
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering strategies for rational polyketide synthase design
Maja
Klaus
 and
Martin
Grininger
*
and
Martin
Grininger
*
Institute of Organic Chemistry and Chemical Biology, Buchmann Institute for Molecular Life Sciences, Cluster of Excellence for Macromolecular Complexes, Goethe University Frankfurt, Max-von-Laue-Str. 15, 60438 Frankfurt am Main, Germany. E-mail: grininger@chemie.uni-frankfurt.de
First published on 25th June 2018
Abstract
Covering: mid 1990s to 2018
Over the last two decades, diverse approaches have been explored to generate new polyketides by engineering polyketide synthases (PKSs). Although it has been proven possible to produce new compounds by designed PKSs, engineering strategies failed to make polyketides available via widely applicable rules and protocols. Still, organic synthetic routes have to be employed whenever new polyketides are needed for applications in medicine, agriculture, and industry. In light of the rising demand for commodity products from feedstock and for fast and cheap access to pharmaceutical compounds, the need for harnessing PKSs to produce such molecules is more urgent than ever before. In this review, we focus on a multitude of approaches to engineer modular PKSs by swapping and replacing PKS modules and domains, which we analyze in the light of recent structural and biochemical data. We conclude with an outlook on possible strategies on how to increase success rates of PKS engineering in future.
 Maja Klaus | Maja Klaus graduated 2015 from the Leibniz University Hannover with a M. Sc. in Life Science. During her Master's thesis with Prof. Chaitan Khosla at Stanford University she started to work on the engineering of protein–protein interactions within polyketide synthases. In summer 2015, she joined Prof. Martin Grininger's lab at the Goethe University Frankfurt to pursue a PhD in chemistry, where she continues her research on harnessing the modularity of polyketide assembly lines in collaboration with Prof. Khosla. |
 Martin Grininger | Martin Grininger studied chemistry at the Universities Linz and Graz (Austria), and conducted his diploma thesis in natural compound synthesis in the lab of Prof. Alois Fürstner. He then worked with Prof. Dieter Oesterhelt at the Max-Planck-Institute of Biochemistry for his PhD, studying flavin sequestration and storage in Archaea and Bacteria. After becoming a project group leader at the same institution, he focused on multidomain fatty acid synthases and contributed to the current structural understanding of these proteins. He also held a visiting professorship at the University Vienna in Biological Chemistry. In 2012, he received a Lichtenberg Professorship of the Volkswagen Foundation hosted by the University Frankfurt. He is co-speaker of the research focus MegaSyn, which develops rules for engineering megasynthases for custom compound synthesis. |
1. Introduction
Polyketide synthases (PKSs) are responsible for the synthesis of polyketide natural products; among them many compounds with high bioactivity, such as environmental toxins (e.g. aflatoxin), antibiotics (e.g. erythromycin and tetracycline), antineoplastics (e.g. daunorubicin) and immunosuppressants (e.g. rapamycin).1–3 PKSs use simple acyl-CoA building blocks and assemble them into complex compounds with molecular weights of up to several kilodaltons (kDa).PKSs can be classified into three different types. Type I PKSs are of highest synthetic capability, and are comprised of large multidomain proteins. Further, type I systems can be subdivided into the iterative (iPKSs) and modular PKSs (modPKSs), whereby the modPKSs occur as cis-AT and trans-AT PKSs.4 In the following, we focus only on type I systems and use the term PKSs for referring to type I cis-AT PKSs. iPKSs perform synthesis in a recursive manner, during which the catalytic domains of a single polypeptide repeatedly condense acyl-CoA precursor units until the specific length of the compound is achieved. In contrast, modPKSs occur as large assembly line-like complexes and successively condense precursor building blocks to the final natural compound. The 6-deoxyerythronolide B synthase (DEBS) is the prototypical example for a large pool of naturally occurring modPKSs, in which the arrangement of modules essentially defines the sequence of functional groups in the final compound (Fig. 1). Each module comprises a set of essential domains for C–C bond formation, namely the catalytic domains ketoacyl synthase (KS) and acyl transferase (AT), as well as the acyl carrier protein (ACP) domain. Within a module, the collaborative function of the AT, ACP, and KS domains results in a non-reduced polyketide chain. Optionally, further processing domains (ketoreductase (KR), dehydratase (DH), and enoylreductase (ER)) are used to catalyze the step-wise reduction of the β-keto group. The flexible ACP domain shuttles the polyketide intermediate both within a module and across modules. Physical linkage between individual modules within a modPKS is either achieved through covalent linkers or non-covalent linker domains (docking domains) at their C- and N-termini.5,6
 | ||
| Fig. 1 Architecture of the 6-deoxyerythronolide B synthase (DEBS). DEBS consists of three polypeptides (DEBS1, DEBS2, and DEBS3) each harboring two modules, which are connected by covalent linkers (colored lines). Modules across polypeptides interact through docking domains at the C- and N-termini (black taps). Additionally, a loading didomain (LDD) for starter unit selection and a thioesterase domain (TE) for product release are present. DEBS uses propionyl-, methylmalonyl-CoA and NADPH to produce 6-deoxyerythronolide B (6-dEB) the precursor for the antibiotic erythromycin. The polyketide intermediates of each module are depicted. Domain annotation: KS – ketosynthase, AT – acyltransferase, KR – ketoreductase, DH – dehydratase, ER – enoylreductase, and ACP – acyl carrier protein. Figure adapted from (ref. 58). | ||
The high pharmaceutical importance of polyketides has initiated intensive research on their biosynthetic generation. Whereas conventional polyketide synthesis through organic chemical routes can be highly intricate, polyketide biosynthesis is remarkably simpler, since it is built on the controlled stepwise condensation of small acyl-CoA substrates and their subsequent chemical modification. The strict correlation of the assembly line architecture of modPKSs and the final polyketide structure, known as the principle of co-linearity, stimulated efforts to engineer modPKSs by swapping entire modules or single domains to generate new polyketides.7–9 Recently, a broadly applicable mix-and-match strategy for engineering the related family of non-ribosomal peptide synthetases (NRPSs) has been presented,10 which uses defined exchange units as interchangeable synthetic modules. Concepts of comparable broad applicability for harnessing PKS modularity for engineering have not yet been established. The extensive scaffold of modPKSs comprises a wealth of permanent domain–domain and module–module interactions and samples a multitude of transient domain–domain interactions during ACP-mediated substrate shuttling. This complexity needs to be further untangled in structural and functional studies before programmable engineering becomes possible.
Besides swapping domains and modules, another PKS engineering strategy is the modification of active sites to induce different substrate specificity or higher substrate promiscuity. Such an approach preserves the structural and conformational properties of the scaffold, and only adapts active sites to the new requirements. PKS engineering by site-directed mutagenesis is covered with just a few examples in literature (for recent examples, see e.g. (ref. 11–13)). It is unclear whether this approach has even lower success rates or whether the chemical biological community largely pursues the enticing mix-and-match and domain-swap approaches to directly harness the inherent modularity of PKSs.
In this review, we will revisit available structural and functional data of the last 20 years to give a concise overview of the current knowledge of interactions and interfaces in modPKSs. On the basis of the current understanding, we will map important interfaces that need to be preserved and others that can function as potential engineering sites. Finally, we suggest a multidisciplinary research approach for collecting quantitative data to enhance engineering successes in the future.
2. The PKS domain–domain and module–module interaction network
Engineering of PKSs requires detailed knowledge about structural arrangements and the interactions of domains and modules. While our understanding of the architecture of PKS modules and their arrangement into assembly lines is still limited, we have insight into structures of subregions or evolutionary related proteins. Recent progress in the characterization of the transient interactions during the catalytic cycle of PKSs and an improved understanding of the vectorial synthetic progress in modPKSs further helps to guide precise engineering approaches.2.1. The PKS structural scaffold
The earliest X-ray structural information on the modPKS, the KS–AT condensing unit and the KR domain, was available from studies of the modPKS DEBS.18–20 When the X-ray crystal structure on porcine FAS was revealed in 2008, it not only gave new insights into mechanisms of fatty acid synthesis, but also spurred significant interest in the field of modPKS (Fig. 2A, mFAS).28 The mammalian fatty acid synthase (mFAS) is evolutionary related to PKSs, and many subregions from mFAS and modPKSs displayed structural consensus, including the non-catalytic linkers in the KS–AT didomain (the KS–AT linker and post-AT linker) and the non-catalytic ΨKR domain.14–17 This comparison led to the appealing model that mFAS is simply a fully reducing PKS, which can accept deletions of modifying domains within the processing wing to build the partially reducing and non-reducing PKS folds. Notwithstanding the still valid tight structural relationship of folds, recent studies revised this view and show decisive structural differences between the processing parts of PKSs and mFAS. Particularly, the first almost complete X-ray structural model of the iterative Mycobacterium smegmatis mycocerosic acid synthase (MAS)-like PKS gave important insight,21 but also earlier models on separate proteins and the KR–ER didomain provided major contributions to the structural understanding of PKSs.20,22–25 The DH domain is most important in the different organizations of the processing part of mFAS and PKSs. In contrast to a V-shaped arrangement in mFAS, DHs of PKSs form an overall elongated dimeric arrangement inducing a different relative positioning of the modifying domains.21,22 The embedment of the ER domain in the PKSs fold is currently disputed. The ER is dimeric in the MAS-like (iterative) PKS, resting upon the extended β-sheet of the DH dimer by forming a small and variable interface (Fig. 2A, MAS-like model). The relevance of a dimeric ER arrangement in modPKSs is unclear,26 since the ER domains from modPKSs, structurally characterized by X-ray crystallography as individual proteins and as part of a KR–ER didomain construct, appeared to be monomeric.23,25 The ER domain may occur in different oligomeric states in PKSs, i.e. as a monomeric ER in modPKSs and dimeric ER in iPKSs, consistent with ER–KR linkers of modPKSs being generally shorter (usually <8 amino acids) than those of iPKSs (>17). A short linker may restrain ER conformational variability in modPKSs and prevent ER dimerization at the twofold axis.26 SAXS data have been reported, which support both dimeric and monomeric ER in modPKSs.21,23 Accordingly, two models for the structural appearance of fully reducing modPKSs exist today: a MAS-like PKS model with the ER domains dimerizing at the C2-axis (Fig. 2A), and a SPNS M2 derived model, in which monomeric ERs are swung out and thereby free space at the protein's central region (Fig. 2B).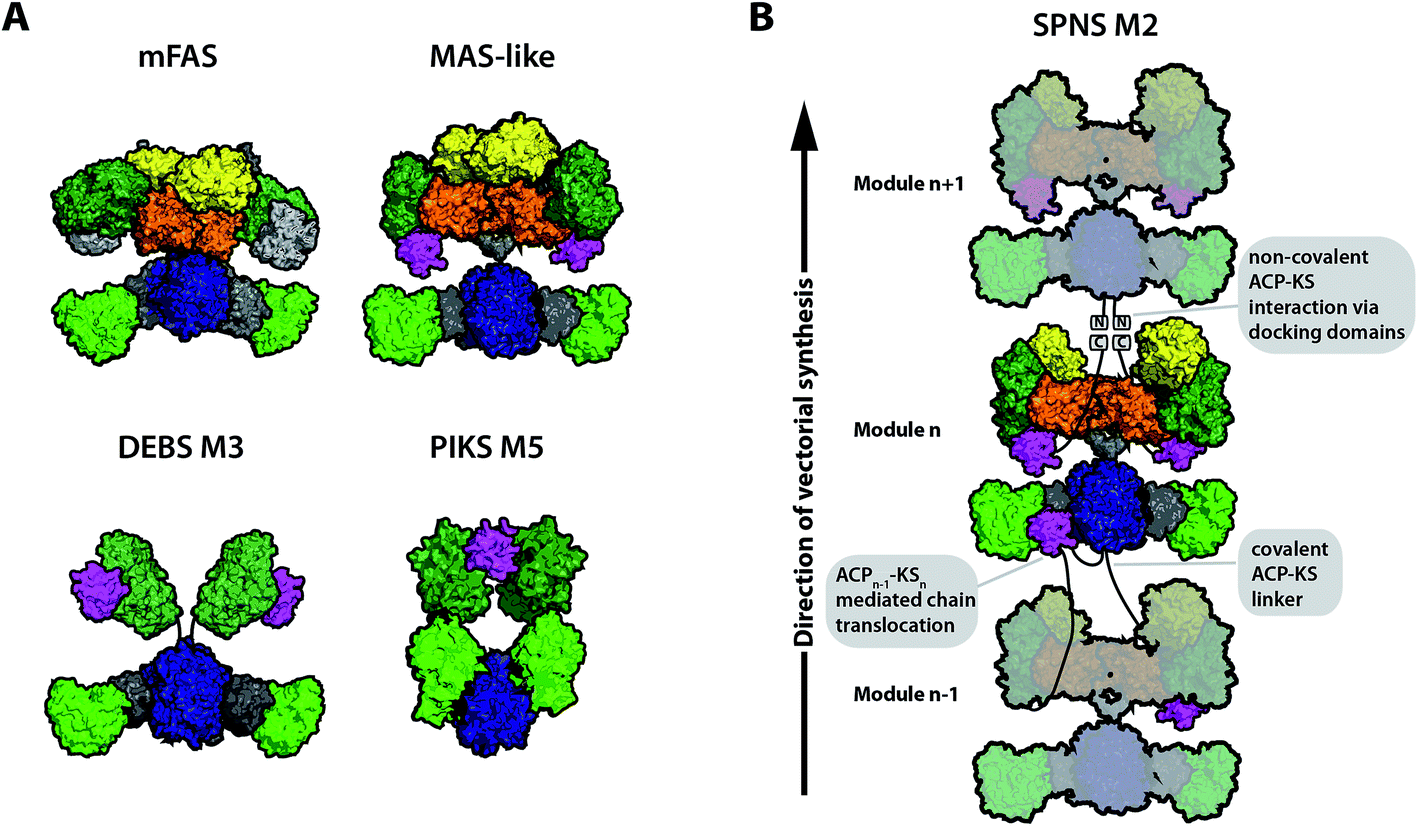 | ||
| Fig. 2 Different models for assembly line PKSs in comparison to mFAS. (A) Schematic representation of mFAS (based on PDB 2VZ8),28 a MAS-like PKS (based on PDB 5BP1 and 5BP4),21 a model for the partially reducing DEBS M3 based on SAXS analysis,33 and a model of the partially reducing pikromycin synthase module 5 (PIKS M5) based on cryo-EM analysis.27 (B) Graphical representation of a PKS module assembly using the example of the proposed model for the spinomycin synthase module 2 (SPNS M2).23 The SPNS M2 model was assembled from PDBs 5BP1 (KS–AT), 3SLK (ER–KR), and 5BP4 (DH) with KR of 3SLK superimposed on KR of 5BP4. PKS modules can either be connected through covalent ACP–KS linkers or through non-covalently interacting docking domains at the C- and N-termini. Additionally, chain translocation is mediated through specific interactions between the upstream ACP (ACPn−1) and the downstream KS (KSn), shown for one of the two ACPs in modulen−1. Domain coloring: KS (blue), AT (light green), KR (dark green), DH (orange), ER (yellow), ACP (magenta), linker regions such as the KS-AT linker (dark gray), and only for mFAS non-catalytic pseudo-methyltransferase domain (light gray). | ||
In order to reach consensus on the appearance of individual PKS modules, structural characterization of a variety of intact modules is required. Until today, detailed structural information on a complete modPKS module is missing, with the exception of medium resolution (7.3–9.5 Å resolution) cryo-electron microscopic (cryo-EM) data on the partially reducing module 5 of picromycin synthase (PIKS M5). The PIKS M5 structure revealed an arched conformation,27 with a relative arrangement of the KS and AT domains that contradicts X-ray crystallographic studies on the mammalian FAS28–30 and PKS KS–AT structures.18,19,21 In all these structures, the AT domains were consistently found to be embedded as bulge of the KS fold framed by a N-terminal KS–AT linker that constitutes most of the KS–AT interface, and a post-AT linker that wraps back to interact with the KS domain.18 In the PIKS M5 model, this structural integrity is dissolved inducing the overall arch-shaped appearance. The PIKS M5 model provides a structural basis that can explain modPKS function; however, before the PIKS M5 model can be accepted as a relevant model for the functional mode of modPKSs, several of its structural features need yet to be reconciled with a large amount of data collected over the last decades.31,32 A low-resolution model of partially reducing PKS modules was additionally derived from SAXS analysis of DEBS.33 Herein, a similar architecture of the KS–AT fold was observed as found in mFAS and PKSs. Thus, two different models for partially reducing modPKS modules can be derived today based on the analysis of PIKS M5 and DEBS modules (Fig. 2A).
While several models can be generated for the overall scaffold of a single PKS module, data on the architecture of a whole assembly line have not been presented to date. In general, modules can be connected by covalent ACP–KS linkers or through non-covalently interacting, α-helical docking domains (Fig. 2B). While several excised docking domains were structurally solved,5,34–36 no high resolution structure of a bimodule was obtained so far.
2.2. Interactions and processes within modular PKSs
ACP domains are responsible for substrate shuttling in PKSs. They are generally loosely attached via unstructured linkers to the catalytic body of (type I) PKSs and interact transiently with the catalytic domains. Owing to the high conformational variability, ACP domains often remain unresolved in structural studies leading to paucity of details to the process of ACP-mediated substrate shuttling. The primary insight into the mode of substrate shuttling in PKSs was again provided by the homologous mFASs. For example, it was shown that swiveling and swinging motions of the condensing and processing part of mFAS occur and presumably assist the mobile ACP domain in substrate shuttling.37–39Despite being loosely attached, the ACP domains are differently constrained in their conformational space in iterative and modular proteins. While in mFAS and iPKSs the ACP is free to move, solely restricted by terminating domains such as TE, the linkage of ACP to the KS of the downstream module constrains its conformational variability in modPKS. In covalently connected modules the average length of an ACP–KS linker is 18 residues in length (in ER containing modules).26 For anticipating a mode of action of ACPs in modPKS assembly lines, the ER arrangement is therefore of decisive impact (see above for the discussion about modPKS scaffolds). A monomeric ER would allow a central positioning of ACP and grant intuitive, easy access to each of the integral catalytic domains, whereas an ER dimer would crowd the C2-axis and force the ACPs toward the peripheral lining of the assembly line (Fig. 2B).
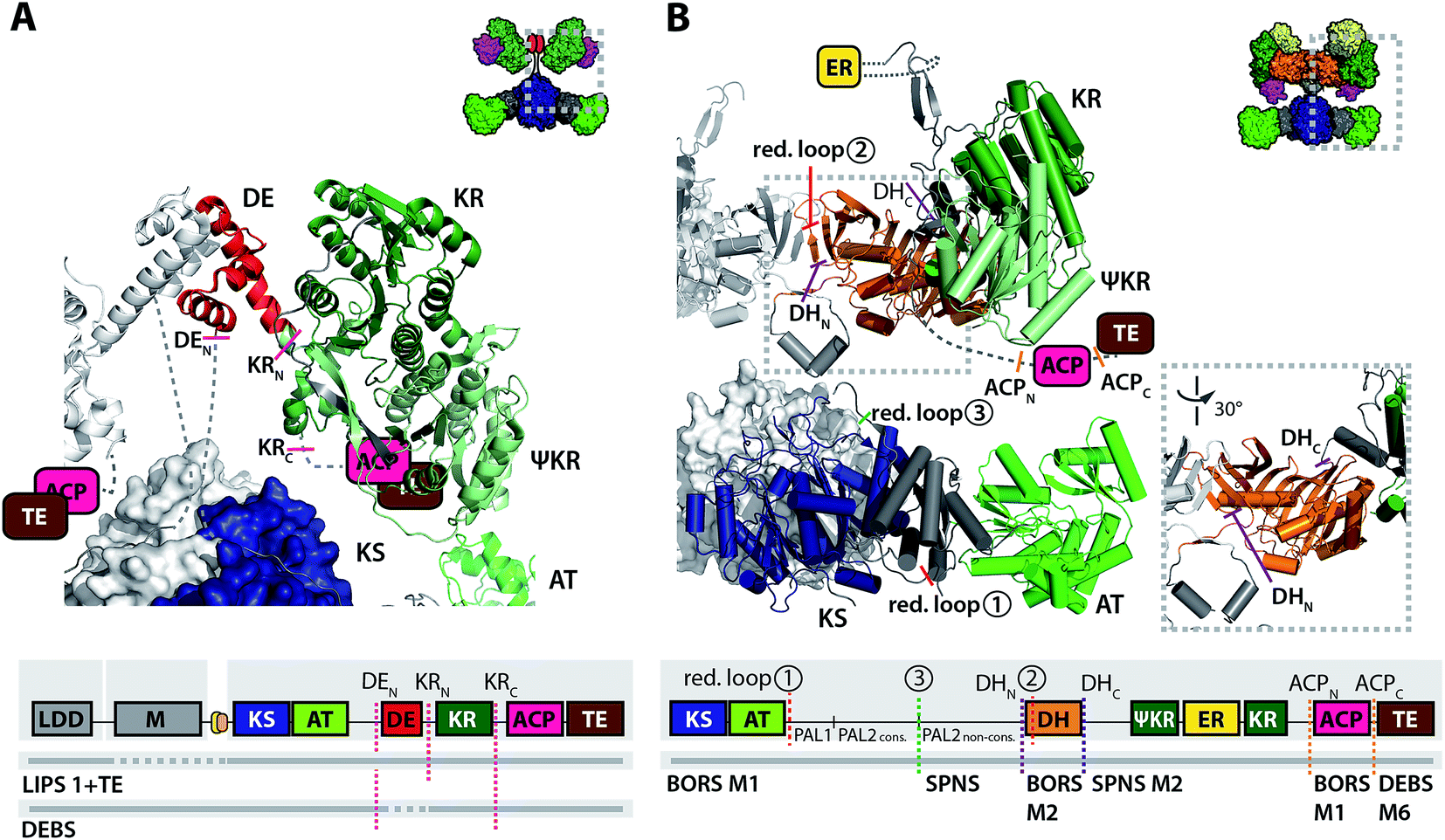
The pivotal steps for catalytic progress along the PKS assembly line are the ACP–KS mediated chain translocation and chain elongation reactions. For a successful chain translocation reaction, two adjacent modules need to interact to bring the upstream ACP and the downstream KS into close proximity. While part of this interaction is mediated through covalent linkers or docking domains, specificity is conferred through the interaction of the ACP and the KS–AT fold itself. Using DEBS as a model system, the ACP–KS interactions were analyzed in a comprehensive study combining in vitro analysis of chimeric ACPs and in silico docking simulations. Generation of different chimeras revealed that during chain elongation, ACPA docks (of protomer A) docks with loop 1 to the KSA–ATA linker, while engaging with KSB (of protomer B) for chain elongation.40 Two distinct recognition sites on the KS–AT linker were found to interact with a minimal epitope in ACP loop 1. In a similar manner, the upstream ACP docks with the first ten N-terminal residues of ACP helix 1 to KSA–ATA linker to mediate chain translocation with KSB.40,41 Although the ACPs dock into the same deep cleft of the KS–AT fragment, the position and orientation is distinct in the event of chain translocation and chain elongation.
2.3. Models for the origin of vectorial synthesis
Vectorial synthesis is the term given to the step-by-step synthetic progress along a directional scaffold; i.e. for modPKSs, the biosynthesis of polyketide compounds in a linear manner. While vectorial synthesis has emerged as a rather robust feature in modPKSs,42,43 its molecular foundation is unclear. Recently, a turnstile mechanism has been postulated, in which a not yet discovered molecular signal prevents the KS domain from iterative use.44 The reopening of the turnstile for reloading necessitates a vacant module as prerequisite. Such a state does not appear during synthesis in iPKSs (and FASs), which implies that vectorial synthesis is a unique feature of modPKSs. An alternative explanation for the origin of vectorial synthesis is the kinetic coupling of catalytic steps along the assembly line, due to the serial arrangement of modules. As iPKSs and modPKSs are tightly evolutionary connected,14 kinetic coupling of catalytic processes would facilitate our understanding of vectorial biosynthesis as a result of the PKS architecture rather than an inherent feature of modPKS. For example, the recently characterized iterative MAS-like PKS, producing an eicosenoic acid derivative, is assigned phylogenetically to modPKSs.21,45 Moreover, modPKSs have been demonstrated to also synthesize in iterative fashion as part of assembly lines (borrelidin synthase (BORS) module 5 (ref. 46) and aureothin producing PKS (AURS)47), and PKS modules have been turned into iteratively synthesizing units by mutation (PIKS M5 (ref. 48) and DEBS M3 (ref. 41)).3. Engineering module–module and domain–domain interactions – early and recent efforts
Engineering of module–module and domain–domain interactions in modPKSs can be classified by different criteria. The often referred to mix-and-match approach considers the swapping of entire PKS modules from one synthase into another, while domain–domain swapping is focusing on single domains e.g. AT domains or KR domains to alter the product spectra of a given PKS. Within those categories, the studies differ in whether they were carried out in vivo, and thus relying solely on differences in product spectra, or in vitro, shedding light on enzyme kinetics and enzyme affinities. In this section, we highlight engineering approaches of modPKSs and evaluate them based on the available structural data.3.1. Module–module exchanges
The discovery of modPKSs inspired the chemical biological community to harness their modularity for the rapid generation of new natural products.49,50 One of the biggest challenges in combining intact PKS modules to chimeric PKS assembly lines is to preserve their catalytic integrity. Early on, the necessity of preserving linker regions between adjacent modules to retain functional proteins was noticed.51–54 Although preservation of native interaction sites helped in generating productive assembly lines, the overall production rates were usually lowered in the engineered systems.The first truly combinatorial approach of generating bimodular chimeric PKSs, consisting of intact modules from different PKS sources, was carried out in an in vivo study recombining 14 modules of 8 PKS clusters.42 Using docking domains derived from DEBS, a total of 154 bimodular chimeric PKSs were assembled. About 50% of the chimeric PKSs yielded detectable product albeit with lower product yields as their natural bimodular reference system.42 Later on, this approach was extended towards chimeric trimodular PKSs with similar results.55
Until now, chimeric PKSs have been constructed by either fusing proteins or by non-covalently connecting individual modules with docking domains derived from natural PKS sources. In vitro characterization of chimeric bimodular PKSs using fusion proteins, in which the non-naturally interacting modules were connected by a covalent linker, revealed similar kcat values for wild type and chimeric PKSs.56 A more thorough analysis showed that linker regions and specific ACP–KS interactions play equal roles for the turnover rate.57 Recently, the finding that linker regions alone are able to connect heterologous modules was challenged in a study creating a library of bimodular and trimodular chimeric PKSs in vitro by employing docking domains to mediate communication between chimeric modules. Surprisingly, and in contrary to the earlier in vivo results, the turnover rates of all chimeric PKSs were drastically diminished, which was assigned to impaired chain translocation due to the non-native ACP–KS interactions.58
As module–module communication is necessary to generate productive chimeric PKS assembly lines, questions arise towards the affinity of these interactions in native proteins. So far only a few studies have addressed this issue. The dissociation constant of docking domains or modules connected by docking domains was found to be KD 70–130 mM (ref. 35) and KD 1–2 mM (ref. 59), respectively. Titration of different DEBS polypeptides and following Michaelis–Menten fitting resulted in K50 values of 2.5–4 μM, indicating that the interaction efficiency of the naturally interacting polypeptides is rather weak.60
Overall, efforts in engineering PKSs via module replacement revealed the importance of linker regions (covalent linkers or matching docking domains) to mediate communication between heterologous modules. The nature of the non-native ACP–KS interaction during chain translocation appears as another factor influencing turnover in chimeric PKSs. A mere module–module exchange employing docking domains to facilitate communication across heterologous modules had limited success. Covalent linking of modules seems to allow the design of catalytically more active PKS chimera, but is done at the expense of modularity.56 Based on the finding that distinct epitopes on the ACP are responsible for KS–AT recognition during chain elongation and chain translocation (Fig. 3A), mix-and-match strategies may be best realized by adapting both the docking domains and chimeric ACP–KS interfaces for productive chain translocation.
 | ||
| Fig. 3 Important interfaces for engineering ACP–KS and AT domain exchanges. (A) Interface of ACP–[KS–AT] according to ref. 40 and 41. Residues of ACP that are responsible for chain translocation in helix 1 are highlighted in blue. Loop 1 (orange) was found as the main determinant in chain elongation. The ACP docks to different positions into the small cleft of the KS–AT fold during chain translocation vs. chain elongation. (B) Sequence alignment of post-AT linkers from partially and fully reducing PKS modules (generated with ClustalW as implemented in JalView 2.9 (ref. 110)). Important segments of the linker sequence are highlighted. Sequences derived from: DEBS – 6-deoxyerythronolide B synthase, BORS – borrelidin synthase, PIKS – pikromycin synthase, EPOS – epothilone synthase, RAPS – rapamycin synthase, RIFS – rifamycin synthase, NIDS – niddamycin synthase, OLEAS – oleandomycin synthase, LIPS – lipomycin synthase, MYCS – mycolactone synthase, and MONENS – monensin synthase. (C) Structure of DEBS KS3–AT3 (PDB 2QO3) with the post-AT linker highlighted. PAL1 (∼N-terminal 35–45 residues) interacts with the AT and the KS–AT linker. The conserved PAL2 sequence (∼13 residues, red) interacts with KS surface residues. The non-conserved part of PAL2 is not solved in structure. Domain coloring as in Fig. 2. (D) Sequence logo of PAL2cons. consensus sequence.111 | ||
3.2. Domain–domain exchanges
Another strategy of engineering PKSs for producing novel compounds is the exchange of individual domains. One of the most common ways of altering the final polyketide product is to insert AT domains with altered specificity to incorporate a different starter or extender unit and thus change the entity at the α-carbon. Altering the stereochemistry of the final product can also result in novel biological activities making KR domain exchanges or reductive loop swaps of particular interest. The other domains (KS, ACP, and DH) were only rarely employed in domain exchanges. In the following, we highlight successful examples of engineering projects and analyze their approaches in the light of available structural information. For a recent comprehensive overview of engineering approaches, we refer to Barajas et al.61AT domain exchanges are a good example for how structural information can guide PKS engineering in defining appropriate exchange sites.
The KR domain has been successfully exchanged in several cases in partly reducing β-modules, particularly for studying the stereochemistry of the reduction reaction that configures the β-hydroxy functionality82,83 and the α-alkyl group in case other units than malonyl-CoA are accepted for elongation.84 These engineering successes can be attributed to the early availability of high resolution 3D-structures,20 which allowed for a better understanding of the KR from a structural point of view. Although KR domains are well described, which has led to a general classification scheme by their reduction and epimerization activity, they have, however, been shown to be inefficient in the simultaneous alteration of alkyl- and hydroxyl-group configurations in a heterologous context.84
More recently, a dimerization element (DE) was identified, which is placed N-terminally to 50% of PKS modules that contain KR as the only processing domain.24,85 The DE turned out to be responsible for the stability of the respective KR domains. As a current rule for KR engineering in partially reducing PKSs (KS–AT–KR–ACP), DE domains of the host PKSs should be preserved in KR exchanges, while a DE–KR unit should replace KR in PKSs not natively carrying a DE structural motif (Fig. 4A).85,86
 | ||
| Fig. 4 Recent examples of PKS engineering by domain swapping in the processing part. (A) KR swapping in a LIPS 1 bimodular (LDD–M1)86 and DEBS trimodular constructs (LDD–M1–M2).85 The success of KR swapping was evaluated by product yields and varied according to co-swapping of DE. The model was assembled from structural models of DEBS KS5-AT5 (PDB 2HG4)18 and SPNS DE3-KR3 (PDB 4IMP).24 A scheme of the domain architecture of the constructs is attached. The AT–DE linking residues (7 amino acids from the PAL2cons. motif to DE in LIPS 1 + TE) are predicted as unstructured by the psipred tool.112 A model of a partly reducing PKS module is shown as inset for clarity. (B) PKS design for adipic acid production by BORS M1. An upstream loading module (not shown), non-covalently interacting with BORS M1, provides succinyl-CoA for condensation and reduction to yield adipic acid.87 Junction sites for domain swapping are indicated in a truncated structural model of MAS-like PKS, and in the attached scheme of the domain architecture (reductive loop swaps 1–3, red dashed line: inactive constructs or constructs with attenuated activity, green solid line: active construct). Note that the AT–DH linking residues (22 amino acids from the PAL2cons. to DH) are predicted as unstructured by the psipred tool.112 Purple dashed line: DH exchange sites (DHN and DHC refer to N- and C-termini), orange dashed line: ACP exchange sites (ACPN and ACPC refer to N- and C-termini). A model of a fully reducing PKS module is shown as inset for clarity. The structures are depicted in cartoon representation (helices shown as cylinders in B). Domain architectures are not shown in correct scale in the attached schemes (e.g. AT–DH linking region enlarged in B). Domains are colored as introduced in Fig. 2. | ||
Swaps of complete processing wings (or reductive loops) were among the earliest achievement of PKS engineering, due to the structural integrity of this unit. A similar strategy has led to the successful production of adipic acid. Here, the processing wing of the borrelidin synthase module 1 (BORS M1), which contains only a KR domain, has been replaced with the fully reducing processing wing of the second module of the spinosyn synthase (SPNS M2). The DH domain turned out to be problematic owing to restricted substrate specificity, but when swapped with the DH-domain of BORS M2 the expected product was produced.87 Not surprisingly, the definition of junction sites for reductive loop insertions underlay the same rules as AT exchanges pointing again at the importance of preserving the KS–PAL2cons. interface (Fig. 4B).
3.3. Non-rational engineering strategies
Besides the aforementioned rational engineering attempts, non-rational engineering can also result in the generation of new polyketides. In nature, homologous recombination is an often used process for the generation of diversity. Based on homologous recombination in S. cerevisiae, chimeras of PIKS M5 and DEBS3 were successfully generated in vivo, of which some had measurable in vitro activity indicative of a stable protein fold.88 Although it was possible to rapidly generate a library of chimeric PKSs, stability of the resulting folds could not be ensured under the experimental conditions. Recently, a similar approach used homologous recombination within the rapamycin synthase (RAPS) to generate several new rapamycin derivatives with altered biological activity.89 Interestingly, the best chimeras revealed junction sites in the KS or AT domain or in the linker upstream of the ACP domain. The approaches use homologous recombination as a tool to mimic evolutionary pressure for receiving chimeric PKSs with sufficient catalytic fitness. Such strategies can identify junction sites that may eventually support rational engineering strategies.4. Roadmap for another 20 years of PKS engineering
Below we highlight several aspects of PKS research that should be considered to come closer to the goal of custom polyketide synthesis by designer PKSs.4.1. Continuing efforts in the structural analysis of PKSs
In recent years, a wealth of structural data at high resolution has given insight into the principles of polyketide biosynthesis.3 There is still the clear need to further analyze PKSs in terms of their structural and conformational dynamic properties with a dedicated focus on understanding module–module interactions in PKS assembly lines. The technological progress in cryo-EM will foster efforts in the characterization of particularly the larger assemblies.90 As a single-particle method, cryo-EM can additionally give insight into conformational dynamics when particles are classified during manual and automated sorting,37,91 or initial electron density maps are analyzed for principle components.92 In addition to the static snapshots provided by cryo-EM or X-ray crystallography, the dynamic character of PKS necessitates the use of spectroscopic methods. These methods can deliver a continuous spectrum of protein conformational dynamics in real-time and add valuable information on the internal dynamics of these domains.93 As conformational motions in proteins are stochastic and synchronization of populations are hardly possible, spectroscopic methods will be particularly powerful when applied at the single-molecule level.4.2. Characterizing selected PKSs in vitro
The access to several PKSs by recombinant production in high yield, high protein quality and by fast purification protocols has been achieved in Escherichia coli and in Streptomyces.94 On the basis of this methodological foundation, selected PKSs have been established as model systems in vitro and characterized to considerable detail to date; i.e. the modPKSs DEBS, PIKS, and SPNS, and the iPKSs PKSA and MSAS.95–99 One needs to further analyze these proteins in quantitative terms in vitro by classical enzymatic methods, as well as to enlarge the toolbox with a set of new enzymes. The carefully selected PKSs can function as testbeds to probe the precise impact of domain swaps and mix-and-match approaches. A tight network of quantitate data on the selected proteins will help understanding structure–function-relationships in PKSs to establish a toolkit of methods and engineering rules that are broadly applicable for the PKS protein family.4.3. Understanding substrate shuttling
Conformational properties of the evolutionarily related mFAS allows to anticipate an overall highly conformationally variable modPKS fold.37,39 The recent X-ray structural study on the MAS-like PKS supports and refines this view, since also revealing substantial conformational variability with the processing wing. The ACP is tethered to the KR domain, which has been identified as the most flexible part of the processing wing.21 Accordingly, in the current perception, substrate shuttling in PKSs is based on the joint mobility of the ACP domains and the PKSs catalytic body. Defining the exact conformational space of ACPs by structural and spectroscopic methods will underpin PKS engineering. For example, in chimeric PKSs, the ACP has to have enough positional variability for translocating the substrates into the downstream KS domain, but should be restricted from accessing the non-cognate, downstream AT to avoid improper loading.Equipped with the necessary conformational freedom, ACP undergoes specific interaction with the catalytic domains. A general problem of any chimeric system is the introduction of non-native interfaces at the junction sites as well as between the cognate ACP and the inserted domain. From the assumption of ACP-mediated substrate shuttling as a finely balanced process, any non-native interfaces between ACP and catalytic domains will lead to a loss of catalytic activity and as such require adaption by surface mutations, assisted by structural information,75,100 and computational methods.101,102
4.4. Redefining module boundaries
Over the last two decades, mix-and-match approaches for PKS engineering were based on the traditional module definition in which the domains are ordered in a KS–AT(–DH–ER–KR)–ACP sequence. Although several studies implied that preservation of native interaction interfaces is crucial with regard to any engineering attempt (e.g. ACP–KS linker,106 ACP–KS interaction52,58), these traditional module boundaries were never challenged as suited junction sites in mix-and-match approaches. Recently, a study on the co-evolution of large PKS assembly lines revealed that the processing domains are more closely related to the downstream than to the upstream KS.107 A unit that may be better exchangeable in mix-and-match approaches than the genetically-encoded, “traditional” module could therefore be of AT(–DH–ER–KR)–ACP–KS domain sequence. From an engineering point of view, an exchangeable unit with such newly defined boundaries may indeed make sense, as it allows to preserve the important interface of the upstream ACP to the downstream KS,58 which was found to be crucial also in the closely related NRPSs,10 as well as in trans-AT PKSs.108 The general applicability of the new module boundaries for PKS engineering remains to be shown.4.5. Controlling vectorial synthesis
In the serial arrangement of modules in PKS assembly lines, reaction progress can occur via re-feeding of the intermediate to the integral KS, leading to repeated condensation and processing as encoded on the integral module (“stuttering”), or the translocation of the intermediate to the KS of the downstream module.1 Engineering of PKS assembly lines requires control of this decision to steer synthesis towards translocation (see Fig. 2B). The means to impose kinetic reaction control include the modulation in kcat/KM, the tuning of electrostatic properties of domain–domain interfaces, and the adjustment of the spatial arrangement of interacting domains.103,104 A recent model study has demonstrated that kinetic engineering can steer synthesis. On the example of the microbial FAS, selected mutations redirected the complex biosynthetic pathways of fatty acid synthesis towards the production of short fatty acids.1054.6. Modeling the PKS reaction networks
In silico modeling of the reaction network underlying polyketide biosynthesis will be necessary to disclose and exploit enzymatic properties of PKSs. Owing to a set of comprehensive enzymatic assays in the labs, selected enzymatic functions have been already experimentally described and many more enzyme kinetic parameters of the catalytic processes will be available during the next years. In silico models need to be implemented using these data to form an knowledgebase, to then, in a computational filtering process, give access to enzyme kinetic properties that are not directly experimentally accessible.109 Once established, reaction networks can be extended to represent effects of engineering, such as the modulated enzymatic constants resulting from the swapped domain and/or changed spatial constraints introduced by binding site design. Computational tools will add a new dimension to the quality of understanding of PKSs that goes beyond a conventional phenomenological analysis, and will eventually inform PKS engineering strategies.5. Conclusions
In this review, we summarize and revise engineering approaches in the light of available structural and functional data. We present several models for PKS architectures and employ sequence and structural analysis to investigate the recent strategies conducted in PKS engineering.There is still a strong demand for a highly detailed structural and biochemical characterization of PKSs especially with improvements in structure determination methods. Inclusion of complementary techniques that provide dynamic information will expand the view on programming PKS biosynthesis. One of the key elements in the rational design of chimeric PKSs is the definition of appropriate junction sites that lead to structurally intact protein folds to guarantee the productive interaction of modules and domains throughout the catalytic cycle. Since many of the important interfaces are still poorly described, we are convinced that a combination of experimental data, evolutionary analysis and in silico modeling will help overcome these shortcomings and pave the way for general applicable engineering rules to PKS systems.
6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
We thank Karthik Paithankar for carefully reading the manuscript. This work was supported by a Lichtenberg grant of the Volkswagen Foundation to M. G. (grant number 85701). Further support was received by the LOEWE program (Landes-Offensive zur Entwicklung wissenschaftlich-ökonomischer Exzellenz) of the state of Hesse conducted within the framework of the MegaSyn Research Cluster.8. References
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- K. J. Weissman, Nat. Chem. Biol., 2015, 11, 660–670 CrossRef PubMed.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- R. W. Broadhurst, D. Nietlispach, M. P. Wheatcroft, P. F. Leadlay and K. J. Weissman, Chem. Biol., 2003, 10, 723–731 CrossRef PubMed.
- J. Dorival, T. Annaval, F. Risser, S. Collin, P. Roblin, C. Jacob, A. Gruez, B. Chagot and K. J. Weissman, J. Am. Chem. Soc., 2016, 138, 4155–4167 CrossRef PubMed.
- P. F. Leadlay, Curr. Opin. Chem. Biol., 1997, 1, 162–168 CrossRef PubMed.
- C. Khosla and R. J. X. Zawada, Trends Biotechnol., 1996, 14, 335–341 CrossRef PubMed.
- R. McDaniel, M. Welch and C. R. Hutchinson, Chem. Rev., 2005, 105, 543–558 CrossRef PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2017, 10, 275–281 CrossRef PubMed.
- A. C. Murphy, H. Hong, S. Vance, R. W. Broadhurst and P. F. Leadlay, Chem. Commun., 2016, 52, 8373–8376 RSC.
- F. Wang, Y. Wang, J. Ji, Z. Zhou, J. Yu, H. Zhu, Z. Su, L. Zhang and J. Zheng, ACS Chem. Biol., 2015, 10, 1017–1025 CrossRef PubMed.
- L. Zhang, J. Ji, M. Yuan, Y. Feng, L. Wang, Z. Deng, L. Bai and J. Zheng, ACS Chem. Biol., 2018, 13, 871–875 CrossRef PubMed.
- H. Jenke-Kodama, A. Sandmann, R. Müller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 2027–2039 CrossRef PubMed.
- S. Smith and S.-C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- T. Maier, M. Leibundgut, D. Boehringer and N. Ban, Q. Rev. Biophys., 2010, 43, 373–422 CrossRef PubMed.
- M. Grininger, Curr. Opin. Struct. Biol., 2014, 25, 49–56 CrossRef PubMed.
- Y. Tang, C. Kim, I. I. Mathews, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11124–11129 CrossRef PubMed.
- Y. Tang, A. Y. Chen, C.-Y. Kim, D. E. Cane and C. Khosla, Chem. Biol., 2007, 14, 931–943 CrossRef PubMed.
- A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 CrossRef PubMed.
- D. A. Herbst, R. P. Jakob, F. Zähringer and T. Maier, Nature, 2016, 531, 533–537 CrossRef PubMed.
- A. Keatinge-Clay, J. Mol. Biol., 2008, 384, 941–953 CrossRef PubMed.
- J. Zheng, D. C. Gay, B. Demeler, M. A. White and A. T. Keatinge-Clay, Nat. Chem. Biol., 2012, 8, 615–621 CrossRef PubMed.
- J. Zheng, C. D. Fage, B. Demeler, D. W. Hoffman and A. T. Keatinge-Clay, ACS Chem. Biol., 2013, 8, 1263–1270 CrossRef PubMed.
- D. Khare, W. A. Hale, A. Tripathi, L. Gu, D. H. Sherman, W. H. Gerwick, K. Håkansson and J. L. Smith, Structure, 2015, 23, 2213–2223 CrossRef PubMed.
- A. T. Keatinge-Clay, Cell Chem. Biol., 2016, 23, 540–542 CrossRef PubMed.
- S. Dutta, J. R. Whicher, D. a. Hansen, W. a. Hale, J. a. Chemler, G. R. Congdon, A. R. H. Narayan, K. Håkansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 512–517 CrossRef PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef PubMed.
- G. Pappenberger, J. Benz, B. Gsell, M. Hennig, A. Ruf, M. Stihle, R. Thoma and M. G. Rudolph, J. Mol. Biol., 2010, 397, 508–519 CrossRef PubMed.
- A. Rittner, K. S. Paithankar, K. V. Huu and M. Grininger, ACS Chem. Biol., 2018, 13, 723–732 CrossRef PubMed.
- A. Rittner and M. Grininger, ChemBioChem, 2014, 15, 2489–2493 CrossRef PubMed.
- K. J. Weissman, Nat. Prod. Rep., 2015, 32, 436–453 RSC.
- A. L. Edwards, T. Matsui, T. M. Weiss and C. Khosla, J. Mol. Biol., 2014, 426, 2229–2245 CrossRef PubMed.
- C. D. Richter, D. Nietlispach, R. W. Broadhurst and K. J. Weissman, Nat. Chem. Biol., 2008, 4, 75–81 CrossRef PubMed.
- T. J. Buchholz, T. W. Geders, F. E. Bartley III, K. a. Reynolds, J. L. Smith and D. H. Sherman, ACS Chem. Biol., 2009, 4, 41–52 CrossRef PubMed.
- J. R. Whicher, S. S. Smaga, D. a. Hansen, W. C. Brown, W. H. Gerwick, D. H. Sherman and J. L. Smith, Chem. Biol., 2013, 20, 1340–1351 CrossRef PubMed.
- E. J. Brignole, S. Smith and F. J. Asturias, Nat. Struct. Mol. Biol., 2009, 16, 190–197 CrossRef PubMed.
- A. K. Joshi, V. S. Rangan, A. Witkowski and S. Smith, Chem. Biol., 2003, 10, 169–173 CrossRef PubMed.
- F. M. C. Benning, Y. Sakiyama, A. Mazur, H. S. T. Bukhari, R. Y. H. Lim and T. Maier, ACS Nano, 2017, 11, 10852–10859 CrossRef PubMed.
- S. Kapur, A. Y. Chen, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22066–22071 CrossRef PubMed.
- S. Kapur, B. Lowry, S. Yuzawa, S. Kenthirapalan, A. Y. Chen, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4110–4115 CrossRef PubMed.
- H. G. Menzella, R. Reid, J. R. Carney, S. S. Chandran, S. J. Reisinger, K. G. Patel, D. A. Hopwood and D. V. Santi, Nat. Biotechnol., 2005, 23, 1171–1176 CrossRef PubMed.
- C. Khosla, D. Herschlag, D. E. Cane and C. T. Walsh, Biochemistry, 2014, 53, 2875–2883 CrossRef PubMed.
- B. Lowry, X. Li, T. Robbins, D. E. Cane and C. Khosla, ACS Cent. Sci., 2016, 2, 14–20 CrossRef PubMed.
- G. Etienne, W. Malaga, F. Laval, A. Lemassu, C. Guilhot and M. Daffé, J. Bacteriol., 2009, 191, 2613–2621 CrossRef PubMed.
- C. Olano, B. Wilkinson, C. Sánchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Braña, C. J. Martin, M. Oliynyk, C. Méndez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87–97 Search PubMed.
- B. Busch, N. Ueberschaar, S. Behnken, Y. Sugimoto, M. Werneburg, N. Traitcheva, J. He and C. Hertweck, Angew. Chem., Int. Ed., 2013, 52, 5285–5289 CrossRef PubMed.
- B. J. Beck, C. C. Aldrich, R. A. Fecik, K. A. Reynolds and D. H. Sherman, J. Am. Chem. Soc., 2003, 125, 4682–4683 CrossRef PubMed.
- S. Donadio, J. B. McAlpine, P. J. Sheldon, M. Jackson and L. Katz, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 7119–7123 CrossRef.
- L. Katz and S. Donadio, Annu. Rev. Microbiol., 1993, 47, 875–912 CrossRef PubMed.
- R. S. Gokhale, S. Y. Tsuji, D. E. Cane and C. Khosla, Science, 1999, 284, 482–485 CrossRef PubMed.
- A. Ranganathan, M. Timoney, M. Bycroft, J. Cortés, I. P. Thomas, B. Wilkinson, L. Kellenberger, U. Hanefeld, I. S. Galloway, J. Staunton and P. F. Leadlay, Chem. Biol., 1999, 6, 731–741 CrossRef PubMed.
- L. Tang, H. Fu and R. McDaniel, Chem. Biol., 2000, 7, 77–84 CrossRef PubMed.
- C. J. Rowe, I. U. Böhm, I. P. Thomas, B. Wilkinson, B. a. M. Rudd, G. Foster, A. P. Blackaby, P. J. Sidebottom, Y. Roddis, A. D. Buss, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 475–485 CrossRef PubMed.
- H. G. Menzella, J. R. Carney and D. V. Santi, Chem. Biol., 2007, 14, 143–151 CrossRef PubMed.
- N. Wu, S. Y. Tsuji, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2001, 123, 6465–6474 CrossRef PubMed.
- N. Wu, D. E. Cane and C. Khosla, Biochemistry, 2002, 41, 5056–5066 CrossRef PubMed.
- M. Klaus, M. P. Ostrowski, J. Austerjost, T. Robbins, B. Lowry, D. E. Cane and C. Khosla, J. Biol. Chem., 2016, 291, 16404–16415 CrossRef PubMed.
- S. Y. Tsuji, D. E. Cane and C. Khosla, Biochemistry, 2001, 40, 2326–2331 CrossRef PubMed.
- B. Lowry, T. Robbins, C. Weng, R. V. O. Brien, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2013, 135, 16809–16812 CrossRef PubMed.
- J. F. Barajas, J. M. Blake-Hedges, C. B. Bailey, S. Curran and J. D. Keasling, Synth. Syst. Biol., 2017, 2, 147–166 Search PubMed.
- A. F. Marsden, B. Wilkinson, J. Cortés, N. J. Dunster, J. Staunton and P. F. Leadlay, Science, 1998, 279, 199–202 CrossRef PubMed.
- S. Kuhstoss, M. Huber, J. R. Turner, J. W. Paschal and R. N. Rao, Gene, 1996, 183, 231–236 CrossRef PubMed.
- P. F. Long, C. J. Wilkinson, C. P. Bisang, J. Cortés, N. Dunster, M. Oliynyk, E. McCormick, H. McArthur, C. Mendez, J. a. Salas, J. Staunton and P. F. Leadlay, Mol. Microbiol., 2002, 43, 1215–1225 CrossRef PubMed.
- B. J. Dunn, D. E. Cane and C. Khosla, Biochemistry, 2013, 52, 1839–1841 CrossRef PubMed.
- M. Oliynyk, M. J. Brown, J. Cortés, J. Staunton and P. F. Leadlay, Chem. Biol., 1996, 3, 833–839 CrossRef PubMed.
- X. Ruan, A. Pereda, D. L. Stassi, D. Zeidner, R. G. Summers, M. Jackson, A. Shivakumar, S. Kakavas, M. J. Staver, S. Donadio and L. Katz, J. Bacteriol., 1997, 179, 6416–6425 CrossRef PubMed.
- D. L. Stassi, S. J. Kakavas, K. a. Reynolds, G. Gunawardana, S. Swanson, D. Zeidner, M. Jackson, H. Liu, a. Buko and L. Katz, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7305–7309 CrossRef.
- J. Lau, H. Fu, D. E. Cane and C. Khosla, Biochemistry, 1999, 38, 1643–1651 CrossRef PubMed.
- R. McDaniel, a. Thamchaipenet, C. Gustafsson, H. Fu, M. Betlach and G. Ashley, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1846–1851 CrossRef.
- C. Y. Kim, V. Y. Alekseyev, A. Y. Chen, Y. Tang, D. E. Cane and C. Khosla, Biochemistry, 2004, 43, 13892–13898 CrossRef PubMed.
- A. Y. Chen, D. E. Cane and C. Khosla, Chem. Biol., 2007, 14, 784–792 CrossRef PubMed.
- S. Yuzawa, S. Kapur, D. E. Cane and C. Khosla, Biochemistry, 2012, 51, 3708–3710 CrossRef PubMed.
- S. Yuzawa, K. Deng, G. Wang, E. E. K. Baidoo, T. R. Northen, P. D. Adams, L. Katz and J. D. Keasling, ACS Synth. Biol., 2017, 6, 139–147 CrossRef PubMed.
- A. Miyanaga, S. Iwasawa, Y. Shinohara, F. Kudo and T. Eguchi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1802–1807 CrossRef PubMed.
- F. T. Wong, A. Y. Chen, D. E. Cane and C. Khosla, Biochemistry, 2010, 49, 95–102 CrossRef PubMed.
- F. T. Wong, X. Jin, I. I. Mathews, D. E. Cane and C. Khosla, Biochemistry, 2011, 50, 6539–6548 CrossRef PubMed.
- C. W. Liew, M. Nilsson, M. W. Chen, H. Sun, T. Cornvik, Z. X. Liang and J. Lescar, J. Biol. Chem., 2012, 287, 23203–23215 CrossRef PubMed.
- Z. Ye, E. M. Musiol, T. Weber and G. J. Williams, Chem. Biol., 2014, 21, 636–646 CrossRef PubMed.
- X. Xie, A. Garg, A. T. Keatinge-Clay, C. Khosla and D. E. Cane, Biochemistry, 2016, 55, 1179–1186 CrossRef PubMed.
- M. P. Ostrowski, D. E. Cane and C. Khosla, J. Antibiot., 2016, 69, 507–510 CrossRef PubMed.
- D. Bedford, J. R. Jacobsen, G. Luo, D. E. Cane and C. Khosla, Chem. Biol., 1996, 3, 827–831 CrossRef PubMed.
- L. Kellenberger, I. S. Galloway, G. Sauter, G. Böhm, U. Hanefeld, J. Cortés, J. Staunton and P. F. Leadlay, ChemBioChem, 2008, 9, 2740–2749 CrossRef PubMed.
- T. Annaval, C. Paris, P. F. Leadlay, C. Jacob and K. J. Weissman, ChemBioChem, 2015, 16, 1357–1364 CrossRef PubMed.
- J. Zheng, S. K. Piasecki and A. T. Keatinge-Clay, ACS Chem. Biol., 2013, 8, 1964–1971 CrossRef PubMed.
- C. H. Eng, S. Yuzawa, G. Wang, E. E. K. Baidoo, L. Katz and J. D. Keasling, Biochemistry, 2016, 55, 1677–1680 CrossRef PubMed.
- A. Hagen, S. Poust, T. De Rond, J. L. Fortman, L. Katz, C. J. Petzold and J. D. Keasling, ACS Synth. Biol., 2016, 5, 21–27 CrossRef PubMed.
- J. A. Chemler, A. Tripathi, D. A. Hansen, M. O. Neil-johnson, R. B. Williams, C. Starks, S. R. Park and D. H. Sherman, J. Am. Chem. Soc., 2015, 137, 10603–10609 CrossRef PubMed.
- A. Wlodek, S. G. Kendrew, N. J. Coates, A. Hold, J. Pogwizd, S. Rudder, L. S. Sheehan, S. J. Higginbotham, A. E. Stanley-Smith, T. Warneck, M. Nur-E-Alam, M. Radzom, C. J. Martin, L. Overvoorde, M. Samborskyy, S. Alt, D. Heine, G. T. Carter, E. I. Graziani, F. E. Koehn, L. McDonald, A. Alanine, R. M. Rodríguez Sarmiento, S. K. Chao, H. Ratni, L. Steward, I. H. Norville, M. Sarkar-Tyson, S. J. Moss, P. F. Leadlay, B. Wilkinson and M. A. Gregory, Nat. Commun., 2017, 8, 1206 CrossRef PubMed.
- W. Kühlbrandt, Science, 2014, 343, 1443–1444 CrossRef PubMed.
- J. R. Whicher, S. Dutta, D. a. Hansen, W. a. Hale, J. a. Chemler, A. M. Dosey, A. R. H. Narayan, K. Håkansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef PubMed.
- L. Ciccarelli, S. R. Connell, M. Enderle, D. J. Mills, J. Vonck and M. Grininger, Structure, 2013, 21, 1251–1257 CrossRef PubMed.
- J. Alfermann, X. Sun, F. Mayerthaler, T. E. Morrell, E. Dehling, G. Volkmann, T. Komatsuzaki, H. Yang and H. D. Mootz, Nat. Chem. Biol., 2017, 13, 1009–1015 CrossRef PubMed.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef PubMed.
- C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef PubMed.
- J. D. Kittendorf and D. H. Sherman, Bioorg. Med. Chem., 2009, 17, 2137–2146 CrossRef PubMed.
- J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243–246 CrossRef PubMed.
- T. Moriguchi, Y. Kezuka, T. Nonaka, Y. Ebizuka and I. Fujii, J. Biol. Chem., 2010, 285, 15637–15643 CrossRef PubMed.
- P. Dimroth, E. Ringelmann and F. Lynen, Eur. J. Biochem., 1976, 68, 591–596 CrossRef PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. L. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S.-C. Tsai and M. D. Burkart, Nature, 2014, 505, 427–431 CrossRef PubMed.
- M. E. Johnson and G. Hummer, PLoS Comput. Biol., 2013, 9, e1003065 CrossRef PubMed.
- M. F. Viegas, R. P. P. Neves, M. J. Ramos and P. A. Fernandes, J. Phys. Chem. B, 2018, 122, 77–85 CrossRef PubMed.
- M. Castellana, M. Z. Wilson, Y. Xu, P. Joshi, I. M. Cristea, J. D. Rabinowitz, Z. Gitai and N. S. Wingreen, Nat. Biotechnol., 2014, 32, 1011–1018 CrossRef PubMed.
- C. Anselmi, M. Grininger, P. Gipson and J. D. Faraldo-Goěmez, J. Am. Chem. Soc., 2010, 132, 12357–12364 CrossRef PubMed.
- J. Gajewski, F. Buelens, S. Serdjukow, M. Janßen, N. Cortina, H. Grubmüller and M. Grininger, Nat. Chem. Biol., 2017, 13, 363–365 CrossRef PubMed.
- R. S. Gokhale, D. Hunziker, D. E. Cane and C. Khosla, Chem. Biol., 1999, 6, 117–125 CrossRef PubMed.
- L. Zhang, T. Hashimoto, B. Qin, J. Hashimoto, I. Kozone, T. Kawahara, M. Okada, T. Awakawa, T. Ito, Y. Asakawa, M. Ueki, S. Takahashi, H. Osada, T. Wakimoto, H. Ikeda, K. Shin-ya and I. Abe, Angew. Chem., Int. Ed., 2017, 56, 1740–1745 CrossRef PubMed.
- D. A. Vander Wood and A. T. Keatinge-Clay, Proteins, 2018, 1–12 Search PubMed.
- D. T. Gillespie, J. Phys. Chem., 1977, 81, 2340–2361 CrossRef.
- A. M. Waterhouse, J. B. Procter, D. M. A. Martin, M. Clamp and G. J. Barton, Bioinformatics, 2009, 25, 1189–1191 CrossRef PubMed.
- G. Crooks, G. Hon, J. Chandonia and S. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef PubMed.
- D. T. Jones, J. Mol. Biol., 1999, 292, 195–202 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |