
Tris functionalized Cu-centered cyclohexamolybdate molecular armor as a bimetallic catalyst for rapid p-nitrophenol hydrogenation†
Qihua
Fang
ab,
Junhong
Fu
b,
Fei
Wang
*a,
Zhaoxian
Qin
b,
Weiguang
Ma
 b,
Jiangwei
Zhang
*b and
Gao
Li
b
b,
Jiangwei
Zhang
*b and
Gao
Li
b
aAdvanced Catalysis and Green Manufacturing Collaborative Innovation Center, Changzhou University, Changzhou 213164, P. R. China. E-mail: wangfei@cczu.edu.cn
bGold Catalysis Research Center, State Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: jwzhang@dicp.ac.cn
First published on 15th November 2018
Abstract
A water-soluble tris functionalized Cu-centered Anderson nanocluster (NH4)4{[NH2C(CH2O)3]2CuMo6O18} with a remote NH2 motif was synthesized for the first time. The bimetallic cluster's structure is identified by single crystal X-ray diffraction, and it is also further well characterized by a combination of technologies, such as XPS, TGA, FT-IR spectroscopy, UV-Vis spectroscopy, ESI-MS, and EA spectroscopy. It can serve as a non-noble metal containing bimetallic catalyst to afford rapid reduction of aqueous p-nitrophenol to p-aminophenol (10 min) with a promising conversion (a decent 100%) and perfect selectivity (ca. 99%) in aqueous solution at room temperature. The catalytic reaction rate constant of the bimetallic {[NH2C(CH2O)3]2CuMo6O18}4− catalyst was one order higher (ca. 9.4 times) than that of the corresponding monometallic [Mo7O24]6− catalyst. The bimetallic cluster shows good catalytic performance and recyclability with an intact structure, confirmed by powder XRD. Cyclic voltammetry investigation indicated that a reversible two one-electron process was observed, in which CuII/CuI and MoVI/MoV were the redox couples and served as an ideal electrochemically stable multi-electron reservoir to promote the reduction process that enabled an obvious bimetallic synergistic catalytic performance during such p-nitrophenol hydrogenation reduction.
Introduction
Amino-aromatics and their corresponding derivatives are important chemical feedstocks, and they are widely used in the synthesis of pharmaceuticals and various fine chemicals.1,2 Among them, the production of p-aminophenol is 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons every year with an increased demand of 8% per year in China. Most consumption (ca. 85%) of p-aminophenol occurs in the pharmaceutical industry, including in the synthesis of paracetamol (4-acetamidophenol), clofibrate, and benorilate (4-acetamidophenyl-2-acetoxybenzoate). In particular, in the analgesic and antipyretic drug fields, paracetamol and benorilate are promising substitutes that possess no side-effects as phenacetin and aspirin do. Thus, they serve as pillar products of non-steroidal anti-inflammatory pharmaceuticals (NSAIPs) worldwide. The industrial routes for their preparation are the reduction of their corresponding nitro derivatives. One route is the reduction of p-nitrophenol, which is generally achieved in the presence of noble metal catalysts under a hydrogen atmosphere3 or a reducing agent.4 The obvious drawbacks of this route are the high cost, poisoning of the catalyst, and excessive hydrogenation leading to 4-amino cyclohexanol by-products due to the high reactivity of noble metals. Another route employs the reduction of nitrobenzene that is accomplished in the presence of iron powder. Although the source of reactant and catalyst is cheap and abundant, the accompanying by-product including its corresponding isomer (o-aminophenol, m-aminophenol, and aniline) causes difficulties and cost in further p-aminophenol purification from various isomeric by-products. Furthermore, the carcinogen aniline as a by-product makes p-aminophenol impractical in the field of pharmaceuticals synthesis.1 Indeed, a more eco-friendly and economically feasible protocol for p-aminophenol production with high purity and selectivity is urgently required.
000 tons every year with an increased demand of 8% per year in China. Most consumption (ca. 85%) of p-aminophenol occurs in the pharmaceutical industry, including in the synthesis of paracetamol (4-acetamidophenol), clofibrate, and benorilate (4-acetamidophenyl-2-acetoxybenzoate). In particular, in the analgesic and antipyretic drug fields, paracetamol and benorilate are promising substitutes that possess no side-effects as phenacetin and aspirin do. Thus, they serve as pillar products of non-steroidal anti-inflammatory pharmaceuticals (NSAIPs) worldwide. The industrial routes for their preparation are the reduction of their corresponding nitro derivatives. One route is the reduction of p-nitrophenol, which is generally achieved in the presence of noble metal catalysts under a hydrogen atmosphere3 or a reducing agent.4 The obvious drawbacks of this route are the high cost, poisoning of the catalyst, and excessive hydrogenation leading to 4-amino cyclohexanol by-products due to the high reactivity of noble metals. Another route employs the reduction of nitrobenzene that is accomplished in the presence of iron powder. Although the source of reactant and catalyst is cheap and abundant, the accompanying by-product including its corresponding isomer (o-aminophenol, m-aminophenol, and aniline) causes difficulties and cost in further p-aminophenol purification from various isomeric by-products. Furthermore, the carcinogen aniline as a by-product makes p-aminophenol impractical in the field of pharmaceuticals synthesis.1 Indeed, a more eco-friendly and economically feasible protocol for p-aminophenol production with high purity and selectivity is urgently required.
In this regard, a one step reaction that employs the corresponding p-nitrophenol as a reactant to afford high purity p-aminophenol is an ideal protocol to address such a predicament elegantly. Meanwhile, considering the pinch and the expense of noble metal catalysts, to achieve the goal regarding an economic protocol for p-aminophenol production, great efforts have been made to employ transition non-noble metal catalysts to substitute these noble metal catalysts in such reactions.1,2 The key factors to achieve this purpose rely on the selection of a suitable reducing agent and non-noble metal catalyst.
Inspired by previous investigations,4 water-soluble NaBH4 is an ideal reducing agent due to its high hydrogen content, stability, and non-toxicity. As a high quality hydrogen source, the dehydrogenation of NaBH4 in water can be effectively catalyzed by transition metals such as Cu and Mo.5,6 Therefore, it is possible to achieve the highly efficient reduction of p-nitrophenol to afford p-aminophenol with high purity by taking advantage of in situ generated hydrogen from NaBH4 in water and the design of a synergistically bimetallic catalyst. Polyoxometalates (POMs) are an exceptional family of inorganic clusters consisting of early transition metal ions (e.g., Mo, W, V, etc.) in their highest oxidation states with structural versatility and a wide range of properties and applications.7–11 Especially, Anderson type hetero-polyoxometalates as the most controllable mediated structural unit can incorporate functional heteroatoms including other transition metals, noble metals, and rare earth metals to enable the design of advanced functional materials that are more accessible and rational.7 Furthermore, the recent direct triol functionalization of Anderson organometallic clusters was intensively conducted to afford Anderson type organic hybrid cluster materials that exhibit stronger stability and fascinating properties ranging from catalysis to biological applications.7,12
Herein, we design a novel bimetallic synergistic tris functionalized Cu-centred Anderson organic hybrid cluster for the first time as a catalyst, (NH4)4{[NH2C(CH2O)3]2CuMo6O18}, to afford effective conversion of p-nitrophenol to p-aminophenol with a promising yield (100%) and selectivity (>99%) in water at room temperature within a rapid reaction time of 10 min in the presence of NaBH4. The reaction rate over the bimetallic cluster was determined to be 9.8 times faster than that on the monometallic cluster [Mo7O24]6−.
Experimental
General methods and materials
(NH4)4[Cu(OH)6Mo6O18] was similarly synthesized according to literature methods.12 All syntheses and manipulations were performed in the open air. All other chemicals, including solvents, were commercially available as reagent grade and used as received without further purification from Adamas-beta®. IR spectra were measured on a Bruker vertex70 FT-IR spectrometer. UV-Vis spectra were measured in H2O using a Shimadzu UV-2100S spectrophotometer. The mass spectra were obtained using an ion trap mass spectrometer (Thermofisher LTQ). Negative mode was chosen for the experiments (capillary voltage 33 V). Sample solution (in H2O) was infused into the ESI source at a flow rate of 300 μL min−1. Elemental analyses of C, H and N were performed using an Elementar Analysensysteme GmbH (vario EL), while the elemental analyses of metallic elements were performed using an X-ray fluorescence (XRF) element analyzer PANalytical Epsilon 5 and the sample was completely dried. X-ray photoelectron spectroscopy (XPS) measurements were performed under ultrahigh vacuum (UHV) with 1.0 × 10−7 Torr, an axis HS monochromatized Al Kα cathode source at 150 W, focused X-ray 100 μm beam, pass energy: 55 eV with 0.1 eV step length and detection angle (take off): 45° on an X-ray microprobe (ULVAC-PHI Quantera SXM). Binding energy was calibrated with C 1s = 284.8 eV. Thermal gravimetric analysis (TGA) curves were recorded on a Mettler Toledo TGA/SDTA851 in flowing air of 50.0 mL min−1 with a heating rate of 20 °C min−1. The powder product was measured using a PANalytical X'Pert Powder X-ray powder diffractometer operating at a voltage of 60 kV and a current of 55 mA with Cu Kα radiation (λ = 1.5406 Å). Gas chromatography mass spectrometry (GC-MS) analysis was performed on an Agilent 7890B single quadrupole mass spectrometer detector used in the electron ionization mode and equipped with a MassHunter Workstation system. The GC column was HP-5 (30 m × 0.32 mm × 0.25 μm). The temperature was programmed from 100 to 270 °C at 10 °C min−1. The other temperatures were 270 °C for the injection port, 290 °C for the jet separator and 310 °C for the ion source. The ionization energy was 70 eV and the spectrum scan was in the range of m/z 10–1000. Cyclic voltammetry (CV) analysis was carried out on a CHI660E electrochemical workstation. The saturated calomel electrode (SCE), platinum wire and glassy carbon (GC) were used as the reference, counter and working electrodes, respectively. An aqueous solution was prepared with 50 mg of (NH4)4[Mo7O24] and compound 1 dissolved in 30 mL of 0.5 M Na2SO4, respectively.Synthesis of (NH4)4{[NH2C(CH2O)3]2CuMo6O18}
2.2 g (2 mmol) of (NH4)4[Cu(OH)6Mo6O18] was dissolved in 20 mL of H2O, then the solution was acidized with 1 M HCl to pH 3.0 and heated to 80 °C for 10 min. 0.484 g (4 mmol) of NH2C(CH2OH)3 was added and the solution was kept at 80 °C for 2 h. Then, the solution was evaporated to dryness. The title compounds could be obtained as bluish green crystalline products (80% yields based on Mo). Suitable single crystals for X-ray diffraction were grown by slow evaporation.C8H32N6CuMo6O24, Mr = 1235.55. H, 2.83; C, 7.91; N, 6.63; Cu, 4.98; Mo, 46.65, while calcd H, 2.61; C, 7.78; N, 6.80; Cu, 5.14; Mo, 46.59. IR (cm−1): 2964, 1633, 1466, 1142, 969, 930, 809, 704, 629. UV-Vis (H2O, nm): λLMCT = 289, λd–d = 694. ESI mass spectrometry: calcd m/z = 582.71 (z = 2), acidized {[NH3C(CH2O)3]2CuMo6O18}2−, found 582.93.
X-ray crystallography
Data collections were performed using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data reduction, cell refinement and experimental absorption correction were performed with the software package Agilent Gemini Ultra CrysAlisPro (Ver 1.171.39.46). The structures were solved by direct methods and refined against F2 by full-matrix least-squares. All non-hydrogen atoms were refined anisotropically. All calculations were carried out using the program package SHELXTL ver 6.2 and Olex2 ver 1.2.10.13,14p-Nitrophenol reduction
In a typical process, 695 mg (5 mmol) of p-nitrophenol was dissolved in 10 mL of H2O and heated to 50 °C to become a bright yellow solution and cooled to room temperature. Then, (NH4)4{[NH2C(CH2O)3]2·CuMo6O18} (1, 2, 5, or 10 mg) (8.1 × 10−4, 1.62 × 10−3, 4.05 × 10−3, 8.1 × 10−3 mmol) and 5 equivalents (945 mg, 25 mmol) of NaBH4 were dissolved in 3 mL of H2O and added to the solution of p-nitrophenol with vigorous stirring. 1 mL of mixed solution was added in a standard quartz cuvette with a 1 cm path length and monitored using UV-Vis spectroscopy to examine the catalytic activity at room temperature. The absorbance of the solution was in situ measured in the scanning range of 250–500 nm to obtain the successive change of the reduction. With the increase of time, the reduction proceeded, and the characteristic absorption peak of p-nitrophenol at 400 nm and a small shoulder accompanying satellite peak at 350 nm gradually decreased, while a new characteristic peak at 300 nm indicated the generation of the p-aminophenol product. The quantitative analysis of p-nitrophenol and p-aminophenol concentrations can be monitored by UV spectroscopy via a standard working curve, using p-nitrophenol and p-aminophenol as the standard substances to afford the corresponding conversion and product selectivity. The final p-aminophenol can be extracted by ethyl acetate from the reaction solution and further be separated using a separating funnel. Note that in order to recover enough catalyst from the first run to perform the next run until the final 5th run, the scale of the catalytic reaction was expanded by 10 times (50 mg of catalyst used). About 80% catalyst can be recovered from each run for the next run. In this method, about 20 mg of recovered catalyst was available for the final 5th run. The catalyst can be recovered and separated from the aqueous phase by rotary evaporation and concentrated by taking advantage of the obvious solubility difference between the catalyst and the corresponding generated sodium borate, since the corresponding product sodium borate is easily dissolved and remained in aqueous solution. In order to eliminate the potential residual NaBH4, the recycled catalyst was acidized using 0.1 M HCl solution, and it was ground and dried in an air-aerated oven for 12 h before the recycling tests. The recycling of the [Mo7O24]6− catalyst in the control experiment was the same as the protocol of the {[NH2C(CH2O)3]2CuMo6O18}4− catalyst. Note that the corresponding reactant conversion and product selectivity can also be obtained based on the material balance combined with the integral area of the GC-MS spectrum of the corresponding reactants and products. This serves as an alternative method to quantitatively verify the reactant conversion and product selectivity in order to mutually evidence the result from UV-Vis monitoring and eliminate experimental error.Results and discussion
Structure elucidation and spectral characterization
Triol ligand functionalized Cu-centred Anderson organometallic derivatives were obtained through the reconstruction protocol reported by Wu.15 They also extended the triol ligands with remote reactive motifs of nitro and hydroxymethyl. However, the extension to amino groups failed due to the stronger coordination interactions between free amino groups and the corresponding heteroatom ions reported previously by Hasenknopf.16 We proved the robustness and generality of the direct parent Anderson functionalization protocol by first central heteroatom encapsulation, thus the coordination interactions between coordination reactive remote moieties including amino and carboxyl in triol ligands and heteroatom ions can be avoided, leading to the failure of triol functionalized Anderson organometallic derivatives.12 However, further post-functionalization using amino groups as the remote reactive site is still a big challenge. The previous reported cases were mainly focused on early transition metal elements, such as Mn and Cr centred Anderson clusters. The investigation regarding the extension to late transition metal elements is just on the way. Recently, it has been achieved by obtaining Ni-centered single-side Anderson triol functionalized derivatives with a remote amino motif reported by Rompel.17 Moreover, to the best of our knowledge, grafting amino groups on Cu-centred Anderson clusters has not been reported yet. The cation exchange of NH4+ with tetra-n-butylammonium leading to a solubility change from aqueous to organic solvent would facilitate the single crystal growth process. But, it is not a necessary option and may restrict applications in aqueous or biological aspects.7 Thus, we also attempted to maintain the NH4+ cation from the parent Cu-centered Anderson clusters to extend its potential applications in future in aqueous or biological situations. We employed tris ligand NH2C(CH2OH)3 to react with the parent Cu-centred Anderson cluster (NH4)4[Cu(OH)6Mo6O18] at 80 °C for 2 h in aqueous media. The blue solution gradually changed to bluish green, and the water-soluble symmetric triol ligand functionalized Cu-centred Anderson organometallic cluster with NH4+ cation (NH4)4{[NH2C(CH2O)3]2CuMo6O18} (referred to as NH2–[CuMo6]–NH2 hereafter) was formed for the first time (Scheme 1, for synthetic details, see the Experimental section).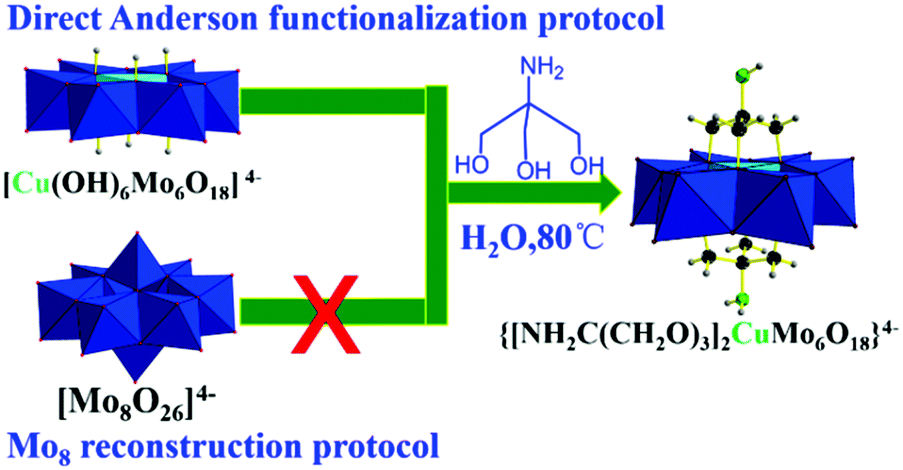 | ||
| Scheme 1 Synthesis of a water-soluble tris functionalized Cu-centred Anderson organic hybrid cluster through the direct parent Anderson alkoxylation protocol. | ||
A suitable bluish green block-like crystal for X-ray diffraction was grown by slow H2O evaporation. Single crystal X-ray diffraction analysis revealed that the NH2–[CuMo6]–NH2 cluster crystallizes in the tetragonal centric space group P42/n.18 As shown in Fig. 1, the geometry of the polyanion is the flat Anderson topology structure (α type) with D3d symmetry: six edge-sharing MoO6 octahedra are arranged around a central CuO6 unit. Tris ligand anchored on three μ3-O atoms on each side of the Anderson cluster. It should be noted that the rigidity of the flat Anderson framework prevents the CuII ion from undergoing marked Jahn–Teller distortions. The coordination octahedron is only very slightly elongated in the bond Cu1–O2 direction. Four cationic NH4+ moieties were inherited from the parent Anderson cluster (NH4)4[Cu(OH)6Mo6O18]. This meets the charge balance. Thus, no protonation of the amino moiety or the remaining μ2-O atoms was observed as previously reported.12 From the 3D framework structure in the direction of the [100] crystal plane (a-axis), the NH2–[CuMo6]–NH2 clusters pack one by one with a rotational angle of 120° forming a quasi 2D structure with atomically dispersed Cu2+ in a uniform distance of 11.76 Å and possessing two shapes of nanopore; one is approximately oval with 5.7 Å × 5.5 Å size and the other is approximately a water droplet with 3.6 Å × 8.6 Å size (Fig. S1 in the ESI†).
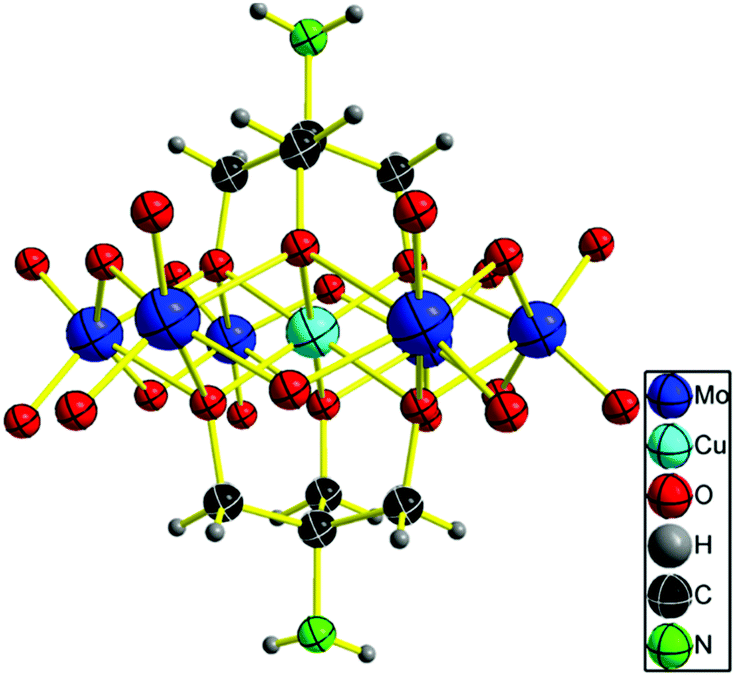 | ||
| Fig. 1 ORTEP drawings of [CuMo6] cluster anions. Thermal ellipsoids are drawn at the 30% probability level. | ||
Further, the NH2–[CuMo6]–NH2 cluster was characterized by XPS, TGA, FT-IR spectroscopy, UV-Vis spectroscopy, ESI-MS, and EA spectroscopy. Thermogravimetric analysis (TGA) was utilized to explore the thermal stability. The NH2–[CuMo6]–NH2 cluster remained intact up to a temperature of 200 °C, as no weight loss was observed. Partial crystal water in the framework wasexpelled with 5% weight loss at the temperature of 200–260 °C. Then, the remaining crystal water and NH4+ counter-ion of 7% weight loss occurred at 260–700 °C. This strongly suggested that the thermal stability of the NH2–[CuMo6]–NH2 cluster is largely improved by the protection of the functionalized triol ligand. It should be noted that the parent Anderson clusters and [Mo7O24]6− with a similar structural topology to the NH2–[CuMo6]–NH2 cluster are not stable at 500 °C, as they are not functionalized with triol ligands (Fig. S2 in the ESI†).12
X-ray photoelectron spectroscopy (XPS) was implemented to investigate the oxidation states of Mo and Cu (Fig. 2 and Fig. S3 in the ESI†). The existence of Mo and Cu elements in NH2–[CuMo6]–NH2 was evidenced by the full spectrum survey. The narrow element spectra of Mo and Co with raw intensity and peak fitting sum using the XPSPEAK41 program are presented. In the narrow spectrum of Mo, the peaks at 235.31 and 232.16 eV were assigned to Mo6+ (3d3/2) and Mo6+ (3d5/2) orbital binding energies, respectively, implying that the Mo oxidation state was the highest valence of MoVI. While in the narrow spectrum of Cu, the peaks at 951.56 and 932.51 eV belonged to Cu2+ (2p1/2) and Cu2+ (2p3/2) orbital binding energies, indicating that the Cu oxidation state was not changed.19
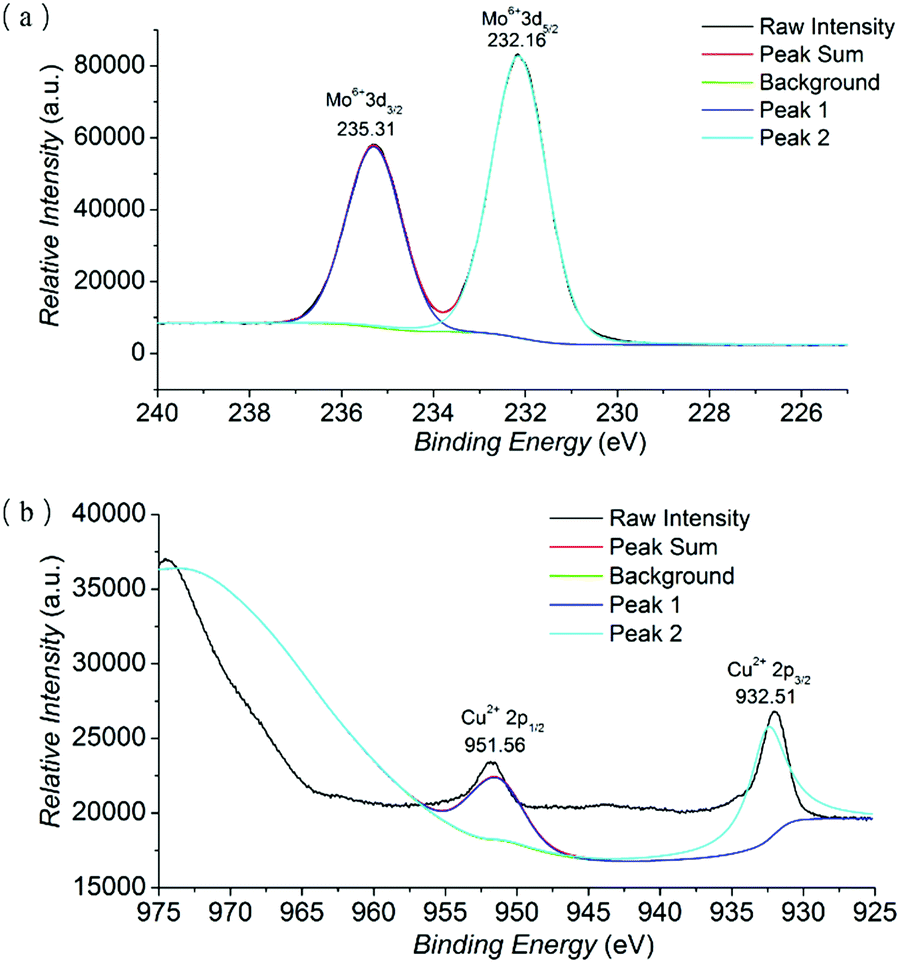 | ||
| Fig. 2 The narrow element XPS spectrum with raw intensity and peak fitting sum of Mo (a) and Cu (b) of the NH2–[CuMo6]–NH2 cluster. | ||
The result was also consistent with the further BVS calculation of Mo and Cu atoms in NH2–[CuMo6]–NH2 using the Bond_Str program; the atomic coordinate was imported from the single crystal cif file of NH2–[CuMo6]–NH2. The bond valence sums of Mo atoms range from 5.94 to 6.54, and the bond valence sum of the Cu atom was 2.12 (the BVS calculation details of NH2–[CuMo6]–NH2 can be found in Table S1 in the ESI†). The combination of XPS spectra, BVS calculation and charge balance consideration definitely confirmed that the Cu and Mo oxidation states were CuII and MoVI, respectively. The bond valence sums of μ2-O atoms (O4–O6) ranging from 1.907 to 2.319 confirmed that no protonation occurred, as compared with the clearly confirmed protonated μ-O atoms in the Anderson cluster, which were obviously smaller and ranged from 1.099 to 1.238.12
In the FT-IR spectra, the characteristic peaks at 969, 930, and 809 cm−1 were assigned to the vibrations of terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O units, and the peaks at 704 and 629 cm−1 belonged to the vibrations of the Mo–O–Mo groups (Fig. S4 in the ESI†). These peaks are consistent with the typical parent Anderson-type structures. The characteristic peak at 1142 cm−1 was assigned to the vibration peak of the C–O bonds, indicating the symmetric grafting of triol onto the surface of the Anderson cluster. ESI-MS analysis shows only one ion peak at m/z = 582.93 with z = −2, which matches well with the anionic cluster of acidized {[NH3C(CH2O)3]2CuMo6O18}2− (theoretical m/z: 582.71, Fig. S5 in the ESI†). UV-Vis spectroscopy of NH2–[CuMo6]–NH2 in H2O was conducted to investigate the LMCT absorption and d–d transition absorption; the band located at 246 nm corresponds to the μ3-O π to metal-centered MoVI t2g* charge transfer transition (LMCT), the d–d transition absorption band located around 687 nm is assigned to the metal centered lowest energy electronic transition from the HOMO t2g* to LUMO eg* of CuII. It should be noted that normally, the substitution of a proton in μ3-O with the triol ligand will not cause the color change of the corresponding triol functionalized Anderson cluster and due to the increase of crystal field splitting energy, a slight hypsochromic shift may occur, as described in our previous report. However, regarding the tris functionalized Cu-centred Anderson cluster, the color was obviously changed from light blue that the parent Cu Anderson cluster possesses to bluish green. And, both the LMCT and d–d transition absorption exhibited a bathochromic shift, as compared with the corresponding LMCT (224 nm) and d–d transition (607 nm) absorption in the parent Cu Anderson cluster (NH4)4[Cu(OH)6Mo6O18]. This may be due to the rigidity of the flat Anderson framework preventing the CuII ion from undergoing marked Jahn–Teller distortions (Fig. S6–S8 in the ESI†).
O units, and the peaks at 704 and 629 cm−1 belonged to the vibrations of the Mo–O–Mo groups (Fig. S4 in the ESI†). These peaks are consistent with the typical parent Anderson-type structures. The characteristic peak at 1142 cm−1 was assigned to the vibration peak of the C–O bonds, indicating the symmetric grafting of triol onto the surface of the Anderson cluster. ESI-MS analysis shows only one ion peak at m/z = 582.93 with z = −2, which matches well with the anionic cluster of acidized {[NH3C(CH2O)3]2CuMo6O18}2− (theoretical m/z: 582.71, Fig. S5 in the ESI†). UV-Vis spectroscopy of NH2–[CuMo6]–NH2 in H2O was conducted to investigate the LMCT absorption and d–d transition absorption; the band located at 246 nm corresponds to the μ3-O π to metal-centered MoVI t2g* charge transfer transition (LMCT), the d–d transition absorption band located around 687 nm is assigned to the metal centered lowest energy electronic transition from the HOMO t2g* to LUMO eg* of CuII. It should be noted that normally, the substitution of a proton in μ3-O with the triol ligand will not cause the color change of the corresponding triol functionalized Anderson cluster and due to the increase of crystal field splitting energy, a slight hypsochromic shift may occur, as described in our previous report. However, regarding the tris functionalized Cu-centred Anderson cluster, the color was obviously changed from light blue that the parent Cu Anderson cluster possesses to bluish green. And, both the LMCT and d–d transition absorption exhibited a bathochromic shift, as compared with the corresponding LMCT (224 nm) and d–d transition (607 nm) absorption in the parent Cu Anderson cluster (NH4)4[Cu(OH)6Mo6O18]. This may be due to the rigidity of the flat Anderson framework preventing the CuII ion from undergoing marked Jahn–Teller distortions (Fig. S6–S8 in the ESI†).
Before the catalytic application in p-nitrophenol reduction, powder XRD was used to check the phase purity of the as-prepared NH2–[CuMo6]–NH2. The experimental powder XRD pattern of NH2–[CuMo6]–NH2 was almost identical to the simulated powder XRD pattern, especially the excellent fitting of the major three peaks at the low angle area of 2θ 5–15° (Fig. S8 in the ESI†). This strongly indicated the phase purity of the NH2–[CuMo6]–NH2 cluster.
p-Nitrophenol hydrogenation reduction
The stable tris functionalized Cu-centred Anderson cluster NH2–[CuMo6]–NH2 was employed as an abundant non-noble metal bimetallic catalyst to afford rapid aqueous p-nitrophenol reduction towards p-aminophenol production. The whole reaction process of p-nitrophenol reduction towards p-aminophenol generation was monitored by UV-Vis spectroscopy. The characteristic absorption peak of p-nitrophenol at 400 nm gradually decreased, while a new characteristic peak of p-aminophenol appeared at 300 nm (Fig. 3).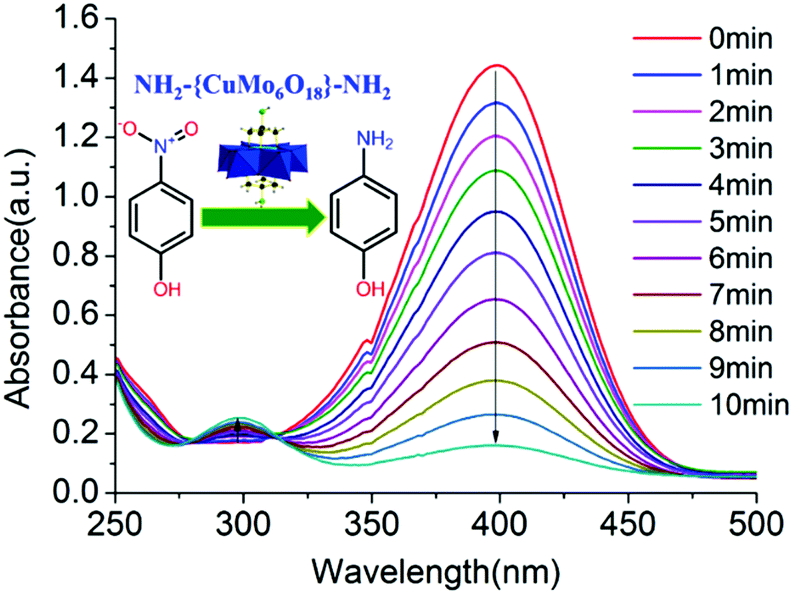 | ||
| Fig. 3 Reaction process of p-nitrophenol reduction towards p-aminophenol generation was monitored by UV-Vis spectroscopy employing 5 mg of bimetallic NH2–[CuMo6]–NH2 as a catalyst. | ||
The p-nitrophenol reduction over the NH2–[CuMo6]–NH2 cluster was rapid. Within 10 min, p-nitrophenol was completely converted with 99% selectivity for p-aminophenol generation. To quantitatively verify the reactant conversion and product selectivity, besides the UV-Vis spectral monitoring, GC-MS spectrum monitoring can serve as an alternative method to eliminate experimental error. The retention time of the reactant p-nitrophenol and the corresponding product p-aminophenol appeared at 10.023 and 8.552 min, respectively. Thus, it was easy to distinguish their corresponding integral area from each other and from other peaks, which indicated the generation of other possible by-products. With the reduction reaction proceeding, the peak intensity and area at 10.023 min gradually diminish, while the peak intensity and area at 8.552 min gradually increase. Considering the fact that at the end of such a catalytic reaction, the peak at 10.023 would almost disappear and become undetectable, to illustrate this clearly, we chose to show the GC-MS monitoring result at the very beginning of such a catalytic reaction at 3.5 min. Through the integral area of these two peaks combined with material balance, it was clear that the corresponding reactant conversion was 31.6% and product selectivity was 100%. The purity of the extracted p-aminophenol was further examined by GC-MS to confirm that no other by-products were produced (Fig. S9 and Table S2 in the ESI†). It should be noted that the bimetallic synergistic effect of NH2–[CuMo6]–NH2 affords the rapid reduction. Control experiments were also conducted under the same reaction conditions with three other control groups: (1) without any metal catalyst; (2) using the bimetallic parent [Cu(OH)6Mo6O18]4− Anderson cluster without triol functionalization and (3) a monometallic [Mo7O24]6− cluster with a similar topology structure as the catalyst, respectively. The quantitative catalytic performance was correlated with the accomplishment of p-nitrophenol conversion time. Without any metal catalyst, after 150 min, the concentration of p-nitrophenol was little changed. For the fresh [Cu(OH)6Mo6O18]4− catalyst, the reaction can fully complete within 10 min, which is somewhat faster than the NH2–[CuMo6]–NH2 cluster. While for [Mo7O24]6−, the reaction was still uncompleted after 130 min. It should be noted that the catalytic reaction can be regarded as completed when the UV absorption of p-nitrophenol reaches 0.02. This implies that both the catalytic reaction rates of the bimetallic [Cu(OH)6Mo6O18]4− and NH2–[CuMo6]–NH2 catalysts were one order higher (∼16 times) than monometallic [Mo7O24]6− (Fig. S10 in the ESI†).
Furthermore, to quantitatively compare their catalytic reaction rate, catalytic reaction kinetics was also investigated. The initial catalytic reaction rate was assumed to be equal to the initial slope (ΔAbs/Δtime), where Abs is the absorbance at 400 nm of p-nitrophenol. For the linear fitting analysis, the logarithm of reactant p-nitrophenol concentration showed a linear dependence versus time in the following equation: lnCt = lnCo − kt with a high Pearson correlation coefficient for these three catalysts (R2 > 0.99, Fig. 4 and Table S3 in the ESI†). It is worth noting that these data were chosen at <40% conversion to investigate the kinetics study. Combined with our previous investigation,20 the reduction reaction process over these three catalysts of NH2–[CuMo6]–NH2, [Cu(OH)6Mo6O18]4− and [Mo7O24]6− was all first order, and the reaction rate was constant as the reaction proceeded. Considering the fact that the reducing agent NaBH4 was in significant excess, the reduction reaction process can be viewed as a pseudo first order reaction. Thus, the catalytic performance regarding the reaction kinetics between them was comparable. The reaction rate constant of the kNH2–[CuMo6]–NH2/k[Mo7O24] value was calculated to be ca. 9.4 and for kNH2–[CuMo6]–NH2/k[Mo7O24]/k[Cu(OH)6Mo6O18], it was determined to be ca. 0.84. The as-prepared [Cu(OH)6Mo6O18]4− cluster exhibited some better catalytic performance than NH2–[CuMo6]–NH2. This indicates that the catalytic reaction rate of the bimetallic catalyst is one order higher than that of the monometallic catalyst. The incorporation of Cu to prepare a bimetallic Anderson organometallic cluster derivative catalyst was essential for such a rapid p-nitrophenol reduction rate.
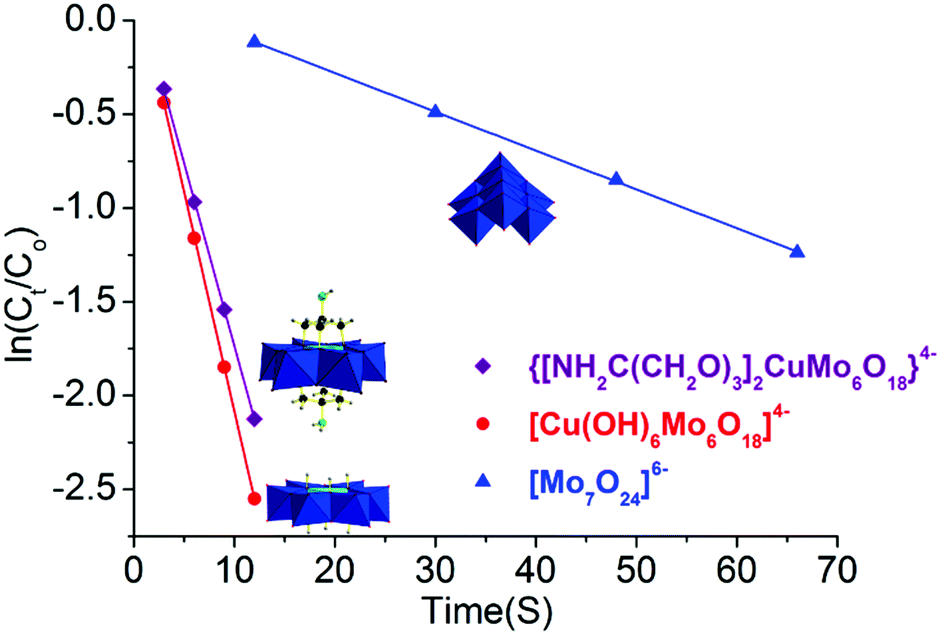 | ||
| Fig. 4 Catalytic reaction kinetics comparison of bimetallic NH2–[CuMo6]–NH2, [Cu(OH)6Mo6O18]4−, and monometallic [Mo7O24]6−. | ||
The amount of bimetallic NH2–[CuMo6]–NH2 catalyst was also optimized. Under the same catalytic reaction conditions, when the dose of bimetallic catalyst was 1 mg, the reaction rate constant was even lower than the monometallic [Mo7O24]6− catalyst. However, the reaction rate constant could dramatically increase to 31.3 times when the bimetallic catalyst amount reached 5 mg. Thus, the dose of bimetallic catalyst was optimized as 5 mg for the p-nitrophenol reduction. The reduction processes with different catalytic doses were confirmed to maintain first order reaction kinetics with a high Pearson correlation coefficient for these three catalysts (R2 > 0.99, Fig. S11 and Table S4 in the ESI†). Note that upon the addition of pure H2 into such a catalytic reaction instead of NaBH4 as the hydrogen source, the p-nitrophenol reduction still works but becomes relatively slow (about 100 min to complete the reaction) because the low solubility of H2 in aqueous reaction systems makes the reduction reaction relatively slow, as previously reported.4,5 Indeed, NaBH4 is more expensive compared to pure H2. However, to balance the price and efficiency of such a catalytic reaction. It was worth employing NaBH4 as an in situ generated hydrogen source for rapid p-nitrophenol hydrogenation as the previous investigation did.4 It should be noted that 5 eq. NaBH4 is required for the p-nitrophenol reduction employing NH2–[CuMo6]–NH2 as the catalyst, based on the Mars–van Krevelen reaction pathway, and for the one molecular p-nitrophenol hydrogenation, at least 1.5 eq. NaBH4 was required, and the hydrogen utilization coefficient of NH2–[CuMo6]–NH2 was 30%. However, our catalyst system was still economically feasible and competitive, considering these two aspects: (i) the relatively cheap price and abundant resource of NaBH4 as compared with the corresponding high-value-added amino-aromatics even though it was still more expensive than pure H2; (ii) to afford amino-aromatics through nitro-aromatics hydrogenation, usually noble metal catalysts4 including Au and Pd with 10–50 eq. NaBH4 in organic solvents (e.g., THF, methanol, etc.) are required to obtain a high yield and selectivity. These catalysts are more expensive, the hydrogen utilization coefficient is about 3–15%, and the use of organic solvents is not sustainable for green production.
The covalent functionalized triol ligand on the Cu-centred Anderson cluster can dramatically improve the thermal stability; and, the incorporation of triol ligand can also adjust the solubility of bimetallic NH2–[CuMo6]–NH2 catalysts in water, which makes it easier for them to be recovered from the aqueous reaction media. Taking these advantages into account, its recyclability was further examined and compared among the NH2–[CuMo6]–NH2, [Cu(OH)6Mo6O18]4− and [Mo7O24]6− catalysts. The recycled catalysts were separated from the reaction system and reused in the reduction reaction under identical conditions. The yield of the p-aminophenol product was defined as selectivity multiplied by conversion (i.e., yield = selectivity × conversion). To examine the robustness of the catalyst in the aspects of recyclability and catalytic reactivity, the conversion, selectivity, and yield of p-nitrophenol were presented and explored, employing {[NH2C(CH2O)3]2CuMo6O18}4− as the catalyst of five cycle runs (Fig. S12 in the ESI†). The recovered cluster catalyst showed no appreciable loss due to its high catalytic performance after five runs. The p-nitrophenol conversion remained at 100% and the p-aminophenol selectivity and yield were only slightly decreased to 98%. It should be noted that the reaction time of these five cycle runs was the same (about 10 min). While employing [Cu(OH)6Mo6O18]4− and [Mo7O24]6− as the catalyst, their catalytic recyclabilities were not stable. For the [Mo7O24]6− clusters, the conversion and the yield could reach 100% and 90%, respectively, in the first run. However, the recycled [Mo7O24]6− cluster irreversibly became “molybdenum blue” after the hydrogenation, indicating that the [Mo7O24]6− catalyst was not stable during the reaction. It was irreversibly and partially reduced and transferred to “molybdenum blue” in the absence of the triol ligand protection.21 And, the recycled [Mo7O24]6− catalyst was obviously depressed with 90% conversion, 79% selectivity, and 71% yield in the second run. For [Cu(OH)6Mo6O18]4−, the conversion and the yield can reach to 100% and 95%, respectively, in the first run. However, a similar “molybdenum blue” effect occurred when [Cu(OH)6Mo6O18]4− was exposed to the BH4− system without the triol ligand protection. And, the recycled [Cu(OH)6Mo6O18]4− catalyst was also partially and permanently became “molybdenum blue” with an obvious catalytic performance depression of 95% conversion, 85% selectivity, and 80.71% yield in the second run (Fig. S13 and S14 in the ESI†). These results indicated that the tris ligands indeed play a key role in sustaining the stability of the CuMo6 Anderson cluster. Furthermore, the recovered catalysts after five cycle tests were characterized by powder XRD. The powder XRD pattern of the recovered catalysts was nearly identical to the original experimental pattern of the as-prepared NH2–[CuMo6]–NH2 cluster. In particular, the excellent fitting of the major three peaks at the low angle area of 2θ ranging from 5 to 15° was possible. Note that the PXRD peak intensity of the recovered catalyst was attenuated due to crystal size reduction, and the crystal lattice defects increase after the catalyst recycling from the homogeneous catalytic reaction process, as in our previous investigation8,9 (Fig. S8 in the ESI†). Thus, the NH2–[CuMo6]–NH2 catalyst exhibited both excellent catalytic reactivity and recyclability, and the NH2–[CuMo6]–NH2 cluster was intact during the p-nitrophenol hydrogenation catalytic reaction process. Although the as-prepared [Cu(OH)6Mo6O18]4− cluster also exhibited high catalytic performance for the first run, however, without triol ligand stabilization, it also exhibited a low catalytic recyclability.
Furthermore, the electrochemical redox properties of NH2–[CuMo6]–NH2 and (NH4)4[Mo7O24] and the tris ligand were also further investigated. The CV curve of NH2–[CuMo6]–NH2 consisted of two one-electron redox processes with the potential range from −0.8 to 0.6 V, while for (NH4)4[Mo7O24] and the tris ligand, only one-electron redox process was observed in the range from −1.2 to −0.2 V and −0.3 to 0.1, respectively (Fig. 5 and Fig. S15). For the (NH4)4[Mo7O24] clusters, obvious defined peak-to-peak separation with a ΔEp of 232 mV between the reduction Epc (−681 mV) and oxidation Epa (−449 mV) with peak potentials I′ and I corresponded to the one-electron reversible redox process with a potential E1/2 of 565 mV, which is attributed to the MoVI/MoV couple. For the tris ligand, obvious defined peak-to-peak separation with a ΔEp of 380 mV between the reduction Epc (−299 mV) and oxidation Epa (81 mV) with peak potentials I′ and I corresponded to the one-electron reversible redox process with a potential E1/2 of 170 mV, which is attributed to the NH2/NH3 couple. This indicates that both the MoVI/MoV couple and the NH2/NH3 couple in the tris ligand contribute to the electrochemical redox process. In NH2–[CuMo6]–NH2, such redox processes of the MoVI/MoV couple and the NH2/NH3 couple were merged, and an ill-defined peak was located at potentials II′ and II with a ΔEp of 273 mV between the reduction Epc (−604 mV) and oxidation Epa (−331 mV) with a potential E1/2 of 467.5 mV. From the characteristic potential E1/2, this one-electron reversible redox process was mainly attributed to the MoVI/MoV couple. The remote NH2 motif electrochemical redox contribution in NH2–[CuMo6]–NH2 was small. The voltage positions of the redox couple of MoVI/MoV in the NH2–[CuMo6]–NH2 cluster were mediated by this remote NH2 motif. Thus, they were a little different from the voltage positions of the redox couple of MoVI/MoV in the (NH4)4[Mo7O24] cluster. The well-defined peak-to-peak separation with a ΔEp of 199 mV between the reduction Epc (−157 mV) and oxidation Epa (42 mV) with peak potentials I′ and I corresponded to the one-electron reversible redox process with a potential E1/2 of 99.5 mV, attributed to the CuII/CuI couple. Thus, the electrochemical redox property of the {[NH2C(CH2O)3]2CuMo6O18}4− cluster showed a reversible two one-electron process, and the CuII/CuI redox couple was the featured electrochemical property. This was reasonable, since the incorporation of a central heteroatom can obviously mediate the Frontier molecular orbitals and HOMO–LUMO energy gaps of the Anderson cluster according to Streb's investigation.22
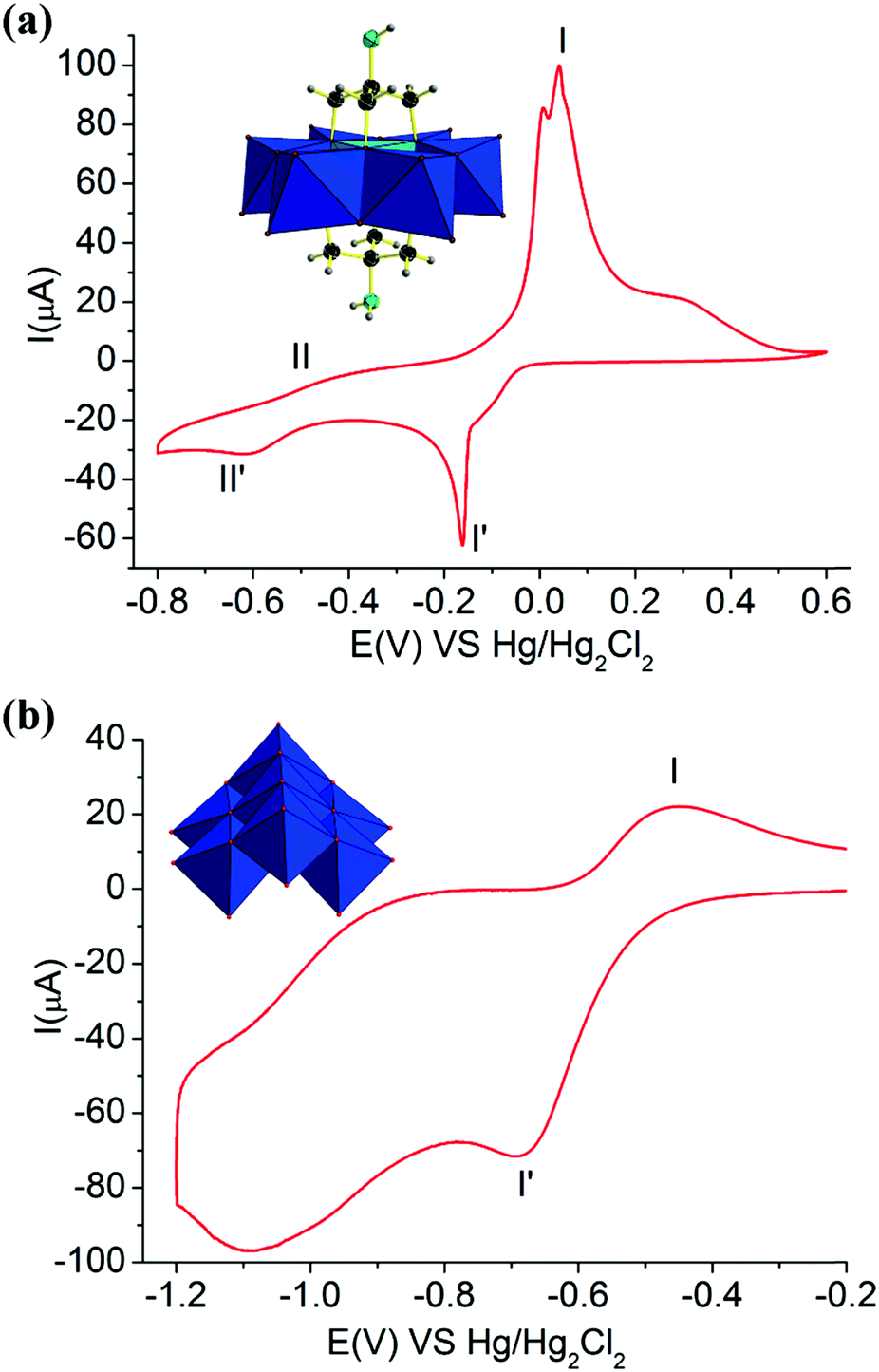 | ||
| Fig. 5 Cyclic voltammetry of compound 1 (a) and (NH4)4[Mo7O24] (b). | ||
Hence, this affected the electrochemical redox behavior of the NH2–[CuMo6]–NH2 cluster and (NH4)4[Mo7O24], resulting in such a difference. The incorporated central heteroatom in polymolybdates usually serves as the first redox couple and the features redox peaks, according to previous studies.8,23 We also discovered the fact that besides the triol ligand that can thermally stabilized the NH2–[CuMo6]–NH2 cluster during the catalytic reaction process, the remote NH2 motif can also mediate and promote the electrochemical redox properties of NH2–[CuMo6]–NH2. Such featured reversible two one-electron redox couples play an important role in the electrochemical stability aspect and served as an ideal multielectron reservoir during such catalytic p-nitrophenol hydrogenation reactions.8 Regarding the reactive site of NH2–[CuMo6]–NH2 in such p-nitrophenol hydrogenation to p-aminophenol, according to the previous hydrogenation reaction mechanism investigation,24 such a catalytic process involved the Mars–van Krevelen reaction pathway, while the μ-O atom serves as the reactive site as follows: the Lewis acidic vacancy is generated by reducing the catalyst surface with NaBH4, and p-nitrophenol binded onto such an oxophilic site through its oxygen atom. The coordinated p-nitrophenol can undergo hydrogenolysis to p-aminophenol. This featured reversible two one-electron redox couples may also promote the reduction process from BH4− to the catalyst surface to enable rapid and effective Lewis acidic vacancy generation.
Conclusions
In conclusion, we have developed a protocol to conveniently obtain a water soluble tris functionalized Cu-centered Anderson organometallic cluster with the remote reactive amino moiety (NH4)4{[NH2C(CH2O)3]2CuMo6O18} by the direct parent Anderson functionalization protocol. The cluster cannot be obtained by the traditional Mo8 reconstruction protocol. The single crystal structure of (NH4)4{[NH2C(CH2O)3]2CuMo6O18} was reported for the first time. XPS and BVS definitely confirm the CuII and MoVI oxidation state. Triol largely improved the thermal stability of the cluster, which remains stable up to 700 °C. Finally, (NH4)4{[NH2C(CH2O)3]2CuMo6O18} can serve as an abundant non noble metal-containing bimetallic catalyst to afford rapid aqueous p-nitrophenol reduction towards p-aminophenol production with high conversion (a decent 100%) and selectivity (ca. 99%) under very mild reaction conditions (e.g., in an aqueous solution at room temperature). The catalytic reaction kinetics curve indicates that such p-nitrophenol reduction was a pseudo first order reaction. The reaction rate constant of the bimetallic {[NH2C(CH2O)3]2CuMo6O18}4− catalyst was one order higher (9.4) than that of the monometallic [Mo7O24]6− catalyst, which was attributed to the bimetallic synergistic effect. The bimetallic cluster maintains excellent catalytic performance and recyclability with an intact structure. However, the [Cu(OH)6Mo6O18]4− cluster without the triol ligand protection cannot maintain catalytic reactivity and recyclability. Tris ligands play a key role in sustaining the stability of the CuMo6 Anderson cluster and the remote NH2 motif can also mediate its electrochemical redox property to maintain high catalytic performance. The cyclic voltammetry investigation indicated that the incorporation of heteroatom Cu mediated the electrochemical redox behaviour of the {[NH2C(CH2O)3]2CuMo6O18}4− cluster. A reversible two one-electron process was observed in which CuII/CuI and MoVI/MoV were the redox couples. This served as an ideal electrochemically stable multielectron reservoir during such p-nitrophenol hydrogenation catalytic reduction. This is a more eco-friendly and economically feasible protocol for p-aminophenol production regarding its important applications in pharmaceutical intermediates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the fund of the National Natural Science Foundation of China (No. 21701168 and 21703235), the Liaoning Natural Science Foundation (No. 20170540897 and 20180510050), Open project Foundation of State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University (No. 201709) and Shanxi Province Hundred Talent Project. The authors acknowledge BL14B and BL17B beamline of National Facility for Protein Science (NFPS), Shanghai Synchrotron Radiation Facility (SSRF) Shanghai, China for providing the beam time.Notes and references
- P. Herves, M. Perez-Lorenzo, L. M. Liz-Marzan, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC.
- P. Zhao, X. Feng, D. Huang, G. Yang and D. Astruc, Coord. Chem. Rev., 2015, 287, 114 CrossRef CAS.
- A. Corma, P. Serna, P. Concepción and J. J. Calvino, J. Am. Chem. Soc., 2008, 130, 8748 CrossRef CAS.
- (a) M.-B. Li, S.-K. Tian, Z. Wu and R. Jin, Chem. Commun., 2015, 51, 4433 RSC; (b) Q. Yang, Y.-Z. Chen, Z. U. Wang, Q. Xu and H.-L. Jiang, Chem. Commun., 2015, 51, 10419 RSC.
- (a) Y. Ma, Y. Ni, F. Guo and N. Xiang, Cryst. Growth Des., 2015, 15, 2243 CrossRef CAS; (b) A. K. Sasmal, S. Dutta and T. Pal, Dalton Trans., 2016, 45, 3139 RSC.
- C. Zhang, J. Lu, M. Li, Y. Wang, Z. Zhang, H. Chen and F. Wang, Green Chem., 2016, 18, 2435 RSC.
- J. Zhang, Y. Huang, G. Li and Y. Wei, Coord. Chem. Rev., 2019, 378, 395 CrossRef CAS.
- Y. Chen, C. Zhang, C. Yang, J. Zhang, K. Zheng, Q. Fang and G. Li, Nanoscale, 2017, 9, 15332 RSC.
- Y. Chen, C. Zhang, J. Zhang, Z. Ye, K. Zheng, Q. Fang and G. Li, Inorg. Chem. Front., 2017, 4, 1917 RSC.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- (a) C. Molitor, A. Bijelic and A. Rompel, Chem. Commun., 2016, 52, 12286–12289 RSC; (b) A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 CrossRef CAS; (c) A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS PubMed.
- (a) J. Luo, Y. Huang, B. Ding, P. Wang, X. Geng, J. Zhang and Y. Wei, Catalysts, 2018, 8, 121–133 CrossRef; (b) J. Zhang, Y. Huang, J. Hao and Y. Wei, Inorg. Chem. Front., 2017, 4, 1215–1218 RSC; (c) J. Zhang, Q. Li, M. Zeng, Y. Huang, J. Zhang, J. Hao and Y. Wei, Chem. Commun., 2016, 52, 2378–2381 RSC; (d) J. Zhang, J. Luo, P. Wang, B. Ding, Y. Huang, Z. Zhao, J. Zhang and Y. Wei, Inorg. Chem., 2015, 54, 2551–2559 CrossRef CAS; (e) J. Zhang, Z. Liu, Y. Huang, J. Zhang, J. Hao and Y. Wei, Chem. Commun., 2015, 51, 9097–9100 RSC; (f) J. Zhang, Z. Zhao, J. Zhang, S. She, Y. Huang and Y. Wei, Dalton Trans., 2014, 43, 17296–17302 RSC; (g) J. Zhang, Y. Huang, J. Zhang, S. She, J. Hao and Y. Wei, Dalton Trans., 2014, 43, 2722–2725 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- (a) Y. Wang, X. Kong, W. Xu, F. Jiang, B. Li and L. Wu, Inorg. Chem., 2018, 57, 3731–3741 CrossRef CAS; (b) Y. Wang, X. Liu, W. Xu, Y. Yue, B. Li and L. Wu, Inorg. Chem., 2017, 56, 7019–7028 CrossRef CAS; (c) Y. Wang, B. Li, H. Qian and L. Wu, Inorg. Chem., 2016, 55, 4271–4277 CrossRef CAS.
- B. Hasenknopf, R. Delmont, P. Herson and P. Gouzerh, Eur. J. Inorg. Chem., 2002, 1081 CrossRef CAS.
- N. I. Gumerova, A. Roller and A. Rompel, Eur. J. Inorg. Chem., 2016, 5507–5511 CrossRef CAS.
- Crystal data and structure refinement for (NH4)4{[NH2C(CH2O)3]2CuMo6O18}·6H2O: tetragonal, space group P42/n, a = b = 16.6347(4) Å, c = 12.9722(5) Å, V = 3589.6(2) Å3, Z = 4, T = 293(2) K, 3715 reflections measured, R1 = 0.0332, wR2 = 0.0823. CCDC 1811030 contains the supplementary crystallographic data for this structure†.
- The reference of standard binding energies of elements in different oxidation states was obtained from National Institute of Standards and Technology (NIST) X-ray Photoelectron Spectroscopy Database at the website: http://srdata.nist.gov/xps/.
- H. Chen, C. Liu, M. Wang, C. Zhang, G. Li and F. Wang, Chin. J. Catal., 2016, 37, 1787–1793 CrossRef CAS.
- T. Yamase, Y. Yano and E. Ishikawa, Langmuir, 2005, 21, 7823 CrossRef CAS.
- S. Schoenweiz, M. Heiland, M. Anjass, T. Jacob, S. Rau and C. Streb, Chem. – Eur. J., 2017, 23, 15370–15376 CrossRef CAS.
- G.-G. Gao, L. Xu, W.-J. Wang, X.-S. Qu, H. Liu and Y.-Y. Yang, Inorg. Chem., 2008, 47, 2325–2333 CrossRef CAS.
- E. Anderson, A. Crisci, K. Murugappan and Y. Roman-Leshkov, ChemSusChem, 2017, 10, 2226–2234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1811030 for compound 1. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c8nj04265a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |