
Thiphenylmethane based structural fragments as building blocks towards solution-processable heteroleptic iridium(III) complexes for OLED use†
Kaspars
Traskovskis
 *a,
Armands
Ruduss
a,
Valdis
Kokars
a,
Igors
Mihailovs
ab,
Natalija
Lesina
b and
Aivars
Vembris
b
*a,
Armands
Ruduss
a,
Valdis
Kokars
a,
Igors
Mihailovs
ab,
Natalija
Lesina
b and
Aivars
Vembris
b
aRiga Technical University, Faculty of Materials Science and Applied Chemistry, 3/7 Paula Valdena Street, Riga LV-1048, Latvia. E-mail: kaspars.traskovskis@rtu.lv; Tel: +371 29148070
bInstitute of Solid State Physics, University of Latvia, 8 Kengaraga Street, Riga LV-1063, Latvia
First published on 12th November 2018
Abstract
A novel structural approach to solution-processable heteroleptic iridium(III) complexes is presented. On the basis of 2-arylbenzo[d]thiazole cyclometalating main ligands and picolinic acid (pic) and acetylacetone (acac) ancillary ligands six new yellow or orange emitting materials were obtained using attached 1,1,1-triphenylmethylpentane substituents as aggregation preventing and solubility enhancing functional fragments. The obtained compounds show high photoluminescence quantum yield values in the range of 0.64 to 0.90. OLEDs with a spin-coated emissive layer were successfully prepared, with the highest achieved external quantum efficiency of 7.9%, current efficiency of 12.4 cd A−1 and power efficiency of 7.8 lm W−1, measured at luminance 6000 cd m−2. Maximal device brightness of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 451 cd m−2 was attained.
451 cd m−2 was attained.
Introduction
During the last few decades the practical use of organic light emitting diodes (OLEDs) has increased significantly in such areas as flat panel displays and lighting devices.1–3 Amongst the compounds used as light emitters, special interest can be attributed to phosphorescent transition metal complexes.4 The inherent ability of these materials to harvest triplet excited states allows electroluminescent devices with 100% internal quantum efficiency to be obtained, in contrast to their fluorescent counterparts that are capped at 25%.5 Amid a wide selection of the studied compounds, cyclometalated iridium(III) complexes can be named as the most extensively researched material class because of their chemical stability, large phosphorescence quantum yields, relatively short phosphorescence lifetimes and wide possibilities of color-tuning through modification of the chemical structure of the attached ligands.6,7Yellow/orange-phosphorescent compounds can be outlined as a distinctive subset of light emitting materials.8 By falling outside of the spectrum of primary colors (red, green and blue), the main proposed application direction of these emitters involves their incorporation in solid state lighting sources. Particularly, two-component white OLEDs, consisting of blue fluorescent and yellow/orange phosphorescent emitters, are considered as a promising light-source device architecture due to a simple design and high efficiency parameters.9–11 Iridium(III) complexes, based on a 2-phenylbenzo[d]thiazole (bt) cyclometalating ligand framework, are the most extensively researched and applied class of yellow phosphorescent materials, with the attained OLED external quantum efficiencies well over the 20% mark for vacuum-deposited devices.12–16
The high production cost of OLEDs is currently limiting a more widespread use of this technology. This is largely attributed to the complicated and energy-demanding vacuum deposition technique that is used to acquire the active layers of the devices. In order to avoid luminescence-quenching that takes place due to the intermolecular interactions, iridium(III) complexes are often mixed in host molecules, with typical concentrations up to 10 weight percent (wt%). As a consequence, this brings additional difficulties during the vacuum-deposition process. It is shown that the material distribution in the acquired films is not homogenous and guest molecules form aggregate networks at concentrations as low as 5 wt%, contributing to a fall in efficiency of OLEDs through the triplet–triplet annihilation process.17 Additionally, a high thermal stability is a requirement for the emitters because chemical transformations can take place during the harsh conditions of thermal evaporation.18 This greatly reduces the number of potentially useful compounds. Solution-processable iridium(III) complexes are being extensively developed in order to overcome these disadvantages, with the dendritic molecular design being the most widespread research direction.19 The iridium(III) complex core is encapsulated by site-isolating and solubility enhancing groups in these compounds, allowing them to be processed with cost-effective solution based methods like spin-coating and ink-jet printing. The research in this direction continues, in order to achieve a right balance between solubility, stability of the solid state morphology, thermal rigidness and charge carrier mobility of materials. This is needed, as the attachment of additional functional fragments to the emitters compromises the efficiency of the devices.20
In this study we explore a novel structural design of solution-processable iridium(III) complexes. On the basis of three different 2-arylbenzo[d]thiazole main ligands, bearing 4-hydroxyphenyl, 4-hydroxynapthyl or carbazole aromatic fragments, and acetylacetone (acac) and 2-picolinic acid (pic) ancillary ligands, we acquired six new solution-processable and glass-forming materials. The phase-behavior of the low-solubility cyclometalated bt based compounds is modified by the introduction of a 1,1,1-triphenylmethylpentane group. Triphenylmethyl-based substituents are known to enhance photo-physical performance characteristics of different organic dye systems by providing an efficient site-isolation and subsequent limitation of intermolecular interactions.21,22 Here we explore the viability of this molecular design towards the acquisition of phosphorescent materials for use in OLEDs by measuring photo-physical, thermoplastic and electroluminescence properties of the acquired compounds.
Results and discussion
Design and synthesis
Introduction of electron donating atoms (O, N) at the 4-position of the benzyl ring of the bt ligand has been shown to be beneficial to the light emission efficiency of the corresponding Ir(III) complexes.23 On the other hand, the polar substituents are known to enhance molecular aggregation processes through electrostatic interactions or by hydrogen bonding, resulting in materials with reduced solubility and increased crystallinity. To overcome this, modified bt-derived ligands containing phenol (Phbt), naphthol (Nphbt) or carbazole (Cbzbt) fragments were acquired, where a phase-behavior-modifying 1,1,1-triphenylpentyl group was attached to the electronegative atoms (Scheme 1). The corresponding cyclometalated heteroleptic iridium(III) complexes were synthesized with satisfactory yields in two steps. First, Nonoyama's procedure24 was applied to obtain μ-chloro-bridged dimers. Then ancillary ligands, acetylacetone (acac) or 2-picolinic acid (pic), were introduced by using 2-ethoxyethanol as the solvent and potassium carbonate as the base. The 1H NMR spectra and the results of elemental analysis are consistent with the proposed structures. The acquired compounds show excellent solubility in volatile, low- to medium-polarity solvents like DCM and chloroform, forming glassy, amorphous mass under minimal solvent exposure. At the same time the compounds do not dissolve in alcohols, showing potential suitability for a sequential solvent-based layer-on-layer deposition during the device fabrication process.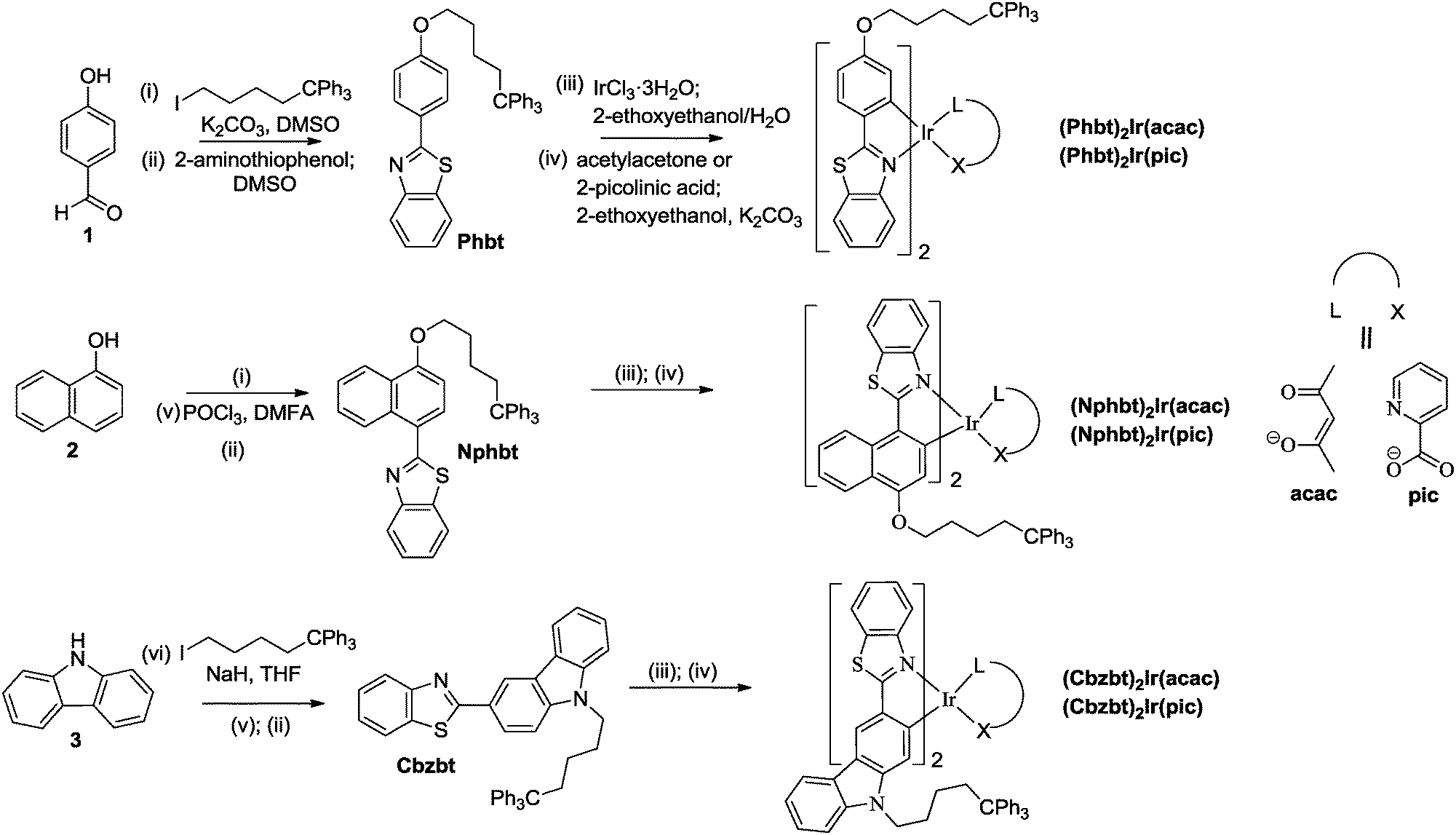 | ||
| Scheme 1 Synthesis of heteroleptic Ir(III) complexes. | ||
Thermal properties and phase behavior
Thermal properties of the materials were examined employing differential scanning calorimetric (DSC) and thermogravimetric (TG) techniques. According to the acquired DSC thermograms (Fig. 1), all the compounds show closely similar phase behavior characteristics. Only one signal, corresponding to a glass transition temperature (Tg), can be detected during the heating process, indicating that the synthesized materials form a highly stable amorphous phase. No signs of inherent crystallinity were detected either in DSC scans of the pristine samples, or during multiple heating–cooling scan cycles. Glassy, transparent films of the materials with high optical quality can be obtained via a spin-coating technique using volatile solvents (chloroform, DCM) with no observable phase changes after months of storage. Regarding the measured Tg values, a notable dispersion of this parameter is evident amongst the studied series, ranging from 124 to 188 °C. Tg increase in the class of low-molecular-weight glass-forming compounds (molecular glasses) can be correlated to larger molecular weight and reduced conformational freedom of molecules.25 A combination of these parameters can partly explain the observed Tg variations, as the compounds based on heavier Nphbt and Cbzbt ligands show higher glass transition temperatures, in contrast to their structural counterparts containing the smaller Phbt ligand. Besides the influence of the main ligand, Tg values of the materials strongly correlate with the structure of the applied ancillary ligand, as the compounds containing a pic moiety show higher glass transition values than those containing an acac group, ranging from 11 °C increase in the case of Cbzbt to 41 °C for Nphbt based complexes. It is known that iridium(III) complexes that contain a pic structural fragment tend to participate in C–H⋯O type hydrogen bonding processes due to the presence of sterically unshielded oxygen atoms of the unbound carboxyl group.26 Thus the existence of solid-phase intermolecular hydrogen bonding is the most likely cause for the observed Tg increase. | ||
| Fig. 1 (a) DSC scans of the materials with outlined Tg values. (b) TG thermograms. | ||
Regarding the thermal stability of the compounds, the results of the performed thermogravimetric analysis (TG) (Fig. 1b) reveal that compounds exhibit decomposition onset in the range 320–350 °C. A steady mass loss of several percent is observed for materials during the heating process that can be attributed to a gradual loss of residual solvent inclusions in the amorphous material bulk. It is worth noting that compounds containing pic ancillary ligands show a tendency of having slightly lower thermal stability in comparison to acac analogues.
Photophysical properties
UV-Vis absorption spectra of the synthesized compounds (Fig. 2 and Table 1) feature a broad absorption region in the 300–500 nm range that is similar to that of bt based close structural analogues.15,23 The most intensive absorption bands, in the 300–350 nm region, can be attributed to the 1π–π* transitions of cyclometalating ligands. Medium strength absorption in the 380–480 nm range marks spin allowed metal-to-ligand charge transitions (1MLCT), while shoulders with a weak intensity stretching to lower-energy wavelengths can be attributed to a spin-forbidden 3MLCT excitation. A bathochromic shift of the lowest energy absorption bands can be observed in the series of Phbt, Cbzt and Nphbt ligand based complexes. This shift can be associated with an extension of the delocalized π-electron system in the corresponding 2-arylbenzo[d]thiazoles that leads to a lowering of local singlet and triplet bandgap values.27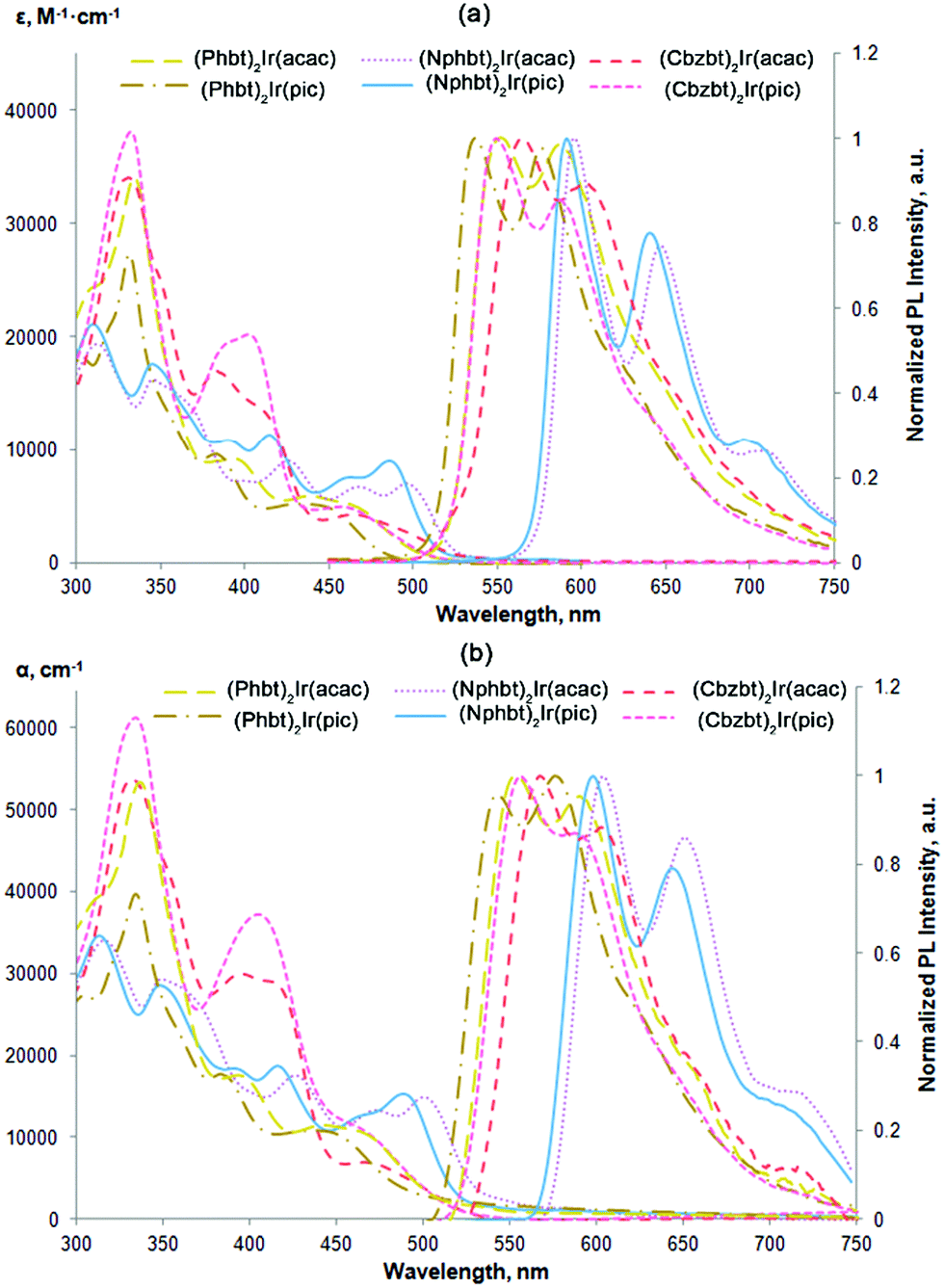 | ||
| Fig. 2 (a) UV-Vis absorption and PL spectra in THF solution, (b) UV-Vis absorption of complexes in amorphous host-free films and PL spectra in the CBP host at 20 wt% emitter concentration. | ||
| Compound | Absorbancea, λmax, nm (lgε) |
Emissionb, λmax, nm | Φ PL (THF) | τ , μs | k r , × 105 s−1 | k nr , × 105 s−1 | ΔES0→S1g, eV | ΔES0→T1g, eV |
|---|---|---|---|---|---|---|---|---|
| a Measured in THF solution. b Values in THF solution (and CBP film at 20 wt% concentration). c Values in deoxygenated THF solution/host-free films/20 wt% PVK film/20 wt% CBP film. d Measured in PMMA film at 1% emitter concentration. e Radiative decay rate kr = ΦPL/τ. f Non-radiative decay rate knr = (1 − ΦPL)/τ. g Calculated excitation energy values of the lowest energy S0 → S1 and S0 → T1 transitions. | ||||||||
| (Phbt)2Ir(acac) | 464(3.7), 437(3.8), 391(4.0), 334(4.5) | ![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif) ![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) , 588 ( , 588 (![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) , 591) , 591) |
0.78/0.02/0.15/0.31 | 3.32 | 1.56 | 1.44 | 3.38 | 2.54 |
| (Phbt)2Ir(pic) | 433(3.7), 382(4.0), 331(4.4) | ![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif) , 576 (544, , 576 (544, ![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) ) ) |
0.87/0.10/0.06/0.39 | 4.48 | 1.78 | 0.45 | 3.41 | 2.56 |
| (Nphbt)2Ir(acac) | 496(4.1), 467(4.1), 425(4.3), 398(4.2), 345(4.5) | ![[9 with combining low line]](https://www.rsc.org/images/entities/char_0039_0332.gif) , 648 ( , 648 (![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif) ![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) , 651) , 651) |
0.64/0.03/0.06/0.27 | 3.75 | 1.15 | 2.49 | 3.19 | 2.21 |
| (Nphbt)2Ir(pic) | 485(4.3), 462(4.2), 414(4.4), 389(4.3), 344(4.5) | ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) , 641 (598, 644) , 641 (598, 644) |
0.64/0.04/0.08/0.50 | 5.63 | 1.10 | 0.68 | 3.21 | 2.22 |
| (Cbzbt)2Ir(acac) | 464(3.9), 410(4.4), 383(4.5), 330(4.8) | ![[8 with combining low line]](https://www.rsc.org/images/entities/char_0038_0332.gif) , 603 (, 603) , 603 (, 603) |
0.69/0.02/0.02/0.12 | 2.61 | 1.34 | 1.52 | 3.26 | 2.52 |
| (Cbzbt)2Ir(pic) | 456(4.0), 402(4.6), 387(4.6), 332(4.9) | , 588 (556, 587) |
0.90/0.05/0.02/0.50 | 3.65 | 2.05 | 0.67 | 3.34 | 2.53 |
Upon a photoexcitation at 450 nm all the compounds show strong phosphorescence (Fig. 2 and Table 1). Emission bands of the compounds feature a vibrionic fine structure, indicating that the emission is strongly associated with ligand centered 3π–π* transitions.13 The observed gap of separation of these vibrational features is in the range of 1020–1370 cm−1 and can be associated with the stretching of aromatic double bonds in the cyclometalated ligands.28 Just like in the case of absorption spectra, the photoluminescence (PL) bands red-shift depending on the present ligand, following the sequence: Phbt, Cbzbt and Nphbt. Compounds based on Phbt and Cbzbt emit yellow light, while in the case of Nphbt derivatives the emission is orange-red. A strong influence on PL characteristics can also be observed in relation to the present ancillary ligand. In the case of acac functionalized compounds a substantial red-shift of PL wavelength is observed in comparison to their pic counterparts. This effect can be explained by a stronger chemical bonding between pic and the iridium atom, resulting in a shorter ligand–metal distance and stronger ligand-induced stabilization of the occupied iridium d-orbitals.29 As a result, bandgap values of d–π*transitions are expected to be larger in pic functionalized compounds.
The synthesized compounds possess relatively large PL quantum yields (ΦPL), with the measured values in dilute degassed THF solutions ranging from 0.64 to 0.90 (Table 1). For reference, the reported PL quantum yield of the unmodified base compound (bt)2Ir(acac) is 0.65.14 As it is expected, larger ΦPL is measured for the yellow Phbt and Cbzbt based emitters in comparison to orange-red Nphbt. The lower lying triplet energy levels for Nphbt increase the probability of nonradiative decay pathways of the excited state.
In the case of Phbt and Cbzbt based complexes the presence of the pic ancillary ligand leads to notably larger ΦPL values. This observation can be explained by the fact that the nonsymmetrical pic ligand changes the electron density of the molecule in such a way that the excited S1 and T1 states in the resulting heteroleptic complex are confined on one single cyclometalating C^N type ligand.30 In the case where symmetrical acac is present, S1 is delocalized on both the primary ligands, while in the emissive T1 state only on one ligand.30 The more pronounced spatial distribution of the excited state orbitals in the case of acac derivatives increases the number of the available vibrational states and, consequently, the probability of the nonradiative decay. Additionally, in order to emit from the T1 state, acac functionalized complexes must undergo an interligand charge transfer that potentially lowers the attainable ΦPL value even more.
In order to validate the previous assumptions a series of density functional theory (DFT) calculations were performed. The optimized geometries and the corresponding highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) for all synthesized complexes are given in Fig. 3. In all cases a distinctive difference in molecular orbital configuration between compounds bearing acac and pic ancillary ligands can be observed. While HOMO placement is fairly similar for all compounds, centered mainly on the electron rich aryl fragments of 2-arylbenzo[d]thiazole ligands, notable deviations can be seen in LUMO distribution. For acac containing compounds, the LUMO is symmetrically distributed over both the main ligands, particularly on the electron deficient benzothiazole ring. For pic, however, the asymmetry is induced by this ancillary group that shifts the LUMO to a single benzothiazole ring. (Cbzbt)2Ir(pic) can be named as an outlier from this trend, as the asymmetry is present not only for the LUMO, but also for the HOMO level. As previously discussed, the more pronounced orbital containment would result in increased ΦPL. This is the case, as the experimental observations follow computational predictions that the presence of the pic ligand leads to increased ΦPL values of the compounds, with (Cbzbt)2Ir(pic) being the most efficient, as its molecular orbitals have the least spatial spread. Additionally, excitation energies for the lowest singlet and triplet excitations were calculated (Table 1). The obtained values correlate well with the experimental absorption wavelengths, as larger calculated bandgaps correspond to lower measured absorption wavelengths.
 | ||
| Fig. 3 DFT optimized structures of the complexes and corresponding HOMO and LUMO visualizations. Hydrogens are omitted for clarity. | ||
In all cases the measured phosphorescence kinetics of the compounds can be expressed as a single exponential, with the lifetimes in the range 2.61–5.63 μs, which is typical for iridium(III) complexes. Radiative decay rates (kr) are notably lower for orange emitting Nphbt derivatives indicating a poorer iridium metal mediated intersystem crossing. For Phbt and Cbzbt based complexes a slight increase in kr is observed if a pic ancillary ligand is present.
The main purpose of the presented molecular design was to acquire materials that are suitable for applications in solution-processable OLED devices. The PL measurements of pure spin-coated films revealed that a strong PL quenching takes place due to the emitter aggregation and the ΦPL values do not exceed 0.10 (Table 1). In some cases almost complete quenching of the emission was observed. This is expected, as the mass concentration (wt%) of the active emitting complex in the compounds, excluding the mass of non-emissive 5,5,5-triphenylpentyl groups, is approximately 50%. The optimal mass concentration of the emitters in the emissive layers of manufactured OLED devices is 8–10%, in order to provide a large enough spatial separation level of individual molecules and limit the negative effects of aggregation-induced emission quenching.31 Two conventional hole transporting materials, polymeric PVK and molecular CBP, were used to prepare guest–host systems with an approximate emitter concentration of 10 wt% (corresponding to 20 wt% of the synthesized compounds). Under these conditions a sufficient level of emitter spatial isolation was achieved, and in CBP ΦPL values of up to 0.50 were measured (Fig. 2 and Table 1). Interestingly, the synthesized materials showed a strong preference for the CBP host in comparison to PVK, as in PVK no notable improvement in solid state ΦPL values was observed in comparison to host-free films. We attribute this behavior to the structure of PVK that features the presence of non-polar aliphatic polymer chains. When exposed to the polar emitter molecules, these lipophilic fragments induce phase separation, creating non-emissive domains of iridium complex aggregates. Nevertheless, this is a convenient property of the compounds, because the use of the molecular host materials results in more efficient OLEDs due to enhanced charge carrier mobilities.32
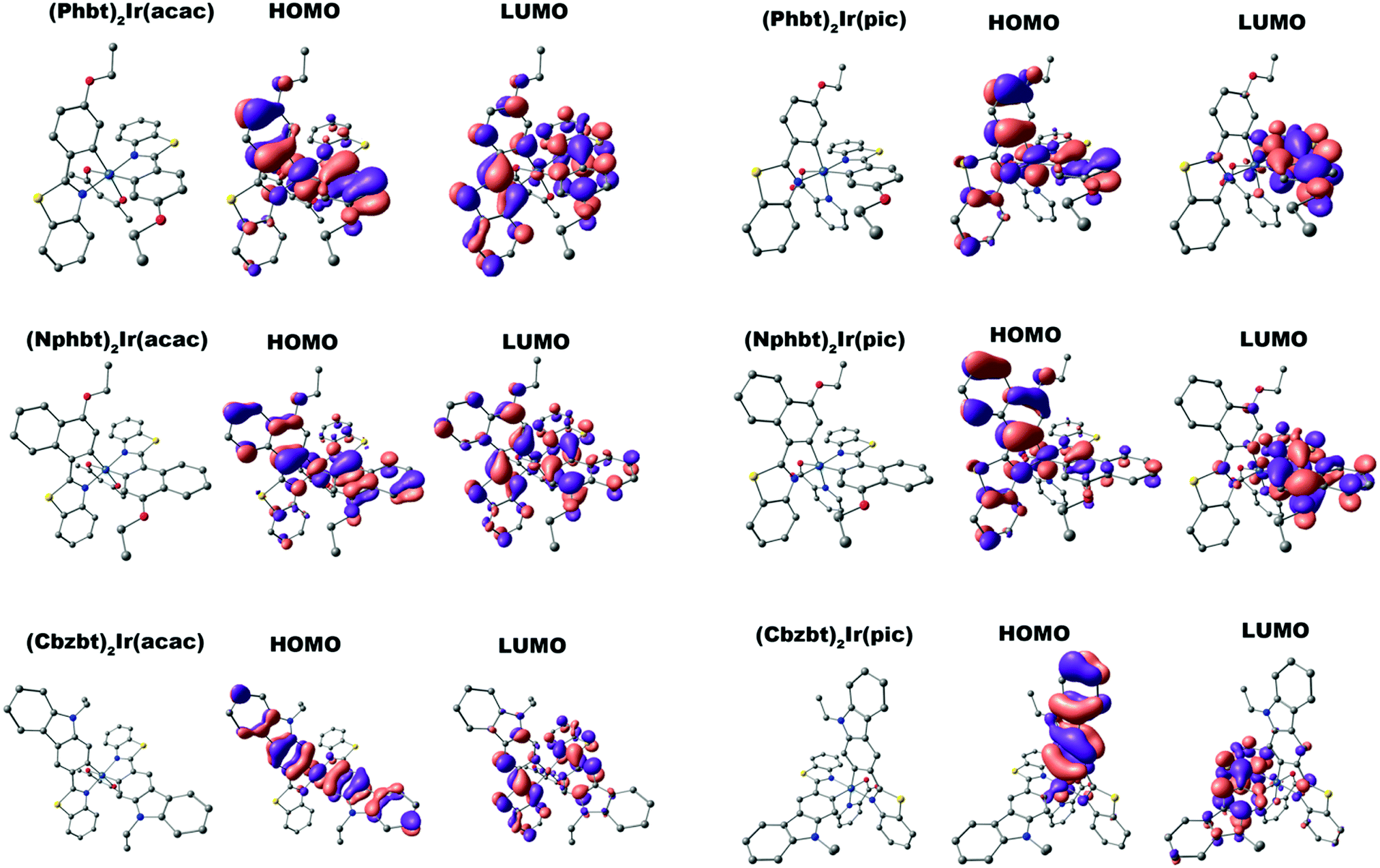
Atomic force microscopy (AFM) was employed in order to closely evaluate the film forming properties of the synthesized compounds. Two spin-coated (from THF solution) samples were examined, where (Cbzbt)2Ir(pic) (20 wt%) was dispersed in PVK and CBP hosts (Fig. 4). The root-mean-square (RMS) roughness values of these films were 2.1 and 1.5 nm. It should be noted that these values are measured by taking into account all surface areas, including the obvious defective sites. By measuring only the flat areas, values of 0.7 and 0.4 nm were acquired, indicating a highly homogenous structure for both samples. Still, a slightly higher overall clustering is detected for the polymeric PVK host. This is in accordance with the measured ΦPL values that indicate much more pronounced emitter–emitter interaction if this host material is used.
 | ||
| Fig. 4 AFM images of spin-coated films, composed of (Cbzbt)2Ir(pic) (20 wt%) and PVK or CBP host. | ||
Another noteworthy observation regarding the solid phase emissive properties of the compounds is a distinctive improvement in measured ΦPL for pic functionalized compounds in comparison to their acac structural analogues, especially in the case of the CBP host. This difference cannot be simply attributed to the intrinsically higher PL efficiency of pic derivatives and indicates alternative solid state packing patterns between the two subtypes of the materials. Taking into consideration the practically identical emission spectra of the compounds in THF and CBP (Fig. 2), but at the same time the notably reduced ΦPL values, the logical explanation for such observation is that in CBP there are two populations of the complex molecules: highly isolated and aggregated. The isolated population behaves as in THF, giving a similar PL band. The aggregated molecules, on the other hand, are weakly emissive, as indicated by the small ΦPL values in the host-free films. The notable difference between ΦPL values of acac and pic derivatives in CBP indicates that the presence of acac induces a more pronounced emitter–emitter aggregation. The following explanation for this can be given. While the molecular surface of the complex core in pic containing compounds is fully aromatic, the presence of an acac ancillary ligand creates a local aliphatic region. The incompatibility between aromatic and aliphatic domains is known to influence solid state packing patterns of iridium complexes. The repulsion between the acac-functionalized face of the complex and the fully aromatic CBP host layer is considered as the origin for a non-isotropic spatial orientation of emitting molecules in vacuum-deposited OLEDs.33 In our case this interaction between aromatic and aliphatic surfaces promotes the aggregation for acac containing compounds, as the complex molecules tend more to interact with each other.
Electroluminescence properties
In order to evaluate the suitability of the synthesized materials as the active electroluminescent components, OLEDs bearing compounds (Cbzbt)2Ir(acac) and (Cbzbt)2Ir(pic) were prepared. (Cbzbt)2Ir(pic) was chosen, as it shows the highest ΦPL value and the fastest radiative decay rate kr amongst the studied series of the compounds. Its structural analogue (Cbzbt)2Ir(acac) was picked to evaluate the difference between acac and pic ancillary ligands. Two series of devices were prepared, where solution-processed emissive layers contain either a polymeric PVK or molecular CBP host: (device-I) ITO/PEDOT:PSS (40 nm)/emitting layer (60 nm, PVK:emitter 20 wt%)/BPhen (20nm)/LiF (1 nm)/Al (100 nm) and (device-II) ITO/PEDOT:PSS (40 nm)/emitting layer (60 nm, CBP:emitter 20 wt%)/TPBi (20 nm)/LiF (1 nm)/Al (100 nm). The obtained results (Fig. 5 and Table 2) are in good agreement with the previously discussed PL measurements that showed a strong ΦPL increase in the cases where the CBP host material was used. The switch from PVK to CBP boosts the OLED's external quantum efficiency (ηext) 2.5 times in the case of (Cbzbt)2Ir(acac) and 1.6 times in the case of (Cbzbt)2Ir(pic) with the corresponding increase in the maximal achievable luminance of 4.3 and 4.7 times, respectively. As expected, the use of a pic functionalized emitter increases the ηext value 1.5 times and the maximal luminance by approximately 2 times in comparison to acac based devices.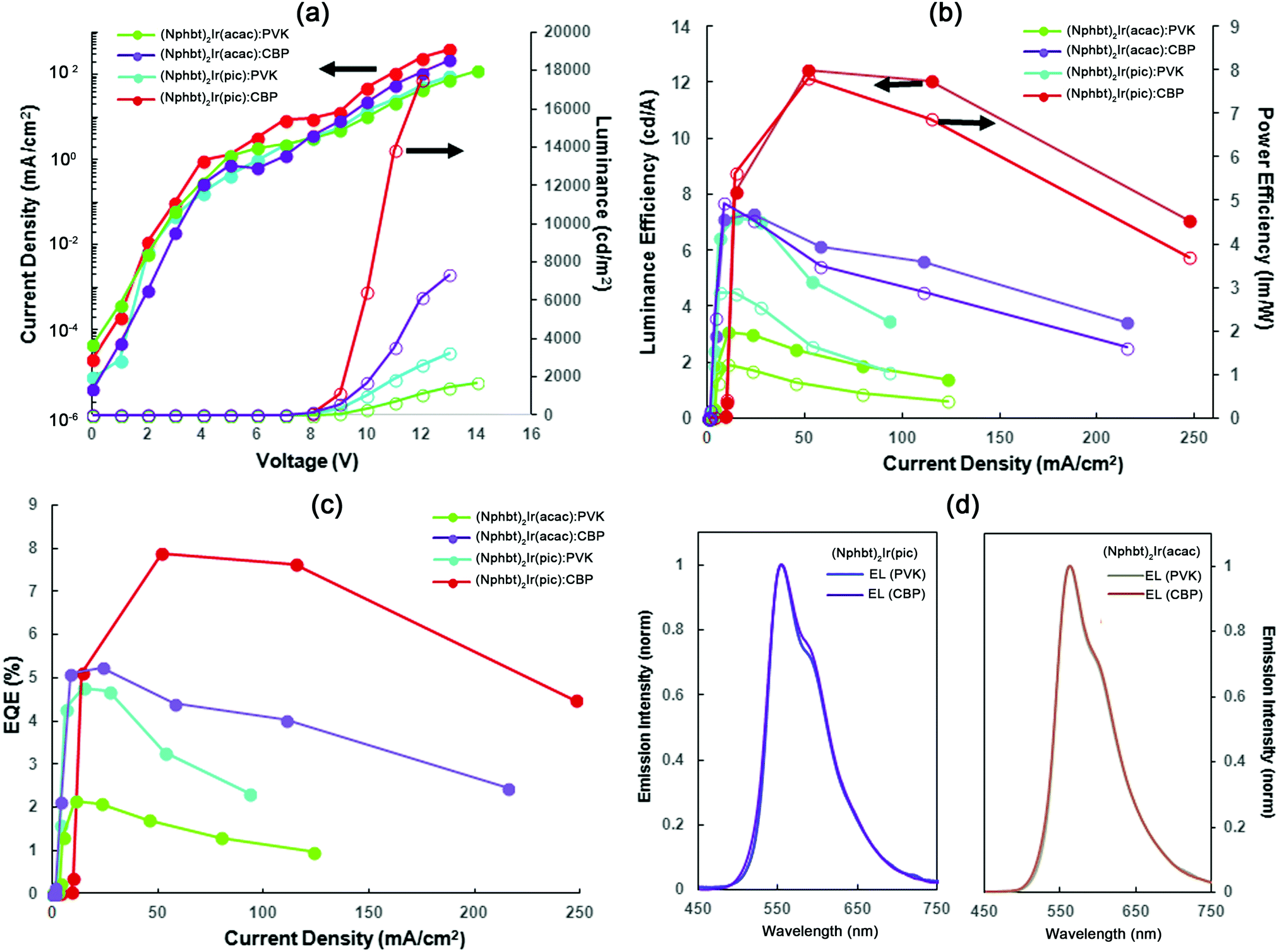 | ||
| Fig. 5 Characteristics of (Cbzbt)2Ir(acac) and (Cbzbt)2Ir(pic) based OLEDs: (a) voltage–current density (filled circle)–luminance (empty circle) plots, (b) luminance (filled circle) and power efficiency (empty circle) versus current density, (c) external quantum efficiency versus current density and (d) EL spectra of OLEDs. | ||
| Emissive layera | V on , V | L max, cd m−2 | η ext , % | η c , cd A−1 | η p , lm W−1 | λ max, nm | CIE (x, y) |
|---|---|---|---|---|---|---|---|
| a The concentration of the iridium(III) complex in the devices is 20 wt%. b Voltage at which luminance of 1 cd m−2 is measured. c OLED's external quantum efficiency. d Current efficiency. e Power efficiency. f Maximal achieved values and values measured at 1000 cd m−2. | |||||||
| (Cbzbt)2Ir(acac):PVK | 7.0 | 1708 | 2.1/1.7f | 3.1/2.4f | 1.9/1.3f | 561 | 0.43, 0.46 |
| (Cbzbt)2Ir(acac):CBP | 6.5 | 7295 | 5.2/5.1 | 7.3/7.1 | 4.9/4.7 | 563 | 0.43, 0.46 |
| (Cbzbt)2Ir(pic):PVK | 6.5 | 3229 | 4.8/4.8 | 7.2/7.2 | 4.5/4.5 | 553 | 0.44, 0.53 |
| (Cbzbt)2Ir(pic):CBP | 5.5 | 17451 |
7.9/5.1 | 12.4/8.0 | 7.8/5.6 | 554 | 0.44, 0.53 |
No notable transformations were detected in the emission spectra of the OLEDs with the variation of either the applied voltage (Fig. S2, ESI†) or the type of host material (Fig. 5d). The emission is closely similar to that seen in the PL spectra of thin films, indicating that the origin of the emitted light is the phosphorescence of iridium(III) complexes. No detectable drop in OLED luminance was detected for the (Cbzbt)2Ir(pic):CBP device after 4 hours of continuous operation at 100 cd m−2, indicating a sufficient stability of the emitting molecule. For all the devices a small diminishment of the current in the range of 6–8 V can be observed in the volt-ampere characteristics (Fig. 5a). The first recombination of the opposite charges in OLEDs can only take place with the reaching of the turn-on voltage; thereby the observed drop of the current is attributed to a small imbalance in the system's charge carrier concentration. This means that the energy level alignment of the layers can be improved to further increase the device efficiency. Systems with CBP as the host material exhibit a higher current density in the voltage region over 8 V, which together with the higher observed brightness points to a better electronic injection in the system compared to PVK based OLEDs.
All the examined devices start to show efficiency roll-off behavior at current densities in the 50–100 mA cm−2 range. While this may be attributed to many factors, like residual solvent inclusions, the efficiency roll-off in OLEDs is mainly ascribed to the aggregation of the emitters. In vacuum-deposited devices the spatial distribution of the complex molecules is more or less determined by the random molecular motion, while in solution there is a higher tendency for them to form aggregates. This process is especially likely to take place during the processing stage that often involves a steep concentration increase on a relatively long time-scale, determined by the slow solvent evaporation speed. Triplet–triplet annihilation is considered as the main mechanism for efficiency roll-off for aggregated molecules. This process takes place if the distance between iridium complex based emitters is less than 2 nm31,34 and governs efficiency roll-off, as the density of triplet excited states in the bulk of the emissive layer increases with the growing current density. Additionally, not only do solution-processed emitter molecules tend to form unwanted aggregates, but this process also proceeds in the case of host materials (CBP).35 As a result this causes a higher charge carrier trapping in the system. Thereby exciton quenching by polarons has high influence on the performance of the device. At higher current density it can dramatically reduce the efficiency of the OLEDs.
The maximal achieved performance parameters of the (Cbzbt)2Ir(pic)/CBP based OLED are the following: ηext of 7.9%, current efficiency of 12.4 cd A−1 and power efficiency of 7.8 lm W−1 at luminance 6000 cd m−2, with the highest measured luminance of 17451 cd m−2. These values exceed the characteristics of the other reported yellow emitting solution-processed devices that contain structurally similar thiazole based iridium(III) complexes, in terms of both the efficiency parameters and brightness.13,36,37 This underlines the viability of the presented molecular design towards the development of large area OLEDs with a lowered production cost.
Conclusions
A novel structural approach to the synthesis of solution-processable yellow and orange light emitting heteroleptic iridium(III) complexes is presented. The materials can be acquired based on O and N functionalized 2-arylbenzo[d]thiazole cyclometalating ligands by an introduction of phase-behavior-modifying 1,1,1-triphenylpentyl substituents in a simple 4 to 5 step-reaction sequence. According to the results of DSC analysis the acquired compounds do not possess inherent crystallinity and form a stable amorphous phase, at the same time showing an excellent solubility in a wide range of organic solvents. The presence of the modifying groups does not affect the emission efficiency, as large ΦPL values in the range of 0.64 to 0.90 were measured for the synthesized complexes. Amongst the investigated compounds a clear improvement in solid-state emissive properties is observed if picolinic acid ancillary ligand is used instead of acetylacetone. Additionally, the presented materials show a distinctive preference for molecular charge transporting host materials in comparison to the polymeric alternatives. The compounds can be successfully used to produce solution-processed OLEDs and the best obtained device showed an external quantum efficiency of 7.9%, current efficiency of 12.4 cd A−1 and power efficiency of 7.8 lm W−1 at luminance 6000 cd m−2, with the highest measured luminance of 17451 cd m−2. According to the observed volt-ampere characteristics, a further device optimization is possible, with the use of more appropriate electron or hole transport layers, in order to improve the charge balance. Despite this, the achieved performance parameters of the solution-processed OLEDs already exceed those reported for their close structural analogues, highlighting the viability of the presented material design.
Experimental
Materials and instruments
Starting materials and solvents were purchased from Alfa Aesar. 5,5,5-Triphenylpentan-1-iodide was prepared according to a known procedure.22 NMR spectra were obtained using a Bruker Avance 300 MHz spectrometer using solvent residue as an internal reference. Mass spectra were acquired using a Waters EMD 1000 MS detector (ESI + mode, cone voltage 30 V). The elemental analysis was carried out using a Costech Instruments ECS 4010 CHNS-O Elemental Combustion System. Differential scanning calorimetry (DSC) thermograms were acquired using a Mettler Toledo DSC-1/200 W apparatus at a scanning rate of 10 °C min−1 while keeping the samples under N2 atmosphere. Decomposition temperatures were obtained using a Perkin Elmer STA 6000 thermal analyzer. Optical measurements in solution were carried out using solutions of typical concentrations 1–4 × 10−5 mol L−1. Solutions for ΦPL measurements were prepared in a glovebox under Ar atmosphere using previously degassed solvents and filled in sealable cuvettes. The UV-Vis spectra were recorded using a Perkin Elmer Lambda 35 spectrometer. Emission spectra, absolute photoluminescence quantum yields (PLQY) and emission lifetimes were determined using a QuantaMaster 40 steady state spectrofluorometer (Photon Technology International, Inc.) equipped with a 6 inch integrating sphere (LabSphere). A Veeco AFM CP-II atomic force microscope was used in tapping mode.Synthesis of Ir(III) complexes
:1) at 130 °C for 24 h. The contents of the reaction vessel were poured into water; the precipitate was collected by filtration and washed with ethanol. Without further purification the acquired dichloro-bridged complex was dissolved in 2-ethoxyethanol and an excessive amount of auxiliary ligand (acetylacetone or 2-picolinic acid) and K2CO3 (in 1:3 molar ratio) was added under argon atmosphere. The reaction mixture was stirred at 100 °C for 2 h and then poured into water. The precipitate was collected by filtration, washed with ethanol and then purified by column chromatography over silica gel using toluene (or CH2Cl2/ethyl acetate 50:1 for compounds containing pic auxiliary ligand) as eluent to yield the desired heteroleptic iridium(III) complex.
(Phbt)2Ir(acac) . Yield: 40%. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.93 (d, J = 7.3 Hz, 2H), 7.65 (d, J = 7.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 7.33 (2H, J = 7.3 Hz, 2H), 7.29–7.03 (m, 32H), 6.28 (dd, J = 8.4 Hz, J = 2.1 Hz, 2H), 5.75 (d, J = 2.1 Hz, 2H), 5.05 (s, 1H), 3.35 (m, 4H), 2.33 (m, 4H), 1.68 (s, 6H), 1.38 (m, 4H), 0.93 (m, 4H). Anal. calcd for C77H67IrN2O4S2: C, 68.98; H, 5.04; N, 2.09. Found: C, 68.96; H, 5.11; N, 2.43.
(Phbt)2Ir(pic) . Yield: 40%. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.35 (d, J = 8.2 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.86 (d, J = 7.7 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 7.65–7.47 (m, 4H), 7.34–7.25 (m, 2H), 7.21–7.06 (m, 32H), 6.80 (t, J = 7.5 Hz, 1H), 6.42 (dd, J = 8.6 Hz, J = 2.2 Hz, 1H), 6.33 (dd, J = 8.6 Hz, J = 2.2 Hz, 1H), 5.99–5.88 (m, 2H), 5.62 (d, J = 2.2 Hz, 1H), 3.51–3.20 (m, 4H), 2.40–2.29 (m, 4H), 1.42–1.29 (m, 4H), 1.05–0.81 (m, 4H). Anal. calcd for C78H64IrN3O4S2: C, 68.70; H, 4.73; N, 3.08. Found: C, 68.41; H, 4.75; N, 3.18.
(Nphbt)2Ir(acac) . Yield: 47%. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.45 (d, J = 8.4 Hz, 2H), 8.08 (d, J = 7.8 Hz, 2H), 7.98 (d, J = 8.1 Hz, 2H), 7.76 (d, J = 7.1 Hz, 2H), 7.58 (t, J = 7.1 Hz, 2H), 7.40–7.02 (m, 36H), 5.84 (s, 2H), 5.15 (s, 1H), 3.43–3.00 (m, 4H), 2.44 (m, 4H), 1.78 (s, 6H), 1.46 (m, 4H), 1.04 (m, 4H). Anal. calcd for C85H71IrN2O4S2: C, 70.86; H, 4.97; N, 1.94. Found: C, 71.14; H, 5.05; N, 2.25.
(Nphbt)2Ir(pic) . Yield: 55%. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.48 (m, 3H), 8.20 (d, J = 7.6 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.92–7.82 (m, 2H), 7.75–7.55 (m, 4H), 7.45–7.35 (m, 2H), 7.35–7.05 (m, 34H), 6.90 (t, J = 8.1 Hz, 1H), 6.18 (d, J = 8.2 Hz, 1H), 6.08 (s, 1H), 5.67 (s, 1H), 3.51–3.01 (m, 4H), 2.43 (m, 4H), 1.56–1.35 (m, 4H), 1.06 (m, 4H). Anal. calcd for C86H68IrN3O4S2: C, 70.56; H, 4.68; N, 2.87. Found: C, 70.54; H, 4.81; N, 3.05.
(Cbzbt)2Ir(acac) . Yield: 31%. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.38 (s, 2H), 8.16 (d, J = 8.2 Hz, 2H), 7.97 (d, J = 7.7 Hz, 2H), 7.62 (d, J = 7.8 Hz, 2H), 7.37–7.15 (m, 42H), 7.10 (t, J = 7.5 Hz, 2H), 7.02 (d, J = 8.1 Hz, 2H), 6.27 (s, 2H), 5.19 (s, 1H), 3.59 (t, J = 7.4 Hz, 4H), 2.48–2.10 (m, 4H), 1.79 (s, 6H), 1.40–1.20 (m, 4H), 0.90 (m, 4H). Anal. calcd for C89H73IrN4O2S2: C, 71.89; H, 4.95; N, 3.77. Found: C, 71.69; H, 5.17; N, 4.04.
(Cbzbt)2Ir(pic) . Yield: 51%. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.55 (d, J = 8.3 Hz, 1H), 8.47 (s, 1H), 8.41 (s, 1H), 8.23 (d, J = 7.6 Hz, 1H), 8.04 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 7.6 Hz, 1H), 7.89–7.80 (m, 2H), 7.55 (d, J = 7.9 Hz, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.44–7.34 (m, 2H), 7.34–7.10 (m, 35H), 7.10–7.01 (m, 3H), 6.90 (t, J = 7.8 Hz, 1H), 6.44 (s, 1H), 6.19–6.12 (m, 2H), 3.69–3.51 (m, 4H), 2.42–2.17 (m, 4H), 1.41–1.19 (m, 4H), 0.98–0.83 (m, 4H). Anal. calcd for C90H70IrN5O2S2: C, 71.59; H, 4.67; N, 4.64. Found: C, 71.46; H, 4.90; N, 4.74.
DFT calculations
DFT calculations were performed using the Gaussian 09, rev. D.01 software package.38 The trans disposition of C^N ligands was assigned for compounds due to the inherent reactivity pattern of iridium cyclometalating reactions.7 The structures of the complexes were simplified by substituting the 1,1,1-triphenylmethylpentane fragment with an ethyl group in order to reduce the computational cost. Frontier molecular orbital levels were calculated under vacuum using the CAM-B3LYP39 density functional, after optimizing the geometry. The basis set both for optimization and for energy calculations was chosen to be D95V(d,p) on C, N, O, and H and D95(2df) on S, with the LANL2DZ basis set and effective core potential being chosen for Ir atoms. Each geometry was at the end optimized with method-wise correct force constants and vibrational frequencies were checked to assure that the obtained structure truly corresponds to the energy minimum. SCF and geometry convergence were left at default values for Gaussian 09 (SCF: 10^(-8) for the commutator of the Fockian and density matrixes, and four different values for maximum and RMS force and coordinate change). The integration grid was also at the default “fine” level, which is a pruned (75302) grid. Singlet–singlet and singlet–triplet excitations were modeled by LR-TD-DFT with the same method and basis set. The triplet excited state geometry was optimized by conventional SCF methods, specifying the spin multiplicity of 3 beforehand.
OLED preparation and characterization
Sandwich type samples with the pixel size of 16 mm2 were prepared with the following structure: ITO/PEDOT:PSS(40 nm)/emitting layer (60 nm)/BPhen or TPBi (20nm)/LiF(1 nm)/Al(100 nm), where poly(3,4-ethylenedioxythiophene)–poly(styrenesulfonate)(PEDOT:PSS), 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1H-benzimidazole) (TPBi) (or 4,7-diphenyl-1,10-phenanthroline (BPhen)) and LiF were used as hole-injection, electron transport and electron-injection layers, respectively. BPhen was used for the PVK matrix and TPBi for the CBP matrix. This choice was made to get the best possible energy level alignment between the host and electron transport layer. Indium tin oxide (ITO) glass (Präzisions Glas & Optik GmbH) with a sheet resistivity of 15 Ω per square was used as a substrate. A 12 mm wide ITO strip line was made by wet etching at the middle of the substrate. Afterwards, ITO substrates were cleaned by the following method: sonicated in CHCl3; sonicated in acetone; 2× rinsed with distilled (DI) water; sonicated in water with 3 vol% of Hellmanex II detergent; rinsed with DI water; sonicated in DI water and isopropyl alcohol. Each sonication lasted for 15 min. Before deposition of PEDOT:PSS the ITO glass was blown dry with nitrogen and treated with UV-ozone for 20 min. The ITO layer was covered with PEDOT:PSS (from H.C. Starck, Al4083) using a Laurell WS650 spin coater. Rotation lasted for 1 minute with speed 2000 rpm. The sample was moved into a glovebox and heated at 200 °C for 10 minutes. The emitting layer consisted of the corresponding synthesized iridium(III) complex in poly(9-vinylcarbazole) (PVK, Sigma Aldrich 368350) or 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) host. In all cases 20 wt% host:guest systems in tetrahydrofuran were prepared. A solution with a concentration of 5 mg ml−1 was spin-coated on the PEDOT:PSS layer with 2000 rpm for 40 sec and heated at 120 °C for 15 minutes afterwards. Furthermore, the samples were moved from the glovebox to a vacuum chamber, without exposure to air, for thermal evaporation of electron transport TPBi (Sigma Aldrich 806781) or BPhen (Sigma Aldrich 133159), and electron-injection of the LiF (Sigma Aldrich 449903) and electrodes at the pressure 6 × 10−6 Torr. The deposition speed was 1 Å s−1, 0.1 Å s−1 and 5 Å s−1 for TPBi and BPhen, LiF and Al, respectively. In the end the samples were encapsulated using a two-component epoxide.The current–voltage characteristics of the OLEDs were measured by a Keithley 2700 multimeter and the voltage source was a Keithley 230 unit. The electroluminescence brightness and CIE 1931 coordinates were measured using a Konica Minolta Luminance and Color Meter CS-150. The electroluminescence spectrum was recorded using a calibrated Ocean Optics HR4000 spectrometer. CIE 1931 coordinates and spectra presented in this work were obtained at the maximum brightness.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the ERDF 1.1.1.1. activity project No. 1.1.1.1/16/A/131 “Design and Investigation of Light Emitting and Solution Processable Organic Molecular Glasses”. The authors are thankful to Rolands Grants for AFM images.References
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585–1595 CrossRef.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Gong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS.
- X. Yang, G. Zhou and W. Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS.
- C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439–6469 RSC.
- P. Chen, W. Xie, J. Li, T. Guan, Y. Duan, Y. Zhao, S. Liu, C. Ma, L. Zhang and B. Li, Appl. Phys. Lett., 2007, 91, 023505 CrossRef.
- B. Zhao, Z. Su, W. Li, B. Chu, F. Jin, X. Yan, F. Zhang, D. Fan, T. Zhang, Y. Gao and J. Wang, Appl. Phys. Lett., 2012, 101, 053310 CrossRef.
- X. Yang, Y. Zhao, X. Zhang, R. Li, J. Dang, Y. Li, G. Zhou, Z. Wu, D. Ma, W. Y. Wong and X. Zhao, J. Mater. Chem., 2012, 22, 7136–7148 RSC.
- M. Li, H. Zeng, Y. Meng, H. Sun, S. Liu, Z. Lu, Y. Huang and X. Pu, Dalton Trans., 2011, 40, 7153–7164 RSC.
- J. Li, R. Wang, R. Yang, W. Zhou and X. Wang, J. Mater. Chem. C, 2013, 1, 4171–4179 RSC.
- C. Fan, L. Zhu, B. Jiang, Y. Li, F. Zhao, D. Ma, J. Qin and C. Yang, J. Phys. Chem. C, 2013, 117, 19134–19141 CrossRef CAS.
- T. Giridhar, W. Cho, Y. H. Kim, T. H. Han, T. W. Lee and S. H. Jin, J. Mater. Chem. C, 2014, 2, 9398–9405 RSC.
- D. Liu, R. Yao, M. Fu, D. Li and S. Zhang, RSC Adv., 2016, 6, 34198–34203 RSC.
- C. Tonnelé, M. Stroet, B. Caron, A. J. Clulow, R. C. Nagiri, A. K. Malde, P. L. Burn, I. R. Gentle, A. E. Mark and B. J. Powell, Angew. Chem., 2017, 56, 8402–8406 CrossRef PubMed.
- E. Baranoff, S. Suarez, P. Bugnon, C. Barolo, R. Buscaino, R. Scopelliti, L. Zuppiroli, M. Graetzel and M. K. Nazeeruddin, Inorg. Chem., 2008, 47, 6575–6577 CrossRef CAS.
- X. Xu, X. Yang, J. Zhao, G. Zhou and W. Y. Wong, Asian J. Org. Chem., 2015, 4, 394–429 CrossRef CAS.
- Y. Wang, S. Wang, N. Zhao, B. Gao, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Polym. Chem., 2015, 6, 1180–1191 RSC.
- K. Traskovskis, I. Mihailovs, A. Tokmakovs, A. Jurgis, V. Kokars and M. Rutkis, J. Mater. Chem., 2012, 22, 11268–11276 RSC.
- K. Traskovskis, K. Lazdovica, A. Tokmakovs, V. Kokars and M. Rutkis, Dyes Pigm., 2013, 99, 1044–1050 CrossRef CAS.
- I. R. Laskar and T. M. Chen, Chem. Mater., 2004, 16, 111–117 CrossRef CAS.
- M. Nonoyama, J. Organomet. Chem., 1975, 86, 263–267 CrossRef CAS.
- K. Naito and A. Miura, J. Phys. Chem., 1993, 97, 6240–6248 CrossRef CAS.
- I. R. Laskar, S. F. Hsu and T. M. Chen, Polyhedron, 2006, 25, 1167–1176 CrossRef CAS.
- D. Kim, J. Phys. Chem. C, 2015, 119, 12690–12697 CrossRef CAS.
- E. Baranoff, S. Fantacci, F. De Angelis, X. Zhang, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2010, 50, 451–462 CrossRef PubMed.
- A. F. Rausch, M. E. Thompson and H. Yersin, J. Phys. Chem. A, 2009, 113, 5927–5932 CrossRef CAS PubMed.
- X. Gu, T. Fei, H. Zhang, H. Xu, B. Yang, Y. Ma and X. Liu, J. Phys. Chem. A, 2008, 112, 8387–8393 CrossRef CAS PubMed.
- Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218–4233 CrossRef CAS.
- M. J. Jurow, C. Mayr, T. D. Schmidt, T. Lampe, P. I. Djurovich, W. Brütting and M. E. Thompson, Nat. Mater., 2016, 15, 85–91 CrossRef CAS.
- W. Staroske, M. Pfeiffer, K. Leo and M. Hoffmann, Phys. Rev. Lett., 2007, 98, 197402 CrossRef CAS.
- Y. J. Cho, S. Taylor and H. Aziz, ACS Appl. Mater. Interfaces, 2017, 9, 40564–40572 CrossRef CAS.
- T. Giridhar, W. Cho, J. Park, J. S. Park, Y. S. Gal, S. Kang, J. Y. Lee and S. H. Jin, J. Mater. Chem. C, 2013, 1, 2368–2378 RSC.
- Y. Wu, H. Xu, J. Yang, J. Li, W. Liang, J. Sun, H. Wang and B. Xu, New J. Chem., 2015, 39, 8908–8914 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj04484h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |