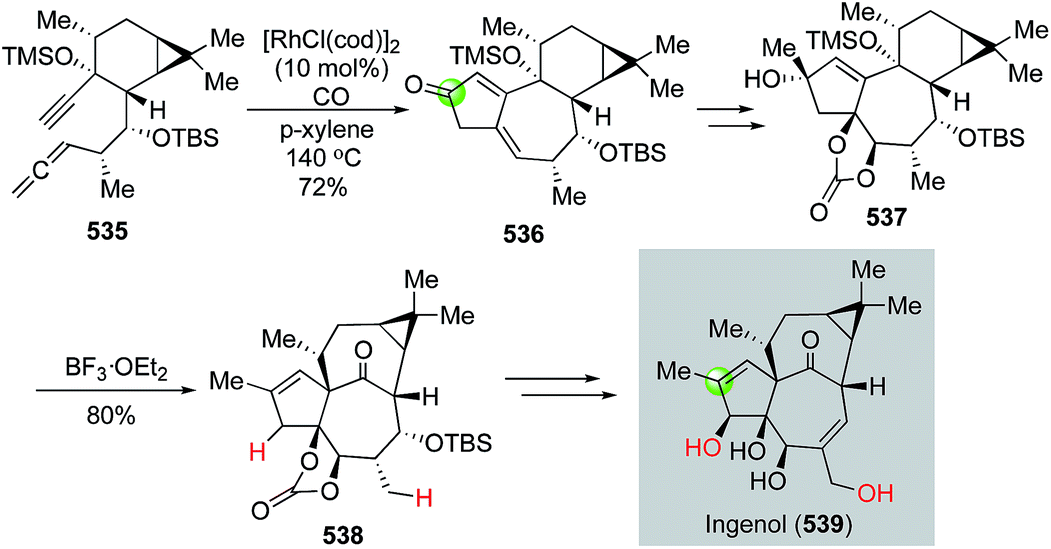 | ||
| Fig. 78 The Baran synthesis of ingenol (2013). | ||
Ileabethoxazole was isolated by Rodriguez and co-workers from the Caribbean octocoral Pseudopterogorgia elisabethae in 2006. This unique but scarce natural product has exhibited potent inhibition activity against Mycobacterium tuberculosis (H37Rv). In 2014, Williams and Shah reported an enantioselective total synthesis of ileabethoxazole (Fig. 79).234 They uncovered an iron carbonyl complex-mediated allenic [2 + 2 + 1] cycloaddition to convert 540 to cyclopentenone 542 under ambient reaction temperatures. Other carbonyl complexes such as Mo(CO)6 and Co2(CO)8 were ineffective mainly because these complexes require elevated reaction temperatures, but substrate 540 was heat sensitive. The iron-mediated allenic [2 + 2 + 1] cycloaddition was proposed to go through three-membered iron metallacycle 541. After converting 542 to 543, an intramolecular aldol condensation was employed to form the central aromatic ring and product 544 was obtained in 93% with KOt-Bu as base. Compound 544 was eventually transformed to ileabethoxazole after introduction of the three chiral centers in the cyclopentane ring and installation of the side chain.
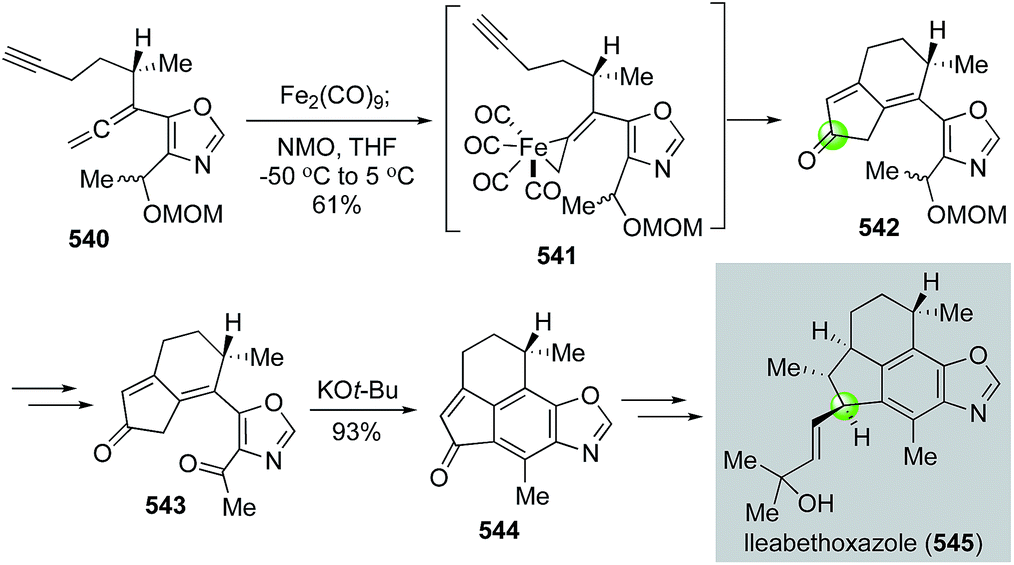 | ||
| Fig. 79 The Williams synthesis of ileabethoxazole (2014). | ||
In 2015, Cramer and Heinz revealed an elegant total synthesis of fijiolide A, a secondary marine metabolite with inhibition activity against transcription factor NFκB (Fig. 80).235 Structurally, fijiolide A is believed to be derived from a putative enediyne precursor pre-fijiolide via a biradical Bergman cyclization followed by chlorination to form the aromatic ring embedded in the 5-5-6 fused tricyclic system. Their synthesis commenced with a boronate-templated [2 + 2 + 2] cyclotrimerization of 546, 547, and 548 catalyzed by Cp*Ru(cod)Cl to yield arylboronate 549 in 59% yield. A subsequent Cu-catalyzed chlorination delivered 550 in 70% yield. After removal of the three silyl groups and isomerization of the propargyl group to an allene with TBAF (cf.551) and protecting the 1,2-diol as TBS-ethers, a Mo(CO)6-mediated allenic Pauson–Khand reaction gave indenylcyclopentenone 552, which was ultimately advanced to fijiolide A (553).
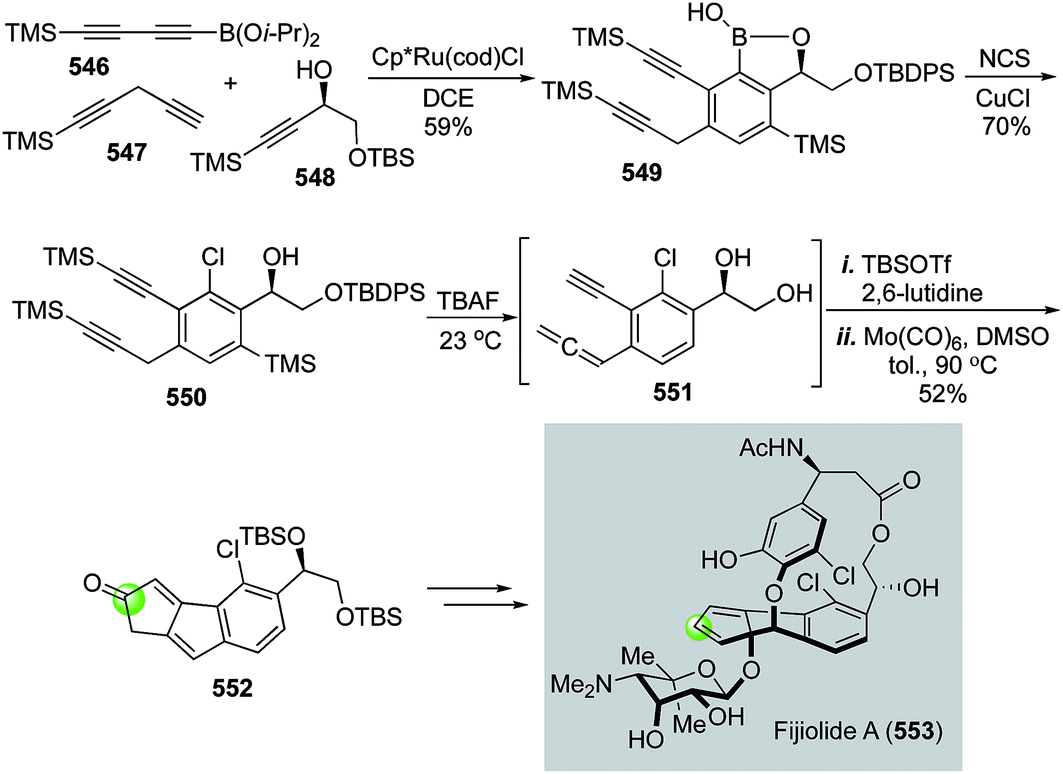 | ||
| Fig. 80 The Cramer synthesis of fijiolide A (2015). | ||
In addition to the aforementioned allene–alkyne [2 + 2 + 1] cycloadditions, allene–alkene [2 + 2 + 1] cycloaddition has been used in natural product synthesis. In 2008, Mukai and co-workers completed the total syntheses of two sesquiterpene natural products with a palladium-catalyzed carbonylative allenoate synthesis and allene–alkene Pauson–Khand reaction as key transformations (Fig. 81).236 Using the procedure developed by Tsuji et al.,237 propargyl carbonate 554 was elaborated to allenoate 555 with Pd(OAc)2/PPh3 as catalyst in MeOH under 10 atm of carbon monoxide. A subsequent [RhCl(cod)]2-catalyzed intramolecular [2 + 2 + 1] cycloaddition led to the formation of 556, which was then converted to sesquiterpenoid 557 and its analog.
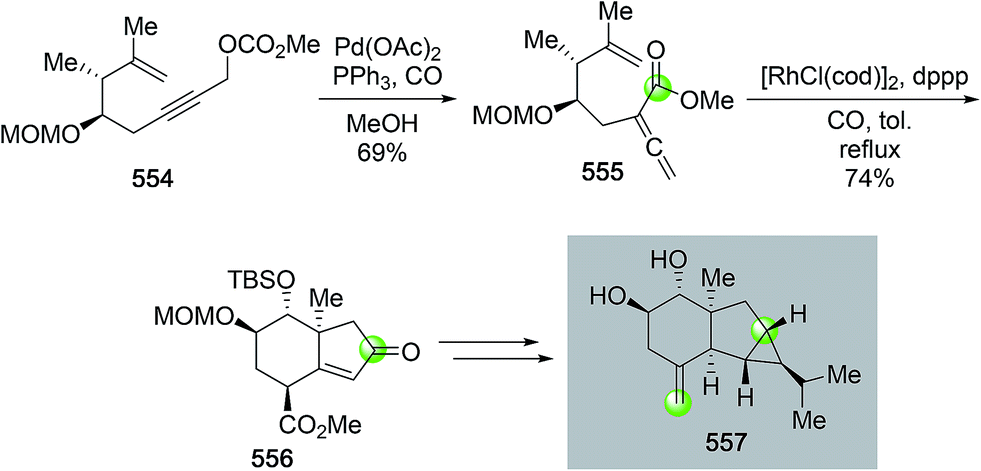 | ||
| Fig. 81 The Mukai synthesis of sesquiterpenoid 557 (2008). | ||
Perforanoid A was isolated from Meliaceae and Simaroubaceae species. Its gross structure was determined by NMR analysis, but the C10 configuration remained unclear. Hao, Yang and co-workers had recourse to total synthesis to solve this structural issue after failing in obtaining a crystal structure of the natural product. Their synthesis features a [Rh(CO)2Cl]2-catalyzed intramolecular [2 + 2 + 1] cycloaddition to build the 5-6-5 fused tricyclic product 559 from 558 in 85% yield (Fig. 82).238 Rapid access to 559 enabled them to prepare both perforanoid A and its C10 epimer and finally elucidate the correct configuration at C10.
 | ||
| Fig. 82 The Hao and Yang synthesis of perforanoid A (2016). | ||
4.3. Rh-catalyzed [n + m + 1] carbonylative cycloaddition
Pioneered by Wender,239–244 Yu,245 Tang,246 Evans,247 Bower248–250 and others, the rhodium-catalyzed [n + m + 1] carbonylative cycloaddition reactions have been effective methods in building cyclic ketones with different ring sizes. In this section, the application of rhodium-catalyzed [3 + 2 + 1], [5 + 1], and [5 + 2 + 1] cycloadditions in natural product synthesis are highlighted.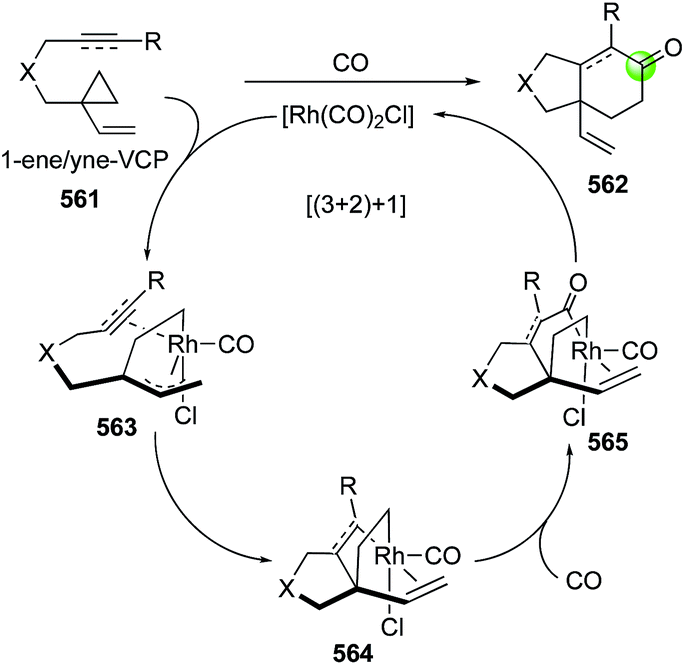 | ||
| Fig. 83 Rhodium-catalyzed [3 + 2 + 1] carbonylative cycloaddition. | ||
Since its discovery, the rhodium-catalyzed 1-yne/ene-vinylcyclopropanes [3 + 2 + 1] carbonylative cycloaddition process has been utilized by Yu and co-workers to synthesize a series of natural products including α-agarofuran, lycoramine,254 galanthamine, gracilamine,255 and cloven-2,9-dione (Fig. 84).256 In general, the yields for the key [3 + 2 + 1] cycloadditions are good to excellent with modest to high diastereoselectivity. For the cases of 566, 573, and 576 containing a 1-yne-vinylcyclopropane moiety, the corresponding cyclohexenones were produced. Substrate 569 contains a 1-ene-vinylcyclopropane, leading to the formation of the cyclohexanone product.
 | ||
| Fig. 84 Rhodium-catalyzed [3 + 2 + 1] cycloaddition in total synthesis by Yu and co-workers. | ||
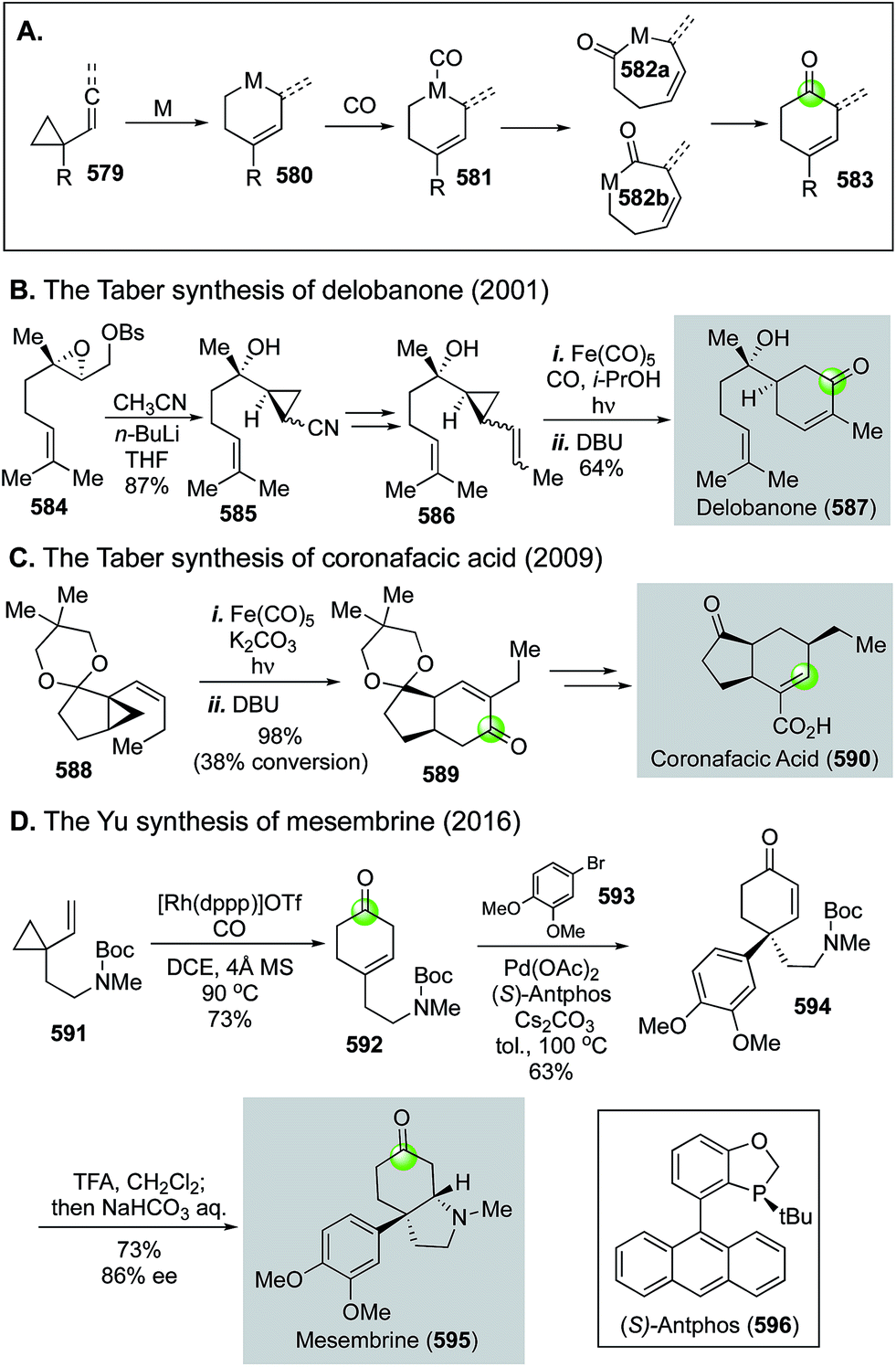 | ||
| Fig. 85 Vinylcyclopropane [5 + 1] cycloaddition. | ||
In addition to using stoichiometric iron carbonyl complexes, de Meijere et al. revealed that the vinylcyclopropane [5 + 1] cycloaddition can be catalyzed by Co2(CO)8 and [Rh(CO)2Cl]2 under carbon monoxide atmosphere.264 This catalytic process was later generalized by Yu and co-workers by using cationic rhodium catalysts.265 They further demonstrated the catalytic vinylcyclopropane [5 + 1] cycloaddition process in an asymmetric total synthesis of mesembrine (Fig. 85D).266 As shown, with a 15 mol% of [Rh(dppp)]OTf, under 1 atm of carbon monoxide in DCE at 90 °C, vinylcyclopropane 591 was converted to 592 in 73% yield. A subsequent Pd(OAc)2/(S)-Antphos-catalyzed enantioselective intermolecular Heck reaction using arylbromide 593 afforded 594 with an all-carbon quaternary center. After removal of the Boc protecting group and an intramolecular aza-conjugate addition, mesembrine 595 was obtained in 73% and 86% ee.
When an allenylcyclopropane is used as substrate in the [5 + 1] carbonylative cycloaddition, a cyclohexenone product with an additional exo-olefin can be prepared. A few transition metal complexes such as Co2(CO)8,267,268 IrCl(CO)(PPh3)2,269 and [Rh(CO)2Cl]2270 have been shown to be able to promote or catalyze this [5 + 1] cycloaddition mode. In 2012, Tang and Zhang utilized the rhodium-catalyzed allenylcyclopropane [5 + 1] cycloaddition strategy to build the key cyclohexenone core of welwitindolinones.271 As shown in Fig. 86A, chlorination of α-diazoester 597 delivered unstable chloro-α-diazoester 598, which upon subjection to the Du Bois' Rh2(esp)2-catalyzed intramolecular cyclopropanation was converted to 599 in 50–60% yield. Compound 599 was then advanced to the key allenylcyclopropane intermediate 600 in six steps. A [Rh(CO)2Cl]2-catalyzed [5 + 1] cycloaddition converted 600 to 601 containing the desired indole and cyclohexenone ring for their future synthesis of welwitindolinones. In 2016, Yu et al. reported their second formal synthesis of galanthamine (Fig. 86B).272 Their new approach features an early stage [Rh(CO)2Cl]2-catalyzed [5 + 1] cycloaddition to convert allenylcyclopropane 603 to cyclohexenone 604. Subsequent CBS-reduction of the resulting ketone gave allylic alcohol 605 in 79% yield and 97% ee. After transforming 605 to 606 in three steps, they used an intramolecular radical cyclization initiated by AIBN/Bu3SnH to provide 607, which was eventually advanced to galanthamine.
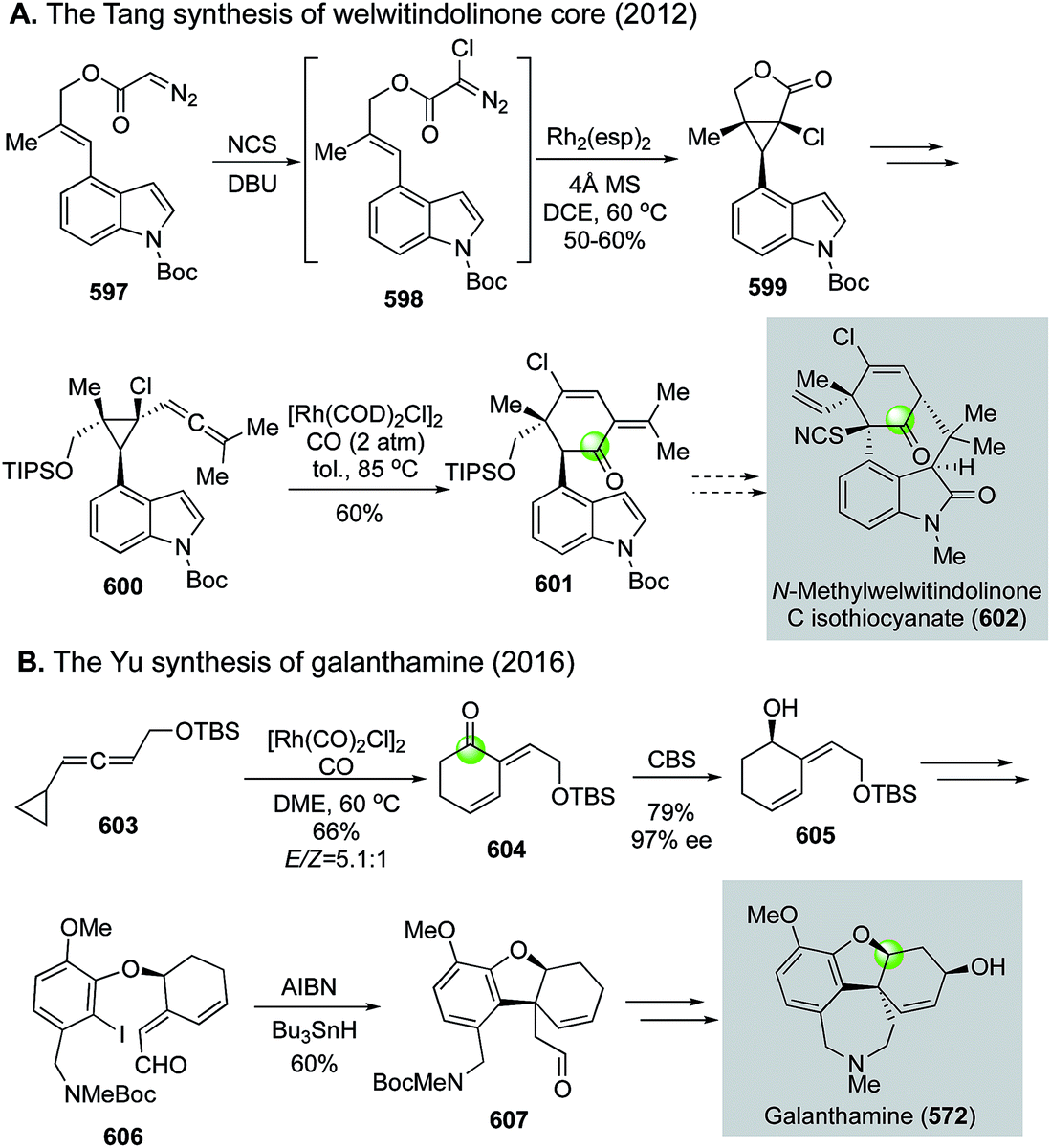 | ||
| Fig. 86 Allenylcyclopropane [5 + 1] cycloaddition. | ||
In 2013, Tang and co-workers revealed a different type of rhodium-catalyzed [5 + 1] cycloaddition involving 3-hydroxy-1,4-enyne as the 5-carbon component instead of using the aforementioned vinyl/allenylcyclopropanes as substrates (Fig. 87).273 As a result, polysubstituted phenols could be produced. They have employed this novel transformation to complete a total synthesis of mahanimbine (610) and a few other carbazole-containing natural products.274 For example, upon the treatment of a catalytic amount of [Rh(CO)2Cl]2 under carbon monoxide atmosphere in DCE at 60 °C, 3-hydroxy-1,4-enyne 608 was converted to carbazole 609 in a single step. The catalytic cycle was proposed to initiate with coordination of enyne substrate on the rhodium metal center to trigger a tandem cyclization process to convert complex 611 to rhodacycle 612. The latter would then undergo a 6π-electrocyclic ring opening reaction to give rhodium carbenoid 613. Carbon monoxide insertion would convert 613 to ketene 614. A subsequent 6π-electrocyclic ring closure process would deliver enone 615, which could tautomerize to carbazole 609. After removal of the Boc-protecting group, a simple heating of the resulting carbazole with citral completed the total synthesis of mahanimbine.
 | ||
| Fig. 87 The Tang synthesis of mahanimbine (2016). | ||
In 2016, Tang and co-workers disclosed a modified rhodium-catalyzed [5 + 1] carbonylative cycloaddition to synthesize highly substituted benzofurans (Fig. 88, 616 → 617).275 In this case, 1-furylpropargylic esters are used as substrates. As shown in Fig. 88, a rhodium-catalyzed 1,2-pivalate migration via618 would convert 616 to 619, which would then undergo a sequence of carbon monoxide migratory insertion, ketene formation, 6π-electrocyclic ring closure and tautomerization to give rise to benzofuran 617. Compound 617 was then converted to a few benzofuran natural products including viniferifuran (622) via two palladium-catalyzed cross coupling reactions to append the styrene and aryl moieties.
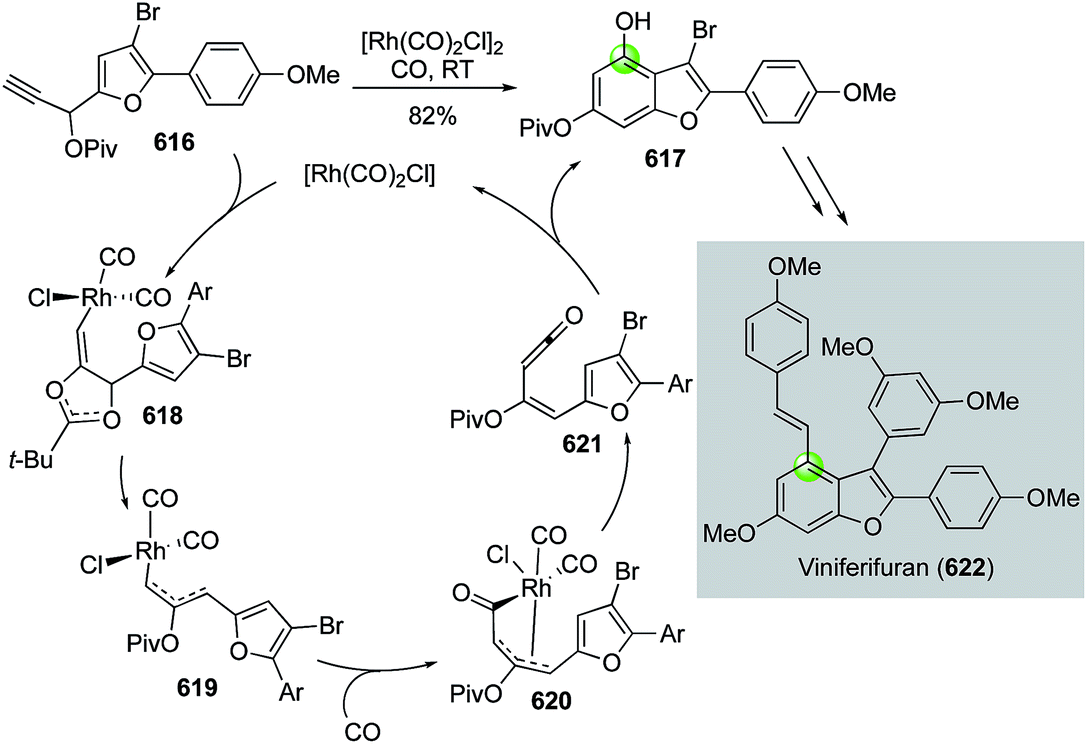 | ||
| Fig. 88 The Tang synthesis of viniferifuran (2017). | ||
 | ||
| Fig. 89 [5 + 2 + 1] cycloaddition in total synthesis. | ||
They first utilized it for a total synthesis of hirsutene and related analogs in 2008 (Fig. 89C).279 Upon the treatment of siloxy-ene-vinylcyclopropane 631 derived from a Simmons–Smith reaction with 5 mol% of [Rh(CO)2Cl]2 catalyst under 0.2 atm of carbon monoxide in dioxane at 80 °C, desired product 632 was obtained, which underwent a subsequent acid-promoted transannular aldol reaction to deliver 633 in 62% yield. After a radical deoxygenation process via oxalate 634 and a Wittig methylenation, a concise total synthesis of hirsutene was completed. In 2009, Yu and co-workers employed a similar strategy to complete formal syntheses of asterisca-3(15),6-diene (639) and pentalenene (640).280 The rhodium-catalyzed [5 + 2 + 1] cycloaddition converted 636 to 637 in 65% yield (Fig. 89D). The latter served as a key intermediate for their asterisca-3(15),6-diene and pentalenene syntheses. The same strategy was also used to build the cyclooctanone moiety of asteriscanolide (646, Fig. 89E).281 After forming cyclooctenone 642 from ene-vinylcyclopropane 641, an intramolecular acyl radical cyclization was employed to build the 5-membered lactone and afford 95% yield of 644, which was later converted to asteriscanolide.
4.4. Macroketonization
Catalytic carbonylation is also capable of forming macrocyclic ketones. One classic example is from the Stille–Hegedus collaborative synthesis of jatrophone (Fig. 90A), where they used the Stille carbonylation reaction to build the macrocyclic dienone system of jatrophone (647 → 648).282 Later in 2001, Rawal and co-workers used stannyl triflate 649 to create the macrocyclic ketone of phomactins C and D (Fig. 90B).283 Under the Stille carbonylative cross coupling conditions, product 650 was produced in modest yield. This carbonylative macrocyclization offers an efficient approach to access the bridged bicyclic core of the phomactins. In general, despite the power of assembling macrocyclic ketones, the carbonylative macroketonization is underused. | ||
| Fig. 90 The Stille carbonylative macroketonization. | ||
5. Miscellaneous catalytic carbonylative cyclizations
The carbonylative benzannulation mediated by Fisher carbenes, the Dötz annulation, is highly effective in synthesizing densely oxygenated arenes (Fig. 91A). The three-component reaction involves an α,β-unsaturated chromium carbene, an alkyne, and a molecule of carbon monoxide and goes through a sequential alkyne insertion and carbon monoxide insertion to form vinylketene 657 for a 6π-electrocyclic ring closure to form phenol product 658. Since its discovery, the Dötz annulation has been frequently used in natural product synthesis.284 Two recent examples are highlighted in this section. | ||
| Fig. 91 The Dötz annulation. | ||
In 2006, Quayle and co-workers utilized the Dötz annulation to convert Fisher carbene 659 and alkyne 660 to tricyclic phenol 661 in 31% yield (Fig. 91B). The bulky TBS group was used to impart high regioselectivity and was removed at a later stage. A formal synthesis of aflatoxin B2 was achieved by using the Dötz annulation strategy.285
The second example is from Nakata, Saikawa and co-workers (Fig. 91C),286 who employed an impressive macrocyclic Dötz annulation developed by Wulff287 and co-workers to construct the cyclophane motif of kendomycin, a potent antibacterial and anticancer natural product isolated from Streptomyces. The macrocyclization precursor 665 was prepared by reacting alcohol 663 with an acetoxychromium carbene complex generated in situ from chromate 664via AcBr treatment. The subsequent macrocyclic Dötz annulation occurred in toluene at 50 °C to provide cyclophane 666 in 58% yield. After protecting the resulting phenol as TBS-ether, a sequence of macrocyclic Claisen rearrangement and acetate formation delivered 667 in 85% yield. The in situ trapping of the newly formed phenol with Ac2O/DMAP was critical for preventing the formation of a dihydrobenzofuran byproduct via a 5-exo-trig cyclization of the phenol on the exo-methylene moiety. Compound 667 was eventually transformed to kendomycin in ten steps.
In 1998, Mitsudo and co-workers developed a hydroquinone synthesis via a Ru3(CO)12-catalyzed [2 + 2 + 1 + 1] cycloaddition of an alkyne, 2-norbornene and two molecules of carbon monoxide.288 This four-component process can lead to unsymmetrically substituted hydroquinones in high yields and has been utilized by Sarpong and co-workers in their formal synthesis of cis-trikentrin A and herbindole B.289 As shown in Fig. 92, the Ru3(CO)12-catalyzed [2 + 2 + 1 + 1] cycloaddition united alkyne 669, strained bicyclic alkene 670 and two carbon monoxide molecules to provide symmetric hydroquinone 671 in 39% yield. The two hydroxyl groups were then differentially converted to a triflate and a tosylate for a sequential palladium-catalyzed Stille cross coupling to install the methyl group and Buchwald–Hartwig amination to introduce the free amine group. After obtaining aniline triflamide 674 from 673, Sarpong and co-workers developed a tandem palladium-catalyzed intramolecular C–H amination and oxidation of the resulting indoline to provide indole 675 in 66% yield. Indole 675 was then converted to a Kerr's intermediate for their herbindole B synthesis.290 Additionally, the triflate group of 674 was reduced via Pd/C-catalyzed hydrogenation. Following a similar process for the herbindole synthesis, the reduced product 676 was then advanced to another Kerr's intermediate to complete a formal synthesis of cis-trikentrin A.
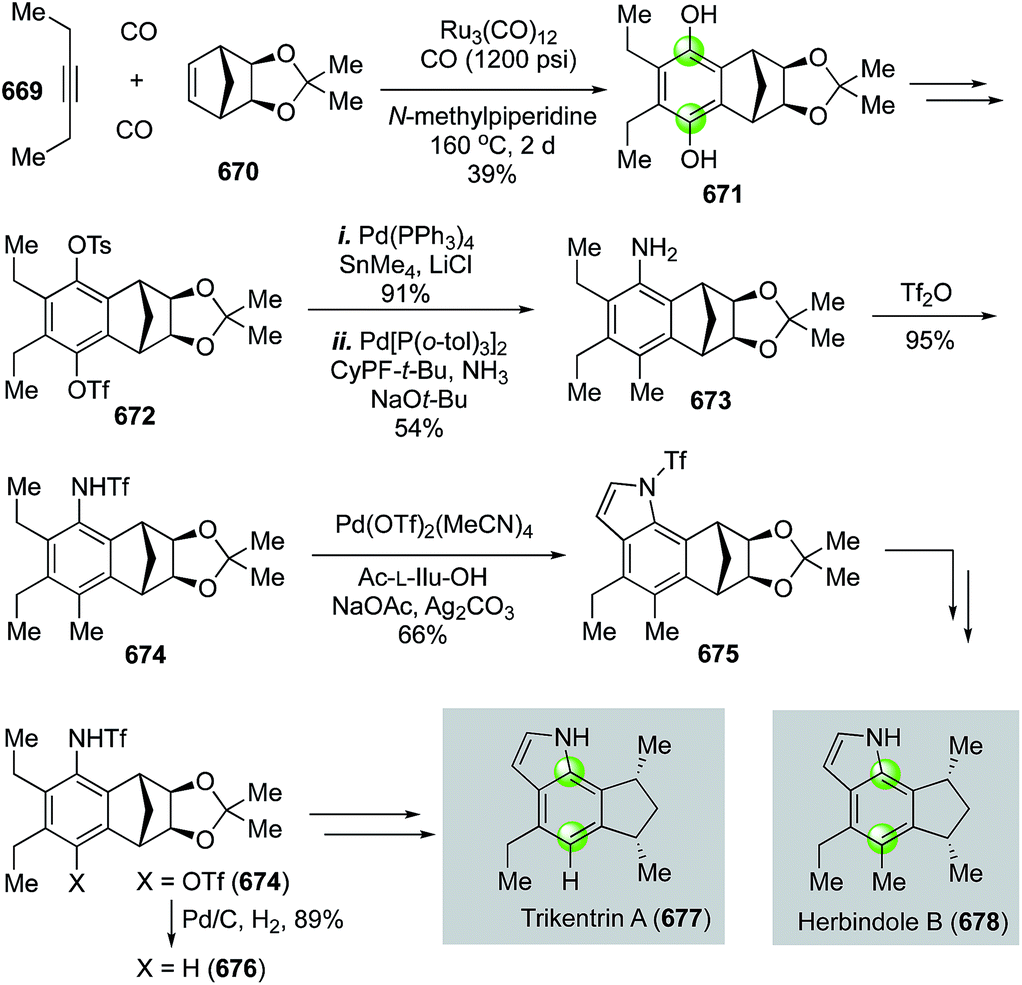 | ||
| Fig. 92 The Sarpong formal synthesis of trikentrin A and herbindole B (2016). | ||
6. Conclusions
In summary, we have summarized recent applications of carbonylative cyclizations in total synthesis of natural products. The highlighted carbonylative cyclization reactions include carbonylation of epoxide, carbonylative (macro)lactonization/lactamization, the Semmelhack reaction, tandem hydroformylation–cyclization, the Pauson–Khand reaction, carbonylative C–H activation cyclization, the Stille/Suzuki carbonylation, the [n + m + 1] carbonylative cycloaddition, the Dötz annulation, and others. In these transformations, carbon monoxide serves as an important one-carbon linchpin to quickly increase structural complexity under either stoichiometric or catalytic conditions. In general, these carbonylative cyclizations are effective in forming 4 to 7-membered ring systems. There are some examples of forming macrocycles, but this area needs further expansion in terms of both reaction type and efficiency. Carbonylative cyclization of alkyl electrophiles and/or nucleophiles,291 enantioselective carbonylative cyclization, carbonylative C–H activation cyclization particularly the non-directed variety are in need of future development as well.7. Conflicts of interest
There are no conflicts to declare.8. Acknowledgements
We thank NSF (CAREER 1553820) and the ACS petroleum research foundation (PRF# 54896-DNI1) for financial support. M. D. thanks Eli Lilly for an unrestricted grant support via the Eli Lilly Grantee Award. K. M. thanks the financial support from the China Scholarship Council (CSC).9. Notes and references
- Y. Bai, D. C. Davis and M. Dai, J. Org. Chem., 2017, 82, 2319 CrossRef CAS.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS.
- S. Quintero-Duque, K. M. Dyballa and I. Fleischer, Tetrahedron Lett., 2015, 56, 2634 CrossRef CAS.
- P. H. Gehrtz, V. Hirschbeck, B. Ciszek and I. Fleischer, Synthesis, 2016, 48, 1573 CrossRef CAS.
- W. Fang, H. Zhu, Q. Deng, S. Liu, X. Liu, Y. Shen and T. Tu, Synthesis, 2014, 46, 1689 CrossRef.
- G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS.
- C. F. J. Barnard, Organometallics, 2008, 27, 5402 CrossRef CAS.
- X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447 CrossRef CAS.
- T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580 CrossRef CAS.
- L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310 CrossRef CAS.
- H. Konishi and K. Manabe, Synlett, 2014, 25, 1971 CrossRef CAS.
- H. W. Yang and D. Romo, Tetrahedron, 1999, 55, 6403 CrossRef CAS.
- J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 6358 CrossRef CAS.
- Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi., Chem. Rev., 2014, 114, 8199 CrossRef CAS.
- R. W. Bates, R. Fernandez-Moro and S. V. Ley, Tetrahedron, 1991, 47, 9929 CrossRef CAS.
- Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174 CrossRef CAS.
- M. Mulzer, B. T. Whiting and G. W. Coates, J. Am. Chem. Soc., 2013, 135, 10930 CrossRef CAS.
- Y. Liang, N. Hnatiuk, J. M. Rowley, B. T. Whiting, G. W. Coates, P. R. Rablen, M. Morton and A. R. Howell, J. Org. Chem., 2011, 76, 9962 CrossRef CAS.
- Q. Chen, M. Mulzer, P. Shi, P. J. Beuning, G. W. Coates and G. A. O'Doherty, Org. Lett., 2011, 13, 6592 CrossRef CAS.
- M. Mulzer, B. J. Tiegs, Y. Wang, G. W. Coates and G. A. O'Doherty, J. Am. Chem. Soc., 2014, 136, 10814 CrossRef CAS PubMed.
- S. N. Goodman and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 4703 CrossRef CAS.
- J. Willwacher, B. Heggen, C. Wirtz, W. Thiel and A. Fürstner, Chem.–Eur. J., 2015, 21, 10416 CrossRef CAS.
- A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS.
- A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS.
- A. Schoenberg and R. F. Heck, J. Am. Chem. Soc., 1974, 96, 7761 CrossRef CAS.
- A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef.
- H. Li, Q. Chen, Z. Lu and A. Li, J. Am. Chem. Soc., 2016, 138, 15555 CrossRef CAS.
- A. M. Camelio, T. C. Johnson and D. Siegel, J. Am. Chem. Soc., 2015, 137, 11864 CrossRef CAS.
- S.-i. Kusaka, S. Dohi, T. Doi and T. Takahashi, Tetrahedron Lett., 2003, 44, 8857 CrossRef CAS.
- H. Herzner, E. R. Palmacci and P. H. Seeberger, Org. Lett., 2002, 4, 2965 CrossRef CAS.
- A. W. Schuppe and T. R. Newhouse, J. Am. Chem. Soc., 2017, 139, 631 CrossRef CAS.
- J. E. Rixson, B. W. Skelton, G. A. Koutsantonis, K. M. Gericke and S. G. Stewart, Org. Lett., 2013, 15, 4834 CrossRef CAS.
- J. A. Marshall and J. Lebreton, Tetrahedron Lett., 1987, 28, 3323 CrossRef CAS.
- A. S. Franklin and L. E. Overman, Chem. Rev., 1996, 96, 505 CrossRef CAS.
- S. Aoyagi, S. Hirashima, K. Saito and C. Kibayashi, J. Org. Chem., 2002, 67, 5517 CrossRef CAS.
- For a review, D. A. Spiegel, J. T. Njardarson, I. M. McDonald and J. L. Wood, Chem. Rev., 2003, 103, 2691 CrossRef CAS.
- For a recent total synthesis: J. C. Leung, A. A. Bedermann, J. T. Njardarson, D. A. Spiegel, G. K. Murphy, N. Hama, B. M. Twenter, P. Dong, T. Shirahata, I. M. McDonald, M. Inoue, N. Taniguchi, T. C. McMahon, C. M. Schneider, N. Tao, B. M. Stoltz and J. L. Wood, Angew. Chem., Int. Ed., 2018, 57, 1991 CrossRef CAS (references 3 and 4 therein).
- M. M. Bio and J. L. Leighton, J. Org. Chem., 2003, 68, 1693 CrossRef CAS.
- T. Yao, D. Yue and R. C. Larock, J. Org. Chem., 2005, 70, 9985 CrossRef CAS PubMed.
- N. A. McGrath, C. A. Lee, H. Araki, M. Brichacek and J. T. Njardarson, Angew. Chem., Int. Ed., 2008, 47, 9450 CrossRef CAS.
- A. C. Tadd, M. R. Fielding and M. C. Willis, Chem. Commun., 2009, 6744 RSC.
- Y. Aoyagi, A. Yamazaki, C. Nakatsugawa, H. Fukaya, K. Takeya, S. Kawauchi and H. Izumi, Org. Lett., 2008, 10, 4429 CrossRef CAS.
- S. Levin, R. R. Nani and S. E. Reisman, J. Am. Chem. Soc., 2011, 133, 774 CrossRef CAS.
- D. F. Taber and C. M. Paquette, J. Org. Chem., 2014, 79, 3410 CrossRef CAS.
- H.-D. Hao and D. Trauner, J. Am. Chem. Soc., 2017, 139, 4117 CrossRef CAS.
- I. T. Chen, I. Baitinger, L. Schreyer and D. Trauner, Org. Lett., 2014, 16, 166 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, Angew. Chem., Int. Ed., 2014, 53, 11351 CrossRef CAS.
- T. R. Hoye and L. Tan, Tetrahedron Lett., 1995, 36, 1981 CrossRef CAS.
- H. Xu, H. Tang, H. Feng and Y. Li, J. Org. Chem., 2014, 79, 10110 CrossRef CAS.
- G. Hikosaka, Y. Hattori and H. Makabe, Tetrahedron: Asymmetry, 2014, 25, 1367 CrossRef CAS.
- Y. Kurogome, Y. Hattori and H. Makabe, Tetrahedron Lett., 2014, 55, 2822 CrossRef CAS.
- V. L. Poral, D. P. Furkert and M. A. Brimble, Org. Lett., 2015, 17, 6214 CrossRef CAS.
- S. G. Davies, P. M. Roberts, P. T. Stephenson, H. R. Storr and J. E. Thomson, Tetrahedron, 2009, 65, 8283 CrossRef CAS.
- G. Nemecek, R. Thomas, H. Goesmann, C. Feldmann and J. Podlech, Eur. J. Org. Chem., 2013, 6420 CrossRef CAS.
- R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS.
- R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS.
- H. Zhang, R. Shi, P. Gan, C. Liu, A. Ding, Q. Wang and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 5204 CrossRef CAS.
- Z. Lian, S. D. Friis and T. Skrydstrup, Chem. Commun., 2015, 51, 1870 RSC.
- X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 7316 CrossRef CAS.
- J. Tjutrins and B. A. Arndtsen, J. Am. Chem. Soc., 2015, 137, 12050 CrossRef CAS.
- Y. Shin, C. Yoo, Y. Moon, Y. Lee and S. Hong, Chem.–Asian J., 2015, 10, 878 CrossRef CAS PubMed.
- D. C. Davis, K. L. Walker, C. Hu, R. N. Zare, R. M. Waymouth and M. Dai, J. Am. Chem. Soc., 2016, 138, 10693 CrossRef CAS PubMed.
- G. Kiss, Chem. Rev., 2001, 101, 3435 CrossRef CAS.
- E. Yoneda, T. Kaneko, S.-W. Zhang, K. Onitsuka and S. Takahashi, Org. Lett., 2000, 2, 441 CrossRef CAS PubMed.
- R. W. Bates and S. Sridhar, J. Org. Chem., 2008, 73, 8104 CrossRef CAS.
- Y. Wang, L. Zhu, Y. Zhang and R. Hong, Angew. Chem., Int. Ed., 2011, 50, 2787 CrossRef CAS.
- Y.-A. Zhang, Q. Liu, C. Wang and Y. Jia, Org. Lett., 2013, 15, 3662 CrossRef CAS PubMed.
- N. M. Kablaoui, F. A. Hicks and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 4424 CrossRef CAS.
- W. E. Crowe and A. T. Vu, J. Am. Chem. Soc., 1996, 118, 1557 CrossRef CAS.
- N. Chatani, T. Morimoto, Y. Fukumoto and S. Murai, J. Am. Chem. Soc., 1998, 120, 5335 CrossRef CAS.
- J. Adrio and J. C. Carretero, J. Am. Chem. Soc., 2007, 129, 778 CrossRef CAS.
- S. K. Kang, K. J. Kim and Y. T. Hong, Angew. Chem., Int. Ed., 2002, 41, 1584 CrossRef CAS.
- P. Gao, P.-F. Xu and H. Zhai, J. Org. Chem., 2009, 74, 2592 CrossRef CAS.
- J. Chen, P. Gao, F. Yu, Y. Yang, S. Zhu and H. Zhai, Angew. Chem., Int. Ed., 2012, 51, 5897 CrossRef CAS.
- C. Tao, J. Zhang, X. Chen, H. Wang, Y. Li, B. Cheng and H. Zhai, Org. Lett., 2017, 19, 1056 CrossRef CAS.
- H.-H. Lu, M. D. Martinez and R. A. Shenvi, Nat. Chem., 2015, 7, 604 CrossRef CAS PubMed.
- M. Ohtawa, M. J. Krambis, R. Cerne, J. M. Schkeryantz, J. M. Witkin and R. A. Shenvi, J. Am. Chem. Soc., 2017, 139, 9637 CrossRef CAS PubMed.
- M. F. Semmelhack and C. Bodurow, J. Am. Chem. Soc., 1984, 106, 1496 CrossRef CAS.
- M. F. Semmelhack, C. Kim, N. Zhang, C. Bodurow, M. Sanner, W. Dobler and M. Meier, Pure Appl. Chem., 1990, 62, 2035 CAS.
- K. J. Light-Wahl, B. E. Winger and R. D. Smith, J. Am. Chem. Soc., 1993, 115, 5869 CrossRef CAS.
- G. A. Kraus and J. Li, J. Am. Chem. Soc., 1993, 115, 5859 CrossRef CAS.
- K. R. Hornberger, C. L. Hamblett and J. L. Leighton, J. Am. Chem. Soc., 2000, 122, 12894 CrossRef CAS.
- H. H. Jung, J. R. Seiders and P. E. Floreancig, Angew. Chem., Int. Ed., 2007, 46, 8464 CrossRef CAS.
- J. D. White, C. L. Kranemann and P. Kuntiyong, Org. Lett., 2001, 3, 4003 CrossRef CAS.
- J. Carpenter, A. B. Northrup, d. Chung, J. J. M. Wiener, S. G. Kim and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2008, 47, 3568 CrossRef CAS.
- J. A. Marshall and M. M. Yanik, Tetrahedron Lett., 2000, 41, 4717 CrossRef CAS.
- L. F. Tietze, R. Fischer, H. J. Guder, A. Goerlach, M. Meumann and T. Krach, Carbohydr. Res., 1987, 164, 177 CrossRef CAS.
- Z. Yang, B. Zhang, G. Zhao, J. Yang, X. Xie and X. She, Org. Lett., 2011, 13, 5916 CrossRef CAS.
- Y. Lu, I. S. Kim, A. Hassan, D. J. Del Valle and M. J. Krische, Angew. Chem., Int. Ed., 2009, 48, 5018 CrossRef CAS.
- S. B. Han, A. Hassan, I. S. Kim and M. J. Krische, J. Am. Chem. Soc., 2010, 132, 15559 CrossRef CAS.
- L. F. Tietze, D. A. Spiegl, F. Stecker, J. Major, C. Raith and C. Große, Chem.–Eur. J., 2008, 14, 8956 CrossRef CAS.
- P. Y. Hayes and W. Kitching, J. Am. Chem. Soc., 2002, 124, 9718 CrossRef CAS.
- Z. Li, Y. Gao, Z. Jiao, N. Wu, D. Z. Wang and Z. Yang, Org. Lett., 2008, 10, 5163 CrossRef CAS.
- Z. Li, Y. Gao, Y. Tang, M. Dai, G. Wang, Z. Wang and Z. Yang, Org. Lett., 2008, 10, 3017 CrossRef CAS.
- J. B. Werness and W. Tang, Org. Lett., 2011, 13, 3664 CrossRef CAS.
- C. L. Nesbitt and C. S. P. McErlean, Tetrahedron Lett., 2009, 50, 6318 CrossRef CAS.
- Q. Xiao, W. W. Ren, Z. X. Chen, T. W. Sun, Y. Li, Q. D. Ye, J. X. Gong, F. K. Meng, L. You, Y. F. Liu, M. Z. Zhao, L. M. Xu, Z. H. Shan, Y. Shi, Y. F. Tang, J. H. Chen and Z. Yang, Angew. Chem., Int. Ed., 2011, 50, 7373 CrossRef CAS.
- M. Dai, B. Liang, C. Wang, J. Chen and Z. Yang, Org. Lett., 2004, 6, 221 CrossRef CAS.
- X.-S. Xu, Z.-W. Li, Y.-J. Zhang, X.-S. Peng and H. N. C. Wong, Chem. Commun., 2012, 48, 8517 RSC.
- C. Ebner and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 11227 CrossRef CAS PubMed.
- E.-i. Negishi, C. Copéret, S. Ma, T. Mita, T. Sugihara and J. M. Tour, J. Am. Chem. Soc., 1996, 118, 5904 CrossRef CAS.
- V. K. Aggarwal, P. W. Davies and W. O. Moss, Chem. Commun., 2002, 972 RSC.
- C. Liu and R. A. Widenhoefer, J. Am. Chem. Soc., 2004, 126, 10250 CrossRef CAS.
- S. Li, F. Li, J. Gong and Z. Yang, Org. Lett., 2015, 17, 1240 CrossRef CAS.
- V. K. Aggarwal, P. W. Davies and A. T. Schmidt, Chem. Commun., 2004, 1232 RSC.
- For a review: R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675 CrossRef CAS.
- Z. A. Kasun, X. Gao, R. M. Lipinski and M. J. Krische, J. Am. Chem. Soc., 2015, 137, 8900 CrossRef CAS PubMed.
- For a review: Y. Li, X. Yin and M. Dai, Nat. Prod. Rep., 2017, 34, 1185 RSC.
- T. Takahashi, S. Kusaka, T. Doi, T. Sunazuka and S. Ōmura, Angew. Chem., Int. Ed., 2003, 42, 5230 CrossRef CAS PubMed.
- B. Schmalzbauer and D. Menche, Org. Lett., 2015, 17, 2956 CrossRef CAS PubMed.
- Y. Bai, D. C. Davis and M. Dai, Angew. Chem., Int. Ed., 2014, 53, 6519 CrossRef CAS.
- F. Manoni, C. Rumo, L. Li and P. G. Harran, J. Am. Chem. Soc., 2018, 140, 1280 CrossRef CAS.
- A. Hassan, I. A. Townsend and M. J. Krische, Chem. Commun., 2011, 47, 10028 RSC.
- I. Shin, S. Hong and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14246 CrossRef CAS.
- S. L. Schreiber, J. Am. Chem. Soc., 1980, 102, 6163 CrossRef CAS.
- Y. Bai, X. Shen, Y. Li and M. Dai, J. Am. Chem. Soc., 2016, 138, 10838 CrossRef CAS.
- B. Yu, Acc. Chem. Res., 2018, 51, 507 CrossRef CAS PubMed.
- R. M. Risi and S. D. Burke, Org. Lett., 2012, 14, 1180 CrossRef CAS PubMed.
- M. Mori, Y. Uozumi, M. Kimura and Y. Ban, Tetrahedron, 1986, 42, 3793 CrossRef CAS.
- K. Orito, M. Miyazawa, R. Kanbayashi, M. Tokuda and H. Suginome, J. Org. Chem., 1999, 64, 6583 CrossRef CAS.
- Y. Onozaki, N. Kurono, H. Senboku, M. Tokuda and K. Orito, J. Org. Chem., 2009, 74, 5486 CrossRef CAS.
- B. M. Trost and M. K. Ameriks, Org. Lett., 2004, 6, 1745 CrossRef CAS.
- O. Zhurakovskyi, Y. E. Türkmen, L. E. Löffler, V. A. Moorthie, C. C. Chen, M. A. Shaw, M. R. Crimmin, M. Ferrara, M. Ahmad, M. Ostovar, J. V. Matlock and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 1346 CrossRef CAS.
- E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378 CrossRef CAS.
- K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342 CrossRef CAS.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS.
- D. Willcox, B. G. N. Chappell, K. F. Hogg, J. Calleja, A. P. Smalley and M. J. Gaunt, Science, 2016, 354, 851 CrossRef CAS.
- W. Li, Z. Duan, Z. Zhang, H. Zhang, M. Wang, R. Jiang, H. Zeng, C. Liu and A. Lei, Angew. Chem., Int. Ed., 2015, 54, 1893 CrossRef CAS.
- Z.-H. Guan, M. Chen and Z.-H. Ren, J. Am. Chem. Soc., 2012, 134, 17490 CrossRef CAS PubMed.
- B. D. Dangel, K. Godula, S. W. Youn, B. Sezen and D. Sames, J. Am. Chem. Soc., 2002, 124, 11856 CrossRef CAS PubMed.
- C. Tsukano, N. Muto, I. Enkhtaivan and Y. Takemoto, Chem.–Asian J., 2014, 9, 2628 CrossRef CAS.
- M. Catellani, E. Motti and N. Della Ca', Acc. Chem. Res., 2008, 41, 1512 CrossRef CAS.
- P.-L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X.-S. Wang, Org. Lett., 2015, 17, 3698 CrossRef CAS.
- J. C. Fox, R. E. Gilligan, A. K. Pitts, H. R. Bennett and M. J. Gaunt, Chem. Sci., 2016, 7, 2706 RSC.
- J. A. Johnson, N. Li and D. Sames, J. Am. Chem. Soc., 2002, 124, 6900 CrossRef CAS.
- C. Mukai, T. Yoshida, M. Sorimachi and A. Odani, Org. Lett., 2006, 8, 83 CrossRef CAS PubMed.
- S. Prateeptongkum, K. M. Driller, R. Jackstell and M. Beller, Chem.–Asian J., 2010, 5, 2173 CrossRef CAS PubMed.
- L. W. Hernandez, J. Pospech, U. Klöckner, T. W. Bingham and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 15656 CrossRef CAS PubMed.
- W. Chiou, A. Schoenfelder, A. Mann and I. Ojima, Pure Appl. Chem., 2008, 80, 1019 CAS.
- H. Ren and W. D. Wulff, Org. Lett., 2013, 15, 242 CrossRef CAS PubMed.
- G. Makado, T. Morimoto, Y. Sugimoto, K. Tsutsumi, N. Kagawa and K. Kakiuchi, Adv. Synth. Catal., 2010, 352, 299 CrossRef CAS.
- R. W. Bates and S. Kasinathan, Tetrahedron, 2013, 69, 3088 CrossRef CAS.
- P. Dübon, A. Farwick and G. Helmchen, Synlett, 2009, 1413 Search PubMed.
- W.-H. Chiou, C.-L. Kao, J.-C. Tsai and Y.-M. Chang, Chem. Commun., 2013, 49, 8232 RSC.
- G. D. Artman and S. M. Weinreb, Org. Lett., 2003, 5, 1523 CrossRef CAS.
- S. E. Denmark, J. H.-C. Liu and J. M. Muhuhi, J. Am. Chem. Soc., 2009, 131, 14188 CrossRef CAS.
- W. Oppolzer, J. Z. Xu and C. Stone, Helv. Chim. Acta, 1991, 74, 465 CrossRef CAS.
- W. Oppolzer, H. Bienayme and A. Genevois-Borella, J. Am. Chem. Soc., 1991, 113, 9660 CrossRef CAS.
- W. Oppolzer and C. Robyr, Tetrahedron, 1994, 50, 415 CrossRef CAS.
- For a review: K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed.
- S. R. Angle, J. M. Fevig, S. D. Knight, R. W. Marquis and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 3966 CrossRef CAS.
- S. D. Knight, L. E. Overman and G. Pairaudeau, J. Am. Chem. Soc., 1995, 117, 5776 CrossRef CAS.
- G. T. Crisp, W. J. Scott and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 7500 CrossRef CAS.
- M. Ishikura, K. Imaizumi and N. Katagiri, Heterocycles, 2000, 53, 553 CrossRef CAS.
- S. Gao, Q. Wang, L. J. S. Huang, L. Lum and C. Chen, J. Am. Chem. Soc., 2010, 132, 371 CrossRef CAS.
- A. Shvartsbart and A. B. Smith III, J. Am. Chem. Soc., 2015, 137, 3510 CrossRef CAS.
- A. Shvartsbart and A. B. Smith, J. Am. Chem. Soc., 2014, 136, 870 CrossRef CAS PubMed.
- H. Yahata, T. Kubota and J. Kobayashi, J. Nat. Prod., 2008, 72, 148 CrossRef PubMed.
- S. K. Murphy, M. Zeng and S. B. Herzon, Science, 2017, 356, 956 CrossRef CAS PubMed.
- T. Esumi, T. Mori, M. Zhao, M. Toyota and Y. Fukuyama, Org. Lett., 2010, 12, 888 CrossRef CAS PubMed.
- L. Xu, C. Wang, Z. Gao and Y.-M. Zhao, J. Am. Chem. Soc., 2018, 140, 5653 CrossRef CAS.
- K. M. Brummond and J. L. Kent, Tetrahedron, 2000, 56, 3263 CrossRef CAS.
- H. Lee and F. Kwong, Eur. J. Org. Chem., 2010, 789 CrossRef CAS.
- B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630 CrossRef CAS PubMed.
- J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680 CrossRef CAS PubMed.
- S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744 CrossRef CAS.
- T. F. Jamison, S. Shambayati, W. E. Crowe and S. L. Schreiber, J. Am. Chem. Soc., 1997, 119, 4353 CrossRef CAS.
- J. Cassayre, F. Gagosz and S. Z. Zard, Angew. Chem., Int. Ed., 2002, 41, 1783 CrossRef CAS.
- T. Kozaka, N. Miyakoshi and C. Mukai, J. Org. Chem., 2007, 72, 10147 CrossRef CAS PubMed.
- M. Ishizaki, Y. Niimi, O. Hoshino, H. Hara and T. Takahashi, Tetrahedron, 2005, 61, 4053 CrossRef CAS.
- G. Stork, R. Mook, S. A. Biller and S. D. Rychnovsky, J. Am. Chem. Soc., 1983, 105, 3741 CrossRef CAS.
- N. Itoh, T. Iwata, H. Sugihara, F. Inagaki and C. Mukai, Chem.–Eur. J., 2013, 19, 8665 CrossRef CAS PubMed.
- M. K. Pallerla and J. M. Fox, Org. Lett., 2007, 9, 5625 CrossRef CAS PubMed.
- K. A. Miller and S. F. Martin, Org. Lett., 2007, 9, 1113 CrossRef CAS PubMed.
- S. J. Min and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 2199 CrossRef CAS.
- G. Mehta, R. Samineni and P. Srihari, Tetrahedron Lett., 2012, 53, 829 CrossRef CAS.
- F. Inagaki, M. Kinebuchi, N. Miyakoshi and C. Mukai, Org. Lett., 2010, 12, 1800 CrossRef CAS PubMed.
- J. S. Clark and C. Xu, Angew. Chem., Int. Ed., 2016, 55, 4332 CrossRef CAS.
- K. Hiroi, T. Watanabe, R. Kawagishi and I. Abe, Tetrahedron: Asymmetry, 2000, 11, 797 CrossRef CAS.
- P. Magnus, M. R. Fielding, C. Wells and V. Lynch, Tetrahedron Lett., 2002, 43, 947 CrossRef CAS.
- R. A. Rodriguez, D. Barrios Steed, Y. Kawamata, S. Su, P. A. Smith, T. C. Steed, F. E. Romesberg and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 15403 CrossRef CAS PubMed.
- S. Su, R. A. Rodriguez and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 13922 CrossRef CAS PubMed.
- Y. Yang, X. Fu, J. Chen and H. Zhai, Angew. Chem., Int. Ed., 2012, 51, 9825 CrossRef CAS PubMed.
- L. Shi and Z. Yang, Eur. J. Org. Chem., 2016, 2356 CrossRef CAS.
- Q. Liu, G. Yue, N. Wu, G. Lin, Y. Li, J. Quan, C. C. Li, G. Wang and Z. Yang, Angew. Chem., Int. Ed., 2012, 51, 12072 CrossRef CAS PubMed.
- J. Huang, L. Fang, R. Long, L. L. Shi, H. J. Shen, C. C. Li and Z. Yang, Org. Lett., 2013, 15, 4018 CrossRef CAS.
- L. You, X. Liang, L. Xu, Y. Wang, J. Zhang, Q. Su, Y. Li, B. Zhang, S. Yang, J. Chen and Z. Yang, J. Am. Chem. Soc., 2015, 137, 10120 CrossRef CAS PubMed.
- J. Zhang, X. Wang, S. Li, D. Li, S. Liu, Y. Lan, J. Gong and Z. Yang, Chem.–Eur. J., 2015, 21, 12596 CrossRef CAS PubMed.
- Z. Zhang, Y. Li, D. Zhao, Y. He, J. Gong and Z. Yang, Chem.–Eur. J., 2017, 23, 1258 CrossRef CAS PubMed.
- D. D. Liu, T. W. Sun, K. Y. Wang, Y. Lu, S. L. Zhang, Y. H. Li, Y. L. Jiang, J. H. Chen and Z. Yang, J. Am. Chem. Soc., 2017, 139, 5732 CrossRef CAS PubMed.
- K. V. Chuang, C. Xu and S. E. Reisman, Science, 2016, 353, 912 CrossRef CAS.
- M. W. Benjamin and D. Trauner, Angew. Chem., Int. Ed., 2016, 55, 2191 CrossRef PubMed.
- G. Volpin, N. A. Vepřek, A. B. Bellan and D. Trauner, Angew. Chem., Int. Ed., 2017, 56, 897 CrossRef CAS.
- N. Zhao, S. Yin, S. Xie, H. Yan, P. Ren, G. Chen, F. Chen and J. Xu, Angew. Chem., Int. Ed., 2018, 57, 3386 CrossRef CAS PubMed.
- B. Darses, I. N. Michaelides, F. Sladojevich, J. W. Ward, P. R. Rzepa and D. J. Dixon, Org. Lett., 2012, 14, 1684 CrossRef CAS PubMed.
- W. Zhang, M. Ding, J. Li, Z. Guo, M. Lu, Y. Chen, L. Liu, Y. H. Shen and A. Li, J. Am. Chem. Soc., 2018, 140, 4227 CrossRef CAS PubMed.
- W. Chen, J.-H. Tay, J. Ying, M. Sabat, X. Q. Yu and L. Pu, Chem. Commun., 2013, 49, 170 RSC.
- J. Ying and L. Pu, Chem.–Eur. J., 2014, 20, 16301 CrossRef CAS PubMed.
- J. D. Winkler, E. C. Y. Lee and L. I. Nevels, Org. Lett., 2005, 7, 1489 CrossRef CAS PubMed.
- J. P. Chen, W. He, Z. Y. Yang and Z. J. Yao, Org. Lett., 2015, 17, 3379 CrossRef CAS PubMed.
- P. A. Wender and F. E. McDonald, Tetrahedron Lett., 1990, 31, 3691 CrossRef CAS.
- L. A. Paquette and S. Borrelly, J. Org. Chem., 1995, 60, 6912 CrossRef CAS.
- G. R. Dake, E. E. Fenster and B. O. Patrick, J. Org. Chem., 2008, 73, 6711 CrossRef CAS PubMed.
- M. E. Krafft, Y. Y. Cheung and C. A. Juliano-Capucao, Synthesis, 2000, 7, 1020 CrossRef.
- C. Mukai, M. Kobayashi, I. J. Kim and M. Hanaoka, Tetrahedron, 2002, 58, 5225 CrossRef CAS.
- T. Ishikawa, H. Ishii, K. Shimizu, H. Nakao, J. Urano, T. Kudo and S. Saito, J. Org. Chem., 2004, 69, 8133 CrossRef CAS PubMed.
- A. Farwick and G. Helmchen, Org. Lett., 2010, 12, 1108 CrossRef CAS PubMed.
- I. Nomura and C. Mukai, J. Org. Chem., 2004, 69, 1803 CrossRef CAS.
- B. Jiang and M. Xu, Angew. Chem., Int. Ed., 2004, 43, 2543 CrossRef CAS PubMed.
- R. M. Moriarty, N. Rani, L. A. Enache, M. S. Rao, H. Batra, L. Guo, R. A. Penmasta, J. P. Staszewski, S. M. Tuladhar, O. Prakash, D. Crich, A. Hirtopeanu and R. Gilardi, J. Org. Chem., 2004, 69, 1890 CrossRef CAS PubMed.
- J. J. Crawford, W. J. Kerr, M. McLaughlin, A. J. Morrison, P. L. Pauson and G. J. Thurston, Tetrahedron, 2006, 62, 11360 CrossRef CAS.
- T. Honda and K. Kaneda, J. Org. Chem., 2007, 72, 6541 CrossRef CAS.
- A. Vázquez-Romero, L. Cárdenas, E. Blasi, X. Verdaguer and A. Riera, Org. Lett., 2009, 11, 3104 CrossRef PubMed.
- A. Nakayama, N. Kogure, M. Kitajima and H. Takayama, Angew. Chem., Int. Ed., 2011, 50, 8025 CrossRef CAS PubMed.
- Y. Hayashi, F. Inagaki and C. Mukai, Org. Lett., 2011, 13, 1778 CrossRef CAS PubMed.
- R. Betík and M. Kotora, Eur. J. Org. Chem., 2011, 3279 CrossRef.
- Y. Hayashi, K. Ogawa, F. Inagaki and C. Mukai, Org. Biomol. Chem., 2012, 10, 4747 RSC.
- K. Fujioka, H. Yokoe, M. Yoshida and K. Shishido, Org. Lett., 2012, 14, 244 CrossRef CAS PubMed.
- N. Itoh, T. Iwata, H. Sugihara, F. Inagaki and C. Mukai, Chem.–Eur. J., 2013, 19, 8665 CrossRef CAS PubMed.
- B. Jiang, M. M. Li, P. Xing and Z. G. Huang, Org. Lett., 2013, 15, 871 CrossRef CAS PubMed.
- H. Yamakoshi, Y. Sawayama, Y. Akahori, M. Kato and S. Nakamura, Org. Lett., 2016, 18, 3430 CrossRef CAS PubMed.
- M. K. Abd El-Gaber, S. Yasuda, E. Iida and C. Mukai, Org. Lett., 2017, 19, 320 CrossRef CAS.
- J. L. Kent, H. Wan and K. M. Brummond, Tetrahedron Lett., 1995, 36, 2407 CrossRef CAS.
- K. M. Brummond and H. Wan, Tetrahedron Lett., 1998, 39, 931 CrossRef CAS.
- K. M. Brummond, H. Wan and J. L. Kent, J. Org. Chem., 1998, 63, 6535 CrossRef CAS.
- K. M. Brummond and D. Chen, Org. Lett., 2008, 10, 705 CrossRef CAS.
- K. M. Brummond, J. Lu and J. Petersen, J. Am. Chem. Soc., 2000, 122, 4915 CrossRef CAS.
- K. M. Brummond, P. C. Sill and H. Chen, Org. Lett., 2004, 6, 149 CrossRef CAS.
- K. M. Brummond and D. Gao, Org. Lett., 2003, 5, 3491 CrossRef CAS PubMed.
- B. Wen, J. K. Hexum, J. C. Widen, D. A. Harki and K. M. Brummond, Org. Lett., 2013, 15, 2644 CrossRef CAS PubMed.
- L. Jørgensen, S. J. McKerrall, C. A. Kuttruff, F. Ungeheuer, J. Felding and P. S. Baran, Science, 2013, 341, 878 CrossRef PubMed.
- S. J. McKerrall, L. Jørgensen, C. A. Kuttruff, F. Ungeheuer and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5799 CrossRef CAS PubMed.
- D. R. Williams and A. A. Shah, J. Am. Chem. Soc., 2014, 136, 8829 CrossRef CAS PubMed.
- C. Heinz and N. Cramer, J. Am. Chem. Soc., 2015, 137, 11278 CrossRef CAS PubMed.
- Y. Hayashi, N. Miyakoshi, S. Kitagaki and C. Mukai, Org. Lett., 2008, 10, 2385 CrossRef CAS PubMed.
- J. Tsuji, T. Sugiura and I. Minami, Tetrahedron Lett., 1986, 27, 731 CrossRef CAS.
- C. Lv, X. Yan, Q. Tu, Y. Di, C. Yuan, X. Fang, Y. Ben-David, L. Xia, J. Gong, Y. Shen, Z. Yang and X. Hao, Angew. Chem., Int. Ed., 2016, 55, 7539 CrossRef CAS PubMed.
- P. A. Wender, M. P. Croatt and N. M. Deschamps, J. Am. Chem. Soc., 2004, 126, 5948 CrossRef CAS PubMed.
- P. A. Wender, N. M. Deschamps and T. J. Williams, Angew. Chem., Int. Ed., 2004, 43, 3076 CrossRef CAS.
- P. A. Wender, M. P. Croatt and N. M. Deschamps, Angew. Chem., Int. Ed., 2006, 45, 2459 CrossRef CAS PubMed.
- W. H. Pitcock Jr, R. L. Lord and M.-H. Baik, J. Am. Chem. Soc., 2008, 130, 5821 CrossRef PubMed.
- M. P. Croatt and P. A. Wender, Eur. J. Org. Chem., 2010, 19 CrossRef CAS.
- P. A. Wender, Tetrahedron, 2013, 69, 7529 CrossRef CAS PubMed.
- Y. Wang and Z.-X. Yu, Acc. Chem. Res., 2015, 48, 2288 CrossRef CAS PubMed.
- D. Shu, X. Li, M. Zhang, P. J. Robichaux, I. A. Guzei and W. Tang, J. Org. Chem., 2012, 77, 6463 CrossRef CAS PubMed.
- S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569 CrossRef CAS PubMed.
- G.-W. Wang and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 2743 CrossRef CAS PubMed.
- G.-W. Wang, N. G. McCreanor, M. H. Shaw, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2016, 138, 13501 CrossRef CAS PubMed.
- For a review: M. H. Shaw and J. F. Bower, Chem. Commun., 2016, 52, 10187 RSC.
- L. Jiao, M. Lin, L. G. Zhuo and Z.-X. Yu, Org. Lett., 2010, 12, 2528 CrossRef CAS PubMed.
- M. Lin, F. Li, L. Jiao and Z.-X. Yu, J. Am. Chem. Soc., 2011, 133, 1690 CrossRef CAS PubMed.
- Y. Koga and K. Narasaka, Chem. Lett., 1999, 7, 705 CrossRef.
- Y. Feng and Z.-X. Yu, J. Org. Chem., 2015, 80, 1952 CrossRef CAS.
- J. Yang, W. Xu, Q. Cui, X. Fan, L. N. Wang and Z.-X. Yu, Org. Lett., 2017, 19, 6040 CrossRef CAS.
- J. Yang, W. Xu, Q. Cui, X. Fan, L. N. Wang and Z.-X. Yu, Org. Lett., 2017, 19, 6040 CrossRef CAS PubMed.
- R. Ben-Shoshan and S. Sarel, J. Chem. Soc., Chem. Commun., 1969, 883 RSC.
- D. F. Taber, K. Kanai, Q. Jiang and G. Bui, J. Am. Chem. Soc., 2000, 122, 6807 CrossRef CAS.
- D. F. Taber, P. V. Joshi and K. Kanai, J. Org. Chem., 2004, 69, 2268 CrossRef CAS PubMed.
- D. F. Taber, P. Guo and N. Guo, J. Am. Chem. Soc., 2010, 132, 11179 CrossRef CAS PubMed.
- D. F. Taber, G. Bui and B. Chen, J. Org. Chem., 2001, 66, 3423 CrossRef CAS PubMed.
- D. F. Taber and R. B. Sheth, J. Org. Chem., 2008, 73, 8030 CrossRef CAS PubMed.
- D. F. Taber, R. B. Sheth and W. Tian, J. Org. Chem., 2009, 74, 2433 CrossRef CAS PubMed.
- T. Kurahashi and A. de Meijere, Synlett, 2005, 17, 2619 Search PubMed.
- G. J. Jian, X. F. Fu, Q. Li and Z.-X. Yu, Org. Lett., 2012, 14, 692 CrossRef.
- L. N. Wang, Q. Cui and Z.-X. Yu, J. Org. Chem., 2016, 81, 10165 CrossRef CAS PubMed.
- N. Iwasawa, Y. Owada and T. Matsuo, Chem. Lett., 1995, 24, 115 CrossRef.
- Y. Owada, T. Matsuo and N. Iwasawa, Tetrahedron, 1997, 53, 11069 CrossRef CAS.
- M. Murakami, K. Itami, M. Ubukata, I. Tsuji and Y. Ito, J. Org. Chem., 1998, 63, 4 CrossRef CAS.
- D. Shu, X. Li, M. Zhang, P. J. Robichaux and W. Tang, Angew. Chem., Int. Ed., 2011, 50, 1346 CrossRef CAS.
- M. Zhang and W. Tang, Org. Lett., 2012, 14, 3756 CrossRef CAS PubMed.
- C. H. Liu and Z.-X. Yu, Org. Biomol. Chem., 2016, 14, 5945 RSC.
- X. Li, W. Song and W. Tang, J. Am. Chem. Soc., 2013, 135, 16797 CrossRef CAS PubMed.
- W. Song, X. Li, K. Yang, X. l. Zhao, D. A. Glazier, B.-m. Xi and W. Tang, J. Org. Chem., 2016, 81, 2930 CrossRef CAS PubMed.
- J. T. Liu, C. J. Simmons, H. Xie, F. Yang, X. l. Zhao, Y. Tang and W. Tang, Adv. Synth. Catal., 2017, 359, 693 CrossRef CAS.
- P. A. Wender, G. G. Gamber, R. D. Hubbard and L. Zhang, J. Am. Chem. Soc., 2002, 124, 2876 CrossRef CAS.
- P. A. Wender, G. G. Gamber, R. D. Hubbard, S. M. Pham and L. Zhang, J. Am. Chem. Soc., 2005, 127, 2836 CrossRef CAS PubMed.
- Y. Wang, J. Wang, J. Su, F. Huang, L. Jiao, Y. Liang, D. Yang, S. Zhang, P. A. Wender and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 10060 CrossRef CAS PubMed.
- L. Jiao, C. Yuan and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 4421 CrossRef CAS PubMed.
- X. Fan, L. G. Zhuo, Y. Q. Tu and Z.-X. Yu, Tetrahedron, 2009, 65, 4709 CrossRef CAS.
- Y. Liang, X. Jiang, X. F. Fu, S. Ye, T. Wang, J. Yuan, Y. Wang and Z.-X. Yu, Chem.–Asian J., 2012, 7, 593 CrossRef CAS.
- A. C. Gyorkos, J. K. Stille and L. S. Hegedus, J. Am. Chem. Soc., 1990, 112, 8465 CrossRef CAS.
- T. J. Houghton, S. Choi and V. H. Rawal, Org. Lett., 2001, 3, 3615 CrossRef CAS PubMed.
- For a review: W. Song, S. A. Blaszczyk, J. Liu, S. Wang and W. Tang, Org. Biomol. Chem., 2017, 15, 7490 RSC and references therein.
- S. A. Eastham, S. P. Ingham, M. R. Hallett, J. Herbert, P. Quayle and J. Raftery, Tetrahedron Lett., 2006, 47, 2299 CrossRef CAS.
- K. Tanaka, M. Watanabe, K. Ishibashi, H. Matsuyama, Y. Saikawa and M. Nakata, Org. Lett., 2010, 12, 1700 CrossRef CAS PubMed.
- H. Wang and W. D. Wulff, J. Am. Chem. Soc., 1998, 120, 10573 CrossRef CAS.
- N. Suzuki, T. Kondo and T. Mitsudo, Organometallics, 1998, 17, 766 CrossRef CAS.
- R. A. Leal, C. Bischof, Y. V. Lee, S. Sawano, C. C. McAtee, L. N. Latimer, Z. N. Russ, J. E. Dueber, J. Q. Yu and R. Sarpong, Angew. Chem., Int. Ed., 2016, 55, 11824 CrossRef CAS.
- S. K. Jackson and M. A. Kerr, J. Org. Chem., 2007, 72, 1405 CrossRef CAS PubMed.
- K. S. Bloome and E. J. Alexanian, J. Am. Chem. Soc., 2010, 132, 12823 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this review article. |
| This journal is © The Royal Society of Chemistry 2019 |