
Dibromination of alkenes with LiBr and H2O2 under mild conditions†
Nayara Silva
Martins
and
Eduardo E.
Alberto
 *
*
Department of Chemistry, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil. E-mail: albertoee@ufmg.br; Tel: +55 31 3409–5696
First published on 20th November 2017
Abstract
Electron-rich and electron-poor alkenes, and alkenes bearing protecting groups can be efficiently and stereoselectively converted to trans-dibromides using LiBr/H2O2 and AcOH as a proton source in 1,4-dioxane. For most substrates addition of 0.1 mol% of PhTeTePh enhances the reaction rate and the yield of the products. Experimental data suggest that the brominating agent prepared in situ is molecular bromine and that LiBr assists the activation of H2O2 allowing bromination to occur using AcOH as a mild proton source in uncatalyzed experiments. Scale-up is feasible: 10.0 mmol of 1-octene was quantitatively converted to 1,2-dibromooctene in one hour of reaction at room temperature.
Introduction
It's widely agreed that halogenated organic compounds represent a paramount class of substances. Among them, brominated substrates find broad applicability as valuable commodities in synthetic transformations, and as pharmaceuticals, agrochemicals, etc.1 The simplest approach to produce brominated products is by the electrophilic addition of elemental bromine to organic substrates (e.g. alkenes and aromatic compounds).2 One alternative to avoid the high vapor pressure of Br2 is the use of ionic liquid tribromide salts.3 Another option is to employ reagents such as N-bromosuccinimide and its derivatives, which usually require the addition of a catalyst for activation.4 However, all of these strategies require the use of elemental bromine (either directly or in an early stage of the process), which is well known for having severe hazards associated with respect to its transport, handling, and storage. In addition, the above-mentioned transformations are usually carried out in highly regulated halogenated solvents.Nature has been producing brominated compounds in a sophisticated manner: promoting the oxidation of bromide salts with peroxides through the action of haloperoxidase enzymes (most commonly found in marine organisms).5 The development of catalytic systems, which can mimic the action of such enzymes, is a promising field of research. Numerous reports in the literature describe the activation of H2O2 by isolated or cloned haloperoxidase enzymes, and metal and metal-free catalysts, typically in aqueous solutions.6 In common, all of these protocols require a source of protons, which is critical to the fate of the oxidized bromide species: under basic conditions the “Br+” species (presumably HOBr) promotes the irreversible disproportionation of H2O2 to oxygen and water faster than it can be captured by an organic substrate.7 Under haloperoxidase-like conditions (H2O2, bromide salt in aqueous acidic solutions) selectivity is problematic. Generally only electron-rich aromatic compounds, ketones or water-soluble functionalized alkenes are appropriate substrates. Attempts to dibrominate simple alkenes under these conditions, often produce a mixture of products, commonly by the addition of water to the bromiranium ion, producing bromohydrins as major products.6,8 Bromination of organic substrates by oxidation of bromide salts using peroxides activated by a catalyst can also be performed in non-aqueous solutions.9 Uncatalyzed reactions are also feasible, but require the addition of strong Brønsted acids (e.g. HCl, HBr, H2SO4) for activation of the peroxide.10 Alternatively, oxidizing agents such as molecular oxygen,11 oxone,12 NaBrO3,13 NaIO414 and DMSO15 have been reported for bromination of organic substrates.
Many efforts have been directed towards achieving approaches to brominate organic compounds employing efficient and reliable methods, also bearing in mind environmental aspects.16 However, although much progress has been achieved in this field, many of the reported protocols suffer from one or more of the following disadvantages (especially in the bromination of alkenes): lack of substrate generality, use of large excess of reagents, harsh reaction conditions (e.g. hazardous reagents, halogenated solvents), undesirable waste formation or low selectivity in the product formation.3,4,6,8–15
Recently the activation of H2O2 by a catalytic amount of diphenyl ditelluride (PhTeTePh) and in situ oxidation of bromide salts to deliver “Br+” species has been disclosed (Scheme 1).6h Water soluble or highly reactive substrates such as 4-pentenoic acid and 1,3,5-trimethoxybenzene could be effectively converted to their respective brominated products in a 1/1 mixture of phosphate buffer (pH 4.2–6.2) and 1,4-dioxane with an excess of NaBr (15.0 equivalents per mmol of substrate). We attempted to use these conditions for the dibromination of cyclohexene 1a. However, the desired product 2a was obtained in only 14% yield (dotted box in Scheme 1).
 | ||
| Scheme 1 Bromination of 1a in aqueous solution of NaBr and H2O2 catalyzed by PhTeTePh. a Yield calculated by CG analysis using undecane as an internal standard. | ||
Therefore, the failure to efficiently promote such transformation prompted us to further investigate this important functional group interconversion. Considering the environmental aspects associated with the bromination of alkenes, we believe that the H2O2/bromide salt combination is very appealing. Bromide salts are innocuous and abundant, while H2O2 is a strong oxidizing agent that produces water as a byproduct.17 Our aim was to promote the oxidation of bromide salts by H2O2in situ with simultaneous bromination of alkenes. Moreover, we were interested in delivering a broad range of brominated products efficiently, both in terms of yield as well as selectivity, employing mild reaction conditions.
Results and discussion
Mass transfer of “Br+” species in aqueous solutions is problematic for bromination of simple alkenes with H2O2/bromide due to the hydrophobic nature of the substrate. Consequently, destruction of H2O2 takes place preferentially unless a strong acid is used. Nevertheless, the solubility of bromide salts in organic solvents should also be considered. So, in order to provide a proton source, necessary to drive the reaction to the formation of organic bromides instead of disproportionation of H2O2 and also to find a balance between solubility of inorganic salts and organic substrates, we decided to use acetic acid as a proton source in combination with an organic solvent.Initially, we focused on the bromination of cyclohexene 1a, using a bromide salt (2.4 equivalents), 8.8 M H2O2 (1.2 equivalents), undecane as an internal standard and PhTeTePh as a catalyst (0.10 mol% related to 1a) in a mixture of acetic acid and 1,4-dioxane at room temperature (Table 1). After 1 hour of reaction, the mixture was quenched and the composition of the organic products was determined by gas chromatography, comparing with authentic samples of pure brominated products. In the first attempt to brominate 1a, NaBr was used as a 5.0 M aqueous solution, along with 0.5 mL of AcOH and 2.0 mL of 1,4-dioxane, resulting in a biphasic system. Dibromide 2a was produced in 65% yield along with bromoacetate 3a in a selectivity of 43/1 favoring the former (entry 1). The introduction of pure NaBr enhanced the reaction yield, but not the selectivity of the reaction (entry 2). It was observed that under these conditions, NaBr was partially soluble in the mixture of solvents. We next evaluated the amount of AcOH necessary to efficiently accomplish the bromination of 1a. Increasing the amount of AcOH to 1.0 mL resulted in a homogeneous solution and formation of 2a was observed in 91% yield. Nevertheless, as it could be anticipated, the product selectivity was considerably reduced (entry 3). Conversely, reduction of AcOH to 0.25 mL promoted a slight increase in the selectivity of the process (54/1) but the reaction yield dropped to 54% (entry 4). Other solvents, including polar, either protic or aprotic, and nonpolar solvents were tested. However, none of them showed better results than the original combination of AcOH and 1,4-dioxane (entries 5–11).18
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | AcOH (mL) | Br− | Catb (mol%) | 2a (%) | 2a/3a |
| a Reaction conditions: cyclohexene 1a (0.5 mmol), bromide salt (1.2 mmol), acetic acid (0.25–1.0 mL), solvent (2.0 mL), undecane (internal standard, 0.25 mmol), 8.8 M H2O2 (0.6 mmol) and PhTeTePh (0.0005 mol). One hour of reaction at room temperature. b The catalyst was added to the reaction mixture as a freshly prepared 12 mM solution in 1,4-dioxane (40 μL). c Determined by GC (average for duplicate runs). d NaBr added to the reaction mixture as a 5.0 M solution in distilled water. e 30 minutes of reaction. f n.d. = not determined. g Aerobic oxidation: H2O2 replaced by a balloon of molecular oxygen. | ||||||
| 1d | 1,4-Dioxane | 0.5 | NaBr | 0.1 | 65 ± 1 | 43/1 |
| 2 | 1,4-Dioxane | 0.5 | NaBr | 0.1 | 86 ± 2 | 46/1 |
| 3 | 1,4-Dioxane | 1.0 | NaBr | 0.1 | 91 ± 2 | 30/1 |
| 4 | 1,4-Dioxane | 0.25 | NaBr | 0.1 | 54 ± 3 | 54/1 |
| 5e | 1,4-Dioxane | 0.5 | NaBr | 0.1 | 81 ± 1 | 41/1 |
| 6e | THF | 0.5 | NaBr | 0.1 | 7 ± 2 | n.d.f |
| 7e | DMF | 0.5 | NaBr | 0.1 | 60 ± 6 | 60/1 |
| 8e | MeCN | 0.5 | NaBr | 0.1 | 10 ± 1 | n.d.f |
| 9e | 2-Propanol | 0.5 | NaBr | 0.1 | 63 ± 3 | 32/1 |
| 10e | AcOEt | 0.5 | NaBr | 0.1 | 16 ± 2 | n.d.f |
| 11e | Toluene | 0.5 | NaBr | 0.1 | 51 ± 1 | 20/1 |
| 12 | 1,4-Dioxane | 0.5 | KBr | 0.1 | 5 ± 1 | n.d.f |
| 13 | 1,4-Dioxane | 0.5 | LiBr | 0.1 | 93 ± 1 | >100/1 |
| 14 | 1,4-Dioxane | 0.5 | NaBr | None | 19 ± 0 | n.d.f |
| 15 | 1,4-Dioxane | 0.5 | LiBr | None | 46 ± 1 | >100/1 |
| 16g | 1,4-Dioxane | 0.5 | LiBr | 0.1 | 10 ± 0 | n.d.f |

Subsequently, we examined the role of the bromide salt in the outcome of the reaction. Swapping NaBr for KBr resulted in a dramatic decrease in the reaction yield, most probably due to the poorest solubility of KBr in organic solvents (entry 12). On the other hand, LiBr, which was entirely solubilized within minutes, increased both the yield and the selectivity for the desired product 2a to a satisfactory level (entry 13). The influence of the catalyst PhTeTePh on the outcome of the reaction was studied. We found out that for the bromination of cyclohexene 1a employing NaBr, addition of 0.10 mol% of PhTeTePh does have a positive impact in the reaction rate. While the catalyzed reaction produces dibromide 2a in 86% yield, under the same reaction conditions, the uncatalyzed experiment delivered the same product in only 19% yield (entries 2 and 14). The impact of the catalyst in the reaction rate is less pronounced when LiBr is the choice of bromide source. Under these conditions, the uncatalyzed reaction responds to nearly half of the formation of the dibrominated product 2a within the same range of selectivity (entries 13 and 15). Increasing the catalyst load doesn’t necessarily mean faster formation of products. In experiments with higher concentrations of PhTeTePh, the rate of bromide oxidation is faster and gas evolution can be observed, most likely molecular oxygen produced by the disproportionation of H2O2 (results not shown). In other words, concentrated solutions of “Br+” can either destroy H2O2 or react with the substrate delivering a bromide product. The former is an unproductive pathway, consuming two equivalents of peroxide, while the concentration of bromide salt and substrate remain unchanged.7 Lastly, in a catalyzed experiment, molecular oxygen was used as an oxidizing agent instead of hydrogen peroxide. Brominated products were produced in only 10% yield, revealing that the aerobic contribution for the oxidation of bromide is negligible compared to the oxidation by H2O2 (entry 16).
Further experiments were performed to help shed light on the reaction mechanism. Firstly, we decided to verify if the oxidation of bromide follows preferentially an ionic or a radical pathway (Scheme 2, set up 1). Reactions conducted in the dark or exposed to daylight with the addition of butylated hydroxytoluene (BHT) as a radical scavenger, produced essentially the same results (yield and selectivity) as those of the optimized reaction conditions (Table 1, entry 13). These results strongly suggest that the oxidation of bromide by H2O2 activated by PhTeTePh is the result of ionic rather than radical species.
 | ||
| Scheme 2 Exploratory experiments for mechanism elucidation. a Yields and selectivity calculated by CG analysis. | ||
Secondly, we were interested in finding out the role of the bromide salt in the reaction outcome. The point to be considered was if the bromide counterion could somehow facilitate the activation of H2O2 or whether the differences observed in the experiments with LiBr, NaBr and KBr were merely a result of their solubility in the mixture of solvents. Therefore, uncatalyzed experiments would be more informative for this purpose. To circumvent the scarce solubility of inorganic bromide salts in organic solvents, we used 1-decyl-3-buthylimidazolium bromide 4, which was completely miscible in the mixture of solvents. After one hour of reaction only 17% of 2a could be detected (Scheme 2, set up 2, experiment 1). This result indicates that oxidation of bromide by H2O2 is not solely a matter of the bromide solubility. We speculate that LiBr showed better results than other bromide salts for bromination of 1a not only because it is more soluble, but also because the lithium ion may assist the activation of H2O2.19 Coordination of Li+ to AcOH may facilitate the protonation of H2O2, which in combination with Br−, would deliver “Br+” species (dotted box in Scheme 2).10 This assumption was reinforced by the prompt formation of “Br+” from the mixture of bromide salt 4 and H2O2 when acetic acid was replaced by a stronger acid, trifluoroacetic acid (TFA). This change in the reaction conditions allowed the production of dibromide 2a in 94% yield and excellent selectivity towards the dibrominated product (Scheme 2, set up 2, experiment 2).
Furthermore, when a catalyst-free solution of H2O2 and LiBr in 1,4-dioxane was treated with AcOH (0.5 mL), after 5.0 minutes the mixture became pale yellow. The color of the solution faded away by the addition of 1a (1.0 equivalent). After 15 minutes the reaction was quenched and product 2a was obtained in 18% yield (Scheme 2, set up 3). This experiment was repeated, but this time in addition to acetic acid, TFA (10.0 equivalents to 1a) was added to the mixture. Rapidly the solution turned bright orange. 1a was added after 5 minutes and the reaction was performed as in the previous experiment. At this time, product analysis indicated that the addition of an acid stronger than AcOH resulted in the formation of 2a in quantitative yield (Scheme 2, set up 3). Although activation of H2O2 by strong Brønsted acids for bromination reactions with bromide salts is a well-known strategy,10 the use of a weaker acid like AcOH would suit better our delineated objectives.
Finally, we were interested in finding out which species would be the best candidate to act as the brominating agent. Considering the oxidation process of bromide salts by H2O2 (either catalyzed or not) a proton source is required. The first oxidized species formed is hypobromous acid (HOBr). This species can either destroy H2O2, or react with another equivalent of acid and bromide producing bromine (Br2), Scheme 3A. However, when our experiments are performed using an excess of AcOH, it is reasonable to consider that acetate would compete with bromide to react with HOBr delivering brominating agents such as acetyl hypobromite (AcOBr).20 Formation of the latter species in higher concentrations should be facilitated by the nucleophilicity of the conjugate base A−. Uncatalyzed bromination reactions of 1a with LiBr/H2O2 and three different acids (Et3N·AcOH, AcOH and TFA) are depicted on Scheme 3B. If species such as AcOBr were indeed produced in the reaction mixture, their concentration would be higher using Et3N·AcOH and AcOH, as the trifluoroacetate anion is less nucleophilic to react with HOBr. The results suggest that the formation of dibrominated product 2a is directly correlated with the pKa of the acid, with the reaction being facilitated by stronger acids. The lack of product formation using Et3N·AcOH may just be due to the fact that this acid is not acidic enough to activate H2O2 and deliver HOBr (Scheme 3A). Interestingly, for both acids that promoted the formation of brominated products (AcOH and TFA), no differences in the selectivity between dibromide 2a and bromoacetate 3a were observed. Formation of a brominating agent such as AcOBr should be accompanied by an increment in the concentration of bromoacetate 3a.
 | ||
| Scheme 3 Acid influence in the oxidation of LiBr by H2O2. a Yields and selectivity calculated by CG analysis. | ||
Perhaps the most important hint to solve this conundrum comes from the heterogeneous experiment (Table 1, entry 1). In a biphasic system, the concentration of bromide would be negligible in the organic phase. Conversely AcOBr, being an organic compound, should be easily transported to the organic phase. Therefore, in this scenario it should be expect that selectivity would favor the formation of bromoacetate 3a. However, the product distributions in the heterogeneous and homogeneous reactions were virtually identical (Table 1, entries 1 and 2)! So, it is reasonable to assume that most likely the brominating agent is elemental bromine (Br2).
For reactions catalyzed by PhTeTePh, using for instance NaBr, formation of the brominating agent is mostly achieved through the action of pertellurinic acid 5 (as discussed in ref. 6h). Uncatalyzed reactions became competitive with the catalyzed process when LiBr is the source of bromide. In this case, formation of the brominating agent is facilitated by acid activation of H2O2, Scheme 4. Formation of bromoacetate 3a should be the result of competition between bromide and acetate in the ring opening of bromiranium ion 1a′.
 | ||
| Scheme 4 Catalyzed or uncatalyzed oxidation of bromide by H2O2 and product distribution. | ||
To verify the substrate scope and limitations, different alkenes were used (Table 2). Reactions were performed either catalyzed by 0.10 mol% of PhTeTePh (condition A) or in catalyst-free reactions (condition B). For most substrates, the addition of the catalyst resulted in better reaction yields in shorter reaction times. Aliphatic alkenes were successfully converted to dibromides 2a–d in a range of 75 to 92% yield in one hour of reaction. Products 2a and 2b were obtained with excellent stereoselectivity: only trans-1,2-dibromides could be detected by 1H NMR.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
:
Representative styrene derivatives were efficiently brominated producing 2e–g. For these compounds, blank experiments showed comparable yields to the catalyzed reactions. Compounds 2h and 2i, prepared from natural products estragole and cinnamyl alcohol, respectively, and highly functionalized 2j were obtained in good yields using both conditions A and B. Substrates bearing common protecting groups were employed to verify if they would survive the reaction conditions. Pleasantly, 2k–m were isolated in reasonable yields. No evidence for deprotection was verified in the course of the experiment. For these compounds an increase in the amount of LiBr and H2O2 was necessary. Lastly, electron-deficient alkenes were examined. Besides the change in the amount of LiBr and H2O2, a substantial increase in the reaction time was required to accomplish the task. In addition to compound 2m, products 2n and 2o were produced in good yields. Product 2o was obtained as a crystalline compound, allowing further characterization by single-crystal X-ray diffraction (Fig. 1).21
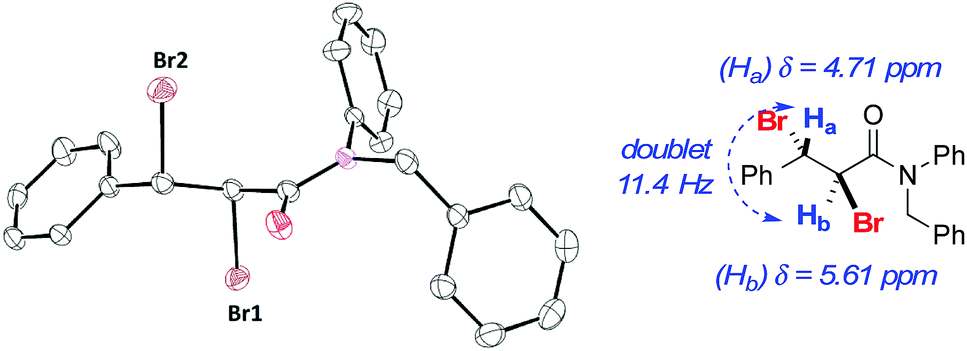 | ||
| Fig. 1 ORTEP and selected 1H NMR data for trans-dibromide 2o. a Hydrogen atoms omitted for clarity; 1H NMR (CDCl3, 400 MHz). | ||
Bromination of 1-octene 1d on a larger scale was also performed. Addition of 0.01 mmol of PhTeTePh (0.10 mol%) in a mixture of 10 mmol of substrate, 24 mmol of LiBr and 12 mmol of H2O2 in a solution of AcOH and 1,4-dioxane produced after one hour 2d in quantitative yield (Scheme 5). This suggests that the developed protocol might become operationally attractive on a preparative scale, as an alternative to replace the undesirable use of Br2.
 | ||
| Scheme 5 Scale-up dibromination of 1-octene 1d. a Yield for a pure isolated compound. | ||
Experimental
General methods and materials
All solvents and reagents were used as received unless otherwise noted. 1,4-Dioxane was distilled under NaBH4 to remove organic peroxides, and acetic acid was refluxed with acetic anhydride and 5% (w/w) of KMnO4 and then fractionally distilled.22 All bromide salts used in this study were anhydrous compounds. The organocatalyst PhTeTePh is commercially available (CAS number: 32294-60-3). Alternatively, it can be prepared according to literature procedures.6h Concentration at reduced pressure was performed on a rotary evaporator. Organic extracts of aqueous solutions were dried over MgSO4, filtered and then concentrated. NMR spectra were recorded on a Bruker AVANCE DRX400 spectrometer (400 MHz for 1H and 100 MHz for 13C). Chemical shifts (δ) referenced to the TMS signal are given in ppm, coupling constants (J) in hertz and multiplicities are given as s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), m (multiplet) or br (broad). All spectra were recorded at 25 °C unless otherwise noted. Elemental analysis was recorded on a Perkin Elmer Series II – CHNS/O Analyzer 2400. MS was recorded on a Shimadzu QP2010 Plus GCMS. HRMS was obtained on a Bruker micrOTOF QII Spectrometer. The progress of the reaction was followed by GC on a Shimadzu GC 2010-Plus, column RTx® – 5MS (30 m × 0.25 mm) using undecane (0.5 equivalent to substrate) as the internal standard. GC product yields were calculated by the determination of the product concentration in the final reaction solutions using calibration curves obtained with authentic product samples and undecane as the internal standard. Melting points are uncorrected.Procedure for bromination of cyclohexene in pH 6 phosphate buffer
A screw capped 3 dram vial was charged with NaBr (772 mg, 7.5 mmol), pH 6.2 phosphate buffer in distilled water (0.2 M in PO4, 5.0 mL) and 1,4-dioxane (5.0 mL). After stirring for five minutes undecane (53 μL, 0.25 mmol) was added followed by cyclohexene 1a (51 μL, 0.5 mmol), 8.8 M H2O2 (85 μL, 0.75 mmol) and PhTeTePh (0.25 mol% related to the substrate: 100 μL of a freshly prepared 12 mM in 1,4-dioxane). No precautions were taken to exclude oxygen or water from the reaction media. After 1 hour the reaction was quenched with 0.25 M NaHSO3 (2.0 mL). The organic phase was separated, dried over MgSO4, filtered, diluted with AcOEt and injected into the GC. Reported yields represent an average of duplicate runs.Optimization of the reaction conditions for bromination of cyclohexene
A screw capped 3 dram vial was charged with bromide salt (LiBr, NaBr, KBr or 1-decyl-3-butylimidazolium bromide 4; 1.2 mmol), acetic acid (0.25–1.0 mL) and solvent (2.0 mL). After stirring for five minutes undecane (53 μL, 0.25 mmol) was added followed by cyclohexene 1a (51 μL, 0.5 mmol), 8.8 M H2O2 (68 μL, 0.6 mmol) and PhTeTePh (0.10 mol% related to the substrate: 40 μL of a freshly prepared 12 mM solution in 1,4-dioxane). When applicable, additives were also added: trifluoroacetic acid (390 μL, 5.0 mmol) or Et3N·AcOH (prepared by the addition of Et3N 8.75 mmol, 1.12 mL to a solution of 0.5 mL of AcOH in 1,4-dioxane and stirred for 5 minutes, followed by the addition of the other reagents). No precautions were taken to exclude oxygen or water from the reaction media. After stirring for the time described in Table 1 the reaction was quenched with 0.25 M NaHSO3 (2.0 mL). The organic phase was separated, dried over MgSO4, filtered, diluted with AcOEt and injected into the GC. Experiments using bromide 4 or Et3N·AcOH were extracted using a solution of hexanes/AcOEt = 9/1. Reported yields represent an average of duplicate runs.Procedure for the reaction in the dark
A screw capped 3 dram vial wrapped in aluminium foil, was charged with LiBr (208 mg, 2.4 mmol), acetic acid (1.0 mL) and 1,4-dioxane (2.0 mL). After stirring for five minutes, undecane (110 μL, 0.5 mmol) was added followed by cyclohexene 1a (102 μL, 1.0 mmol), 8.8 M H2O2 (136 μL, 1.2 mmol) and PhTeTePh (0.10 mol% related to the substrate: 80 μL of a freshly prepared 12 mM solution in 1,4-dioxane). No precautions were taken to exclude oxygen or water from the reaction media. After 1 hour, the reaction was quenched with 0.25 M NaHSO3 (2.0 mL). The organic phase was separated, dried over MgSO4, filtered, diluted with AcOEt and injected into the GC. Reported yields represent an average of duplicate runs.Procedure for the reaction with a radical inhibitor
A clear screw capped 3 dram vial was charged with LiBr (208 mg, 2.4 mmol), acetic acid (1.0 mL) and 1,4-dioxane (4.0 mL). After stirring for five minutes undecane (110 μL, 0.5 mmol) was added followed by cyclohexene 1a (102 μL, 1.0 mmol), butylated hydroxytoluene – BHT as the radical inhibitor (44 mg, 0.2 mmol), 8.8 M H2O2 (136 μL, 1.2 mmol) and PhTeTePh (0.10 mol% related to the substrate: 80 μL of a freshly prepared 12 mM solution in 1,4-dioxane). No precautions were taken to exclude oxygen or water from the reaction media. After 1 hour, the reaction was quenched with NaHSO3 0.25 M (2.0 mL). The organic phase was separated, dried over MgSO4, filtered, diluted with AcOEt and injected into the GC. Reported yields represent an average of duplicate runs.Procedure for aerobic oxidation of LiBr
A screw capped 3 dram vial was charged with LiBr (208 mg, 2.4 mmol), acetic acid (1.0 mL) and 1,4-dioxane (4.0 mL). After stirring for five minutes undecane (110 μL, 0.5 mmol) was added followed by cyclohexene 1a (102 μL, 1.0 mmol), and PhTeTePh (0.10 mol% related to the substrate: 80 μL of a freshly prepared 12 mM solution in 1,4-dioxane). The reaction was performed under aerobic conditions (balloon filled with molecular oxygen attached to the reaction vessel). After 1 hour, the reaction was quenched with 0.25 M NaHSO3 (2.0 mL). The organic phase was separated, dried over MgSO4, filtered, diluted with AcOEt and injected into the GC. Reported yields represent an average of duplicate runs.General procedure for bromination of alkenes
Procedure for the scale-up bromination of 1-octene
A 100 mL one-neck round-bottom flask was charged with LiBr (2.08 g, 24 mmol), 1,4-dioxane (40.0 mL), glacial acetic acid (10.0 mL) and 1d (98% purity) (1.6 mL, 10.0 mmol). After stirring for five minutes, 8.8 M H2O2 (1.36 mL, 12.0 mmol) was added followed by PhTeTePh (0.10 mol% related to the substrate: 0.8 μL of a freshly prepared 12 mM solution in 1,4-dioxane). No precautions were taken to exclude oxygen, water or sun light from the reaction media. The mixture was then stirred at room temperature for one hour. At the end of the reaction, it was diluted with distilled water (50 mL) and the product extracted with a 9/1 solution of hexanes/AcOEt (3 × 25 mL). The organic phase was washed with Na2CO3 1 M (1 × 50 mL), dried over MgSO4, filtered and the solvents removed under reduced pressure. Purification was performed by silica gel chromatography (hexanes/AcOEt = 98/2) to afford 2d as a pale yellow oil (2.71 g, quantitative yield).Conclusions
Dibromination of alkenes could be efficiently and stereoselectively achieved by oxidation of bromide salts with hydrogen peroxide, an inexpensive and potent oxidizing agent, which produces H2O as a by-product. Acetic acid, a mild organic acid, could be employed satisfactorily as a proton source for this transformation in 1,4-dioxane as a solvent. The issue of product selectivity in bromination reactions of alkenes employing bromide salts and H2O2 in acidic media has been successfully addressed: the ratio of dibromide/side-products (e.g. bromohydrins, bromoacetate) observed was greater than 100/1, favoring the former. Electron-rich and electron-deficient alkenes and alkenes bearing protecting groups could be successfully converted to their respective dibromides. For most substrates used, the addition of a very small amount of commercially available catalyst PhTeTePh (0.10 mol%) is not necessarily required. Yet, catalyzed experiments often resulted in better yields in shorter reaction times. The experimental data suggest that the brominating agent prepared in situ from the bromide/H2O2 mixture is molecular bromine, both under catalyzed or uncatalyzed conditions. Activation of H2O2 in the uncatalyzed process is well correlated with the acidity of Brønsted acids tested. For weaker acids such as AcOH, assistance by coordination with cationic lithium is inferred. Finally, scale-up of the reaction using 1-octene as a substrate was examined. The desired product, 1,2-dibromooctane, was produced in quantitative yield after only one hour of reaction at room temperature.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to CNPq (grant number 303727/2014-4), CAPES and FAPEMIG. We are also thankful to prof. Bernardo L. Rodrigues (UFMG) for the X-ray analysis of dibromide 2o, and Luana Bettanin and prof. Antonio L. Braga (UFSC) for HRMS analysis. Prof. Maria H. de Araújo (UFMG) is acknowledged for providing bromide 4.Notes and references
- D. Yoffe, R. Frim, S. D. Ukeles, M. J. Dagani, H. J. Barda, T. J. Benya and D. C. Sanders, Bromine Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, 2013, pp. 1–31 Search PubMed.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS PubMed.
- (a) K. Tanaka, R. Shiraishi and F. Toda, J. Chem. Soc., Perkin Trans. 1, 1999, 3069–3070 RSC; (b) J. Salazar and R. Dorta, Synlett, 2004, 1318–1320 CrossRef CAS; (c) V. Kavala, S. Naik and B. K. Patel, J. Org. Chem., 2005, 70, 4267–4271 CrossRef CAS PubMed.
- (a) C. Ye and J. M. Shreeve, J. Org. Chem., 2004, 69, 8561–8563 CrossRef CAS PubMed; (b) L. X. Shao and M. Shi, Synlett, 2006, 1269–1271 CrossRef CAS; (c) S. D. F. Tozetti, L. S. de Almeida, P. M. Esteves and M. C. S. de Mattos, J. Braz. Chem. Soc., 2007, 18, 675–677 CrossRef CAS; (d) G. Hernández-Torres, B. Tan and C. F. Barbas III, Org. Lett., 2012, 14, 1858–1861 CrossRef PubMed; (e) M. Stodulski, A. Goetzinger, S. V. Kohlhepp and T. Gulder, Chem. Commun., 2014, 50, 3435–3438 RSC; (f) Y. Kitazawa, R. Takita, K. Yoshida, A. Muranaka, S. Matsubara and M. Uchiyama, J. Org. Chem., 2017, 82, 1931–1935 CrossRef CAS PubMed; (g) L. S. Pimenta, E. V. Gusevskaya and E. E. Alberto, Adv. Synth. Catal., 2017, 359, 2297–2303 CrossRef CAS.
- (a) A. Butler and J. V. Walker, Chem. Rev., 1998, 93, 1937–1944 CrossRef; (b) L. C. Blasiak and C. L. Drennan, Acc. Chem. Res., 2009, 42, 147–155 CrossRef CAS PubMed; (c) A. Butler and M. Sandy, Nature, 2009, 460, 848–854 CrossRef CAS PubMed.
- Selected examples: (a) G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469–3478 CrossRef CAS; (b) J. V. Walker, M. Morey, H. Carlsson, A. Davidson, G. D. Stucky and A. Butler, J. Am. Chem. Soc., 1997, 119, 6921–6922 CrossRef CAS; (c) M. D. Drake, F. V. Bright and M. R. Detty, J. Am. Chem. Soc., 2003, 125, 12558–12566 CrossRef CAS PubMed; (d) S. M. Bennett, Y. Tang, D. McMaster, F. V. Bright and M. R. Detty, J. Org. Chem., 2008, 73, 6849–6852 CrossRef CAS PubMed; (e) D. Wischang, J. Hartung, T. Hahn, R. Ulber, T. Stumpf and C. Fecher-Trost, Green Chem., 2011, 13, 102–108 RSC; (f) M. Sandy, J. N. Carter-Franklin, J. D. Martiny and A. Butler, Chem. Commun., 2011, 47, 12086–12088 RSC; (g) E. E. Alberto, A. L. Braga and M. R. Detty, Tetrahedron, 2012, 68, 10476–10481 CrossRef CAS; (h) E. E. Alberto, L. M. Muller and M. R. Detty, Organometallics, 2014, 33, 5571–5581 CrossRef CAS; (i) C. M. Gatley, L. M. Muller, M. A. Lang, E. E. Alberto and M. R. Detty, Molecules, 2015, 20, 9616–9639 CrossRef CAS PubMed.
- (a) R. R. Everett and A. Butler, Inorg. Chem., 1989, 28, 393–395 CrossRef CAS; (b) R. R. Everett, J. R. Kanofskyen and A. Butler, J. Biol. Chem., 1990, 265, 4908–4914 CAS.
- (a) U. Bora, G. Bose, M. K. Chaudhuri, S. S. Dhar, R. Gopinath, A. T. Khan and B. K. Patel, Org. Lett., 2000, 2, 247–249 CrossRef CAS PubMed; (b) B. F. Sels, D. E. Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350–8359 CrossRef CAS PubMed; (c) R. K. Sharma and C. Sharma, Tetrahedron Lett., 2010, 51, 4415–4418 CrossRef CAS; (d) F. Mendoza, R. Ruíz-Guerrero, C. Hernández-Fuentes, P. Molina, M. Norzagaray-Campos and E. Reguera, Tetrahedron Lett., 2016, 57, 5644–5648 CrossRef CAS; (e) S. P. Dash, A. K. Panda, S. Dhaka, S. Pasayat, A. Biswas, M. R. Maurya, P. K. Majhi, A. Crochet and R. Dinda, Dalton Trans., 2016, 18292–18307 RSC.
- Selected examples: (a) F. Romano, A. Linden, M. Mba, C. Zonta and G. Licini, Adv. Synth. Catal., 2010, 352, 2937–2942 CrossRef CAS; (b) K. Yonehara, K. Kamata, K. Yamaguchi and N. Mizuno, Chem. Commun., 2011, 47, 1692–1694 RSC; (c) E. Palmajumder, S. Patra, M. G. B. Drew and K. K. Mukherjea, New J. Chem., 2016, 8696–8703 RSC; (d) E. Badetti, F. Romano, L. Marchio, S. Taşkesenlioğlu, A. Daştan, C. Zonta and G. Licini, Dalton Trans., 2016, 45, 14603–14608 RSC.
- (a) N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron, 1999, 55, 11127–11142 CrossRef CAS; (b) A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212–1218 RSC; (c) T. Moriuchi, M. Yamaguchi, K. Kikushima and T. Hirao, Tetrahedron Lett., 2007, 48, 2667–2670 CrossRef CAS.
- (a) G. Zhang, R. Liu, Q. Xu, L. Ma and X. Liang, Adv. Synth. Catal., 2006, 348, 862–866 CrossRef CAS; (b) A. Molinari, G. Varani, E. Polo, S. Vaccari and A. Maldotti, J. Mol. Catal. A: Chem., 2007, 262, 156–163 CrossRef CAS; (c) A. Podgorsek, M. Eissen, J. Fleckenstein, S. Stavber, M. Zupana and J. Iskra, Green Chem., 2009, 11, 120–126 RSC; (d) Z. Huang, F. Li, B. Chen, T. Lu, Y. Yuan and G. Yuan, ChemSusChem, 2013, 6, 1337–1340 CrossRef CAS PubMed; (e) J. Naapuri, J. D. Rolfes, J. Keil, C. M. Sapu and J. Deska, Green Chem., 2017, 19, 447–452 RSC.
- (a) K. Moriyama, Y. Izumisawa and H. Togo, J. Org. Chem., 2011, 76, 7249–7255 CrossRef CAS PubMed; (b) M. Z. Zhang, W. B. Sheng, Q. Jiang, M. Tian, Y. Yin and C. C. Guo, J. Org. Chem., 2014, 79, 10829–10836 CrossRef CAS PubMed; (c) J. Xu and R. Tong, Green Chem., 2017, 19, 2952–2956 RSC.
- S. Adimurthy, G. Ramachandraiah, A. V. Bedekar, S. Ghosh, B. C. Ranu and P. K. Ghosh, Green Chem., 2006, 8, 916–922 RSC.
- G. K. Dewkar, S. V. Narina and A. Sudalai, Org. Lett., 2003, 5, 4501–4509 CrossRef CAS PubMed.
- (a) M. Karki and J. Magolan, J. Org. Chem., 2015, 80, 3701–3707 CrossRef CAS PubMed; (b) S. Song, X. Li, X. Sun, Y. Yuan and N. Jiao, Green Chem., 2015, 17, 3285–3289 RSC.
- For reviews concerning environmental aspects of bromination reactions see: (a) M. Eissen and D. Lenoir, Chem. – Eur. J., 2008, 14, 9830–9841 CrossRef CAS PubMed; (b) A. Podgorsek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef CAS PubMed.
- One can argue that H2O2 may impose more safety concerns (depending on the conditions it can violently decompose producing H2O and O2) than Oxone (potassium monopersulfate). Even though manufactured Oxone is indeed safer to handle than concentrated solutions of H2O2, this inorganic salt is generated from Caro's acid (persulfuric acid), which can decompose explosively. Additionally, Caro's acid is prepared by the addition of H2O2 to hazardous chlorosulfonic acid.
- We expected that THF would produce a result comparable to the reaction carried out with 1,4-dioxane. We rationalized that the better result obtained with the latter may be due to the fact that this solvent produces a stable complex with Br2, which seems to be beneficial for the outcome of the reaction. To date, there is no report describing the formation of such complexes with THF. For the 1,4-dioxane·Br2 complex see: O. Hassel and J. Hvolsef, Acta Chem. Scand., 1954, 8, 873 CrossRef CAS.
- Y. Kitazawa, R. Takita, K. Yoshida, A. Muranaka, S. Matsubara and M. Uchiyama, J. Org. Chem., 2017, 82, 1931–1935 CrossRef CAS PubMed.
- S. G. Levine and M. E. Wall, J. Am. Chem. Soc., 1959, 81, 2826–2829 CrossRef CAS.
- The crystal structure of compound 2o has been deposited at the CCDC, with the assigning number: CCDC 1568276†.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth/Heinemann (Elsevier Science), Bodmin, UK, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data of the starting material and products (including 1H, 13C NMR and HRMS copies) and crystallographic data for compound 2o. CCDC 1568276. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj04300g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |