
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetracarboxylic acids on a thiacalixarene scaffold: synthesis and binding of dopamine hydrochloride†
O. A.
Mostovaya
a,
P. L.
Padnya
 a,
A. A.
Vavilova
a,
D. N.
Shurpik
a,
B. I.
Khairutdinov
ab,
T. A.
Mukhametzyanov
a,
A. A.
Khannanov
a,
M. P.
Kutyreva
a and
I. I.
Stoikov
*a
a,
A. A.
Vavilova
a,
D. N.
Shurpik
a,
B. I.
Khairutdinov
ab,
T. A.
Mukhametzyanov
a,
A. A.
Khannanov
a,
M. P.
Kutyreva
a and
I. I.
Stoikov
*a
aKazan Federal University, A.M. Butlerov Chemistry Institute, 420008, Kremlevskaya Street, 18, Kazan, Russian Federation. E-mail: Ivan.Stoikov@mail.ru; Fax: +7-8432-752253; Tel: +7-8432-337463
bKazan Institute of Biochemistry and Biophysics, 420111, Lobachevsky Street, 2/31, Kazan, Russian Federation
First published on 15th November 2017
Abstract
For the first time thiacalix[4]arene derivatives in 1,3-alternate conformation simultaneously containing amide, carboxyl and hydroxyl groups capable of forming 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry complexes with dopamine hydrochloride were obtained. The efficiency of dopamine hydrochloride binding was evaluated by a number of spectral methods. Using the methods of fluorescent, UV-Vis and NMR spectroscopy, the mechanism of interaction of the synthesized macrocycles with dopamine has been studied. It was shown that quenching of dopamine fluorescence by the studied macrocycles is carried out through a static mechanism.
:1 stoichiometry complexes with dopamine hydrochloride were obtained. The efficiency of dopamine hydrochloride binding was evaluated by a number of spectral methods. Using the methods of fluorescent, UV-Vis and NMR spectroscopy, the mechanism of interaction of the synthesized macrocycles with dopamine has been studied. It was shown that quenching of dopamine fluorescence by the studied macrocycles is carried out through a static mechanism.
Introduction
Dopamine is one of the most widely known neurotransmitters and hormones, which are necessary for human and animal vital activity. It plays a key role during various physiological processes, such as adaptation to stress, and serves as an important part of the brain reward system.1,2 At present, excess dopamine is associated with such diseases as schizophrenia (its positive symptoms), restless legs syndrome, myofascial syndrome, tics, and deficiency with parkinsonism, depressive states, and dementia,3–8 and therefore the design of compounds that selectively bind dopamine is essential.Currently, various synthetic compounds capable of selective binding with dopamine through multiple non-covalent interactions, such as electrostatic interactions, hydrogen bonding, π–π-stacking, and hydrophobic forces, have been proposed as host molecules. The organophosphorus compounds,9 calix[n]arenes (n = 4, 5, 6),10–16 calix[4]resorcinarenes,17 and pillar[5]arenes18 with acidic,10,12 quinone11 and amide8 groups, and charged fragments14,18 as binding sites, show complexation ability toward dopamine.
Nevertheless, despite the abundance of information on dopamine binding with different compounds, the mechanism of its interaction with host molecules and biopolymers has not been sufficiently studied. Thus, for the same biomacromolecules, different mechanisms of association are proposed in different publications. For example, studying the interaction of dopamine with BSA by fluorescence method, opposing extinguishing mechanisms are described: dynamic and static.19,20 At the same time, knowledge of the mechanism enables the possibility of its further application in sensory systems (dynamic quenching) or mass transfer and, in particular, targeted drug delivery in the case of static quenching (stable adduct formation).19,21 Thus, detailed analysis of the interaction mechanism is important.
In our opinion, a very promising direction is design of the new host molecules for dopamine. (Thia)calixarenes have well established themselves as a class of compounds widely used as the basis for constructing various extractants, carriers, electrode ionophores, drug delivery devices, etc.22–30 In addition, the thiacalixarene macrocyclic platform is easily functionalized. Moreover, they differ favorably from the classical calixarenes platform by the possibility of obtaining various spatial isomers with given position of binding groups relative to the cyclophane platform by using the template synthesis method.31 It should be noted that at this time there are no data on host molecules for dopamine on the basis of thiacalix[4]arene. However, one paper was published where a thiacalix[4]arene derivative functionalized by a dopamine fragment in the stereochemical conformation of 1,3-alternate was proposed for the electrochemical determination of dopamine.32
So, our work is devoted to the preparation of new thiacalix[4]arene derivatives in 1,3-alternate conformation simultaneously containing amide, carboxyl and hydroxyl groups providing hydrogen bonding with dopamine, as well as studying the mechanism of their interaction with dopamine by fluorescent, UV-Vis and NMR spectroscopy methods.
Results and discussion
Synthesis of the p-tert-butylthiacalix[4]arene derivatives 2–6
The reaction of cyclic anhydrides (succinic, maleic, citraconic, diglycolic) and L-lactide with tetraamine 1 in 1,3-alternate conformation was studied in order to obtain tetrasubstituted thiacalix[4]arene derivatives containing terminal carboxyl and hydroxyl groups. Polar aprotic solvents are generally used for the acylation reaction. Tetraamine 1, selected cyclic anhydrides and L-lactide are readily soluble in two such solvents, pyridine and tetrahydrofuran. However, synthesis in pyridine, according to 1H NMR spectroscopy, results in formation of hardly separable mixtures of partially substituted products. Therefore, tetrahydrofuran was chosen for this work. The reaction with all anhydrides studied proceeds for 15 hours at the solvent boiling point, with the exception of succinic anhydride, which reacts more slowly (25 hours) (Scheme 1). Apparently, this is due to the low solubility of succinic anhydride in THF. The reaction of tetraamine 1 with L-lactide also lasts for 25 hours. This can be explained by the lower reactivity of esters compared to carboxylic anhydrides in nucleophilic substitution reactions. As expected,33,34 when the lactide fragment is introduced to the macrocyclic platform, its conformation is kept, which is expressed in the saving of the negative Cotton effect characteristic for L-lactide in the CD spectrum of compound 6 (ESI,† Fig. S29). Compounds 2–6 were synthesized with high yields of 81–90%. The structures of the obtained p-tert-butylthiacalix[4]arene derivatives were characterized by (1D) 1H NMR, 13C NMR, 2D 1H–1H NOESY NMR, IR spectroscopy, and mass spectrometry, and the composition was confirmed by elemental analysis data.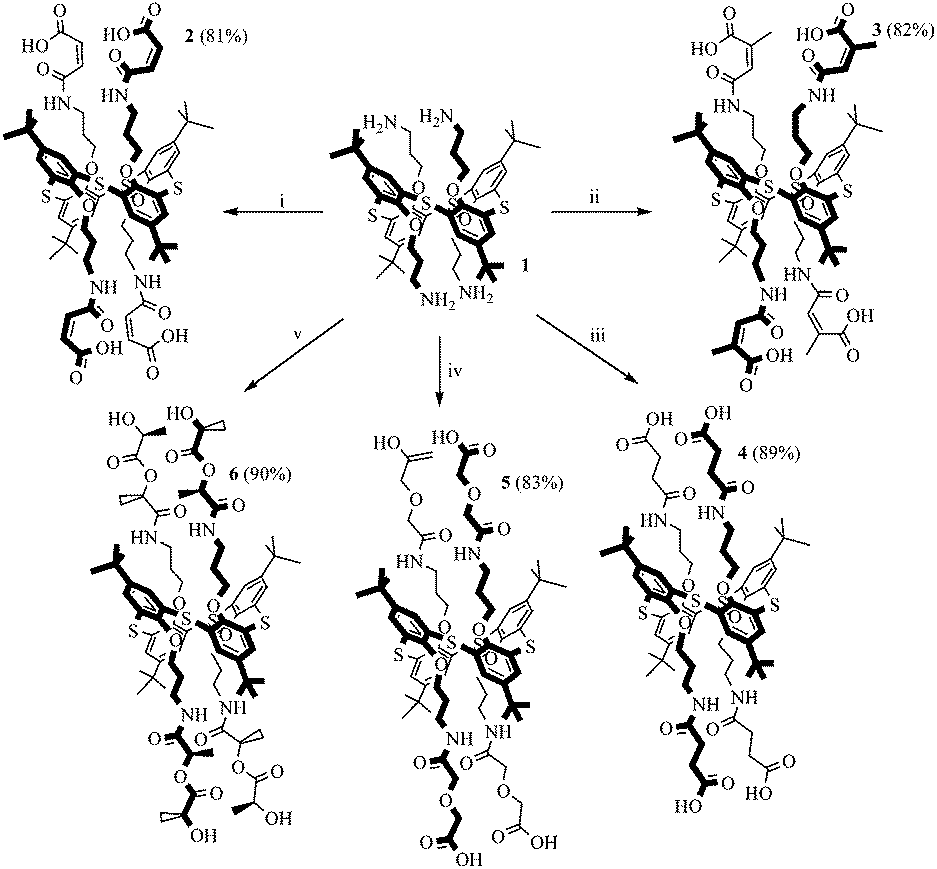 | ||
| Scheme 1 Reagents and conditions: (i) maleic anhydride, THF, reflux, 15 hours; (ii) citraconic anhydride, THF, reflux, 15 hours; (iii) succinic anhydride, THF, reflux, 25 hours; (iv) anhydride of diglycolic acid, THF, reflux, 15 hours; (v) (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione, THF, reflux, 25 hours. | ||
If the structures of compounds 2 and 4–6 are uniquely determined, for compound 3 the formation of two structures is possible due to the asymmetry of the citraconic anhydride molecule. When ring opening occurs, it is possible to form a mixture of two products with a methyl group in the geminal (product A) or vicinal (product B) positions relative to the amide group (Scheme 2).
 | ||
| Scheme 2 Possible structures of products of interaction of citraconic anhydride with amines. | ||
It was shown previously35 that the interaction of amines with citraconic anhydride under mild conditions produces a mixture of isomers with the predominance of structure A. However, in some cases, there is further isomerization of product A into the more thermodynamically stable product B.35 The reaction with reflux led to the isolation of the single product with the assumed structure B. The position of the characteristic signal of the proton near the double bond in citraconamic acid can unambiguously assert which carbonyl group is attacked by the nucleophilic nitrogen atom of the amino group.35 According to ref. 35 in the 1H NMR spectrum (DMSO-d6) the proton signal is geminal relative to the carboxyl group and appears in the range of 5.7–5.9 ppm, while the signal is geminal relative to the amide group in the 6.0–6.2 ppm region. In the 1H NMR spectrum of compound 3 in DMSO-d6, there is a 6.18 ppm signal corresponding to the proton near the double bond. To confirm the structure, homo- and hetero-correlation NMR spectra were recorded: 1H–1H TOCSY, 1H–13C HSQC, 1H–13C HMBC (Fig. 1 and ESI,† Fig. S16–S18).
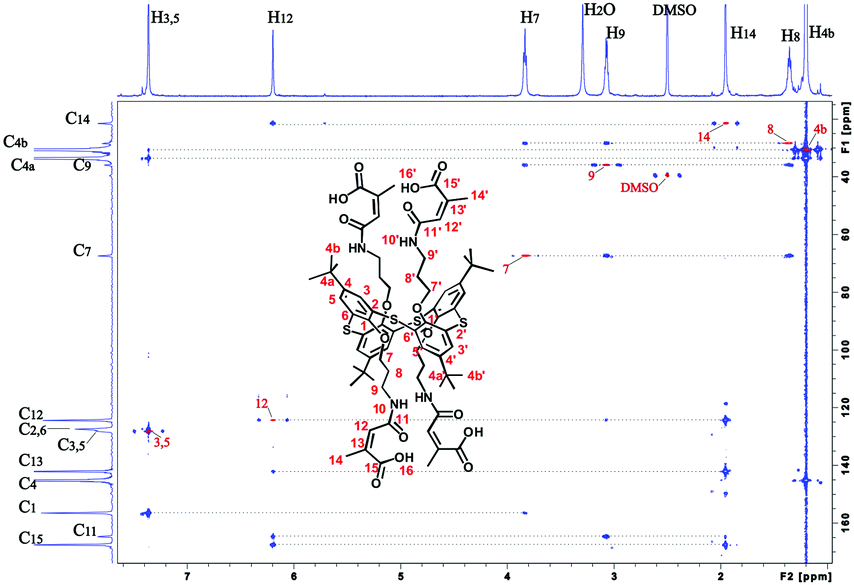 | ||
| Fig. 1 Overlay of 1H–13C HSQC (red) and 1H–13C HMBC (blue) spectra of compound 3 (600 MHz, DMSO-d6, 303 K). The red peaks of the 1H–13C HSQC spectrum are marked with red text. | ||
A TOCSY experiment contains cross-peaks due to protons that can be combined in two spin system. The first spin system is large and contains the resonance signals of the four protons H7–H10. The second spin system is small and combines the signals of the two spectral lines: methyl H14 (δ 1.95 ppm) and methine protons H12 (δ 6.20 ppm) (ESI,† Table S1). Consideration of the correlation signals from the protons H14 and H12 in the HMBC spectrum help to assign carbon atom C13, as well as C11 and C15. In this case, methine carbon atom C13 is assigned unambiguously, as it falls into the characteristic region of 105–145 ppm. The signals of carbon atoms C11 and C15 are close. However, the signal of the carbon atom of the amide group C11 (δ 164.91 ppm) can be assigned based on the cross-peak from the proton H9 (δ 3.07 ppm). Therefore, the remaining carbon signal is the carboxyl group C15 (δ 167.72 ppm). The correct assignment of C15 is confirmed by the higher intensity of the correlation peak from the H14 protons per carbon atom C15, rather than the carbon atom C11, because of the proximity of the methyl group to the carboxyl fragment.
The proximity of the methine carbon atom C12 to the amide group was verified by the correlation peak from H9 protons (δ 3.07 ppm), which belongs to the first spin group in the TOCSY spectrum. Thus, it can be affirmed that the interaction of citraconic anhydride with tetraamine 1 under reflux in THF produces only one product with a methyl group in the vicinal position relative to the amide fragment (Scheme 1).
Thus, synthesis techniques for new polyfunctional p-tert-butylthiacalix[4]arene derivatives 2–6 in 1,3-alternate conformation simultaneously containing amide, ester, alcohol and carboxyl groups with high yields were developed.
Binding of dopamine
One of the most widely used methods for studying dopamine binding is fluorescence spectroscopy.36–40 It is known that dopamine has significant fluorescence upon excitation at 285 nm. The experiment was carried out in methanol, because in other solvents the compounds proved to be insoluble. Unfortunately, it was not possible to study the interaction of dopamine with compounds 2 and 3 because of their extremely low solubility practically in all organic solvents and water.It turned out that when all three studied thiacalix[4]arenes 4–6 were added to the solution of dopamine, quenching of its fluorescence with a maximum at 315 nm was observed (ESI,† Fig. S30–S32). However, it remained unclear how the quenching of luminescence was caused: by the formation of the complex or by impingement with the macrocycles molecules. To confirm the complexation, we recorded UV/visible spectra. Dopamine has three absorption maxima with λmax at 203, 225, and 285 nm, corresponding to π–π* transitions of the aromatic ring, somewhat shifted to the red region of the spectrum compared to the parent benzene due to substituents effect.41,42 It turned out that when the dopamine interacts with all thiacalixarenes, the hypochromic effect is observed only at 203 nm, which indicates the complex formation and, accordingly, the static character of fluorescence quenching (Fig. 2 and ESI,† Fig. S33).
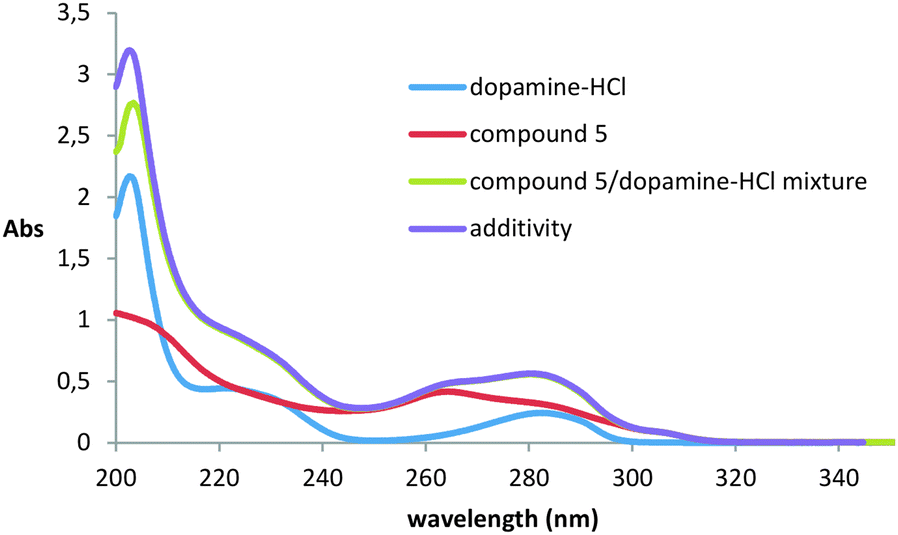 | ||
| Fig. 2 UV/Vis absorption spectra of thiacalixarene 5 (0.01 mM) in the absence and presence of 0.1 mM dopamine-HCl at 293 K. | ||
An attempt to confirm or confute this type of quenching mechanism was also undertaken by using the fluorescence method. It is known that in a single type of quenching (dynamic or static) in the Stern–Volmer coordinates, the quenching value is linearly related to the quencher concentration, and for a combination of types, linearity is disturbed.43 The recording of emission spectra at different temperatures (283 and 303 K) showed the linear dependence in the whole range of concentrations studied. However, it turned out unexpectedly that the slope of the curves at different temperatures is constant (ESI,† Fig. S34). The absence of the growth in the quenching constant with temperature increasing unambiguously testifies against the dynamic mechanism of quenching.
The binding constants were determined from analysis of binding isotherms (obtained by fluorescence spectroscopy in the absence of dynamic quenching) and fitted to a 1:1 binding model.44–46 The Bindfit application, which was developed for supramolecular systems,47 was used to process the results. To confirm the proposed stoichiometry, the titration data were also processed by the binding model at the ratio “host:guest” = 1:2. However, in this case the constants are determined with much greater uncertainty (ESI,† Fig. S30–S32).
The determination of the association constants showed that all compounds bind well enough to dopamine hydrochloride and still have similar values of association constants. Compound 6 most strongly binds dopamine-HCl (Ka = 1.91 × 104 M−1), while for others the bonding is weaker: (Ka = 1.50 × 104 M−1 for 5 and 0.95 × 104 M−1 for 4).
It is known that amide fragments are often used as binding sites for anionic substrates.48–50 The presence of amide moieties in the structures of compounds 4–6 may lead to the fact that binding of the anion is carried out (Cl−). However, the study of the UV spectra of the obtained thiacalixarenes in the presence of tetrabutylammonium chloride indicates the absence of this kind of interaction, as evidenced by the absence of both hypo- and hyper-chromic effects in the electronic spectra of these systems (ESI,† Fig. S35). Thus, catecholamine binding, rather than its anionic fragment, is observed, which makes it possible to extend the proposed mechanism of interaction not only to the salt, but also to the free amine.
To further confirm the complexation and establish the structure of the complex, we also employed the 1H NMR spectroscopy method (ESI,† Fig. S36). In the spectrum of free dopamine hydrochloride, signals of aromatic protons are observed as a multiplet (6.56–6.74 ppm), as well as two triplets of methylene protons (3.41 and 2.98 ppm). When thiacalixarene 5 is added, a significant upfield shift of the proton signals of the methylene groups of amine into the region of 3.11 and 2.79 ppm is observed, and a small shift of the singlet in the weak fields (6.69 ppm) indicates dopamine binding with a macrocycle.
The formation of the complex between macrocycle 6 and dopamine hydrochloride and its spatial structure were also confirmed by 2D 1H–1H NOESY NMR spectroscopy (Fig. 3). The presence of cross-peaks between the protons of the pyrocatechol and ethylene fragments of dopamine, as well as aromatic protons and methylene protons bound with amide groups of 6 indicates the formation of an endohedral complex between the compounds studied.
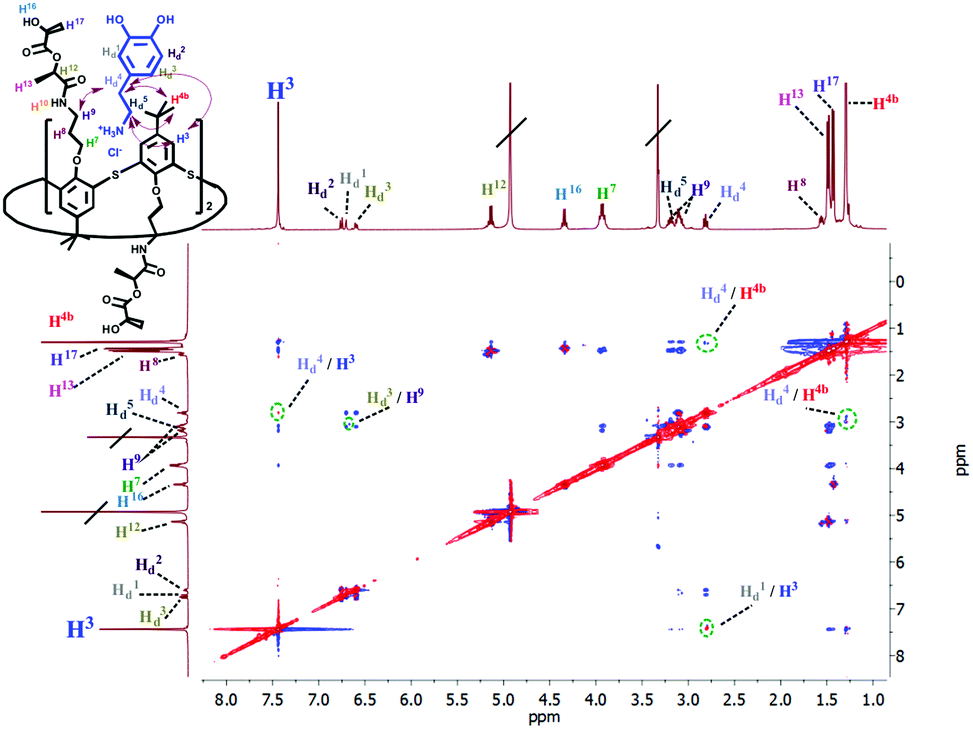 | ||
| Fig. 3 Partial NOESY spectrum (400 MHz, CD3OD, 298 K) of 6 (5 mM) and dopamine hydrochloride (5 mM). | ||
The two-dimensional DOSY spectroscopy method also testifies in favor of complex formation of 6 with dopamine-HCl. The diffusion coefficients of 6, dopamine-HCl and mixture of dopamine-HCl–6 at 298 K (1 mM) were determined (Table 1). The DOSY spectrum for the mixture of catecholamine with 6 in 1:1 ratio (1 mM) indicates the presence of only one type of particles with a diffusion coefficient lower than for the individual compounds. The greatest decrease in the diffusion rate was shown by dopamine-HCl in the presence of 6 (Table 1), which indicates the formation of guest–host complexes. During complex formation a decrease in the diffusion of thiacalix[4]arene 6 molecules was also observed. It indicates the decrease in the mobility of guest (dopamine-HCl) and host molecules as a result of the complexation (ESI,† Fig. S37).
| Compounds | D (10−10 m2 s−1) |
|---|---|
| Dopamine-HCl | 21.60 |
| 1,3-alternate-6 | 3.68 |
| 1,3-alternate-6 with dopamine-HCl | 3.35 |
Conclusions
Thus, new p-tert-butylthiacalix[4]arene derivatives 2–6 in 1,3-alternate conformation simultaneously containing amide, carboxyl and hydroxyl groups were synthesized. The ability of thiacalix[4]arenes 4–6 to bind dopamine hydrochloride was established in methanol by spectral methods (UV, fluorescence, NMR spectroscopy). By fluorescence spectroscopy, it was shown that quenching of dopamine fluorescence by the studied macrocycles is carried out through a static mechanism. All the complexes formed have 1:1 stoichiometry, while for all studied compounds the association constants were close (in the order of (1–1.9) × 104 M−1). Unfortunately, at this stage, the possibility of using the synthesized compounds to determine dopamine is not revealed, but the obtained results can be useful for the development of new synthetic dopamine receptors.
Experimental
General
The 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer (400.17 MHz for H-atoms) for 3–5% solutions in DMSO-d6, CDCl3 and methanol-d4. The residual solvent peaks were used as an internal standard. Homo- and hetero-correlation NMR spectra were recorded at 30 °C on a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5 mm TXI Probe. Assignments were done using 2D 1H–1H TOCSY, 1H–13C HSQC and 1H–13C HMBC experiments. Repetition delay d1 was set to 2 s for all 2D correlation spectra. The mixing time for 1H–1H TOSCY experiment was 80 ms. All NMR data were processed and analyzed using Topspin 3.5 software.The IR spectra were recorded using a Spectrum 400 (PerkinElmer) IR spectrometer.
Elemental analysis was performed on a PerkinElmer 2400 Series II instrument.
Electrospray ionization mass spectra (ESI) were obtained on an AmazonX mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany). The measurements were carried out in the positive ions registration regime in the m/z range from 100 to 2800. The voltage on the capillary was −4500 V. Nitrogen was used as the drying gas with a temperature of 300 °C and a flow rate of 10 L min−1. The compounds were dissolved in acetonitrile to a concentration of 10−6 g L−1. Data were processed using DataAnalysis 4.0 (Bruker Daltonik GmbH, Bremen, Germany).
The MALDI mass spectra were recorded on an Ultraflex III mass spectrometer. p-Nitroaniline was used as the matrix.
Melting points were determined using Boetius Block apparatus. The purity of the compounds was monitored by melting points, 1H NMR and TLC on 200 μm UV 254 silica gel plates.
The electronic absorption spectra were recorded using a Shimadzu UV-3600 spectrometer in methanol in quartz cuvettes with the thickness of the transmissive layer at 10 mm. The mixtures of thiacalixarenes (10 μM) and dopamine-HCl (100 μM) in methanol were incubated for 5 minutes. The experiment was carried out at 293 K.
The CD spectra were recorded using a Jasco-1500 spectrophotometer in quartz cuvettes with the thickness of the transmissive layer at 1 mm. The spectra were measured with a scan rate of 15 nm min−1, spectral range of 210–260 nm, slit width of 1 nm, sampling step of 1 nm and 5 scans co-addition.
Fluorescence spectra were recorded on a Fluorolog 3 luminescent spectrometer (Horiba Jobin Yvon). The excitation wavelength was selected as 285 nm. The emission scan range was 300–540 nm. Excitation and emission slits were 3 nm. Quartz cuvettes with optical path length of 10 mm were used. The cuvette was located in the front face position. Emission spectra were automatically corrected by the Fluoressence program. The fluorescence spectra were recorded in methanol solutions with a dopamine concentration of 10 μM. The concentrations of thiacalix[4]arenes ranged from 0 to 90 μM. The experiment was carried out at 293 K. The temperature dependences of fluorescence for all compounds were determined at 283 and 303 K on a LS-55 Fluorescence Spectrometer (PerkinElmer) using a Peltier RTR1 cuvette thermostatic holder.
General procedure for the synthesis of compounds 2–4
A mixture of 5,11,17,23-tetra-tert-butyl-25,26,27,28-tetrakis(3-aminopropoxy)-2,8,14,20-tetrathiacalix[4]arene (1)24 (0.50 g, 0.53 mmol) and the corresponding anhydride (4.20 mmol) in 7 ml of dry tetrahydrofuran was refluxed for 15 hours (25 hours in the case of compound 4). After cooling the reaction mixture, the solvent was removed under reduced pressure. 20 ml of chloroform was added to the residue and then stirred for 1.5 hours. It was then heated to the boiling point of the solvent and the insoluble precipitate was filtered hot. The resulting precipitate was washed with 30 ml of hot methanol and dried in a vacuum desiccator over P2O5.![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–CH2), 3.12 (dt, 8H, –2–NH–, 3JHH = 5.6 Hz, 3JHH = 6.6 Hz), 3.86 (t, 8H, O–
2–CH2), 3.12 (dt, 8H, –2–NH–, 3JHH = 5.6 Hz, 3JHH = 6.6 Hz), 3.86 (t, 8H, O–![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) –, 3JHH = 7.4 Hz), 6.27 (d of AB system, 4H, –CH
–, 3JHH = 7.4 Hz), 6.27 (d of AB system, 4H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, 3JHH = 12.8 Hz), 6.43 (d of AB system, 4H, –CHCH–, 3JHH = 12.8 Hz), 7.37 (s, 8H, Ar–H), 9.11 (br.t, 4H, NH), 15.13 (br.s, 4H, –OH). 13C NMR (DMSO-d6, δ, ppm): 28.46, 30.86, 33.85, 36.35, 67.48, 127.64, 128.62, 131.64, 133.26, 145.57, 156.87, 165.30, 165.50. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H8′, H4b/H9′, H4b/H10′, H4b/H12′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H10′. IR (ν/cm−1): 3272 (NH); 3092 (CH–); 2962, 1008 (OH); 2909, 2870, 1708, 1443, 1241 (COOH); 1632, 1570, 1264 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1341.7, found [M + H]+m/z = 1342.5, [M + Na]+m/z = 1363.5, [M + K]+m/z = 1380.5. El. anal. calcd for C68H84N4O16S4 (%): C, 60.88; H, 6.31; N, 4.18; S, 9.56. Found (%): C, 60.46; H, 5.95; N, 3.95; S, 9.23.
2–CH2–, 3JHH = 7.2 Hz, 3JHH = 7.2 Hz), 1.95 (s, 12H, –CHC(3)–), 3.05 (dt, 8H, –2–NH–, 3JHH = 5.4 Hz, 3JHH = 6.0 Hz), 3.82 (t, 8H, O––, 3JHH = 7.2 Hz), 6.18 (s, 4H, –C(CH3)–), 7.35 (s, 8H, Ar–H), 8.61 (br.t, 4H, NH), 14.49 (br.s, 4H, –OH). 13C NMR (DMSO-d6, δ, ppm): 21.72, 28.64, 30.88, 33.87, 36.16, 67.56, 124.49, 127.62, 128.40, 142.44, 145.58, 156.78, 164.92, 168.02. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H9′, H4b/H10′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H10′, H3/H13′, H10/H12. IR (ν/cm−1): 3253 (NH); 3075 (CH–); 2964, 1007 (OH); 2870, 1777, 1698, 1436, 1308, 1241 (COOH); 1633, 1533, 1581, 1263 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1397.8, found [M]+m/z = 1397.7, [M + Na]+m/z = 1420.4, [M + K]+m/z = 1436.4. El. anal. calcd for C72H92N4O16S4 (%): C, 61.87; H, 6.63; N, 4.01; S, 9.17. Found (%): C, 61.46; H, 6.82; N, 3.88; S, 8.96.
2–CH2–, 3JHH = 7.0 Hz, 3JHH = 7.2 Hz), 2.31 (AB part of AA′BB′ system, 8H, –2–CH2–C(O), 3JHAHB = 6.8 Hz, 2JHAHA′ = 13.6 Hz), 2.41 (A′B′ part of AA′BB′ system, 8H, –CH2–2–C(O), 3JHAHB = 6.8 Hz, 2JHAHA′ = 13.6 Hz), 2.95 (dt, 8H, –2–NH–, 3JHH = 6.0 Hz, 3JHH = 6.2 Hz), 3.79 (t, 8H, O–2–, 3JHH = 7.2 Hz), 7.35 (s, 8H, Ar–H), 7.77 (t, 4H, NH, 3JHH = 5.2 Hz), 12.03 (br.s, 4H, –OH). 13C NMR (DEPT) (DMSO-d6, δ, ppm): 29.05, 29.28, 30.07, 30.91, 33.89, 35.86, 67.80, 127.68, 128.54, 145.43, 156.88, 170.89, 173.82. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H8′, H4b/H9′, H4b/H10′, H4b/H12′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H12′, H3/H13′. IR (ν/cm−1): 3350, 3206 (NH); 2960, 954 (OH); 2903, 2867, 1718, 1708, 1407, 1245, 1239 (COOH); 1652, 1537, 1263 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1349.7, found [M + H]+m/z = 1351.0, [M + Na]+m/z = 1372.6, [M + K]+m/z = 1388.6. El. anal. calcd for C68H92N4O16S4 (%): C, 60.51; H, 6.87; N, 4.15; S, 9.50. Found (%): C, 59.97; H, 6.55; N, 3.86; S, 8.99.
CH–, 3JHH = 12.8 Hz), 6.43 (d of AB system, 4H, –CHCH–, 3JHH = 12.8 Hz), 7.37 (s, 8H, Ar–H), 9.11 (br.t, 4H, NH), 15.13 (br.s, 4H, –OH). 13C NMR (DMSO-d6, δ, ppm): 28.46, 30.86, 33.85, 36.35, 67.48, 127.64, 128.62, 131.64, 133.26, 145.57, 156.87, 165.30, 165.50. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H8′, H4b/H9′, H4b/H10′, H4b/H12′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H10′. IR (ν/cm−1): 3272 (NH); 3092 (CH–); 2962, 1008 (OH); 2909, 2870, 1708, 1443, 1241 (COOH); 1632, 1570, 1264 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1341.7, found [M + H]+m/z = 1342.5, [M + Na]+m/z = 1363.5, [M + K]+m/z = 1380.5. El. anal. calcd for C68H84N4O16S4 (%): C, 60.88; H, 6.31; N, 4.18; S, 9.56. Found (%): C, 60.46; H, 5.95; N, 3.95; S, 9.23.
2–CH2–, 3JHH = 7.2 Hz, 3JHH = 7.2 Hz), 1.95 (s, 12H, –CHC(3)–), 3.05 (dt, 8H, –2–NH–, 3JHH = 5.4 Hz, 3JHH = 6.0 Hz), 3.82 (t, 8H, O––, 3JHH = 7.2 Hz), 6.18 (s, 4H, –C(CH3)–), 7.35 (s, 8H, Ar–H), 8.61 (br.t, 4H, NH), 14.49 (br.s, 4H, –OH). 13C NMR (DMSO-d6, δ, ppm): 21.72, 28.64, 30.88, 33.87, 36.16, 67.56, 124.49, 127.62, 128.40, 142.44, 145.58, 156.78, 164.92, 168.02. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H9′, H4b/H10′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H10′, H3/H13′, H10/H12. IR (ν/cm−1): 3253 (NH); 3075 (CH–); 2964, 1007 (OH); 2870, 1777, 1698, 1436, 1308, 1241 (COOH); 1633, 1533, 1581, 1263 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1397.8, found [M]+m/z = 1397.7, [M + Na]+m/z = 1420.4, [M + K]+m/z = 1436.4. El. anal. calcd for C72H92N4O16S4 (%): C, 61.87; H, 6.63; N, 4.01; S, 9.17. Found (%): C, 61.46; H, 6.82; N, 3.88; S, 8.96.
2–CH2–, 3JHH = 7.0 Hz, 3JHH = 7.2 Hz), 2.31 (AB part of AA′BB′ system, 8H, –2–CH2–C(O), 3JHAHB = 6.8 Hz, 2JHAHA′ = 13.6 Hz), 2.41 (A′B′ part of AA′BB′ system, 8H, –CH2–2–C(O), 3JHAHB = 6.8 Hz, 2JHAHA′ = 13.6 Hz), 2.95 (dt, 8H, –2–NH–, 3JHH = 6.0 Hz, 3JHH = 6.2 Hz), 3.79 (t, 8H, O–2–, 3JHH = 7.2 Hz), 7.35 (s, 8H, Ar–H), 7.77 (t, 4H, NH, 3JHH = 5.2 Hz), 12.03 (br.s, 4H, –OH). 13C NMR (DEPT) (DMSO-d6, δ, ppm): 29.05, 29.28, 30.07, 30.91, 33.89, 35.86, 67.80, 127.68, 128.54, 145.43, 156.88, 170.89, 173.82. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H8′, H4b/H9′, H4b/H10′, H4b/H12′, H4b/H13′, H3/H7′, H3/H8′, H3/H9′, H3/H12′, H3/H13′. IR (ν/cm−1): 3350, 3206 (NH); 2960, 954 (OH); 2903, 2867, 1718, 1708, 1407, 1245, 1239 (COOH); 1652, 1537, 1263 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1349.7, found [M + H]+m/z = 1351.0, [M + Na]+m/z = 1372.6, [M + K]+m/z = 1388.6. El. anal. calcd for C68H92N4O16S4 (%): C, 60.51; H, 6.87; N, 4.15; S, 9.50. Found (%): C, 59.97; H, 6.55; N, 3.86; S, 8.99.
Yield: 0.62 g (83%). M.p.: 241 °C. 1H NMR (DMSO-d6, δ, ppm, J/Hz): 1.21 (s, 36H, (CH3)3C), 1.25 (tt, 8H, –CH2–2–CH2–, 3JHH = 6.4 Hz, 3JHH = 7.2 Hz), 3.00 (dt, 8H, –2–NH–, 3JHH = 6.0 Hz, 3JHH = 6.0 Hz), 3.80 (t, 8H, O–2–, 3JHH = 7.2 Hz), 3.95 (s, 8H, (–CH2–C(O))), 4.12 (s, 8H, (–CH2–C(O))), 7.35 (s, 8H, Ar–H), 7.60 (br.t, 4H, NH), 12.81 (br.s, 4H, OH). 13C NMR (DMSO-d6, δ, ppm): 29.00, 30.87, 33.90, 35.20, 67.17, 67.78, 70.01, 127.60, 128.12, 145.57, 156.65, 168.62, 171.38. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H16′, H4b/H10′, H4b/H12′, H4b/H14′, H4b/H7′, H4b/H9′, H3/H7′, H3/H8′, H3/H9′, H3/H10′, H3/H12′. IR (ν/cm−1): 3282 (NH); 2952, 1000 (OH); 2907, 2870, 1734, 1441, 1235 (COOH); 1635, 1547, 1263 (C(O)–NH). MS (MALDI-TOF): calculated [M+] m/z = 1413.7, found [M]+m/z = 1413.3, [M + Na]+m/z = 1436.2, [M + K]+m/z = 1452.3. El. anal. calcd for C68H92N4O20S4 (%): C, 57.77; H, 6.56; N, 3.96; S, 9.07. Found (%): C, 57.82; H, 6.68; N, 4.01; S, 9.23.
Yield: 0.26 g (90%). M.p.: 175 °C. [α]25D = −58.97 (CH3OH, 1 × 10−2 M). 1H NMR (CDCl3, δ, ppm, J/Hz): 1.18 (m, 8H, –CH2–2–CH2–), 1.24 (s, 36H, (CH3)3C), 1.47 (d, 12H, –CH3, 3JHH = 6.6 Hz), 1.49 (d, 12H, –CH3, 3JHH = 7.0 Hz), 3.00 (m, 8H, –2–NH–), 3.82 (m, 8H, –O–2–), 4.38 (q, 4H, –(CH3), 3JHH = 6.6 Hz), 5.23 (q, 4H, –(CH3), 3JHH = 7.0 Hz), 6.53 (br.t, 4H, NH), 7.32 (s, 8H, Ar–H). 13C NMR (DEPT) (CDCl3, δ, ppm): 18.09, 20.65, 29.05, 31.33, 34.43, 36.60, 66.34, 67.31, 71.23, 127.83, 128.15, 146.52, 156.48, 170.45, 173.90. 1H–1H NOESY NMR spectrum (most important cross-peaks are presented): H4b/H7′, H4b/H10′, H4b/H9′, H3/H7′, H3/H8′, H3/H13′. IR (Nujol, ν/cm−1): 3300 (OH, NH); 1742, 1656, 1536, 1264 (C(O)–NH). MS (ESI): calculated [M+] m/z = 1525.9, found [M + H]+m/z = 1525.8, [M + Na]+m/z = 1547.8. El. anal. calcd for C76H108N4O26S4 (%): C, 59.82; H, 7.13; N, 3.67; S, 8.40. Found (%): C, 60.03; H, 6.99; N, 3.78; S, 8.25.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Russian Science Foundation (no. 16-13-00005). Study of the spatial structure of compounds by NMR spectroscopy was funded by a subsidy of the Russian Government to support the Program of Competitive Growth of Kazan Federal University among World's Leading Academic Centers.Notes and references
- C. M. Cameron, R. M. Wightman and R. M. Carelli, Neuropharmacology, 2016, 111, 223 CAS.
- M. Leyton, Curr. Opin. Behav. Sci., 2017, 13, 130 Search PubMed.
- P. Li, G. L. Snyder and K. E. Vanover, Curr. Top. Med. Chem., 2016, 16, 3385 CrossRef CAS PubMed.
- K. Kamińska, K. Noworyta-Sokołowska, A. Jurczak, A. Górska, Z. Rogóż and K. Gołembiowska, Pharmacol. Rep., 2017, 69, 13 CrossRef PubMed.
- In vivo Neuropharmacology and Neurophysiology, Neuromethods, ed. A. Philippu, 2017, 121, p. 428 Search PubMed.
- B. J. Jongkees and L. S. Colzato, Neurosci. Biobehav. Rev., 2016, 71, 58 CrossRef PubMed.
- A. Dawson, N. Stensson, B. Ghafouri, B. Gerdle, T. List, P. Svensson and M. Ernberg, J. Headache Pain, 2016, 17, 65 CrossRef PubMed.
- F. H. Khan, C. D. Ahlberg, C. A. Chow, D. R. Shah and B. B. Koo, J. Neurol., 2017, 264, 1634 CrossRef CAS PubMed.
- P. Młynarz, E. Rudzińska, Ł. Berlicki and P. Kafarski, Curr. Org. Chem., 2007, 11, 1593 CrossRef.
- T. Oshima, K. Oishi, K. Ohto and K. Inoue, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 79 CrossRef CAS.
- S. M. Santos, P. J. Costa, M. D. Lankshear, P. D. Beer and V. Félix, J. Phys. Chem. B, 2010, 114, 11173 CrossRef CAS PubMed.
- S. Le Gac, J.-F. Picron, O. Reinaud and I. Jabin, Org. Biomol. Chem., 2011, 9, 2387 CAS.
- E. Makrlík, P. Vaňura and P. Selucký, J. Radioanal. Nucl. Chem., 2013, 295, 2077 CrossRef.
- G. Gattuso, A. Notti, S. Pappalardo, M. F. Parisi, I. Pisagatti and S. Patanè, New J. Chem., 2014, 38, 5983 RSC.
- F. Zou, B. Wu, X. Wang, Y. Chen, K. Koh, K. Wang and H. Chen, Sens. Actuators, B, 2017, 241, 160 CrossRef CAS.
- G. Arena, A. Pappalardo, S. Pappalardo, G. Gattuso, A. Notti, M. F. Parisi, I. Pisagatti and C. Sgarlata, J. Therm. Anal. Calorim., 2015, 121, 1073 CrossRef CAS.
- P. Vitovič, D. P. Nikolelis and T. Hianik, Biochim. Biophys. Acta, 2006, 1758, 1852 CrossRef PubMed.
- X.-D. Xiao, L. Shi, L.-H. Guo, J.-W. Wang and X. Zhang, Spectrochim. Acta, Part A, 2017, 173, 6 CrossRef CAS PubMed.
- Q. Zhang, Y. Ni and S. Kokot, Spectrosc. Lett., 2012, 45, 85 CrossRef CAS.
- J. Li, H. Duan, W. Wei and Sh. Luo, Phys. Chem. Liq., 2012, 50, 453 CrossRef CAS.
- M. Shi, J. Lu and M. S. Shoichet, J. Mater. Chem., 2009, 19, 5485 RSC.
- B. Mokhtari, K. Pourabdollah and N. Dalali, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 1 CrossRef CAS.
- Y. Zhou, H. Li and Y.-W. Yang, Chin. Chem. Lett., 2015, 26, 825 CrossRef CAS.
- O. A. Mostovaya, M. N. Agafonova, A. V. Galukhin, B. I. Khayrutdinov, D. Islamov, O. N. Kataeva, I. S. Antipin, A. I. Konovalov and I. I. Stoikov, J. Phys. Org. Chem., 2014, 27, 57 CrossRef CAS.
- I. I. Stoikov, A. Y. Zhukov, M. N. Agafonova, R. R. Sitdikov, I. S. Antipin and A. I. Konovalov, Tetrahedron, 2010, 66, 359 CrossRef CAS.
- P. Zlatušková, I. Stibor, M. Tkadlecová and P. Lhoták, Tetrahedron, 2004, 60, 11383 CrossRef.
- P. L. Padnya, E. A. Andreyko, O. A. Mostovaya, I. K. Rizvanov and I. I. Stoikov, Org. Biomol. Chem., 2015, 13, 5894 CAS.
- O. A. Mostovaya, P. L. Padnya, D. N. Shurpik, A. A. Vavilova, V. G. Evtugyn, Yu. N. Osin and I. I. Stoikov, Macroheterocycles, 2017, 10, 154 CrossRef.
- A. A. Vavilova, R. V. Nosov, A. N. Yagarmina, O. A. Mostovaya, I. S. Antipin, A. I. Konovalov and I. I. Stoikov, Macroheterocycles, 2012, 5, 396 CrossRef CAS.
- E. A. Andreyko, P. L. Padnya, R. R. Daminova and I. I. Stoikov, RSC Adv., 2014, 4, 3556 RSC.
- N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed.
- G. A. Evtugyn, R. V. Shamagsumova, R. R. Sitdikov, I. I. Stoikov, I. S. Antipin, M. V. Ageeva and T. Hianik, Electroanalysis, 2011, 23, 2281 CrossRef CAS.
- K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015, 54, 14858 CrossRef CAS PubMed.
- K. Kan, M. Fujiki, M. Akashi and H. Ajiro, ACS Macro Lett., 2016, 5, 1014 CrossRef CAS.
- A. E. Baydar, G. V. Boyd, J. Aupers and P. F. Lindley, J. Chem. Soc., Perkin Trans. 1, 1981, 2890 RSC.
- H. Y. Wang, X. G. Feng, M. Zhang and H. Zhao, Anal. Sci., 2007, 23, 1297 CrossRef CAS PubMed.
- S. Garabagiu, AIP Conference Proceedings, 1565, 215.
- Y. J. Jang, J. H. Jun, K. M. K. Swamy, K. Nakamura, H. S. Koh, Y. J. Yoon and J. Yoon, Bull. Korean Chem. Soc., 2005, 26, 2041 CrossRef CAS.
- Y. Suzuki, Sens. Actuators, B, 2017, 239, 383 CrossRef CAS.
- S. Niu, Y. Fang, K. Zhang, J. Sun and J. Wan, Sci. Technol., 2017, 45, 101 CAS.
- R. P. Rava and T. G. Spiro, J. Phys. Chem., 1985, 89, 1856 CrossRef CAS.
- W. J. Barreto, S. Ponzoni and P. Sassi, Spectrochim. Acta, Part A, 1999, 55, 65 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, US, 2006, p. 954 Search PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792 RSC.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Org. Lett., 2017, 19, 2933 CrossRef PubMed.
- Bindfit v0.5 (Open Data Fit, 2016); http://supramolecular.org/bindfit/.
- Anion Sensing (Top. Curr. Chem., 255), ed. I. Stibor, Springer, Berlin, 2005, p. 238 Search PubMed.
- A. A. Vavilova and I. I. Stoikov, Beilstein J. Org. Chem., 2017, 13, 1940 CrossRef CAS PubMed.
- L. S. Yakimova, D. N. Shurpik and I. I. Stoikov, Chem. Commun., 2016, 52, 12462 RSC.
- P. Dais, M. Misiak and E. Hatzakis, Anal. Methods, 2015, 7, 5226 RSC.
- A. K. Sundaresan, C. L. Gibb, B. C. Gibb and V. Ramamurthy, Tetrahedron, 2009, 65, 7277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nj03953k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |