
The effect of alkyl chain tethers on the kinetics and mechanistic behaviour of bifunctional dinuclear platinum(II) complexes bearing N,N′-dipyridylamine ligands†
Panyako Asman
Wangoli
 *a and
Grace
Kinunda
b
*a and
Grace
Kinunda
b
aSchool of Chemistry and Physics, University of KwaZulu-Natal, 3209, Scottsville, Pietermaritzburg, South Africa. E-mail: pwangoli@yahoo.com
bDepartment of Chemistry, University of Dar es Salaam, P.O. Box 35061, Dar es Salaam, Tanzania
First published on 13th November 2017
Abstract
In the current paper, we report the kinetics of bifunctional dinuclear platinum(II) complexes viz., 1,2-N,N′-di-(2,2-dipyridylamine)ethanetetraaquaplatinum(II), PtL2, 1,3-N,N′-di-(2,2-dipyridylamine)propanetetraaquaplatinum(II), PtL3, 1,4-N,N′-di-(2,2-dipyridylamine)butanetetraaquaplatinum(II), PtL4, 1,5-N,N′-di-(2,2-dipyridylamine)pentanetetraaquaplatinum(II), PtL5 and 1,6-N,N′-di-(2,2-dipyridylamine)hexanetetraaquaplatinum(II), PtL6. The substitution reactions were carried out on tetraaqua complexes with thiourea nucleophiles under pseudo-first-order conditions as a function of nucleophile concentration and temperature by stopped-flow and UV-vis spectrophotometric techniques. An experimental study was conducted with the aim of determining the influence of alkyl chains on the steric and electronic structure of dinuclear platinum(II) complexes. The reactivity of these complexes was dependent on the length of the alkyl spacer. The results obtained herein demonstrate the intriguing odd–even effects induced by the alkyl chain on the complexes. Artificial constraints imposed by the alkyl chain significantly affect their conformational structure to be either synperiplanar (syn-) or antiperiplanar (anti-) characterized by the odd and even effect. The kinetic, mechanistic and conformational behaviour was influenced by the size of the alkyl chain in accordance with odd–even alterations of the spacer. Computational modeling using density functional theory (DFT) calculations supplemented experimental findings that structural features and the reactivity pattern of these organometallic complexes are governed by both steric and electronic effects arising from the flexibility and inductive nature of the alkyl spacer. The strong σ-donicity of longer alkyl chains favours sufficient accumulation of electron density at the metal centre and stabilizes a 14-electron intermediate. The study shows the HOMO–LUMO energy (ΔE) is affected by the length of the spacer. Kinetic and DFT data indicate electron donation by the alkyl spacer. The low positive values of enthalpy of activation and significantly large negative values of entropy of activation indicate an associative mechanism of substitution.
Introduction
Although platinum based-drugs, such as cisplatin, oxaliplatin and carboplatin have become the mainstays of cancer chemotherapy, problems of resistance and side effects that include severe ototoxicity, neurotoxicity,1–5 nausea, vomiting and nephrotoxicity6,7 have limited their clinical use. This has motivated the development of other platinum anticancer drugs that are less toxic, with less cross resistance and with a broader spectrum of activity.8–10 Novel platinum(II) drugs, distinctive from cisplatin and its analogues in terms of the structure and nature of Pt–DNA adducts, are viewed to improve the biological profile of platinum anticancer agents by circumventing or complementing cisplatin specific biological processes in DNA repair. This study is in search for new multinuclear platinum(II) complexes which represent a completely new paradigm shift in the development of novel anticancer agents.11–13Multinuclear complexes contain two or more platinum centres that can bind to DNA forming long range flexible and non-directional DNA adducts that induce DNA conformational changes.14–17 This has been shown by complexes bridged by aliphatic diamines showing higher cytotoxic activity in cisplatin-resistant cell-lines.10,12,14,18–20 Complexes of this class have overcome both acquired and intrinsic resistance due to their ability to form long range DNA adducts.9–12 The amphiphilic nature of the amine linkers, coupled with charge dispersion along their carbon backbone, results in an enhanced cellular uptake of the anticancer drug. This cytotoxic potency of these anticancer agents is strictly regulated by their electronic and structural conformations.21,22 These characteristic variations are attributed to the coordinating ligands that connect platinum nuclei.23–27 The best activity for such a class of complexes has been reported for BBR3464, [{trans-PtCl(NH3)2}2{μ-trans-Pt(NH3)2(H2N(CH2)6NH2)2}]4+, (1,0,1/t,t,t), the first trinuclear bifunctional DNA binding agent to undergo phase II clinical trials. Furthermore, it is also the first and only non-cisplatin analogue introduced into the human body.28–32 Although multinuclear complexes have been reported to prevent tumour growth, their exact underlying molecular and kinetic mechanisms remain unexplored.
To provide information on the substitution mechanisms of dinuclear Pt(II) complexes and to estimate their reactivity inside the cell, determination of substitution kinetics remains vital. Furthermore, kinetic studies are of interest to shed light on the complex metabolism and predict treatment response in cancer patients.33 For the first time, the current study investigates the substitution reactions of tetraaqua dinuclear Pt(II) complexes having a [cis-Pt(N,N′)(H2O)2] molecular structure with an alkyl backbone that mimic cisplatin. This is with the view that Pt(II) complexes with cis geometry have a higher efficacy in antitumour cell lines than those with trans-geometry. To obtain these complexes, a series of 2,2′-dipyridylamine chelates appended to alkyl chain tethers of variable lengths (n = 2–6) were used. The only variable was the –(CH2)n– moiety in the spacer which allowed the effect of the alkyl chain on the behaviour of the complexes with cis-geometry to be probed.
These compounds may be useful to gain more insights into how these Pt(II) complexes interact with bio-relevant molecules and enable them to be applied as antitumour agents. Their substitution reaction with thiourea nucleophiles was investigated. This class of nucleophiles was of interest due to their role as chemoprotective agents.34–37 The nucleophiles viz. thiourea (TU), N,N′-dimethylthiourea (DMTU) and N,N,N′N′-tetramethylthiourea (TMTU) were chosen as representatives of the targets and competitors38 of platinum drugs in the cellular environment. They have strong affinity towards the platinum centre and good solubility. Also their relatively fast reactivity with Pt(II) complexes makes this set of nucleophiles convenient for preliminary experiments.39–41aScheme 1 shows the complexes investigated while a mononuclear dipyridylamine complex PtL1*, from the literature,41b was included for comparison purposes.
 | ||
| Scheme 1 Chemical structures of the investigated Pt(II) complexes. | ||
Experimental section
Materials and methods
All starting materials were of analytical grade and were obtained from commercial sources and used without further purification. The nucleophiles thiourea (TU, 99%), N,N′-dimethylthiourea (DMTU, 99%) and N,N,N′N′-tetramethylthiourea (TMTU, 98%) and the starting materials 1,2-diiodoethane, 99%, 1,3-diiodopropane, 99%, 1,4-diiodobutane, 99%, 1,5-diiodopentane, 97%, 1,6-diiodohexane, 97%, and AgClO4 99.99% were obtained from Sigma Aldrich. K2PtCl4 and 2,2′-dipyridylamine (dpa) were obtained from Strem Chemicals. De-ionized water was used in all experiments.Synthesis of the ligands
1,2-N,N′-Di-(2,2′-dipyridylamine)ethane (L2), 1,3-N,N′-di-(2,2′-dipyridylamine)propane (L3), 1,4-N,N′-di-(2,2′-dipyridylamine)butane (L4), 1,5-N,N′-di-(2,2′-dipyridylamine)pentane (L5) and 1,6-N,N′-di-(2,2′-dipyridylamine)hexane (L6) were synthesized following a reported procedure by Krebs et al.42 To a suspension of KOH (0.023 mol, 1.30 g) in 30 mL of DMSO, 2,2′-dipyridylamine (dpa) (5.00 mmol, 0.856 g) was added. The suspension was stirred at room temperature for 24 h and the respective linker, α,ω-iodoalkane (2.5 mmol), was added drop-wise. The mixture was stirred for another 5 hours and quenched with 50 mL of water thereafter. The desired products were extracted with chloroform (4 × 30 mL) and the organic layer was dried over anhydrous magnesium sulphate. The solvent was removed under reduced pressure and the product was recrystallized from a dichloromethane/hexane mixture to obtain pure solids or oil. These ligands were characterized using 1H, 13C, NMR, mass spectroscopy and elemental analysis.L2, yield: 548.3 mg, 60% (white needle crystalline powder). 1H NMR (400 MHz, CDCl3): δ 8.26 (dd, 4H), 7.60 (dd, 4H), 7.55 (td, 4H), 6.85 (dd, 4H), 1.78 (brs, 4H). 13C NMR (400 MHz, CDCl3): 154.0, 147.8, 137.8, 116.4, 111.7 and 41.0. Anal. calcd for C22H20N6: C 71.74, H 5.43, N 22.83; found: C 72.12, H 5.28, N 23.18, TOF MS ES+: m/z = [M + H]+ = 369.452.
L3, yield: 577.8 mg, 60% (yellow needle crystalline powder). 1H NMR (400 MHz, CDCl3): δ 8.24 (dd, 4H), 7.58 (dd, 4H), 7.54 (t, 4H), 6.83 (dd, 4H), 4.19 (brs, 2H), 2.04 (brs, 4H). 13C NMR (400 MHz, CDCl3): 154.4, 148.3, 138.3, 116.9, 112.2, 41.5 and 25.8. Anal. calcd for C23H22N6: C 72.25, H 5.76, N 21.99; found: C 72.52, H 5.56, N 21.72, TOF MS ES+: m/z = 383.1987 [M + H]+.
L4, yield: 601.9 mg, 61% (yellow cubic like crystals). 1H NMR (400 MHz, CDCl3): δ 8.30 (dd, 4H), 7.48 (dd, 4H), 7.03 (t, 4H), 6.82 (dd, 4H), 4.19 (m, 4H), 1.76 (brs, 4H). 13C NMR (400 MHz, CDCl3): 157.5, 148.4, 137.1, 116.9, 114.9, 48.1 and 25.8. Anal. calcd for C24H24N6: C 72.73, H 6.06, N 21.21; found: C 73.11, H 6.40, N 21.17, TOF MS ES+: m/z = 419.1791 [M + Na]+.
L5, yield: 628.3 mg, 61% (yellow oil). 1H NMR (400 MHz, CDCl3): δ 8.18 (dd, 4H), 7.37 (dd, 4H), 6.94 (d, 4H), 6.70 (dd, 4H), 4.03 (m, 4H), 1.62 (m, 2H), 1.27 (m, 4H). 13C NMR (400 MHz, CDCl3): 157.4, 148.3, 137.1, 116.8, 114.6, 48.4, 28.1 and 24.6. Anal. calcd for C25H26N6: C 73.17, H 6.34, N 20.49; found: C 73.13, H 5.93, N 20.87, TOF MS ES+: m/z = 411.2292 [M + H]+.
L6, yield: 653.0 mg, 63% (yellow oil). 1H NMR (400 MHz, CDCl3): δ 8.13 (dd, 4H), 7.33 (dd, 4H), 6.88 (d, 4H), 6.66 (dd, 4H), 4.25 (m, 4H), 1.49 (m, 4H), 1.40 (m, 4H). 13C NMR (400 MHz, CDCl3): 157.4, 148.2, 137.0, 116.8, 114.6, 48.2, 28.1 and 26.8. Anal. calcd for C26H28N6: C 73.58, H 6.60, N 19.81; found: C 73.24, H 6.34, N 20.44, TOF MS ES+: m/z = 425.2441 [M + H]+.
Preparation of the platinum(II) complexes
The chloro forms of the Pt(II) complexes were synthesized using a general procedure reported by Krebs et al.42 To a stirred solution of K2PtCl4 (0.5 mmol, 0.2075 g) in 50 mL of water, a solution of alkyl diamine (0.25 mmol) of the bridging spacer dissolved in 10 mL of ethanol was added dropwise. The reaction mixture was then refluxed at 50 °C for 24 h. The resulting yellow precipitates were filtered off, washed with ultrapure water, ethanol and diethyl ether and dried in a vacuum.PtL2, yield: 165.5 mg, 74%. 1H NMR (400 MHz, DMSO-d6): δ 8.77 (dd, 4H), 7.97(dd, 4H), 7.29 (dd, 4H), 7.11 (ddd, 4H), 2.53 (brs, 4H). 13C NMR (400 MHz, DMSO-d6): 151.6, 151.0, 141.9, 119.6, 120.4 and 19.7. Anal. calcd for C22H20Cl4N6Pt2: C 29.33, H 2.22, N 9.33; found: C 29.64, H 2.81, N 9.15.
PtL3, yield: 159.2 mg, 70%. 1H NMR (400 MHz, DMSO-d6): δ 8.24 (dd, 4H), 7.58 (dd, 4H), 7.54 (t, 4H), 6.83 (dd, 4H), 4.19 (brs, 2H), 2.04 (brs, 4H). 13C NMR (400 MHz, DMSO-d6): 154.4, 148.3, 138.3, 116.9, 112.2, 41.5 and 25.8. Anal. calcd for C23H22Cl4N6Pt2: C 30.20, H 2.41, N 9.19; found: C 29.94, H 2.25, N 8.99.
PtL4, yield: 152 mg, 66%. 1H NMR (400 MHz, DMSO-d6): δ 8.78 (dd, 4H), 8.05 (dd, 4H), 7.51 (d, 4H), 7.24 (dd, 4H), 4.23 (brs, 4H), 1.96 (brs, 4H). 13C NMR (400 MHz, DMSO-d6): 153.9, 152.6, 142.4, 122.7, 117.9, 50.5 and 25.1. Anal. calcd for C24H24Cl4N6Pt2: C 31.03, H 2.59, N 9.05; found: C 30.90, H 2.76, N 8.18.
PtL5, yield: 156.5 mg, 66%. 1H NMR (400 MHz, DMSO-d6): δ 8.81 (dd, 4H), 8.03 (dd, 4H), 7.49 (d, 4H), 7.27 (dd, 4H), 4.16 (t, 4H), 1.91 (m, 2H), 1.72 (m, 4H). 13C NMR (400 MHz, DMSO-d6): 153.4, 151.8, 141.6, 121.9, 117.1, 50.2, 31.3 and 26.5. Anal. calcd for C25H26Cl4N6Pt2: C 31.85, H 2.76, N 8.92; found: C 32.27, H 2.80, N 8.17.
PtL6, yield: 145.7 mg, 61%. 1H NMR (400 MHz, DMSO-d6): δ 8.79 (dd, 4H), 8.06 (dd, 4H), 7.54 (d, 4H), 7.26 (dd, 4H), 4.15 (br, 4H), 1.65 (m, 8H). 13C NMR (400 MHz, DMSO-d6): 154.2, 152.6, 142.4, 122.4, 118.1, 50.8, 32.1 and 28.3. Anal. calcd for C26H28Cl4N6Pt2: C 32.63, H 2.93, N 8.79; found: C 32.95, H 2.89, N 8.34.
Instrumentation and physical measurements
NMR characterization of the ligands and complexes was performed on a Bruker Avance III 500 or a Bruker Avance 400 spectrometer at frequencies of 400 MHz and 125 MHz/100 MHz using either a 5 mm BBOZ probe or a 5 mm TBIZ probe at 30 °C with chemical shifts referenced to the relevant solvent signal. Mass analysis was done on a Waters Micro-mass LCT Premier Spectrometer and elemental analysis on a Thermo Scientific Flash 2000. Kinetic studies for fast reactions were monitored using an Applied Photophysics SX20 Stopped-Flow spectrophotometer coupled to an online data acquisition system, and a Varian Cary 100 Bio UV-visible spectrophotometer thermostated using a Varian Peltier temperature controller to within ±0.05 °C was used for the slow kinetic measurements. The kinetic traces were analyzed using the Origin 7.5® software package.43 The pH of the solutions was recorded on a Jenway 4330 conductivity/pH meter. The pH meter was calibrated using standard buffer solutions at pH values of 4.0, 7.0 and 10.0.Preparation of solutions for kinetic analysis
Due to the low solubility of the chloro complexes in kinetic solutions, they were converted to their aqua analogues according to a literature procedure.44–47 The solutions of the aqua complexes were prepared by the reaction of a known amount of the chloro Pt(II) complex with AgClO4 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.98 in 0.1 M HClO4. The mixture was stirred at 40–50 °C for 48 h in the dark to protect it from light. After cooling the AgCl precipitate was removed by filtration through a 0.45 μm pore membrane filter. The filtrate was diluted with 0.1 M HClO4 solution to final concentrations of the respective complex solutions.
:3.98 in 0.1 M HClO4. The mixture was stirred at 40–50 °C for 48 h in the dark to protect it from light. After cooling the AgCl precipitate was removed by filtration through a 0.45 μm pore membrane filter. The filtrate was diluted with 0.1 M HClO4 solution to final concentrations of the respective complex solutions.
pKa determination of the aqua complexes
The acidity of the aqua ligands coordinated to the metal centre was determined by spectrophotometric titration to obtain pKa values. This method was preferred due to its ability to give a titration curve that enables the estimation of the pKa values. Spectrophotometric measurements were performed on a Varian Cary 100 Bio UV-visible spectrophotometer thermostated using a Varian Peltier temperature controller to within ±0.05 °C in the wavelength range 200–800 nm (visible spectrum). The pH measurements were recorded on a Jenway 4330 conductivity/pH meter. Large volumes of the platinum aqua complex (300 mL) were used during titration to avoid absorbance corrections due to dilution. NaOH was used as the base for spectrophotometric titration in the pH range of 1–10 at 25 °C. Crushed NaOH pellets were used in the pH range of 1–3 after which Pasteur pipettes were used for dropwise addition of 0.5 M, 0.1 M, 0.05 M and 0.001 M of NaOH solution. After addition of the base, samples of about 500 μL were taken in ampoules and their pH was recorded. After determining the pH the samples in the ampoules were discarded to avoid possible chloride ion contamination from the pH electrode. 2.0 mL solution of the platinum aqua complex was placed in a cuvette for UV/vis spectrophotometric pH titration with NaOH as a base. The plots of absorbance versus pH at a specific wavelength were used to determine the pKa values of the coordinated aqua ligands. The data obtained were fitted to separate single sigmoidal functions by the Boltzmann equation using the Origin 7.5® software. A classic example of a sigmoid curve plotted as absorbance versus pH to determine the pKa values is illustrated in Fig. S41 in the ESI.† Fig. S39 and S40 (ESI†) shows typical sigmoid curves obtained from the complexes. The inflection point corresponds to the pKa value reported in the study. The titration data for the complexes were fitted to the sigmoid curve using the Boltzmann eqn (2) that showed four stepwise deprotonation values for dinuclear complexes as reported in Table 3.Kinetic measurements
All kinetic measurements were performed under pseudo-first-order conditions. The nucleophile concentration was at least a 40-fold excess compared to that of the Pt(II) complexes in all reactions. This ensured at least 10-fold of nucleophile concentration at each platinum centre and hence forced the reaction to completion. Spectral changes resulting from mixing the dinuclear Pt(II) complex and nucleophile solutions were recorded over the wavelength range 200 to 800 nm to establish a suitable wavelength at which kinetic measurements could be performed. A summary of the wavelengths used for each nucleophile is presented in Table S1 in the ESI.† The temperature was controlled throughout all kinetic experiments within an accuracy of ±0.05 °C. For the determination of the activation parameters (enthalpies and entropies) the rate of the reaction was measured as a function of temperature, over a temperature range of 15 to 40 °C at intervals of 5 °C. The rate constants reported represent an average value of at least six to eight independent kinetic runs for reactions performed on the stopped flow spectrophotometer and of triplicate runs for those followed on the UV-visible spectrophotometer. The kobs values were obtained from nonlinear least-squares by fitting the exponential function in eqn (1) to the time dependent absorbance.48| At = A∞ + (A0 − A∞)exp(−kobst) | (1) |
Computational calculations
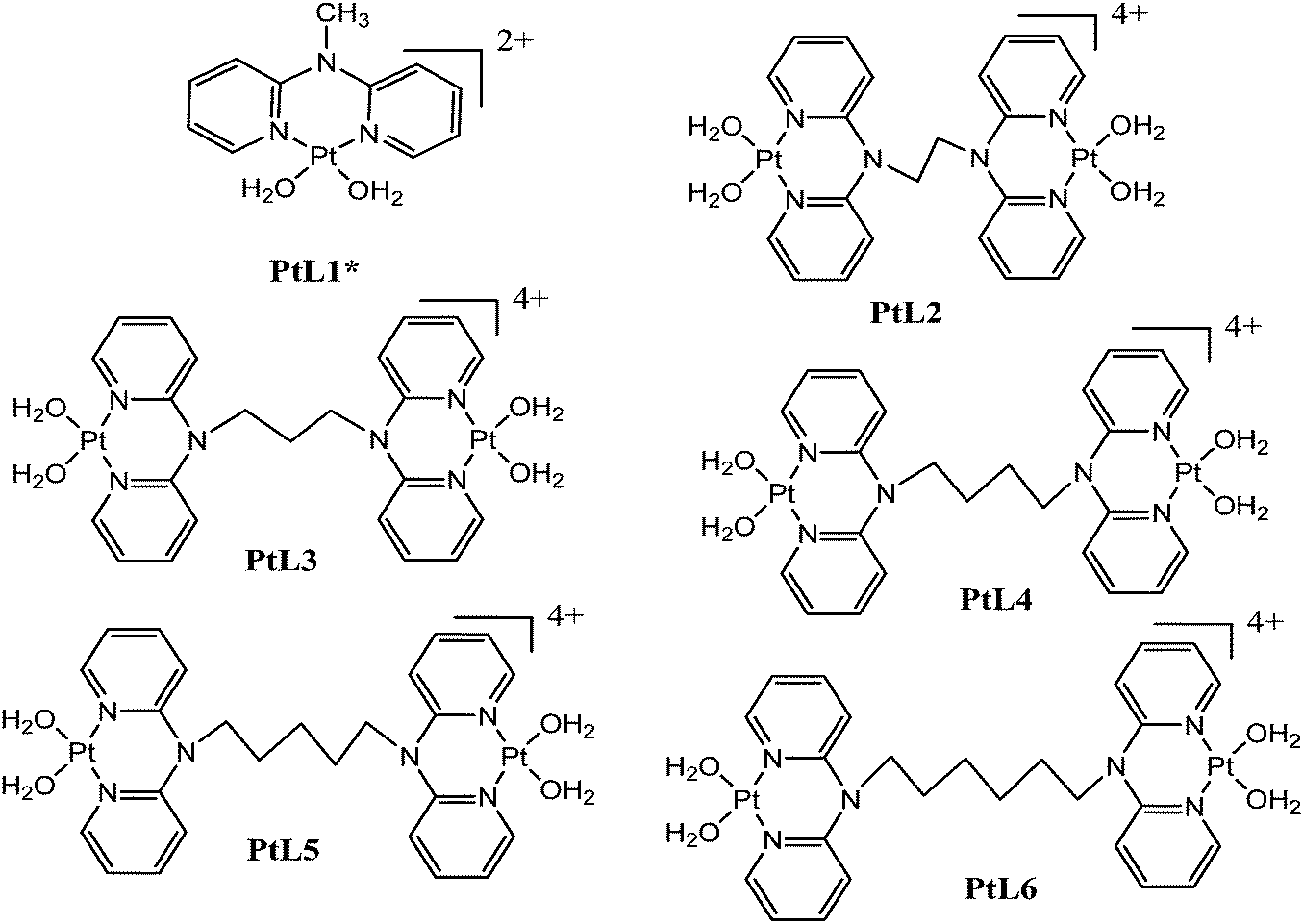
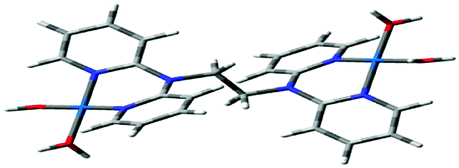
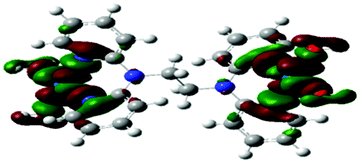
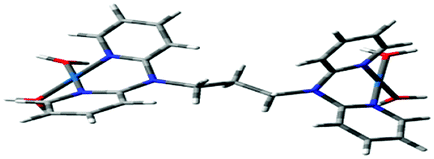
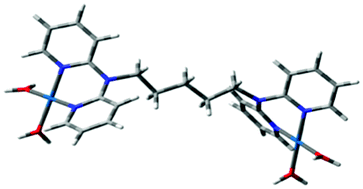
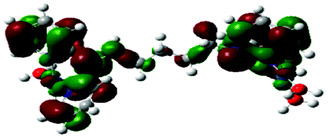
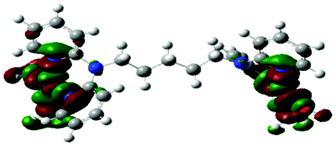
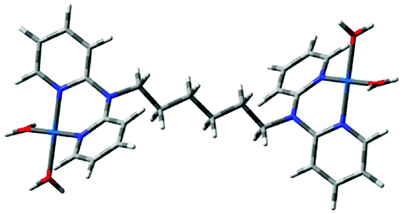
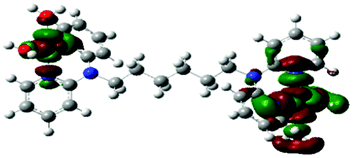
Computational calculations for parameters such as the chemical potential, chemical hardness and electrophilicity indices, using density functional theory (DFT), were performed with the Gaussian 09 programme suite.49 Geometrical optimizations were carried at the B3LYP/LanL2DZ level of theory. B3LYP refers to a three parameter functional hybrid exchange of Becke50 with the functional correlation gradient of Lee, Yang and Parr,51 whereas LanL2DZ refers to the Los Alamos National Laboratory 2 Double ζ basis set.52 The singlet states were used due to the low electronic spin of Pt(II) complexes. The aqua complexes were modelled as cations with an overall charge of +4. The optimized structures of frontier molecular orbitals and other key geometrical data are presented in Tables 1 and 2 respectively. The dihedral and basal angles of the complexes were obtained from the optimized structures of the respective complexes as indicated in Fig. 1 and the values are summarized in Table 2.| Complex | Planarity | HOMO map | LUMO map |
|---|---|---|---|
| PtL2 |
|

|
|
| PtL3 |
|
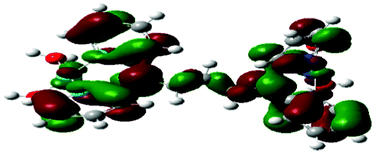
|
|
| PtL4 |

|
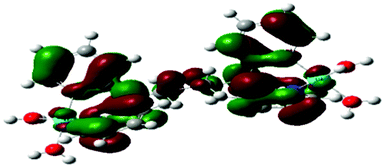
|
|
| PtL5 |
|
|
|
| PtL6 |
|

|
|
|
|
PtL2 | PtL3 | PtL4 | PtL5 | PtL6 |
|---|---|---|---|---|---|
| HOMO–LUMO energy | |||||
| LUMO/eV | −12.33 | −11.99 | −11.63 | −11.37 | −11.13 |
| HOMO/eV | −16.66 | −16.23 | −15.85 | −15.52 | −15.26 |
| ΔE/eV | 4.34 | 4.24 | 4.22 | 4.15 | 4.13 |
| Electrophilicity index (ω) (eV) | 48.44 | 46.96 | 44.74 | 43.49 | 42.09 |
| NBO charges | |||||
| Pt1 | 0.776 | 0.771 | 0.770 | 0.767 | 0.767 |
| Pt2 | 0.776 | 0.771 | 0.770 | 0.767 | 0.767 |
| N1 | −0.517 | −0.516 | −0.518 | −0.518 | −0.518 |
| N2 | −0.515 | −0.516 | −0.517 | −0.517 | −0.518 |
| N3 | −0.536 | −0.513 | −0.513 | −0.509 | −0.509 |
| Dipole moment (Debye) | 0.5785 | 5.5062 | 3.3977 | 4.6459 | 1.3105 |
| Bond length (Å) | |||||
| N3–R | 1.4972 | 1.5004 | 1.5043 | 1.5050 | 1.5067 |
| Pt1–Pt2 | 9.44 | 9.70 | 11.58 | 12.47 | 14.03 |
| Bond angles (°) | |||||
| Dihedral | 50.55 | 50.47 | 50.28 | 50.06 | 50.30 |
| Basal | 129.45 | 129.53 | 129.72 | 129.94 | 129.70 |
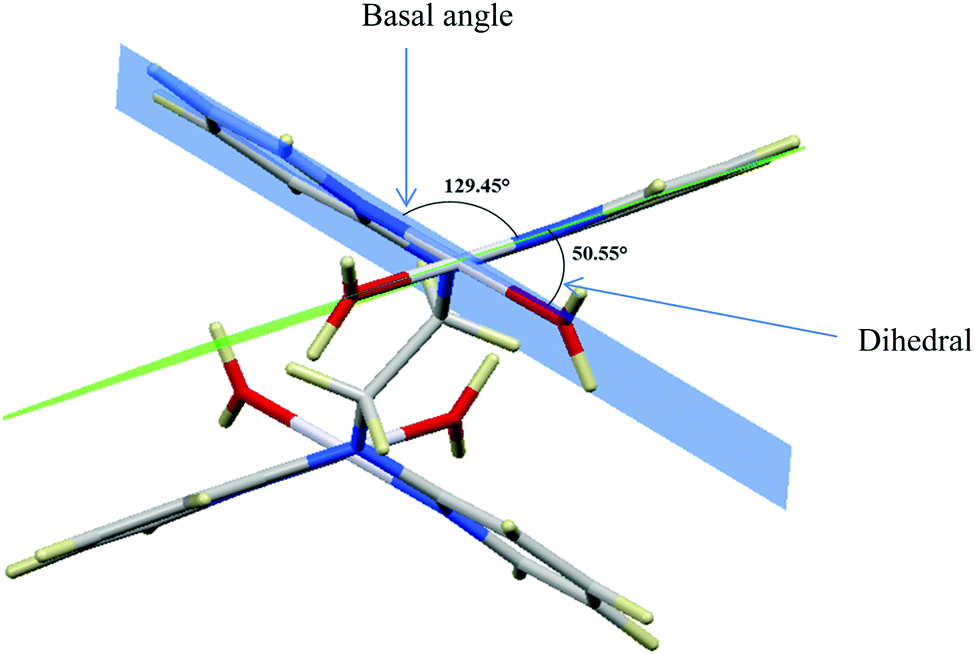 | ||
| Fig. 1 DFT-optimized structure of PtL2 showing the dihedral and basal angles between boat conformed pyridyl groups. | ||
The two pyridyl rings of each dpa have similar dihedral angles because of similar spatial repulsions. The values of these angles for the complexes are summarized in Table 2. Thus, preorganization is shown by the alkyl chain to impart conformational stability. The preorganization is achieved by the alkyl chain to impose electronic and steric effects that restrict the dihedral angles of dipyridylamines. Although longer alkyl chains are expected to introduce more hindrance to the complexes, similar dihedral and basal angles show that an increase in alkyl chain length provides little steric effect in the immediate vicinity of the platinum atoms. This is in agreement with recent studies on dipyridylamine complexes by Asman and Jaganyi.53–55 Both current and previous studies give an overview of kinetics and substitution mechanisms of dipyridylamine backbone Pt(II) complexes with sulphur-donor biomolecules to show that they have similar behaviour. The complexes investigated have similarities in structure to cisplatin. Both reviews correlate kinetic, thermodynamic and structural properties of dipyridylamine Pt(II) complexes appended to different chelating ligands. The current and previous studies have contributed to a better understanding of these dipyridylamine Pt(II) complexes with important biomolecules and most likely improve their antitumour activity.
Acidity and pKa titrations of the aqua Pt(II) complexes
The acidity of the complexes in this study is discussed based on the first deprotonation. A typical spectrum obtained during the pH titrations is shown in Fig. 2. From the first deprotonation, the trend for the pKa1 values shows that the acidity of the coordinated aqua ligands depends on the length of the alkyl bridge between the metal centres and decreases in acidity as the alkyl chain increases. As was mentioned in the experimental section, the pKa values were determined from the Boltzmann eqn (2) by fitting a characteristic sigmoid curve and locating the inflection point using the Origin 7.5® software.43 The pKa values of the deprotonated complexes are summarized in Table 3 while the proposed deprotonation mechanism is represented in the equilibrium reaction in Scheme 2.| y = A2 + (A1 − A2)/(1 + exp(x − x0)/∂x) | (2) |
 | ||
| Fig. 2 UV/vis spectral changes for the titration of PtL2 within the pH range of 1 to 10; the inset shows the titration curve at λ = 245 nm. | ||
| Complex | PtL1* | PtL2 | PtL3 | PtL4 | PtL5 | PtL6 |
|---|---|---|---|---|---|---|
| pKa values for PtL1* were obtained from ref. 41b. | ||||||
| pKa1 | 5.24 ± 0.07* | 2.11 ± 0.01 | 2.27 ± 0.01 | 2.34 ± 0.01 | 2.47 ± 0.03 | 2.71 ± 0.06 |
| pKa2 | 6.19 ± 0.04* | 3.74 ± 0.03 | 3.60 ± 0.01 | 3.39 ± 0.04 | 3.44 ± 0.03 | 3.49 ± 0.01 |
| pKa3 | — | 4.89 ± 0.04 | 4.58 ± 0.02 | 4.88 ± 0.01 | 4.68 ± 0.05 | 4.74 ± 0.04 |
| pKa4 | — | 6.45 ± 0.01 | 6.38 ± 0.01 | 6.53 ± 0.02 | 6.33 ± 0.06 | 7.04 ± 0.06 |
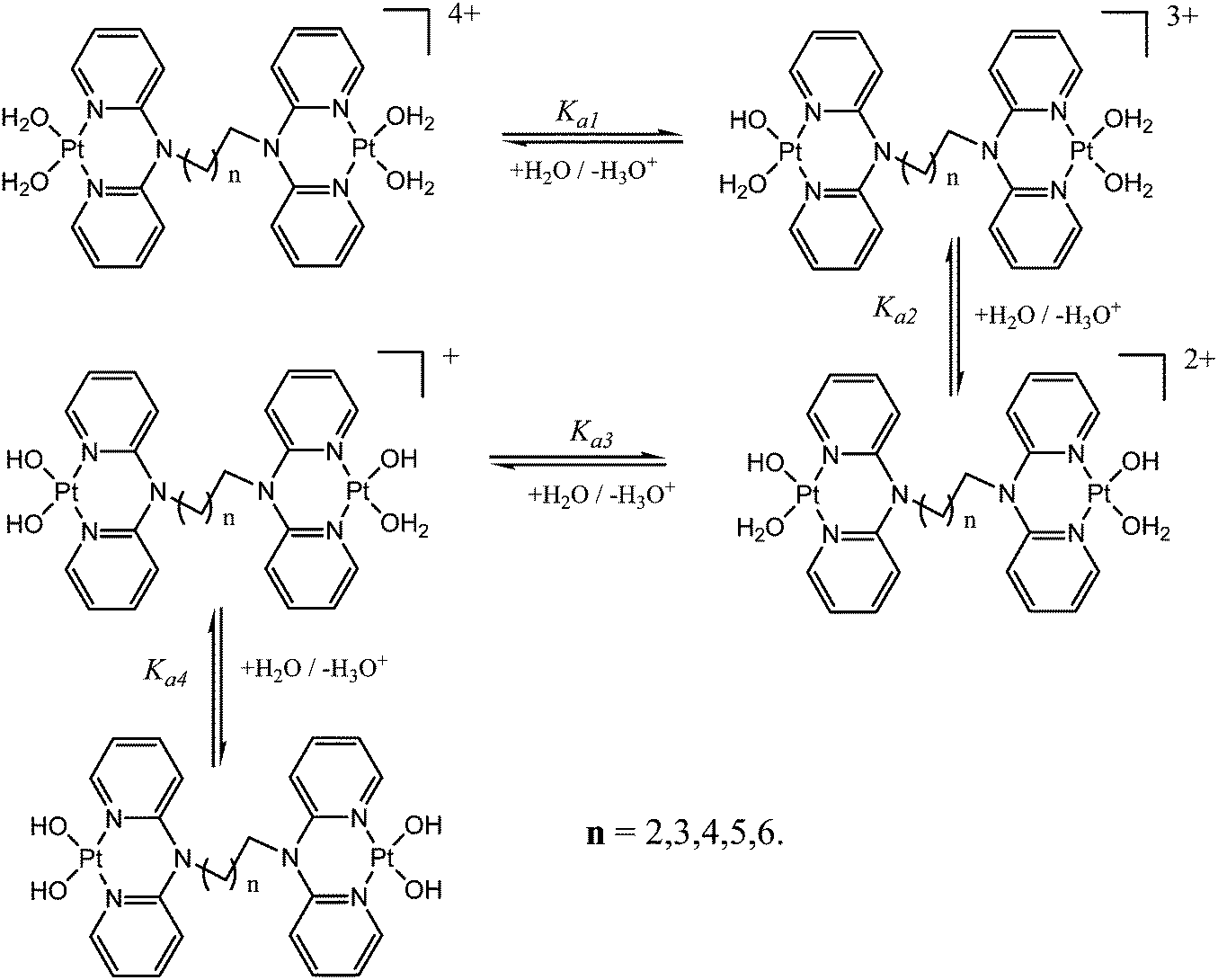 | ||
| Scheme 2 Proposed stepwise deprotonation of the four aqua ligands on dinuclear Pt(II) complexes. | ||
The difference in the acidity of dinuclear complexes is attributed to the metal–metal distance through charge addition and also to the inductive effects of the alkyl chain. Complexes with shorter distances between the platinum(II) centres undergo addition of single charges that enhances electrophilicity and acidity due to the close proximity of Pt2+ ions.56 The mutual charge addition of each Pt(II) centre becomes weaker as the distance between the metal centres becomes longer as shown by the current study. This is evident from the pKa1 trend in the acidity of the complexes PtL2 > PtL3 > PtL4 > PtL5 > PtL6. The inductive effect of the alkyl group on the pyridine rings is proposed to increase the electron density on the pyridine nitrogen atoms making them stronger donors. The study also suggests that the strength of the inductive effect is dependent on the length of the alkyl chain. The trend shows the longer alkyl chains to stabilize the platinum centres due to their increased inductive effect which is also supported by the slight increase in N3–R bond lengths across the series as shown in Table 2. As a result, the aqua ligands become more basic and hence have high pKa values. An increase in electron density around the platinum atom leads to a stronger σ bond and increased intrinsic basicity of the complexes. This increased inductive effect of alkyl moieties enhances the negative charge around platinum centres that results in their stability. This study suggests the inductive effects to be a key factor in the acidity of the complexes. In addition it shows that the dinuclear complexes are more acidic than the mononuclear ones. This difference is attributed to the difference in the overall charge of the complexes which is +2 for the mononuclear and +4 for the dinuclear complexes. Coordination of a second Pt(II) atom results in addition of charges that make the dinuclear platinum complexes more electrophilic that favours aqua deprotonation. Because of the overall charge of +4 on the dinuclear Pt(II) complexes the aqua ligands are more acidic compared to the reference mononuclear complex PtL1*.
The stability of these complexes is due to each metal centre possessing a delocalized electronic structure with a boat conformation.42 This makes the Pt centres to be in a conjugated six membered ring that causes the electron density to be delocalized into the ring, hence resulting in the shielding of alkyl protons. The inductive effect of the alkyl group is reported to be transmitted to the Pt(II) centres via the lone pair on nitrogen being delocalized over the amide group. The electron lone pair of the nitrogen atom affects cross conjugation of the complexes affecting the stability of Pt(II) centres. As such the NBO charges on the Pt centres do not change significantly since the surrounding environment has identical atomic and conjugative connectivity.57a In such complexes, the NBO charge on the metal is not readily quantified due to being in a delocalized system.57b Due to the chelating effect the metal ions with a positive charge in the complexes are stabilized through resonance. This behaviour is supported by the raising of frontier orbitals in Table 1. The positively charged Pt2+ ion becomes stabilized as the alkyl chain increases indicating increased inductive effect as the alkyl chain length increases.58,59 This observation is in agreement with the negative charge being stabilized as one moves from sp3 → sp2 closer to the positively charged nucleus. This is also supported by the general decrease in the electrophilicity index across the complexes as the alkyl chain increases. The results also show that the subsequent pKa values increase with subsequent deprotonation; this observation is attributed to the lowering of the overall charge from +4 to +1. This decrease in the overall charge decreases the electrophilicity and the acidity of the metal centres making it more difficult to deprotonate the consequent coordinated aqua ligand due to the formation of hydroxo species. The pKa studies show that the complexes undergo four stepwise deprotonation, an observation reported in our previous studies53–55 and investigations by van Eldik et al.47,60
Kinetic results
The substitution kinetics of the coordinated aqua ligands in the complexes was followed spectrophotometrically by the change in the absorbance at suitable wavelengths as a function of nucleophile concentration and time. Fig. 3 and 4 show typical spectral changes of absorbance versus wavelength for the slow reaction steps of complexes with odd and even number of carbons in the alkyl chain of PtL3 and PtL6 respectively. The inset is a kinetic trace of absorbance versus time from which the pseudo-first-order rate constants kobs were obtained by fitting the single exponential function.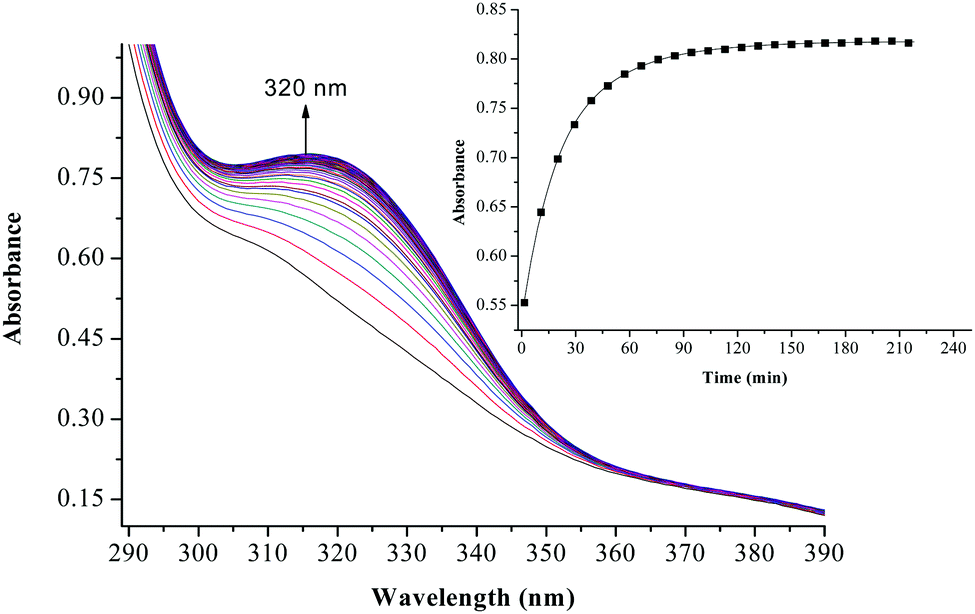 | ||
| Fig. 3 UV/vis spectral changes observed during the reaction between PtL3 and TU; the inset is a typical kinetic trace of absorbance versus time at 320 nm, T = 298.15 K, pH = 2.0 and I = 0.1 M NaClO4. | ||
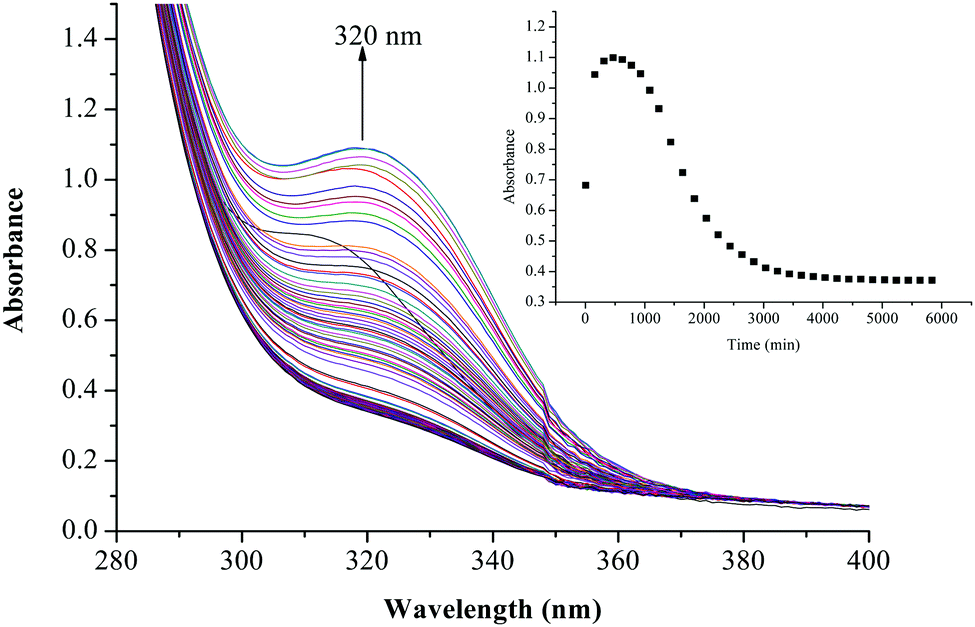 | ||
| Fig. 4 UV/vis spectral changes observed during the reaction between PtL6 and TU; the inset is a typical kinetic trace of absorbance versus time at 320 nm, T = 298.15 K, pH = 2.0 and I = 0.1 M NaClO4. | ||
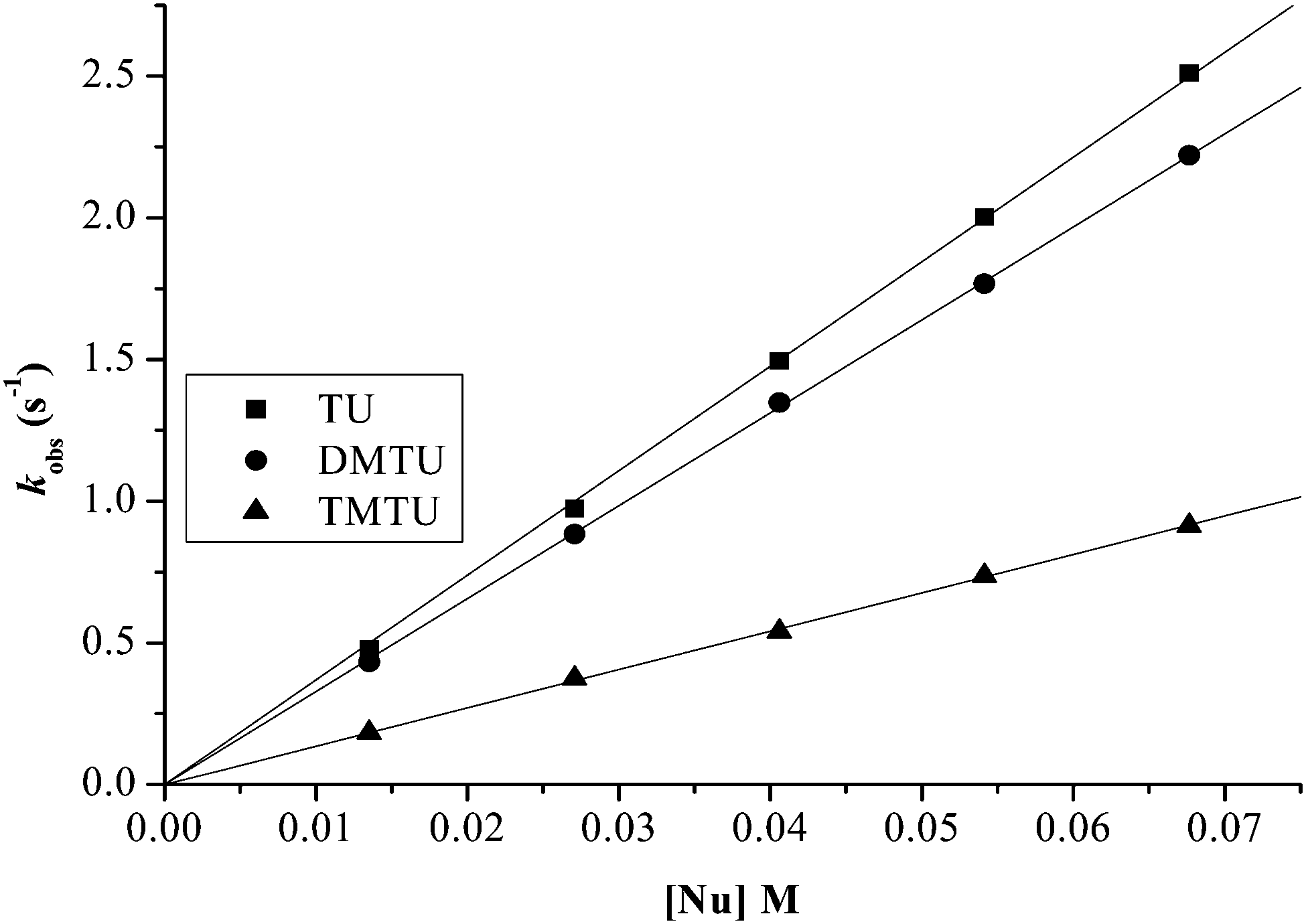
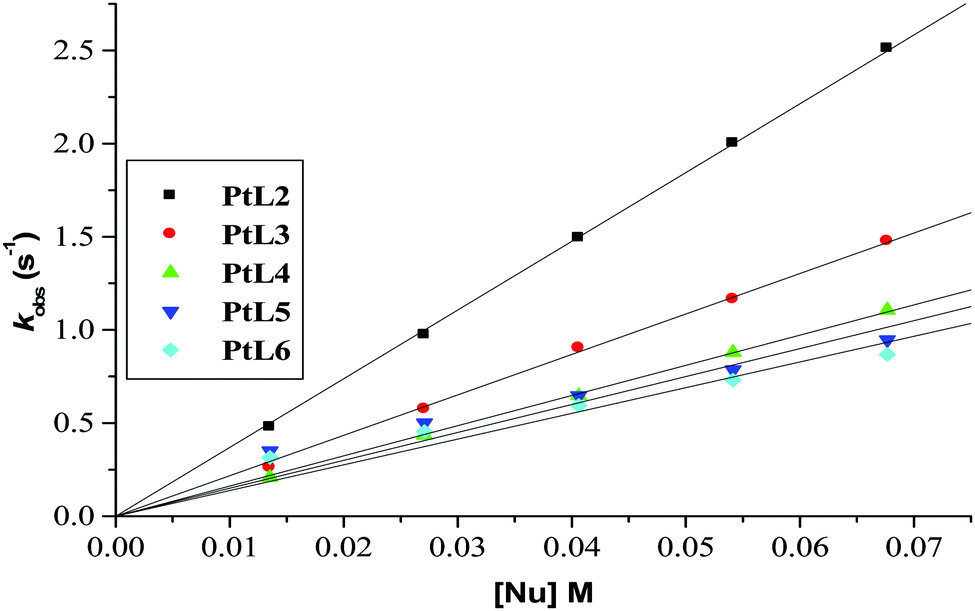
Plots of kobs against nucleophile concentrations resulted in linear dependence second-order rate constants, k2, with no significant intercepts as shown in Fig. 5, 6 and Fig. S1–S3 in the ESI.† The summary of the second order rate constants is shown in Table 4. Based on this observation, eqn (3) represents the corresponding rate laws for the consecutive reaction steps.
| kobs(1,2,3) = k−2 + k2[Nu] ≈ k2[Nu] | (3) |
 | ||
| Fig. 5 Plots of kobs(1)versus nucleophile concentration for the reaction of PtL2 at pH = 2.0, T = 298.15 K, I = 0.1 M NaClO4. | ||
 | ||
| Fig. 6 Plots of kobs(1)versus thiourea concentration for the reaction of the dinuclear platinum(II) complexes at pH = 2.0, T = 298.15 K, I = 0.1 M NaClO4. | ||
| Complex | Nu | Second-order rate constant/M−1 s−1 | ||
|---|---|---|---|---|
| k 2(1st) | k 2(2nd) × 10−2 | k 2(3rd) × 10−3 | ||
| Data for PtL1* extracted from ref. 41. | ||||
| PtL1* | TU | 38.70 ± 0.60 | 106 ± 3 | — |
| DMTU | 48.80 ± 0.50 | 2 ± 0.1 | — | |
| TMTU | 22.80 ± 0.40 | — | — | |
| PtL2 | TU | 36.90 ± 0.18 | 4 ± 0.2 | — |
| DMTU | 32.79 ± 0.11 | 7 ± 0.1 | — | |
| TMTU | 13.53 ± 0.06 | 1 ± 0.1 | — | |
| PtL3 | TU | 21.71 ± 0.22 | 7 ± 0.1 | — |
| DMTU | 17.43 ± 0.18 | 3 ± 0.1 | — | |
| TMTU | 9.71 ± 0.16 | 3 ± 0.1 | — | |
| PtL4 | TU | 16.20 ± 0.10 | 11 ± 0.6 | 5 ± 0.09 |
| DMTU | 13.57 ± 0.02 | 6 ± 0.1 | 3 ± 0.03 | |
| TMTU | 7.04 ± 0.08 | 4 ± 0.06 | Too slow | |
| PtL5 | TU | 10.92 ± 0.14 | 5 ± 0.02 | — |
| DMTU | 13.45 ± 0.24 | 3 ± 0.05 | — | |
| TMTU | 6.28 ± 0.04 | 0.8 ± 0.01 | — | |
| PtL6 | TU | 10.21 ± 0.03 | 8 ± 0.09 | 0.5 ± 0.02 |
| DMTU | 12.60 ± 0.23 | 3 ± 0.04 | 0.7 ± 0.02 | |
| TMTU | 5.10 ± 0.04 | 2 ± 0.04 | Too slow | |
Activation parameters
The values of activation parameters ΔH# and ΔS# are calculated from Fig. 7, 8 and Fig. S4–S7 in the ESI† using eqn (4) below.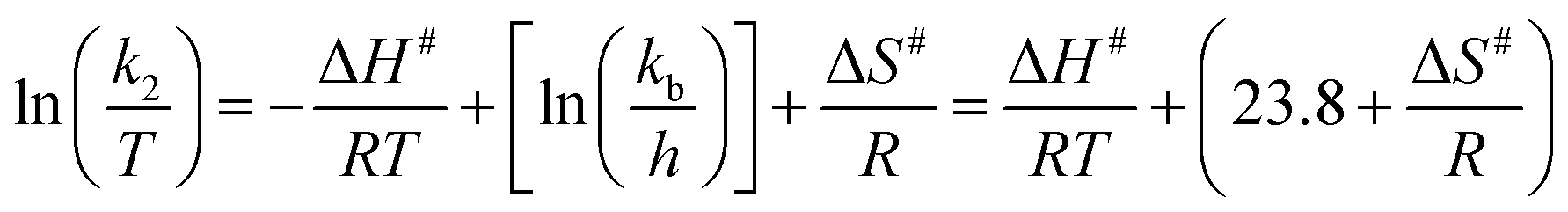 | (4) |
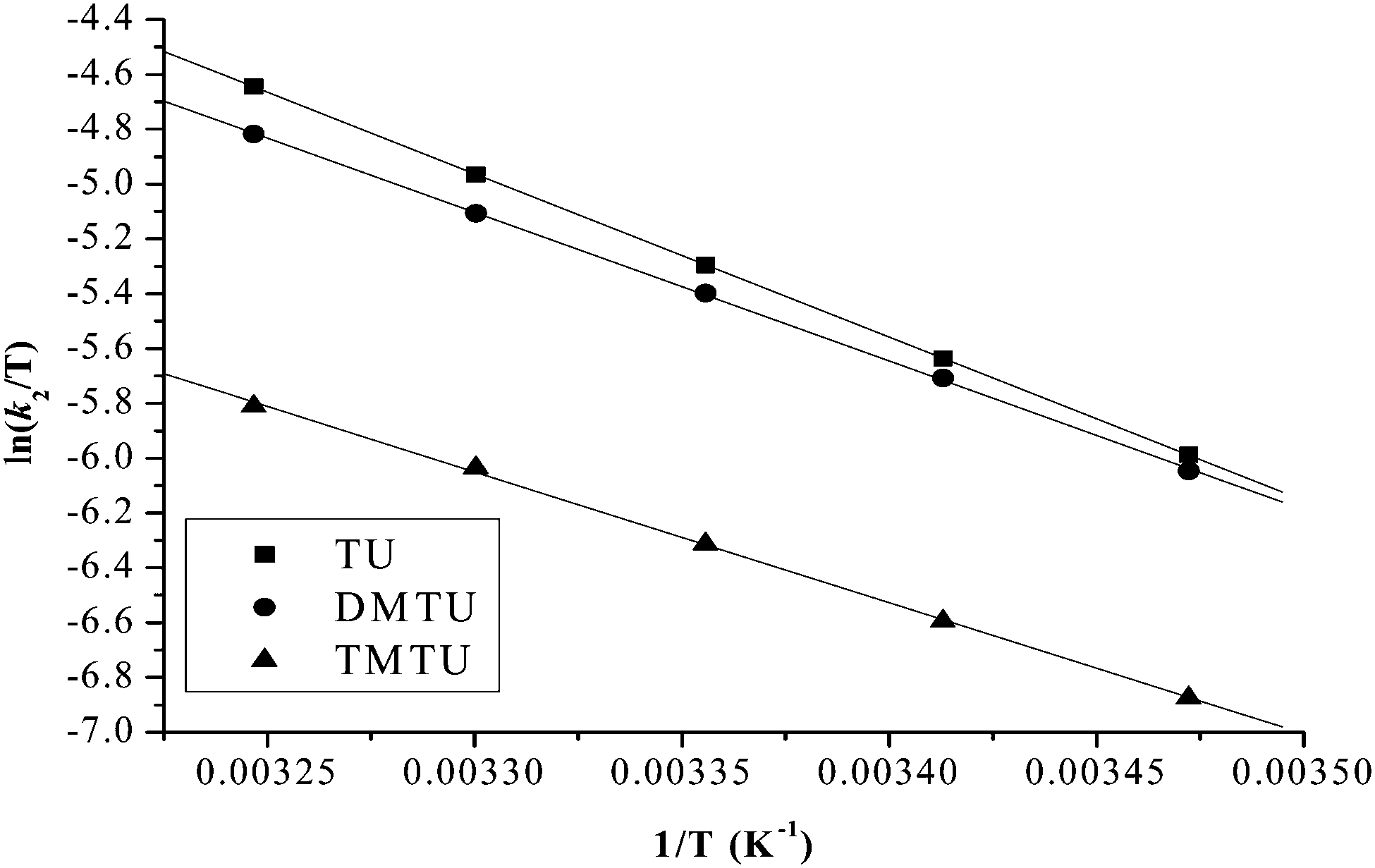 | ||
| Fig. 7 Eyring plots for the first substitution step of PtL2 with different nucleophiles at pH = 2.0, I = 0.1 M NaClO4. | ||
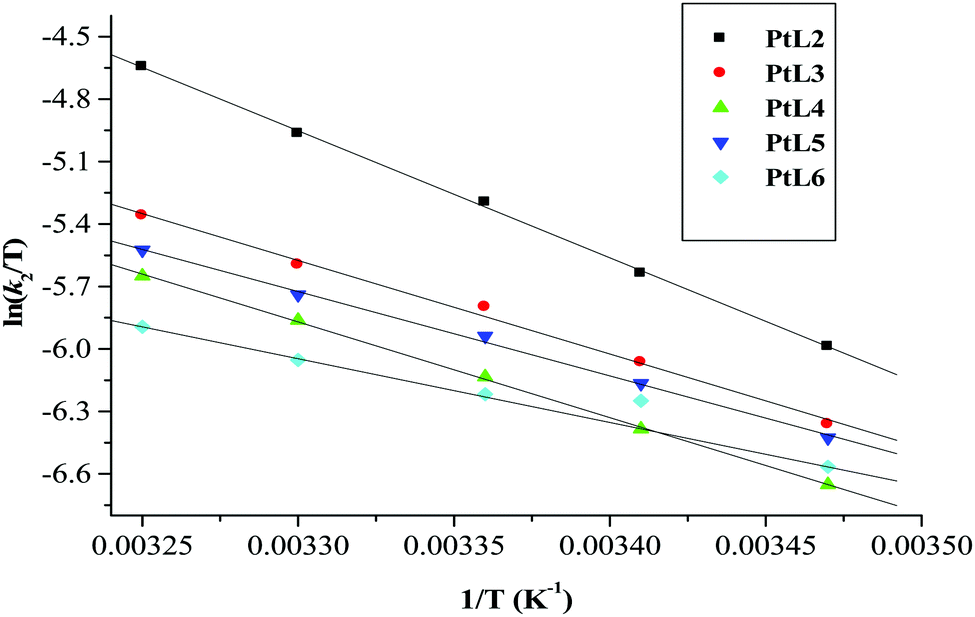 | ||
| Fig. 8 Eyring plots for the first substitution step of dinuclear platinum(II) complexes with thiourea at pH = 2.0, I = 0.1 M NaClO4. | ||
| Complex | Nu | Activation enthalpy/kJ mol−1 | Activation entropy/J mol−1 K−1 | ||||
|---|---|---|---|---|---|---|---|
| ΔH#1 | ΔH#2 | ΔH#3 | ΔS#1 | ΔS#2 | ΔS#3 | ||
| PtL2 | TU | 49.53 ± 0.1 | 24.41 ± 0.3 | — | −76 ± 1 | −226 ± 1 | — |
| DMTU | 45.09 ± 0.4 | 32.52 ± 0.3 | — | −92 ± 1 | −206 ± 1 | — | |
| TMTU | 39.69 ± 0.6 | 46.12 ± 1.6 | — | −117 ± 2 | −155 ± 5 | — | |
| PtL3 | TU | 36.53 ± 1.1 | 23.05 ± 0.1 | — | −123 ± 4 | −219 ± 1 | — |
| DMTU | 21.01 ± 0.5 | 40.08 ± 0.5 | — | −177 ± 2 | −170 ± 2 | — | |
| TMTU | 47.39 ± 0.7 | 37.21 ± 0.6 | — | −94 ± 2 | −180 ± 2 | — | |
| PtL4 | TU | 37.30 ± 0.6 | 23.37 ± 0.3 | 24.16 ± 0.9 | −124 ± 2 | −218 ± 1 | −232 ± 3 |
| DMTU | 39.60 ± 0.4 | 29.70 ± 0.3 | 29.57 ± 1.2 | −117 ± 1 | −205 ± 1 | −220 ± 4 | |
| TMTU | 39.58 ± 0.5 | 29.40 ± 1.1 | — | −125 ± 2 | −201 ± 4 | ||
| PtL5 | TU | 32.90 ± 0.6 | 44.62 ± 0.5 | — | −137 ± 2 | −149 ± 2 | — |
| DMTU | 49.54 ± 1.1 | 46.61 ± 0.8 | — | −82 ± 4 | −146 ± 3 | — | |
| TMTU | 48.93 ± 0.3 | 30.65 ± 0.4 | — | −92 ± 1 | −208 ± 1 | — | |
| PtL6 | TU | 24.74 ± 0.2 | 30.26 ± 0.4 | 11.55 ± 0.1 | −167 ± 1 | −194 ± 1 | −293 ± 0.1 |
| DMTU | 18.44 ± 0.1 | 27.02 ± 0.2 | 22.51 ± 0.3 | −189 ± 0.4 | −211 ± 1 | −255 ± 1 | |
| TMTU | 71.15 ± 1.4 | 43.57 ± 0.5 | — | −19 ± 5 | −153 ± 2 | — | |
The small but positive ΔH# and large negative values of ΔS# support an associative mechanism. The low enthalpy values are attributed to bond formation and participation of the entering ligand in the transition state. The large negative values of entropies signify a more compact transition state than that of the reactants.61
Substitution reaction
The number of substitution steps was dependent on the “odd or even” effect of the alkyl spacer.62–64 Complexes with even number of methylene groups [n = 4, 6], PtL4 and PtL6, showed anti-conformations (Fig. S32, ESI†) and three kinetic steps were observed except for [n = 2] PtL2 which showed two reaction steps. On the other hand complexes with odd groups [n = 3, 5], PtL3 and PtL5, showed syn conformations with only two reaction steps. These conformations are supported by Albrecht and others who also observed a similar odd–even effect.65 As such different substitution behaviour is expected due to the different architectural framework. The syn conformation restricts Pt(II) centres in the same plane making them kinetically equivalent. This equivalency at Pt(II) centres results in simultaneous substitution at the two Pt(II) centres as suggested in this study. The second slow step involves a dechelation after the second water molecule at each Pt(II) centre is substituted. The study shows the displacement of the labilized amine spacer from two Pt(II) ions to occur concurrently. Similar behaviour of a single kinetic step release of two metal ions from the spacer in dinuclear complexes has also been reported by Basollote et al.66However, in the anti-conformational configuration, the study proposes the first step to be the fast substitution of the water molecule at each Pt(II) centre followed by a slow displacement of the second water molecule that involves the stepwise displacement of the labilized alkyl spacer as a result of the strong trans effect of thiourea ligands. However, PtL2 showed two reaction steps instead of the expected three steps due to its ability to exhibit either syn- or anti-conformation (Scheme 3). This is partly attributed to its ability to interconvert by rotations around the single bond by approximately 6 × 109 s−1 at 25 °C67 and also due to the shorter spacer between the two Pt(II) centres. Due to the proximity of the metal centres, the study suggests a single kinetic step to release the two metal centres from the spacer.
 | ||
| Scheme 3 Proposed conformational shifts of PtL2 as either trans or gauche conformation.67 | ||
To show that the complexes undergo a dechelation process, a substitution reaction of the chloride complexes with 6 equivalents of TU was carried out. The reaction products were analyzed using 1H and 195Pt NMR spectroscopy. The 195Pt NMR spectrum of PtL6 in DMSO-d6 at −2175 ppm indicated that the platinum centres were coordinated to two nitrogen atoms (PtN2Cl2).68 Addition of 6 equivalents of TU to the chloride complex solution leads to the shifting of the PtN2Cl2 peak from −2175 ppm to −3909.1 ppm. This confirmed the formation of Pt(TU)42+ complex69–71 as expected from the cis isomer of (PtNNCl2) when reacted with thiourea. The complex reacts completely with thiourea to give Pt(TU)42+ because of the large trans effect as proposed in Scheme 4. The 1H NMR study shows a σ effect that reduces the electron density from the ligand that leads to a downfield shift. In the 1H NMR spectrum of complex PtL6 the alkyl protons (Ha) shift to 1.65 ppm compared to Hb at 1.40 ppm of the ligand (Fig. 9). This downfield shift indicates deshielding of alkyl protons attributed to reduced electron density upon coordination to the metal centre. This decrease in electron density causes alkyl protons to resonate downfield. These shifts support the presence of σ inductive effects causing the shifts of protons of free ligands and their coordinated forms. Due to the cis configuration of this class of complexes, the strong trans-effect upon coordination to thiourea labilizes the linker that leads to the dechelation as supported by Fig. 9 and 10.
 | ||
| Scheme 4 Proposed substitution mechanism for the odd numbered alkyl chain (n = 3, 5). The k2(3rd) in the even numbered alkyl chain (n = 4, 6) is due to stepwise displacement of the alkyl diamine linker. | ||
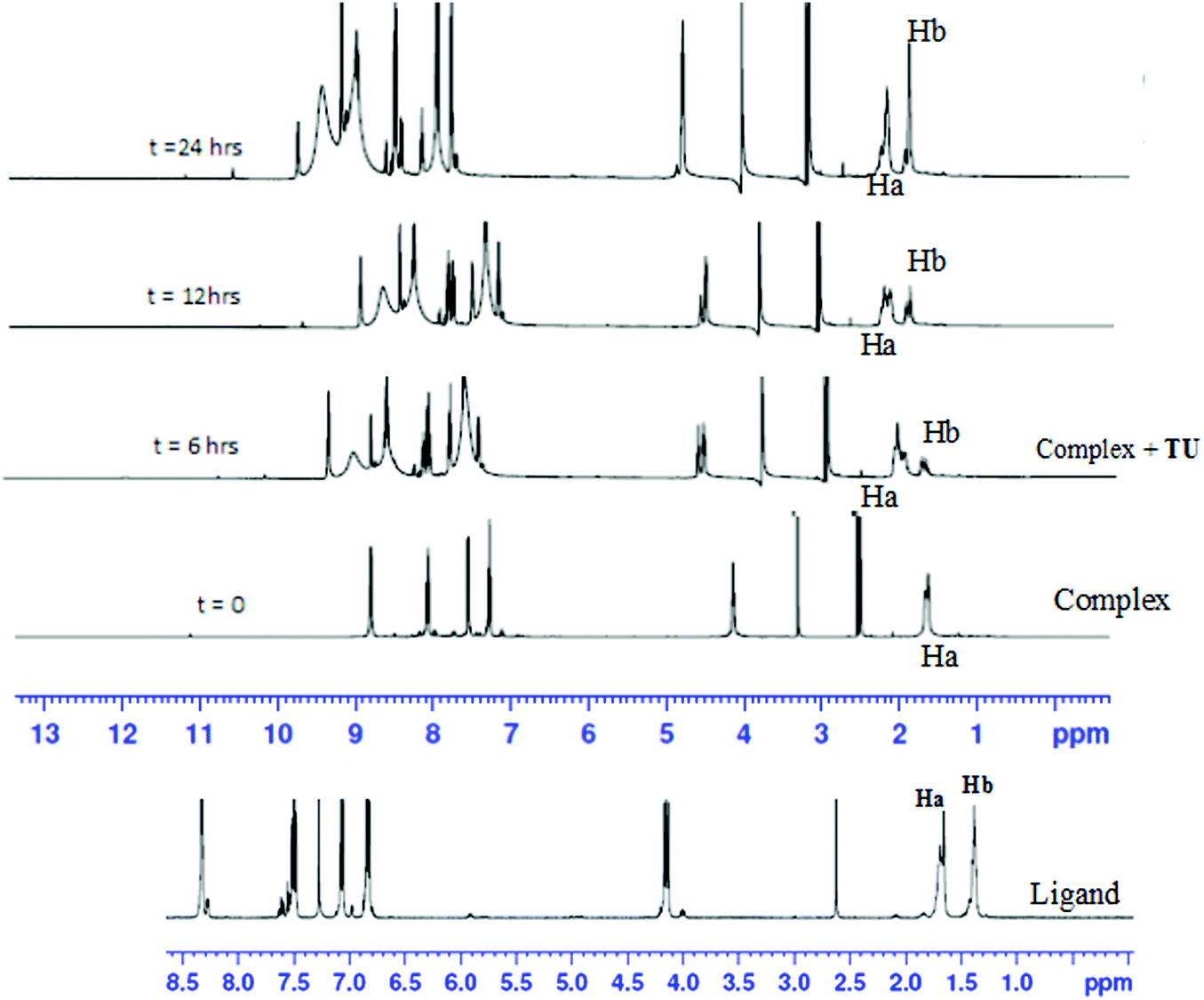 | ||
| Fig. 9 Time-dependent changes in the 1H NMR spectrum of PtL6 upon addition of 6 equiv. of TU in DMSO-d6. The complex undergoes dechelation to form the free ligand (starting material) after 24 h. | ||
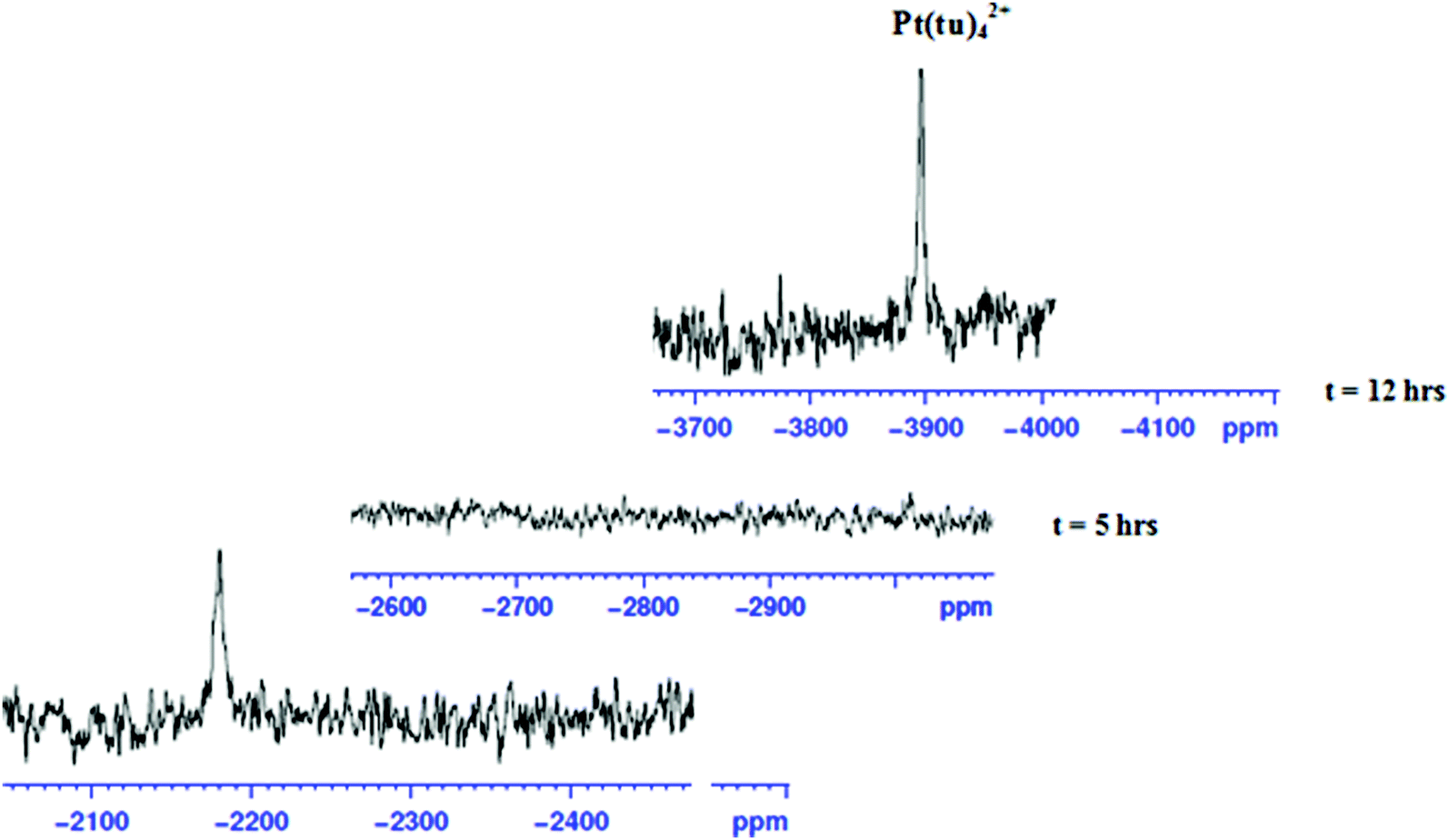 | ||
| Fig. 10 Time-dependent changes in the 195Pt NMR spectrum of PtL6; upon addition of 6 equiv. of TU in DMSO-d6 the peak at −2175 ppm disappears and reappears at −3909.1 ppm indicating the formation of the [Pt(TU)42+] complex.69 | ||
Discussion
The substitution behaviour of dinuclear dipyridylamine Pt(II) complexes with varying alkyl chain lengths was investigated. The kinetic results show that mononuclear 2,2′-dipyridylamine complexes react faster than their dinuclear counterparts. The slow reactivity of dinuclear analogues is due to their increased steric demand compared to mononuclear complexes. In dinuclear complexes, results indicate that the bridging alkyl chain influences the reactivity of the complexes towards the nucleophiles. For the first substitution step, the trend is PtL1 > PtL2 > PtL3 > PtL4 > PtL5 > PtL6 supporting our view on the influence of the chain length. This trend is attributed to the stronger σ-donation as the alkyl chain increases that leads to the stabilization of the metal centres through increased σ-donicity.72 The longer alkyl groups increase the electron density around the Pt(II) centres through an inductive process. This is enhanced by the lone pair of electrons on the amine nitrogen making them more σ electron donating leading to a decrease in electrophilicity at the metal centres. The electrophilicity of the complexes, PtL2 (48.44) > PtL3 (46.96) > PtL4 (44.74) > PtL5 (43.49) > PtL6 (42.09) eV, is in agreement with the reactivity trend. The electronic effect in these complexes is also supported by DFT calculations that shows raised frontier orbitals as the alkyl chain is increased (Table 2). The low lying LUMO orbitals in the short alkyl spacers indicate less σ effect to the dipyridyl rings. This makes complexes with shorter alkyl bridging ligands more reactive towards nucleophilic attack.73However, the HOMO–LUMO energy gap, ΔE, reflects a contradicting trend shown by kinetic data. The Pt(II) complex with the least reactivity, PtL6, has the smallest HOMO–LUMO energy gap (ΔE = 4.13 eV) while the most reactive complex, PtL2, has the highest energy gap (ΔE = 4.34 eV). Increased inductive electron donation raises both the HOMO and the LUMO due to the increasing chain length as shown in Fig. 11. This increase in energy is in line with the expectation of electron donating groups to raise the energy of molecular orbitals.
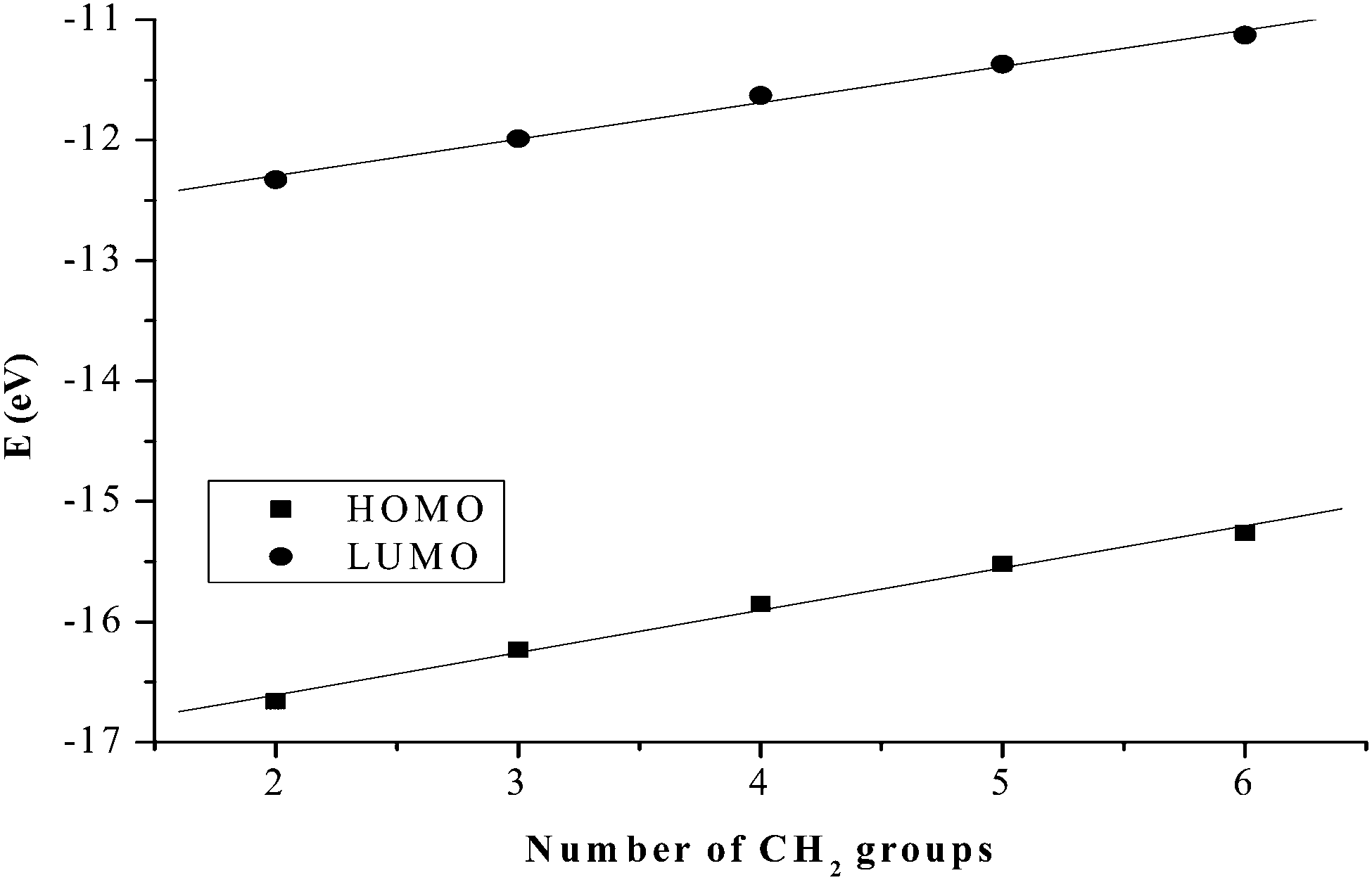 | ||
| Fig. 11 Linear correlation between the chain length and the HOMO/LUMO orbital energy levels. | ||
The results obtained from the current study were compared to those of previous works74,75 where the bis(2-pyridylmethyl)amine (bpma) head group was appended to alkyl bridges of variable length. The findings in this paper show an opposite trend to those previously reported by Jaganyi and Mambanda.72 Although both studies used flexible alkyl linkers, the difference is attributed to the different head functionalities at the termini viz. bis(2-pyridylmethyl)amine and 2,2′-dipyridylamine. A decrease in the reactivity of 2,2′-dipyridylamine Pt(II) complexes was observed as the –(CH2)n– moiety increased due to the linker being in the cis-position relative to the leaving group. The trend in the reactivity of this class of bidentate Pt(II) complexes is attributed to the strong σ-ability of the –(CH2)n– moiety in the cis-position to the leaving group that causes accumulation of electron density around the metal centre. However, this trend is opposite to the kinetic studies reported on the ligand substitution of dinuclear Pt(II) complexes with flexible linkers having bis(2-pyridylmethyl)amine functionality. This head group forms monodentate dinuclear Pt(II) complexes whose reactivity increases with an increase in chain length due to the trans-effect. The high trans-effect76–78 of bis(2-pyridylmethyl)amine linked platinum(II) complexes enhances the rate of substitution through ground state destabilization. This shows the stronger effect of the trans ligand to accommodate the excess electronic charge added to the central metal by the entering ligand. By comparing these two studies, they suggest that cis ligands are not as effective as trans ligands in lowering the activation energy of the ligand → metal σ bonding. This observation is in agreement with classical explanation; the trans effect causes electrostatic destabilization of the ligand in the trans position. As such, the study supports the fact that the electronic properties of the alkyl spacer dictate the strength of its trans effect in the bis(2-pyridylmethyl)amine functionality as the chain length increases. From the two reactivity trends it follows that the cis and trans effects have an opposite net effect. While the trans effect accelerates the rate of substitution through ground state destabilization, the cis effect decelerates substitution rates through accumulation of electron density around the metal centre.
This study has shown that the reactivities of dinuclear Pt(II) complexes may be manipulated by the geometry of the complexes ranging from the relationship of the diamine bridge with the leaving group to the nature of the diamine bridge. It further shows that the investigated complexes are stabilized as the alkyl chain increases, a trend that has been previously reported by van Eldik66 and Jaganyi.79 The stability of the complexes was attributed to stronger σ inductive effects in the cis configuration due to the accumulation of electron density around the Pt(II) centre as the alkyl chain increases. From these findings it is concluded that the structural features and reactivity pattern of the investigated dinuclear Pt(II) complexes are governed by both steric and electronic effects arising from the methylene bridge.
Steric factors were influenced by the size of the complex and the preferential conformations of the complexes. Increasing –(CH2)n– groups increases the size and flexibility of the complexes resulting in an increased steric hindrance. The slow reactivity of large complexes may partially be attributed to their large size. The study also investigated the reactivity of the entering nucleophiles based on their steric and electronic demands. This investigation shows reactivity to be dependent on the steric influence of the entering nucleophile. The reactivity order of the nucleophiles followed the trend TU > DMTU > TMTU. However, in some cases where DMTU is faster than TU, the enhanced reactivity is attributed to the inductive effect by the two methyl groups.80 The second and third steps are much slower since they are governed by the steric hindrance caused by coordination of the first nucleophile. In cases where the kinetic data are not reported, it was due to the slow reactivity of the nucleophile involved.
Conclusion
This paper has provided a comprehensive investigation on the effect of alkyl chains on the mechanistic substitution reactions of cis-dinuclear Pt(II) complexes with tetraaqua ligands. The study shows that the length and parity of the flexible linker are influential parameters on the transitional properties of the metal complexes. The study supports literature findings that the length of the alkyl chain has a significant influence on the structural and electronic properties of the complexes. The kinetic behaviour of the complexes is controlled by the conformations and electronic effect of the alkyl spacers. An increase in alkyl chain length into the Pt(II) coordination sphere results in a decrease in acidity of the coordinated tetraaqua ligands and hence higher stability of the complexes. The results also show the number of substitution steps to be controlled by the odd–even effect of the alkyl spacer. The absence of separate kinetic steps for the release of both Pt(II) ions in the odd numbered alkyl chain suggests an apparent single process of dechelation. The mechanism of substitution is associative in nature as shown by small but positive values of enthalpy and large negative values of entropy. The ability of these complexes to undergo ligand exchange makes them potential candidates for building chemotherapeutic agents for drug delivery. However, the displacement of the labilized chain linker due to the strong trans effect by S-donor nucleophiles may limit the application of this family of complexes as anticancer agents. This work has provided a useful approach for tuning the reactivity of new but promising dinuclear platinum anticancer candidates. These results are beneficial in understanding the antitumour activity and toxicity of dinuclear bifunctional Pt(II) complexes. Elucidation of this multilevel mechanism of dipyridylamine Pt(II) complexes with sulphur-donor bio-relevant molecules will greatly increase our understanding of the mechanism of action of bifunctional dinuclear Pt(II) complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge the financial support from the University of KwaZulu Natal. The authors also thank the School of Chemistry and Physics at the University of KwaZulu Natal Pietermaritzburg Campus for support. The authors extend their sincere thanks to Dr Allen Mambanda for his positive criticism and his suggestions to this paper. Last but not least is our appreciation to Mr Craig Grimmer and Miss Janse van Resenburg for NMR, mass and elemental analyses.References
- (a) P. Gamez, P. de Hoog, O. Roubeau, M. Lutz, W. L. Driessen, A. L. Spek and J. Reedijk, Chem. Commun., 2002, 1488–1498 RSC; (b) P. Gamez, P. de Hoog, M. Lutz, A. L. Spek and J. Reedijk, Inorg. Chim. Acta, 2003, 351, 319–326 CrossRef CAS.
- P. de Hoog, P. Gamez, M. Leuken, O. Roubeau, B. Krebs and J. Reedijk, Inorg. Chim. Acta, 2004, 357, 213–218 CrossRef CAS.
- S. Demeshko, G. Leibeling, S. Dechert and F. Meyer, Dalton Trans., 2004, 3782–3787 RSC.
- C. Seward, W. I. Jia, R. Y. Wang and S. Wang, Inorg. Chem., 2004, 43, 978–985 CrossRef CAS PubMed.
- W. I. Jia, D. R. Bai, T. McCormick, Q. D. Liu, M. R. Motala, R. Y. Wang, C. Seward, Y. Tao and S. Wang, Chem. – Eur. J., 2004, 10, 994–1006 CrossRef CAS PubMed.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS PubMed.
- N. J. Wheate and J. G. Collins, Coord. Chem. Rev., 2003, 241, 133–145 CrossRef CAS.
- N. Farrell, Y. Qu, U. Bierbach, M. Valsecchi and E. Menta, in Chemistry and Biochemistry of Leading Anticancer Drug, ed. Lippert, B., Verlag Helvetica Chimica Acta, Wiley-VCH, Zurich, Weinheim, 1999, pp. 479–496 Search PubMed.
- (a) N. Farrell, Comments Inorg. Chem., 1995, 16, 373–389 CrossRef CAS; (b) N. Farrell, in Platinum and Other Metal Coordination Compounds in cancer Chemotherapy, ed. S. B. Howell, Plenum Press, New York, 1991, pp. 81–82 Search PubMed.
- E. Raymond, S. Faivre, S. Chaney, J. Woynarowski and E. Cvitkovic, Mol. Cancer Ther., 2002, 1, 227–235 CAS.
- D. Wong and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef PubMed.
- J. Kasparkova, J. Novakova, O. Vrana, N. Farrell and V. Brabec, J. Biol. Inorg. Chem., 1999, 38, 10997–11005 CAS.
- J. Kasparkova, J. Zehnulova, N. Farrell and V. Brabec, J. Biol. Inorg. Chem., 2002, 277, 48076–48086 CAS.
- J. W. Cox, S. J. Berners Price, M. S. Davies, W. Barlage, Y. Qu and N. Farrell, Inorg. Chem., 2000, 39, 1710–1715 CrossRef.
- M. S. Ali, K. H. Whitmire, T. Toyomasi, Z. H. Siddik and A. R. Khokhar, J. Inorg. Biochem., 1999, 77, 231–238 CrossRef CAS PubMed.
- S. Komeda, M. Lutz, L. A. Spek, M. Chikuma and J. Reedijk, Inorg. Chem., 2000, 39, 4230–4236 CrossRef CAS PubMed.
- A. J. Kraker, J. D. Hoeschele, W. L. Elliot, H. D. Showalter, A. D. Sercel and N. Farrell, J. Med. Chem., 1992, 35, 4526–4532 CrossRef CAS PubMed.
- Y. Qu and N. Farrell, J. Am. Chem. Soc., 1991, 113, 4851–4857 CrossRef CAS.
- Y. Qu and N. Farrell, Inorg. Chem., 1992, 31, 930–932 CrossRef CAS.
- U. Bierbach, Y. Qu, T. W. Hambley, J. Peroutka, H. L. Nguyen, M. Doedee and N. Farrell, Inorg. Chem., 1999, 38, 3535–3542 CrossRef CAS PubMed.
- N. Farrell, Y. Qu and M. P. Hacker, J. Med. Chem., 1990, 33, 2179–2184 CrossRef CAS PubMed.
- A. Gund and B. K. Keppler, Angew. Chem., 1994, 106, 198–2005 CrossRef CAS.
- R. Mital, T. S. Srivastata, H. K. Parekh and M. P. Chitnis, J. Inorg. Biochem., 1991, 41, 93–103 CrossRef CAS PubMed.
- H. M. Mansuri-Torshizi, T. S. Srivastata, H. K. Parekh and M. P. Chitnis, J. Inorg. Biochem., 1992, 45, 135–148 CrossRef CAS PubMed.
- Y. Qu, H. Rauter, A. P. Soares Fontes, R. Bandarage, L. R. Kelland and N. Farrell, J. Med. Chem., 2000, 43, 3189–3192 CrossRef CAS PubMed.
- R. A. Ruhayel, S. Janina, J. S. Langner, M. J. Oke, S. J. Berners-Price, Z. I. Ibrahim and N. Farrell, J. Am. Chem. Soc., 2012, 134, 7135–7146 CrossRef CAS PubMed.
- N. Farrell, Met. Ions Biol. Syst., 2004, 41, 252–296 Search PubMed.
- S. Komeda, Unique platinum-DNA interactions may lead to more effective platinum-based antitumour drugs, Metallomics, 2011, 3, 650–655 RSC.
- J. B. Mangrum and N. Farrell, Chem. Commun., 2010, 46, 6640–6650 RSC.
- P. Perego, C. Caserini, L. Gatti, N. Carenini, S. Romanelli, R. Supino, D. Colangelo, I. Viano, R. Leone, S. Spinelli, G. Pezzoni, C. Manzotti, N. Farrell and F. Zunino, Mol. Pharmacol., 1999, 55, 528–534 CAS.
- R. Jarzynka, A. Topolski, M. Uzarska and R. Czajkowski, Inorg. Chim. Acta, 2014, 413, 60–67 CrossRef.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS PubMed.
- W. E. Curtis, M. E. Muldrow, N. B. Parker, R. Barkley, S. L. Linas and J. E. Repine, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 3422–3428 CrossRef CAS.
- A. A. Shoukry, J. Chem. Sci., 2013, 125, 643–651 CrossRef CAS.
- K. Matsumoto, H. Aizawa, H. Inoue, H. Koto, S. Fukuyama and N. Hara, Eur. J. Pharmacol., 2000, 403, 267–275 CrossRef.
- P. Pil and S. J. Lippard, Specific Binding of Chromosomal Protein HMGI to DNA Damaged by the Anticancer Drug Cisplatin, Science, 1992, 256, 234–236 CAS.
- B. Petrović, Ž. D. Bugarčić, A. Dees, I. Ivanović-Burmazović, F. W. Heinemann, R. Puchta, S. N. Steinmann, C. Corminboeuf and R. van Eldik, Inorg. Chem., 2012, 1516–1529 CrossRef PubMed.
- M. T. Ashby, Comments Inorg. Chem., 1990, 10, 297–313 CrossRef CAS.
- (a) S. G. Murray and F. R. Hartley, Chem. Rev., 1981, 81, 365–441 CrossRef CAS; (b) G. Kinunda and D. Jaganyi, Transition Met. Chem., 2016, 41, 235–248 CrossRef CAS.
- (a) S. Fakih, W. C. Tung, W. D. Eierhoff, C. Mock and B. Krebs, Z. Anorg. Allg. Chem., 2005, 631, 1397–1402 CrossRef CAS; (b) M. J. Rauterkus, S. Fakih, C. Mock, I. Puscasu and B. Krebs, Inorg. Chim. Acta, 2003, 350, 355–365 CrossRef CAS.
- V.B. Origin7.5™ SRO in Origin Lab Corporation, Northampton, One, Northampton, MA, 01060, USA, 2003.
- J. Bogojeski, Ž. D. Bugarčić, R. Puchta and R. van Eldik, Eur. J. Inorg. Chem., 2010, 5439–5445 CrossRef CAS.
- S. Jovanović, B. Petrović, D. Čanović and Ž. D. Bugarčić, Int. J. Chem. Kinet., 2011, 43, 99–106 CrossRef.
- N. Summa, W. Schiessl, R. Puchta, R. van Eikema and R. van Eldik, Inorg. Chem., 2006, 45, 2948–2959 CrossRef CAS PubMed.
- N. Hochreuther, S. T. Nandibewoor, R. Puchta and R. van Eldik, Dalton Trans., 2012, 41, 512–522 RSC.
- J. D. Atwood, Inorganic and Organometallic Reaction Mechanisms, John Wiley & Sons, Canada, 1985, pp. 82–83 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (b) P. J. Stephens, F. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 1623–1627 CrossRef.
- (a) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. W. Asman and D. Jaganyi, Int. J. Chem. Kinet., 2017, 49, 545–561 CrossRef.
- P. W. Asman, Inorg. Chim. Acta, 2018, 469, 341–352 CrossRef CAS.
- P. W. Asman, J. Coord. Chem., 2017 DOI:10.1080/00958972.2017.1371702.
- H. Ertürk, A. Hofmann, R. Puchta and R. van Eldik, Dalton Trans., 2007, 2295–2301 RSC.
- (a) V. Rai and I. N. N. Namboothiri, Eur. J. Org. Chem., 2006, 4693–4703 CrossRef CAS; (b) F. Weinhold and R. L. Clark, Discovering Chemistry with Natural Bond Orbitals, John Wiley & Sons, New Jersey, 2012, pp. 132–133 Search PubMed.
- R. Tandon, T. A. Nigst and H. Zipse, Eur. J. Org. Chem., 2013, 5423–5430 CrossRef CAS.
- (a) O. Exner, J. Phys. Org. Chem., 1999, 12, 265–274 CrossRef CAS; (b) M. Charton, J. Phys. Org. Chem., 1999, 12, 275–282 CrossRef CAS; (c) V. Galkin, J. Phys. Org. Chem., 1999, 12, 283–288 CrossRef CAS; (d) O. Exner, M. Charton and V. Galkin, J. Phys. Org. Chem., 1999, 12, 289–290 CrossRef CAS.
- H. Stephanie, R. Puchta and R. van Eldik, Inorg. Chem., 2011, 50, 8984–8996 CrossRef PubMed.
- A. Mandal, P. Karmakar, S. Mallick, B. K. Bera, S. Mondal, S. Ray and A. K. Ghosh, J. Chem. Sci., 2012, 124, 801–807 CrossRef CAS.
- G. Pistolis, A. K. Andreopoulou, A. Malliaris and J. K. Kallitsis, J. Phys. Chem. B, 2005, 109, 11538–11543 CrossRef CAS PubMed.
- Y. Qu, N. J. Scarsdale, M. C. Tran and N. Farrell, J. Biol. Inorg. Chem., 2003, 8, 19–28 CrossRef CAS PubMed; Y. Qu, N. J. Scarsdale, M. C. Tran and N. Farrell, J. Inorg. Biochem., 2004, 98, 1585–1590 CrossRef PubMed; Y. Qu, M. C. Tran and N. Farrell, J. Biol. Inorg. Chem., 2009, 14, 969–977 CrossRef PubMed.
- M. J. Birenne, J. Gabard, M. Leclercq, J. M. Lehn, M. Cesario, C. Pascard, M. Cheve and G. Dutruc-Rosset, Tetrahedron Lett., 1994, 35, 8157–8167 CrossRef.
- (a) M. Albrecht and S. Kotila, Angew. Chem., Int. Ed. Engl., 1995, 34, 2134–2137 CrossRef CAS; (b) M. Albrecht, Chem. Soc. Rev., 1998, 27, 281–288 RSC; (c) M. Albrecht, I. Janser, H. Houjou and R. Frohlich, Chem. – Eur. J., 2004, 10, 2839–2850 CrossRef CAS PubMed; (d) M. Albrecht and R. Frohlich, Bull. Chem. Soc. Jpn., 2007, 80, 797–808 CrossRef CAS; (e) C. T. Imrie and P. A. Henderson, Chem. Soc. Rev., 2007, 36, 2096–2124 RSC.
- M. G. Basallote, J. Durán, M. J. Fernández-Trujillo and M. A. Máñez, J. Chem. Soc., Dalton Trans., 1999, 3817–3823 RSC.
- E. Eliel and H. Wilen, Stereochemistry of Organic Compounds, Wiley, New York, 1994, pp. 596–599 Search PubMed.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- H. Ertürk, R. Puchta and R. van Eldik, Eur. J. Inorg. Chem., 2009, 1331–1338 CrossRef.
- K. Zamani, A. MobiniKhaledi, N. Foroughifar, K. Faghihi and V. Mahdavi, Turk. J. Chem., 2003, 27, 71–75 CAS.
- H. Ertürk, J. Magut, R. Puchta and R. van Eldik, Dalton Trans., 2008, 2759–2766 RSC.
- (a) D. Reddy and D. Jaganyi, Int. J. Chem. Kinet., 2011, 43, 161–174 CrossRef CAS; (b) A. Hofmann, L. Dahlengburg and R. van Eldik, Inorg. Chem., 2003, 42, 6528–6538 CrossRef CAS PubMed.
- O. Exner and S. Böhm, Eur. J. Org. Chem., 2007, 2870–2876 CrossRef CAS.
- A. Mambanda and D. Jaganyi, Dalton Trans., 2011, 40, 79–91 RSC.
- A. Mambanda, D. Jaganyi, S. Hochreuther and R. van Eldik, Dalton Trans., 2010, 39, 3595–3608 RSC.
- B. Pinter, V. van Speybroeck, M. Waroquier, P. Geerlings and F. de Proft, Phys. Chem. Chem. Phys., 2013, 15, 17354–17365 RSC.
- (a) C. H. Langford and H. B. Gray, Ligand Substitution Processes, W. A. Benjamin, Inc., New York, 1966, vol. 27-33, pp. 79–81 Search PubMed; (b) F. Basolo, Mechanisms of Inorganic Reactions, American Chemical Society, 1965, pp. 23–32 Search PubMed.
- (a) B. Coe and S. Glenwright, Coord. Chem. Rev., 2000, 203, 5–80 CrossRef CAS; (b) E. M. Shustorovich, M. Porai-Koshits and Y. Buslaev, Coord. Chem. Rev., 1975, 17, 1–98 CrossRef CAS.
- P. O. Ongoma and D. Jaganyi, Transition Met. Chem., 2014, 39, 407–420 CrossRef CAS; D. Jaganyi, V. M. Munisamy and D. Reddy, Int. J. Chem. Kinet., 2006, 38, 202–210 CrossRef.
- D. Jaganyi, F. Tiba, O. Q. Munro, B. Petrović and Ž. D. Bugarcić, Dalton Trans., 2006, 2943–2949 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Selected wavelengths used for kinetic measurements, tables of observed pseudo-first-order rate constants, Eyring plots, mass spectrometry: mass spectral data showing respective molecular ions and their fragmentation patterns, NMR data for 1H NMR, 13C NMR and 195Pt NMR, pKa titration spectra of the complexes. See DOI: 10.1039/c7nj03021e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |