
Antibacterial properties and clinical potential of pleuromutilins
Olivia
Goethe
 a,
Abigail
Heuer
a,
Xiaoshen
Ma
a,
Zhixun
Wang
a and
Seth B.
Herzon
*ab
a,
Abigail
Heuer
a,
Xiaoshen
Ma
a,
Zhixun
Wang
a and
Seth B.
Herzon
*ab
aDepartment of Chemistry, Yale University, New Haven, Connecticut 06520, USA. E-mail: seth.herzon@yale.edu
bDepartment of Pharmacology, Yale School of Medicine, New Haven, Connecticut 06520, USA
First published on 6th July 2018
Abstract
Covering: up to 2018
Pleuromutilins are a clinically validated class of antibiotics derived from the fungal diterpene (+)-pleuromutilin (1). Pleuromutilins inhibit bacterial protein synthesis by binding to the peptidyl transferase center (PTC) of the ribosome. In this review we summarize the biosynthesis and recent total syntheses of (+)-pleuromutilin (1). We review the mode of interaction of pleuromutilins with the bacterial ribosome, which involves binding of the C14 extension and the tricyclic core to the P and A sites of the PTC, respectively. We provide an overview of existing clinical agents, and discuss the three primary modes of bacterial resistance (mutations in ribosomal protein L3, Cfr methylation, and efflux). Finally we collect structure–activity relationships from publicly available reports, and close with some forward looking statements regarding future development.
 Olivia Goethe | Olivia Goethe grew up on her parents' farm in Kalamazoo, Michigan and received her BSc degree in Biochemistry from Michigan State University in 2016. In college, she worked on enzymatic reactions in the lab of Professor Kevin D. Walker and held medicinal and process chemistry internships at Zoetis VMRD and Kalexsyn, Inc. In 2016, she joined the lab of Seth Herzon at Yale University to pursue her PhD. She is working towards new ways to access pleuromutilin antibiotics through total synthesis. |
 Abigail Heuer | Abi Heuer was born and raised in West Bend, WI. She obtained her BSc degree in Biochemistry in 2015 from the University of St. Thomas where she studied the synthesis of novel GLP-1 stimulants and oxazolidinone antibiotics with Professor J. Thomas Ippoliti. She is currently pursuing a PhD at Yale University under the supervision of Professor Seth Herzon, focusing on the synthesis of novel pleuromutilin antibiotics. |
 Xiaoshen Ma | Xiaoshen Ma was born and raised in Beijing, China. He received his Bachelor's degree in 2013 from the College of Chemistry and Molecule Engineering, Peking University under the supervision of Professor Jianbo Wang working on copper mediated cross coupling reactions between diazo compounds and acetylene. In 2018, he received his PhD degree in organic chemistry from Yale University under the supervision of Professor Seth B. Herzon. His PhD research focused on alkene hydrogenation and hydrofunctionalization by cobalt-mediated hydrogen atom transfer, direct C–H hydroxylation of pleuromutilin analogs, and biophysical characterization and molecular assignment of “host release factor” in marine symbioses. |
 Zhixun Wang | Zhixun Wang was born in Danyang, Jiangsu, China. He received his BS degree in chemistry from Fudan University in 2012. His undergraduate research focused on palladium catalyzed cross-coupling reactions under the direction of Professor Tao Tu. In 2017, he received PhD degree in organic chemistry at the University of California, Santa Barbara under the supervision of Professor Liming Zhang. His graduate research focused on designing novel bi-functional phosphine ligands for gold-catalyzed reactions of alkynes and allenes. After graduation, he joined Professor Seth Herzon's group in Yale University and started his post-doctoral study on developing novel pleuromutilin antibiotics. |
 Seth B. Herzon | Seth B. Herzon completed his undergraduate studies at Temple University, obtained a PhD from Harvard University in 2006 under the guidance of Andrew G. Myers, and was an NIH postdoctoral fellow with John F. Hartwig at the University of Illinois, Urbana-Champaign. He began his independent career at Yale in 2008 and is currently the Milton Harris ’29 Ph.D. Professor of Chemistry and a Professor of Pharmacology at the Yale School of Medicine. Herzon's research focuses on synthetic and translational studies of genotoxic natural products, human gut microbiome metabolites, and antibiotic development. |
1. Introduction
1.1 Background
(+)-Pleuromutilin (1, Fig. 1) is a diterpene fungal metabolite that was first isolated in the early 1950s.1,2 (+)-Pleuromutilin (1) displays potent activity against Gram-positive bacteria, and many large pharmaceutical companies had programs aimed at developing novel pleuromutilin-based antibiotics (referred to as “pleuromutilins” hereafter) by semisynthesis. As outlined in Section 3, these efforts resulted in the two veterinary agents tiamulin (2) and valnemulin (3), and the clinical agent retapamulin (4). | ||
| Fig. 1 Structures of (+)-pleuromutilin (1), tiamulin (2), valnemulin (3), retapamulin (4), and lefamulin (5). Tiamulin (2) and valnemulin (3) are veterinary agents. Retapamulin (4) is a clinical agent used to treat skin infections. Lefamulin (5) recently passed a Phase III trial for the treatment of community-acquired bacterial pneumonia by oral and intravenous administration. | ||
While major pharmaceutical companies have historically been the primary engine driving antibiotic development, by the mid 2000s most large companies had discontinued their antibiotic programs. This is due to a confluence of economic and evolutionary pressures coupled with stringent regulatory hurdles, which situate antibiotics near the bottom of the financial hierarchy of commercial pharmaceuticals. Nabriva Therapeutics, a spinoff of Sandoz Pharmaceuticals, is the only company (to our knowledge) actively investigating pleuromutilins. These researchers developed lefamulin (5), which recently a passed Phase III trial for the treatment of community-acquired bacterial pneumonia by oral and intravenous administration.3 As many pleuromutilins have notoriously poor bioavailability [retapamulin (4), is administered as a topical ointment], lefamulin's success constitutes a significant milestone for the class. As discussed in Section 5.4, the Nabriva scientists also recently disclosed that extension of the C12 pseudoequatorial position with diamine fragments increases the spectrum of activity to include drug-resistant Gram-negative bacteria.4–11 The activities of these “extended spectrum pleuromutilins” (ESPs) as well as the progress of lefamulin (5) suggest additional useful agents await discovery.
(+)-Pleuromutilin (1) contains an unusual carbon skeleton bearing three fused rings and eight contiguous stereocenters. Consequently, it has received significant attention from the synthetic community. The first total synthesis of (±)-pleuromutilin was completed by E. Grant Gibbons, working in the laboratory of the late R. B. Woodward.12 The Gibbons route made use of a strategic doubly diastereoselective tandem Michael addition to rapidly assemble the hydrindane core. An unusual bromonium ion-initiated Grob fragmentation was employed to construct the eight-membered ring. Shortly thereafter, Boeckman reported the second route to the target, which utilized an anionic oxy-Cope rearrangement to form the eight-membered ring.13 In 2015, Procter and co-workers reported the first enantioselective synthetic route to (+)-pleuromutilin (1).14 Their approach features a powerful diastereoselective cascade cyclization reaction to forge the five- and eight-membered rings in one step. In 2017, our laboratory developed an enantioselective synthetic route to (+)-pleuromutilin (1) that relies on the convergent union of a complex hydrindane derivative with a bifunctional C11–C13 synthon.15,16 Finally, Reisman and co-workers reported a distinct enantioselective synthetic route to (+)-pleuromutilin (1) that employs an allylboronate addition to establish the C12 stereocenter.17 Many synthetic studies toward (+)-pleuromutilin (1) have been disclosed,18–26 and certain simplified, fully synthetic analogs have been shown to possess activity against Mycobacterium tuberculosis.27
Pleuromutilins have been the subject of many reviews,28–36 and we place emphasis here on topics that have not been covered. In Section 1.3 we summarize recent genetic studies that have led to a complete elucidation of pleuromutilin biosynthesis. In Section 1.4 we review synthetic work that has appeared since 2014. For earlier efforts in this area, including reviews of the Gibbons, Boeckman, and Procter syntheses, the reader is directed to Procter's35 skillful review. In Section 2 we summarize the mode of interaction of (+)-pleuromutilin (1) with its biological target, the bacterial ribosome. In Section 3 we provide an overview of existing clinical agents, their indications, and advanced clinical agents. In Section 4 we discuss mechanisms of pleuromutilin resistance. In Section 5 we collect publicly available structures of pleuromutilin derivatives and discuss structure–activity relationships and bacterial susceptibilities. Finally, we close with some forward looking statements and suggestions for future development.
1.2 Discovery and structure elucidation
(+)-Pleuromutilin (1) was first isolated in 1951 by Kavanagh, Hervey, and Robbins1,2 from cultures of the fungi Pleurotus mutilus (aka Clitopilus scyphoides) and Pleurotus passeckerianus. The metabolite was found to possess activity against Gram-positive bacteria, particularly Staphylococcus aureus with a minimal inhibitory concentration (MIC) = 0.25 μg mL−1. The results of in vivo studies conducted at that time were modest. Only 63% of mice infected with Streptococcus hemolyticus survived after 7 days of treatment with (+)-pleuromutilin (1; 50 mg kg−1), vs. 100% survival rate in mice treated with penicillin. The full structure of (+)-pleuromutilin (1) was not known at that time, but the authors noted that the antibacterial activity was eliminated by boiling the isolate in 0.1 N sodium hydroxide. This treatment most likely removed the glycolic ester residue, which was later determined to be essential for antimicrobial activity. As discussed in Section 2.2, pleuromutilins inhibit bacterial protein synthesis by binding the peptidyl transferase center (PTC) of the ribosome. The glycolic acid residue extends into the P-site of the PTC and engages in essential hydrogen bonding interactions with the target.Initial degradation studies of (+)-pleuromutilin (1) were conducted by Anchel.37 These efforts defined (+)-pleuromutilin (1) as a neutral substance with two hydroxyl groups. Shortly thereafter, Arigoni and Birch independently arrived at the complete structure of (+)-pleuromutilin (1), as shown in Fig. 1.38,39 The structure was later confirmed by X-ray analysis.40
1.3 Biosynthesis
Arigoni and Birch recorded the first insights into the biosynthesis of (+)-pleuromutilin (1).38,39 Based on feeding studies, they proposed that geranylgeranyl diphosphate (6) undergoes a π-cation cyclization to the trans-decalin 7 (Scheme 1). 1,2-Hydride migration then gives the cation 8, which undergoes 1,2-shift of a methyl substituent to form 9. 1,2-Hydride migration then yields the tertiary cation 10 and establishes the C4 stereocenter of (+)-pleuromutilin (1). Ring contraction of 10via 1,2-alkyl migration (10 → 11), followed by elimination of a proton, then provides the hydrindane 12. The eight-membered ring of (+)-pleuromutilin (1) is established via SN′ displacement of pyrophosphate with concomitant generation of the C11–C12 bond and formation of a carbocation at C10 (see 13). A remarkable transannular hydride shift from C14 (13 → 14), followed by trapping of the resulting C14 cation by water, then forms the alcohol 15. | ||
| Scheme 1 Biosynthesis of (+)-pleuromutilin (1). | ||
In 2016 Foster and co-workers identified the pleuromutilin gene cluster in Clitopilus passeckerianus.41 Remarkably, the molecule is biosynthesized using only seven enzymes. This advance subsequently allowed the full biosynthetic pathway to be elucidated (Scheme 1).42 To achieve this, the researchers utilized stepwise heterologous expression of the pleuromutilin gene cluster in Aspergillus oryzae. In this approach, A. oryzae transformants differing by the incremental addition of a single gene in the pleuromutilin cluster were prepared, and their metabolic products were characterized. Co-expression of the geranylgeranyl diphosphate synthase pl-ggs and the pathway-specific cyclase pl-cyc resulted in production of the alcohol 15. The structure of 15 was confirmed by independent synthesis from (+)-pleuromutilin (1). Addition of the putative cytochrome P450 monooxygenase gene pl-p450-1 resulted in production of the C11 oxidation product 16. Further addition of both pl-p450-1 and pl-p450-2 resulted in formation of the C3 oxidation product 17. Addition of the short-chain dehydrogenase/reductase pl-sdr generated the ketone 18. Finally, introduction of the acetyl transferase pl-atf and the cytochrome P450 monooxygenase pl-p450-3 provided the acetylated derivative 19 and the natural product 1, respectively. The latter was derived from oxidation at C22.
1.4 Chemical synthesis
As noted earlier, synthetic work in this area was reviewed in detail in 2014 by Procter and co-workers.35 Accordingly, we limit discussion to reports following that publication. In 2017, we reported a convergent synthesis of (+)-pleuromutilin (1) and 12-epi-pleuromutilins (Scheme 2).15,16,25,26 Beginning with cyclohex-2-ene-1-one (20), the α-methyl-β-ketoester 21 was obtained in one step by an enantio- and diastereoselective four-component coupling reaction (70%, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 97:3 er).26 1,2-Addition of the magnesium acetylide derived from methyl propargyl ether formed the alcohol 22 (97%). Treatment of 22 with methanesulfonic acid induced a Rupe rearrangement–Nazarov cyclization cascade to deliver the hydrindenone 23 (71%). 1,4-Hydrocyanation43 of 23 (diethylaluminum cyanide) provided the tertiary nitrile 24 (53%) with 3:1 diastereoselectivity in favour of the C9 isomer shown. Protection of the ketone function (ethylene glycol, p-toluenesulfonic acid) generated the dioxolane 25 (83%). The tetracyclic eneimide 26 was then formed by 1,2-addition of methyllithium to the nitrile, in situ attack of the resulting anion upon the adjacent ester function, and acylation of the eneamide with di-tert-butyldicarbonate (80%). The C11–C13 synthon 27 (derived from alkylation of an Evans imide) was treated with t-butyllithium to effect lithium–iodine exchange; addition of the resulting neopentyllithium reagent to 26, followed by in situ hydrolysis of the resulting acylimine intermediate (not shown), formed 28 (48%). Treatment of 28 with excess potassium hexamethyldisilazide and Comins' reagent44 resulted in vinyl triflate formation and in situ elimination to form the alkyne 29 (81%). Cleavage of the p-methoxybenzyl ether and oxidation of the resulting alcohol produced the alkynyl aldehyde 30 (95%). Nickel-catalyzed exo-selective reductive cyclization of 30,45–48 followed by cleavage of the resulting silyl ether, generated the allylic alcohol 31 (60%). Oxidation of the C11 alcohol followed by reduction of the resulting enone (not shown) provided the ketone 32 (98%). Finally, diastereoselective reduction of the C11 and C14 ketones, followed by dioxolane cleavage, produced (+)-12-epi-mutilin (34). 12-epi-Mutilin (34) was elaborated to 12-epi-pleuromutilin (36) by stepwise acylation, followed by deprotection. (+)-Pleuromutilin (1) was obtained by stepwise acylation, C12 epimerization (vide infra) and deprotection.
:1 dr, 97:3 er).26 1,2-Addition of the magnesium acetylide derived from methyl propargyl ether formed the alcohol 22 (97%). Treatment of 22 with methanesulfonic acid induced a Rupe rearrangement–Nazarov cyclization cascade to deliver the hydrindenone 23 (71%). 1,4-Hydrocyanation43 of 23 (diethylaluminum cyanide) provided the tertiary nitrile 24 (53%) with 3:1 diastereoselectivity in favour of the C9 isomer shown. Protection of the ketone function (ethylene glycol, p-toluenesulfonic acid) generated the dioxolane 25 (83%). The tetracyclic eneimide 26 was then formed by 1,2-addition of methyllithium to the nitrile, in situ attack of the resulting anion upon the adjacent ester function, and acylation of the eneamide with di-tert-butyldicarbonate (80%). The C11–C13 synthon 27 (derived from alkylation of an Evans imide) was treated with t-butyllithium to effect lithium–iodine exchange; addition of the resulting neopentyllithium reagent to 26, followed by in situ hydrolysis of the resulting acylimine intermediate (not shown), formed 28 (48%). Treatment of 28 with excess potassium hexamethyldisilazide and Comins' reagent44 resulted in vinyl triflate formation and in situ elimination to form the alkyne 29 (81%). Cleavage of the p-methoxybenzyl ether and oxidation of the resulting alcohol produced the alkynyl aldehyde 30 (95%). Nickel-catalyzed exo-selective reductive cyclization of 30,45–48 followed by cleavage of the resulting silyl ether, generated the allylic alcohol 31 (60%). Oxidation of the C11 alcohol followed by reduction of the resulting enone (not shown) provided the ketone 32 (98%). Finally, diastereoselective reduction of the C11 and C14 ketones, followed by dioxolane cleavage, produced (+)-12-epi-mutilin (34). 12-epi-Mutilin (34) was elaborated to 12-epi-pleuromutilin (36) by stepwise acylation, followed by deprotection. (+)-Pleuromutilin (1) was obtained by stepwise acylation, C12 epimerization (vide infra) and deprotection.
 | ||
| Scheme 2 Synthesis of (+)-pleuromutilin (1) and 12-epi-pleuromutilin (36) by Herzon and co-workers.15,16,25,26 | ||
In 2018, Reisman and co-workers reported a distinct synthetic route to (+)-pleuromutilin (1, Scheme 3).17trans-Dihydrocarvone (37) was obtained by chromatographic purification of a commercial 4:1 mixture of diastereomers. Oxidative elimination of the isopropenyl substituent (57%)49 followed by silyl-accelerated50 copper-catalyzed 1,4-addition of the Grignard reagent 38 provided the enoxysilane 39. Saegusa–Ito–Larock oxidation51,52 then formed the enone 40 (91%, two steps). Copper-catalyzed 1,4-addition of iso-propenyl magnesium bromide generated the ketone 41 as a 4.8:1 mixture of diastereomers. Allylic chlorination followed by removal of the acetal and in situ condensation provided the hydrindenone 43 (40%, two steps). Cerium-promoted53 1,2-addition of methyl magnesium bromide, followed by oxidative rearrangement,54 generated the enone 45 (78%, two steps). Kornblum oxidation55 then formed the unsaturated aldehyde 46 (69%). Crotylboration of 46 using the allylic boronate 47 in the presence of (R)-3,3′-Br2-BINOL56,57 generated the desired addition product 48 as well as its 11,12-di-epi-diastereomer (not shown; 41% yield of 48). Protection of the C11 hydroxyl (methoxymethyl chloride, Hünig's base, 79%) followed by removal of the trityl ether and oxidation58 generated the aldehyde 51 (81%, two steps). Reductive cyclization of 51 using samarium iodide provided the tricycle 52 in 93% yield and with 23:1 diastereoselectivity at C14. The ketone was then protected as the corresponding enoxysilane 53 (76%) and the resulting product was subjected to alkene reduction under hydrogen atom transfer conditions.59 While the C10–C17 alkene was successfully reduced, the C14 alcohol, surprisingly, underwent oxidation under these conditions. The C14 ketone was reduced with lithium in ammonia to regenerate the C14 alcohol (61%). Finally, stepwise acylation followed by deprotection generated (+)-pleuromutilin (1, 80%). By employing the (E)-isomer of 47, 12-epi-pleuromutilin (36, Scheme 2) was also prepared.
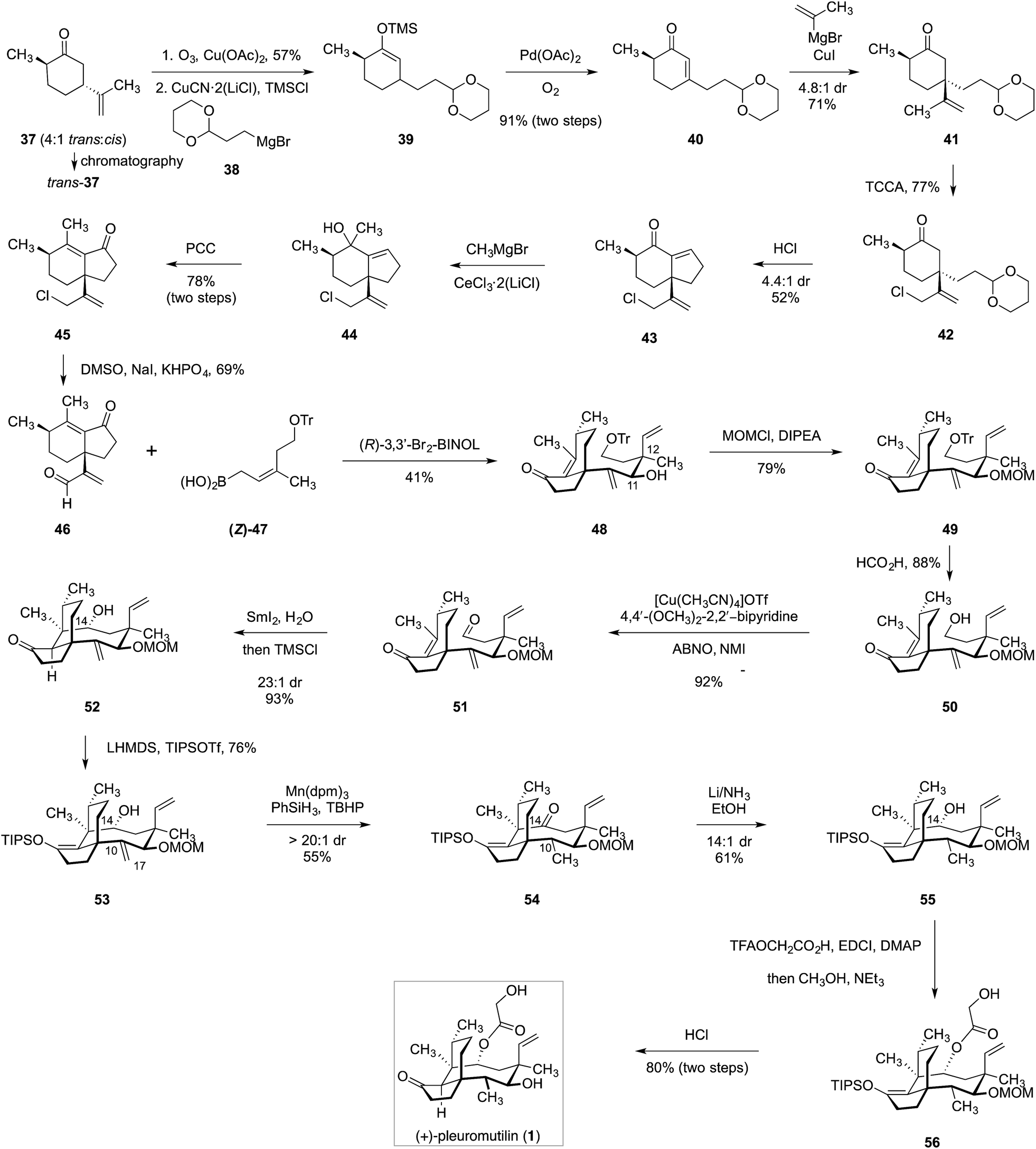 | ||
| Scheme 3 Synthesis of (+)-pleuromutilin (1) by Reisman and co-workers.17 | ||
2. Target binding
2.1 Overview of protein assembly
The ribosome is a complex organelle comprised of ribosomal proteins (r-proteins) and ribosomal rRNA (rRNA) that polymerizes amino acids into proteins using information encoded in messenger RNA (mRNA). A schematic of protein translation is shown in Fig. 2. | ||
| Fig. 2 Schematic of protein translation.60 | ||
First, the initiation complex, comprising the small ribosomal subunit, mRNA, and initiator tRNA, binds to an AUG start codon and initiation factors. The amino acid monomers are transported by tRNA and aminoacylated at the 3′ end. Three binding sites exist for tRNA: the aminoacyl tRNA site (A site), peptidyl side (P site), and exit site (E site). The first tRNA binds to the P site. A second tRNA enters the A site and the anticodon loops of both associate with the mRNA. The peptide bond is formed in the space of the peptidyl transferase center (PTC). As the ribosome moves down the mRNA chain, the A site opens to receive a new tRNA substrate, while the newly formed protein chain grows in the exit tunnel of the 50S subunit. Disruption of any of these processes can impair protein synthesis, resulting in growth inhibition and in some cases, cell death.
Crystallization of prokaryotic ribosomes revealed that the PTC exists in the center of the ribosome and is made up of 180 highly conserved rRNA nucleotides.61 All ribosome crystal structures to date share this catalytic center homology, and this region of the ribosome is resistant to mutations.62 The PTC provides positional catalysis for the formation of the peptide bond between two tRNA molecules. The PTC also includes space for the translocation of tRNA, thereby enabling continuous polymerization. No fewer than five widely used antibiotic classes inhibit protein synthesis by interacting with the PTC: phenicols, lincosamides, streptogramins A, oxazolidinones, and pleuromutilins.63
2.2 Mechanism of action of (+)-pleuromutilin (1)
The first clues to the mechanism of action of (+)-pleuromutilin (1) were obtained when the water-soluble derivative tiamulin (2) was found to inhibit polyphenylalanine synthesis in ribosomes isolated from E. coli.64 Competition experiments suggested that tiamulin (2) competes with chloramphenicol, which binds to the A site, and the 3′ tRNA terminus analogs puromycin and CpCpA oligonucleotide. Later, experiments by Högenauer and co-workers revealed that pleuromutilins selectively bind to the 50S ribosome subunit.65 Other pleuromutilin derivatives displaced tritiated tiamulin (2) from the 50S subunit with abilities that correlated with their MIC values. It was also found that pleuromutilin binding to the 50S ribosomal subunit does not interfere with the assembly of the 70S ribosome complex. Additionally, pleuromutilins do not interfere with ribosome function once peptide elongation has been initiated.64By studying their effects on amide bond formation between Phe-tRNA and puromycin (which mimics acceptor-tRNA in the P site), tiamulin (2) and valnemulin (3) were found to completely inhibit the peptidyl transferase reaction.66 They were also shown to footprint in the central part of domain V of 23S RNA at nucleotides A2058-9, U2506, and U2584-5, as observed with other peptidyl transferase inhibitors.66 In 2004, the 3.5 Å structure of tiamulin (2) complexed with the 50S ribosome subunit of Deinococcus radiodurans was solved (PDB 1XBP, Fig. 3).67
 | ||
| Fig. 3 Partial structure of tiamulin (2) bound to the 50S ribosomal subunit of D. radiodurans.67 | ||
Consistent with previous findings, tiamulin (2) is located in the PTC. The tricyclic core of the molecule resides in the A site, while the C14 side chain extends into the P site. The crystal structure confirmed the results of the footprinting studies, revealing hydrophobic interactions between tiamulin (2) and nucleotides G2061, A2062, C2063, A2451, C2452, A2503, U2504, G2505, U2506, U2585 and C2586 in domain V and hydrogen bonds with G2061 and U2585. Once bound, the core is surrounded by G2061, A2451, C2452, A2503, U2504, G2505, and U2506. The cyclopentanone engages in hydrophobic interactions with A2451 and C2452. The C3 ketone, known to be essential for bioactivity, is located near the carbohydrate of A2503. The C16 methyl substituent interacts with A2451 and U2506. The C12 vinyl substituent, as well as the C17, C18 and C12 methyl groups, share hydrophobic interactions with the nucleotides G2061, C2063, A2451, A2503, U2504, G2505 and C2586. A hydrogen bond exists between the C11 hydroxyl group and O1P of G2505. G2061 hydrogen bonds from N1 and N2 to the C21 ketone. N2 also hydrogen bonds to the sulfur atom in the C14 side chain. The C14 side chain also experiences hydrophobic interactions with A2062, U2585, and C2586 nucleotides in the P site.67
Davidovich et al. crystallized D50S with three additional pleuromutilin derivatives: SB-571519, SB-280080, and retapamulin (4) (PDB 2OGM, 2OGN, 2OGO, Fig. 4).68 As with the crystal structure of tiamulin (2), all three bound the PTC and engaged in similar hydrophobic and hydrogen bonding interactions. Conformational rearrangements of the rRNA were also reported following the binding of these pleuromutilins, including a 40° rotation of U2506 and a 180°-flipped rearrangement of U2585 (PDB 2OGO, 1NKWFig. 5).
 | ||
| Fig. 4 Partial structure of retapamulin (4) bound to the 50S ribosomal subunit of D. radiodurans.68 | ||
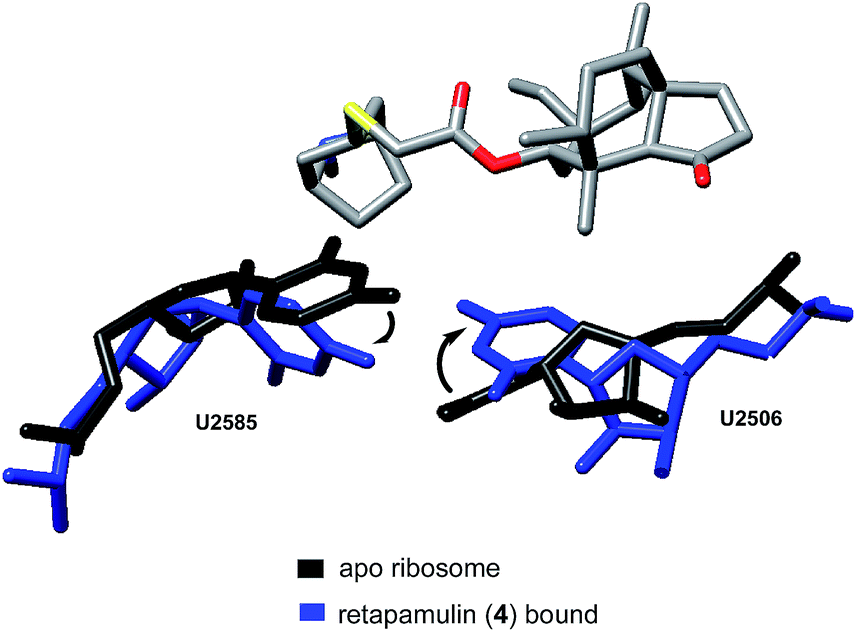 | ||
| Fig. 5 Conformational rearrangements of U2585 and U2506.68 | ||
The rotation of U2506 closes the binding pocket around the pleuromutilins; this mode of interaction is referred to as induced-fit binding. U2585 is known to be a flexible base and, based on previously solved structures, should exist in a position to engage the C14 side chains of these derivatives. However, presumably due to steric hindrance, U2585 shifts upon binding of retapamulin and other C22-modified pleuromutilins. In the case of SB-571519 and SB-280080, antibiotic binding causes U2585 to hydrogen bond with U2506. In no cases was U2585 observed to interact with the C14 extension.68
The crystal structure of tiamulin (2) bound to the 50S ribosome subunit is consistent with previous findings that tiamulin (2) competes for binding with chloramphenicol, puromycin, and carbomycin A (Fig. 6).64,65 The binding site of tiamulin (2) can be partially overlaid with chloramphenicol, clindamycin and the disaccharide extension of carbomycin A, and is nearly the same as the streptogramin antibiotics dalfopristin and virginiamycin A, despite their very different structures. The galactose ring of clindamycin forms hydrogen bonds to A2058, A2059, G2505, and A2503 in the 50S subunit, overlapping with the tiamulin (2) C11 hydroxyl group and G2505. Clindamycin induces a shift of U2506. Chloramphenicol also hydrogen bonds to G2505.69 As with the pleuromutilin class, dalfopristin engages in hydrogen bonds with G2061 and G2050.70
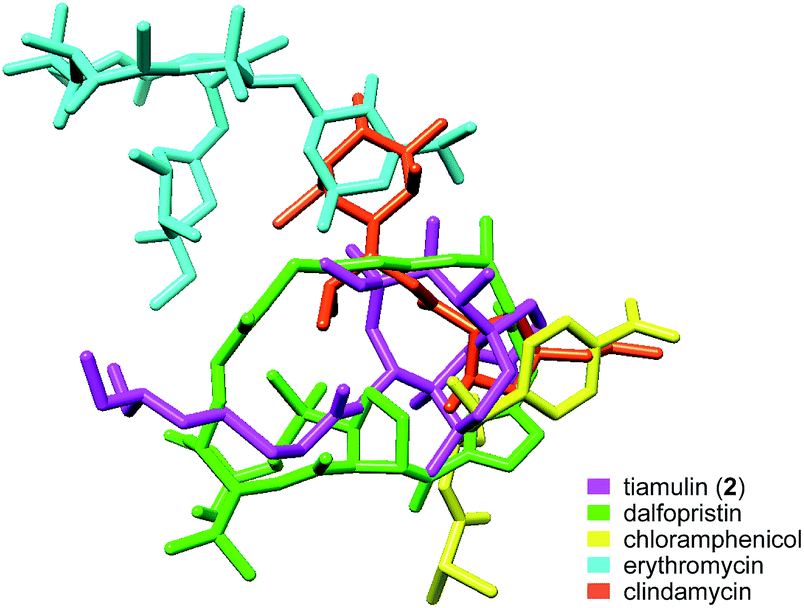 | ||
| Fig. 6 Overlay of ribosome-targeting antibiotics. | ||
Eyal et al. reported the crystal structure of lefamulin (5) bound to the ribosome of S. aureus in 2016 (PDB 5HL7, Fig. 7).71 Consistent with the crystal structures of tiamulin (2) and retapamulin (4), hydrogen bonding interactions between C21 carbonyl and G2061 and between C11 hydroxyl and G2505 phosphate are observed. In lefamulin (5), an additional hydrogen bond is formed between the primary amine within the C14 extension and the O2 of A2062 ribose. U2585 is stacked with the cyclohexyl ring of the C14 extension. This interaction is further stabilized by U-U-4-carbonyl N3 symmetric interactions between U2585 and U2506 and leads to a tighter binding pocket, consistent with lower IC50 values observed for lefamulin (5) than for tiamulin (2) and BC-3205 (60).71
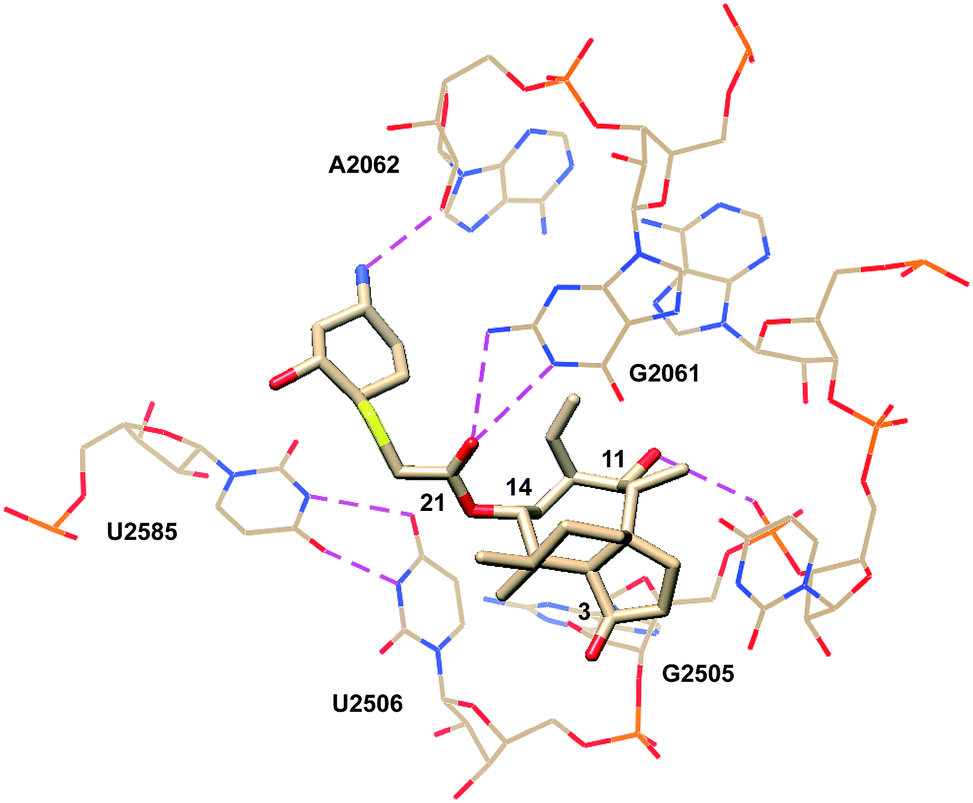 | ||
| Fig. 7 Partial structure of lefamulin (5) bound to the 50S ribosomal subunit of S. aureus.71 | ||
Resistance mechanisms are discussed in Section 4 but are mentioned here briefly as they relate to ribosome structure and binding. Resistance to PTC-binding antibiotics is slow to develop and in line with this, pleuromutilins have spontaneous mutation frequencies of 10−9 to 10−11 in S. aureus and other organisms.72 Resistance to PTC antibiotics is attributed to modifications of distal nucleotides that alter the conformation of the induced-fit binding.
Accounts of tiamulin (2) resistance have been reported across Europe and have provided a basis to study the mechanisms of clinical resistance. Groups of Brachyspira spp. that had shown increased resistance to tiamulin (2) in pig herds were analyzed and found to possess mutations in genes encoding for the ribosomal protein L3 and a nucleotide proximal to the PTC. In one of the isolates, amino acid 148 of the L3 protein was the only mutation observed. As expected, tiamulin (2) displayed reduced binding to ribosomal subunits in footprinting assays conducted on these strains. It was suggested that the mutations in the L3 protein induce a change in the position of the binding pocket by altering the interaction of tiamulin (2) with G2576, U2506 and G2505.73 Davidovich et al. showed that L3 Arg:144 interacts with U2506 phosphate but does not make any interactions with the pleuromutilins. An alternative resistance mechanism involves methylation of A2503 by the methyltransferase Cfr. This methylation results in steric hindrance that prevents pleuromutilins and other PTC antibiotics from binding. Overall, incidents of resistance to pleuromutilin antibiotics remain relatively low with just 0.18% reported for S. aureus treated with lefamulin (5) in 2010.36
The C14 side chain has been the subject of extensive derivatization with the aim of improving the potencies and pharmacokinetics of pleuromutilins.28–36 Docking studies have suggested that chemical modification of the side chain may engender additional interactions with the ribosome. C14 extensions containing a 2-aminothiazole residue can possibly achieve an additional hydrogen bond between the NH2 extending from the thiazole and the hydroxyl group of C2565.74 Pyrimidine derivatives such as 57 developed by Yi et al. achieved a hydrogen bond between the NH2 and C2046, and piperidine derivatives such as 58 achieved a hydrogen bond interaction between a hydroxyl group and A2418 (Fig. 8).75 Compared to tiamulin (2), these compounds showed some improvements in potency and efficacy against infections in mice. The C14 side chain may also be important to overcoming resistance.31 While the L3 mutation diminishes the antibiotic activities of (+)-pleuromutilin (1) and tiamulin (2), it is thought that through its side chain, valnemulin (3) is able to make additional favorable interactions with the ribosome that allow it to accommodate the changed binding pocket induced by the L3 mutation.76
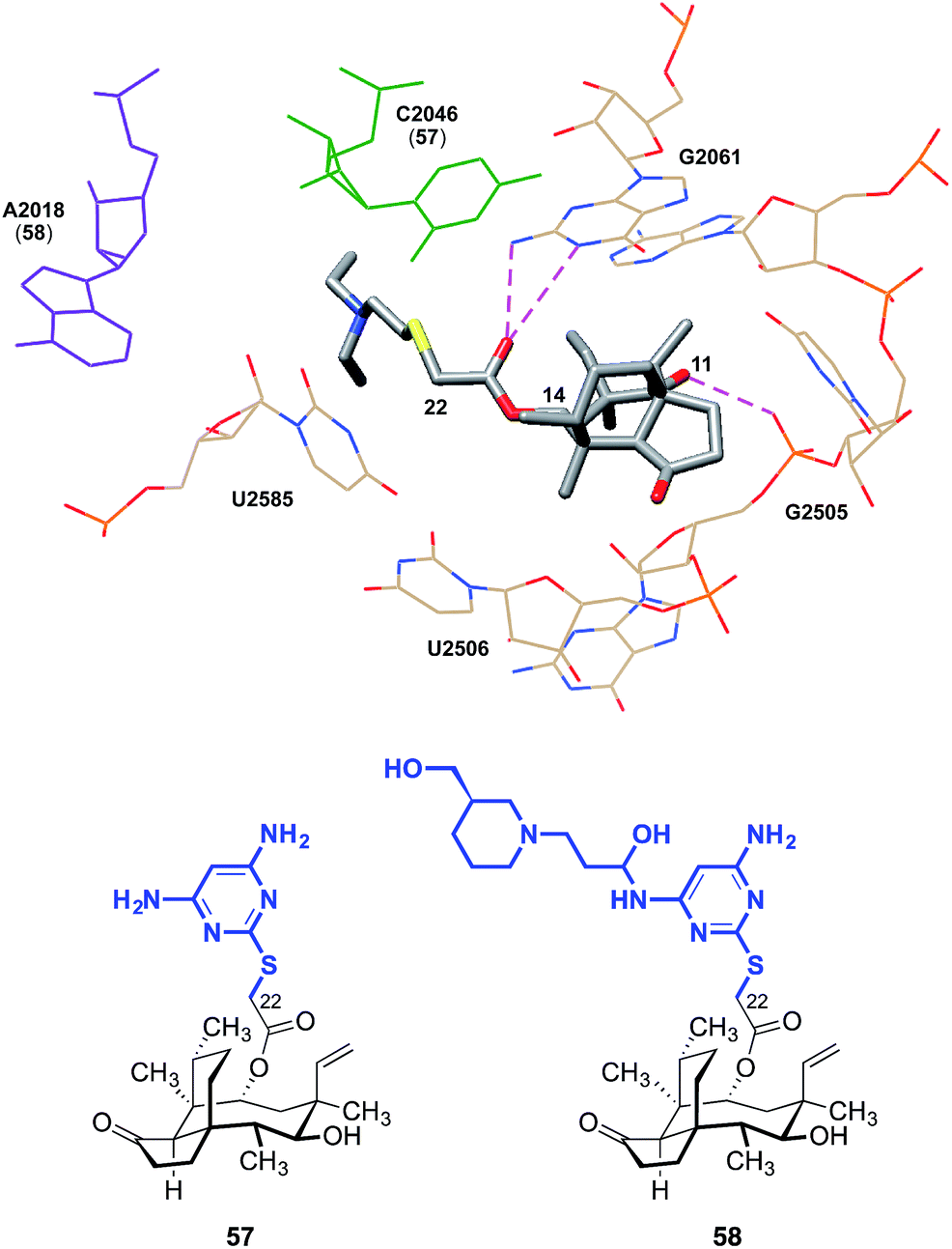 | ||
| Fig. 8 The structure of tiamulin (2) bound to the 50S subunit of D. radiodurans (top). The pleuromutilin derivatives 57 and 58 (bottom) engage in additional interactions with the PTC (C2046 and A2018 for 57 and 58, respectively). | ||
2.3 Selectivity of the pleuromutilin class for the bacterial ribosome
While many ribosome characteristics are conserved across all life, differences between the eukaryotic and bacterial ribosome rRNA sequence exist, making the latter a viable anti-infective target. As seen in pleuromutilin-bound prokaryotic ribosome crystal structures, a double layer of nucleotides creates a closed conformation around the pleuromutilins, keeping them in the binding pocket (PDB 5HL7, 4UG0, Fig. 9).67,68 This is accomplished in part by interactions between the nucleotides U2504 and C2452; in addition to the hydrophobic interactions they achieve with the tricyclic core of pleuromutilin derivatives, in bacteria, U2504 and C2452 interact with each other through non-Watson–Crick C:U 4-carbonyl-amino N3–N3 interactions.77 The corresponding nucleotides in eukaryotes and archaea are positioned differently and cannot interact with one another. U2504 forms a π-stacking interaction with A2055 in eukaryotes, stabilizing the open conformation and preventing the binding site from closing around the pleuromutilins.71 The closed conformation is further stabilized in bacteria by the PTC second shell base pair A2543-U2500, which stacks to U2504 and C2452. In eukaryotes, the corresponding nucleotides 2453 and 2500 are both uridines and do not form a base pair, and in archaea, the base pairing between A2543 and U2500 is hindered by the presence of stacking between U2504 and A2055.71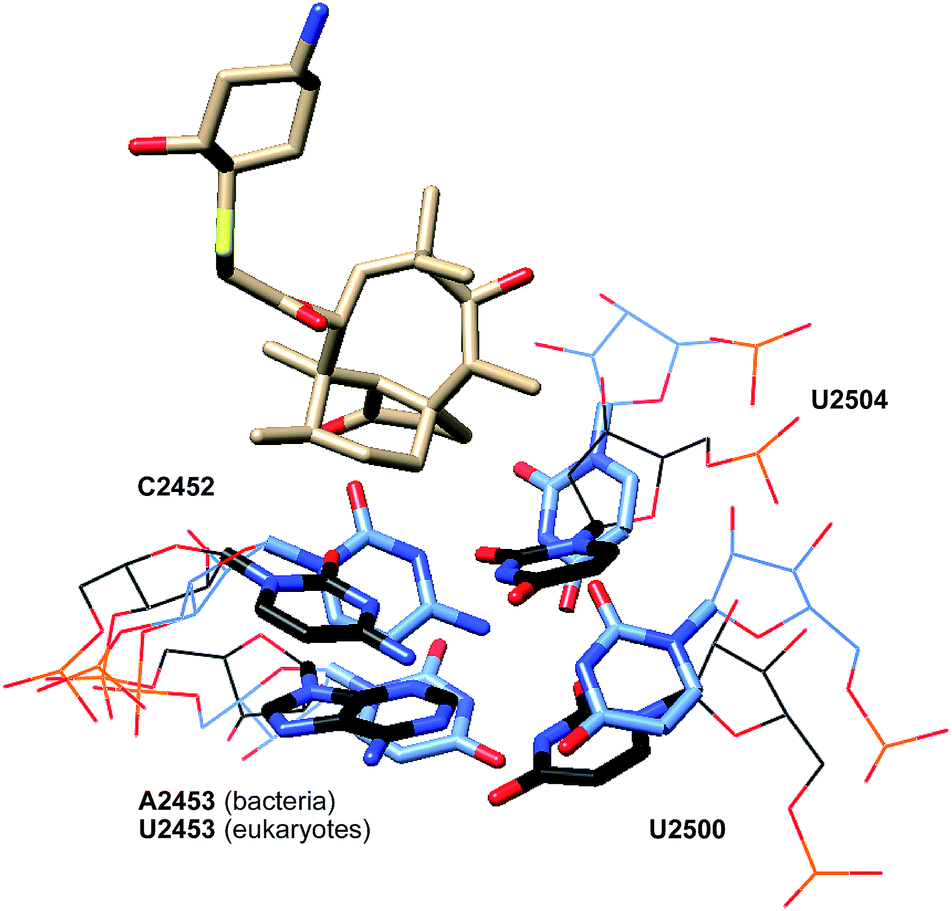 | ||
| Fig. 9 Partial structure of lefamulin (5) bound to the 50S ribosomal subunit of S. aureus (black) superimposed onto the partial structure of the human 80S ribosomal subunit (blue). | ||
3. Clinical and pre-clinical pleuromutilins
While thousands of pleuromutilin derivatives have been prepared by modification of the side chain or tricyclic core,28–36 only sulfanylacetyl derivatives, most of which contain a basic amine residue on the C14 extension, have advanced into or beyond Phase I clinical studies (Fig. 10).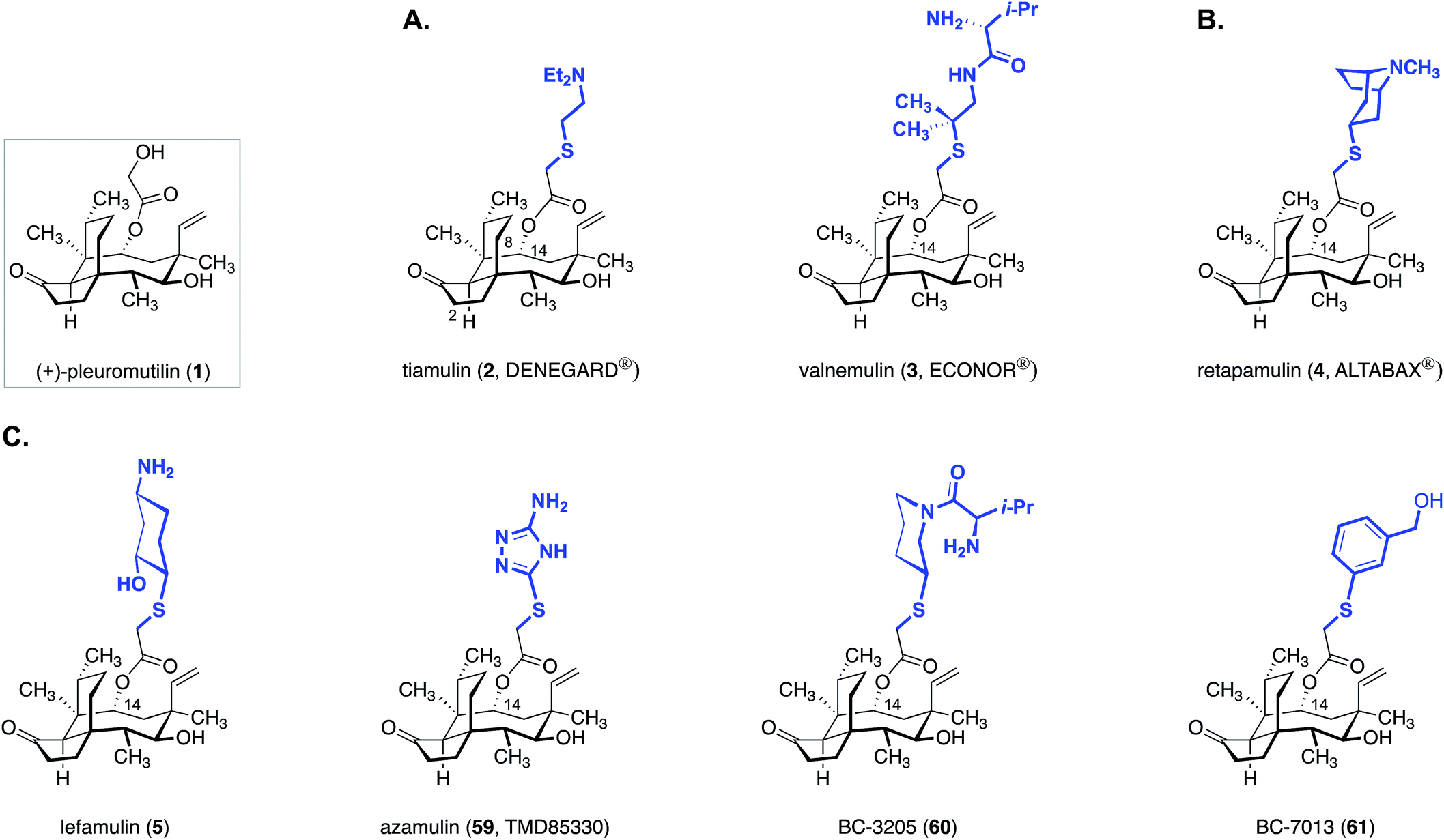 | ||
| Fig. 10 Clinical and preclinical pleuromutilin derivatives: (A) the veterinary antibiotics tiamulin (2) and valnemulin (3). (B) The human topical antibiotic retapamulin (4). (C) The preclinical agents lefamulin (5), azamulin (59), BC-3205 (60), and BC-7013 (61). | ||
Tiamulin (2, DENEGARD®), the first pleuromutilin antibiotic employed in veterinary medicine, was developed by the Sandoz group in the 1970s.78,79 Although tiamulin (2) is rapidly metabolized in vivo by cytochrome P450 oxidation at C2 and C8, its high antimicrobial activity against many Gram-positive bacteria renders it effective as both a prophylactic and therapeutic treatment for bacterial infections in farm animals.80 It is given to swine81–83 to treat dysentery caused by Brachyspira hyodysenteriae, porcine colonic spirochaetosis (colitis) caused by B. pilosicoli, porcine proliferative enteropathy (ileitis) caused by Lawsonia intracellularis, and enzootic pneumonia caused by Mycoplasma hyopneumoniae. It is given to chickens to treat chronic respiratory disease (CRD) and airsacculitis caused by M. gallisepticum, and infectious synovitis caused by M. synoviae. It is given to turkeys to treat infectious sinusitis and airsacculitis caused by M. gallisepticum, synoviae and meleagridis. It is given to rabbits to treat epizootic rabbit enterocolitis (ERE). Pharmacokinetic studies in swine and chicken reveal that tiamulin (2) has high oral bioavailability (70–95%) and is widely distributed throughout the body. Once absorbed, tiamulin (2) accumulates in the lungs (a target tissue), liver (site of metabolism and excretion) and kidneys (site of excretion). In swine, unabsorbed or unmetabolized tiamulin (2) passes through the intestines and concentrates in the colon, a target tissue. Due to the rapid metabolism and excretion of tiamulin (2), the withdrawal period for meat and offal are generally short (0–6 days depending on the animal species and dosage).80
In 1999, Novartis launched valnemulin (3, ECONOR®), the second veterinary antibiotic derived from (+)-pleuromutilin (1).84–86 Valnemulin (3) has broad spectrum activity against Gram-positive pathogens such as S. aronson, S. aureus, M. arthritidis, M. bovigenitalium, M. bovimastitidis, M. bovirhinis, M. canis, M. felis, M. fermentans, M. gallinarum, M. gallisepticum, Acholeplasma granularum (A. granularum), M. hominis, M. hyorhinis, A. laidlawii, M. meleagridis, M. neurolyticum, M. pneumonia and M. hyopneumoniae.85 Against many of these strains, valnemulin (3) has superior antimicrobial activity than tiamulin (2) in vitro87,88 and in vivo.88 Valnemulin (3) is used as prophylactic or cure for swine dysentery, ileitis, colitis and pneumonia, and epizootic rabbit enterocolitis. It is also reported to reduce or eliminate lung infections caused by M. bovis in calves.89 Although it is primarily used in veterinary medicine, valnemulin (3) has been used in humans to treat life-threatening drug-resistant Mycoplasma infections in immuno-compromised patients.90
The crisis of antibiotic resistance91–93 has motivated enormous efforts to develop pleuromutilin derivatives for human use. However it was not until 2007 that the first pleuromutilin, retapamulin (4, sold under the tradename ALTABAX® in the US94 and ALTARGO® in the EU95), was introduced. Retapamulin (4) shows excellent potency against S. aureus, S. pyogenes, S. agalactiae95 and several erythromycin, mupirocin, oxacillin, or methicillin-resistant strains in vitro (MIC90 ≤ 0.12 μg mL−1).96,97 Retapamulin (4) is used as a topical, short-term treatment for skin and soft tissue infections (SSTIs) including impetigo and small infected lacerations, abrasions, or sutured wounds.95,98
However, despite equal in vitro antibacterial activity against methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA), low clinical efficacy was observed in the treatment of patients with secondarily infected open wound (SIOW) infections caused by MRSA.95 Retapamulin (4) is a substrate for P-glycoprotein (Pgp) pumps and inhibits Pgp-mediated digoxin transport (IC50 = 28.2 μM). It is also a CYP3A4 substrate and inhibitor; it is extensively oxidized in vivo by microsomes and inhibits oxidation of midazolam (IC50 = 1.1 μM) and nifedipine (IC50 = 6.2 μM).94 Nonetheless, when administered topically, these effects are not clinically significant.99
Perhaps the greatest impediment to developing systemic pleuromutilin antibiotics for human use is their poor metabolic stability. In the 1990s, azamulin (59, aka TMD85330), an azole derivative of (+)-pleuromutilin (1) intended for human use, was disclosed by GlaxoSmithKline. Azamulin (59) was reported to have good oral bioavailability at dosage levels appropriate for clinical use, despite its extensive metabolism by cytochrome P450 oxidation.100,101 However, azamulin (59) did not progress beyond a Phase I clinical trial because it was found to be a strong and irreversible inhibitor of CYP3A (IC50 = 30–240 nM).102–104 It seems that this CYP inhibition is specific to azamulin (59), rather than a general property of pleuromutilins, which encouraged further pursuit of new derivatives suitable for systemic administration.100
Nabriva Therapeutics has extensively investigated C14 derivatives of (+)-pleuromutilin (1). These researchers identified lefamulin (5, BC-3781) as potentially the first pleuromutilin drug suitable for systemic use in humans. Lefamulin (5) has shown promising activity for a number of clinical indications105 including community acquired bacterial pneumonia (CABP, currently in Phase 3), acute bacterial skin and skin structure infections (ABSSSIs, Phase 2 completed), pediatric indications (pre-clinical), sexually transmitted infections (STIs, pre-clinical), hospital-acquired bacterial pneumonia (HABP)/ventilator-associated bacterial pneumonia (VABP, pre-clinical), osteomyelitis (pre-clinical), and prosthetic joint infections (pre-clinical). Lefamulin (5) exhibits excellent potency against a broad spectrum of Gram-positive bacteria,31 especially drug- and multidrug-resistant isolates.106–110 Although lefamulin (5) inhibits CYP3A in vitro, these effects were not problematic in a clinical setting.111 Lefamulin (5) is the second compound Nabriva has pursued as a systemic agent. BC-3205 (60), advanced to Phase I studies but was discontinued.112,113
In addition to the systemic agents lefamulin (5) and BC-3205 (60), Nabriva also announced the development of a novel topical pleuromutilin, BC-7013 (61),114 which recently completed Phase 2 clinical trial for the treatment of uncomplicated skin and skin structure infections (uSSSIs).105 BC-7013 (61) is highly active against Gram-positive bacteria that cause skin and ocular infections. The MIC90 values for BC-7013 (61) against MRSA strains are up to 20-fold lower than those of mupirocin and 8-fold lower than those of retapamulin (4). Studies of BC-7013 (61) have also demonstrated potent activity against Chlamydia trachomatis, the leading cause of blindness in the world, and Propionibacterium acnes, the causative agent of acne.115
4. Resistance
One of the most valuable aspects of pleuromutilins is that they are associated with low rates of resistance development. Additionally, they display minimal cross-resistance with other antibiotics that target the bacterial ribosome.36 To date, three mechanisms of resistance to pleuromutilin have been identified. These include mutations in 23S rRNA and rplC genes encoding the ribosomal protein L3, methylation of the nucleotide A2503 by Cfr methyltransferase, and drug efflux by ATP-binding cassette (ABC) transporters.4.1 Mutations in ribosomal protein L3
Investigations with tiamulin (2) against laboratory-selected Brachyspira spp. strains and clinical B. hyodysenteriae field isolates revealed that mutations in ribosomal protein L3 proceed in a slow and stepwise manner.62 More recently, studies of tiamulin (2) and retapamulin (4) show that single and double mutations in rplC only slightly diminish susceptibility, while a third mutation is required for a significant increase of resistance.116 However, the frequency of the three-step mutation is low (<10−9) and all third-step mutants exhibited severe growth defects and were frequently outcompeted by faster-growing variants.116 These data provide an explanation for the low rates of clinical resistance following treatment.314.2 Nucleotide methylation by Cfr methyltransferase
Resistance to pleuromutilins can also arise from methylation of the nucleotide A2503 by Cfr methyltransferase, which impedes pleuromutilin binding. The cfr gene was primarily detected in livestock-associated Staphylococci but has also been found in human Staphylococci isolates.36 The cfr gene is located on the chromosome and various plasmids and as a result, is potentially transferable. In vitro, a cfr-carrying plasmid was found to be transferable between Enterococcus faecalis (E. faecalis) by conjugation117 and from S. epidermidis to MRSA by conjugation or transduction.118 Outbreaks of cfr-encoding strains have been observed in clinical settings.1194.3 Pleuromutilin efflux
Diminished antibiotic activities of pleuromutilin derivatives were detected in ABC transporter-containing bacteria. The transporters are encoded by vga(A)120 [and its variant vga(A)v121], vga(C),120vga(E),122,123lsa(C),124 and lsa(E),125 which are hosted by transposons and plasmids. However, the frequency of drug-efflux induced resistance remains low. A study of 5676 S. aureus clinical isolates drawn from data from eight preclinical studies, one global surveillance study, and five Phase III clinical trials, revealed that only six vga(A) or vga(A)v containing strains are detected with retapamulin (4) resistance.126Despite the detection of resistant strains and the identification of possible resistance mechanisms, the overall rates of resistance to pleuromutilins remain low. In the SENTRY antimicrobial surveillance program conducted with lefamulin (5) in 2010, only 0.18% of S. aureus and 3.4% of coagulase-negative Staphylococcus spp. exhibited resistance. Among the S. aureus isolates, 0.05% had mutated rplC, 0.018% harbored the cfr gene, and 0.11% harbored the vga(A) gene. Among coagulase-negative Staphylococcus spp., the incidences for rplD, cfr, and vga(A) alterations were 0.34%, 2.5%, and 0.11%, respectively.36
5. Key structure–activity relationships (SARs)
Since its isolation in the 1950s, continuous efforts have been made to improve the pharmacological properties of (+)-pleuromutilin (1). As (+)-pleuromutilin (1) can be obtained in large quantities via industrial fermentation, these efforts focused on semisynthesis, and thousands of analogs have now been prepared.28–36 Due to their ease of synthesis, C14 glycolic ester derivatives constitute the vast majority of derivatives.29,34,35 Although analogs with C2, C8, and C12 modifications have also been examined, all derivatives that have advanced to clinical trials contain the unmodified tricyclic core. This section discusses the key structure–activity relationships (SAR) and presents selected examples from the major classes of pleuromutilin derivatives.5.1 Modification at C22
A straightforward functionalization strategy involving activation of the C22 hydroxyl group by sulfonylation, followed by SN2 displacement, has been commonly employed to access C22 derivatives (Scheme 4).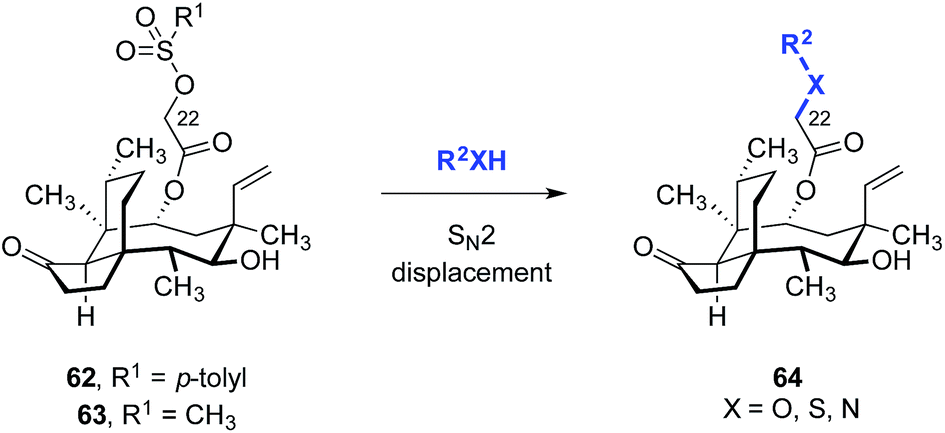 | ||
| Scheme 4 General strategy for C22 modification. | ||
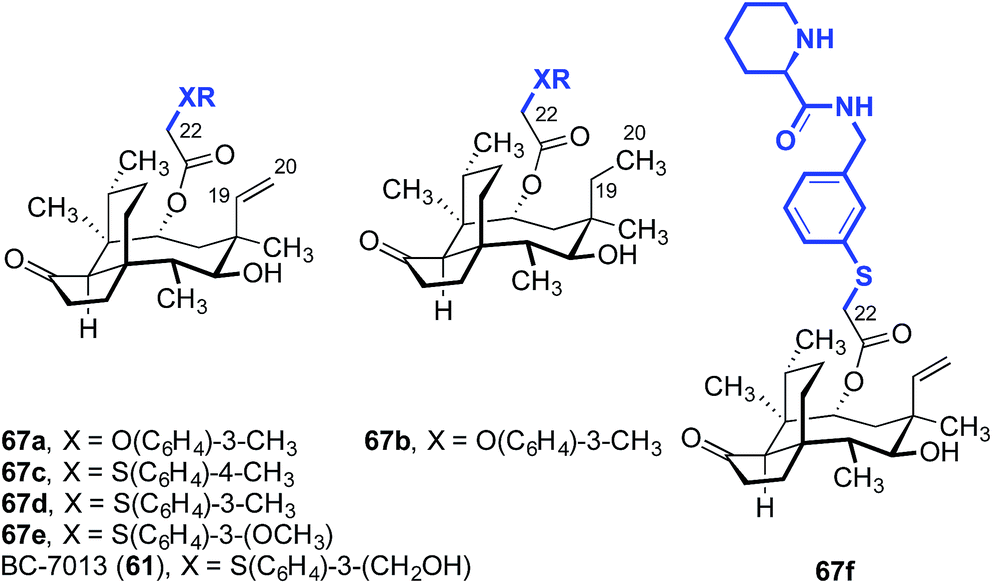
This strategy was used in the earliest SAR studies of semisynthetic pleuromutilin analogs reported by Biochemie in 1976 (Table 1).127–129 In the original report, which led to the discovery of tiamulin (2), a number of valuable insights emerged.129 The negligible difference in activities between natural (+)-pleuromutilin (1, entry 1) and 19,20-dihydropleuromutilin (65, entry 2) indicated that the C19–C20 alkene was disposable. Installation of a neutral aryl sulfide at C22 slightly improved antimicrobial activities (66a, entry 3). Compounds containing an acidic C12 side chain, such as a C-terminal glutamic acid, were not active (66b, entry 4). Compounds containing a basic C14 substituent, such as a diethylamino group, had some activity (66c, entry 5). Although the potency of 66c was lower than (+)-pleuromutilin (1), the introduction of a basic residue was recognized as a potential strategy to increase polarity and hydrophilicity, which could address the limited bioavailability of the natural product. Combining the benefit of enhanced activity induced by the sulfide and the polarity increase from the addition of a basic residue resulted in the discovery of tiamulin (2, entry 6), which contains a 2-(diethylamino)ethylsulfide residue. Tiamulin (2) was several orders of magnitude more potent than (+)-pleuromutilin (1). Increasing the distance between the sulfide and the basic amine groups by either inserting an aryl group (66d, entry 7) or extending the alkyl chain to five methylene units (66e, entry 8) led to analogs with further improved bioactivities. However, further increasing the basicity of the terminal substituent by switching to a guanidyl group diminished activity (66f, entry 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |||
| S. aureus | M. hominis | M. gallisepticum | M. hyorhinis | ||
| 1 | (+)-Pleuromutilin (1) | 0.5 | 0.3 | 0.3 | 2 |
| 2 | 65 | 0.3 | 0.8 | 0.3 | 3 |
| 3 | 66a | 0.02 | 0.04 | 0.02 | 0.8 |
| 4 | 66b | >10 | >10 | >10 | >100 |
| 5 | 66c | 1.4 | 5 | 5 | 3 |
| 6 | Tiamulin (2) | 0.05 | 0.01 | 0.006 | 0.03 |
| 7 | 66d | 0.01 | 0.001 | 0.003 | 0.2 |
| 8 | 66e | 0.02 | 0.001 | 0.006 | 0.04 |
| 9 | 66f | 5 | 0.08 | 0.2 | 1 |

BC-7013 (61), a clinical candidate for the topical treatment of uncomplicated skin and skin structure infections (uSSSIs, see Section 3) was discovered in an effort to probe the effects of an aryl sulfide linkage (Table 2).114,130,131 The two aryl ethers 67a and 67b, which differ only by the presence or absence of the C19–C20 alkene, respectively, had comparable activities (entries 1 and 2). This confirms the studies outlined in Table 1, which suggested the alkene was not important for activity. Also in accord with the data in Table 1, the thioethers 67c–67e and 61 (BC-7013; entries 3–6) exhibited greater potencies than the structurally similar ethers 67a and 67b (entries 1 and 2). Among the aryl thioethers studied, the meta-substituted derivatives 67d–67e and 61 (BC-7013; entries 4–6) were more potent than the para-substituted derivative 67c (entry 3). Although the addition of a hydrogen-bond acceptor on the arene, as in 67e (entry 5), did not improve the efficacy, introduction of a hydrogen-bond donor significantly increased activity leading to the discovery of BC-7013 (61, entry 6). A further increase in potency was also observed when an additional hydrogen-bond donor was introduced to the side chain (67f, entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |||
| S. aureus | E. faecalis | M. catarrhalis | E. coli | ||
| 1 | 67a | 0.05 | >6.4 | 0.0125 | >6.4 |
| 2 | 67b | 0.05 | >6.4 | 0.025 | >6.4 |
| 3 | 67c | 0.025 | >25.6 | ≤0.0125 | >25.6 |
| 4 | 67d | 0.025 | >6.4 | 0.0125 | >6.4 |
| 5 | 67e | 0.025 | >25.6 | ≤0.0125 | >25.6 |
| 6 | BC-7013 (61) | ≤0.0125 | 6.4 | ≤0.0125 | 12.8 |
| 7 | 67f | ≤0.0125 | 1.6 | ≤0.0125 | 12.8 |
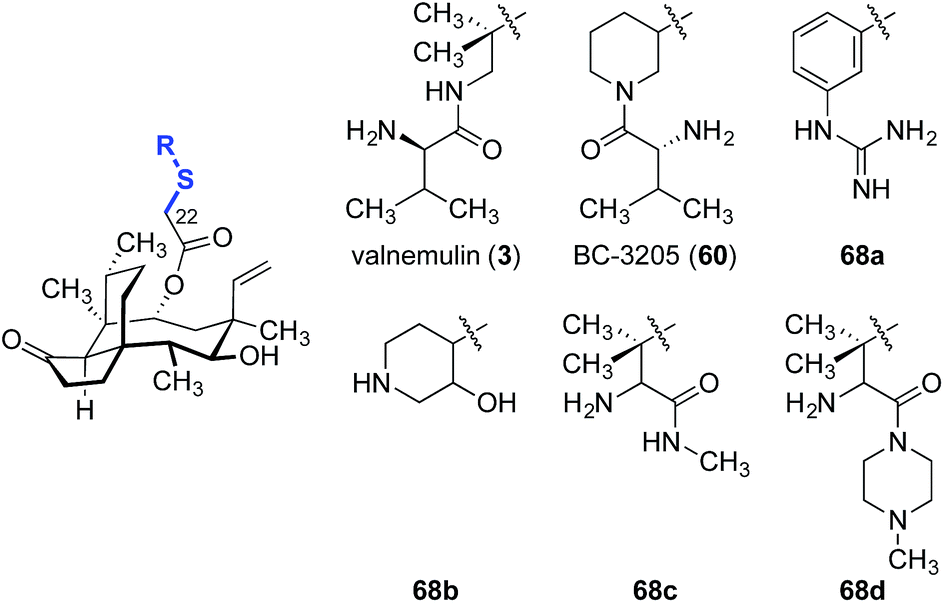
Valnemulin (3) and related derivatives have been evaluated against strains of M. tuberculosis (Table 3).132 Both M. tuberculosis strains employed (denoted “strain A” and “strain B”) are sensitive to isoniacid, rifampicin, streptomycin and valnemulin (3, entry 1). A two-fold improvement in potency was observed when the conformationally constrained analog BC-3205 (60, entry 2) was evaluated, suggesting the spatial orientation of the amine residue may be important for binding. Substituting the ammonium functionality with an aryl guanidinium group (68a, entry 3) created a better hydrogen-bond donor that is conformationally restricted, and led to a 2–4-fold increase in potencies. Introduction of a weaker hydrogen bond donor (68b, entry 4) or increasing the steric hindrance around the ammonium functionalities (68c and 68d, entries 5 and 6, respectively) diminished activity.
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| Strain A | Strain B | ||
| 1 | Valnemulin (3) | 4 | 4 |
| 2 | BC-3205 (60) | 2 | 2 |
| 3 | 68a | 1 | 0.5 |
| 4 | 68b | 10 | 4 |
| 5 | 68c | 32 | 32 |
| 6 | 68d | 32 | 32 |
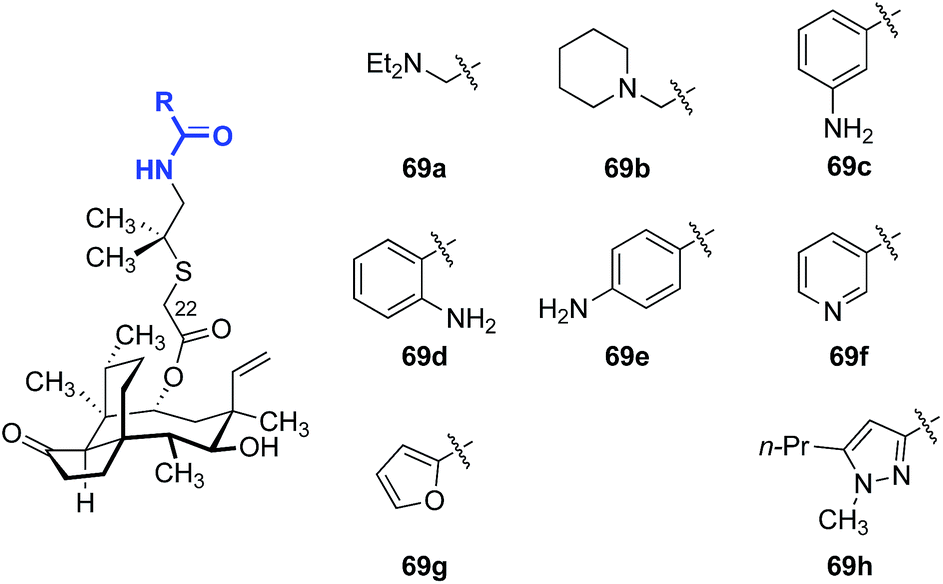
As valnemulin (3) has been used successfully in veterinary medicine for decades, extensive studies were carried out to adapt this side chain for use in humans (Table 4).133–136 Attempts to combine the advantageous features of tiamulin (2) and valnemulin (3) led to analog 69a, which showed moderate activity against E. coli (entry 1).133 Moreover, the acyclic derivative 69a was 4-fold more potent than the structurally-related cyclic derivative 69b (entry 2).133 Aromatic amides bearing an amine on the aryl ring (69c–69e) also demonstrated modest antimicrobial activities (entries 3–5). The ortho- and para-substituted derivatives 69d and 69e were 4-fold more potent than the meta-substituted derivative 69c. The nicotinamide derivative 69f showed the greatest potency among the analogs studied (entry 6).135,136 Furan (69g, entry 7) and pyrazole (69h, entry 8) derivatives were not active in E. coli.135 These data suggested that addition of positive charge on the far end of the C22 side chain was beneficial to the antibiotic activities of the analogs. To the best of our knowledge, only one report of human usage of valnemulin (3) was disclosed, by Webster and co-workers in 2001.90 Although valnemulin (3) has never gone through human clinical trials, three patients with Common Variable Immunodeficiency (CVID) infected by M. pneumonia, Ureaplasma urealyticum (U. urealyticum), and M. hominis were successfully treated with orally administered valnemulin (3). Although no major side effects were observed in these three patients, the dosage was carefully monitored. Doses of 20 mg kg−1 daily induced anaemia and thrombocytopenia in one case. Moreover, similar to tiamulin (2), valnemulin (3) depresses the hepatic CYP450, which could increase toxic effects of other drugs administered simultaneously.
|
|
||
|---|---|---|
| Entry | Compound | MIC (μg mL−1), E. coli |
| 1 | 69a | 4 |
| 2 | 69b | 16 |
| 3 | 69c | 8 |
| 4 | 69d | 2 |
| 5 | 69e | 2 |
| 6 | 69f | 0.25 |
| 7 | 69g | >128 |
| 8 | 69h | >128 |
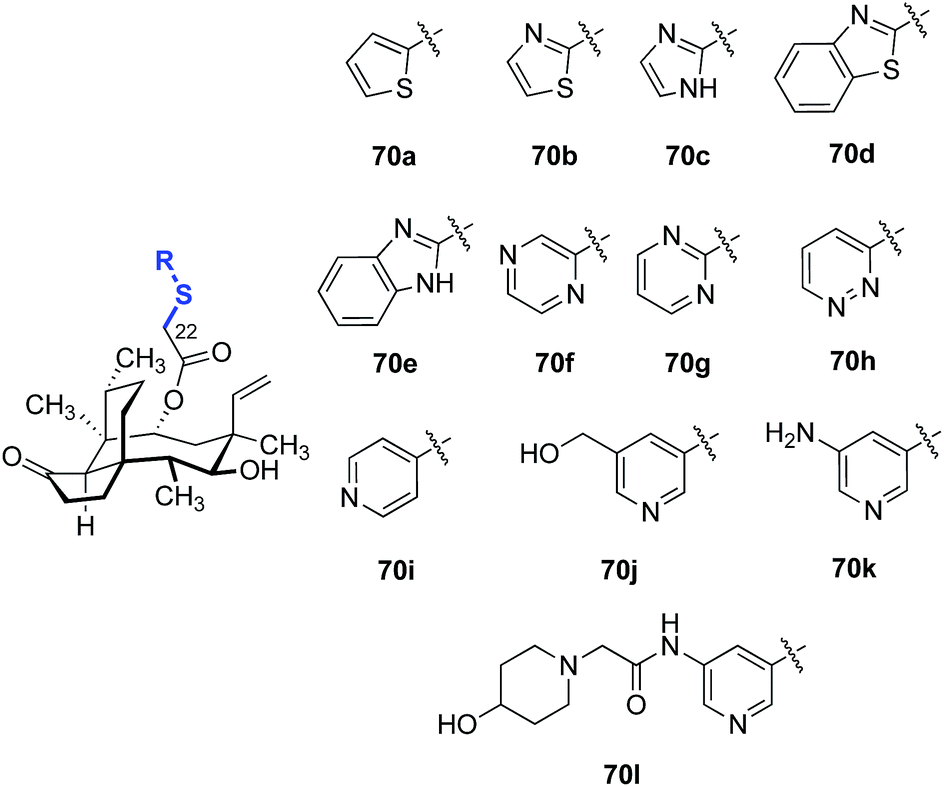
A survey of various heteroaromatic thiol substituents yielded improvements in activity against Gram-positive strains (Table 5).137,138 Among the examples selected, the pyridine derivatives 70i–l (entries 9–12) were 2–4-fold more potent against MRSA and methicillin-resistant S. epidermidis (MRSE) than other five- (70a–e, entries 1–5) and six-membered (70f–h, entries 6–8) heterocyclic derivatives evaluated. The addition of polar substituents to the pyridine ring (70j–l, entries 10–12) further improved the potencies of the compounds (c.f. the unsubstituted pyridine derivative 70i, entry 9).
|
|
||||
|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | ||
| S. aureus | MRSA | MRSE | ||
| 1 | 70a | 0.5 | 0.5 | 0.25–1 |
| 2 | 70b | 0.5 | 0.5 | 0.25–2 |
| 3 | 70c | 1 | 0.5 | 1 |
| 4 | 70d | 0.5 | 0.25 | 0.5 |
| 5 | 70e | 0.25 | 0.25 | 0.25 |
| 6 | 70f | 0.25 | 0.125–0.25 | 0.125 |
| 7 | 70g | 0.125 | 0.063–0.125 | 0.125 |
| 8 | 70h | 0.25 | 0.25 | 0.063–16 |
| 9 | 70i | 0.031 | 0.063–0.125 | 0.063–0.25 |
| 10 | 70j | 0.031 | 0.031–0.063 | 0.031 |
| 11 | 70k | 0.031 | 0.031–0.063 | 0.031 |
| 12 | 70l | 0.031 | 0.063 | 0.125–0.5 |
Although most of the discussion in this section is limited to studies of the relationship between structure and in vitro potencies, the reader is reminded that the pharmacokinetic changes are equally important (Table 6). For example, analogs 70j and 70l possess comparable in vitro potencies (Table 5). However, 70j possesses more acceptable plasma exposure levels (defined as the area under curve (AUC)) and serum half-life (T1/2) compared to 70l (entries 1 and 2, respectively). Both analogs possessed comparable mean residence times (MRT), clearance levels (CL), and volumes of distribution (Vss).
| Entry | Compound | AUC0–t (h ng mL−1) | T 1/2 (h) | MRT (h) | CL (L h−1 kg−1) | V ss (L kg−1) |
|---|---|---|---|---|---|---|
| 1 | 70j | 3482 ± 441 | 6.00 ± 2.71 | 0.54 ± 0.15 | 2.88 ± 0.33 | 1.56 ± 0.48 |
| 2 | 70l | 650 ± 138 | 0.67 ± 0.17 | 0.61 ± 0.05 | 3.2 ± 0.66 | 1.9 ± 0.30 |
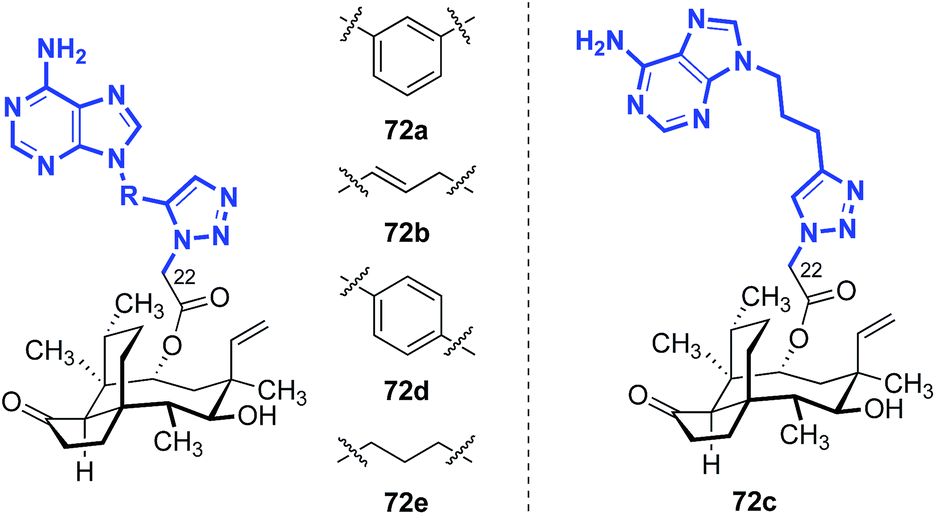
Five other classes of heteroarene-containing C22 thioethers have been examined in detail. In 2008, Hirokawa and co-workers evaluated the influence of introduction of a purine ring to the C22 side chain, to generate nucleoside-like analogs (Table 7).139–141 These compounds were evaluated against MSSA, MRSA, and vancomycin-resistant Enterococci (VRE) in vitro, and MSSA in mice. A comparison between compounds 71a and 71b showed that the site of nitrogen substitution in the purine ring had a modest impact on the in vitro efficacy (entries 1 and 2). Installation of an amino substituent at the 2-position of the purine ring, as in 71c (entry 3), provided similar in vitro efficacy against MSSA but improved in vivo efficacy (c.f.71a, entry 1). The opposite trends in in vitro and in vivo efficacies with or without the C-6 piperazine ring and with or without the C-2 amino group demonstrated by 71d (entry 4) and 71e (entry 5) led to the design of the 6-(3-aminopyrrolidino)purine derivative 71f (entry 6), which possessed excellent activities in vitro and in vivo. Attempts to increase the efficacy by modifying the linker between the glycolic acid and the purine ring afforded complex results. Although the piperazine linker analog 71i (entry 9) was the most potent analog in vivo, the in vitro efficacy was diminished compared to 71f. Of note, increasing the nitrogen substitution or steric hindrance on the pyrrolidine ring yielded a progressive decrease in in vivo efficacy {71i (NH2) > 71j [NH(CH3)] > 71k [NH(CH2CH3)] > 71l [N(CH3)2]}, though these compounds were comparable in vitro (entries 9–12). Additional efforts using the piperazine linker in 71i–l with modified N-substituents have also been reported; however, only minor improvements in in vitro potencies were observed.142–144
|
|
|||||
|---|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | MSSA, ED50 (mg kg−1) | ||
| MSSA | MRSA | VRE | |||
| 1 | 71a | 0.05 | 0.05 | 0.05 | >3.13 |
| 2 | 71b | 0.1 | 0.1 | 0.1 | >3.13 |
| 3 | 71c | 0.05 | 0.05 | 0.05 | 2.89 |
| 4 | 71d | 0.25 | 0.5 | 0.125 | 1.86 |
| 5 | 71e | 0.063 | 0.063 | 0.043 | 2.94 |
| 6 | 71f | 0.063 | 0.125 | 0.032 | 1.47 |
| 7 | 71g | 0.25 | 0.5 | 0.125 | 2.04 |
| 8 | 71h | 0.5 | 2 | 1 | 1.79 |
| 9 | 71i | 0.25 | 1 | 0.5 | 0.80 |
| 10 | 71j | 0.25 | 1 | 0.25 | 1.30 |
| 11 | 71k | 0.25 | 0.5 | 0.25 | 2.21 |
| 12 | 71l | 0.25 | 0.25 | 0.125 | 2.94 |

Nielsen and co-workers also disclosed a series of pleuromutilin analogs with purine rings on the side chain, introduced via [3 + 2] cycloaddition between an alkynyl nucleotide and a C22-azido pleuromutilin derivative (Table 8).145–147 The 6-aminopurine fragments were attached to the pleuromutilin core via an azide–alkyne cycloaddition with a C22-azide substituent.145–147 The in vitro potencies of these compounds against E. coli (AS 19) were modest.
|
|
||
|---|---|---|
| Entry | Compound | MIC (μg mL−1), E. coli (AS 19) |
| 1 | Valnemulin (3) | <0.125 |
| 2 | 66a | 0.5–1 |
| 3 | 72a | 1 |
| 4 | 72b | 8 |
| 5 | 72c | 8–16 |
| 6 | 72d | 16–32 |
| 7 | 72e | >32 |
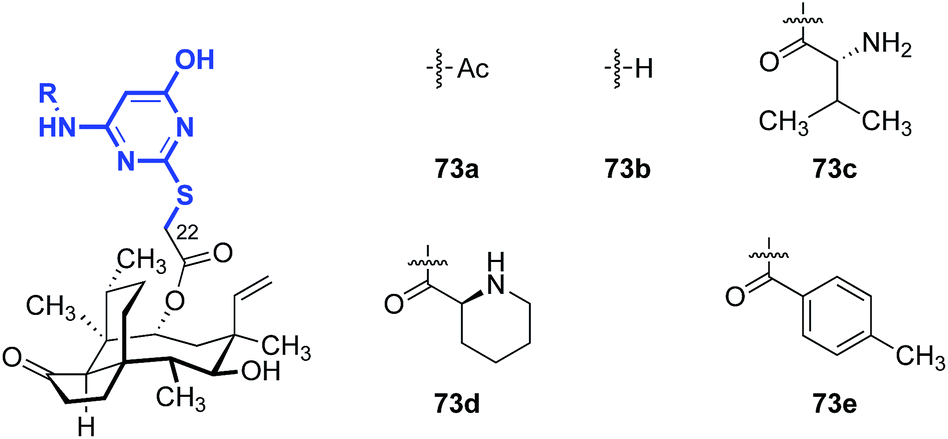
Shang and co-workers reported a survey of 2-mercaptopyrimidine derivatives against MRSA and MRSE (Table 9).148 With the exception of 73a (entry 1), analogs that contained primary amine substituents (73b–c, entries 2 and 3) were more active than analogs that lacked this substituent (73d–e, entries 4 and 5).
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| MRSA | MRSE | ||
| 1 | 73a | 0.125 | 0.125 |
| 2 | 73b | 0.25 | 0.125 |
| 3 | 73c | 1 | 0.125 |
| 4 | 73d | 4 | 1 |
| 5 | 73e | 4 | 1 |
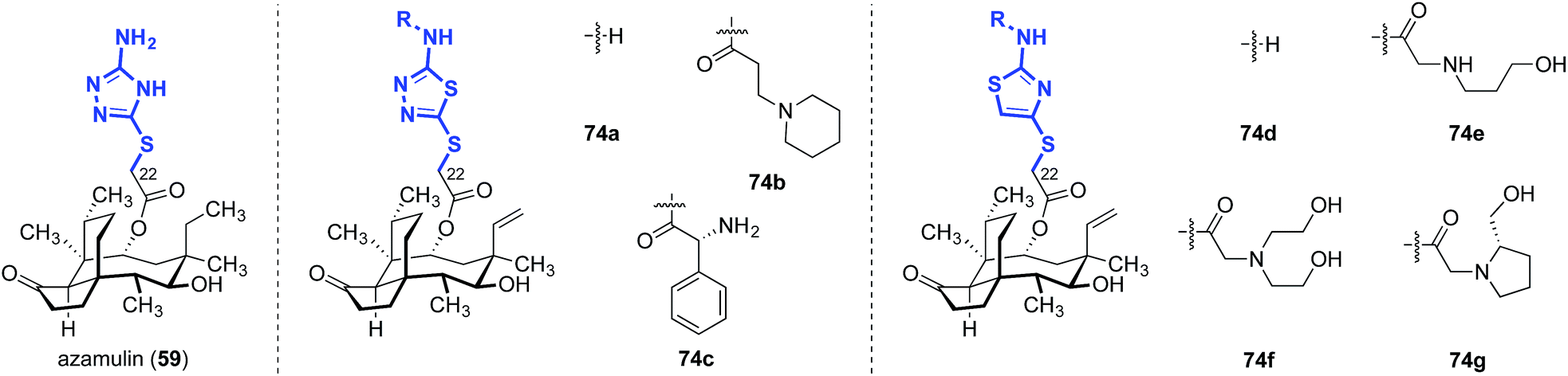
The influence of amino thiazole and amino thiadiazole rings at the C22 position have been evaluated against MRSA and MRSE in vitro by Ling, Liang and Liu (Table 10).74,149–152 These two heterocycles were selected due to their structural similarity to azamulin (59, entry 1), the first pleuromutilin analog introduced into human clinical trials. Although substitution of the 1,2,4-triazone with a thiadiazole did not lead to significant improvements in potencies (74a–c, entries 2–4),151,152 thiazole derivatives such as 74d–74g possessed two- to eight-fold greater potencies (entries 5–8). The progressive improvement of in vitro activities on going from 74d to 74f, then to the conformationally restricted analog 74g suggests a specific orientation of the terminal hydroxyl group is important for ribosome binding. This was corroborated by docking studies,149 which revealed two unexpected but important putative interactions with the ribosome.74,149,151 First, a π–π interaction between the heteroarene and A2045 residue was observed. Second, a hydrogen bonding interaction between the amide carbonyl group and C2565 residue was also discovered. Both were suggested to play crucial roles in the interaction between the glycolic ester side chain and the ribosome.74,149,151 Several other classes of pleuromutilin analogs have also been reported, such as analogs with bicycloamine (75)153 and carbohydrate residues (76, Fig. 11).154 However, due to the limited biological data disclosed, no trends can be confidently inferred.
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| MRSA | MRSE | ||
| 1 | Azamulin (59) | 0.5 | 0.5 |
| 2 | 74a | 0.5 | 1 |
| 3 | 74b | 0.5 | 1 |
| 4 | 74c | 0.5 | 2 |
| 5 | 74d | 0.5 | 0.125 |
| 6 | 74e | 0.5 | 0.5 |
| 7 | 74f | 0.125 | 0.125 |
| 8 | 74g | 0.063 | 0.063 |
 | ||
| Fig. 11 The bicycloamine and carbohydrate derivatives 75 and 76. | ||
5.2 Modification at C14
Replacement of the C14 ester with carbamate and imide substituents has also been studied (Table 11). One of the most active C14 analogs was the GSK compound SB-222734 (77f),155 which showed strong activity against Gram-negatives such as E. coli and Haemophilus influenzae.|
|
||||
|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | ||
| S. aureus | H. influenzae | E. coli | ||
| 1 | 77a | ≤0.06 | >64 | >64 |
| 2 | 77b | ≤0.06 | >64 | >64 |
| 3 | 77c | 0.5 | 8 | 4 |
| 4 | 77d | 4 | >64 | >64 |
| 5 | 77e | 0.5 | 4 | 2 |
| 6 | 77f | ≤0.06 | 1 | 1 |

Mono alkyl (77c, entry 3) and cyclic dialkyl carbamates (77d, entry 4) exhibited moderate activity against S. aureus with MICs in the 0.5–4 μg mL−1 range. The activities of 77c and 77d against Gram-negative strains were lower (MICs = 4–64 μg mL−1). Increasing the polarity of the structures by introducing a primary carbamate (77e, entry 5) resulted in a 2–4 fold improvement in activities against Gram-negative pathogens while maintaining good activities against Gram-positive pathogens (MIC = 0.5 μg mL−1). Further development of SB-222734 (77f) was impeded by the hydrophobicity of the structure.
Further optimization with the same carbamate or imide scaffold failed to afford compounds with higher potencies and/or higher hydrophilicities, based on the data disclosed.156–163 The poor aqueous solubility of 77f could be solved by preparing the phosphate prodrug (vide infra); however, the general PK profile of the API was not improved.164 Attempts to replace the carbamoyl group with isosteric β-diketone functionality were disclosed. However, no bioactivity data was reported.165
5.3 Core modifications
Due a lack of general methods to functionalize the tricyclic mutilin scaffold, the number of core-modified derivatives prepared and evaluated is relatively small when compared to derivatives at C14 or C22.The major metabolic pathway that deactivates (+)-pleuromutilin (1) involves cytochrome P450 oxidation at C2 and C8 (vide supra). The analog 78a which bears a C8 ketone substituent was prepared (Table 12, entry 1) and found to be up to five orders of magnitude less potent than the corresponding C8 methylene analog 78b (entry 2) against S. aureus, Moraxella catarrhalis (M. catarrhalis), and H. influenzae.166
|
|
||||
|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | ||
| S. aureus | M. catarrhalis | H. influenzae | ||
| 1 | 78a | 32 | 64 | 64 |
| 2 | 78b | 0.003 | 0.125 | 4 |

C2-oxidized derivatives have also been prepared and studied (Table 13). The C2-hydroxylated pleuromutilin analogs 79b and 79c (entries 2 and 3) were four- to eight-fold less active than the non-oxidized analog 79a (entry 1).167,168 However, the C2-fluorinated analogs 79d and 79e (entries 4 and 5) possessed up to a two-fold improvement in antimicrobial activities.168,169 Unfortunately, neither of these analogs appear to have advanced further.
|
|
||||
|---|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | ||
| MSSA | MRSA | MRSE | ||
| 1 | 79a | 0.125 | 0.125 | 0.125 |
| 2 | 79b | 1 | 1 | 1 |
| 3 | 79c | 0.5 | 0.5 | 0.5 |
| 4 | 79d | 0.0625 | 0.0625 | 0.125 |
| 5 | 79e | 0.0625 | 0.0625 | 0.0625 |

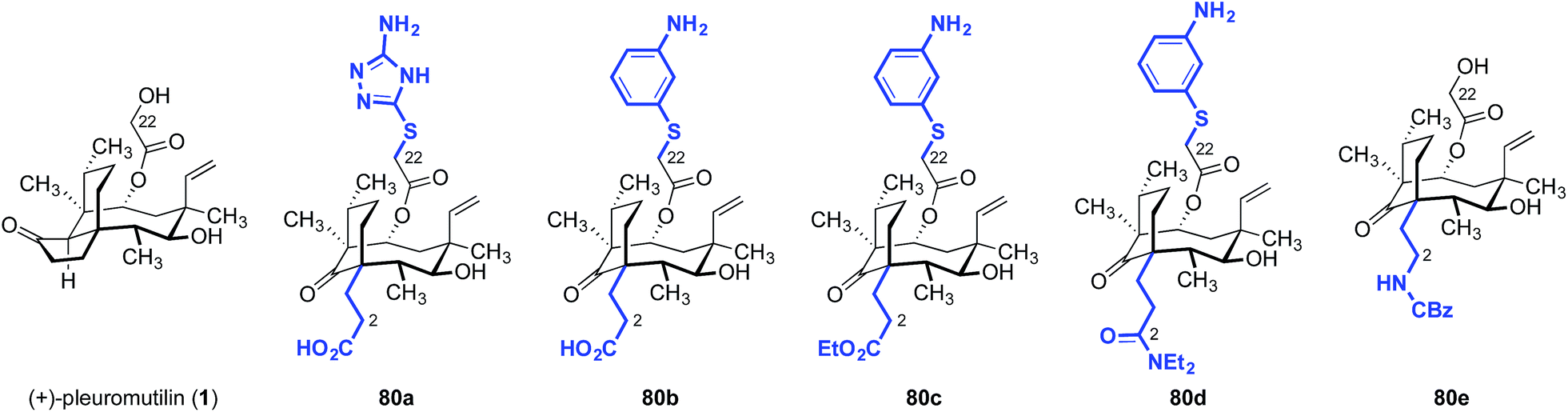
Although hydrophobic interactions between the tricyclic core and the ribosome may be important for binding, analogs derived from cleavage of the cyclopentanone ring were found to be potent as well (Table 14).170 Of the compounds evaluated, the carboxylic acid derivative 80b had the greatest potency against Streptococcus pneumoniae and S. aureus in vitro (MIC = 0.125, 0.25 μg mL−1, respectively).
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| S. pneumoniae | S. aureus | ||
| 1 | (+)-Pleuromutilin (1) | 0.5 | 0.5 |
| 2 | 80a | >64 | >128 |
| 3 | 80b | 0.125 | 0.25 |
| 4 | 80c | 1 | 1 |
| 5 | 80d | 4 | 32 |
| 6 | 80e | 0.25 | 16 |
5.4 Modifications at C12
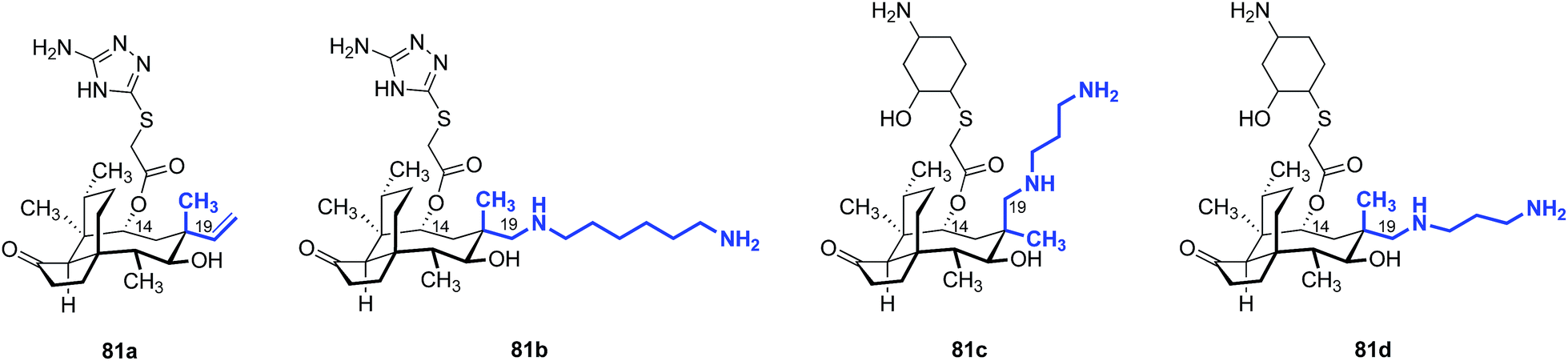
In 1986, Berner and colleagues reported a method to epimerize the C12 quaternary position of (+)-pleuromutilin (1) by an unusual retroallylation–allylation cascade.171 Due to the rigidity of the tricyclic core, the spatial orientation of the resulting pseudoequatorial alkene is distinct from the pseudoaxial alkene in natural material. Nabriva has explored substituent effects in these “12-epi-mutilins” (Tables 15 and 16).172,173 Oxidative cleavage followed by reductive amination or Wittig olefination (C19 functionalization), or hydroboration–oxidation (C20 functionalization) provided access to a wide array of short- and long-chain primary and secondary amine, alcohol, and guanidinium derivatives. Many of these analogs possessed greater in vitro potencies against Gram-positive bacteria and, in the case of 81b and 81d, useful levels of activity against E. coli (Table 15).172 The differential potencies of 81c and 81d underscore the difference in spatial orientiation of the pseudoaxial and pseudoequatorial C12 substituents.|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| S. aureus (ATCC49951) | E. coli (ATCC25922) | ||
| 1 | 81a | 0.5 | 128 |
| 2 | 81b | 0.5 | 4 |
| 3 | 81c | 16 | >32 |
| 4 | 81d | 0.25 | 4 |
| 5 | (+)-Pleuromutilin (1) | 1 | >32 |
| 6 | Tiamulin (2) | 0.5 | >32 |
| 7 | Valnemulin (3) | ≤0.03 | 32 |
| 8 | Retapamulin (4) | ≤0.03 | 32 |
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| S. aureus (ATCC49951) | E. coli (ATCC25922) | ||
| 1 | 82a | ≤0.03 | 0.5 |
| 2 | 82b | ≤0.03 | 0.25 |
| 3 | 82c | 0.06 | 0.5 |
| 4 | 82d | 0.125 | 4 |
| 5 | 82e | 0.25 | 4 |
| 6 | 82f | ≤0.03 | 1 |
| 7 | 82g | ≤0.03 | 0.5 |
| 8 | 82h | ≤0.03 | 0.25 |
| 9 | 82i | ≤0.03 | 0.5 |
| 10 | (+)-Pleuromutilin (1) | 1 | >32 |
| 11 | Tiamulin (2) | 0.5 | >32 |
| 12 | Valnemulin (3) | ≤0.03 | 32 |
| 13 | Retapamulin (4) | ≤0.03 | 32 |
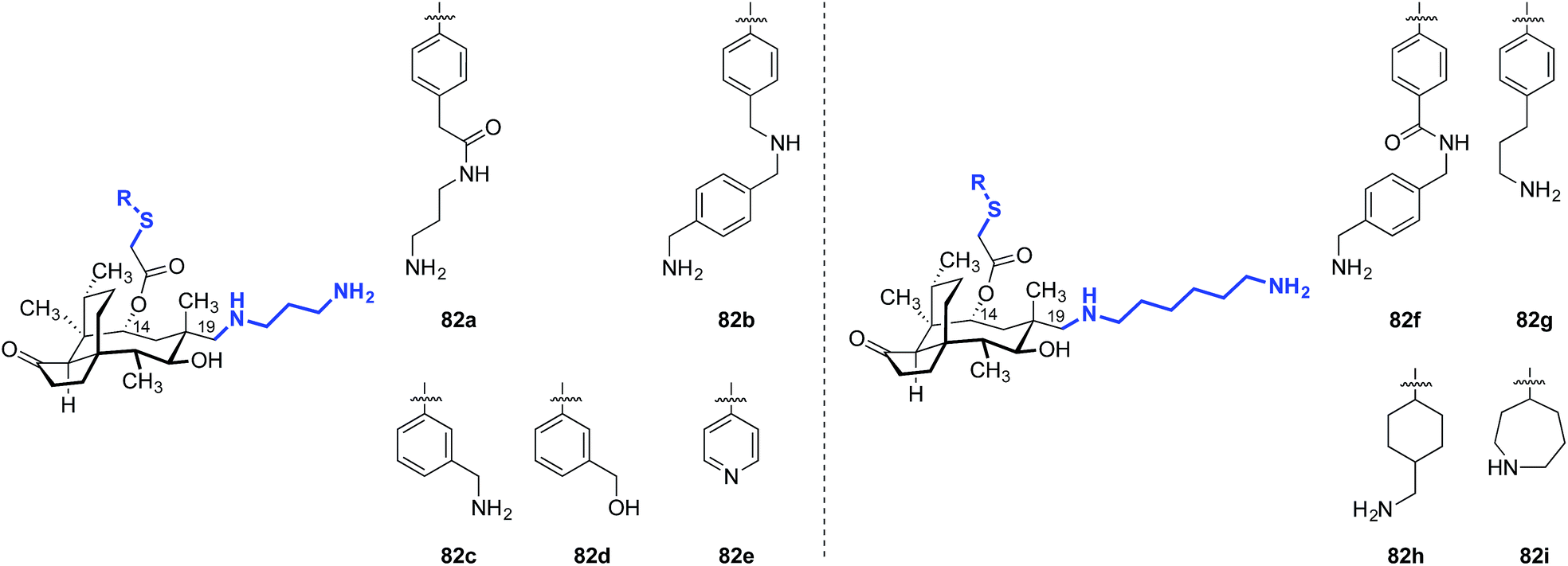
Nabriva has also reported the activities of amine-functionalized 12-epi-mutilins with 6- or 8-carbon linkers.9 Although the structures were not disclosed, these derivatives possessed activity against Gram-negative Klebsiella pneumoniae (MIC90 = 2–4 μg mL−1), Citrobacter freundii (MIC90 = 0.5–1 μg mL−1), and Enterobacter cloacae (MIC90 = 0.5–1 μg mL−1) while maintaining high potency for Gram-positive MRSA (MIC90 = 0.06–0.12 μg mL−1) and S. pneumoniae (MIC90 = 0.12–0.5 μg mL−1). C19 amine-functionalized 12-epi-mutilins with 3-carbon linkers were showed low activity towards K. pneumoniae (MIC90 = >32 μg mL−1), but achieved similar potency to the C6 and C8 linker compounds against the other bacterial species tested.
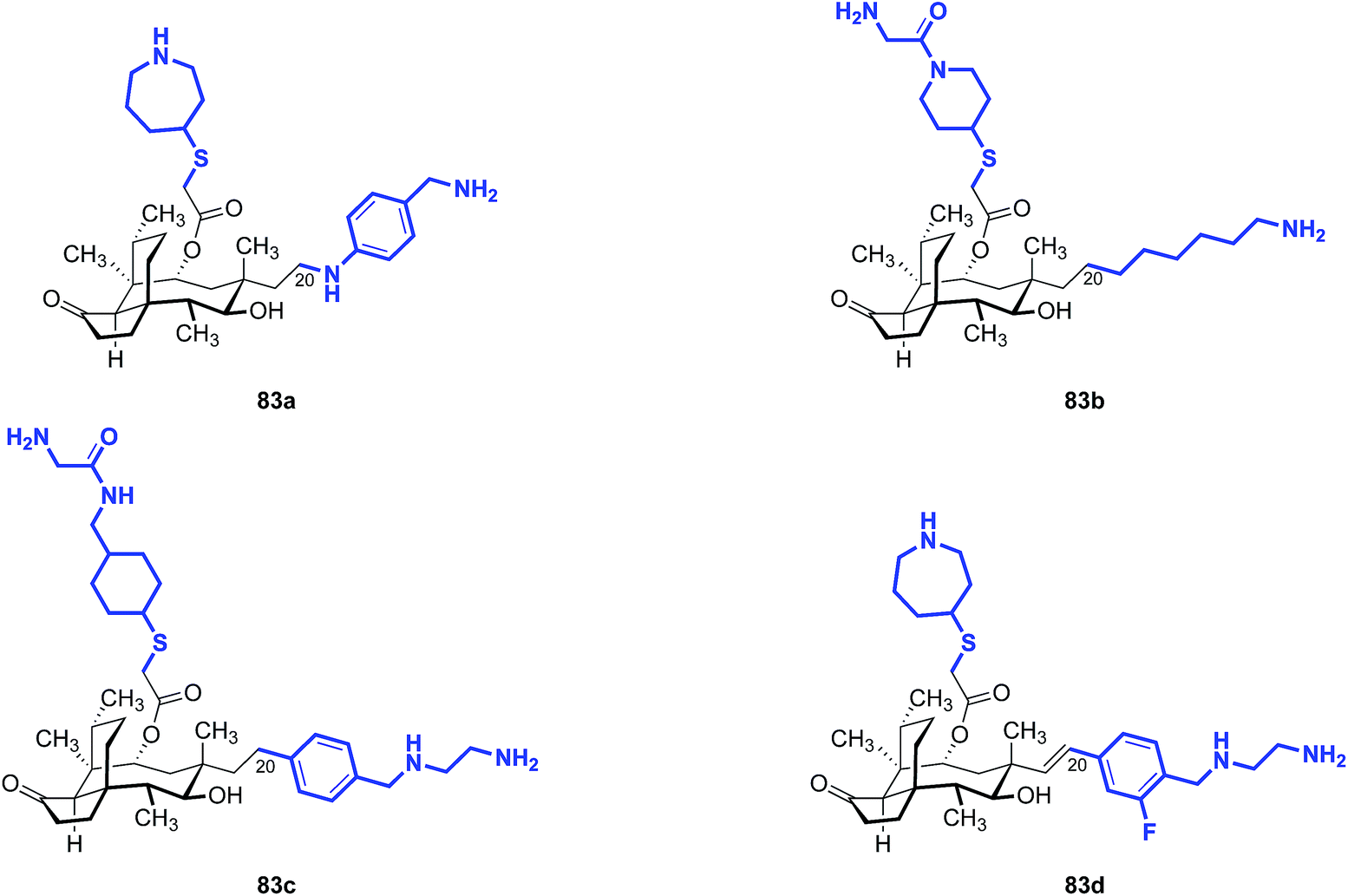
Note that all of the functionalized compounds in Tables 15 and 16 are modified at C19, the internal carbon atom of the vinyl group in the natural product. Compounds containing functionalization at C20, the terminal carbon atom of the vinyl group, have also been prepared and studied (Table 17). These derivatives consistently yielded MIC values in the 4–8 μg mL−1 range against E. coli in vitro, markedly higher than that of the C19-functionalized analogs. Nabriva did not report any compounds with non-nitrogen heteroatoms at this “C20” position, although the apparent need for this heteroatom suggests that ether and thioether analogs merit investigation.
|
|
|||
|---|---|---|---|
| Entry | Compound | MIC (μg mL−1) | |
| S. aureus (ATCC49951) | E. coli (ATCC25922) | ||
| 1 | 83a | 0.125 | 4 |
| 2 | 83b | ≤0.03 | 8 |
| 3 | 83c | 0.25 | 4 |
| 4 | 83d | 0.25 | 4 |
| 5 | (+)-Pleuromutilin (1) | 1 | >32 |
| 6 | Tiamulin (2) | 0.5 | >32 |
| 7 | Valnemulin (3) | ≤0.03 | 32 |
| 8 | Retapamulin (4) | ≤0.03 | 32 |
5.5 Prodrug modifications
The aqueous solubilities of pleuromutilins are poor. For example, the clogP of retapamulin (4) is ∼4.4. Several groups have attempted to increase the water solubility of pleuromutilin derivatives by conversion to a prodrug.137,164,174 This strategy has been successfully employed for a wide variety of insoluble pharmaceuticals.175–180 One strategy commonly employed involves phosphorylation of the active drug to effectively solubilize it in the body. This is followed by phosphate ester cleavage in vivo by alkaline phosphatases, liberating the active form of the drug.Fu and co-workers used this approach to evaluate the change in in vivo metabolic activity between C14 carbamate analogs with high in vitro activity and their phosphorylated prodrug forms (Fig. 12).164 The C11-phosphate derivatives 84a and 84b were prepared by phosphorylation of the corresponding alcohol. The aryl phosphate derivative 84c was prepared by site-selective phosphorylation of the corresponding phenol. Phosphorylation at C11 improved the in vitro stability, but this derivative still had relatively low aqueous solubility (57.6 mg mL−1). However, phosphorylation at the C14 phenol side chain increased the aqueous solubility to 96.8 mg mL−1.
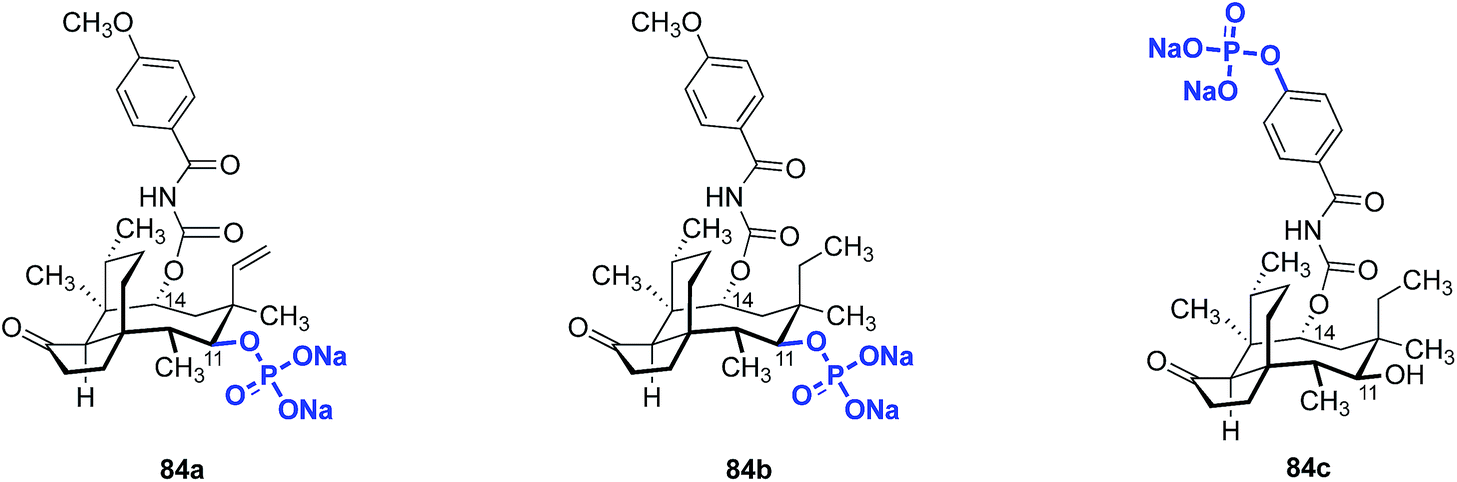 | ||
| Fig. 12 Structures of the pleuromutilin prodrugs 84a–84c. | ||
Surprisingly, pharmacokinetic studies of the prodrugs 84a and 84b indicated they were not dephosphorylated to the active products, an observation attributable to the steric hindrance surrounding C11. Plasma levels of the phosphorylated prodrug remained high (84a, Cmax = 30650 ng mL−1) while only very low levels of the corresponding dephosphorylated compound were detected (Cmax = <5 ng mL−1 for the dephosphorylated compound derived from 84a). However, the C14 side chain-phosphorylated product 84c was readily dephosphorylated to the active phenol (Cmax = 3.39 ± 0.4 μg mL−1), with a maximum plasma concentration of the dephosphorylated drug being reached within five minutes. The activity of 84cin vivo was comparable to vancomycin against MSSA and MRSA (Table 18). Thus, conversion of pleuromutilins to prodrug forms appears to be a promising avenue to increase the in vivo efficacy of otherwise potent but poorly water-soluble compounds.
| Entry | Compound | MIC (μg mL−1) | |
|---|---|---|---|
| MSSA | MRSA | ||
| 1 | 84c | 17.38 | 20.89 |
| 2 | Vancomycin | 13.49 | 15.85 |
5.6 Chimeric pleuromutilins
Covalent fusion of pleuromutilin with fragments of unrelated antibacterial agents could provide both a unique mechanism of action and a strategy to combat resistance to currently available drugs. Chimeric pleuromutilin–sulfonamide derivatives exemplify this approach (Fig. 13).181 MIC data for six chimeric analogs against a series of Gram-positive bacteria indicated that the chimeric molecules possessed greater potencies than the sulfonamide or valnemulin (3) alone. The chimeric molecules bearing the unsubstituted (85a), pyridine- (85b), or thiazole- (85c) substituted benzenesulfonamides displayed the highest activity against S. aureus, MRSA, Streptococcus equi, and M. gallisepticum, surpassing the potency of the parent benzoxypleuromutilin derivative (85d) and sulfanilamide. The chimeric molecules were not, however, active against Gram-negative bacteria (MICs = 12 to >128 μg mL−1 against E. coli in vitro). | ||
| Fig. 13 Structures of the chimeric pleuromutilin–sulfonamide derivatives 85a–c and the parent C14-substituted pleuromutilin 85d.181 | ||
5.7 De novo synthesis
De novo synthesis of pleuromutilins constitutes a promising strategy to access derivatives not attainable by semisynthesis.182 Sorensen and co-workers have successfully employed ring-closing metathesis to access a modified pleuromutilin core24 (Fig. 14) and Nozaki–Hiyama–Kishi (NHK) cyclization to construct novel C5, C6, C10-des-methyl pleuromutilin derivatives (Table 19).27 The NHK-derived cores contained either an exocyclic methylene or endocyclic alkene at C12 (as opposed to the C12 vinyl functionality in the native core).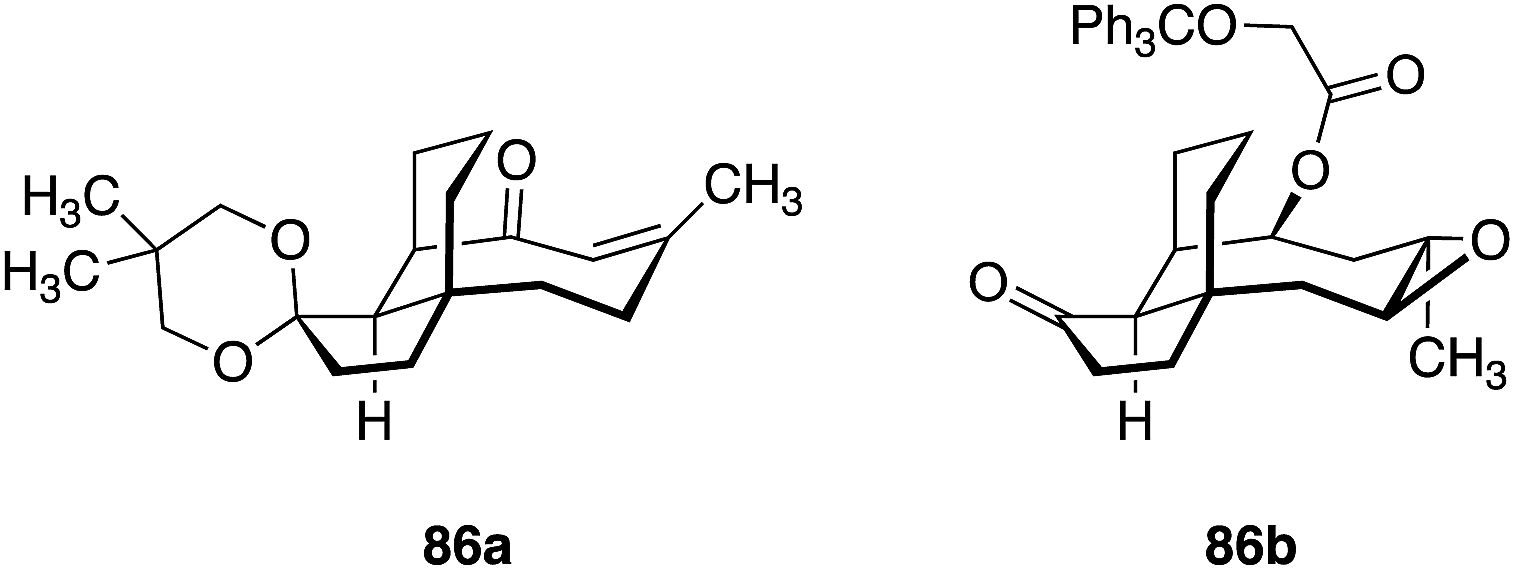 | ||
| Fig. 14 Structures of modified pleuromutilin cores accessed by diastereoselective 1,4 conjugate addition, followed by ring-closing metathesis.24 | ||
|
|
||
|---|---|---|
| Entry | Compound | M. tuberculosis mc27000 MIC (μg mL−1) |
| 1 | 87a | 50–100 |
| 2 | 87b | 12–25 |
| 3 | 87c | >200 |
| 4 | 87d | 50 |
| 5 | Pleuromutilin (1) | 25–50 |
| 6 | Tiamulin (2) | 12–25 |
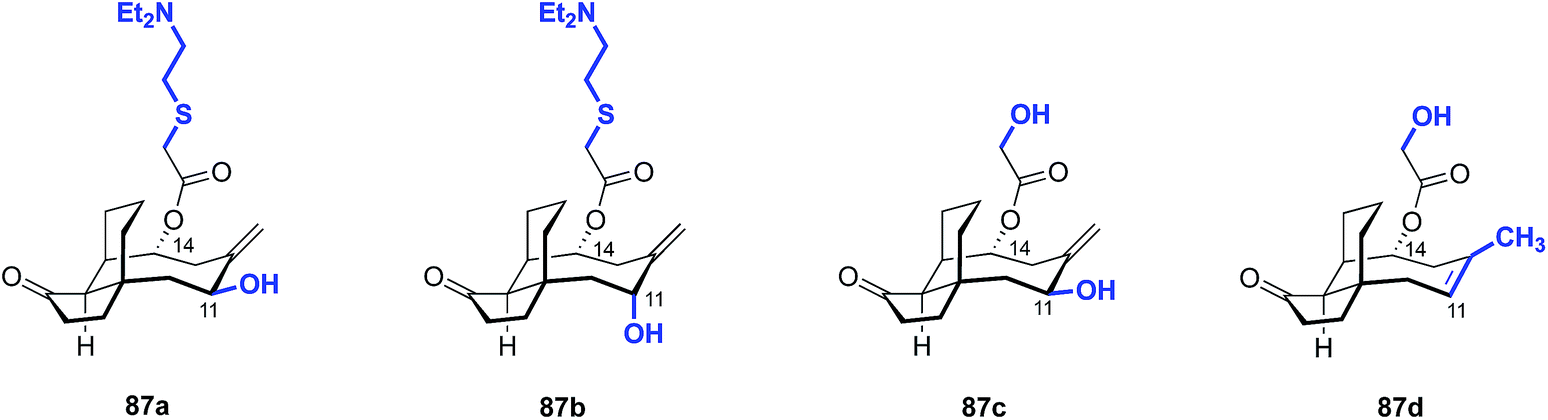
Although these core modifications are relatively minor, a slight variance in MIC values was observed between 87b and 87d (Table 19). Functionalization of C14 to the glycolic acid or amine substituted thioether [as seen in tiamulin (2)] and subsequent antimicrobial testing against M. tuberculosis mc27000 revealed that the tiamulin-type thioethers were most potent, regardless of core type. C11 epimers 87a and 87b displayed a 2–3 fold difference in potency (87a MIC = 12–25 μg mL−1, 87b MIC = 50–100 μg mL−1). Unsurprisingly, the glycolic acid derivative (87c) was the least potent (MIC > 200 μg mL−1), as thioether C14 functionality and terminal basicity has been shown to be one of the key modulators of potency (vide infra). Further modification of the core by de novo synthesis using NHK or other methods could have significant effects on potency, especially when combined with thoughtful C14 functionalization.
5.8 C–H oxidation
Although C–H functionalization of complex molecules and drug candidates have found use in medicinal chemistry,183–185 applications of these methodologies to pleuromutilins have been limited. The first C–H functionalization of pleuromutilins was reported by Hanson and co-workers in 2002 using whole cell cultures of Streptomyces griseus strains (Fig. 15).186 The C2, C7, and C8 oxidized mutilin derivatives 88a, 88b, and 88c were obtained in 1.0%, 22%, and 9.8% yields, respectively. White and co-workers investigated oxidation of modified pleuromutilins using electrophilic iron-based oxidants.187 The C7 oxidation products 89a and 89b were formed in 42% and 20% yields, respectively. Amination products have been prepared by Uccello188 (90), White189 (91), and Du Bois190 (92) using insertion of an electrophilic nitrenoid to cleave the C–H bond. Hartwig and co-workers reported that iridium-catalyzed hydrogen–deuterium exchange of tiamulin (2) selectively provided the trans-deuteration product 2-d.191 Among these examples, 91 was the only instance involving oxidation of a primary C–H bond. | ||
| Fig. 15 Oxidation products formed from various pleuromutilin derivatives. | ||
Our laboratory recently reported a strategy to oxidize the methyl substituents of (+)-pleuromutilin (1) by hydroxyl-directed iridium-catalyzed C–H silylation,192 followed by Tamao–Fleming oxidation (Fig. 16).193 18-Hydroxy-19,20-dihydropleuromutilin (93), 12-epi-17-hydroxy-19,20-dihydropleuromutilin (94), 17,18-dihydroxy-19,20-dihydropleuromutilin (95), the C16-oxidized mutilin derivative 96, the C16 oxidation 4-epi-mutilin derivative 97, and the C15, C16 dioxidized mutilin derivative 98 were prepared by this approach. Additionally, synthetic routes to the C17-amino derivatives 99a–d and 6-normethyl-19,20-dihydropleuromutilin (100) were developed (Fig. 17).
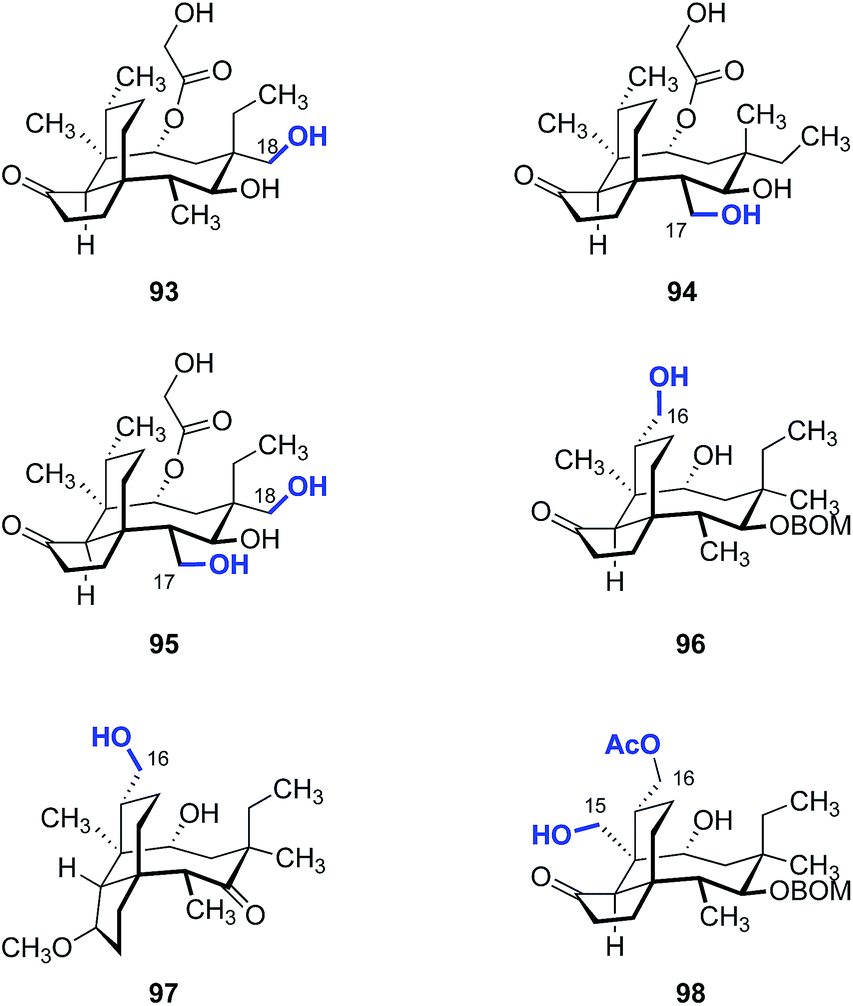 | ||
| Fig. 16 Products derived from hydroxyl-directed iridium-catalyzed C–H silylation, followed by Tamao–Fleming oxidation. | ||
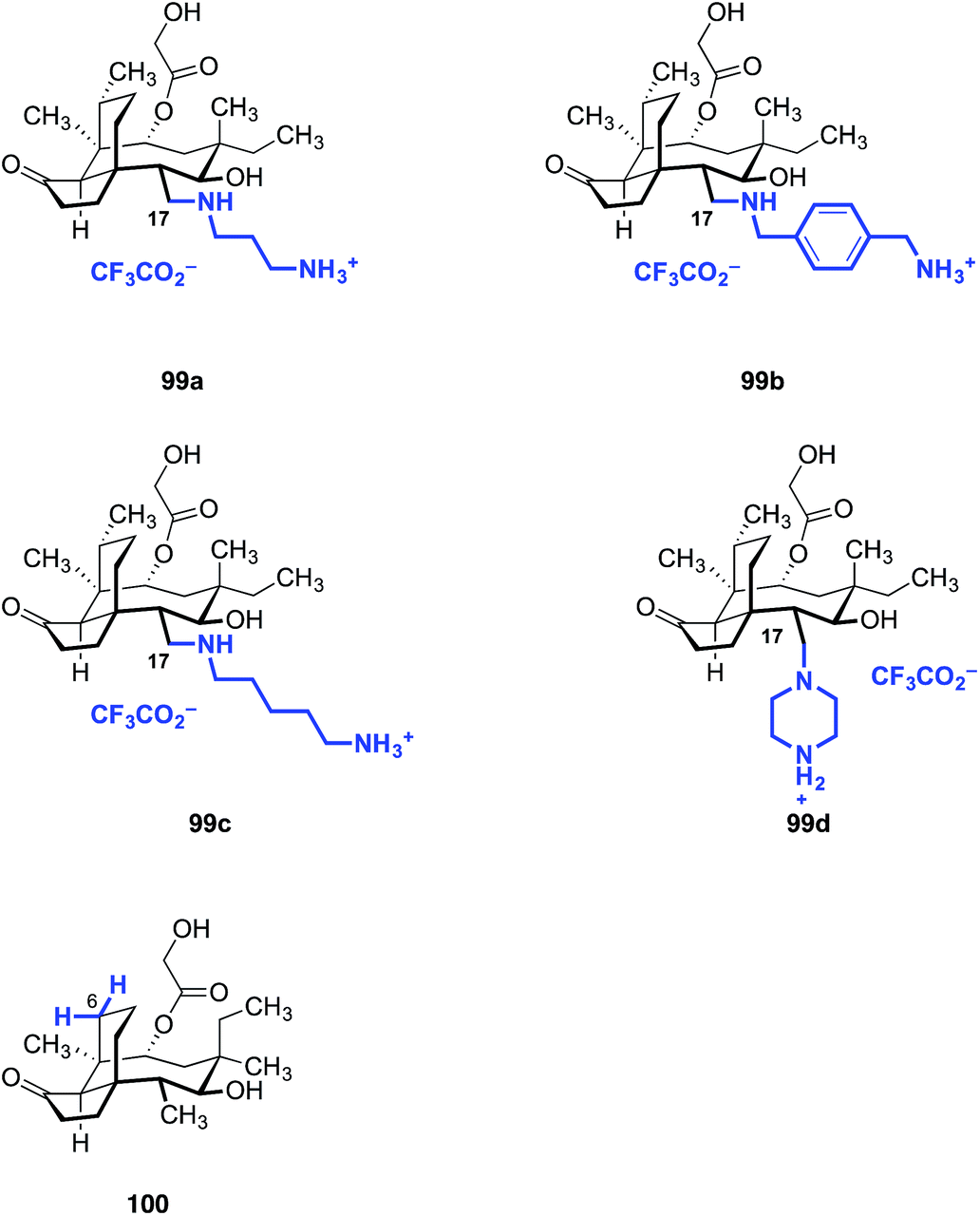 | ||
| Fig. 17 Amine derivatives 99a–d prepared from the C17 oxidation product 94 and the C6-normethyl derivative 100 prepared form the C16 oxidation product 96. | ||
In conclusion, some valuable trends can be gleaned from the available SAR data. First, the vast majority of the analogs synthesized so far vary only at the C22 position, likely due to the ease of synthesis. All of the analogs in the markets or clinical trials share the similarity of an intact pleuromutilin core structure, a C22 thioether, and a basic amine on the sulfide extension. Second, minor changes on the C22 side chain not only alter the antimicrobial activities of the analogs, they have a significant impact on the in vivo activities, pharmacokinetic, and pharmacodynamic profiles of the resulting derivatives. Third, replacement of the glycolic ester with carbamates or imides led to analogs with improved antimicrobial activities, which is consistent with the known interactions of the C14 carbonyl and the ribosome. However, to our knowledge, none of these analogs have advanced to clinical trials. Fourth, although saturation of the C19–20 alkene had a negligible effect on the antimicrobial activities of the analogs, epimerization of the C12 position followed by functionalization provides ESPs with activity against drug-resistant Gram-negative pathogens. Finally, modifications on the core structure have thus far been met with limited success.
6. Conclusion and outlook
In this review we have attempted to summarize what is known about the pleuromutilins. Extensive efforts to modify the C22 position of the metabolite have led to the discovery of several clinical and advanced preclinical agents. Through studies of 12-epi-pleuromutilin derivatives, researchers at Nabriva have taught us that the spectrum of activity of pleuromutilins can be manipulated to encompass drug-resistant Gram-negative strains. Given the small number of core-modified pleuromutilins that have been prepared (as compared to C22 derivatives), the Nabriva discoveries provide (in our minds) compelling evidence suggesting additional useful antibiotics await creation. We anticipate that a combination of total synthetic and semisynthetic approaches to pleuromutilins incorporating structure-based drug design will emerge as the most efficient path forward.It is perhaps instructive to look backward to understand the path forward in antibiotic development. The year 2018 marks the 90th anniversary of the discovery of penicillin by the English scientist Sir Alexander Fleming. By the mid 1940s, the problem of large-scale production of the antibiotic (by fermentation) was solved, leading to a sea change in the way common infections were treated. In 1957, John Sheehan at MIT completed the first total chemical synthesis of penicillin, which had eluded dozens of research groups and was widely-regarded at that time as an impossible feat. Sheehan's efforts led to the development of ampicillin, which is still in use today, and provided access to aminopenicillanic acid, which served as a precursor to thousands of novel penicillins.
In retrospect, the development of penicillin might be framed as being driven primarily by two factors. First, the existential threat of World War II led the United States and the United Kingdom to marshal an incredible number of scientists from academia and industry (notably, Merck, Pfizer, and Squibb), and to put in place the vast resources required to solve the problem of penicillin production. Second, the doggedness and fortitude of researchers such as Sheehan (and many others) ultimately unlocked synthetic access to penicillin and paved the way for the production of new derivatives with improved activities.
In 2018 humanity faces a new existential menace, in the form of drug-resistant bacteria. Unlike World War II, this threat is state agnostic. It portends entry to a post-antibiotic era, where deaths from everyday infections are (once again) common. Comparable amounts of resources are needed to address this threat, and numerous researchers skilled in the art will need to be recruited. The former is a complex problem that is still largely unsolved, but progress has been made on the latter front.182 Total syntheses of the vancomycin,194 tetracycline,195 macrolide,196 and streptogramin197 classes of antibiotics have now been recorded, establishing platforms for the exploration and industrial production of new derivatives with deep-seated structural modifications. Additionally, the recent breath-taking advances in C–H oxidation chemistry, particularly Csp3–H activation185,193 (as exemplified in the pleuromutilin studies outlined in Section 5.8) and the site-selective modification of natural products198 suggest new opportunities for semisynthetic optimization that were not conceivable a decade or even five years ago. The addition of many more scientists to the ranks of the global antibiotic effort is desperately needed, and addressing the meagre financial incentives associated with antibiotic development will be required to re-engage industrial partners.
7. Conflicts of interest
Provisional patent applications covering work in this area from the Herzon laboratory have been filed.8. Acknowledgements
Financial support from Yale University is gratefully acknowledged.9. Notes and references
- F. Kavanagh, A. Hervey and W. J. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 570–574 CrossRef CAS.
- F. Kavanagh, A. Hervey and W. J. Robbins, Proc. Natl. Acad. Sci. U. S. A., 1952, 38, 555–560 CrossRef CAS.
- Lefamulin Evaluation Against Pneumonia (LEAP 2) Phase 3 Topline Results, https://investors.nabriva.com/static-files/5c34b447-99cc-4739-b9d6-d4ea4c7d13b9, accessed 2018/06/20.
- S. Paukner, A. Gruss, T. R. Fritsche, Z. Ivezic-Schoenfeld and R. N. Jones, presented in part at the 53rd Interscience Conference on Antimicrobial Agents and Chemotherapy, Denver, CO, 2013 Search PubMed.
- S. Paukner, D. B. Strickmann and Z. Ivezic-Schoenfeld, presented in part at the 24th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Barcelona, Spain, 2014 Search PubMed.
- W. W. Wicha and Z. Ivezic-Schoenfeld, presented in part at the 24th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Barcelona, Spain, 2014 Search PubMed.
- K. Thirring, W. Heilmayer, R. Riedl, H. Kollmann, Z. Ivezic-Schoenfeld, W. Wicha, S. Paukner and D. Strickmann, World Pat., WO2015110481A1, 2015.
- S. Paukner, H. Kollmann, R. Riedl and Z. Ivezic-Schoenfeld, presented in part at the 25th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Copenhagen, Denmark, 2015 Search PubMed.
- S. Paukner, W. W. Wicha, W. Heilmayer, K. Thirring and R. Riedl, presented in part at the Interscience Conference of Antimicrobial Agents and Chemotherapy/International Congress of Chemotherapy and Infection (ICAAC/ICC), San Diego, CA, 2015 Search PubMed.
- S. Paukner, W. W. Wicha, K. Thirring, H. Kollmann and Z. Ivezic-Schoenfeld, presented in part at the 25th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Copenhagen, Denmark, 2015 Search PubMed.
- W. W. Wicha, S. Paukner, D. B. Strickmann, K. Thirring, H. Kollmann, W. Heilmayer and Z. Ivezic-Schoenfeld, presented in part at the25th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Copenhagen, Denmark, 2015 Search PubMed.
- E. G. Gibbons, J. Am. Chem. Soc., 1982, 104, 1767–1769 CrossRef CAS.
- R. K. Boeckman, D. M. Springer and T. R. Alessi, J. Am. Chem. Soc., 1989, 111, 8284–8286 CrossRef CAS.
- N. J. Fazakerley, M. D. Helm and D. J. Procter, Chem.–Eur. J., 2013, 19, 6718–6723 CrossRef CAS.
- S. K. Murphy, M. Zeng and S. B. Herzon, Science, 2017, 356, 956–959 CrossRef CAS.
- M. Zeng, S. K. Murphy and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 16377–16388 CrossRef CAS.
- E. P. Farney, S. S. Feng, F. Schäfers and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 1267–1270 CrossRef CAS.
- M. Kahn, Tetrahedron Lett., 1980, 21, 4547–4548 CrossRef CAS.
- L. A. Paquette, P. C. B. Page, P. D. Pansegrau and P. E. Wiedeman, J. Org. Chem., 1988, 53, 1450–1460 CrossRef CAS.
- L. A. Paquette, P. D. Pansegrau, P. E. Wiedeman and J. P. Springer, J. Org. Chem., 1988, 53, 1461–1466 CrossRef CAS.
- L. A. Paquette, P. E. Wiedeman and P. C. B. Page, J. Org. Chem., 1988, 53, 1441–1450 CrossRef CAS.
- E. Bacque, F. Pautrat and S. Z. Zard, Org. Lett., 2003, 5, 325–328 CrossRef CAS.
- T. J. K. Findley, D. Sucunza, L. C. Miller, M. D. Helm, M. Helliwell, D. T. Davies and D. J. Procter, Org. Biomol. Chem., 2011, 9, 2433–2451 RSC.
- J. Liu, S. D. Lotesta and E. J. Sorensen, Chem. Commun., 2011, 47, 1500–1502 RSC.
- S. K. Murphy, M. Zeng and S. B. Herzon, Org. Lett., 2016, 18, 4880–4883 CrossRef CAS.
- S. K. Murphy, M. Zeng and S. B. Herzon, Org. Lett., 2017, 19, 4980–4983 CrossRef CAS.
- S. D. Lotesta, J. Liu, E. V. Yates, I. Krieger, J. C. Sacchettini, J. S. Freundlich and E. J. Sorensen, Chem. Sci., 2011, 2, 1258–1261 RSC.
- O. A. Phillips and L. H. Sharaf, Expert Opin. Ther. Pat., 2007, 17, 429–435 CrossRef CAS.
- C. Hu and Y. Zou, Mini-Rev. Med. Chem., 2009, 9, 1397–1406 CrossRef CAS.
- R. Jarvest, Functional Molecules from Natural Sources, RSC, 2010 Search PubMed.
- R. Novak, Ann. N. Y. Acad. Sci., 2011, 1241, 71–81 CrossRef CAS.
- S. S. Stokes, M. Morningstar, H. Kocis and J. C. Verheijen, Pharm. Pat. Anal., 2012, 1, 601–620 CrossRef CAS.
- Y. Z. Tang, Y. H. Liu and J. X. Chen, Mini-Rev. Med. Chem., 2012, 12, 53–61 CrossRef CAS.
- R. Shang, J. Wang, W. Guo and J. Liang, Curr. Trends Med. Chem., 2013, 13, 3013–3025 CrossRef CAS.
- N. J. Fazakerley and D. J. Procter, Tetrahedron, 2014, 70, 6911–6930 CrossRef CAS.
- S. Paukner and R. Riedl, Cold Spring Harbor Perspect. Med., 2017, 7, a027110 CrossRef.
- M. Anchel, J. Biol. Chem., 1952, 199, 133–139 CAS.
- D. Arigoni, Gazz. Chim. Ital., 1962, 92, 884–901 CAS.
- A. J. Birch, C. W. Holzapfel and R. W. Rickards, Tetrahedron, 1966, 22, 359–387 CrossRef.
- G. Bonavia, Pleuromutilin: Stereochemie und detaillierte Biosynthese, PhD Thesis, ETH Zürich, 1968.
- A. M. Bailey, F. Alberti, S. Kilaru, C. M. Collins, K. de Mattos-Shipley, A. J. Hartley, P. Hayes, A. Griffin, C. M. Lazarus, R. J. Cox, C. L. Willis, K. O'Dwyer, D. W. Spence and G. D. Foster, Sci. Rep., 2016, 6, 25202 CrossRef CAS.
- F. Alberti, K. Khairudin, E. R. Venegas, J. A. Davies, P. M. Hayes, C. L. Willis, A. M. Bailey and G. D. Foster, Nat. Commun., 2017, 8, 1831 CrossRef.
- W. Nagata and M. Yoshioka, Org. React., 2005, 255–476 Search PubMed.
- D. L. Comins and A. Dehghani, Tetrahedron Lett., 1992, 33, 6299–6302 CrossRef CAS.
- R. M. Moslin, K. Miller-Moslin and T. F. Jamison, Chem. Commun., 2007, 4441–4449 RSC.
- A.-R. Shareef, D. H. Sherman and J. Montgomery, Chem. Sci., 2012, 3, 892–895 RSC.
- H. Wang, S. Negretti, A. R. Knauff and J. Montgomery, Org. Lett., 2015, 17, 1493–1496 CrossRef CAS.
- E. P. Jackson, H. A. Malik, G. J. Sormunen, R. D. Baxter, P. Liu, H. Wang, A.-R. Shareef and J. Montgomery, Acc. Chem. Res., 2015, 48, 1736–1745 CrossRef CAS.
- J. D. White, U. M. Grether and C.-S. Lee, Org. Synth., 2005, 82, 108–114 CrossRef CAS.
- S. Matsuzawa, Y. Horiguchi, E. Nakamura and I. Kuwajima, Tetrahedron, 1989, 45, 349–362 CrossRef CAS.
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011–1013 CrossRef CAS.
- R. C. Larock, T. R. Hightower, G. A. Kraus, P. Hahn and D. Zheng, Tetrahedron Lett., 1995, 36, 2423–2426 CrossRef CAS.
- A. Krasovskiy, F. Kopp and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 497–500 CrossRef CAS.
- W. G. Dauben and D. M. Michno, J. Org. Chem., 1977, 42, 682–685 CrossRef CAS.
- N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562 CrossRef CAS.
- S. Lou, P. N. Moquist and S. E. Schaus, J. Am. Chem. Soc., 2006, 128, 12660–12661 CrossRef CAS.
- R. Alam, T. Vollgraff, L. Eriksson and K. J. Szabó, J. Am. Chem. Soc., 2015, 137, 11262–11265 CrossRef CAS.
- J. E. Steves and S. S. Stahl, J. Org. Chem., 2015, 80, 11184–11188 CrossRef CAS.
- K. Iwasaki, K. K. Wan, A. Oppedisano, S. W. Crossley and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 1300–1303 CrossRef CAS.
- Schematic of protein synthesis by the ribosome from mRNA, http://www.drdiagram.com/wp-content/uploads/2017/01/dna-translation-diagram-design-dna-translation-diagram-dna-translation-diagram-labeled-dna-translation-diagram-dna-transcription-and-translation.jpg, accessed 2018/04/17.
- I. Agmon, A. Bashan, R. Zarivach and A. Yonath, Biol. Chem., 2005, 386, 833–844 CAS.
- M. Pringle, J. Poehlsgaard, B. Vester and K. S. Long, Mol. Microbiol., 2004, 54, 1295–1306 CrossRef CAS.
- D. N. Wilson, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 393–433 CrossRef CAS.
- L. A. Hodgin and G. Högenauer, Eur. J. Biochem., 1974, 47, 527–533 CrossRef CAS.
- G. Högenauer and C. Ruf, Antimicrob. Agents Chemother., 1981, 19, 260–265 CrossRef.
- S. M. Poulsen, M. Karlsson, L. B. Johansson and B. Vester, Mol. Microbiol., 2001, 41, 1091–1099 CrossRef CAS.
- F. Schlünzen, E. Pyetan, P. Fucini, A. Yonath and J. M. Harms, Mol. Microbiol., 2004, 54, 1287–1294 CrossRef.
- C. Davidovich, A. Bashan, T. Auerbach-Nevo, R. D. Yaggie, R. R. Gontarek and A. Yonath, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4291–4296 CrossRef CAS.
- J. A. Dunkle, L. Xiong, A. S. Mankin and J. H. D. Cate, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17152–17157 CrossRef CAS.
- J. M. Harms, F. Schlünzen, P. Fucini, H. Bartels and A. Yonath, BMC Biol., 2004, 2 DOI:10.1186/1741-7007-2-4.
- Z. Eyal, D. Matzov, M. Krupkin, S. Paukner, R. Riedl, H. Rozenberg, E. Zimmerman, A. Bashan and A. Yonath, Sci. Rep., 2016, 39004 CrossRef CAS.
- K. Kosowska-Shick, C. Clark, K. Credito, P. McGhee, B. Dewasse, T. Bogdanovich and P. C. Appelbaum, Antimicrob. Agents Chemother., 2006, 50, 765–769 CrossRef CAS.
- Z. Eyal, D. Matzov, M. Krupkin, I. Wekselman, S. Paukner, E. Zimmerman, H. Rozenberg, A. Bashan and A. Yonath, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5805 CrossRef CAS.
- X. Wang, Y. Ling, H. Wang, J. Yu, J. Tang, H. Zheng, X. Zhao, D. Wang, G. Chen, W. Qiu and J. Tao, Bioorg. Med. Chem. Lett., 2012, 22, 6166–6172 CrossRef CAS.
- Y. Yi, X. Xu, Y. Liu, S. Xu, X. Huang, J. Liang and R. Shang, Eur. J. Med. Chem., 2017, 126, 687–695 CrossRef CAS.
- K. S. Long, L. H. Hansen, L. Jakobsen and B. Vester, Antimicrob. Agents Chemother., 2006, 50, 1458–1462 CrossRef CAS.
- D. Söll and U. RajBhandary, tRNA: Structure, Biosynthesis, and Function, ASM Press, 1995 Search PubMed.
- J. Drews, A. Georgopoulos, G. Laber, E. Schütze and J. Unger, Antimicrob. Agents Chemother., 1975, 7, 507–516 CrossRef CAS.
- H. Egger and H. Reinshagen, US Pat., US4208326 A, 1980.
- Tiamulin - Article 34 Referral - Annex I, II, III, http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Tiamutin_34/WC500094801.pdf, accessed 2018/01/26.
- L. D. Olson, J. Am. Vet. Med. Assoc., 1986, 188, 1165–1170 CAS.
- R. W. Pickles, Vet. Rec., 1982, 110, 403–405 CAS.
- D. G. Burch, Vet. Rec., 1982, 110, 244–246 CAS.
- K. Koller and F. Schwarz, US Pat., US20010021693 A1, 2001.
- K. Koller and F. Schwarz, World Pat., WO2001041758 A2, 2001.
- D. G. S. Burch, P. H. Ripley and E. Zeisl, World Pat., WO1998001127 A1, 1998.
- P. C. Hannan, H. M. Windsor and P. H. Ripley, Res. Vet. Sci., 1997, 63, 157–160 CrossRef CAS.
- F. T. W. Jordan, C. A. Forrester, P. H. Ripley and D. G. S. Burch, Avian Dis., 1998, 42, 738–745 CrossRef CAS.
- L. Stipkovits, P. H. Ripley, M. Tenk, R. Glávits, T. Molnár and L. Fodor, Res. Vet. Sci., 2005, 78, 207–215 CrossRef CAS.
- C. Heilmann, L. Jensen, J. S. Jensen, K. Lundstrom, D. Windsor, H. Windsor and D. Webster, J. Infect., 2001, 43, 234–238 CrossRef CAS.
- H. C. Neu, Science, 1992, 257, 1064–1073 CrossRef CAS.
- C. L. Ventola, Pharmacol. Ther., 2015, 40, 277–283 Search PubMed.
- E. Martens and A. L. Demain, J. Antibiot., 2017, 70, 520 CrossRef CAS.
- Altabax (Retapamulin) Ointment - FDA Drug Approval Package, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2007/022055s000TOC.cfm, accessed 2018/01/28.
- Altargo: EPAR - Product Information, http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000757/WC500024409.pdf, accessed 2018/01/27.
- R. N. Jones, T. R. Fritsche, H. S. Sader and J. E. Ross, Antimicrob. Agents Chemother., 2006, 50, 2583–2586 CrossRef CAS.
- F. J. Candel, G. Morales and J. J. Picazo, Rev. Esp. Quimioter., 2011, 24, 127–130 CAS.
- D. A. Williamson, G. P. Carter and B. P. Howden, Clin. Microbiol. Rev., 2017, 30, 827–860 CrossRef.
- Australian Public Assessment Report for Retapamulin, https://www.tga.gov.au/sites/default/files/auspar-retapamulin-131111.pdf, accessed 2018/01/28.
- P. Brown and M. J. Dawson, in Progress in Medicinal Chemistry, eds. G. Lawton and D. R. Witty, Elsevier, 2015, vol. 54, pp. 135–184 Search PubMed.
- A. Bryskier, in Antimicrobial Agents, American Society of Microbiology, 2005, DOI:10.1128/9781555815929.ch50.
- D. M. Stresser, M. I. Broudy, T. Ho, C. E. Cargill, A. P. Blanchard, R. Sharma, A. A. Dandeneau, J. J. Goodwin, S. D. Turner, J. C. Erve, C. J. Patten, S. S. Dehal and C. L. Crespi, Drug Metab. Dispos., 2004, 32, 105–112 CrossRef CAS.
- S. E. Clarke, Xenobiotica, 1998, 28, 1167–1202 CrossRef CAS.
- H. K. Lim, N. Duczak Jr, L. Brougham, M. Elliot, K. Patel and K. Chan, Drug Metab. Dispos., 2005, 33, 1211–1219 CrossRef CAS.
- Current Pipeline of Nabriva, https://www.nabriva.com/pipeline-research, accessed 2018/01/28.
- H. S. Sader, R. E. Mendes, P. R. Rhomberg, S. Paukner, R. K. Flamm and D. J. Farrell, Antimicrobial Activity of Lefamulin, an Investigational Pleuromutilin Antibiotic against Staphylococcus aureus Strains with Decreased Susceptibility to Vancomycin, Poster C-559, ICAAC, 2015 Search PubMed.
- H. S. Sader, S. Paukner, Z. Ivezic-Schoenfeld, D. J. Biedenbach, F. J. Schmitz and R. N. Jones, J. Antimicrob. Chemother., 2012, 67, 1170–1175 CrossRef CAS.
- S. Paukner, H. S. Sader, Z. Ivezic-Schoenfeld and R. N. Jones, Antimicrob. Agents Chemother., 2013, 57, 4489–4495 CrossRef CAS.
- R. E. Mendes, D. J. Farrell, R. K. Flamm, G. H. Talbot, Z. Ivezic-Schoenfeld, S. Paukner and H. S. Sader, Antimicrob. Agents Chemother., 2016, 60, 4407–4411 CrossRef CAS.
- S. Jacobsson, S. Paukner, D. Golparian, J. S. Jensen and M. Unemo, Antimicrob. Agents Chemother., 2017, 61 DOI:10.1128/AAC.01497-17.
- U. Schmidt, W. W. Wicha, F. Obermayr, R. Novak and W. Prince, BC-3781: Evaluation of the CYP3A Interaction Potential, Poster P1524, ECCMID, 2011 Search PubMed.
- J. E. Ross, H. S. Sader, Z. Ivezic-Schoenfeld, S. Paukner and R. N. Jones, J. Clin. Microbiol., 2012, 50, 3361–3364 CrossRef CAS.
- D. J. Biedenbach, R. N. Johns, Z. Ivezic-Schoenfeld, S. Paukner and R. Novak, In Vitro Antibacterial Spectrum of BC-3205, a Novel Pleuromutilin Derivative for Oral Use in Humans, Poster F1–1513, ICAAC, 2009 Search PubMed.
- K. Thirring, G. Ascher, S. Paukner, W. Heilmayer and R. Novak, World Pat., WO2007079515 A1, 2007.
- 2016 Nabriva Annual Report, https://investors.nabriva.com/static-files/2910a335-ccd6-445c-95d1-29f7a67771bb, accessed 2018/01/28.
- D. R. Gentry, S. F. Rittenhouse, L. McCloskey and D. J. Holmes, Antimicrob. Agents Chemother., 2007, 51, 2048–2052 CrossRef CAS PubMed.
- L. Diaz, P. Kiratisin, R. E. Mendes, D. Panesso, K. V. Singh and C. A. Arias, Antimicrob. Agents Chemother., 2012, 56, 3917–3922 CrossRef CAS PubMed.
- F. Cafini, L. T. T. Nguyen, M. Higashide, F. Román, J. Prieto and K. Morikawa, J. Antimicrob. Chemother., 2016, 71, 587–592 CrossRef CAS PubMed.
- H. Bonilla, M. D. Huband, J. Seidel, H. Schmidt, M. Lescoe, S. P. McCurdy, M. M. Lemmon, L. A. Brennan, A. Tait-Kamradt, L. Puzniak and J. P. Quinn, Clin. Infect. Dis., 2010, 51, 796–800 CrossRef.
- K. Kadlec, C. F. Pomba, N. Couto and S. Schwarz, J. Antimicrob. Chemother., 2010, 65, 2692–2693 CrossRef CAS PubMed.
- D. R. Gentry, L. McCloskey, M. N. Gwynn, S. F. Rittenhouse, N. Scangarella, R. Shawar and D. J. Holmes, Antimicrob. Agents Chemother., 2008, 52, 4507–4509 CrossRef CAS.
- S. Schwendener and V. Perreten, Antimicrob. Agents Chemother., 2011, 55, 4900–4904 CrossRef CAS.
- T. Hauschild, A. T. Feßler, K. Kadlec, C. Billerbeck and S. Schwarz, J. Antimicrob. Chemother., 2012, 67, 503–504 CrossRef CAS.
- B. Malbruny, A. M. Werno, D. R. Murdoch, R. Leclercq and V. Cattoir, Antimicrob. Agents Chemother., 2011, 55, 1470–1474 CrossRef CAS.
- S. Wendlandt, C. Lozano, K. Kadlec, E. Gómez-Sanz, M. Zarazaga, C. Torres and S. Schwarz, J. Antimicrob. Chemother., 2013, 68, 473–475 CrossRef CAS.
- D. R. Gentry, L. McCloskey, M. N. Gwynn, S. F. Rittenhouse, N. Scangarella, R. Shawar and D. J. Holmes, Antimicrob. Agents Chemother., 2008, 52, 4507–4509 CrossRef CAS.
- K. Riedl, J. Antibiot., 1976, 29, 132–139 CrossRef CAS.
- H. Egger and H. Reinshagen, J. Antibiot., 1976, 29, 915–922 CrossRef CAS.
- H. Egger and H. Reinshagen, J. Antibiot., 1976, 29, 923–927 CrossRef CAS.
- K. Thirring and W. Heilmayer, World Pat., WO2009009812 A1, 2009.
- K. Thirring and W. Heilmayer, Europe Pat., EP2176211 B1, 2013.
- G. Ascher and J. Hildebrandt, US Pat., US20050131023 A1, 2005.
- Y. Y. Zhang, K. P. Xu, D. Ren, S. R. Ge, Y. L. Wang and Y. Z. Wang, Chin. Chem. Lett., 2009, 20, 29–31 CrossRef CAS.
- P. Xu, Y.-Y. Zhang, Y.-X. Sun, J.-H. Liu, B. Yang, Y.-Z. Wang and Y.-L. Wang, Chem. Biol. Drug Des., 2009, 73, 655–660 CrossRef CAS.
- R. Shang, S. Wang, X. Xu, Y. Yi, W. Guo, Y. Liu and J. Liang, PLoS One, 2013, 8, e82595 CrossRef.
- R. Shang, Y. Liu, Z. Xin, W. Guo, Z. Guo, B. Hao and L. Jianping, Eur. J. Med. Chem., 2013, 63, 231–238 CrossRef CAS.
- C. Ling, L. Fu, S. Gao, W. Chu, H. Wang, Y. Huang, X. Chen and Y. Yang, J. Med. Chem., 2014, 57, 4772–4795 CrossRef CAS.
- Y. Yi, Y. Fu, P. Dong, W. Qin, Y. Liu, J. Liang and R. Shang, Molecules, 2017, 22, 996 CrossRef.
- Y. Hirokawa, H. Kinoshita, T. Tanaka, T. Nakamura, K. Fujimoto, S. Kashimoto, T. Kojima and S. Kato, Bioorg. Med. Chem. Lett., 2008, 18, 3556–3561 CrossRef CAS.
- Y. Hirokawa, H. Kinoshita, T. Tanaka, T. Nakamura, K. Fujimoto, S. Kashimoto, T. Kojima and S. Kato, Bioorg. Med. Chem. Lett., 2009, 19, 170–174 CrossRef CAS.
- Y. Hirokawa, H. Kinoshita, T. Tanaka, T. Nakamura, K. Fujimoto, S. Kashimoto, T. Kojima and S. Kato, Bioorg. Med. Chem. Lett., 2009, 19, 175–179 CrossRef CAS PubMed.
- H. Liu, S. Xiao, D. Zhang, S. Mu, L. Zhang, X. Wang and F. Xue, Biol. Pharm. Bull., 2015, 38, 1041–1048 CrossRef CAS.
- J. Luo, Q.-E. Yang, Y.-Y. Yang, Y.-Z. Tang and Y.-H. Liu, Chem. Biol. Drug Des., 2016, 88, 699–709 CrossRef CAS.
- M.-L. Gao, J. Zeng, X. Fang, J. Luo, Z. Jin, Y.-H. Liu and Y.-Z. Tang, Eur. J. Med. Chem., 2017, 127, 286–295 CrossRef CAS.
- L. Lolk, J. Pøhlsgaard, A. S. Jepsen, L. H. Hansen, H. Nielsen, S. I. Steffansen, L. Sparving, A. B. Nielsen, B. Vester and P. Nielsen, J. Med. Chem., 2008, 51, 4957–4967 CrossRef CAS.
- I. Dreier, S. Kumar, H. Søndergaard, M. L. Rasmussen, L. H. Hansen, N. H. List, J. Kongsted, B. Vester and P. Nielsen, J. Med. Chem., 2012, 55, 2067–2077 CrossRef CAS PubMed.
- I. Dreier, L. H. Hansen, P. Nielsen and B. Vester, Bioorg. Med. Chem. Lett., 2014, 24, 1043–1046 CrossRef CAS PubMed.
- Y. Yi, G. Yang, C. Zhang, J. Chen, J. Liang and R. Shang, Eur. J. Med. Chem., 2015, 101, 179–184 CrossRef CAS.
- Y. Ling, X. Wang, H. Wang, J. Yu, J. Tang, D. Wang, G. Chen, J. Huang, Y. Li and H. Zheng, Arch. Pharm., 2012, 345, 638–646 CrossRef CAS.
- S. Mu, H. Liu, L. Zhang, X. Wang, F. Xue and Y. Zhang, Biol. Pharm. Bull., 2017, 40, 1165–1173 CrossRef CAS.
- R.-F. Shang, G.-H. Wang, X.-M. Xu, S.-J. Liu, C. Zhang, Y.-P. Yi, J.-P. Liang and Y. Liu, Molecules, 2014, 19, 19050–19065 CrossRef.
- R. Shang, X. Pu, X. Xu, Z. Xin, C. Zhang, W. Guo, Y. Liu and J. Liang, J. Med. Chem., 2014, 57, 5664–5678 CrossRef CAS.
- S. Dabbs, S. Davies, D. K. Dean, C. H. Frydrych, A. Gaiba, S. Howard, E. Hunt, F. D. King, A. Naylor and A. K. Takle, World Pat., WO2000027790 A1, 2000.
- R. Nagarajan, US Pat., US4130709 A, 1978.
- G. Brooks, W. Burgess, D. Colthurst, J. D. Hinks, E. Hunt, M. J. Pearson, B. Shea, A. K. Takle, J. M. Wilson and G. Woodnutt, Bioorg. Med. Chem., 2001, 9, 1221–1231 CrossRef CAS.
- A. K. Takle, E. Hunt and A. Naylor, US Pat., US6121281 A, 2000.
- J. D. Hinks, A. K. Takle and E. Hunt, US Pat., US6239175 B1, 2001.
- J. Yang, World Pat., WO2007062334 A2, 2007.
- A. M. Diederich, J. Sisko and F. G. Vogt, World Pat., WO2007062335 A3, 2007.
- D. H. Igo and B. A. Norton, US Pat., US20080269342 A1, 2008.
- Y. Fukuda, M. Takadoi, Y. Asahina, T. Sato, H. Kurasaki, H. Ebisu, M. Takei and H. Fukuda, US Pat., US20100197909 A1, 2010.
- Y. Fukuda, M. Takadoi, Y. Asahina, T. Sato, H. Kurasaki, H. Ebisu, M. Takei and H. Fukuda, US Pat., US8222407 B2, 2012.
- L. Fu, X. Liu, C. Ling, J. Cheng, X. Guo, H. He, S. Ding and Y. Yang, Bioorg. Med. Chem. Lett., 2012, 22, 814–819 CrossRef CAS PubMed.
- L. Fu, Z. Jiang, Z. Cai, X. Liu, H. He and Y. Yang, Bioorg. Med. Chem. Lett., 2009, 19, 5407–5410 CrossRef CAS.
- D. K. Dean, A. Naylor and A. K. Takle, US Pat., US7045524 B2, 2006.
- D. M. Springer, M. E. Sorenson, S. Huang, T. P. Connolly, J. J. Bronson, J. A. Matson, R. L. Hanson, D. B. Brzozowski, T. L. LaPorte and R. N. Patel, Bioorg. Med. Chem., 2003, 13, 1751–1753 CrossRef CAS.
- G. Brooks, E. Hunt and S. Howard, US Pat., US20030114674 A1, 2003.
- L. Q. Fu, X. S. Guo, X. Liu, H. L. He, Y. L. Wang and Y. S. Yang, Chin. Chem. Lett., 2010, 21, 507–510 CrossRef CAS.
- G. Brooks and E. Hunt, World Pat., WO2000007974 A1, 2000.
- D. M. Springer, A. Bunker, B.-Y. Luh, M. E. Sorenson, J. T. Goodrich, J. J. Bronson, K. DenBleyker, T. J. Dougherty and J. Fung-Tomc, Eur. J. Med. Chem., 2007, 42, 109–113 CrossRef CAS.
- H. Berner, H. Vyplel, G. Schulz and H. Schneider, Monatsh. Chem., 1986, 117, 1073–1080 CrossRef CAS.
- K. Thirring, W. Heilmayer, R. Riedl, H. Kollmann, Z. Ivezic-Schoenfeld, W. Wicha, S. Paukner and D. Strickmann, World Pat., WO2015110481 A1, 2015.
- D. Dean, A. Naylor and A. Takle, US Pat., US20050143393A1, 2005.
- I. Macher, World Pat., WO1993022288A1, 1993.
- A. Bentley, M. Butters, S. P. Green, W. J. Learmonth, J. A. MacRae, M. C. Morland, G. O'Conno and J. Skuse, Org. Process Res. Dev., 2002, 6, 109–112 CrossRef CAS.
- H. Gilzad Kohan, K. Kaur and F. Jamali, PLoS One, 2015, 10, e0126786 CrossRef.
- H.-K. Han and G. L. Amidon, AAPS PharmSci, 2000, 2, 48–58 CrossRef.
- V. J. Stella and K. W. Nti-Addae, Adv. Drug Delivery Rev., 2007, 59, 677–694 CrossRef CAS.
- D. W. Siemann, D. J. Chaplin and P. A. Walicke, Expert Opin. Invest. Drugs, 2009, 18, 189–197 CrossRef CAS.
- L. M. Rorick-Kehn, E. J. Perkins, K. M. Knitowski, J. C. Hart, B. G. Johnson, D. D. Schoepp and D. L. McKinzie, J. Pharmacol. Exp. Ther., 2006, 316, 905–913 CrossRef CAS PubMed.
- L. Chen, D. Yang, Z. Pan, L. Lai, J. Liu, B. Fang and S. Shi, Chem. Biol. Drug Des., 2015, 86, 239–245 CrossRef CAS PubMed.
- P. M. Wright, I. B. Seiple and A. G. Myers, Angew. Chem., Int. Ed., 2014, 53, 8840–8869 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- J. F. Hartwig, Acc. Chem. Res., 2017, 50, 549–555 CrossRef CAS PubMed.
- R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed.
- R. L. Hanson, J. A. Matson, D. B. Brzozowski, T. L. LaPorte, D. M. Springer and R. N. Patel, Org. Process Res. Dev., 2002, 6, 482–487 CrossRef CAS.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS.
- D. P. Uccello, S. M. Miller, N. A. Dieterich, A. F. Stepan, S. Chung, K. A. Farley, B. Samas, J. Chen and J. I. Montgomery, Tetrahedron Lett., 2011, 52, 4247–4251 CrossRef CAS.
- S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. Christina White, Nat. Chem., 2015, 7, 987–994 CrossRef CAS.
- N. D. Chiappini, J. B. C. Mack and J. D. Bois, Angew. Chem., Int. Ed., 2018, 130, 5050–5053 CrossRef.
- J. Zhou and J. F. Hartwig, Angew. Chem., Int. Ed., 2008, 47, 5783–5787 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS.
- X. Ma, R. Kucera, O. F. Goethe, S. K. Murphy and S. B. Herzon, J. Org. Chem., 2018 DOI:10.1021/acs.joc.8b00462.
- A. Okano, N. A. Isley and D. L. Boger, Chem. Rev., 2017, 117, 11952–11993 CrossRef CAS PubMed.
- F. Liu and A. G. Myers, Curr. Opin. Chem. Biol., 2016, 32, 48–57 CrossRef CAS PubMed.
- I. B. Seiple, Z. Zhang, P. Jakubec, A. Langlois-Mercier, P. M. Wright, D. T. Hog, K. Yabu, S. R. Allu, T. Fukuzaki, P. N. Carlsen, Y. Kitamura, X. Zhou, M. L. Condakes, F. T. Szczypiński, W. D. Green and A. G. Myers, Nature, 2016, 533, 338–345 CrossRef CAS PubMed.
- Q. Li and I. B. Seiple, J. Am. Chem. Soc., 2017, 139, 13304–13307 CrossRef CAS PubMed.
- C. R. Shugrue and S. J. Miller, Chem. Rev., 2017, 117, 11894–11951 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |