
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe architectures of iterative type I PKS and FAS
Dominik A.
Herbst
 a,
Craig A.
Townsend
b and
Timm
Maier
*a
a,
Craig A.
Townsend
b and
Timm
Maier
*a
aDepartment Biozentrum, University of Basel, Klingelbergstrasse 50/70, 4056 Basel, Switzerland. E-mail: timm.maier@unibas.ch
bDepartment of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 23rd August 2018
Abstract
Covering: up to mid of 2018
Type I fatty acid synthases (FASs) are giant multienzymes catalyzing all steps of the biosynthesis of fatty acids from acetyl- and malonyl-CoA by iterative precursor extension. Two strikingly different architectures of FAS evolved in yeast (as well as in other fungi and some bacteria) and metazoans. Yeast-type FAS (yFAS) assembles into a barrel-shaped structure of more than 2 MDa molecular weight. Catalytic domains of yFAS are embedded in an extensive scaffolding matrix and arranged around two enclosed reaction chambers. Metazoan FAS (mFAS) is a 540 kDa X-shaped dimer, with lateral reaction clefts, minimal scaffolding and pronounced conformational variability. All naturally occurring yFAS are strictly specialized for the production of saturated fatty acids. The yFAS architecture is not used for the biosynthesis of any other secondary metabolite. On the contrary, mFAS is related at the domain organization level to major classes of polyketide synthases (PKSs). PKSs produce a variety of complex and potent secondary metabolites; they either act iteratively (iPKS), or are linked via directed substrate transfer into modular assembly lines (modPKSs). Here, we review the architectures of yFAS, mFAS, and iPKSs. We rationalize the evolution of the yFAS assembly, and provide examples for re-engineering of yFAS. Recent studies have provided novel insights into the organization of iPKS. A hybrid crystallographic model of a mycocerosic acid synthase-like Pks5 yielded a comprehensive visualization of the organization and dynamics of fully-reducing iPKS. Deconstruction experiments, structural and functional studies of specialized enzymatic domains, such as the product template (PT) and the starter-unit acyltransferase (SAT) domain have revealed functional principles of non-reducing iterative PKS (NR-PKSs). Most recently, a six-domain loading region of an NR-PKS has been visualized at high-resolution together with cryo-EM studies of a trapped loading intermediate. Altogether, these data reveal the related, yet divergent architectures of mFAS, iPKS and also modPKSs. The new insights highlight extensive dynamics, and conformational coupling as key features of mFAS and iPKS and are an important step towards collection of a comprehensive series of snapshots of PKS action.
 Dominik A. Herbst | Dominik A. Herbst studied biochemistry in Tübingen, Germany, and obtained his diploma in biochemistry in 2012, for studies on the interaction of MbtH-like proteins with nonribosomal peptide synthetases. Afterwards he joined the group of Timm Maier at the Biozentrum in Basel, Switzerland, and obtained his PhD in structural biology for his work on the functional architectures of polyketide synthases in 2017. Until July 2018 he continued his research in Basel and recently joined the group of Eva Nogales as postdoctoral fellow at the University of California in Berkeley. |
 Craig A. Townsend | Craig A. Townsend is the Alsoph H. Corwin Professor of Chemistry at The Johns Hopkins University. He holds joint appointments in the Departments of Biology and Biophysics, and has close associations and collaborations in the School of Medicine. The research of his group is broadly in bioorganic chemistry, with specific interests in natural product biosynthesis, the enzymology and molecular biology of secondary metabolism, structures of complex biosynthetic enzymes, biomimetic synthesis and the engineering of biosynthetic enzymes, and the application of new beta-lactams against tuberculosis, other mycobacterial infections and Gram-negative pathogens. |
 Timm Maier | Timm Maier studied biochemistry at the University of Tübingen, Germany, and completed his doctorate in structural biology with Wolfram Saenger at Freie Universität Berlin, Germany, in 2003. He then joined the lab of Nenad Ban as Postdoc at ETH Zurich, where he was promoted to a team leader and lecturer position in 2006. In 2011, Timm Maier moved to Biozentrum, University of Basel, Switzerland, as tenure track assistant professor in Structural Biology and is associate professor at Biozentrum since 2016. He and his team are best known for their studies on the architecture of giant multienzymes in primary and secondary metabolism, in particular fatty acid and polyketide synthases, as well as on metabolic regulation and mTOR complexes. |
I. Introduction
Fatty acids are central molecules for life. They serve as core components of membrane lipids and act as energy storage compounds in the form of triacylglycerols. Fatty acids further act as signalling molecules and are attached to proteins to serve as membrane anchors. Biological synthesis of fatty acids is based on variations of a conserved pathway for the iterative elongation of carbohydrate-derived precursors by two-carbon units provided by coenzyme A (CoA)-activated carboxylic acids. In the late 1950s and early 1960s the principal mechanism of fatty acid biosynthesis was established (reviewed in ref. 1): acetyl-CoA is the initial substrate for fatty acid biosynthesis. It is iteratively elongated in multiple reaction steps by two-carbon units from malonyl-CoA, which is obtained by energy-dependent carboxylation of acetyl-CoA. The growing intermediate is tethered to a small acyl carrier protein (ACP) by covalent thioester linkage to the terminal thiol group of a phosphopantetheine (Ppant) cofactor bound to a conserved ACP serine. In the late 1960s, individual enzymes had been isolated from bacteria catalysing all reactions of fatty acid biosynthesis. In the 1970s, it was discovered that eukaryotic fatty acid biosynthesis is carried out by large, multifunctional proteins, which had been termed type I fatty acid synthases (FASs) in contrast to bacterial dissociated type II FASs. Sequences of eukaryotic FASs finally demonstrated in the late 1980s that yeast FAS and metazoan FAS (mFAS) have a completely different domain organisation.Sequence analysis of the gene cluster for the biosynthesis of the antibiotic erythromycin in Saccharopolyspora erythraea demonstrated in 1990 that the enzymatic domains and their linear organization in microbial polyketide synthases (PKSs), which produce complex and potent secondary metabolites, is related to those of mFASs.2 Here, we review the structural organization of the iterative yeast-type (yFASs) and mFASs, together with those of iterative PKSs (iPKSs), which also catalyse multiple rounds of precursor elongation by the same set of enzymatic domains in one multienzyme. The architecture of modular PKSs (modPKSs), which hand-over intermediates after a single round of elongation to a downstream module and include the erythromycin biosynthesis system will be reviewed elsewhere in this issue.
II. Yeast-type fatty acid synthase (yFAS)
In yFAS, the acetyl moiety of acetyl-CoA is loaded by a specific acetyltransferase (AT) onto ACP prior to transfer to the active site cysteine of the condensing ketosynthase (KS) (Fig. 1A). The malonyl-moiety of malonyl-CoA is transferred by the malonyl-palmitoyl-transferase (MPT) to ACP. KS catalyses the decarboxylative Claisen condensation of the acetyl and malonyl groups to yield a β-ketoacyl intermediate bound to the ACP. This intermediate is modified by the sequential action of an NADPH-dependent ketoreductase (KR), water elimination by a dehydratase (DH) and reduction by an FMN-containing NADPH-dependent enoylreductase (ER) to yield a fully saturated acyl intermediate elongated by two carbon atoms. This intermediate serves as the initial substrate for another round of elongation. After seven or eight iterative elongation cycles, the resulting acyl-group is transferred back to CoA by MPT to produce the products palmitoyl- and stearoyl-CoA, respectively; for an extended discussion of individual enzymatic functions see ref. 1, 3 and 4.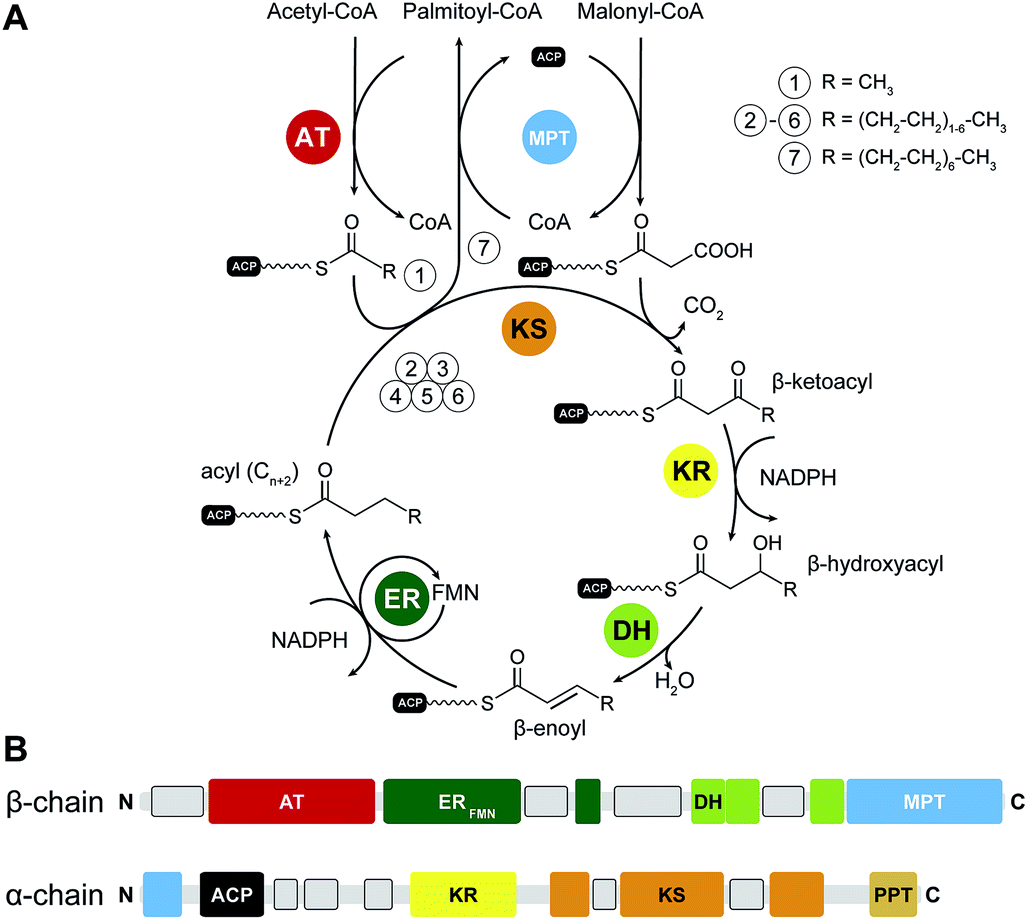 | ||
| Fig. 1 Catalytic reaction cycle and domain organization of yFAS. (A) Fatty acid biosynthesis is initiated by loading acetyl-CoA onto the ACP, which is subsequently elongated by the KS in iterative cycles of decarboxylative condensations using the extender unit malonyl-CoA. Fully saturated acyl products are generated by sequential reduction and water elimination (KR, DH, ER) at the β-carbon position in each cycle and serve as substrate for further elongation reactions by the KS. While the initial substrate is loaded by the AT, the MPT has a dual function for extender-unit loading and unloading the final product. (B) Linear domain organization of yeast FAS. Linker regions with tertiary structure are indicated as grey boxes. KS: ketosynthase, AT: acetyltransferase, MPT: malonyl-palmitoyl transferase, DH: dehydratase, ER: enoylreductase, KR: ketoreductase, ACP: acyl-carrier protein, PPT: phosphopantetheinyltransferase. | ||
II.1 The architecture of yFAS
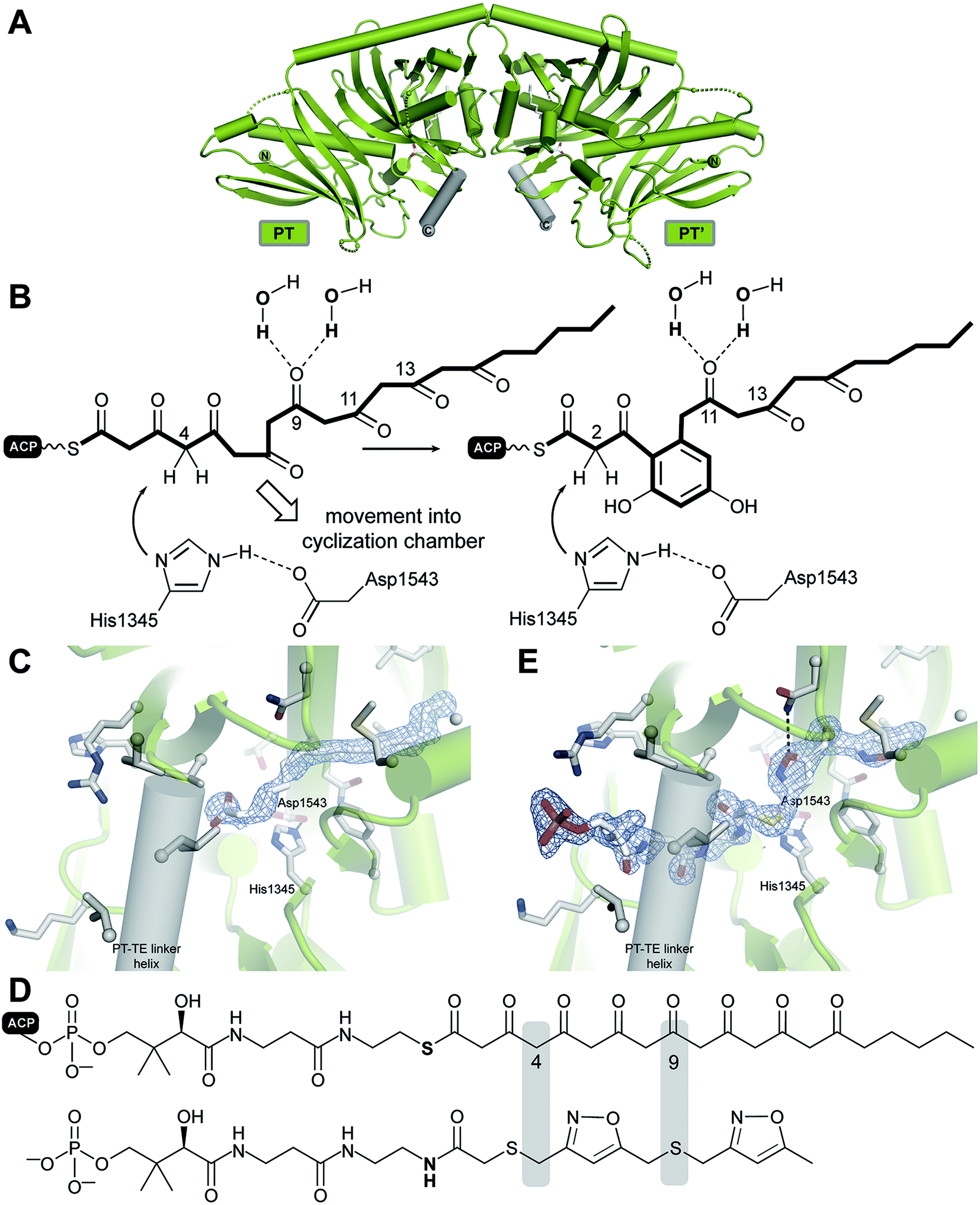
Yeast FAS was characterized at low resolution beginning in the 1960s by electron microscopy (EM) and solution scattering techniques, but detailed insights into the structure of yeast and other fungal FASs were only obtained between 2006 and 2008 by X-ray crystallography: the structure of Thermomyces lanuginosus FAS was first published at 5 Å resolution,5 and then at 3.1 Å resolution,6 simultaneously with a structure of yeast FAS at the same resolution.7 Importantly, in the yeast FAS structure the ACP was fortuitously trapped under the crystallization conditions at the KS active site. A concurrent 4 Å resolution crystal structure8 misinterpreted chain topology in the MPT region, but provided the localization of the integral phosphopantetheinyltransferase (PPT) domain, which catalyses the Ppant-ylation of the integral ACP domain of yFAS, in agreement with a subsequent 4 Å crystal structure of yeast FAS inhibited by the covalent KS inhibitor cerulenin.9 Further insights into PPT function were obtained by the structure determination and functional characterization of the excised PPT domain.10 Finally, a cryo-EM reconstruction at 5.9 Å resolution provided a solution structure of yeast FAS and an initial suggestion for the localization of ACP in various steps of the catalytic cycle.11 In the following paragraph, we will discuss the architecture of yeast FAS itself based on the above, before turning to variations and evolution of yFAS.The catalytic domains of yeast FAS are distributed over two protein chains (Fig. 1B). The FAS1 gene encodes the β-chain, which comprises (in sequence order) the AT, ER, DH and the majority of the split MPT domain. The α-chain is encoded by the downstream FAS2 gene, and contains the smaller part of the MPT domain, and the ACP, KR, KS and PPT domains. The two chains assemble into a heterododecameric 2.6 MDa α6β6 complex with D3 symmetry and a barrel shape with overall dimensions of 250 Å × 270 Å. Conserved regions of enzymatic domains in fungal FAS provide only roughly 50% of the total polypeptide length. The other half of the sequence is composed of non-catalytic rigid scaffolding elements and a few flexible linking regions (Fig. 2A and B).
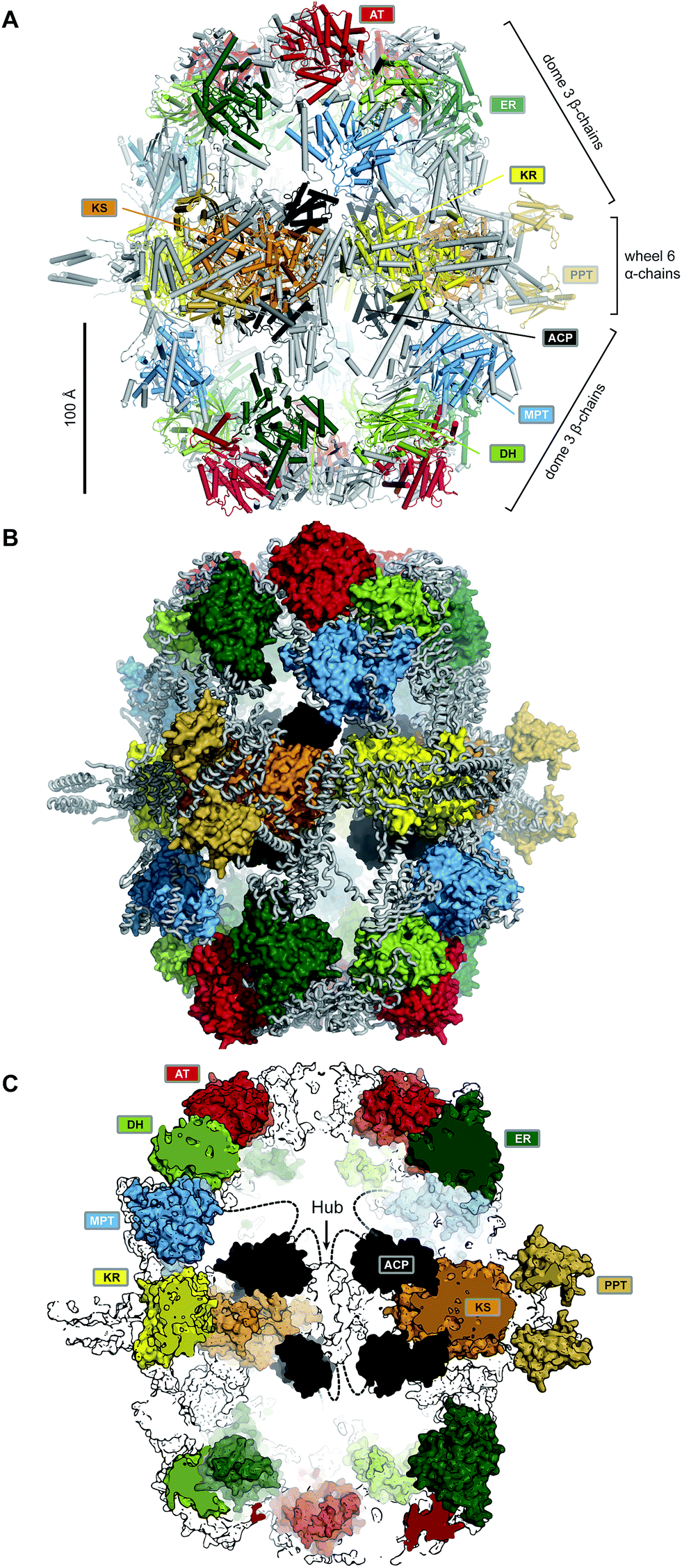 | ||
| Fig. 2 The barrel-like architecture of yeast FAS. (A) Cartoon representation of the heterododecameric yeast FAS shows a central wheel of six α-chains and two domes of three β-chains on each side. (B) Approx. 50% of the ∼2.6 MDa architecture consist of scaffolding linkers (grey loops) that embed 42 catalytic domains and six ACPs (colored surfaces). (C) Cut-open view of the barrel. Each side of the barrel contains two reaction chambers with three double-tethered ACPs, which are organized by a central hub. Domain names correspond to Fig. 1. | ||
Six α-chains form a wheel-like structure with D3 symmetry, with a central helical hub, connected via helical spokes to the rim. The dimeric KS and KR domains together with linking elements form the tire. The ACP is double tethered; a 47 amino acid (aa) long, alanine and proline rich linker connects its N-terminus to the MPT fragment, and the ACP C-terminus is bridged to a scaffolding region preceding the KR by a 27 aa long, proline-free linker. The PPT domain is connected to the outside of the tire, without access to ACP and in an inactive monomeric form, indicating that Ppant-ylation of ACP occurs prior to complete yeast FAS assembly catalysed by a PPT oligomer formed between different α-chains.10
Two domes with C3 symmetry formed by three β-chains cap the α-chain wheel on either side. The β-chains include the structurally and functionally related MPT and AT acyltransferases, and a double-hotdog fold DH extended by another hot-dog-fold linker element, as well as an FMN-containing TIM-barrel ER. In contrast to the KR and KS domains, all domains of the β-chain are monomeric. The β-chain domes define two large reaction chambers (Fig. 2C) at either face of the wheel, each containing three full sets of enzymatic domains required for fatty acid biosynthesis as well as three mobile ACP domains, which are double tethered to the central hub and the chamber walls. The openings in the chamber wall are small, presumably restricting access of other proteins to the reaction chambers.
The ACP domain of yeast FAS is extended relative to the canonical bacterial four-helix bundle ACP by a second four-helix bundle. Based on NMR studies of the yeast FAS ACP domain, the canonical four-helix bundle, and not the additional subdomain of ACP, interacts with covalently attached acyl chains, however, only very weakly with about 15% sequestration of acyl chains to ACP.12 In the yeast FAS crystal structure, ACP binds symmetrically to both active sites of the KS dimer. The canonical four-helix bundle of ACP primarily interacts with the KS domain, while the extension bundle mediates further interactions to the non-catalytic scaffolding spoke region. The environment for substrate shuttling by restricted ACP diffusion appears to be well adapted to the catalytic cycle of fungal FAS. KS, KR, DH, and ER are located on a closed path. MPT, which is required in each elongation cycle and for product release is located closer to the condensing KS than AT, which is only required in the starting cycle. Chain-length determination is mediated by kinetic competition between substrates and intermediates for MPT and possibly also for KS.3,13 The product spectrum depends on the substrate conditions; under physiological conditions palmitoyl-CoA and stearoyl-CoA are produced in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.
:3 ratio.
II.2 Bacterial and secondary-metabolism yFAS
Yeast FAS is the most thoroughly studied member of the yFAS family. Nevertheless, as early as 1969, multienzyme yFAS had been discovered as co-occurring with dissociated type II FAS systems in Mycobacterium phlei.14 Based on the availability of genomic sequences, yFAS systems have now been discovered as components for fatty acid biosynthesis15–18 across three genera of the phylum Actinobacteria, the Corynebacteria, Mycobacteria and Nocardia, also known as the CMN group of Actinobacteria. In Mycobacteria, yFAS contributes to providing precursors for the biosynthesis of complex cell wall lipids. A secondary metabolic yFAS specialized in the production of hexanoic acid (HexS) has been found in the filamentous fungus Aspergillus parasiticus.19 Its hexanoic acid product is directly transferred as a starter unit to an iterative non-reducing PKS (NR-PKS) for the synthesis of aflatoxin B1.20 Due to the inherent difficulties in aligning the less-conserved non-enzymatic sequence regions, understanding of the structural and functional differences between various bacterial and fungal yFAS has been linked closely to experimental structure determination.Structures of mycobacterial yFAS have been resolved via cryo-EM at resolutions of 7.5 Å under application of D3 symmetry21 and in three distinct conformational states without applying symmetry at 17.5 Å to 27.0 Å resolution.22 Interpretation of the low-resolution maps was achieved using models derived on the basis of the yeast FAS structure. The central wheel region is similar between mycobacterial and yeast FAS. The dome regions are slightly smaller and have larger lateral or top openings due to the absence of some of the non-catalytic regions. Such reduced extent of scaffolding is likely linked to the observed higher structural variability of yFAS from Mycobacteria. Conformational changes may well be coupled to substrate turnover.22
II.3 The evolution of yFAS
Supported by structural insights, the evolution from early bacterial yFASs to yeast FAS can be traced along gene splitting, domain deletion and domain duplication events (Fig. 3A).17,18 A primordial bacterial yFAS was encoded by a contiguous gene encompassing the enzymatic domains for fatty acid biosynthesis as in the yeast FAS1 and FAS2 genes, but lacking several scaffolding regions. In bacterial yFASs, the PPT domain is not part of the contiguous single gene, but it is encoded as a separate protein by the directly adjacent downstream acpS gene in Mycobacteria. The considerably more accessible reaction chambers of bacterial yFAS may provide access of an external PPT or other proteins directly interacting with ACP-bound substrates, resembling the direct substrate transfer suggested for HexS.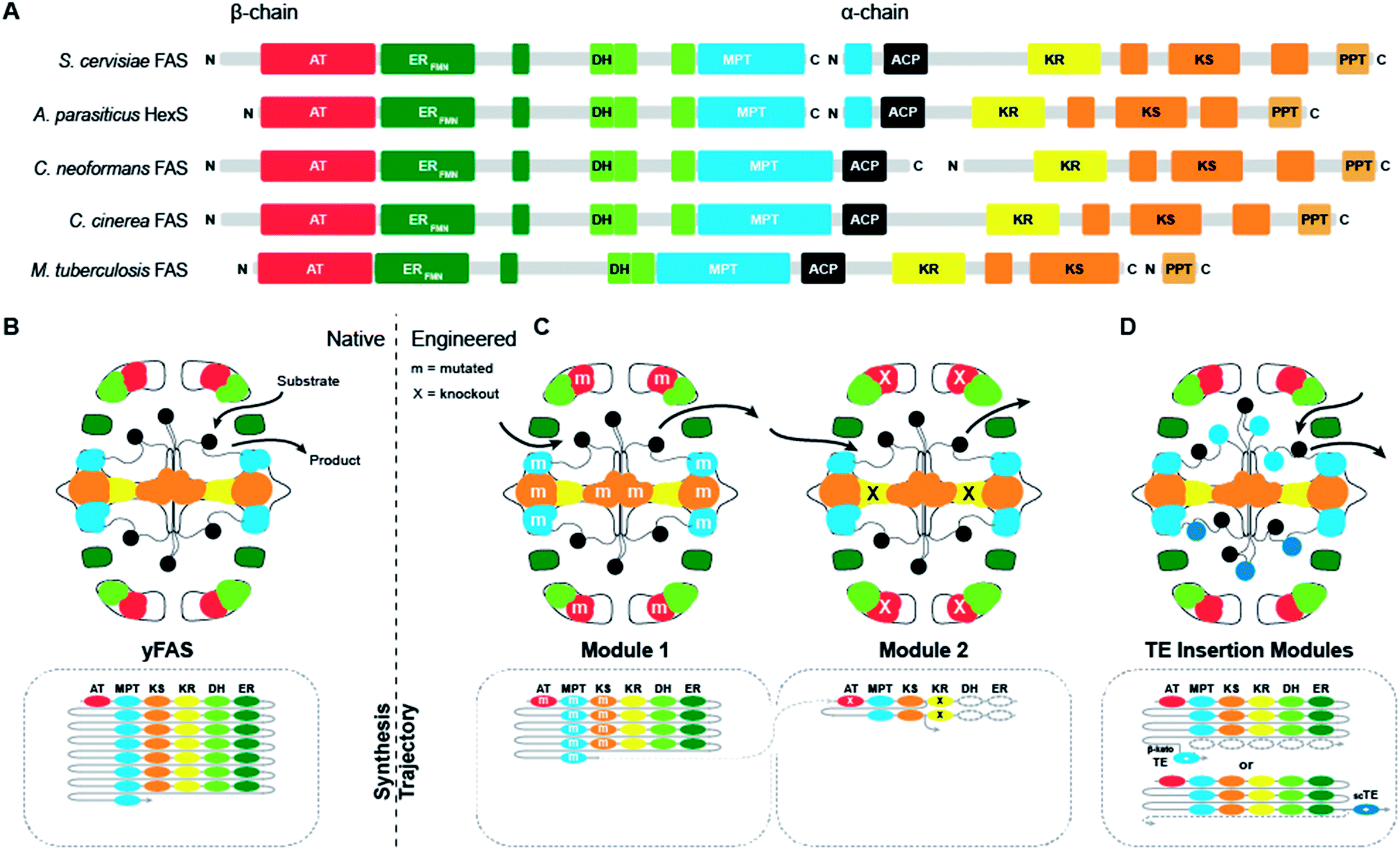 | ||
| Fig. 3 Domain organization and engineering approaches for yFASs. (A) Domain representation in sequence of evolutionally related yFAS. (B) Schematic representation of yFAS architecture and synthesis trajectory. Domains are colored according to (A). The sequential domain utilization of each domain in the reaction cycle is indicated below the barrel. (C) Two engineered yFAS assembly lines according to Gajweski et al. Domain mutagenesis modified the biosynthetic trajectory (m: mutation, X: knockout) and yielded novel products. (D) Engineered yFAS according to Zhu et al. Two separate yFAS variants are indicated in each reaction chamber of the barrel that harbor a TE insertion for the production of short chain fatty acids. TEs can be inserted either into the C-terminal (indicated for the upper chamber) or N-terminal (indicated for the lower chamber) ACP linker. Domain names correspond to Fig. 1. scTE: short chain TE. | ||
Early fungal FASs are encoded by a single gene, which includes the PPT encoding region. In fungal evolution, splitting of the contiguous FAS gene occurred at different positions, leading to splitting into two consecutive FAS genes. Splitting never occurs directly in the ACP linker, so that the ACP remains double-tethered. Segment duplication presumably has contributed to the extension of fungal FAS, as indicated by the occurrence of a third, non-catalytic hotdog-fold next to the catalytic double hotdog fold of the DH domain, or the duplication of the ACP domain in the oleaginous yeast Rhodosporidium toruloides FAS, which has also been structurally characterized by cryo-EM at 7.8 Å resolution.23
The evolution from early bacterial to late fungal yFAS is mapped out by a large number of intermediate sequences. The origins, however, of a primordial contiguous FAS gene encoding the entire yFAS are enigmatic. Due to the low level of sequence conservation in non-enzymatic linker regions, early evolutionary intermediates are difficult to detect by sequence analysis alone. Crystallographic structure determination, however, has confirmed the presence of early precursors of docking and scaffolding elements from yFAS already in two bacterial proteins involved in type II FASs and polyketide biosynthesis:17 FabY, a non-canonical starter KS for fatty acid biosynthesis in Pseudomonas aeruginosa is a close homolog of yFASs KS domains. Structure determination revealed that it already encompasses structural elements contributing to the spoke and hub structure of yFASs, while in FabY they presumably are stabilizing the dimeric KS structure. The ER domain of the dual-function trans-acting AT-ER protein DfnA, part of the trans-AT PKS for difficidin biosynthesis, and likely also ER proteins in PUFA biosynthesis, contain insertion regions relative to the bacterial TIM-barrel FabK ER, which are ancestors of ER–KS interacting regions in yFASs. The regions of bacterial yFASs that presumably have evolved from the insertion regions of DfnA and FabY mediate a core contact between the FAS wheels and domes, qualifying the two proteins as potential early precursors for partial assembly of a bacterial yFAS.
II.4 Engineering yFAS for alternate product synthesis
The chemical versatility of multienzymes renders them interesting targets for re-engineering of product formation, either via mutation of individual active sites or via deletion, addition or swapping of functional domains. In contrast to other families of multienzymes, such as PKSs, naturally occurring yFASs only produce a single type of product, saturated fatty acids, albeit with different chain length. The extensive scaffolding and tight domain integration in yFAS massively complicate the exchange of enzymatic domains. Consistently, functional exchange of whole domains has not been reported for yFAS.Nevertheless, two successful routes to the production of alternate products have recently been discovered (Fig. 3B–D). The first approach24 exploits kinetic competition between enzymatic domains based on comprehensive simulation of product formation in yFAS and specific active site mutations (Fig. 3C). A first reaction module was engineered based on Corynebacterium ammoniagenes FAS and mutations in KS, MPT and AT, that increase the production of short-chain fatty acids. A second module designed on the same FAS by a mutation in KR loads the short chain products from the first module via its MPT domain and catalyses one round of condensation with malonyl-CoA, before an inactivating mutation in KR prevents normal β-carbon modification. The β-keto-intermediate is condensed in a side reaction to the normal elongation cycle one more time with malonyl-CoA. Spontaneous cyclization results in the release of a new lactone product. In the second approach,25 the complications of inserting domains into the rigid yFAS barrel were elegantly circumvented (Fig. 3D). Here, the large reaction chambers and flexible nature of the ACP linkers were exploited to insert ectopic thioesterase domains with alternate specificity into the ACP linking regions in yeast FAS. This approach enabled production of short-chain fatty acids or methyl ketones by a single type of designed yeast FAS.
III. Metazoan fatty acid synthase
Fatty acid biosynthesis by mFASs uses a variation of the synthetic scheme of yFAS. The acyl groups of the starter substrate acetyl-CoA and the elongation substrate malonyl-CoA are loaded both by a single dual-specific malonyl-acetyl transferase (MAT) onto ACP (Fig. 4A). The ER of mFAS is a member of the medium-chain dehydrogenase family, different from the FMN-dependent TIM barrel ERs of yFASs6 and bacterial FabK26 or the canonical bacterial short-chain dehydrogenase ERs. The main product of mFAS is palmitate, which is released from the ACP by an integral C-terminal thioesterase domain.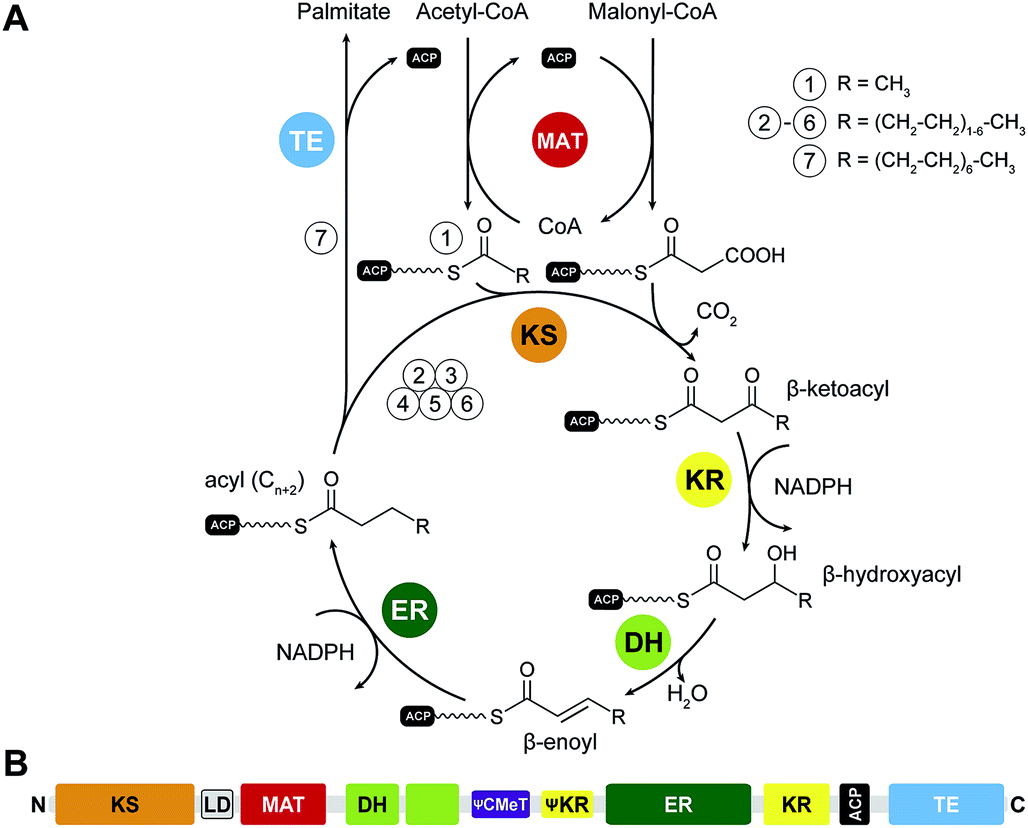 | ||
| Fig. 4 Catalytic reaction cycle and domain organization of mFAS. (A) Fatty acid biosynthesis is initiated by loading an acetyl-unit from acetyl-CoA onto the ACP, which is subsequently elongated by the KS in iterative cycles of decarboxylative condensations using the extender unit malonyl-CoA. All building blocks are loaded by the MAT. Fully saturated acyl products are generated by sequential reduction and water elimination (KR, DH, ER) at the β-carbon position in each cycle and serve as substrate for further elongation reactions by the KS. (B) Linear domain organization of mFAS. KS: ketosynthase, MAT: malonyl-acetyl transferase, DH: dehydratase, CMeT: C-methyltransferase, ER: enoylreductase, KR: ketoreductase, ACP: acyl-carrier protein, TE: thioesterase. Inactive domains are indicated by leading Ψ (pseudo) symbols. | ||
III.1 The structural organization of mFAS
The FASN gene encodes the single protein chain of human mFAS (Fig. 4B), which forms a 540 kDa homodimer. Initial insights into the dimeric organization of mFASs were obtained by careful biochemical, mutational and complementation analysis.1,27,28 Early EM analysis has been considerably more challenging than for yFAS due to the smaller size, less characteristic shape and higher conformational variability of mFAS, but yielded a shape description at a resolution of 19 Å.29 Crystallographic analysis of an intact mammalian mFAS finally provided an atomic structural model at 3.2 Å resolution5,30 (Fig. 5A). The crystal structure did not resolve the flexibly tethered and mobile C-terminal ACP and TE domains, but structural data on the isolated domains are available (Fig. 5B).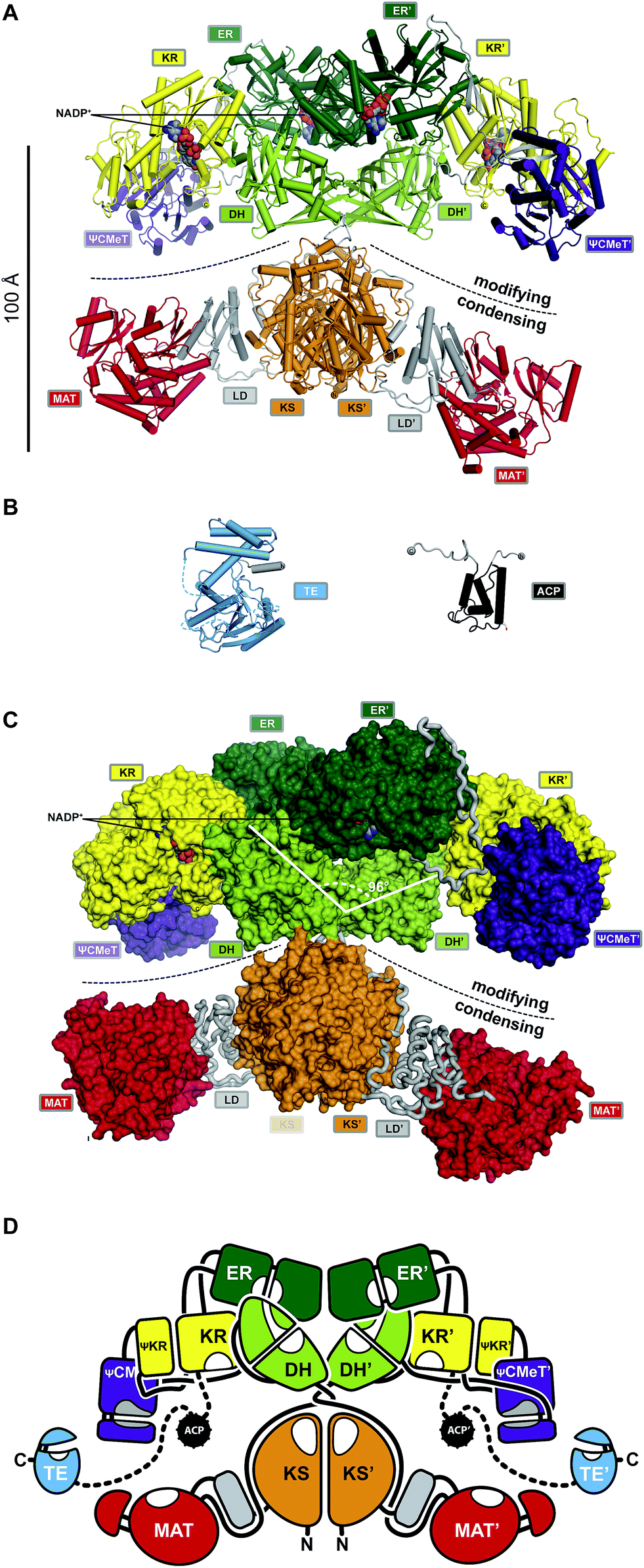 | ||
| Fig. 5 The architecture of mFASs. (A) Cartoon representation of the asymmetric porcine FAS crystal structure reveals a structural separation into a modifying and condensing regions with a central hinge. (B) Crystal structure of human FAS TE and NMR structure of rat FAS ACP in cartoon representation. (C) In contrast to yFAS, mFAS invests only 9% of the total polypeptide into scaffolding. In the modifying region all domains (surface representation) are connected by direct domain–domain interfaces. The DH dimer has a V-shaped organization. (D) Schematic architectural drawing of the mFAS domain connectivity. Dotted lines indicate linkers that have not been experimentally observed. Domain names correspond to Fig. 4. | ||
The ACP domain of rat FAS has been analysed by NMR.31 The ACP domain adopts a canonical four-helix bundle fold. In contrast to bacterial FAS, mFAS ACP does not sequester the covalently linked acyl moieties. The monomeric α/β hydrolase fold TE has been resolved by X-ray crystallography.32 A structure of TE in complex with the inhibitor Orlistat further revealed a mechanism for chain-length selectivity based on distinct binding sites for acyl chains of different length.33 The resulting discrimination of chain lengths together with the chain length-dependence of the mFAS KS activity provides the basis for the palmitate product specificity of mFAS.
The ACP linkers are not enriched in Ala–Pro residues as observed for yFAS. The properties of the flexible linker leading into the ACP domain of mFAS have not been studied in detail. The longer linker between ACP and TE, typically 20 residues or more, is remarkably permissive for modifications: substitution with a considerably longer linker from PKS had no effect, and mFAS variants with harsh or almost complete deletions in the linker region maintained about 30% of activity.34
The structurally resolved core of porcine mFAS adopts an X-shape structure with dimensions of approx. 200 Å × 170 Å × 110 Å (Fig. 5C and D). The structure is segregated into two subregions centrally connected via a narrow link of extended polypeptides. The dimeric condensing KS domain in the centre and laterally disposed monomeric loading MAT domains define the “condensing region”. The N-terminal KS domain is linked via a small α/β-fold linker domain (LD) to the MAT, and a post-MAT linker winds through the linker domain and around the KS to the central connection of the two regions. The second subregion comprises the domains required for β-carbon modification, the KR, DH and ER domains, and is thus referred to as the “modifying region”. The modifying and condensing region hardly form any direct contact other than the covalent peptide linkage. Asymmetry of the crystal structure of dimeric mFAS suggests pronounced hinge-like flexibility around the central connection.
In the dimeric mFAS, the two DH domains arrange in a V-shape with few contacts between the DH domains, which is mostly stabilized by interactions with the dimeric ER domain. The KR domains are laterally linked to the ER domains. While related bacterial KR domains are true oligomers, the fold of the mFAS KR domain is completed by a non-catalytic fragment of a KR domain, termed the pseudo-KR (ΨKR). A second non-catalytic domain is found in the modifying region. This domain resembles S-adenosyl-methionine (SAM) dependent C-methyltransferases (CMeTs), but lacks conserved active site residues, and is here referred to as pseudo-C-methyltransferase (ΨCMeT).
The domains in the modifying region are linked by a minimal amount of linking sequences, either extended peptides or short β-strands. Overall, only 9% of the total sequence is invested into linking regions and an additional 16% in the two non-catalytic domains, the pseudo-KR (ΨKR) and ΨCMeT (Fig. 5C) in porcine mFAS.
The X-shaped fold of mFAS exposes all enzymatic domains to the environment. In contrast to yFAS, the ACP is not confined into a specific reaction chamber, but is accessible by other proteins. The accessibility of mFAS TE is exploited by thioesterase 2 (TE2),35 a short chain-specific thioesterase, which is highly expressed in lactating mammary glands, but also in breast cancer. TE2 cleavage of intermediates from mFAS–ACP results in an enrichment of short-chain acyl products.
The X-shape of porcine mFAS defines two lateral reaction clefts each surrounded by a complete set of enzymatic domains required for fatty acid biosynthesis (Fig. 5D). Based on the static crystal structure, the tether that links ACP to KR is just sufficient to reach all active sites of the proximal reaction cleft, but too short to transfer substrates into the distal cleft. However, biochemical data indicated that also substrate transfer between the two clefts occurs albeit at a lower rate.28,36
III.2 Structural variability of mFAS
The analysis of contacts and asymmetry in the crystal structure of mFAS already suggested the possibility for large-scale conformational changes,30,37 in particular a hinge motion around the central connection. Experimental evidence for such large-scale conformational changes was provided by EM analysis, which indeed identified a variety of modes of motions in mFAS.38 Sorting of particles observed in negative stain EM at low resolution into conformational classes in 2D and 3D depicted large-scale swinging and rotation motions around the central connection, which behaved like a ball-and-socket joint. Little structural variation was observed in the condensing region, in line with a close structural agreement of the structure of this region in the entire mFAS and as an excised fragment.4,39 Considerably larger variability was detected in the modifying region. The close match between the structures of an engineered human ΨCMeT–ΨKR–KR fragment bound to an inhibitor and the corresponding region in intact mFAS40 suggest, that a large contribution to the variability in the modifying region may originate from the motion of ER– and ΨCMeT–ΨKR–KR relative to DH.The EM analysis of mFAS trapped by mutation at various stages of the catalytic cycle revealed some correlation between the functional state and the conformational equilibrium of mFAS.38 Still, multiple conformational states were observed in each state. The structural and mechanistic basis for the coupling of conformational changes to enzymatic turnover remains unknown. To resolve this issue, novel approaches for studying single mFAS molecules during turnover may be required, e.g. based on single-molecule fluorescence. Most recently, it has also been demonstrated that high-speed atomic force microscopy is able to resolve large-scale conformational changes in mFAS at a spatial and temporal resolution permitting detailed analysis of the transitions of single mFAS molecules between different conformational states.41 However, further work will be required to apply this technique for a comprehensive analysis of mFAS structural dynamics.
IV. From FAS to iterative polyketide synthases (PKS)
While FASs are highly optimized molecular machines specifically for the production of fatty acids, many multienzymes with an overlapping set of functional domains and a related synthetic scheme are not involved in primary fatty acid metabolism. In contrast to mFAS (Fig. 6A), these machines do not necessarily produce fully reduced products and reveal Cα as well as Cβ modifications that originate from incomplete reduction cycles caused by the absence of specific modifying domains, by catalytically inactive domains, or by programmed domain skipping (Fig. 6B–E). Because of their chemically diverse products, these synthases are named according to their primary condensation product: polyketide synthases (PKSs). The principal catalytic domains of PKSs are closely related to those of mFAS. They include KS, DH, ER, KR as well as CMeT domains. Substrate loading is catalyzed by acyltransferases, which are specific for the particular substrate of the PKS and are commonly also abbreviated as AT, but should not be confused with the acetyl specific acyltransferase of yFASs.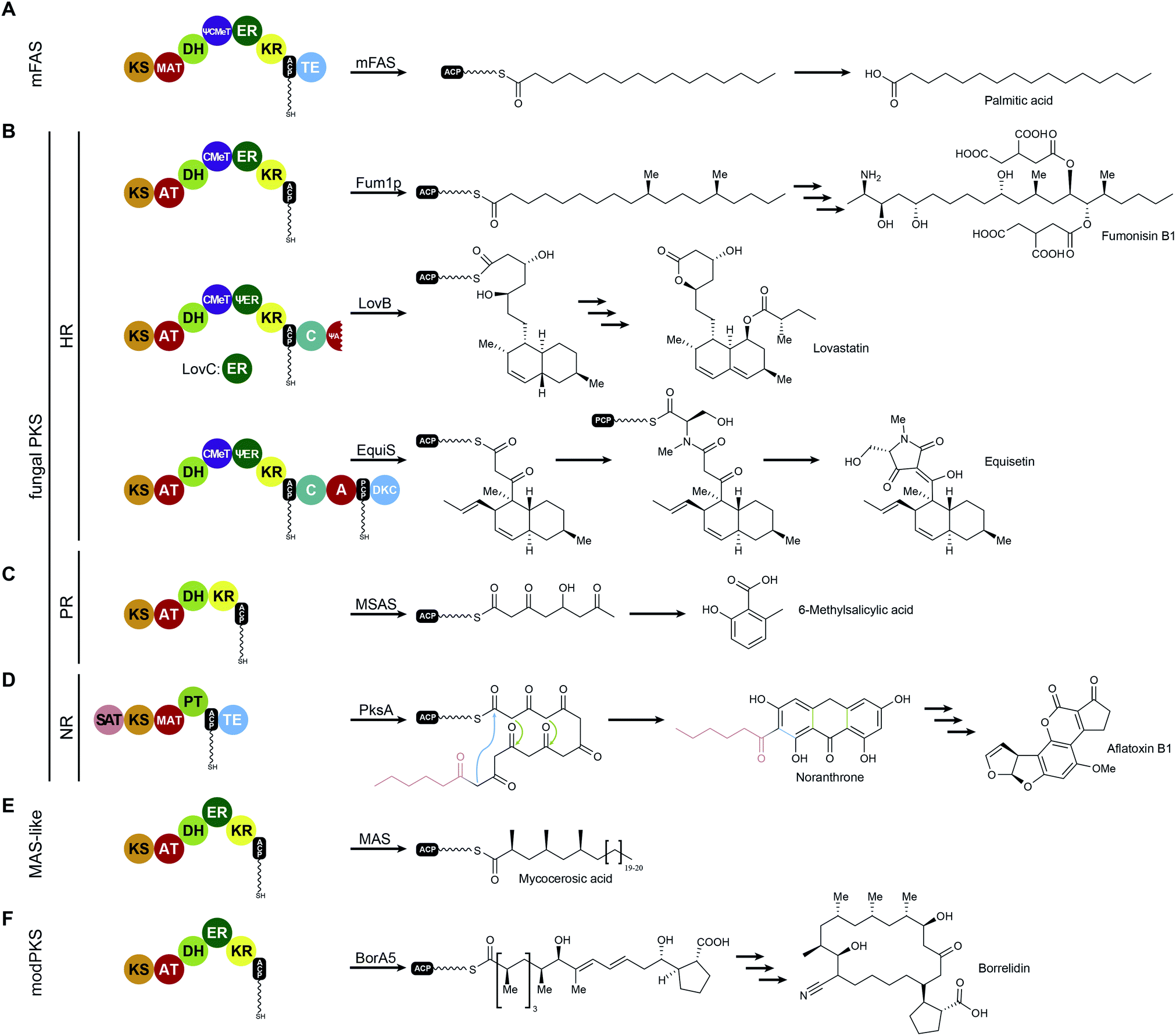 | ||
| Fig. 6 Examples for iteratively acting mFAS and PKS modules. The domain organization and the corresponding biosynthetic products are indicated for mFASs (A), HR-PKSs (B), PR-PKSs (C), NR-PKSs (D), MAS-like PKS (E), and modPKSs (F). KS: ketosynthase, AT: acyltransferase, MAT: malonyl-acetyl transferase (mFAS context)/malonyl-specific acyltransferase (NR-PKS context), DH: dehydratase, CMeT: C-methyltransferase, ER: enoylreductase, KR: ketoreductase, ACP: acyl-carrier protein, TE: thioesterase, C: condensing domain (NRPS), A: adenylation domain (NRPS), PCP: peptidyl-carrier protein (NRPS), DKC: Diekmann cyclase, SAT: starter-unit acyltransferase, PT: product template domain, D: docking domain (modPKS). Inactive domains are indicated by leading Ψ (pseudo) symbols. | ||
All PKSs that make use of dissociated domains (type II), the self-contained type III PKSs, as well as all members of the multienzyme (type I) superfamily work iteratively,42 except for modular PKSs (modPKSs) that function linearly through linked modules in an assembly line with vectorial transfer of intermediates.43 The iterative type I PKSs (iPKS) include the distinct families of bacterial,44 mycocerosic acid synthase-like PKSs (MAS-like PKSs),15,45,46 and fungal iPKSs.47 Fungal iPKS are further classified by their degree of reductive behavior into highly reducing (HR-), partially reducing (PR-), and non-reducing (NR-) PKSs. In the past years, substantial progress has been made in characterization of the architecture of MAS-like, HR-, and NR-PKSs. This work has provided new concepts of PKS assembly with relevance also for other types of PKSs, in particular modPKSs, and will be reviewed in the following sections of this article. Progress towards the depiction of intact synthetic assemblies is less advanced for partially-reducing PKSs as well as for other specialized iPKSs, such as endiyne synthases,48 polycyclic tetramate macrolactam synthases (PTMs)49 and polyunsaturated fatty acid synthases (PUFAs),50 which are not covered in this review.
V. Fully-reducing iterative PKS
MAS-like as well as HR-PKSs contain a fully reducing modifying region, such as mFASs (Fig. 6 A, B and E), and are capable of stepwise reduction of the primary β-carbonyl condensation product to form a fully saturated carbon–carbon bond using KR, DH, and ER activities. These single module PKSs are involved in the synthesis of secondary metabolites and often produce precursors or unusual fatty acids that are channeled to other PKSs or downstream processing enzymes. In Chapters V.1 and V.2 these two enzyme families will be discussed in more detail. In contrast to HR-PKSs, NR-PKRs lack all reducing/modifying domains and use the primary polyketide chain for aromatic cyclization reactions (Fig. 6D), which will be discussed in Chapter VI. Despite integration into modPKS assembly lines, several modPKSs modules have been found to work iteratively51,52 (Fig. 6F), which will be discussed in the context of a common architecture with MAS-like PKSs.V.1 Fungal highly reducing PKSs (HR-PKSs)
Fungi are well known for their extraordinary repertoire of complex secondary metabolites including many toxins. Usually a fungal secondary metabolic gene cluster contains between one to two PKSs and five to twenty accessory enzymes, respectively. Table 1 lists a selection of HR-PKS pathways that often channel their products to downstream HR-PKSs, NR-PKSs, or non-ribosomal peptide synthetases for further modifications and production of fungal toxins, such as fumonisin,53 important drugs like the cholesterol lowering agent lovastatin,54 or the HIV integrase inhibitor equisetin55,56 (Fig. 6B).| Pathway product | PKS name | Organism | Reference | Remark |
|---|---|---|---|---|
| Lovastatin | LovB, LovF | Aspergillus terreus | 71 | LovF: diketide synthase |
| Desmethylbassianin | DMBS | Beauveria bassiania 992.05 | 138 | Hybrid HR-PKS–NRPS |
| Tenellin | TENS | Beauveria bassiania 110.25 | 139 | Hybrid HR-PKS–NRPS |
| Fumonisin | Fum1p | Gibberella fujikuroi | 53 and 140 | |
| Equisetin | EquiS | Fusarium equiseti | 73 and 141 | Hybrid HR-PKS–NRPS |
| Cladosporin | Cla2 | Cladosporium cladosporioides | 142 | Transfer to Cla3 (NR) |
| Hypothemycin | Hmp8 | Hypomyces subiculosus | 143 | Transfer to Hmp3 (NR) |
| Radicicol | Rdc5 | Pochonia chamydosporia | 143 | Transfer to Rdc1 (NR) |
| Zearaleone | Pks4 | Gibberella zeae | 144 | Transfer to Pks13 (NR) |
| 10,11-Dehydrocurvularin | CURS1 | Aspergillus terrus | 145 | Transfer to CURS2 (NR) |
| Asperfuranone | AfoG | Aspergillus nidulans | 146 | Transfer to AfoE (NR) |
| Chaetomugilin/chaetoviridin | CazF | Chaetomium globosum | 147 | Transfer to CazM (NR) |
| Cytochalasin | CheA | Penicillium expansum | 69 | Hybrid HR-PKS–NRPS |
| Prosolanapyrone | Sol1 | Alternaia solani | 148 | |
| Squalestatin | SQTKS | Phoma sp. C2932 | 149 | |
| T-Toxin | Pks1, Pks2 | Cochliobolus heterostrophus | 150 | |
| Fusarin C | Fus1 | Fusarium moniliforme | 151 | Hybrid HR-PKS–NRPS |
| Aspyridone | AdpA | Aspergillus nidulans | 152 | Hybrid HR-PKS–NRPS |
| Brefeldin A | Bref-PKS | Eupenecillium brefeldianum | 153 | |
| Compactin | MlcA, MlcB | Penicillium citrinum | 154 | MlcB: diketide synthase |
Among the various patterns the most common is programmed methylation. Most HR-PKSs contain either an inactive ΨCMeT or, in contrast to mFASs, a fully active SAM-dependent CMeT, which is capable of methylating the Cα-methylene. CMeT catalyzed methylation must precede β-carbonyl reduction by the KR (Fig. 7A). For the lovastatin PKS LovB, Cacho et al. have shown that the specific intermediate of elongation cycle three binds with exceptional specificity to the CMeT and thereby outcompetes the KR.58 This programmed methylation event indicated a gatekeeping function, which is likely to result in off-loading reactions after deficient reaction cycles.58 An equivalent function has also been observed for the citrinin producing NR-PKS PksCT, which relies on methylation for complete polyketide elongation and also indicated a contribution of the KS in programmed chain length control.59 In other cases, such as the closely related HR-PKSs for the production of desmethylbassianin (DMBS) and tenellin (TENS) (Table 1), domain swapping experiments indicated chain length control by the KR.60
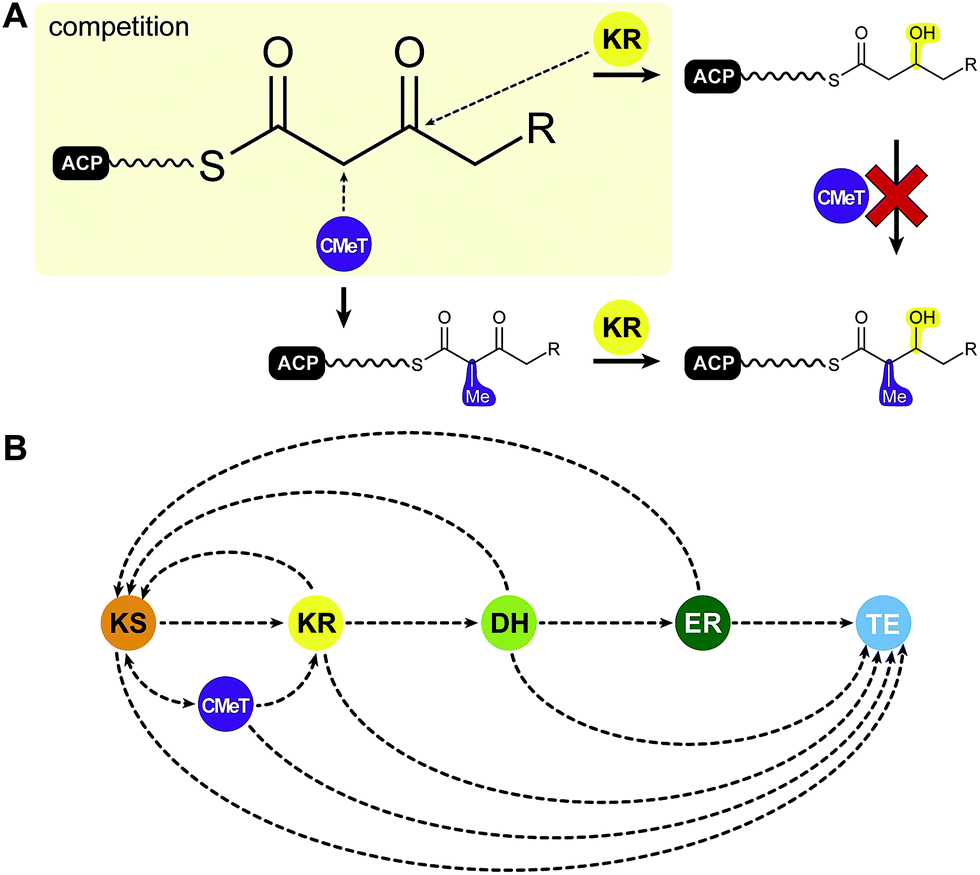 | ||
| Fig. 7 Putative programming trajectory in fungal iPKS. (A) CMeTs and KRs compete for their substrates: CMeT domains require Cα acidity for methylation, which is lost after β-keto reduction. (B) Programming trajectory in fungal iPKS. The main programmed path appears to be described by specific ACP-substrate interactions with each target domain in every elongation and modification cycle. Domain names correspond to Fig. 6. | ||
Special programming functions have also been observed for ERs. The cis acting ER of the squalestatin HR-PKS (SQTKS) was shown to contribute to sequestering the final product and preventing further elongation.61 In the lovastatin HR-PKS LovB, the inactive ΨER is functionally substituted by the trans acting LovC ER, which is only active in two out of eight elongation cycles.
In summary, current data on HR-PKSs suggest that programming is primarily controlled by kinetic competition of all enzymes with all ACP-bound intermediates, which describes the trajectory for selective domain usage in each elongation cycle58,61 (Fig. 7B).
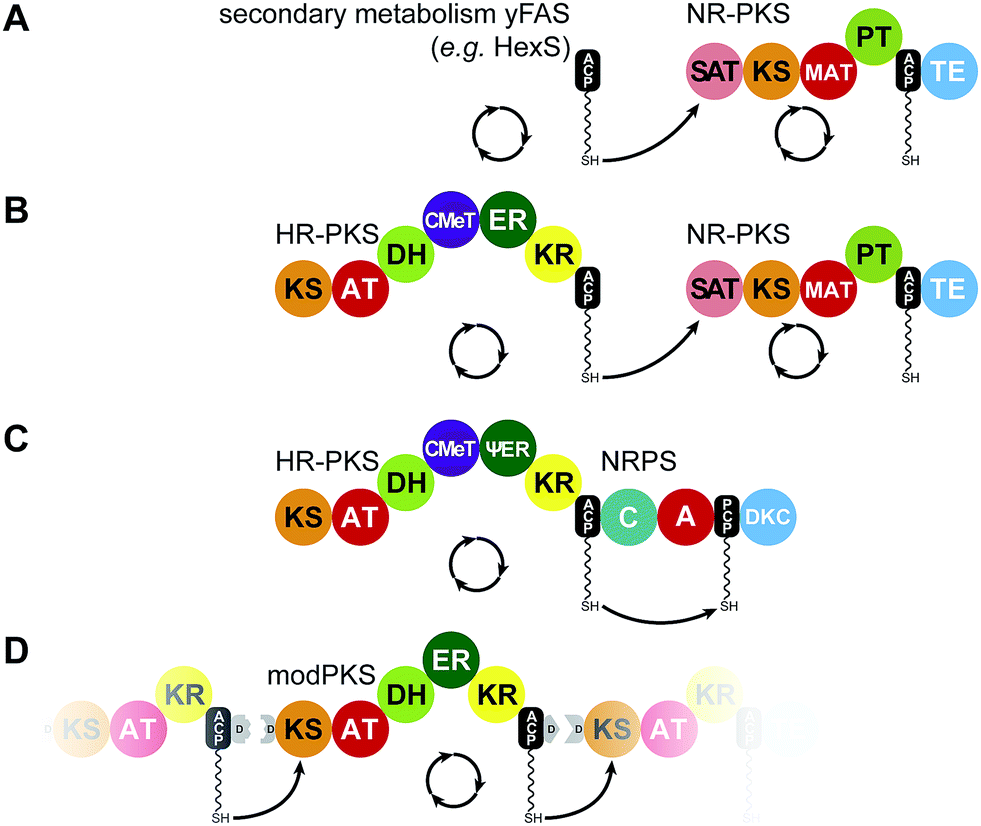 | ||
| Fig. 8 Iterative PKS assembly lines. Direct transfer from iteratively acting PKS and secondary metabolic yFAS ACPs to downstream acting modules has been observed for the pairs of yFAS:NR-PKS (A), HR-PKS:NR-PKS (B), HR-PKS:NRPS (C), and modPKS:modPKS (D). Domain names correspond to Fig. 6. | ||
The organization in the modifying region is likely to be particularly similar between HR-PKSs and mFASs30,40 too: while HR-PKSs and mFASs maintain degraded non-functional CMeT remnants (ΨCMeTs), NR-, MAS-like, and modPKSs mostly do not contain such an inactive domain. The conservation of mFAS architecture in HR-PKSs is further supported by the dimeric character of the cis acting ER domains of the squalestatin HR-PKS (SQTKS).61
The integration of (Ψ)CMeT domains in HR-PKSs between the DH and ΨKR resembles mFAS, but low sequence conservation and deletions in inactive ΨCMeTs made detailed linking analysis difficult. For the same reason the mFAS ΨCMeT was initially annotated as unknown interdomain region two (ID2)37 until higher resolution data became available.30 After crystal structures of active CMeTs from the modPKS CurJ79 and NR-PKS PksCT59 were obtained, identification of ΨCMeT domain remnants has become more reliable.
The recent CMeT structures revealed that deletions mostly occur in the N-terminal subdomain, which is exposed to the lateral reaction cleft of the synthase (based on the mFAS structure), while the C-terminal subdomain is structurally more conserved. It remains unclear, however, why ΨCMeTs have not been totally deleted from mFASs. They may serve additional relevant functions e.g. in restricting ACP diffusion in the lateral reaction clefts.
The two structurally characterized CMeT domains are integrated into different sequence contexts in their full-length PKSs. CurJ CMeT shares the mFAS integration pattern, while the PksCT CMeT is inserted downstream of the ACP. nevertheless, both CMeT structures are highly similar and exhibit a belt-helix in the N-terminal linker, which is not present in mFAS ΨCMeT. The linker ends in a different relative position, which might indicate a divergent CMeT organization of cis-AT modPKSs and NR-PKSs relative to mFAS. In summary, the organization of HR-PKS modifying regions remains unclear, but current data suggest a considerable similarity to mFAS.
V.2 Mycobacterial mycocerosic acid synthase (MAS)-like PKSs
Mycobacteria are well known for their complex cell envelope and their huge repertoire of multienzymes including PKSs, NRPSs, and various FASs.15,46 These multienzymes contribute to the biosynthesis of unusual cell wall lipids with very long and branched chain fatty acids46,80 as reviewed in detail by Quadri.81 The prototype of mycobacterial PKSs is the mycocerosic acid synthase (MAS) from Mycobacterium tuberculosis, which produces mycocerosic acids. These branched chain fatty acids are the main component of dimycocerosates, which are involved in immune evasion,82 contribute to cell wall impermeability,45,83 and determine lipid-specific replication of M. tuberculosis in lung tissue.84 Due to the high sequence identity (up to ∼60%) and functional similarity to the canonical MAS, related mycobacterial PKSs have been named MAS-like PKSs.45While many HR-PKSs and mFASs use acetyl starter- and malonyl-extender units, MAS-like PKSs can use methylmalonyl extender units and larger starters, such as C12–C20 fatty acids, which are presumably loaded by the AT domain or a member of the fatty acyl-AMP ligase family (FAAL).81,85 Complex programming patterns, as described for fungal HR-PKSs, are generally not observed, although programming events cannot be entirely ruled out.86 For MAS itself, a trans-acting polyketide associated acyltransferase (PapA5)87 directly links the product mycocerosic acid to the product of the PpsA-E modPKSs to form phthiocerol or phenolpthtiocerol lipids. Such fusion steps of two PKS products by trans-acting acyltransferase domains are common, have been proposed to be involved in related pathways,88 and can also be found in the biosynthesis of lovastatin (LovD).89
Although MAS-like PKSs produce fatty acids and contain a fully reducing modifying region, they are distinct from mFAS and HR-PKSs. Unlike HR-PKSs and mFASs, MAS-like PKSs lack a (Ψ)CMeT domain and are more closely related to modPKSs (∼27–35% seq. id.) than to HR-PKSs (20–22% seq. id.) or mFASs (∼17–19% seq. id.),77 which is reflected in phylogenetic analysis of individual domains as well as larger multidomain regions77 (Fig. 9). While mFASs may serve as a prototype for HR-PKS architecture, it is likely not the correct template for MAS-like or modPKSs, as confirmed by the hybrid structure of the MAS-like Pks5,77 which is involved in lipooligosaccharide (LOS) biosynthesis86,90 in M. smegmatis. Due to its high sequence identity of 60%, Pks5 was initially annotated as MAS in databases; it is representative of the general architecture of MAS-like and modPKSs fully reducing modules.
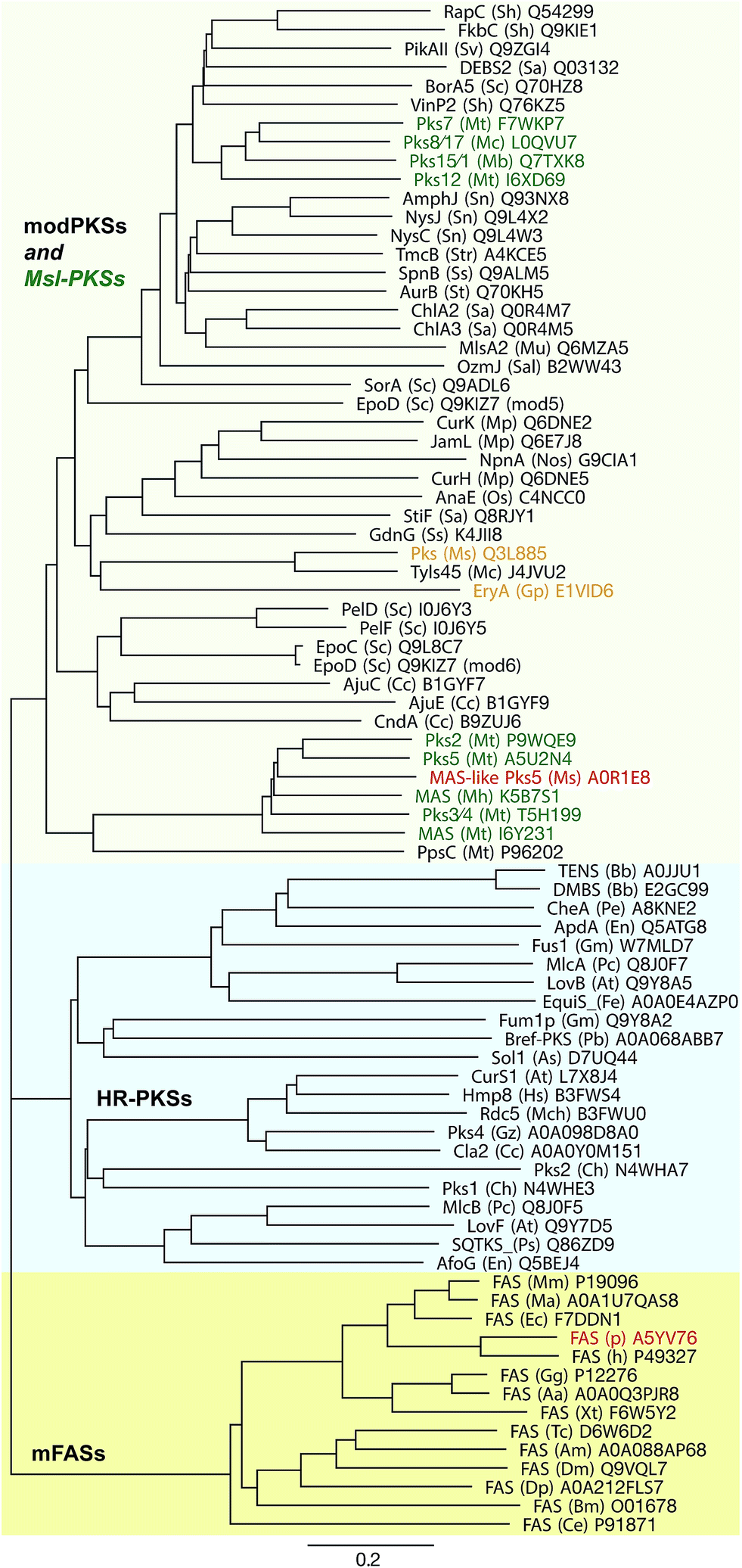 | ||
| Fig. 9 Phylogeny of fully reducing modules of mFASs, HR-PKSs, and modPKSs/MAS-like PKSs. The three groups are indicated in colored boxes. Green: MAS-like PKSs; red: known structures of complete modules; orange: representative modPKSs with short ER–KR linker, low ER interface conservation and same architecture as MAS-like Pks5. All modules are labeled as: protein name (organism abbr.) Uniprot number. Units are given as amino acid substitutions per site. | ||
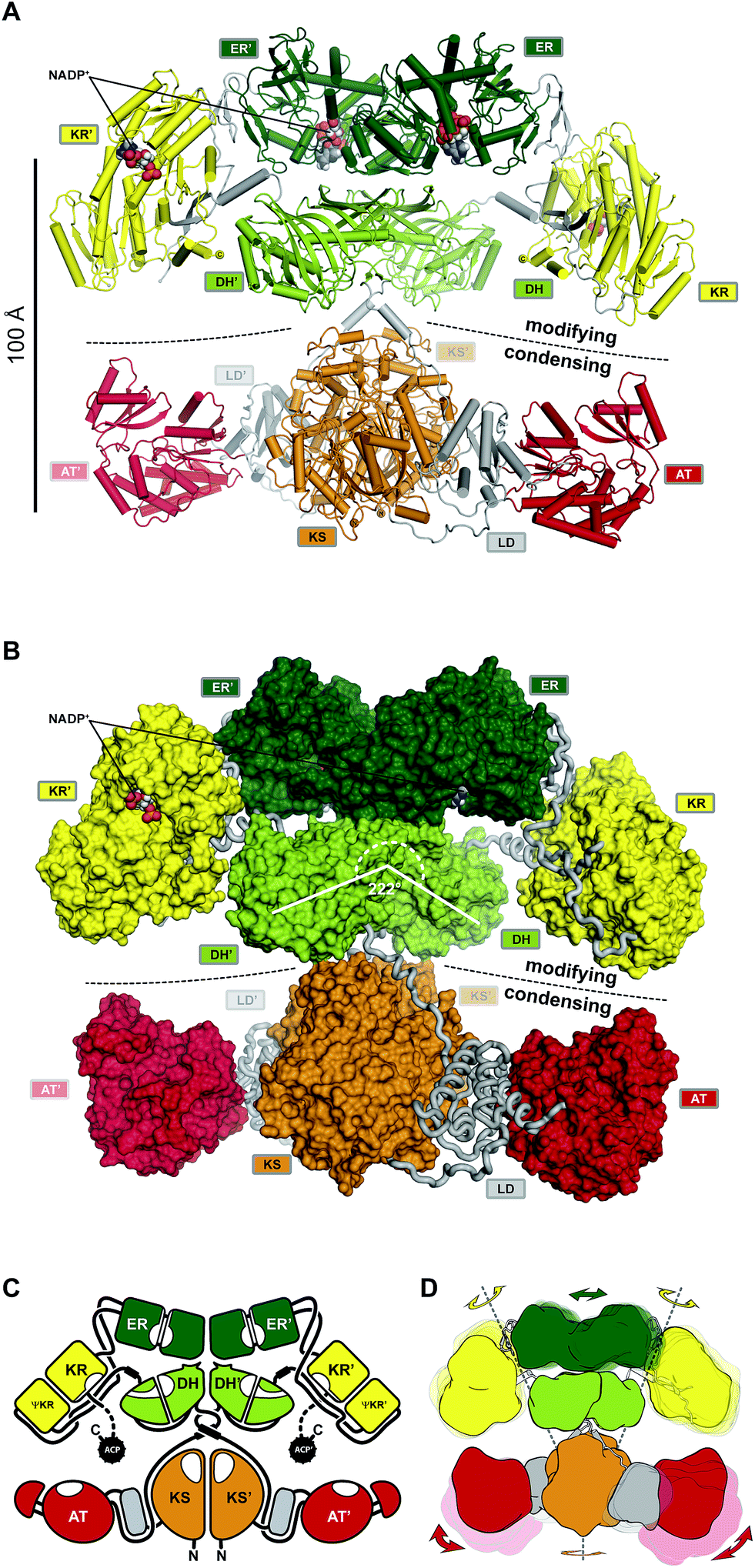 | ||
| Fig. 10 The architecture of MAS-like PKSs. (A) Cartoon representation of the hybrid crystal structure of MAS-like Pks5 showing the canonical separation into a modifying and condensing regions. (B) In contrast to mFAS, the modifying region reveals a linker-based architecture. The KR is double tethered by linkers (loops) to the DH and ER without a direct interface. The DH–ER interface is small and highly variable. Compared to mFAS the DH dimer angle is inverted. (C) Schematic representation of the MAS-like architecture. Dotted lines indicate linkers that have not been experimentally observed. (D) Scheme indicating the conformational variability in fully reducing PKS modules. Domain movements in the modifying region are conformationally coupled across the dimeric synthase. Domain names correspond to Fig. 6. | ||
The MAS-like PKS condensing region77 revealed the same conserved architecture as all other mFAS,30,39 NR-PKS,78 and modPKS67,75 condensing regions, which, however, differs from those observed in the modPKS PikAIII module76,92 at low resolution by cryo-EM. The high conservation of the condensing region (25–30% seq. id.) is not surprising, because it catalyzes the universally conserved elongation step.
The architecture of the modifying region, a main evolutionary contributor to product diversity, is less conserved between mFASs and PKSs. Differences between mFASs and modPKSs modifying regions were already indicated by the first structure of a modPKS DH domain from module four of the 6-deoxyerythronolide B synthase (DEBS).93 While the DH domains in porcine mFAS form a V-shaped arrangement at an angle of 96° in the center of the modifying region (Fig. 5A and C), the DEBS DH and other PKS DHs93–96 (unpublished PDBs: 5J6O, 5IL5, 5IL6, 5HU7, 5HQW) or DH-like domains97 are rather linear dimers at angles between 170–200°. Further support for an alternate PKS architecture arose from ΨKR/KR structures that revealed an N-terminal β–α–β–α extension of the ΨKR domain77,98–100 relative to mFAS.30,40,101 Finally, an excised, non-naturally occurring ER–ΨKR/KR fragment of the spinosyn modPKS SpnB, which lacked the DH domain, was crystallized as a domain-swapped monomer with a completely distinct disposition of the ER relative to the KR as compared to mFAS. This structure was the basis for the prediction of an alternative PKS modifying region architecture with a monomeric ER.100
The hybrid crystal structure of the MAS-like PKS modifying region77 rationalizes most of the predicted mFAS–PKS differences, such as the ΨKR extension, the DH dimer angle, and helices in linker fragments of individual modPKSs domain structures, which connect the condensing region with the DH93,96,97 as well as the DH94 with the ΨKR/KR.99 Relative to mFAS, the opening angle of the DH dimer is rather inverted at 222° (Fig. 10B) and resembles angles of recently determined trans-AT modPKS structures (unpublished PDBs: 5J6O, 5IL5, 5IL6, 5HU7, 5HQW).
However, the predicted PKS architecture based on the SpnB fragment with a monomeric ER was not reflected in the MAS-like PKS structure. The ER forms a similar dimer as in mFAS and rests on top of the DH platform with a small and highly variable interface. While ER dimers have been observed here and for the modPKS PpsC (PDB: 1PQW), some excised ER domains have been crystallized as monomers.100,102 Whether this difference is caused by reduced avidity due to the deletion of dimeric partner domains in the SpnB construct or indicative of an alternative architecture remains unclear. The ΨKR/KR is laterally attached to the DH and ER in the MAS-like PKS structural model, but, in contrast to mFAS, reveals a different tilt angle and has no interface with neighboring domains.
In fact, the entire modifying region architecture in MAS-like PKSs is based on linker-mediated interactions rather than on direct domain–domain interfaces. The DH connects the ΨKR via a linker containing a spacer helix, interactions with the ΨKR/KR surface and the ER–KR linker, and a small disordered segment that is probably used for CMeT integration in homologous PKSs. The ER is connected to the ΨKR via linkers that interact with the ER–KR linker. Thus, the ΨKR/KR is connected via a double tethered hinge with its two partner domains.
Such a linker-mediated architecture provides a framework for evolutionary domain shuffling without a requirement for adjustment of direct domain interfaces; it rationalizes the evolutionary modularity and variability of modifying regions in PKS.
The observation of nine independent dimers in the crystallographic asymmetric unit of the MAS-like Pks5 modifying region revealed conformational variability and coupling of the lateral ΨKR/KR domain hinge-bending (up to 40° rotation) via a screw motion of the ER dimer (translation of 8.5 Å) on the DH platform (Fig. 10D). This coupling creates an entirely dynamic architecture, which affords additional room to compensate for ACP linker constraints and might provide a mechanism for coordinated asymmetric polyketide biosynthesis in the two lateral MAS-like PKS reaction clefts. The condensing region presumably also contributes to the overall dynamics of MAS-like PKSs, because a comparison with homologous structures indicates a hinge in the LD. Conformational coupling has also been observed for mFAS at low resolution,38 which supports the general relevance of a dynamic architecture, rather than a static scaffold, in ACP-dependent multienzymes. Notably, in MAS-like Pks5, domain flexibility increases laterally with the ΨKR/KR domain being the most flexible catalytic domain and the ACP the most flexible non-catalytic part.
While phylogenetic data indicate that the architecture of mFAS is representative for HR-PKSs, the MAS-like architecture appears to be more related to modPKSs. The SpnB fragment-based prediction of a modPKS architecture with outward facing monomeric ERs100 was supported by differences in mFAS and modPKS linkers. Also the MAS-like Pks5 reveals a relatively long ER–KR linker of 20 aa, while many, but not all, modPKSs have short linkers (5–10 aa) as well as low sequence conservation in the putative ER dimerization interface; however, the short linkers are tolerated in a MAS-like Pks5-based modPKS model.
In order to experimentally validate a MAS-like PKS based architecture of modPKS, two fully reducing modPKS modifying regions with short ER–KR linkers and low sequence conservation in the ER dimerization interface were analyzed by small angle X-ray scattering (SAXS), which is suited to distinguish the two principal architectures. Both modPKS modifying regions conformed to the MAS-like architecture, supporting a general relevance of this architecture in modPKSs. However, the existence of an alternative architecture in other modPKSs cannot be completely ruled out.
Despite some HR-PKS–NRPS hybrids as well as HR-PKSs that pass their substrates on to NR-PKSs, most iterative PKS lack integration into an assembly line that imposes vectorial transport of intermediates. In modPKSs, the shuttling direction is imposed by ACP–KS recognition52,103,104 as well as kinetic mechanisms.105 In some cases, modPKS modules, such as BorA5 (ref. 51) (Fig. 6F), were also found to act/function iteratively (Fig. 8D). Additionally, modPKSs modules such as PikAIII, can be switched to an iterative mode of action when isolated from the assembly line106 or by engineering ACP recognition.104
Altogether, the structural data on MAS-like PKSs and the functional intermixing of iPKSs and modPKSs create close links between members of the two families. MAS-like iPKSs may actually be viewed as an iterative version of a modPKS module.
VI. Non-reducing iterative polyketide synthases
Although aromatic metabolites isolated from fungi are among the earliest characterized natural products, the nature and mechanisms of the underlying polyketide synthases remained a mystery and lagged behind the rapid advances that occurred after 1990 (ref. 2 and 107) in understanding modPKSs.108 In no small measure this comparative ignorance owed not only to technical barriers to experiment but also to the long-held belief based on sequence comparisons to mFASs that the NR-PKSs contained four catalytic domains, a KS, a malonyl-specific AT (MAT; not to be confused with the dual-specific mFAS MAT), ACP and TE. An extended N-terminus and internal region were thought to be dimerization interfaces to support α2-structures akin to mFASs. A quite different picture emerged, however, when the predictive algorithm UMA was applied to PksA, the NR-PKS that lies at the heart of aflatoxin biosynthesis, and the limited number of other NR-PKS sequences known at the time (Fig. 11).109 This bioinformatics tool can be used to analyse a set of related protein sequences to identify linker regions between presumably compactly folded domains. While the KS, MAT, ACP and TE were correctly discerned, the elongated N-terminus and the previously undefined internal region both appeared to be well-structured.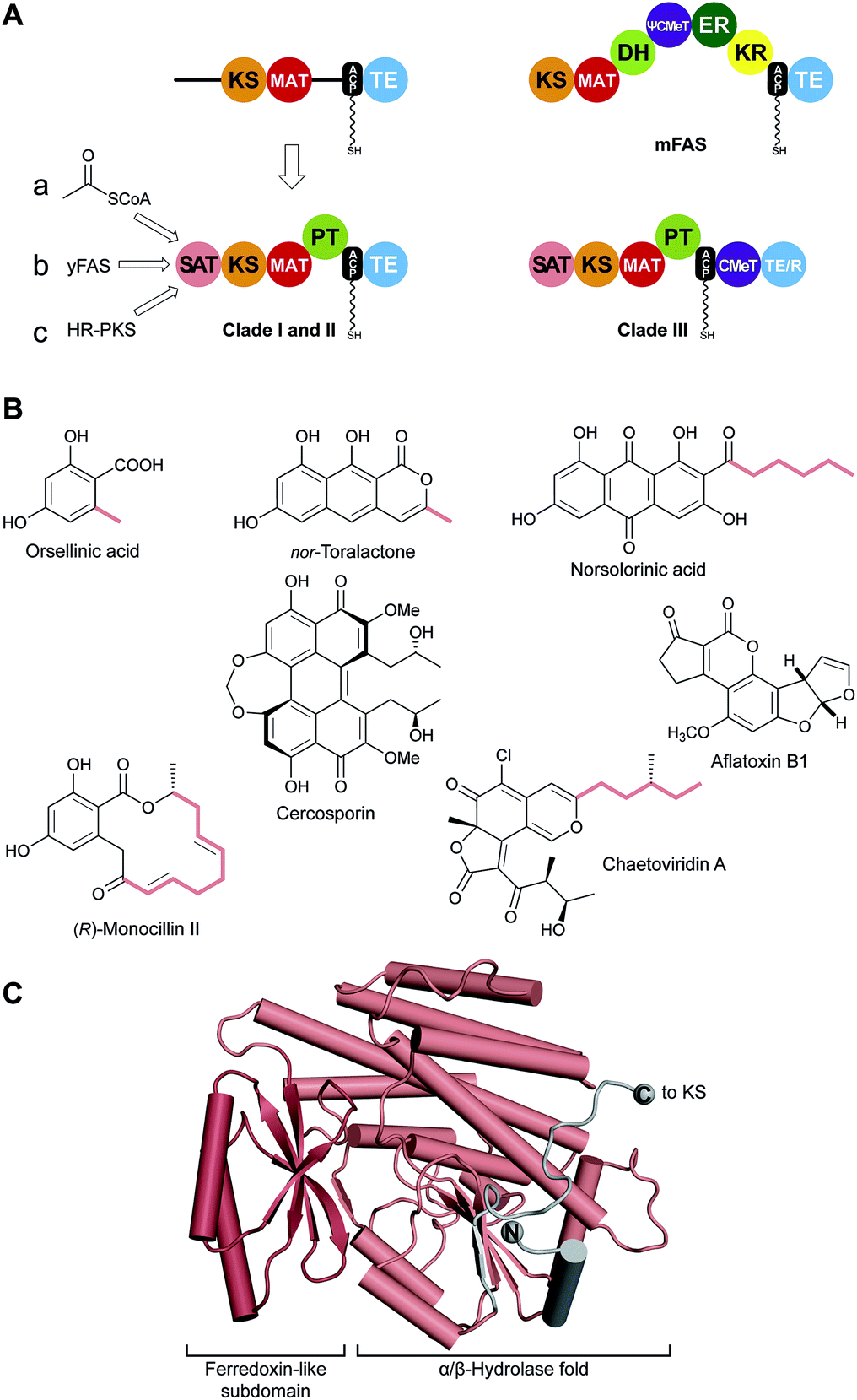 | ||
| Fig. 11 Identification of SAT and PT domains in NR-PKS. (A) SAT and PT domains of NR-PKSs were identified by the UMA algorithm and deconstruction experiments. SATs can accept substrates from three different pathways (a–c). Only clade III NR-PKS contain CMeT domains. (B) Products of pathways involving NR-PKSs. The identity of some substrates loaded by the SAT are indicated (colored). (C) Crystal structure of the CazM SAT domain. Domain names correspond to Fig. 6. R: reduction domain. | ||
Unique to the NR-PKS family, the N-terminus was identified as a starter-unit acyltransferase (or transacylase) SAT domain, which loads a primer for polyketide extension and accounts for the classical “starter unit effect”, a phenomenon long known for the anomalous incorporation level of radiolabeled precursors at this site.20,110 Strikingly absent are the three reduction/dehydration domains of FASs or HR-PKSs. Highly reactive poly-β-ketone chains are synthesized, but controlled in length and cyclization behaviour. The internal product template (PT) domain dictates a defined cyclization pattern, as detailed below, of the polyketide intermediate to particular monocyclic or fused ring systems. The coupled loss of water and resonance stabilization of the aromatic ring(s) generated provide enormous driving force to product.
VI.1 The starter-unit acyltransferase (SAT) domain
Using the simple, original phylogenetic designations of Kroken111 clade I and II NR-PKSs have the domain organization depicted in Fig. 11A, although two, even three, ACP domains in row can occur.112 Members of the related clade III NR-PKSs carry out C-methylation(s) coupled to polyketide elongation,59 and can terminate in either a TE or NADPH-dependent reduction (R) domain to release the product typically as an aldehyde.113C-Methylation does not take place in every cycle of polyketide extension, but at “programmed” sites/intervals determined primarily by the CMeT domain itself.59In most NR-PKSs (Fig. 11A, path a) the SAT domain is modestly selective for acetyl-CoA, a fundamental metabolic building block of the cell.114 There are many examples (Fig. 11B) beginning with molecules as simple as orsellinic acid to the tricyclic structure of nor-toralactone in which the formation of the terminal pyrone is catalyzed by the TE domains.115 This cyclization is an exception among TE-catalyzed reactions, but sets the pathway in motion to the biosynthesis of the widespread phytotoxin cercosporin.115
The substrate adaptability of the SAT domain, however, considerably expands the synthetic capabilities of NR-PKSs. For example, PksA accepts a hexanoyl starter unit from a specialized yFAS pair of HexA and HexB, together also referred to as HexS (Fig. 11A, path b), and carries out seven malonyl extensions to give the C20-anthraquinone norsolorinic acid, the first committed intermediate in a ∼15 step reaction sequence to the environmental toxin aflatoxin B1.116 With an eye to redirecting synthesis to non-natural products, the substrate tolerance of the PksA SAT domain has been tested and >20 alternative starter units were successfully processed by the SAT–KS–MAT + PT–ACP to correctly cyclized products.117
Finally, a HR-PKS (Fig. 11A, path c) can be functionally linked to a NR-PKS to generate, for example, the extended primer shown bolded in the resorcylic acid lactone (R)-monocillin II, the precursor of radicicol, a potent inhibitor of heat shock protein 90 (Hsp90).118 Similarly, a HR-PKS creates the (S,E)-4-methyl-hex-2-enoyl triketide starter unit in chaetoviridin A (bolded carbons).65
The SAT domain of the clade III NR-PKS CazM transfers this initiating component into the rest of the biosynthetic machinery. A crystal structure of the isolated CazM SAT domain has been obtained with a simpler hexanoyl surrogate of the native starter bound to the active site cysteine.65 The isolated CazM SAT closely resembles the general fold of PKS AT domains (Fig. 11C). These proteins share an α/β-hydrolase fold with a small ferredoxin-like subdomain forming a palm and thumb motif with the active site at the base of a cleft between the two. The active sites are largely hydrophobic corresponding to the generally hydrophobic nature of starter unit substrates. This SAT domain and the related SAT domain of PksA serve as adapter proteins recognized in trans by a donor ACP from a HR-PKS or FAS and an in cis ACP acceptor through a single-access transfer site.63
VI.2 Product template (PT) domain
Transfer of the starter unit from the SAT to the KS domain initiates polymerization of successive malonyl units to form a polyketide intermediate whose programmed length is dictated mainly by the KS.119–121 PT domains are known that specifically catalyze C2–C7, C4–C9 and C6–C11 first-ring cyclizations as shown schematically and exemplified in Fig. 12.122 Guided by UMA, it has been possible to dissect various NR-PKS proteins to give a library of mono-, di- and tridomain fragments. Reconstitution of native components has faithfully restored wild-type synthetic activity.20,114,119,123 Similarly, heterocombinations of these domains have enabled synthesis of non-native products and, in so doing, given a detailed picture of the division of labor shared among the domains and the extent of control each exerts on product outcome.121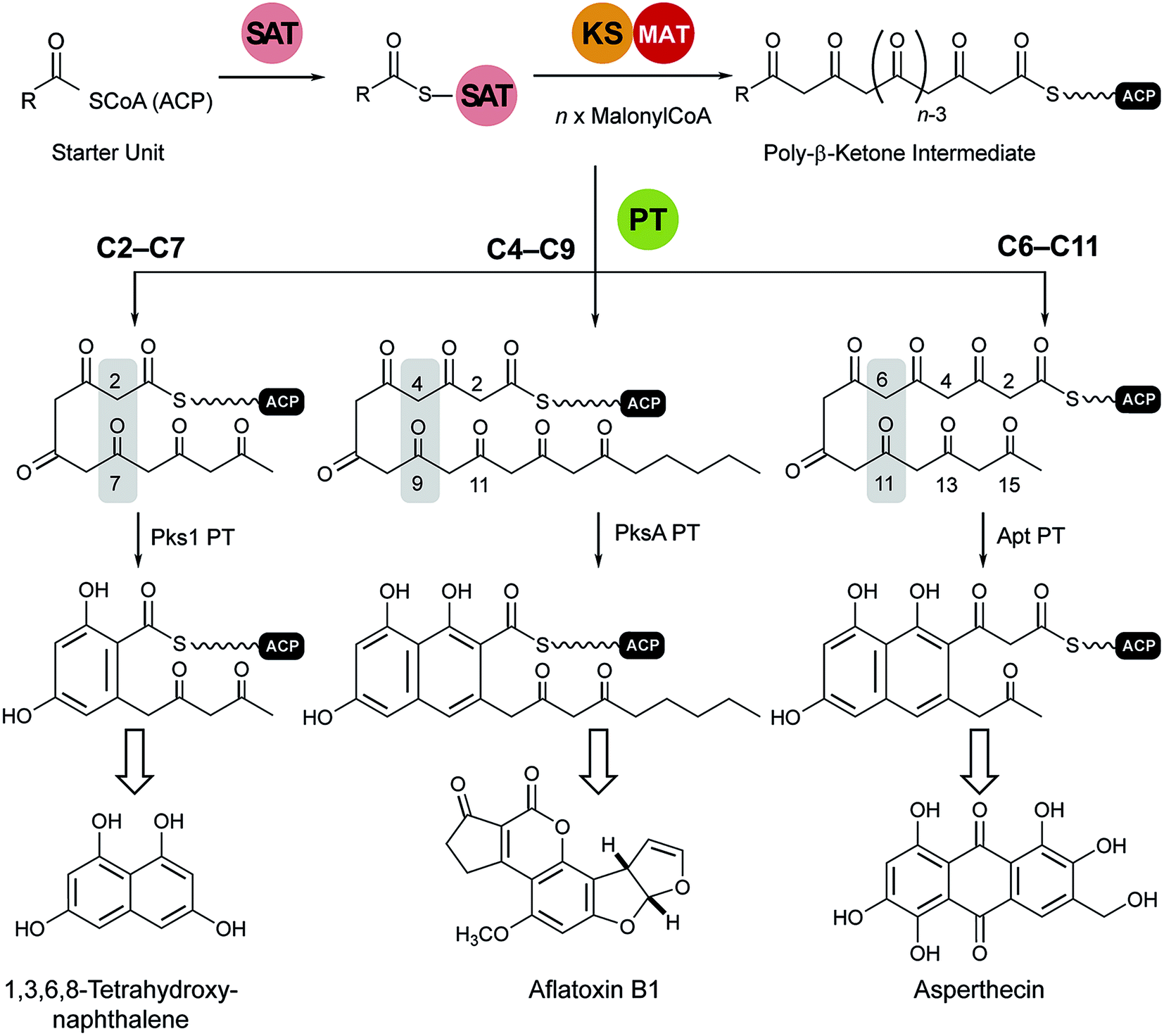 | ||
| Fig. 12 Cyclization reactions catalyzed by PT domains. The product of the loading/condensing region (SAT–KS–MAT) can be cyclized via three different routes. Domain names correspond to Fig. 6. | ||
Analysis of the catalytic cycle of PksA by mass spectrometry revealed that the time-average population of species bound to the ACP was dominated by building blocks (hexanoyl, malonyl and acetyl, presumably arising from decarboxylation of malonyl units) and lower intensity mass signatures in keeping with the fully extended polyketide intermediate and two successive dehydrations (cyclizations) enroute to product release (Fig. 13A). Importantly no intermediate chain lengths were detectable despite the introduction of mutations intended to slow polyketide extension.119,124 We attribute these observations to rapid, processive extension and, in contrast to the behavior of clade III NR-PKSs, the extending chain probably never leaves the KS, shuttling rapidly back and forth between the active site Cys and transient reactions with malonyl-ACP (Fig. 13A).
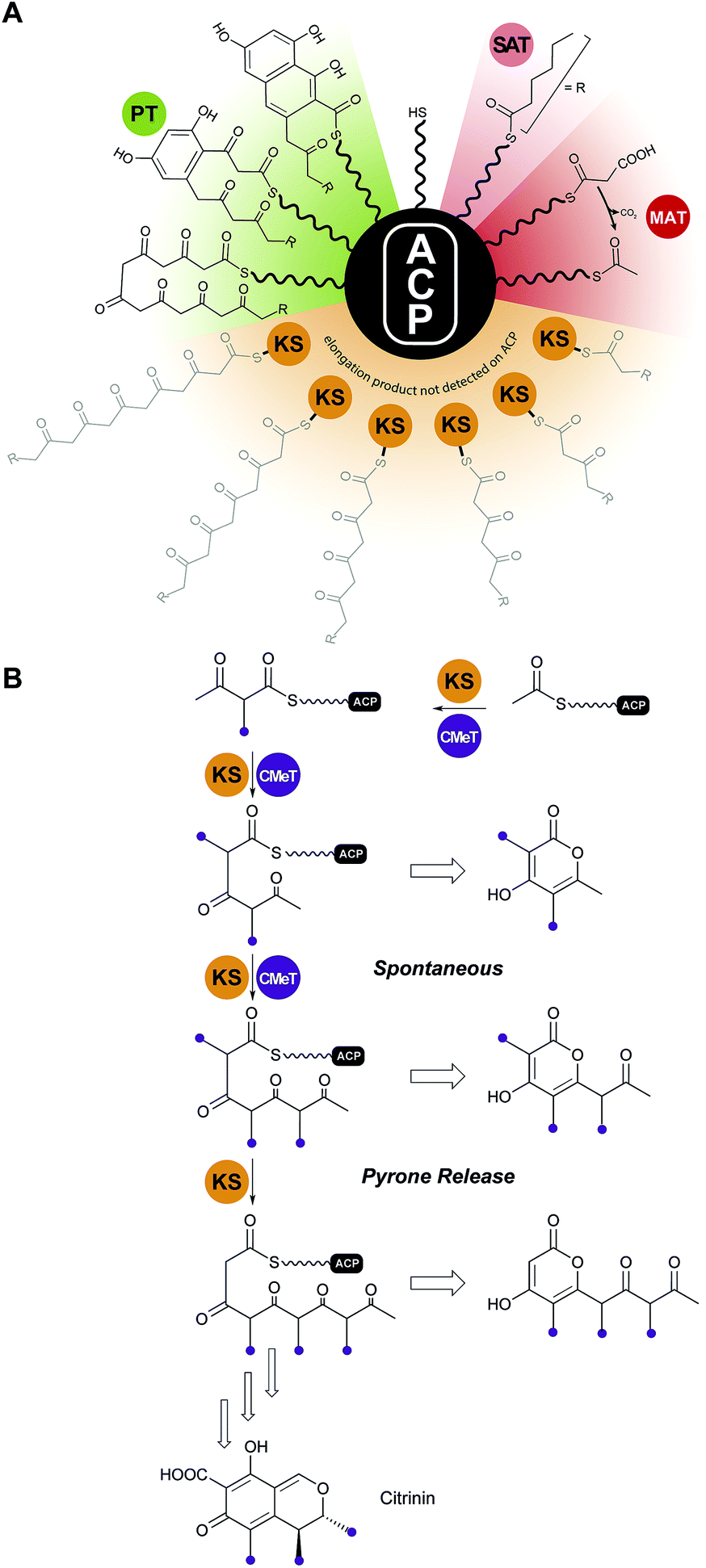 | ||
| Fig. 13 Protection of the nascent polyketide and programming. (A) In clade I and II NR-PKS, intermediates of the nascent polyketide prior to achieving full length (shown in grey) could not be detected on the ACP, indicating that the intermediates stay tethered to the KS during the chain elongation cycles. (B) In clade III NR-PKS the nascent polyketide chain has to leave the KS active site for C-methylation, which is a programmed event and required for complete chain elongation, but allows derailed pyrone products to be observed. Domain names correspond to Fig. 6. | ||
For clade III enzymes polyketides of intermediate length leave the KS to the CMeT in a programmed manner for α-methylation, which gives fleeting exposure of this reactive ACP-bound species outside of either protein. The opportunity presents in reconstitution experiments for spontaneous intramolecular pyrone formation and the appearance of derailed, immature side products in competition with return to the KS and completion of programmed polyketide elongation.59 This contrasting behavior is illustrated for PksCT, which lies at the beginning of citrinin biosynthesis (Fig. 13B).59
The X-ray structure of only one PT domain is known, that for PksA. While at the time bioinformatics tools did not recognize the evolutionary relationship of PT and DH domains, the 1.8 Å crystal structure showed the PksA PT as a homodimer of unmistakable double “hot dog” fold monomers125 (Fig. 14A). Close to the dimer interface is the entrance to a deep active site pocket extending 30 Å into the interior. A fairly linear 14 Å channel binds the Ppant of ACP to deliver the C20-polyketide intermediate inward to an 8 × 13.5 Å reaction chamber housing a His–Asp catalytic dyad seen in DH domains (Fig. 14B). The deepest extent of the active site is then reached by a largely hydrophobic tunnel, which was designated the “hexyl binding” region to accommodate the hexyl starter of the polyketide intermediate. In support for this picture was the fortuitous co-crystallization of palmitate with its hydrocarbon tail deep in this pocket and arrayed in a fully extended conformation to place the carboxylate in the reaction chamber proximal to, but not binding with, the His–Asp dyad (Fig. 14C). Using the well-resolved C16 chain of palmitate as a template, the C20-polyketide intermediate was overlaid and docked in the active site such that C4 was constrained to be nearby the catalytic His. The energy minimized structure that emerged revealed dual hydrogen bonds specifically to the C9 carbonyl from crystallographic (static) water molecules on the “wet” side of the reaction chamber. This unanticipated outcome of the computational model gave sudden insight into how PT domains might control the pluripotent reactivity of an octa-carbonyl intermediate and guide it to a single cyclization product.125 Generation of a nucleophilic anion or enol at C4 by the catalytic His can now partner with the C9 carbonyl, which has been specifically activated by dual hydrogen bonds126 as the most reactive electrophile for intramolecular aldol reaction (Fig. 14D). Dehydration, the primordial DH function, seals first-ring formation and aromatization. The PT cyclization chamber is large enough to accommodate a second ring. One can visualize that after formation of the first ring, C2 is drawn inward to the catalytic His and the C11 carbonyl is correspondingly displaced and activated by the water network. Analogous aldol ring closure takes place to the trihydroxynaphthalene, which had been detected in earlier experiments by mass spectrometry and UV-vis spectroscopic comparison.119
 | ||
| Fig. 14 Cyclization by the PksA PT domain. (A) Structure of the dimeric PksA PT domain. (B) The proposed cyclization mechanism involves a movement of the polyketide chain into the PT cyclization chamber. Fo–Fc shaked omit electron density maps at 3.0σ of the palmitate (C) and a polyketide substrate analog (E) bound to the PT active site. (D) Derivation of the crystallized substrate analog. | ||
In support of this proposal for PT function, a bisisoxazole model of the experimentally inaccessible PksA polyketide intermediate was synthesized with thioether linkers and N,O-heteroatoms intended to mimic the alternating pattern of keto/enol sites in the native substrate. A 1.8 Å structure was obtained (Fig. 14E), which gratifyingly revealed the well-resolved mimic in an extended conformation where the Ppant stretched from the terminal phosphate interaction with a PT surface Arg to deliver the critical C4 methylene precisely to the catalytic His, in keeping with the computational model. The short hydrophobic tail is positioned as predicted in the “hexyl binding” region.127 The rigid heterocycles and unavoidable 90° C–S–C bond angles of the bridging thioethers give a congested, highly bent presentation of the carbonyl surrogates in the reaction chamber. While the mimetic accuracy of their interactions with the ordered active site water molecules is uncertain, in sum, the structure is a major advance and provides important experimental support for the in silico model of PT function proposed above.125
VI.3 Thioesterase (TE) domain
With no further ring formation possible in the PT cyclization chamber, the significantly more stable naphthaloyl aldol product is transferred to the Ser of the catalytic triad of the TE domain, where it undergoes not hydrolysis to the carboxylic acid, but a final Dieckmann cyclization and concomitant release of norsolorinic acid anthrone, which spontaneously oxidizes to the corresponding anthraquinone (Fig. 15A). This interesting variant of TE behavior is commonly seen among fungal NR-PKSs.128 The overall fold is of an α/β-hydrolase in which the active site is closed at each end, but a large lid region opens from the side to admit the substrate and release the ring-closed product most like the macrocycling NRPS TE domains129 (Fig. 15B). It has been proposed that as the ACP–Ppant departs the TE active site after transacylation, the thiolate removes the triad histidinium proton (Fig. 15A) to create a base for the final condensation. However, for the large TE lid to close over the substrate, the β-diketone of the substrate rotates into the ACP–Ppant binding channel to be oriented now proximal to the His for final Dieckmann reaction.130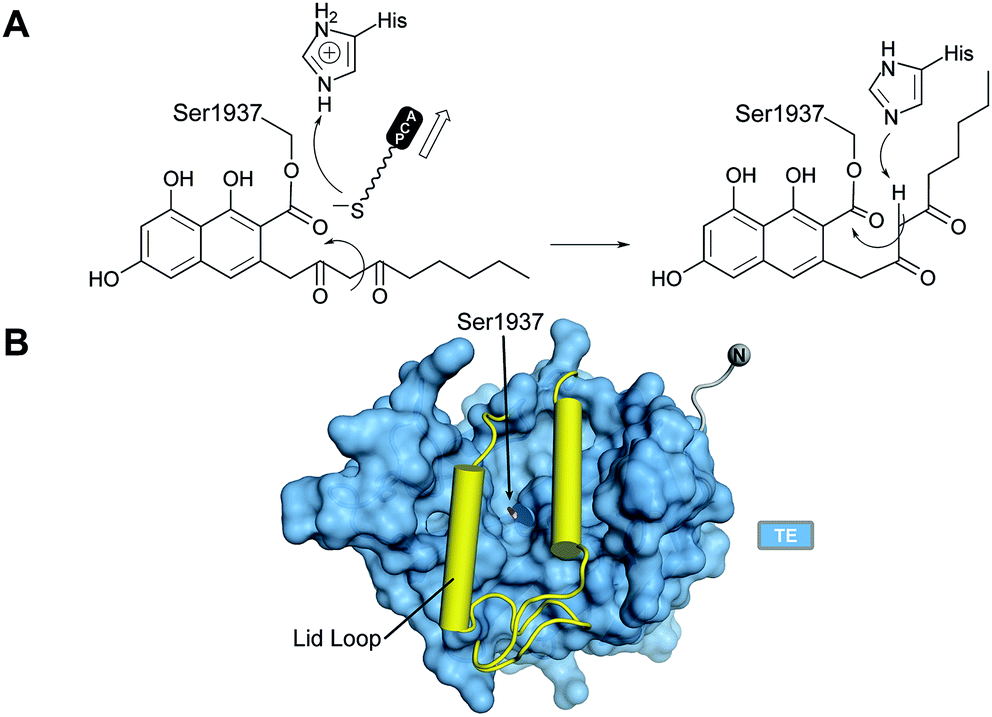 | ||
| Fig. 15 The PksA TE domain. (A) Proposed reaction mechanism and product release. (B) TE domains have a lid loop that closes the active site. | ||
VI.4 Substrate loading architecture of NR-PKSs
Enormous progress has been made in the last dozen years toward understanding the iterative catalytic cycle of NR-PKS mega-enzymes. The discovery and functional characterization of SAT and PT domains, which are unique to this family of PKSs, made it possible to assign individual synthetic tasks to all domains. Structural studies of individual domains have greatly amplified detailed understanding of key mechanistic steps. While some sense of the overall orchestration of iterative synthesis has been gained from these experiments, it has been an underlying goal for more than a decade since the mFAS and yFAS structures were reported5,37 to obtain comparable structural information about iterative PKS enzymes. Recently a major step in that direction has been made with a 2.8 Å crystal structure of the dimeric CTB1 loading/condensing region (SAT–KS–MAT) together with trapping of the KS in its substrate-loading state by mechanism-based chemical crosslinking to the ACP and analysis by cryo-EM at 7.1 Å resolution.78The crystal structure reveals a symmetrical, compact dimer of overall rhomboidal shape (Fig. 16). The integrated SAT domain closely resembles the CazM SAT and has N- and C-termini on opposite sides as oriented along the central dimer axis as in modPKSs. The two CTB1 SAT domains reveal an intertwined integration, where each SAT interfaces the KS–MAT condensing region of the neighboring protomer. The SATs form a weak dimer and are held in place by extensive interactions with the MATs and stabilization via an extensive SAT–KS linker, which binds to the surface of each neighboring SAT (Fig. 16B). As compared to the monomeric character of the excised SAT of CazM this represents another example for the relevance of avidity in multienzymes for oligomerization (see above). The condensing region (KS–MAT) reveals the same conserved organization as observed in other PKSs and mFASs,4,30,39,67,75,77,131 except for PikAIII.76 The cavity formed by the S-shaped condensing region (view along the dimeric axis) is closed at the bottom by the SATs. This arrangement forms a surface channel that connects the active site entrances to the SAT and KS active sites and might guide ACP translocation.
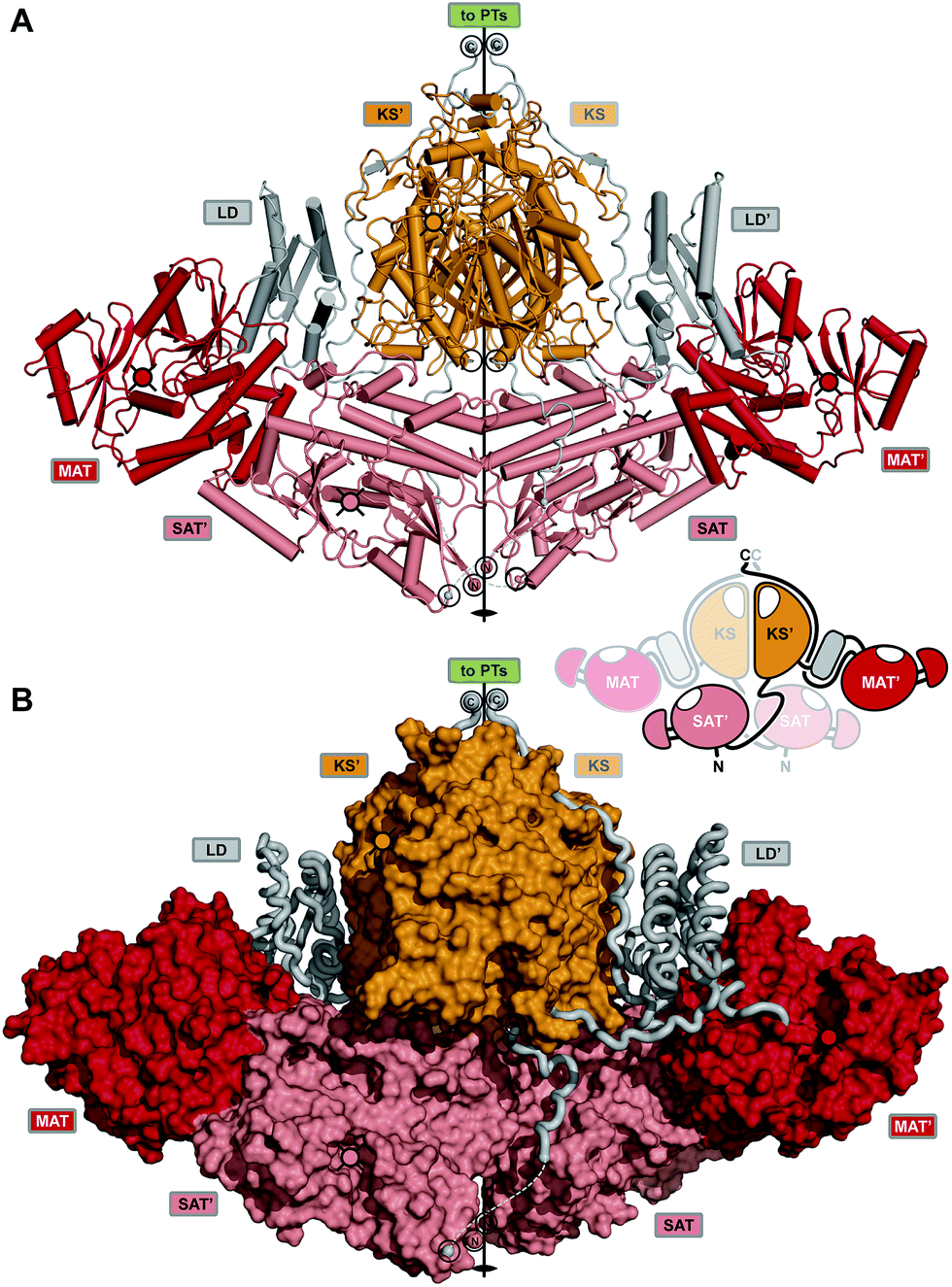 | ||
| Fig. 16 Crystal structure of the CTB1 NR-PKS loading/condensing region. (A) The SAT domains are integrated into the canonical condensing region architecture and align the N- and C-termini along the central two-fold axis (black circles) together with the termini of the KS. The inset representation shows the intertwined organization where the SAT mainly interfaces with the neighboring protomer. (B) Linker representation of (A) showing an extended SAT-linker interaction with its neighboring partner SAT domain. Active site locations and entrances, respectively, are indicated by cross-hatched filled-in circles. Domain names correspond to Fig. 6. | ||
Structural and functional analysis of productive ACP interactions with its target in most cases require a mechanism-based crosslinking approach owing to the transient nature of this interaction. This approach was developed in the Burkart Laboratory132 and has become indispensable for functional and structural studies on carrier protein-based molecular machines. So far, this approach has been employed for structural studies of the bacterial type II FAS DH133 and for the NR-PKS CTB1 loading/condensing region (SAT–KS–MAT).78
Upon crosslinking of the holo-ACP to the KS of the CTB1 loading/condensing region, conformational changes are evident that propagate throughout the dimeric structure. While crosslinking was carried out to >90% at both KS and KS′, the ACP is plainly resolved at only one KS but completely disordered at the other (Fig. 17A). Both SAT domains move toward the central KS·KS′ and the KS–MAT linker domain (LD) and MAT itself shift upward 4.4 and 8.4 Å, respectively. The high asymmetry of the domain movements gives a dramatic glimpse of an important step in the iterative catalytic cycle where C2-extension occurs. It would appear that coordinated conformational changes block reaction at KS′ on the companion protomer. Such a view is in accord with the kinetics of analogous tridomain crosslinking experiments using two other NR-PKS systems, but where a 1:2 stoichiometry (ACP:KS) was observed as rapidly as for CTB1. To achieve a full 1:1 ratio required excess ACP and prolonged reaction times.78,134 These results indicate a mechanism that mediates asymmetric coupling of polyketide biosynthesis in the two reaction clefts of the dimeric synthase (Fig. 17B and C). Notably, conformational asymmetry and coupling has also been observed during the reaction cycle of mFAS38 and for the modifying region of the MAS-like Pks5,77 pointing towards a general biosynthetic strategy.
 | ||
Fig. 17 Proposed mode of conformational coupling in NR-PKS substrate loading. (A) Asymmetric cryo-EM structure of crosslinked ACP to the KS-active site of the loading/condensing region CTB1. (B) Structures of all NR-PKSs catalytic domains have been elucidated. The structures of CTB1 SAT–KS–MAT![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ACP2 (, crosslinked to KS) and the structures of PksA TE and PT are shown. (C) Schematic representation of the NR-PKS architecture in a catalytically relevant asymmetric state. Conformational changes upon ACP binding are indicated with arrows. Domain names correspond to Fig. 6. ACP2 (, crosslinked to KS) and the structures of PksA TE and PT are shown. (C) Schematic representation of the NR-PKS architecture in a catalytically relevant asymmetric state. Conformational changes upon ACP binding are indicated with arrows. Domain names correspond to Fig. 6. | ||
VII. Conclusion
For a decade, high-resolution structural data have been available for the two distinct types of FAS multienzymes, yFAS6 and mFAS.30 These data provided a new foundation for conceptual understanding of the architecture, evolution and function of the FAS and PKS families of ACP-containing multienzymes.For yFAS, the structural insights revealed the previously enigmatic route of evolution to yeast FAS from early single-functional ancestors in bacterial primary and secondary metabolism via bacterial yFAS.17,18 Structure-guided re-engineering of yFAS has most recently succeeded in creating novel reaction modules for the synthesis of new product families.24,25
For mFAS, the structural model of the core elongation domains has enabled rational dissection into crystallizable fragments.39,40,135 These fragments have successfully been employed in screening small-molecule FAS inhibitors as potential leads against cancer and metabolic disorders. mFAS continues to serve as a test-bed for functional studies of ACP-containing multienzymes, as exemplified by attempts towards analysing mFAS dynamics at the single molecule level via fluorescence methods136 or high-speed atomic force microscopy.41
Structural insights into PKS architectures massively lagged behind studies on mFAS, despite their related domain organization. Meanwhile, different conceptual models have been discussed based on structures of isolated domains or limited size fragments of PKS. The intermediate resolution visualization of a minimal reducing modPKS module, PikAIII, in different conformational states served as a first example of PKS organization76,137 until the hybrid structural model of a MAS-like PKS was obtained with demonstrated relevance also for modPKSs.77 These structures depict different conformations and states, with additional data supporting the functional relevance of either of the structures.
A key aim of ongoing structural biology work on PKS is to conclusively reveal the principal architecture of different classes of PKSs as well as the ensemble of conformational states contributing to catalytic efficiency. The available data have reinforced the notion that overall conformational dynamics, and linker-mediated interactions are crucial for PKS function.
Obtaining a “blueprint” of the organization of different types of PKSs remains a key aim in natural product research. Site specific crosslinking of ACPs to their catalytic partner domains has proven to provide crucial “snapshots” of catalytic states not seen before. They are of fundamental value to visualize and understand the dynamic, coordinated function of sophisticated PKS molecular machines and will guide their reprogramming to new synthetic objectives.
VIII. Conflicts of interest
There are no conflicts to declare.IX. Acknowledgements
This work has been supported by the Swiss National Science Foundation grants 159696 and 179323 to TM, DAH has been supported by a Fellowship for Excellence from the Werner Siemens Foundation. CAT gratefully acknowledges support of the National Institutes of Health grant RO1 ES001670.X. Notes and references
- T. Maier, M. Leibundgut, D. Boehringer and N. Ban, Q. Rev. Biophys., 2010, 43, 373–422 CrossRef PubMed.
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176–178 CrossRef PubMed.
- M. Leibundgut, T. Maier, S. Jenni and N. Ban, Curr. Opin. Struct. Biol., 2008, 18, 714–725 CrossRef PubMed.
- A. Rittner, K. S. Paithankar, K. V. Huu and M. Grininger, ACS Chem. Biol., 2018, 13, 723–732 CrossRef PubMed.
- S. Jenni, M. Leibundgut, T. Maier and N. Ban, Science, 2006, 311, 1263–1267 CrossRef PubMed.
- S. Jenni, M. Leibundgut, D. Boehringer, C. Frick, B. Mikolasek and N. Ban, Science, 2007, 316, 254–261 CrossRef PubMed.
- M. Leibundgut, S. Jenni, C. Frick and N. Ban, Science, 2007, 316, 288–290 CrossRef PubMed.
- I. B. Lomakin, Y. Xiong and T. A. Steitz, Cell, 2007, 129, 319–332 CrossRef PubMed.
- P. Johansson, B. Wiltschi, P. Kumari, B. Kessler, C. Vonrhein, J. Vonck, D. Oesterhelt and M. Grininger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12803–12808 CrossRef PubMed.
- P. Johansson, B. Mulinacci, C. Koestler, R. Vollrath, D. Oesterhelt and M. Grininger, Structure, 2009, 17, 1063–1074 CrossRef PubMed.
- P. Gipson, D. J. Mills, R. Wouts, M. Grininger, J. Vonck and W. Kuhlbrandt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9164–9169 CrossRef PubMed.
- D. R. Perez, M. Leibundgut and G. Wider, Biochemistry, 2015, 54, 2205–2213 CrossRef PubMed.
- M. Sumper, C. Riepertinger and F. Lynen, FEBS Lett., 1969, 5, 45–49 CrossRef PubMed; J. Gajewski, F. Buelens, S. Serdjukow, M. Janssen, N. Cortina, H. Grubmuller and M. Grininger, Nat. Chem. Biol., 2017, 13, 363–365 CrossRef PubMed.
- D. N. Brindley, S. Matsumur and K. Bloch, Nature, 1969, 224, 666–669 CrossRef.
- E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68, 501–517 CrossRef PubMed.
- G. Gago, L. Diacovich, A. Arabolaza, S. C. Tsai and H. Gramajo, FEMS Microbiol. Rev., 2011, 35, 475–497 CrossRef PubMed.
- H. S. T. Bukhari, R. P. Jakob and T. Maier, Structure, 2014, 22, 1775–1785 CrossRef PubMed.
- M. Grininger, Curr. Opin. Struct. Biol., 2014, 25, 49–56 CrossRef PubMed.
- D. W. Brown, T. H. Adams and N. P. Keller, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14873–14877 CrossRef.
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728–16733 CrossRef PubMed.
- D. Boehringer, N. Ban and M. Leibundgut, J. Mol. Biol., 2013, 425, 841–849 CrossRef PubMed.
- L. Ciccarelli, S. R. Connell, M. Enderle, D. J. Mills, J. Vonck and M. Grininger, Structure, 2013, 21, 1251–1257 CrossRef PubMed.
- M. Fischer, D. Rhinow, Z. Zhu, D. J. Mills, Z. K. Zhao, J. Vonck and M. Grininger, Protein Sci., 2015, 24, 987–995 CrossRef PubMed.
- J. Gajewski, R. Pavlovic, M. Fischer, E. Boles and M. Grininger, Nat. Commun., 2017, 8, 14650 CrossRef PubMed.
- Z. Zhu, Y. J. Zhou, A. Krivoruchko, M. Grininger, Z. K. Zhao and J. Nielsen, Nat. Chem. Biol., 2017, 13, 360–362 CrossRef PubMed.
- J. Saito, M. Yamada, T. Watanabe, M. Iida, H. Kitagawa, S. Takahata, T. Ozawa, Y. Takeuchi and F. Ohsawa, Protein Sci., 2008, 17, 691–699 CrossRef PubMed.
- A. Witkowski, A. Ghosal, A. K. Joshi, H. E. Witkowska, F. J. Asturias and S. Smith, Chem. Biol., 2004, 11, 1667–1676 CrossRef PubMed.
- S. Smith, A. Witkowski and A. K. Joshi, Prog. Lipid Res., 2003, 42, 289–317 CrossRef PubMed.
- J. Brink, S. J. Ludtke, C. Y. Yang, Z. W. Gu, S. J. Wakil and W. Chiu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 138–143 CrossRef PubMed.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef PubMed.
- E. Ploskon, C. J. Arthur, S. E. Evans, C. Williams, J. Crosby, T. J. Simpson and M. P. Crump, J. Biol. Chem., 2008, 283, 518–528 CrossRef PubMed.
- B. Chakravarty, Z. Gu, S. S. Chirala, S. J. Wakil and F. A. Quiocho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15567–15572 CrossRef PubMed.
- C. W. Pemble, L. C. Johnson, S. J. Kridel and W. T. Lowther, Nat. Struct. Mol. Biol., 2007, 14, 704–709 CrossRef PubMed.
- A. K. Joshi, A. Witkowski, H. A. Berman, L. Zhang and S. Smith, Biochemistry, 2005, 44, 4100–4107 CrossRef PubMed.
- M. K. Ritchie, L. C. Johnson, J. E. Clodfelter, C. W. Pemble, B. E. Fulp, C. M. Furdui, S. J. Kridel and W. T. Lowther, J. Biol. Chem., 2016, 291, 3520–3530 CrossRef PubMed.
- A. K. Joshi, V. S. Rangan, A. Witkowski and S. Smith, Chem. Biol., 2003, 10, 169–173 CrossRef PubMed.
- T. Maier, S. Jenni and N. Ban, Science, 2006, 311, 1258–1262 CrossRef PubMed.
- E. J. Brignole, S. Smith and F. J. Asturias, Nat. Struct. Mol. Biol., 2009, 16, 190–197 CrossRef PubMed.
- G. Pappenberger, J. Benz, B. Gsell, M. Hennig, A. Ruf, M. Stihle, R. Thoma and M. G. Rudolph, J. Mol. Biol., 2010, 397, 508–519 CrossRef PubMed.
- M. A. Hardwicke, A. R. Rendina, S. P. Williams, M. L. Moore, L. Wang, J. A. Krueger, R. N. Plant, R. D. Totoritis, G. Zhang, J. Briand, W. A. Burkhart, K. K. Brown and C. A. Parrish, Nat. Chem. Biol., 2014, 10, 774–779 CrossRef PubMed.
- F. M. C. Benning, Y. Sakiyama, A. Mazur, H. S. T. Bukhari, R. Y. H. Lim and T. Maier, ACS Nano, 2017, 11, 10852–10859 CrossRef PubMed.
- C. Hertweck, Angew. Chem., Int. Ed. Engl., 2009, 48, 4688–4716 CrossRef PubMed; S. Smith and S. C. Tsai, Nat. Prod. Rep., 2007, 24, 1041–1072 RSC.
- K. J. Weissman, Nat. Chem. Biol., 2015, 11, 660–670 CrossRef PubMed; K. J. Weissman, Nat. Prod. Rep., 2015, 32, 436–453 RSC.
- T. Bretschneider, G. Zocher, M. Unger, K. Scherlach, T. Stehle and C. Hertweck, Nat. Chem. Biol., 2011, 8, 154–161 CrossRef PubMed.
- T. D. Sirakova, A. K. Thirumala, V. S. Dubey, H. Sprecher and P. E. Kolattukudy, J. Biol. Chem., 2001, 276, 16833–16839 CrossRef PubMed.
- R. S. Gokhale, P. Saxena, T. Chopra and D. Mohanty, Nat. Prod. Rep., 2007, 24, 267–277 RSC.
- K. J. Weissman, ChemBioChem, 2010, 11, 485–488 CrossRef PubMed.
- S. G. Van Lanen, S. Lin and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 494–499 CrossRef PubMed; W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef PubMed.
- J. A. Blodgett, D. C. Oh, S. Cao, C. R. Currie, R. Kolter and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11692–11697 CrossRef PubMed; Y. Li, H. Chen, Y. Ding, Y. Xie, H. Wang, R. L. Cerny, Y. Shen and L. Du, Angew. Chem., Int. Ed. Engl., 2014, 53, 7524–7530 CrossRef PubMed.
- U. Kaulmann and C. Hertweck, Angew. Chem., Int. Ed. Engl., 2002, 41, 1866–1869 CrossRef PubMed.
- C. Olano, B. Wilkinson, C. Sanchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Brana, C. J. Martin, M. Oliynyk, C. Mendez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87–97 Search PubMed.
- S. J. Moss, C. J. Martin and B. Wilkinson, Nat. Prod. Rep., 2004, 21, 575–593 RSC.
- R. H. Proctor, D. W. Brown, R. D. Plattner and A. E. Desjardins, Fungal Genet. Biol., 2003, 38, 237–249 CrossRef PubMed.
- L. Hendrickson, C. R. Davis, C. Roach, D. K. Nguyen, T. Aldrich, P. C. McAda and C. D. Reeves, Chem. Biol., 1999, 6, 429–439 CrossRef PubMed.
- C. D. Campbell and J. C. Vederas, Biopolymers, 2010, 93, 755–763 CrossRef PubMed.
- S. B. Singh, D. L. Zink, M. A. Goetz, A. W. Dombrowski, J. D. Polishook and D. J. Hazuda, Tetrahedron Lett., 1998, 39, 2243–2246 CrossRef.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. Van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519–11520 CrossRef; W. L. Kelly, Org. Biomol. Chem., 2008, 6, 4483–4493 RSC.
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef PubMed.
- P. A. Storm, D. A. Herbst, T. Maier and C. A. Townsend, Cell Chem. Biol., 2017, 24, 316–325 CrossRef PubMed.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. S. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef PubMed.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2017, 8, 1116–1126 RSC.
- T. S. Hitchman, E. W. Schmidt, F. Trail, M. D. Rarick, J. E. Linz and C. A. Townsend, Bioorg. Chem., 2001, 29, 293–307 CrossRef PubMed.
- J. Foulke-Abel and C. A. Townsend, ChemBioChem, 2012, 13, 1880–1884 CrossRef PubMed.
- L. L. Bedard and T. E. Massey, Cancer Lett., 2006, 241, 174–183 CrossRef PubMed.
- J. M. Winter, D. Cascio, D. Dietrich, M. Sato, K. Watanabe, M. R. Sawaya, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 9885–9893 CrossRef PubMed.
- R. W. Broadhurst, D. Nietlispach, M. P. Wheatcroft, P. F. Leadlay and K. J. Weissman, Chem. Biol., 2003, 10, 723–731 CrossRef PubMed; T. J. Buchholz, T. W. Geders, F. E. Bartley III, K. A. Reynolds, J. L. Smith and D. H. Sherman, ACS Chem. Biol., 2009, 4, 41–52 CrossRef PubMed; J. Dorival, T. Annaval, F. Risser, S. Collin, P. Roblin, C. Jacob, A. Gruez, B. Chagot and K. J. Weissman, J. Am. Chem. Soc., 2016, 138, 4155–4167 CrossRef PubMed; J. Zeng, D. T. Wagner, Z. Zhang, L. Moretto, J. D. Addison and A. T. Keatinge-Clay, ACS Chem. Biol., 2016, 11, 2466–2474 CrossRef PubMed; C. D. Richter, D. Nietlispach, R. W. Broadhurst and K. J. Weissman, Nat. Chem. Biol., 2008, 4, 75–81 CrossRef PubMed.
- J. R. Whicher, S. S. Smaga, D. A. Hansen, W. C. Brown, W. H. Gerwick, D. H. Sherman and J. L. Smith, Chem. Biol., 2013, 20, 1340–1351 CrossRef PubMed.
- D. P. Dowling, Y. Kung, A. K. Croft, K. Taghizadeh, W. L. Kelly, C. T. Walsh and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 12432–12437 CrossRef PubMed.
- J. Schumann and C. Hertweck, J. Am. Chem. Soc., 2007, 129, 9564–9565 CrossRef PubMed.
- W. X. Jiao, Y. J. Feng, J. W. Blunt, A. L. J. Cole and M. H. G. Munro, J. Nat. Prod., 2004, 67, 1722–1725 CrossRef PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef PubMed.
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. K. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef PubMed.
- J. W. Sims, J. P. Fillmore, D. D. Warner and E. W. Schmidt, Chem. Commun., 2005, 186–188 RSC.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W. Li, J. C. Vederas, Y. Tang and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef PubMed.
- Y. Tang, A. Y. Chen, C. Y. Kim, D. E. Cane and C. Khosla, Chem. Biol., 2007, 14, 931–943 CrossRef PubMed; Y. Tang, C. Y. Kim, I. I. Mathews, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11124–11129 CrossRef PubMed.
- S. Dutta, J. R. Whicher, D. A. Hansen, W. A. Hale, J. A. Chemler, G. R. Congdon, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 512–517 CrossRef PubMed.
- D. A. Herbst, R. P. Jakob, F. Zahringer and T. Maier, Nature, 2016, 531, 533–537 CrossRef PubMed.
- D. A. Herbst, C. R. Huitt-Roehl, R. P. Jakob, J. M. Kravetz, P. A. Storm, J. R. Alley, C. A. Townsend and T. Maier, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef PubMed.
- M. A. Skiba, A. P. Sikkema, W. D. Fiers, W. H. Gerwick, D. H. Sherman, C. C. Aldrich and J. L. Smith, ACS Chem. Biol., 2016, 11, 3319–3327 CrossRef PubMed.
- T. Chopra and R. S. Gokhale, Methods Enzymol., 2009, 459, 259–294 Search PubMed; L. Brown, J. M. Wolf, R. Prados-Rosales and A. Casadevall, Nat. Rev. Microbiol., 2015, 13, 620–630 CrossRef PubMed.
- L. E. N. Quadri, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 179–211 CrossRef PubMed.
- C. Astarie-Dequeker, L. Le Guyader, W. Malaga, F. K. Seaphanh, C. Chalut, A. Lopez and C. Guilhot, PLoS Pathog., 2009, 5, e1000289 CrossRef PubMed; C. J. Cambier, K. K. Takaki, R. P. Larson, R. E. Hernandez, D. M. Tobin, K. B. Urdahl, C. L. Cosma and L. Ramakrishnan, Nature, 2014, 505, 218–222 CrossRef PubMed; M. B. Reed, P. Domenech, C. Manca, H. Su, A. K. Barczak, B. N. Kreiswirth, G. Kaplan and C. E. Barry III, Nature, 2004, 431, 84–87 CrossRef PubMed.
- L. R. Camacho, P. Constant, C. Raynaud, M. A. Laneelle, J. A. Triccas, B. Gicquel, M. Daffe and C. Guilhot, J. Biol. Chem., 2001, 276, 19845–19854 CrossRef PubMed.
- J. S. Cox, B. Chen, M. McNeil and W. R. Jacobs Jr, Nature, 1999, 402, 79–83 CrossRef PubMed.
- Y. Rombouts, L. Alibaud, S. Carrere-Kremer, E. Maes, C. Tokarski, E. Elass, L. Kremer and Y. Guerardel, J. Biol. Chem., 2011, 286, 33678–33688 CrossRef PubMed.
- G. Etienne, W. Malaga, F. Laval, A. Lemassu, C. Guilhot and M. Daffe, J. Bacteriol., 2009, 191, 2613–2621 CrossRef PubMed.
- J. Buglino, K. C. Onwueme, J. A. Ferreras, L. E. Quadri and C. D. Lima, J. Biol. Chem., 2004, 279, 30634–30642 CrossRef PubMed; O. A. Trivedi, P. Arora, A. Vats, M. Z. Ansari, R. Tickoo, V. Sridharan, D. Mohanty and R. S. Gokhale, Mol. Cell, 2005, 17, 631–643 CrossRef PubMed.
- K. C. Onwueme, J. A. Ferreras, J. Buglino, C. D. Lima and L. E. Quadri, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4608–4613 CrossRef PubMed.
- X. K. Xie, K. Watanabe, W. A. Wojcicki, C. C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef PubMed.
- E. C. Boritsch, W. Frigui, A. Cascioferro, W. Malaga, G. Etienne, F. Laval, A. Pawlik, F. Le Chevalier, M. Orgeur, L. Ma, C. Bouchier, T. P. Stinear, P. Supply, L. Majlessi, M. Daffe, C. Guilhot and R. Brosch, Nat. Microbiol., 2016, 1, 15019 CrossRef PubMed.
- A. L. Edwards, T. Matsui, T. M. Weiss and C. Khosla, J. Mol. Biol., 2014, 426, 2229–2245 CrossRef PubMed.
- A. Rittner and M. Grininger, ChemBioChem, 2014, 15, 2489–2493 CrossRef PubMed.
- A. Keatinge-Clay, J. Mol. Biol., 2008, 384, 941–953 CrossRef PubMed.
- D. L. Akey, J. R. Razelun, J. Tehranisa, D. H. Sherman, W. H. Gerwick and J. L. Smith, Structure, 2010, 18, 94–105 CrossRef PubMed.
- W. D. Fiers, G. J. Dodge, D. H. Sherman, J. L. Smith and C. C. Aldrich, J. Am. Chem. Soc., 2016, 138, 16024–16036 CrossRef PubMed; A. Faille, S. Gavalda, N. Slama, C. Lherbet, L. Maveyraud, V. Guillet, F. Laval, A. Quemard, L. Mourey and J. D. Pedelacq, J. Mol. Biol., 2017, 429, 1554–1569 CrossRef PubMed.
- D. Gay, Y. O. You, A. Keatinge-Clay and D. E. Cane, Biochemistry, 2013, 52(49), 8916–8928 CrossRef PubMed.
- T. Bretschneider, J. B. Heim, D. Heine, R. Winkler, B. Busch, B. Kusebauch, T. Stehle, G. Zocher and C. Hertweck, Nature, 2013, 502, 124–128 CrossRef PubMed.
- S. A. Bonnett, J. R. Whicher, K. Papireddy, G. Florova, J. L. Smith and K. A. Reynolds, Chem. Biol., 2013, 20, 772–783 CrossRef PubMed; M. Schafer, C. E. Stevenson, B. Wilkinson, D. M. Lawson and M. J. Buttner, Cell Chem. Biol., 2016, 23, 1091–1097 CrossRef PubMed.
- A. T. Keatinge-Clay, Chem. Biol., 2007, 14, 898–908 CrossRef PubMed; A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 CrossRef PubMed; J. Zheng and A. T. Keatinge-Clay, J. Mol. Biol., 2011, 410, 105–117 CrossRef PubMed; J. Zheng, S. K. Piasecki and A. T. Keatinge-Clay, ACS Chem. Biol., 2013, 8, 1964–1971 CrossRef PubMed; J. Zheng, C. A. Taylor, S. K. Piasecki and A. T. Keatinge-Clay, Structure, 2010, 18, 913–922 CrossRef PubMed.
- J. Zheng, D. C. Gay, B. Demeler, M. A. White and A. T. Keatinge-Clay, Nat. Chem. Biol., 2012, 8, 615–621 CrossRef PubMed.
- T. Lu, C. Schubert, M. D. Cummings, G. Bignan, P. J. Connolly, K. Smans, D. Ludovici, M. H. Parker, C. Meyer, C. Rocaboy, R. Alexander, B. Grasberger, S. De Breucker, N. Esser, E. Fraiponts, R. Gilissen, B. Janssens, D. Peeters, L. Van Nuffel, P. Vermeulen, J. Bischoff and L. Meerpoel, Bioorg. Med. Chem. Lett., 2018, 28, 2159–2164 CrossRef PubMed.
- D. Khare, W. A. Hale, A. Tripathi, L. Gu, D. H. Sherman, W. H. Gerwick, K. Hakansson and J. L. Smith, Structure, 2015, 23(12), 2213–2223 CrossRef PubMed.
- B. Busch, N. Ueberschaar, S. Behnken, Y. Sugimoto, M. Werneburg, N. Traitcheva, J. He and C. Hertweck, Angew. Chem., Int. Ed. Engl., 2013, 52, 5285–5289 CrossRef PubMed.
- S. Kapur, B. Lowry, S. Yuzawa, S. Kenthirapalan, A. Y. Chen, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4110–4115 CrossRef PubMed.
- B. Lowry, X. Li, T. Robbins, D. E. Cane and C. Khosla, ACS Cent. Sci., 2016, 2, 14–20 CrossRef PubMed.
- B. J. Beck, C. C. Aldrich, R. A. Fecik, K. A. Reynolds and D. H. Sherman, J. Am. Chem. Soc., 2003, 125, 4682–4683 CrossRef PubMed.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Science, 1991, 252, 675–679 CrossRef PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC; C. Khosla, Y. Tang, A. Y. Chen, N. A. Schnarr and D. E. Cane, Annu. Rev. Biochem., 2007, 76, 195–221 CrossRef PubMed.
- D. W. Udwary, M. Merski and C. A. Townsend, J. Mol. Biol., 2002, 323, 585–598 CrossRef PubMed.
- W. B. Turner and D. C. Aldridge, Fungal Metabolites II, Academic Press, London, 1983 Search PubMed.
- S. Kroken, N. L. Glass, J. W. Taylor, O. C. Yoder and B. G. Turgeon, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15670–15675 CrossRef PubMed.
- R. E. Bradshaw and S. Zhang, Mycopathologia, 2006, 162, 201–213 CrossRef PubMed.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, J. Chem. Soc., Chem. Commun., 2007, 4053–4055 RSC; R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- J. M. Crawford and C. A. Townsend, Nat. Rev. Microbiol., 2010, 8, 879–889 CrossRef PubMed.
- A. G. Newman and C. A. Townsend, J. Am. Chem. Soc., 2016, 138, 4219–4228 CrossRef PubMed.
- R. E. Minto and C. A. Townsend, Chem. Rev., 1997, 97, 2537–2555 CrossRef PubMed.
- C. R. Huitt-Roehl, E. A. Hill, M. M. Adams, A. L. Vagstad, J. W. Li and C. A. Townsend, ACS Chem. Biol., 2015, 10, 1443–1449 CrossRef PubMed.
- H. Zhou, K. J. Qiao, Z. Z. Gao, J. C. Vederas and Y. Tang, J. Biol. Chem., 2010, 285, 41412–41421 CrossRef PubMed.
- J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243–246 CrossRef PubMed.
- Y. R. Li, W. Xu and Y. Tang, J. Biol. Chem., 2010, 285, 22762–22771 Search PubMed.
- A. G. Newman, A. L. Vagstad, P. A. Storm and C. A. Townsend, J. Am. Chem. Soc., 2014, 136, 7348–7362 CrossRef PubMed; A. L. Vagstad, A. G. Newman, P. A. Storm, K. Belecki, J. M. Crawford and C. A. Townsend, Angew. Chem., Int. Ed. Engl., 2013, 52, 1718–1721 CrossRef PubMed.
- H. Zhou, Y. Li and Y. Tang, Nat. Prod. Rep., 2010, 27, 839–868 RSC.
- S. M. Ma, K. Watanabe, X. Xie, W. Zhang, C. C. C. Wang and Y. Tang, J. Am. Chem. Soc., 2007, 129, 10642–10643 CrossRef PubMed.
- A. L. Vagstad, S. B. Bumpus, K. Belecki, N. L. Kelleher and C. A. Townsend, J. Am. Chem. Soc., 2012, 134, 6865–6877 CrossRef PubMed.
- J. M. Crawford, T. P. Korman, J. W. Labonte, A. L. Vagstad, E. A. Hill, O. Kamari-Bidkorpeh, S. C. Tsai and C. A. Townsend, Nature, 2009, 461, 1139–1143 CrossRef PubMed.
- J. Hine and K. Ahn, J. Org. Chem., 1987, 52, 2083–2086 CrossRef; F. Hibbert and J. Emsley, Adv. Phys. Org. Chem., 1990, 26, 255–379 CrossRef.
- J. F. Barajas, G. Shakya, G. Moreno, H. Rivera Jr, D. R. Jackson, C. L. Topper, A. L. Vagstad, J. J. La Clair, C. A. Townsend, M. D. Burkart and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4142–E4148 CrossRef PubMed.
- I. Fujii, A. Watanabe, U. Sankawa and Y. Ebizuka, Chem. Biol., 2001, 8, 189–197 CrossRef PubMed.
- S. D. Brunner, T. Weber, R. H. Kohli, D. Schwarzer, M. A. Marahiel, C. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310 CrossRef; S. A. Samel, B. Wagner, M. A. Marahiel and L.-O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef PubMed.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S. C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246–6251 CrossRef PubMed.
- X. Li, N. Sevillano, F. La Greca, L. Deis, Y. C. Liu, M. C. Deller, I. I. Mathews, T. Matsui, D. E. Cane, C. S. Craik and C. Khosla, J. Am. Chem. Soc., 2018, 140, 6518–6521 CrossRef PubMed.
- J. L. Meier, A. C. Mercer, H. Rivera Jr and M. D. Burkart, J. Am. Chem. Soc., 2006, 128, 12174–12184 CrossRef PubMed; A. S. Worthington and M. D. Burkart, Org. Biomol. Chem., 2006, 4, 44–46 RSC; A. S. Worthington, H. Rivera, J. W. Torpey, M. D. Alexander and M. D. Burkart, ACS Chem. Biol., 2006, 1, 687–691 CrossRef PubMed.
- C. Nguyen, R. W. Haushalter, D. J. Lee, P. R. Markwick, J. Bruegger, G. Caldara-Festin, K. Finzel, D. R. Jackson, F. Ishikawa, B. O'Dowd, J. A. McCammon, S. J. Opella, S. C. Tsai and M. D. Burkart, Nature, 2014, 505, 427–431 CrossRef PubMed.
- J. Bruegger, R. W. Haushalter, A. L. Vagstad, G. Shakya, N. Mih, C. A. Townsend, M. D. Burkart and S. C. Tsai, Chem. Biol., 2013, 20, 1135–1146 CrossRef PubMed.
- K. H. Sippel, N. K. Vyas, W. Zhang, B. Sankaran and F. A. Quiocho, J. Biol. Chem., 2014, 289, 33287–33295 CrossRef PubMed.
- C. S. Heil, A. Rittner, B. Goebel, D. Beyer and M. Grininger, bioRxiv, 2018 DOI:10.1101/282525.
- J. R. Whicher, S. Dutta, D. A. Hansen, W. A. Hale, J. A. Chemler, A. M. Dosey, A. R. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef PubMed.
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- A. G. McInnes, D. G. Smith, J. A. Walter, L. C. Vining and J. L. C. Wright, J. Chem. Soc., Chem. Commun., 1974, 282–284 RSC; K. L. Eley, L. M. Halo, Z. S. Song, H. Powles, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2007, 8, 289–297 CrossRef PubMed.
- R. H. Proctor, A. E. Desjardins, R. D. Plattner and T. M. Hohn, Fungal Genet. Biol., 1999, 27, 100–112 CrossRef PubMed.
- H. R. Burmeister, G. A. Bennett, R. F. Vesonder and C. W. Hesseltine, Antimicrob. Agents Chemother., 1974, 5, 634–639 CrossRef PubMed.
- R. V. K. Cochrane, R. Sanichar, G. R. Lambkin, B. Reiz, W. Xu, Y. Tang and J. C. Vederas, Angew. Chem., Int. Ed. Engl., 2016, 55, 664–668 CrossRef PubMed.
- C. D. Reeves, Z. H. Hu, R. Reid and J. T. Kealey, Appl. Environ. Microbiol., 2008, 74, 5121–5129 CrossRef PubMed.
- Y. T. Kim, Y. R. Lee, J. M. Jin, K. H. Han, H. Kim, J. C. Kim, T. Lee, S. H. Yun and Y. W. Lee, Mol. Microbiol., 2005, 58, 1102–1113 CrossRef PubMed; G. I. Gaffoor and F. Trail, Appl. Environ. Microbiol., 2006, 72, 1793–1799 CrossRef PubMed.
- Y. Q. Xu, P. Espinosa-Artiles, V. Schubert, Y. M. Xu, W. Zhang, M. Lin, A. A. L. Gunatilaka, R. Sussmuth and I. Molnar, Appl. Environ. Microbiol., 2013, 79, 2038–2047 CrossRef PubMed.
- Y. M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef PubMed.
- J. M. Winter, M. Sato, S. Sugimoto, G. Chiou, N. K. Garg, Y. Tang and K. Watanabe, J. Am. Chem. Soc., 2012, 134, 17900–17903 CrossRef PubMed.
- K. Kasahara, T. Miyamoto, T. Fujimoto, H. Oguri, T. Tokiwano, H. Oikawa, Y. Ebizuka and I. Fujii, ChemBioChem, 2010, 11, 1245–1252 CrossRef PubMed.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. M. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261, 10.1039/b411973h.
- G. Yang, M. S. Rose, B. G. Turgeon and O. C. Yoder, Plant Cell, 1996, 8, 2139–2150 CrossRef PubMed; S. E. Baker, S. Kroken, P. Inderbitzin, T. Asvarak, B. Y. Li, L. Shi, O. C. Yoder and B. G. Turgeon, Mol. Plant-Microbe Interact., 2006, 19, 139–149 CrossRef PubMed.
- Z. S. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef PubMed.
- S. Bergmann, J. Schumann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef PubMed.
- A. O. Zabala, Y. H. Chooi, M. S. Choi, H. C. Lin and Y. Tang, ACS Chem. Biol., 2014, 9, 1576–1586 CrossRef PubMed.
- Y. Abe, T. Suzuki, C. Ono, K. Iwamoto, M. Hosobuchi and H. Yoshikawa, Mol. Genet. Genomics, 2002, 267, 636–646 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |