
Single X-ray crystal structure, DFT studies and topoisomerase I inhibition activity of a tailored ionic Ag(I) nalidixic acid–piperazinium drug entity specific for pancreatic cancer cells†
Imtiyaz
Yousuf
a,
Mohammad
Usman
a,
Musheer
Ahmad
b,
Sartaj
Tabassum
 a and
Farukh
Arjmand
*a
a and
Farukh
Arjmand
*a
aDepartment of Chemistry, Aligarh Muslim University, Aligarh 202002, Uttar Pradesh, India. E-mail: farukh_arjmand@yahoo.co.in
bDepartment of Applied Chemistry, Aligarh Muslim University, Aligarh 202002, Uttar Pradesh, India
First published on 5th December 2017
Abstract
Novel ionic Ag(I)−piperazinium nalidixic acid conjugate 1 was synthesized as a potential antitumor agent and was thoroughly characterized by elemental analysis, FT-IR, 1H and 13C NMR and single X-ray crystal diffraction studies. Complex 1 crystallized in the triclinic space group P1 and comprises a dipiperazinium–Ag(I) cationic unit, two nalidixate (naI−) anionic moieties and a nitrate ion. The Ag(I) ion adopted a linear configuration upon coordination with two nitrogen atoms of piperazinium cations (pipzH+) arranged in a trans fashion. The density functional theory (DFT) studies of 1 revealed the HOMO and LUMO to be localized on the metal center viz., the dx2−y2 orbital and partially localized on the C27, C28, C29, C30, C31, C32, N7 and N6 atoms of the piperazinium moiety. Non covalent interaction (NCI) calculations were carried out to identify the weak non-covalent interactions from the topological analysis and reduced gradient of the electron density of complex 1. Our results revealed significant inter- and intramolecular non-covalent interactions between the naI− and [Ag(pip)2]2+ units. Furthermore, an analysis of Hirshfeld surfaces and fingerprint plots were carried out to ascertain a comparison between intermolecular interactions which provide interesting supramolecular architectures involving combinations of N–H⋯O, O–H⋯O and C–H⋯O linkages into a two-dimensional framework. In vitro binding studies of 1 with ct-DNA and tRNA revealed higher binding propensity for tRNA which was evidenced from its higher intrinsic binding constant, Kb and binding constant, K values and the mode of binding was found to be groove binding in nature. The catalytic activity of topoisomerase I enzyme in the presence of complex 1 was ascertained by gel electrophoretic assay which demonstrated significant inhibitory effects at a low concentration of 25 μM. The cytotoxicity activity of 1 was determined by SRB assay on MIA-PA-CA-2, HepG2, HeLa and MCF7 human cancer cell lines; these results exhibited specific and selective antitumor activity for the MIA-PA-CA-2 cancer cell line with a GI50 value <10.
Introduction
Pancreatic cancer is the most aggressive form of cancer characterized by early metastasis, and poor prognosis with an exceptionally high mortality rate.1,2 Being one of the four most common cancer phenotypes in developing countries with a marginal 5 year survival rate of less than 5%, there is serious concern regarding the cure and prevention of these cancers.3 Although general factors (long standing diabetes, alcohol consumption, etc.) pose high risk for developing pancreatic cancer, most of the cancer cases are known to occur by somatic mutations.4,5 Regarding the treatment modalities, surgery at an earlier stage in combination with chemotherapy and radiotherapy may help to shrink the tumor cells enough so that they can be removed by other invasive methods. The chemotherapeutic intervention for pancreatic cancers involves drugs viz., Nab-paclitaxel (Abraxane),6 oxaliplatin (Eloxatin)7 and fluorouracil (5-FU)8etc. which are used alone or in combination. Despite enormous research efforts undertaken to improve the clinical responses of these drugs towards pancreatic cancers, no substantial success has been achieved so far. Furthermore, adverse side effects, viz., peripheral neuropathy, gastrointestinal problems, mouth sores, vomiting, diarrhoea, and loss of appetite and patient compliance, often arise.9 To overcome such issues, extensive efforts are being undertaken to develop and design transition metal-based target specific anticancer agents having maximal curative potential and minimal side effects.Cisplatin and other PtII based metallo-drugs have been acclaimed globally for clinical use against testicular, ovarian and colorectal cancers.10 However, their remarkable success is hindered by their severe limitations viz., acquired or intrinsic resistance problems, limited spectrum of activity, and “off-target” side effects leading to high toxicity.11 Among the transition metal ions, beyond the chemistry of platinum, coinage metals, viz., gold, silver and copper have received considerable attention in the area of chemotherapeutics. Whilst copper and gold complexes have been extensively examined for their anticancer activities, silver complexes have remained far less exploited.12–14 Elemental silver and its salts have been known for a long time to possess pharmacological benefits from antibacterial activity to anti-inflammatory activity, and difficult-to-heal wounds have been treated with ‘silver magical bullets’.15–17 Silver-based FDA approved drugs viz., silver sulphadiazine (Silvadene® and Flamazine®) have been used in wound therapy while a dilute solution of AgNO3 is being used for the prophylaxis of bacterial conjunctivitis among infants.18,19 The pharmacological activity of silver is predominantly attributed to its interference with the electron transport system or cell membranes and partly to its interaction with thiol groups of the vital enzymes.20 In recent years, many research groups have explored silver(I) complexes as efficient anticancer agents exhibiting lower systemic toxicity, and interestingly some of them demonstrated antiproliferative activity greater in magnitude than cisplatin itself.20–23 Furthermore, Ag(I) complexes have been found to be selective towards various types of cancer cells and the mode of selectivity was found to be dependent on the nature of the coordinated ligand.24
RNA targeted therapeutics have gained much impetus as RNA is located upstream from the proteins in the gene expression pathway, and thus by blocking RNA molecules, inhibition of diverse functioning of proteins can be envisioned. A few notable metal complexes have shown selective and efficient targeting of 3-dimensional non-duplex RNA (tRNA).25 Herein, we report a novel tailored RNA targeted quinolone drug derivative Ag(I) nalidixic acid–piperazine drug entity (1) which is highly water soluble and has structural novelty as a potent drug entity against a human pancreatic cancer cell line with GI50 < 10. Complex 1 is the first demonstration of an ionic molecular entity which possesses a dicationic piperazinium Ag(I) moiety and a dianionic nalidixic drug pharmacophore as a counter ion. Nalidixic acid belongs to the quinolone drugs that are targeted to DNA gyrases and intervene selectively via topoisomerase enzyme inhibitory pathway mechanisms.26 Topoisomerase I (Topo I) is a ubiquitous cellular enzyme that is overexpressed in many forms of cancers and represents one of the most potential biotargets in the area of cancer chemotherapeutics.27 Topoisomerase inhibitors are selectively chosen for “targeted therapies” against an array of malignancies as they cause permanent DNA damage by triggering a series of cellular events leading to cell cycle arrest and/or apoptotic cell death.28 On the other hand, the piperazine template forms an essential molecular backbone capable of inhibiting cell cycle progression in tumour cells causing DNA damage possibly via induction of apoptosis.29,30 Our synthetic design could provide a novel therapeutic intervention for pancreatic cancers, and will hopefully overcome patient compliance as it is expected to show mute toxicity and efficacious treatment modality.
Results and discussion
Synthesis and structural characterization
Complex 1 was prepared from an ion pair, {(pipzH)+(naI)−} as a result of the in situ proton transfer reaction between nalidixic acid (HnaI) and piperazine (pipz), and its subsequent reaction with silver(I) nitrate (Scheme 1). The soft Ag(I) ion prefers to coordinate with an N atom of pipz rather an O− (hard) ion of naI− consistent with the hard soft acid base (HSAB) principle. The synthesized complex was found to be remarkably soluble in H2O, MeOH and other non-polar solvents viz., DMF and DMSO. The molecular structure of 1 was established by X-ray diffraction studies which corroborated well with elemental analysis and other spectroscopic measurements (UV-vis, FT-IR, NMR and ESI-MS).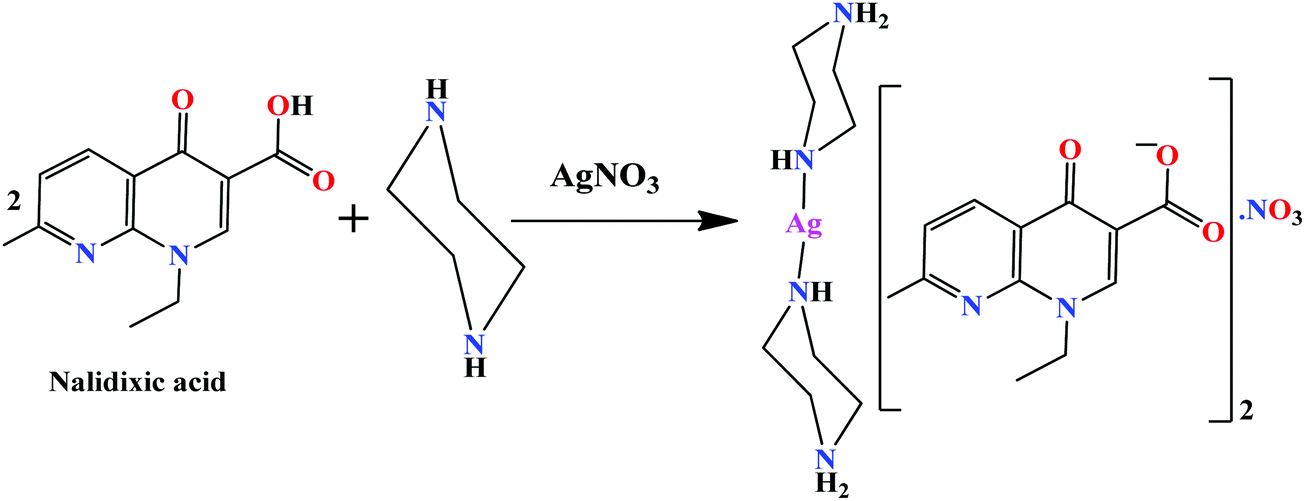 | ||
| Scheme 1 Synthetic route for complex 1. | ||
Complex 1 crystallized in the triclinic space group P1 and the asymmetric unit comprises a piperazinediium cationic unit, two nalidixate anionic moieties, a nitrate ion and solvent molecules (Fig. 1). The silver(I) ion adopts a linear configuration upon coordination with two nitrogen atoms of piperazinediium cations having Ag1–N6 = 2.145(8) Å, Ag1–N7 = 2.130(8) and a bond angle ∠N6–Ag1–N7 of 178.17°. The two piprazinium moieties are arranged in a trans fashion with the usual stable chair conformation.
 | ||
| Fig. 1 Asymmetric unit of complex 1 (lattice water molecules have been omitted for clarity). | ||
Intricate hydrogen bonding between the lattice water molecules, oxygen atoms of the carboxylate anions of nalidixate (naI−) (O–H⋯O−) and NH2 of pipzH+ (HN–H⋯O) play an important role in the 3D supramolecular formation and stabilization of the crystal structure. The crystal structure cohesion is further reinforced by considerable π–π stacking and –CH⋯π interactions between two aromatic rings with an average distance of 3.15–3.50 Å (Fig. 2 and Fig. S1, ESI†). Selected bond lengths and bond angles are listed in Table S1 (ESI†).
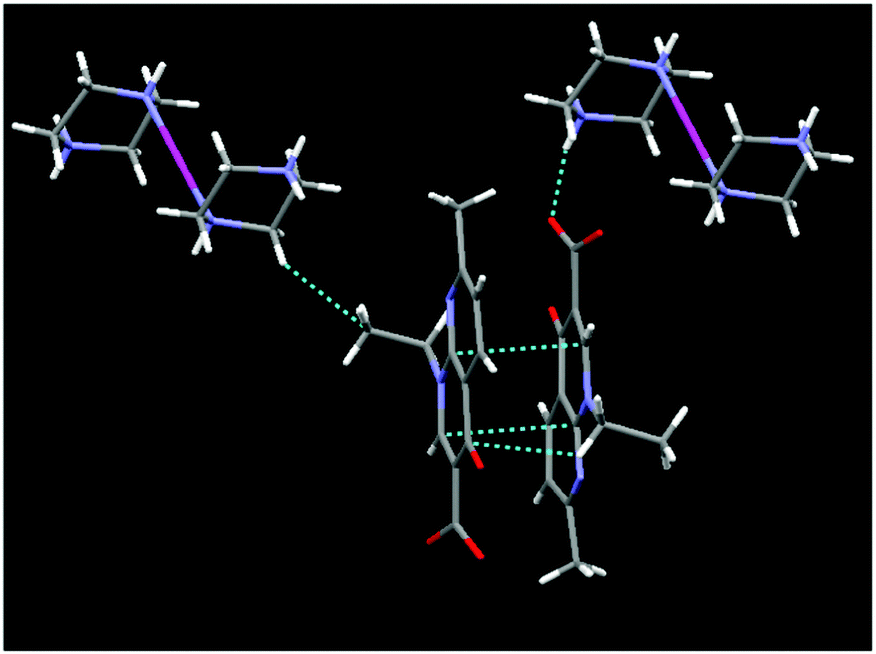 | ||
| Fig. 2 Diagrammatic representation of π⋯π and –CH⋯π interactions. | ||
The IR spectrum of nalidixic acid (HnaI) displayed absorption bands at 3385, 1718 and 1366 cm−1 that can be attributed to the ν(O–H), νasym(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)carboxylic and νsym(CO)carboxylic stretching vibrations, respectively of the carboxylic acid group (–COOH). However, in the IR spectrum of complex 1, the absence of a peak at 3385 cm−1 confirmed its deprotonation, and the absence of any shift in the ν(CO) bands implicated a non-cordinating behaviour of the –COOH group. Two important IR vibrations of the complex corresponding to ν(N–H) and ν(CH2) group vibrations of the piprazinium moiety were observed at 3127 and 2922 cm−1, respectively. A broad band at ca. 3450 cm−1 suggested the presence of lattice water in complex 1. In addition, the appearance of medium intensity frequency bands at 445 cm−1 in the far IR region of 1 was attributed to (M–N) streching vibrations.
O)carboxylic and νsym(CO)carboxylic stretching vibrations, respectively of the carboxylic acid group (–COOH). However, in the IR spectrum of complex 1, the absence of a peak at 3385 cm−1 confirmed its deprotonation, and the absence of any shift in the ν(CO) bands implicated a non-cordinating behaviour of the –COOH group. Two important IR vibrations of the complex corresponding to ν(N–H) and ν(CH2) group vibrations of the piprazinium moiety were observed at 3127 and 2922 cm−1, respectively. A broad band at ca. 3450 cm−1 suggested the presence of lattice water in complex 1. In addition, the appearance of medium intensity frequency bands at 445 cm−1 in the far IR region of 1 was attributed to (M–N) streching vibrations.
The 1H NMR spectrum of complex 1 recorded in DMSO-d6 displayed no resonance peak for the O–H proton of HnaI (ca. 10–13 ppm) confirming its deprotonation. The characteristic aromatic signatures of naI− were displayed as two doublets and one singlet in the region 8.99–7.33 ppm (H2, H5, and H6).31 A quartet and a triplet centered at 4.53 and 1.39 ppm were assigned to CH2 and CH3 protons of the –NCH2CH3 group, respectively. A sharp signature at 4.03 ppm and a singlet at 3.79 ppm were attributed to [NH2]+ and CH2 protons of the piprazinium moiety, respectively.32 Moreover a singlet corresponding to methyl protons of the naI− ligand was observed at 2.55 ppm. 13C NMR spectra of the complex 1 revealed carbonyl and carboxylate carbons resonating at 186.71 and 177.22 ppm, respectively. The signals corresponding to aromatic carbons were displayed as a broad envelope in the range of 149–119 ppm. Two resonance signatures of CH2 carbons of piperazinium and naI− (–NCH2CH3) moieties were found in the range of 67.96 and 57.91 ppm, respectively. Additionally, a resonance signal corresponding to CH3 carbons of the –NCH2CH3 group was found at 11.16 ppm.33
Density functional theory
The frontier molecular orbital analysis
The LUMO and HOMO orbital energy parameters are significantly important for the charge transfer, chemical reactivity and kinetic/thermodynamic stability of a molecule.34 Metal complexes with a small energy gap (ΔE) between the HOMO and LUMO are more polarizable, and thereby act as soft molecules with higher chemical reactivity. However, complexes with a large energy gap often offer greater stability and lower chemical reactivity than those with small HOMO–LUMO energy gaps.35 The FMOs of complex 1 were calculated using B3LYP/DFT methods in the gas phase. The DFT results revealed that the HOMO and LUMO were localized on the metal center, particularly on the dz2 orbital, and partial localization was observed at the C27, C28, C29, C30, C31, C32, N7 and N6 atoms of the pipz ligand (Fig. 3). It could be speculated that the HOMO and LUMO are mainly composed of σ(Ag–Npip) orbitals, except for a small contribution from the pipz molecular fragment, whereas the HOMO−1 is mostly localized on the solvated NO3− ion and the LUMO+1 is localized over co-crystalized naI− moieties. | ||
| Fig. 3 The HOMO and LUMO frontier orbital surfaces of complex 1 along with their respective energy values and the band gap. | ||
The HOMO–LUMO gap of complex 1 was calculated to be 1.671 eV and the frontier molecular orbital energies of the complex, EHOMO and ELUMO were found to be −3.584 and −1.913 eV, respectively (Table 1).
| FMO | Energy (eV) |
|---|---|
| LUMO+3 | −1.473 |
| LUMO+2 | −1.489 |
| LUMO+1 | −1.730 |
| LUMO | −1.913 |
| HOMO | −3.584 |
| HOMO−1 | −4.406 |
| HOMO−2 | −5.399 |
| HOMO−3 | −5.565 |
The time dependent density functional theory (B3LYP/TDDFT) calculations were also performed with complex 1 to get better insight into the electronic interactions of the complex and simulate the spectral assignments of the UV-vis spectra.36 The theoretical analysis of electronic structure and molecular orbitals demonstrated that the low-lying absorption bands in the UV-vis spectrum of 1 were mainly ascribed to π–π* and n–π* transitions, consistent with the spectroscopic results (Fig. 4).
 | ||
| Fig. 4 B3LYP/TDDFT simulated electronic absorption spectra of complex 1. | ||
Non-covalent interaction index (NCI) approach
The NCI method is a theoretical strategy used to visualize the weak non-covalent interactions from the topological analysis of the electron density and of its reduced gradient (RDG) as originally proposed by Yang et al.37,38 The approach is based on the analysis and graphical investigation of the electronic density (ρ) and its derivatives, which measure the RDG or deviation from a homogeneous electron distribution s(ρ) asThe NCI index analysis was accomplished and built on electronic density of the molecules to anticipate both attractive (hydrogen bonding, van der Waals) and repulsive (steric) interactions and the electronic clouds located on interacting groups which essentially perturb each other thereby influencing the local densities and associated properties.39,40 It has been reported that the inter- or intramolecular interactions lead to a substantial transformation in the reduced density gradient (S or RDG) in between interacting atom pairs and interacting fragments within the crystal lattice, resulting in density critical points.41 To analyze the non-covalent attractive or repulsive interactions, the sign of the eigenvalue λ2 of the electronic density Hessian matrix, ∇2ρ = λ1 + λ2 + λ3 (λ1 < λ2 < λ3) was utilized. In the case of hydrogen bonding interactions the λ2 exhibits a negative value which is characterized by an accumulation of the electron density perpendicular to the attractive non-covalent bonding interaction. In contrast, a positive value of λ2 indicates non-bonding interactions (steric repulsions) which result in electron density depletion. Thus, the RDG is used to recognize non-covalent interactions, and at the point at which the value of S is approaching zero, the strength of the interaction is defined by quantity sign (λ2)ρ, i.e., the higher the value (−ve: attractive interaction vs. +ve: repulsive interaction), the stronger the interaction will be.42

In our experiments, the NCI index calculations of complex 1 revealed significant inter- and intramolecular non-covalent interactions between the coordination sphere [Ag(pip)2]2+ and the two counter ions (naI−) which are responsible for the stabilization of geometric conformation within the crystal lattice. These interactions were identified from the 2D plots as spikes with a cut off value (RDG 2 and (λ2)ρ − 0.25) and their corresponding 3D electronic density map with color coding. The 3D electronic density map of complex 1 not only revealed the attractive non-covalent interactions but also displayed valuable information about the destabilization factor such as steric clashes which are repulsive in nature and responsible for the geometrical distortion in the molecular entity. These stabilization and destabilization contacts differ in the shape of the isosurfaces whose color code reveals the nature of the interaction as red, blue and green for destabilizing, stabilizing and weak interactions, respectively. The RDG vs. sign(λ2)ρ in complex 1 exhibited six characteristic spikes with density ((λ2)ρ) −0.09, −0.03, −0.015, and −0.01 and 0.006, 0.015, and 0.005 which correspond to the strong N–H⋯O, O–H⋯O, π⋯π, and CH⋯π interactions (attractive or stabilizing) and steric clashes (repulsive or destabilizing), respectively (Fig. 5).
 | ||
| Fig. 5 A 2D plot of the reduced density gradient vs. the electronic density multiplied by the sign of the second Hessian eigenvalue of complex 1. | ||
Noticeably, in complex 1 various sites were observed where electronic density was distributed between the nalidixate and [Ag(pip)2]2+ moieties revealing significant dispersive forces that are responsible for the stabilization of the geometrical conformation of complex 1 (Fig. 6). Therefore, on the basis of NCI index analysis it can be speculated that there is a drastic accumulation of electronic density in between the two naI− and [Ag(pip)2]2+ units, thereby providing ample evidence of existing interactions between the two moieties.
 | ||
| Fig. 6 The NCI electronic density distribution in complex 1 (a) with isovalue 0.7 and (b) with isovalue 0.3. Electronic density inside the red circle is responsible for synergistic interaction between naI− and [Ag(pip)2]2+ moieties. | ||
Hirshfeld surface analysis
The Hirshfeld surface is defined as the volume of space where the molecule electron density exceeds that from all neighboring molecules. This study offers a three-dimensional (3D) view of the close contacts in a crystal structure that can be summarized in a 2D fingerprint plot.43 In our Hirshfeld surface analysis, the dnorm surface was used to study the surface characteristics and identify close intermolecular interactions (Table 2).| Surface contact | % Contribution |
|---|---|
| C–C | 4.0 |
| C–H | 3.5 |
| C–N | 0.5 |
| C–O | 0.4 |
| H–H | 40 |
| H–N | 1.0 |
| H–O | 46.8 |
| N–O | 0.7 |
| O–O | 2.3 |
| Ag–O | 0.7 |
In complex 1, the dnorm values mapped in the range of 0.5 to 1.5 Å onto the Hirshfeld surface displayed a three-color scheme; red regions corresponding to closer intermolecular contacts other than van der Waals interactions, blue regions corresponding to longer intermolecular contacts; and white regions corresponding to cases where the distance of the intermolecular contacts is exactly the same (Fig. 7).
 | ||
| Fig. 7 Hirshfeld surface map representing (a) dnorm, (b) curvedness and (c) shape index of complex 1. | ||
The deep red regions on the dnorm surface indicated the close-contact interactions, which are mainly responsible for the significant hydrogen bonding contacts. The small red regions on the dnorm surfaces correspond to the significant C–H interactions.44 The 2D fingerprint plot was formed by the combination of the distances from the Hirshfeld surface to the nearest nucleus inside and outside the surface (Fig. 8).
 | ||
| Fig. 8 Fingerprint plots of complex 1: full (left) and resolved into C⋯C, C⋯H, C⋯N, C⋯O, H⋯H, H⋯N, H⋯O, N⋯O, and O⋯O contacts showing the percentage of contacts contributing to the total Hirshfeld surface area of the molecules. | ||
In vitro DNA/RNA binding studies
The UV-vis spectra of complex 1 exhibited intense absorption bands centered at 276 and 331 nm attributable to π–π* and n–π* intraligand (IL) transitions of the naI− ligand. Upon progressive addition of ct-DNA/tRNA (0–5.0 × 10−5 M) to a fixed concentration of complex 1 (1 × 10−5 M), significant hyperchromism of 43% and 64% was observed along with blue shifts of 4 and 6 nm, respectively (Fig. S2, ESI†). Hyperchromic effects are usually associated with the breakage of the hydrogen bonds that stabilize the secondary structure of nucleic acids.45 Nevertheless, these binding interactions could be further stabilized by hydrogen-bonding interactions involving –CO2− groups of naI− or –NH2+ groups of piperazinium with DAT groups positioned on the edge of the DNA/RNA nucleobases. The quantitative assessment of the binding strength of 1 towards ct-DNA and yeast tRNA was ascertained by calculating intrinsic binding constant, Kb values using the Wolfe–Shimer equation.46 The intrinsic binding constant Kb values for 1 with ct-DNA and yeast tRNA were found to be 4.11(±0.11) × 104 and 7.96(±0.07) × 104 M−1, respectively.The emission spectra of complex 1 displayed a weak luminescence at 370 nm in Tris–HCl buffer at room temperature in the absence of DNA/RNA when excited at 280 nm. The concomitant addition of ct-DNA/tRNA (0.00–1.20 × 10−6 M) to a fixed concentration (0.2 × 106 M) of complex 1, resulted in a significant enhancement in the fluorescence emission intensity with no apparent change in the shape and position of the emission bands (Fig. S3, ESI†). The observed enhancement in emission intensity is related to the extent to which the complexes are protected inside the hydrophobic environment of the DNA/RNA helix. The binding strength of 1 with DNA/RNA was quantified from the values of binding constant, K using the Scatchard equation47 and was found to be 3.93 × 104 and 6.81 × 104 M−1, respectively, consistent with absorption spectral studies.
Circular dichroic (CD) spectroscopy was further used to investigate the conformational changes caused by 1 with respect to ct-DNA and tRNA (Fig. 9a and b). The CD spectrum of B-DNA exhibited a positive band at 275 nm (base stacking) and a negative band at 245 nm (right handed-helicity) which demonstrated significant perturbations upon interaction with 1.48 Upon incubation of ct-DNA with 1 ([complex]/[DNA] = 1) a prominent decrease in intensity of both the positive (helicity) and negative (ellipticity) bands was observed. The decrease in the intensity in the positive and negative bands by complex 1 suggested unwinding of the DNA helix that lead to the loss of DNA helicity.49
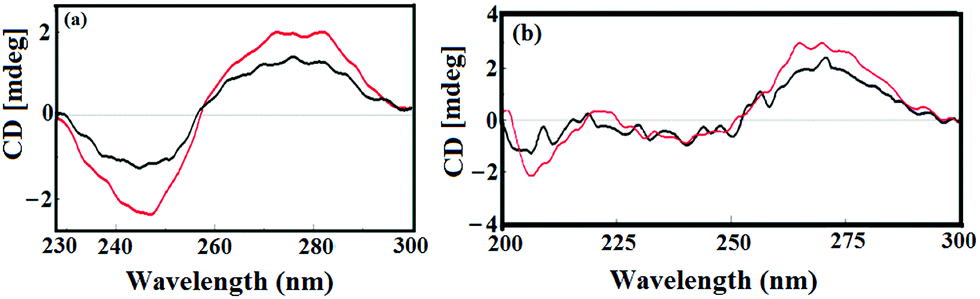 | ||
| Fig. 9 CD spectra of (a) ct-DNA alone (black) and in the absence and presence of 1 (red) and (b) yeast tRNA alone (black) and in the absence and presence of 1 (red). [Complex 1] = [DNA] = [RNA] = 10−4 M. | ||
The CD spectrum of tRNA depicted the characteristic four major peaks at 210 and 240 nm (negative), and 225 and 270 nm (positive) consistent with the A-conformation of tRNA. Upon incubation of 1 with tRNA solution ([complex]/[DNA] = 1) a major increase in the 210 and 270 nm bands was observed while the positive bands at 221 and 240 nm demonstrated relatively less amplification of their bands with no prominent band shifting. Furthermore, the prominent alterations in the intensity of the ellipticity band at 270 nm could be due to reduction in base stacking interactions upon addition of complex 1, which is typical of the A-form RNA conformation. Moreover, the absence of any shift in the CD bands at 210 and 270 nm confirmed that tRNA still remains in the A-conformation.50,51
The binding of complex 1 with ct-DNA was further studied by measuring the changes in the DNA melting temperature (Tm) that characterize the transition from a double-stranded DNA to a single-stranded one. The observed Tm value is the temperature where 50% of double-stranded DNA denatures into single-stranded DNA, and was calculated from the mid-point of the DNA melting curve (λmax = 260 nm). It is documented that groove binding or electrostatic interactions with DNA reveal a smaller change in Tm value, while intercalation leads to a significant increase of the same due to the stabilization of the DNA duplex.52 Our experiments demonstrated a small change in the ΔTm value of 3 °C thereby suggesting a groove binding mode of interaction between the complex and ct-DNA (Fig. 10).53
 | ||
| Fig. 10 Thermal denaturation melting curves of free DNA (black) and DNA in the presence of complex 1 (red). | ||
Topoisomerase I inhibitory assay
Topoisomerase enzymes (Topo I and II) are essential for cellular events viz., DNA replication, recombination, chromosome segregation and gene transcription.54 Topo I initiates a nucleophilic attack of the hydroxyl group of an active tyrosine residue on a phosphodiester linkage in DNA, thereby displacing the 5′-end of DNA and gets covalently attached to the 3′-end of DNA, thus forming a ‘cleavage complex’.55 The religation process occurs faster than cleavage so the equilibrium favors the uncleaved DNA. However, in the presence of Topo I inhibitors that act by stabilizing the cleavable Topo I–DNA complex, Topo I can become stalled in the DNA cleavage complex, and thereby the rate of religation is inhibited or reduced. A standard cleavage assay was employed to investigate the effects of complex 1 on Topo I activity by gel electrophoresis (Fig. 11). If Topo I catalytic activity is intact there will be the disappearance of SC DNA (fastest band) and the appearance of bands for NOC DNA (slowest band) and R DNA (migrates between SC and NOC band). In contrast, no R or NOC DNA bands are observed if the activity of the enzyme is inhibited.56 It was observed that supercoiled DNA was fully relaxed with the Topo I enzyme in the absence of complex 1 (Lane 2). However, upon increasing the concentration of complex 1 (5–25 μM), the levels of relaxed form were inhibited (Lanes 3–6) and the relaxation effect caused by Topo I was totally inhibited at a concentration of 25 μM (Lane 7). Interestingly, the Topo I inhibitory activity of complex 1 was significantly higher than some of the classical Topo I inhibitors used as antitumor drugs in the clinic.57 Topo I inhibition activity by 1 can be attributed to either its direct interference with the enzyme (stabilization of Topo–DNA cleavable complexes) or its strong binding with DNA, which also prevents the relaxation process by the enzyme. | ||
| Fig. 11 Agarose gel electrophoresis showing effect of different concentrations of complex 1 on the activity of DNA topoisomerase I (Topo I); Lane 1, DNA control; Lane 2, Topo I + DNA; Lane 3, 5 μM of 1 + DNA + Topo I; Lane 4, 10 μM of 1 + DNA + Topo I; Lane 5, 15 μM of 1 + DNA + Topo I; Lane 6, 20 μM of 1 + DNA + Topo I; Lane 7, 25 μM of 1 + DNA + Topo I. | ||
Molecular docking studies
Molecular docking studies have been instrumental in the design and synthesis of drug molecules that selectively target nucleic acids and proteins. Complex 1 was shown to fit inside the curved contour of the narrow and slightly deeper GC-rich sequences of the minor groove and was found in close proximity to C-3, G-4, C-21, G-2, G-22, C-24, G-24 and A-5 DNA base pairs (Fig. S4, ESI†).The closer proximity of the complex in the minor groove allows more contact surface area for the complex to bind strongly and facilitate the intermolecular H-bond formation between the O atoms (H-bonding acceptor groups) of complex 1 and H atoms of N2 (H-bonding donor groups) of C21 and G4 DNA base pairs; DC21:N5⋯O2 of complex 1 (pipzH+) = 3.0245 Å; DG4:N2⋯O3 of complex 1 = 2.99 Å and DG4:N2⋯O1 of complex 1 (naI−) = 3.06 Å. Additional stabilization interactions could be offered by the van der Waals and hydrophobic contacts of the naI− ring skeleton with the DNA functional groups inside the groove.
Yeast tRNA has well defined 3D structures including regions like the D arm, acceptor stem, T arm, ψ loop and anticodon arm which offer specific target sites for small molecules during the specific recognition process.58 The resulting docked model illustrated in Fig. 12 implied that 1 was inserted into the active pocket of the anticodon arm in close proximity to C-32, A-31, A-36, A-38, G-30, C-40, U-41, C-28, C-27 and A-29.
 | ||
| Fig. 12 Molecular docked model of complex 1 located inside the active pocket of the anticodon arm of yeast tRNA shown inside the full-length structure of yeast tRNAPhe (PDB ID: 6TNA). | ||
The optimal binding conformation of complex 1 inside the RNA target offers various stablization interactions viz., intermolecular H-bonds, van der Waals etc. H-bond interactions were observed between the O atoms of a H-bonding acceptor group of the complex (naI− ligand) and the H (H-bonding donor group) atom of G30, such that G30:O2′⋯O2 of complex 1 = 2.991 Å and C27:O2′⋯O2 of complex 1 = 2.962 Å. The resulting binding energies calculated for the best docked pose of 1 with the DNA and yeast tRNA targets were found to be −263 and −281 kJ mol−1, respectively which correlated well with the results of in vitro DNA and RNA binding studies.
Further validation of Topo I inhibitory activity of the complex was ascertained by carrying out docking study experiments of 1 with the active-site of human-DNA–Topo I complex (PDB ID1SC7).59 Speculations regarding the detailed binding pattern of complex 1 with Topo I were made for the best docked conformation which revealed that 1 approached towards the DNA cleavage site of the enzyme involving significant interactions in the AT region viz., A15, A16 A17 (+1) and C111, T110, T109 (−1) in the minor groove of the scissile strand (Fig. 13).
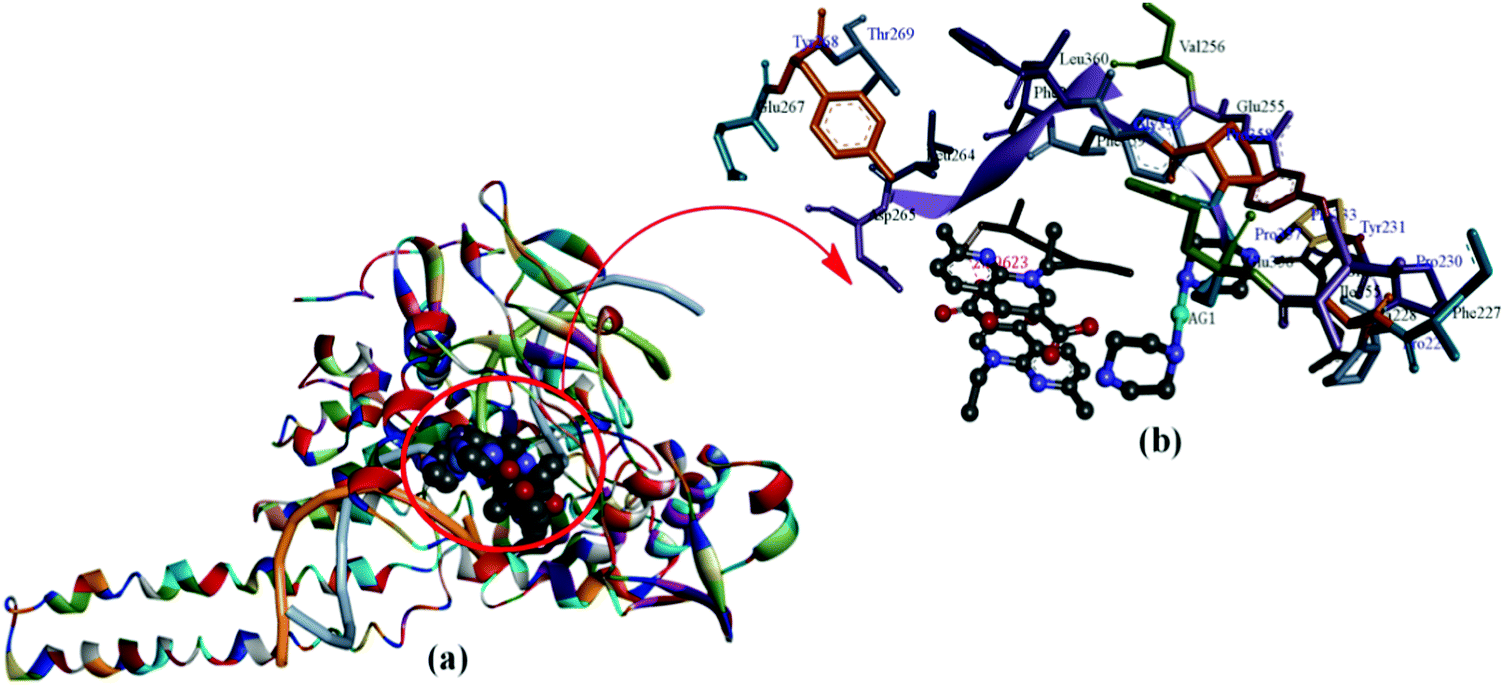 | ||
| Fig. 13 (a) Molecular docked model of complex 1 with human-DNA–Topo-I (PDB ID: 1SC7). (b) Occupying the chemically significant site, adjacent to G–C base pairs of the minor groove of the DNA along with its H-bonding interaction(s). | ||
The insertion of 1 between the DNA base pairs at the cleavage site was followed by an increase in the physical distance between the cleaved DNA termini which resulted in a consequent inhibition of the religation step of Topo I. Moreover, a carbonyl O atom of a nalidixate moiety acts as a hydrogen bond acceptor and forms a H-bond with the Lys 262 residue (2.296 Å). These interactions are quite significant with complex 1 at the DNA–Topo I active site, as it forms the basis for the inhibitory effect on Topo I. The resulting calculated binding energy of Topo I docked complex 1 was found to be −288 kJ mol−1.
Cytotoxic activity
The in vitro cytotoxic activity of complex 1 was evaluated in terms of GI50 (concentration of drug that produces 50% inhibition of the cells), TGI (concentration of the drug that produces total inhibition of the cells) and LC50 (concentration of the drug that kills 50% of the cells) values by semi-automated sulforhodamine-B (SRB) assay, against a panel of human carcinoma cell lines, viz., Hop62 (lung), HeLa (cervix), MIA-PA-CA-2 (pancreas) and MCF7 (breast) (Fig. 14). | ||
| Fig. 14 Growth curve showing % control growth vs. drug concentration (μg mL−1) against different human carcinoma cell lines: Hep-G2 (liver), HCT15 (colon), MIA-PA-CA-2 (pancreas), HeLa (cervix) and MCF-7 (breast). | ||
Interestingly, our results revealed a selective preference of 1 towards the pancreatic cancer cell line with GI50 values <10 μg mL−1. Thus, complex 1 acts as a potent selective antitumor agent with pronounced GI50 value evidenced from antitumor screening data against different human cancer cell lines (Table 3).
| Cell line | Hep-G2 | MIA-PA-CA-2 | HeLA | MCF7 | |
|---|---|---|---|---|---|
| GI50 = growth inhibition of 50% (GI50) calculated from [(Ti–Tz)/(C–Tz)] × 100 = 50, drug concentration that results in a 50% reduction in the net protein increase. ADR = Adriamycin (taken as a positive control compound). TGI = tumor growth inhibition. LC50 = lethal concentration of 50% (LC50). | |||||
| GI50 | Complex 1 | >80 | <10 | >80 | 35.0 |
| ADR | <10 | <10 | <10 | <10 | |
| TGI | Complex 1 | >80 | 22.4 | >80 | 57.0 |
| ADR | <10 | <10 | <10 | 39.0 | |
| LC50 | Complex 1 | >80 | 72.3 | >80 | 49.2 |
| ADR | <10 | <10 | 27.1 | 80.0 | |
Conclusions
A novel tailored RNA targeted quinolone drug derivative, silver(I) nalidixic acid–piperazinium molecular entity (1) was synthesized, and characterized by various spectroscopic techniques (FT-IR, NMR, ESI-MS) and single X-ray diffraction crystallography. The DFT studies of complex 1 revealed that the HOMO and LUMO were localized particularly on the metal center. The HOMO–LUMO gap of complex 1 was calculated to be 1.671 eV and the frontier molecular orbital energies of the complex, EHOMO and ELUMO were found to be −3.584 and −1.913 eV, respectively. Time dependent DFT calculations were also performed to get better insight into the electronic interactions of complex 1 and simulate the spectral assignments which further complemented our spectroscopic results. Non covalent interaction analysis was carried out to identify the weak non-covalent interactions from the topological analysis and its reduced gradient of the electron density which further revealed accumulation of electronic density in between the naI− and [Ag(pip)2]2+ units, thereby providing evidence of existing synergistic interactions between the two moieties. Analysis of Hirshfeld surfaces and fingerprint plots were carried out to ascertain a comparison between intermolecular interactions that involve N–H⋯O, O–H⋯O and CH⋯O linkages into a two-dimensional framework. In vitro comparative binding studies of 1 with ct-DNA and yeast tRNA were carried out by various biophysical methods and on the basis of intrinsic binding constant Kb and binding constant K values, 1 revealed higher binding propensity towards RNA as compared to DNA and the binding mode was found to be groove binding. Topoisomerase I enzyme inhibitory activity of complex 1 was ascertained by gel electrophoretic assay in a concentration dependent manner which showed significant inhibitory effects of 1 at a low concentration of 25 μM. In vitro cytotoxic activity on a panel of four human cancer cell lines was determined, which revealed selective activity specifically against the MIA-PA-CA-2 (pancreatic) cancer cell line with GI50 value <10. Thus complex 1 acts as a potential water soluble cytotoxic agent which warrants further thorough in vivo anticancer investigation.Experimental
General methods and instrumentation
Commercially obtained chemicals were used as received without further purification, including nalidixic acid (Sigma Aldrich), AgNO3, and tris-(hydroxymethyl)aminomethane (tris-buffer) (Merck). Calf thymus DNA (ct-DNA) and yeast tRNA (Type IX from Torula yeast) were purchased from Sigma Co. and were stored at 4 °C, and 6× loading dye (Fermental Life Science), supercoiled plasmid DNA pBR322 (Genei) and human topoisomerase I enzyme (CalBioChem) were used.Elemental analysis was carried out on aCarlo Erba Analyser Model 1106. Fourier-transform infrared (FT-IR) spectra were recorded on Spectrum Two (Pelkin Elmer) IR spectrometers. ESI-MS spectra were recorded on a Micromass Quattro II triple quadrupole mass spectrometer. NMR (1H and 13C) spectra were obtained on a Bruker DRX-400 spectrometer with Me2SO-d6 as a solvent. Electronic spectra were recorded on a Perkin-Elmer Lambda 25 using cuvettes of 1 cm path length and data were reported in λmax/nm. Emission spectra were acquired on a Shimadzu RF–5301PC Spectrofluorometer. All the experiments involving the interaction of the complex with ct-DNA and the yeast tRNA were carried out in aerated buffer (5 mM Tris–HCl, 50 mM NaCl, pH = 7.2). The concentration per base pair for both DNA and RNA was determined spectrophotometrically by assuming the ε260 values to be 6600 and 7700 M−1 cm−1 respectively.60,61 The viscosity measurements were carried out using an Oswald capillary viscometer maintained at 25 °C. CD spectra were measured on a Jasco J-815-CD spectropolarimeter at room temperature.
M.p. 273 °C. FT IR (KBr, νmax/cm−1): 3450 ν(H2O); 3127 ν(N–H); 2922 ν(–CH2); 1718 νasym(CO); 445 ν(Ag–N). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.99 (s, 1H, H2), 8.44 (d, 1H, H5), 7.33 (d, 1H, H6), 4.03 (s, 2H, NH2, pipzH+), 3.79 (q, 2H, NCH2CH3, 16H, CH2, pipzH+), 2.55 (s, 3H, ArCH3). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 186.71 (CO), 177.22 (COO−), 162.73 (C-7), 149.50 (C-2), 147.60 (C-10), 135.86 (C-5), 120.79 (C-6), 119.42 (C-3), 57.91 (−CH2 of pipzH+ C-13), 49.25 (C-12), 15.96 (C-11 and C-13). CCDC: 1547115.†
| Parameter | Complex 1 |
|---|---|
| Empirical formula | C32H60N9O17Ag |
| Formula wt. | 950.76 |
| Crystal system | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a, Å | 7.607(5) |
| b, Å | 11.845(5) |
| c, Å | 12.098(5) |
| α (°) | 85.314(5) |
| β (°) | 76.322(5) |
| γ (°) | 84.973(5) |
| U, Å3 | 1053.0(9) |
| Z | 1 |
| ρ calc, Mg m−3 | 1.499 |
| μ, mm−1 | 0.560 |
| Temperature (K) | 293(2) |
| Reflections collected | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 077 077 |
| Independent reflections | 3971 [Rint = 0.0368, Rsigma = 0.0424] |
| Data/restraints/parameters | 3971/0/348 |
| Goodness-of-fit on F2 | 1.070 |
| Final R indexes [I ≥ 2σ (I)] | R 1 = 0.0294, wR2 = 0.0701 |
| Final R indexes [all data] | R 1 = 0.0319, wR2 = 0.0713 |
| Largest diff. peak/hole/e Å−3 | 0.45/−0.39 |
Density functional theory
Computational methodology
The molecular structure used in the theoretical studies of complex 1 was taken from the X-ray diffraction results. Keeping all distances, angles and dihedral angles frozen, single-point DFT calculations were performed on the model. The single-point ground-state calculations were carried out using the hybrid B3LYP functional as implemented in ORCA.69,70 The present calculations were performed using the additional approximation that the coulomb integrals are approximated by the sum of atom centered s, p, d… functions, of the auxiliary (or fitting) basis set.71 This allows for efficient treatment of the coulomb interactions, and hence reduces the time for given calculations. The Def2-TZVP main, and Def2-TZVP/J auxiliary, basis set were used.72 The main basis set is of [5s3p2d] quality for Ag, [5s2p1d] for C, N, O and [2s] for H. In addition, the 28 electron relativistic effective core potential (ECP) of Andrae et al. was used for silver in the whole calculations and was obtained from the Turbo mole basis set library: http://ftp.chemie.uni-karlsruhe.de/pub/basen/.73 The structure was further calculated using the densities obtained from the def2-TZVP basis to calculate the electronic density for NCI-index analysis.37,38 The Hirshfeld surface was mapped using Crystal Explorer74 using crystal structure coordinates of CIF files.In vitro DNA/RNA binding
DNA/RNA binding experiments including absorption spectral traces, emission spectroscopy, viscosity measurements and CD conformed to the standard methods and practices previously adopted by our laboratory.75–77 While measuring the absorption spectra an equal amount of DNA/RNA was added to both the compound solution and the reference solution to eliminate the absorbance of the ct-DNA/tRNA itself and tris buffer was subtracted through base line correction. DNA melting experiments were carried out by monitoring the absorption (260 nm) of ct-DNA (1 × 10−4 M) at various temperatures in the absence and presence of complexes (5 × 10−5 M) in buffer (50 mM tris–HCl/50 mM NaCl, pH 7.4). The melting temperature, Tm (temperature at which 50% of double-stranded DNA denatures into single-stranded DNA) was calculated by plotting the temperature vs. relative absorption intensity (A/A0), and measurements were carried out using a Perkin-Elmer Lambda 850 spectrophotometer equipped with a Peltier temperature controlling programmer.Topoisomerase I inhibition assay
One unit of the enzyme was defined as the amount of enzyme required to completely relax 1 μg of negatively supercoiled pBR322 DNA in 30 min at 310 K under the standard assay conditions. The reaction mixture (30 μL) contained 35 mM Tris–HCl (pH 8.0), 72 mM KCl, 5 mM MgCl2, 5 mM DTT, 2 mM spermidine, 0.1 mg mL−1 BSA, 0.25 μg pBR322 DNA, 2 Unit Topo-I and complex 1. These reaction mixtures were incubated at 310 K for 30 min, and the reaction was terminated by addition of 4 μL of 5× buffer solution consisting of 0.25% bromophenol blue, 4.5% SDS and 45% glycerol. The samples were electrophoresed through 1% agarose in TAE at 30 V for 8 h.Molecular docking studies
Molecular docking studies were performed by using HEX 8.0 software78 which is an interactive molecular graphics program for calculating and displaying feasible docking modes of proteins and DNA molecules. The crystal structure of the B-DNA dodecamer d(CGCGAATTCGCG)2 (PDB ID: 1BNA), RNA (PDB ID: 6TNA) and human-DNA-Topo I (PDB ID: 1SC7) were downloaded from the protein data bank (http://www.rcsb.org./pdb). Human-DNA–Topo I was retrieved from the Protein Data Bank in which Topo I is bound to the oligonucleotide sequence 5′-AAAAAGACTTsX-GAAAATTTTT-3′, where ‘s’ is the 5′-bridging phosphorothiolate of the cleaved strand and ‘X’ represents any of the four bases A, G, C or T. The phosphodiester bond of G12 in 1SC7 was rebuilt and SH of G11 on the scissile strand was changed to OH. Visualization of the docked pose has been carried out by using CHIMERA (http://www.cgl.ucsf.edu/chimera), PyMol (http://pymol.sourceforge.net/) and Discovery Studio molecular graphics program.Cytotoxic studies
The in vitro antitumor screening of 1 was carried out on the following human cancer lines of different histological origin viz., Hop62 (lung), HeLa (cervix), MIA-PA-CA-2 (pancreas) and MCF7 (breast). Human malignant cell lines were procured and grown in RPMI–1640 medium supplemented with 10% Fetal Bovine Serum (FBS) and antibiotics to study the growth pattern of these cells. The proliferation of the cells upon treatment with chemotherapy was determined by means of the sulphorhodamine-B (SRB) semi-automated assay.79 All cell lines were seeded into 96 well plates and the cells were counted and the cell count was adjusted according to the titration readings so as to give optical density in the linear range (0.5 to 1.8) and then the cells were incubated at 37 °C in a CO2 incubator for 24 h. The stock solution of the complexes were prepared as 100 mg mL−1 in DMSO and four dilutions i.e. 10 μg mL−1, 20 μg mL−1, 40 μg mL−1, and 80 μg mL−1, in triplicate were tested, each well receiving 90 μL of cell suspension. The plates were labeled properly and were incubated for 48 h. Termination of the experiment was carried out by gently layering the cells with 50 μL of chilled 30% TCA (in the case of adherent cells) and 50% TCA (in the case of suspension cell lines) for cell fixation and kept at 4 °C for 1 h. The plates were stained with 50 μL of 0.4% SRB for 20 min. All experiments were performed in triplicate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to SAIF and CIL Panjab University, Chandigarh for providing NMR and ESI-MS and elemental analysis facilities respectively. The authors are grateful to ACTREC, Mumbai for carrying out the cytotoxicity studies. The authors also acknowledge the generous financial support from DST-PURSE program and DRS-1 (SAP) from UGC, New Delhi.References
- (a) I. Garrido-Laguna and M. Hidalgo, Nat. Rev. Clin. Oncol., 2015, 12, 319–334 CrossRef CAS PubMed; (b) A. Vincent, J. Herman, R. Schulick, R. H. Hruban and M. Goggins, Lancet, 2011, 378, 607–620 CrossRef PubMed.
- (a) L. Zhu, C. Staley, D. Kooby, B. El-Rays, H. Mao and L. Yang, Cancer Lett., 2017, 388, 139 CrossRef CAS PubMed; (b) Z. Wang, Y. Li, A. Ahmad, S. Banerjee, A. S. Azmi, D. Kong and F. H. Sarkar, Nat. Rev. Gastroenterol. Hepatol., 2011, 8, 27–33 CrossRef CAS PubMed.
- M. Ilic and I. Ilic, World J. Gastroenterol., 2016, 22(44), 9694–9705 CrossRef PubMed.
- S. Raimondi, P. Maisonneuve and A. B. Lowenfels, Nat. Rev. Gastroenterol. Hepatol., 2009, 6, 699–708 CrossRef PubMed.
- D. Yadav and A. B. Lowenfels, Gastroenterology, 2013, 144, 1252–1261 CrossRef PubMed.
- K. K. Frese, A. Neesse, N. Cook, T. E. Bapiro, M. P. Lolkema, D. Jodrell and D. A. Tuveson, Cancer Discovery, 2012, 2, 260–269 CrossRef CAS PubMed.
- A. Stathis and M. J. Moore, Nat. Rev. Clin. Oncol., 2010, 7, 163–172 CrossRef CAS PubMed.
- H. Oettle, H. Riess, J. M. Stieler, G. Heil, I. Schwaner, J. Seraphin, M. Görner, M. Mölle, T. F. Greten, V. Lakner, S. Bischoff, M. Sinn, B. Dörken and U. Pelzer, J. Clin. Oncol., 2014, 32, 2423–2429 CrossRef CAS PubMed.
- A. Scott Paulson, H. S. T. Cao, M. A. Tempero and A. M. Lowy, Gastroenterology, 2013, 144, 1316–1326 CrossRef PubMed.
- (a) B. Rosenberg, L. V. Camp and T. Krigas, Nature, 1969, 205, 698–699 CrossRef; (b) S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 23–24, 1089–1097 CrossRef PubMed; (c) L. Ronconi and D. Fregona, Dalton Trans., 2009, 10670–10680 RSC.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- M. Wehbe, A. W. Y. Leung, M. J. Abrams, C. Orvig and M. B. Bally, Dalton Trans., 2017, 46, 10758–10773 RSC.
- S. S. Al-Jaroudi, M. Altaf, A. A. Seliman, S. Yadav, F. Arjmand, A. Alhoshani, H. M. Korashy, S. Ahmad and A. A. Isab, Inorg. Chim. Acta, 2017, 464, 37–48 CrossRef CAS.
- I. Yousuf, F. Arjmand, S. Tabassum, L. Toupet, R. A. Khan and M. A. Siddiqui, Dalton Trans., 2015, 44, 10330–10342 RSC.
- (a) G. McDonnell and A. D. Russell, Clin. Microbiol. Rev., 1999, 12, 147–179 CAS; (b) H. J. Klasen, Burns, 2000, 26, 131–138 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- (a) B. S. Atiyeh, M. Costagliola, S. N. Hayek and S. A. Dibo, Burns, 2007, 33, 139–148 CrossRef PubMed; (b) S. Silver, L. T. Phung and G. Silver, Ind. Microbiol. Biotechnol., 2006, 33, 627–634 CrossRef CAS PubMed.
- A. B. Lansdown, Curr. Probl. Dermatol., 2006, 33, 17–34 CAS.
- A. B. G. Lansdown, Silver in Healthcare: its Antimicrobial Efficacy and Safety in Use, 6, Royal Society of Chemistry Publishing, Cambridge, 2010 Search PubMed.
- C. N. Banti, A. D. Giannoulis, N. Kourkoumelis, A. M. Owczarzak, M. Kubicki and S. K. Hadjikakou, J. Inorg. Biochem., 2015, 142, 132–144 CrossRef CAS PubMed.
- (a) C. N. Banti and S. K. Hadjikakou, Metallomics, 2013, 5, 569–596 RSC; (b) E. Fourie, E. Erasmus, J. C. Swarts, A. Tuchscherer, A. Jakob, H. Lang, G. K. Joone and C. E. J. Van Rensburg, Anticancer Res., 2012, 32, 519–522 CAS.
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042–15053 CrossRef CAS PubMed.
- C. Pettinari, F. Marchetti, G. Lupidi, L. Quassinti, M. Bramucci, D. Petrelli, L. A. Vitali, M. Fatima, C. Guedes da Silva, L. M. D. R. S. Martins, P. Smolenski and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 11173–11183 CrossRef CAS PubMed.
- (a) H.-L. Zhu, X.-M. Zhang, X.-Y. Liu, X.-J. Wang, G.-F. Liu, A. Usman and H.-K. Fun, Inorg. Chem. Commun., 2003, 6, 1113–1116 CrossRef CAS; (b) C. Santini, M. Pellei, G. Papini, B. Morresi, R. Galassi, S. Ricci, F. Tisato, M. Porchia, M. P. Rigobello and V. Gandin, J. Inorg. Biochem., 2011, 105, 232–240 CrossRef CAS PubMed; (c) M. J. McKeage, P. Papathanasiou, G. Salem, A. Sjaarda, G. F. Swiegers, P. Waring and S. B. Wild, Met.-Based Drugs, 1998, 5, 217–223 CrossRef CAS PubMed.
- (a) H. Xu, Y. Liang, P. Zhang, F. Du, B.-R. Zhou, J. Wu, J.-H. Liu, Z.-G. Liu and L.-N. Ji, J. Biol. Inorg. Chem., 2005, 10, 529–538 CrossRef CAS PubMed; (b) M. Cusumano, M. Letizia, D. Pietro, A. Giannetto and P. A. Vainiglia, Inorg. Chem., 2007, 46, 7148–7153 CrossRef CAS PubMed.
- (a) L. A. Mitscher, Chem. Rev., 2005, 105, 559–592 CrossRef CAS PubMed; (b) L. L. Shen, J. Baranowski and A. G. Pernet, Biochemistry, 1989, 28, 3879–3885 CrossRef CAS PubMed; (c) M. Gellert, K. Mizuuchi, M. H. O'dea, T. Itoh and J.-I. Tomizawa, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4772–4776 CrossRef CAS PubMed.
- (a) F. M. Deane, E. C. O’Sullivan, A. R. Maguire, J. Gilbert, J. A. Sakoff, A. McCluskey and F. O. McCarthy, Org. Biomol. Chem., 2013, 11, 1334–1344 RSC; (b) Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- (a) P. Katkar, A. Coletta, S. Castelli, G. L. Sabino, R. A. A. Couto, A. M. da Costa Ferreira and A. Desideri, Metallomics, 2014, 6, 117–125 RSC; (b) T. X. Nguyen, A. Morrell, M. Conda-Sheridan, C. Marchand, K. Agama, A. Bermingam, A. G. Stephen, A. Chergui, A. Naumova, R. Fisher, B. R. O’Keefe, Y. Pommier and M. Cushman, J. Med. Chem., 2012, 55, 4457–4478 CrossRef CAS PubMed.
- M. Yarim, M. Koksa, I. Durmaz and R. Atalay, Int. J. Mol. Sci., 2012, 13, 8071–8085 CrossRef CAS PubMed.
- A. Chopra, A. Anderson and C. Giardina, J. Biol. Chem., 2014, 289, 2978–2991 CrossRef CAS PubMed.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039–9052 CrossRef CAS PubMed.
- H. Aghabozorg, E. Motieiyan, A. R. Salimi, M. Mirzaei, F. Manteghi, A. Shokrollahi, S. Derki, M. Ghadermazi, S. Sheshmani and H. Eshtiagh-Hosseini, Polyhedron, 2010, 29, 1453–1464 CrossRef CAS.
- G. Mendoza-Diaz and J. Ireta-Moreno, J. Inorg. Biochem., 1994, 54, 235–246 CrossRef CAS.
- (a) J. Aihara, Phys. Chem. Chem. Phys., 2000, 2, 3121–3125 RSC; (b) R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS; (c) A. A. G. Moreira, P. D. Lima-Neto, E. W. S. Caetano, I. L. Barroso-Netoa and V. N. Freire, New J. Chem., 2016, 40, 10353–10362 RSC.
- M. Usman, F. Arjmand, M. Ahmad, M. S. Khan, I. Ahmad and S. Tabassum, Inorg. Chim. Acta, 2016, 453, 193–201 CrossRef CAS.
- R. A. Khan, M. Usman, R. Dhivya, P. Balaji, A. Alsalme, H. Al Lohedan, F. Arjmand, K. Al Farhan, M. A. Akbarsha, F. Marchetti, C. Pettinari and S. Tabassum, Sci. Rep., 2017, 7, 45229, DOI:10.1038/srep45229.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-Garcıa, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- (a) J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef CAS PubMed; (b) M. Usman, F. Arjmand, R. A. Khan, A. Alsalme, M. Ahmad and S. Tabassum, RSC Adv., 2017, 7, 47920–47932 RSC.
- R. Chaudret, B. de Courcy, J. Contreras-Garcia, E. Gloaguen, A. Zehnacker-Rentien, M. Mons and J.-P. Piquemal, Phys. Chem. Chem. Phys., 2014, 16, 9876–9891 RSC.
- (a) S. Tsuzuki and A. Fujii, Phys. Chem. Chem. Phys., 2008, 10, 2584–2594 RSC; (b) M. P. D. Hatfield, N. Y. Palermo, J. Csontos, R. F. Murphy and S. Lovas, J. Phys. Chem. B, 2008, 112, 3503–3508 CrossRef CAS PubMed.
- J. A. Harrison, M. A. Sajjad, P. Schwerdtfeger and A. J. Nielson, Cryst. Growth Des., 2016, 16, 4934–4942 CAS.
- N. Han, Y. Zeng, X. Li, S. Zheng and L. Meng, J. Phys. Chem. A, 2013, 117, 12959–12968 CrossRef CAS PubMed.
- S. K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar and S. Mukhopadhyay, Cryst. Growth Des., 2011, 11, 3250–3265 CAS.
- H.-Y. Zhang, W. Wang, H. Chen, S.-H. Zhang and Y. Li, Inorg. Chim. Acta, 2016, 453, 507–515 CrossRef CAS.
- C. N. Banti, A. D. Giannoulis, N. Kourkoumelis, A. M. Owczarzak, M. Poyraz, M. Kubicki, K. Charalabopoulos and S. K. Hadjikakou, Metallomics, 2012, 4, 545–560 RSC.
- A. Wolfe, G. H. Shimer, Jr. and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- E. F. Healy, J. Chem. Educ., 2007, 84, 1304–1307 CrossRef CAS.
- R. Loganathan, S. Ramakrishnan, E. Suresh, M. Palaniandavar, A. Riyasdeen and M. A. Akbarsha, Dalton Trans., 2014, 43, 6177–6194 RSC.
- (a) P. U. Maheswari and M. Palaniandavar, J. Inorg. Biochem., 2004, 98, 219–230 CrossRef; (b) F. Bisceglie, S. Pinelli, R. Alinovi, M. Goldoni, A. Mutt, A. Camerini, L. Piola, P. Tarasconi and G. Pelosi, J. Inorg. Biochem., 2014, 140, 111–125 CrossRef CAS PubMed.
- (a) J. Kypr and M. Vorlickova, Biopolymers, 2002, 67, 275–277 CrossRef PubMed; (b) M. Vorlickova, Biophys. J., 1995, 69, 2033–2043 CrossRef CAS PubMed.
- (a) C. N. N. Soukpoe-Kossi, C. Descoteaux, E. Asselin, J. Bariyanga, H.-A. Tajmir-Riahi and G. Berube, DNA Cell Biol., 2008, 27, 337–343 CrossRef PubMed; (b) E. Froehlich, J. S. Mandeville, D. Arnold, L. Kreplak and H. A. Tajmir-Riahi, Biomacromolecules, 2012, 13, 282–287 CrossRef CAS PubMed.
- S. Bhattacharyya, A. Sarkar, S. K. Dey, G. P. Jose, A. Mukherjee and T. K. Sengupta, Dalton Trans., 2013, 42, 11709–11719 RSC.
- A. Ghosh, A. Mandoli, D. K. Kumar, N. S. Yadav, T. Ghosh, B. Jha, J. A. Thomas and A. Das, Dalton Trans., 2009, 9312–9321 RSC.
- F. M. Deane, E. C. O’Sullivan, A. R. Maguire, J. Gilbert, J. A. Sakoff, A. McCluskey and F. O. McCarthy, Org. Biomol. Chem., 2013, 11, 1334–1344 CAS.
- P. Katkar, A. Coletta, S. Castelli, G. L. Sabino, R. A. Alves Couto, A. M. da Costa Ferreira and A. Desideri, Metallomics, 2014, 6, 117–125 RSC.
- T. X. Nguyen, A. Morrell, M. Conda-Sheridan, C. Marchand, K. Agama, A. Bermingam, A. G. Stephen, A. Chergui, A. Naumova, R. Fisher, B. R. O’Keefe, Y. Pommier and M. Cushman, J. Med. Chem., 2012, 55, 4457–4478 CrossRef CAS PubMed.
- (a) K. Suzuki, F. Shono and M. Uyeda, Biosci., Biotechnol., Biochem., 1998, 62, 2073–2075 CrossRef CAS; (b) K. Suzuki and M. Uyeda, Biosci., Biotechnol., Biochem., 2002, 66, 1706–1712 CrossRef CAS; (c) J. M. Fortune, L. Velea, D. E. Graves, T. Utsugi, Y. Yamada and N. Osheroff, Biochemistry, 1999, 38, 15580–15586 CrossRef CAS PubMed; (d) F. Gao, H. Chao, J. Q. Wang, Y. X. Yuan, B. Sun, Y. F. Wei, B. Peng and L. N. Ji, J. Biol. Inorg. Chem., 2007, 12, 1015–1027 CrossRef CAS PubMed.
- D. Agudelo, P. Bourassa, M. Beauregard, G. Berube and H.-A. Tajmir-Riahi, PLoS One, 2013, 8, e69248 CAS.
- H. T. M. Van and W.-J. Cho, Bioorg. Med. Chem. Lett., 2009, 19, 2551–2554 CrossRef PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- K. A. Meadows, F. Liu, J. Sou, B. P. Hudson and D. R. McMillin, Inorg. Chem., 1993, 32, 2919–2923 CrossRef CAS.
- International Tables for X-ray Crystallography, Kynoch Press, Birmingham, England, 1970, vol. III, p. 257 Search PubMed.
- SMART & SAINT Software Reference manuals, Version 6.45, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2003.
- G. M. Sheldrick, SADABS, software for empirical absorption correction, Ver. 2.05, University of Göttingen, Göttingen, Germany, 2002 Search PubMed.
- XPREP, version 5.1, Siemens Industrial Automation Inc., Madison, WI, 1995.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, J. Appl. Crystallogr., 2007, 40, 609–613 CrossRef CAS.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS; (b) F. Neese, Orca. An ab Initio, Density Functional and Semiempirical Program Package version, 2009; (c) F. Neese, Orca. An ab Initio, Density Functional and Semiempirical Program Package version, 2009; (d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- (a) M. Dennehya, R. M. Ferullo, O. V. Quinzani, S. D. Mandolesi, N. Castellani and M. Jennings, Polyhedron, 2008, 27, 2243–2250 CrossRef; (b) V. T. Yilmaz, S. Hamamci and S. Gumus, Chem. Phys. Lett., 2006, 425, 361–366 CrossRef CAS; (c) B. L. Schottel, H. T. Chifotides, M. Shatruk, A. Chouai, L. M. Perez, J. Bacsa and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 5895–5912 CrossRef CAS PubMed; (d) C.-S. Liu, P.-Q. Chen, E.-C. Yang, J.-L. Tian, X.-H. Bu, Z.-M. Li, H.-W. Sun and Z. Lin, Inorg. Chem., 2006, 45, 5812–5821 CrossRef CAS PubMed.
- A. Schaefer, H. Horn and R. J. Ahlrichs, Chem. Phys., 1992, 97, 2571–2577 CAS.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS; (c) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- (a) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. R. Lakowicz and G. Webber, Biochemistry, 1973, 12, 4161–4170 CrossRef CAS PubMed.
- A. Wolfe, G. H. Shimer Jr. and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- F. Arjmand, I. Yousuf, Y. Zaidi and L. Toupet, RSC Adv., 2015, 5, 16250–16264 RSC.
- (a) D. Mustard and D. W. Ritchie, Proteins: Struct., Funct., Bioinf., 2005, 60, 269–274 CrossRef CAS PubMed; (b) G. Macindoe, L. Mavridis, V. Venkatraman, M.-D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, 445–449 CrossRef PubMed.
- T. Lammers, P. Peschke, V. Ehemann, J. Debus, B. Slobodin, S. Lavi and P. Huber, Mol. Cancer, 2007, 6, 65–78 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1547115. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj03602g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |