
Three in situ-synthesized novel inorganic–organic hybrid materials based on metal (M = Bi, Pb) iodide and organoamine using one-pot reactions: structures, band gaps and optoelectronic properties†
Zhi Shuo
Yan
a,
Ji Ying
Long
a,
Yun
Gong
 *a and
Jian
Hua Lin
*ab
*a and
Jian
Hua Lin
*ab
aDepartment of Applied Chemistry, College of Chemistry and Chemical Engineering, Chongqing University, Chongqing 401331, P. R. China. E-mail: gongyun7211@cqu.edu.cn; Tel: +86-023-65678932
bState Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: jhlin@pku.edu.cn; Tel: +86-010-62753541
First published on 29th November 2017
Abstract
Based on metal iodide, three novel inorganic–organic hybrid materials formulated as (N-ethyl-pyridine)2(N-ethyl-1,3,4-triazole)(Bi2I9) (1), [(N-ethyl-pyridine)(PbI3)]n (2) and [(Hpyridine)(PbI3)]n (3) were in situ – synthesized solvothermally and they have been characterized by single-crystal X-ray diffraction. In compound 1, the in situ-formed (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+ cations are linked with a zero-dimensional (0D) (Bi2I9)3− cluster via strong H bonds to form a three-dimensional (3D) supramolecular architecture. While the in situ-formed (N-ethyl-pyridine)+ cations in compound 2 and (Hpyridine)+ cations in compound 3 are linked with infinite one-dimensional (1D) Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains via strong H bonds. Density functional theory (DFT) calculations demonstrate that the three compounds have different band structures. And among them, compound 1 possesses the narrowest band gap and it produces the largest photocurrent density under visible light illumination (350 nm < λ < 650 nm). Moreover, compounds 1 and 2 possess good water-proof properties in aqueous solution, which is probably due to hydrophobic aromatic cations such as (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+ in their structures.
Introduction
Recently, organic–inorganic hybrid materials including metal–metalloid clusters or chains have drawn great interest not only due to their low cost and easy preparation, but also due to their tunable physicochemical properties.1 To date, lead halide perovskites such as CH3NH3PbI3, as one of the most typical organic–inorganic hybrid materials, have been widely studied. The perovskite CH3NH3PbI3 is the most suitable solar cell absorber due to its proper optical band gap (1.5 eV), high extinction coefficient, small exciton binding energy as well as relative high power conversion efficiencies (PCEs) exceeding the 20% efficiency milestone.2 However, the perovskite CH3NH3PbI3 exhibits severe degradation in ambient air and is vulnerable to environmental stresses and humidity, which is ascribed to its inherent vulnerability to moisture and heat.3 Thus, their long-term operational stability remains a bottleneck toward their practical applications.Because hydrophilic CH3NH3+ in the three-dimensional (3D) Pb–I–Pb skeleton of CH3NH3PbI3 can be easily replaced by bulky H2O molecules in the presence of aqueous solution, further deteriorating the physicochemical properties of CH3NH3PbI3,4 in order to solve the moisture stability of the perovskite CH3NH3PbI3, hydrophilic CH3NH3+ can be replaced with hydrophobic aromatic cations with a higher molecular weight. On the other hand, it is well known that a low-dimensional framework displays better charge transfer ability with respect to the 3D framework.5 If the 3D Pb–I–Pb skeleton of the perovskite CH3NH3PbI3 was altered to a low-dimensional framework, such as metal–metalloid clusters or a one-dimensional (1D) chain, it is expected that the photophysical and electronic properties may be improved.
Based on this, we utilize three organic precursors, 3-(5-(5-(5-(pyridin-3-yl)-2H-1,2,4-triazol-3-yl) thiophen-2-yl)-1H-1,2,4-triazol-3-yl) pyridine (L1), 2,6-di (1H-imidazol-1-yl) pyridin-4-ol (L2), and 2-(5-(5-bromopyridin-3-yl)-2H-1,2,4-triazol-3-yl) pyridine (L3) (Scheme 1), for the in situ synthesis of three metal halide-based inorganic–organic hybrid materials formulated as (N-ethyl-pyridine)2(N-ethyl-1,3,4-triazole)(Bi2I9) (1), [(N-ethyl-pyridine)(PbI3)]n (2) and [(Hpyridine)(PbI3)]n (3) via one-pot reactions (Scheme 2). Compound 1 has a zero-dimensional (0D) (Bi2I9)3− cluster while compounds 2 and 3 possess an infinite one-dimensional (1D) Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chain, in the structure of compounds 1–3, the hydrophobic aromatic organoamine cations were all included. Density functional theory (DFT) calculations reveal that the three compounds possess different band structures. Their UV-vis absorption and diffuse reflectance spectra (DRS), photoluminescence properties, electrochemical behaviors, photocurrent responses as well as thermal stabilities have been investigated.
 | ||
| Scheme 1 Schematic views of the organic precursors L1 (a), L2 (b), and L3 (c). | ||
 | ||
| Scheme 2 The nucleophilic reactions concerning the in situ-formed aromatic organoamine cations derived from the organic precursors L1 (a), L2 (b), and L3 (c). | ||
Experimental section
General considerations
All the starting chemicals and reagents were purchased from commercial resources and without further purification. 3-(5-(5-(5-(Pyridin-3-yl)-2H-1,2,4-triazol-3-yl) thiophen-2-yl)-1H-1,2,4-triazol-3-yl) pyridine (L1), 2,6-di(1H-imidazol-1-yl) pyridin-4-ol (L2), and 2-(5-(5-bromopyridin-3-yl)-2H-1,2,4-triazol-3-yl) pyridine (L3) were obtained from Jinan Henghua Sci. & Tec. Co., Ltd. The melting point was determined using an uncorrected X-4 melting point apparatus of Beijing Kaifu Company. Infrared spectra (IR) were recorded using KBr pellets in the range 4000–400 cm−1 on a Nicolet iS50 FT-IR spectrometer. Powder X-ray diffraction (PXRD) data were collected on a Tongda TD-3500 diffractometer using Cu Kα radiation (λ = 0.154056 nm). UV-vis absorption and diffuse reflectance spectra (DRS) were measured on a Beijing Puxi TU-1901 UV-vis spectrometer. Solid-state emission spectra were measured on an Agilent Cary Eclipse fluorescence spectrometer at room temperature. The morphologies of the samples were observed by scanning electron microscopy (SEM, JSM-7600F, JEOL). Thermogravimetric analyses (TGA) were implemented on a NETZSCHSTA 449C instrument from room temperature (∼25 °C) to 800 °C under steady N2 flowwith a heating rate of 10 °C min−1.Computational methods
The band structures and density of states (DOS) of compounds 1–3 and the perovskite CH3NH3PbI3 were calculated based on density functional theory (DFT) through the CASTEP program in the Materials Studio 7.0. And a two-step methodology was employed for the calculation. The first step was to geometrically optimize the crystal structures based on the crystallographic data. The second step was to calculate the band structures and density of states (DOS) for the optimized structures. To simplify the calculation, the disordered atom was removed from the crystal structure of compound 3. All the calculations were done using the GGA-PBE functional and ultrasoft pseudo-potentials based on reciprocal space with a 500 eV of kinetic energy cutoff and 1.0 × 10−5 eV per atom of SCF tolerance thresholds. The sets of k points chosen for compounds 1–3 and CH3NH3PbI3 are 2 × 2 × 2, 3 × 3 × 2, 3 × 3 × 2 and 2 × 2 × 2, respectively.Electrochemical measurements
The electrochemical measurements were carried out in a three-electrode cell system with a platinum foil and a saturated calomel electrode (SCE) as the counter and reference electrodes, respectively. The working electrode was modified using a drop-coating method. Typically, 2 mg of sample was uniformly dispersed in 2 mL of anhydrous ethanol and 0.05 mL of nafion solution (5%) by ultrasonication to form homogeneously mixed suspension, and then 0.4 mL of the well-dispersed suspension were deposited on a well-polished glassy carbon electrode (GCE) and dried using an IR lamp to obtain a sample-modified working electrode with a geometrical area of 0.2 cm2, and a loading density of 0.4 mg cm−2. 0.2 M Na2SO4 neutral aqueous solution (50 mL) was used as the electrolyte and all the electrochemical measurements were performed on a CHI660E electrochemical workstation (Shanghai, China).Photoelectrochemical measurements
The photoelectrochemical measurements were performed in a three-electrode cell with an Ag/AgCl electrode and platinum foil as the reference and counter electrodes, respectively. The working electrode was also prepared using a drop-coating method. First of all, a fluorine doped tin oxide (FTO)-coated glass substrate (12–14 μS m−1) was washed with acetone, isopropyl alcohol and deionized water successively to remove the impure chemicals. Secondly, 2 mg of the sample was dispersed well in 2 mL of anhydrous ethanol by ultrasonication to obtain a homogeneous ink, and 0.4 mL of the homogeneous ink were dropped carefully on the surface of an FTO conductive glass substrate with a geometrical area of 1 × 1 cm2 to obtain the working electrode (also denoted as photoelectrode), whose loading density was 0.4 mg cm−2. All the electrodes were immersed in a neutral aqueous solution (50 mL) of 0.2 M Na2SO4 and a CHI660E electrochemical workstation was used for photoelectrochemical measurements. The transient photocurrent responses of the photoelectrodes were measured at 0 V vs. Ag/AgCl under intermittent visible light irradiation from the back side using a 300 W xenon lamp (CEL-HXF300, Beijing Aulight Co., 350 nm < λ < 650 nm) with a light irradiation interval of 100 seconds in the whole test time of 1800 seconds. Electrochemical impedance spectroscopy (EIS) measurements were conducted in the frequency range of 0.01 Hz–1 MHz at the potential of 0 V vs. Ag/AgCl.Syntheses
X-ray crystallographic study
Single-crystal X-ray diffraction data collection for compounds 1–3 were performed on a Bruker-APEX CCD area detector-equipped diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at room temperature. All the structures were solved using direct methods and refined by full-matrix least-squares on F2 using the SHELX-97 program. All the non-hydrogen atoms were refined anisotropically, and all the hydrogen atoms were generated geometrically. The crystal data and structure refinements for compounds 1–3 are summarized in Table 1. Selected bond lengths and angles for compounds 1–3 are listed in Table 2. The CCDC 1524687 for compound 1, 1524688 for compound 2, and 1524689 for compound 3.† The crystal data and structural information for the perovskite CH3NH3PbI3 can be found in the previous literature.7| Compounds | 1 | 2 | 3 |
|---|---|---|---|
| a R 1 = ∑(||Fo| − |Fc||)/∑|Fo|. b wR2 = ∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]1/2. | |||
| Empirical formula | C18H28N5I9Bi2 | C7H10NI3Pb | C5H6NI3Pb |
| Formula weight | 1874.51 | 696.05 | 668.00 |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | Pnma | Pnma | Pnma |
| a (Å) | 16.7661(15) | 18.36(2) | 14.9571(12) |
| b (Å) | 22.860(2) | 7.910(10) | 8.0960(8) |
| c (Å) | 11.4060(11) | 19.67(2) | 9.8979(9) |
| α (°) | 90.00 | 90.00 | 90.00 |
| β (°) | 90.00 | 90.00 | 90.00 |
| γ (°) | 90.00 | 90.00 | 90.00 |
| V (Å3) | 4371.7(7) | 2857(6) | 1198.56(19) |
| Z | 4 | 8 | 4 |
| D c (g cm−3) | 2.848 | 3.237 | 3.702 |
| μ (mm−1) | 14.410 | 18.259 | 21.750 |
| F(000) | 3256 | 2400 | 1136 |
| θ range (°) | 2.34–25.01 | 2.35–25.02 | 2.47–25.02 |
| R (int) | 0.0000 | 0.0918 | 0.1676 |
| R 1,a wR2b (I > 2σ(I)) | 0.0859, 0.2283 | 0.0400, 0.0824 | 0.0900, 0.2185 |
| GOF on F2 | 1.025 | 1.044 | 1.115 |
| Symmetry transformations used for generating equivalent atoms: #1: x, −y + 1/2, z; #2: x, −y + 5/2, z; #3: x, −y + 3/2, z; #4: −x, −y, −z + 1. | |||
|---|---|---|---|
| Compound 1 | |||
| Bi(1)–I(5) | 2.940(3) | Bi(1)–I(4) | 2.952(3) |
| I(1)–Bi(1)#1 | 3.255(2) | I(3)–Bi(1)#1 | 3.282(2) |
| Bi(1)–I(3)–Bi(1)#1 | 80.68(7) | Bi(1)#1–I(1)–Bi(1) | 81.51(8) |
| I(1)–Bi(1)–I(3) | 81.16(6) | I(4)–Bi(1)–I(1) | 173.38(7) |
| Compound 2 | |||
| I(1)–Pb(1)#2 | 3.209(3) | Pb(1)–I(5) | 3.220(3) |
| Pb(1)–I(4) | 3.240(3) | Pb(1)–I(2) | 3.273(3) |
| Pb(1)–I(2)–Pb(1)#3 | 73.83(9) | I(4)–Pb(1)–I(2) | 84.29(8) |
| I(4)–Pb(1)–I(3) | 172.98(3) | I(1)–Pb(1)–I(2) | 176.28(3) |
| Compound 3 | |||
| Pb(1)–I(3) | 3.2184(15) | Pb(1)–I(1)#4 | 3.2595(16) |
| I(2)#4–Pb(1)–I(1) | 84.19(4) | I(3)–Pb(1)–I(3)#4 | 180.00(4) |
Results and discussion
Syntheses
Compounds 1–3 were synthesized solvothermally employing one-pot reactions. Interestingly, it is found that the organic precursors L1, L2 and L3 have been transformed into (N-ethyl-pyridine)+/(N-ethyl-1,3,4-triazole)+, (N-ethyl-pyridine)+ and (Hpyridine)+ during the syntheses of compounds 1, 2 and 3, respectively (Scheme 2). The possible mechanism for the in situ formed organoamine is as follows. Firstly, the organic precursor was decomposed into a single aromatic ring such as pyridine or triazole group under the solvothermal conditions. In the syntheses of compounds 1 and 2, in the presence of H+, the solvent molecule EtOH combined with H+ to form CH3CH2+, then the CH3CH2+ intermediate reacted with single aromatic rings nucleophilically to form N-ethylated organoamine cations (Scheme 2a and b). Herein, the injection of dilute HNO3 (∼1 M) into the reaction mixture plays an important role in the in situ reaction, which can enhance the dissociation of the –OH group from EtOH to form the CH3CH2+ group.Crystal structure of (N-ethyl-pyridine)2(N-ethyl-1,3,4-triazole)(Bi2I9) (1)
Based on single-crystal X-ray diffraction analysis, compound 1 crystallizes in the orthorhombic crystal system with the space group Pnma (Table 1). The asymmetric unit contains one (BiI6)3− ion, one uncoordinated (N-ethyl-pyridine)+ cation and a half uncoordinated (1-N-ethyl-1,3,4-triazole)+ cation. As shown in Fig. 1a, Bi1 displays a slightly distorted octahedral coordination geometry, occupied by three μ2-I− and three terminal I− ions [Bi–I distance: 2.940(3)–3.282(2) Å; ∠I–Bi–I: 81.16(6)–173.38(7)°] (Table 2). The valence sum calculation shows that the crystallographically independent Bi ion is in 3+ oxidation state.8 | ||
| Fig. 1 Coordination environment of the Bi(III) ion in compound 1, which is constructed by two (N-ethyl-pyridine)+, one (1-N-ethyl-1,3,4-triazole)+ and one (Bi2I9)3− (H atoms omitted for clarity) (a); 3D supramolecular architecture of compound 1 constructed by the H bonds between the organoamine and the (Bi2I9)3− clusters (H bonds denoted by dotted lines) (b). | ||
In compound 1, two Bi(III) ions are linked by three μ2-I− ions to form a zero-dimensional (0D) (Bi2I9)3− cluster with a Bi⋯Bi separation of 4.250 Å, in which another six I− ions act as terminal ligands. The uncoordinated organoamines, (N-ethyl-pyridine)+ and (1-N-ethyl-1,3,4-triazole)+ are linked with the (Bi2I9)3− clusters via H bonds (Table 3) into a 3D supramolecular architecture (Fig. 1b).
| D | H | A | D⋯A distance | H⋯A distance | ∠D–H⋯A |
|---|---|---|---|---|---|
| Symmetry transformations used for generating equivalent atoms: #1: 1 − x, −1/2 + y, 1 − z; #2: 3/2 − x, 1 − y, −1/2 + z; #3 1 + x, 1 + y, −1 + z; #4 1/2 + x, 1 + y, 1/2 − z. | |||||
| Compound 1 | |||||
| C6 | H6 | I5 | 4.31 | 3.22 | 156 |
| C5 | H5 | I4 | 4.28 | 3.10 | 144 |
| Compound 2 | |||||
| C3 | H3 | I4#1 | 4.03 | 3.27 | 167 |
| C4 | H4 | I3#2 | 4.42 | 3.37 | 164 |
| Compound 3 | |||||
| C2 | H2 | I1#3 | 4.18 | 3.16 | 139 |
| N1 | H1 | I3#4 | 4.22 | 3.37 | 121 |
Crystal structure of [(N-ethyl-pyridine)(PbI3)]n (2) and [(Hpyridine)(PbI3)]n (3)
Compounds 2 and 3 both crystallize in the orthorhombic crystal system with the space group Pnma according to the single-crystal X-ray diffraction analyses (Table 1). In compound 2, the asymmetric unit contains one (PbI6)4− ion and two half uncoordinated (N-ethyl-pyridine)+ (Fig. 2a). Whereas in compound 3, the asymmetric unit contains a half (PbI6)4− ion and a half uncoordinated protonated pyridine cation (Hpyridine)+ (Fig. 3a). In the two compounds, both the crystallographically independent Pb1 ions display a slightly distorted octahedral coordination geometry, occupied by six μ2-I− ions (Fig. 3a and 4a) [Pb–I distance: 3.209(3)–3.273(3) Å in 2 and 3.2184(15)–3.2595(16) Å in 3; ∠I–Pb–I: 84.29(8)–176.28(3)° in 2 and 84.19(4)–180.00(4)° in 3] (Table 2). Valence sum calculations display the crystallographically independent Pb ions in the two compounds both in +2 oxidation state.8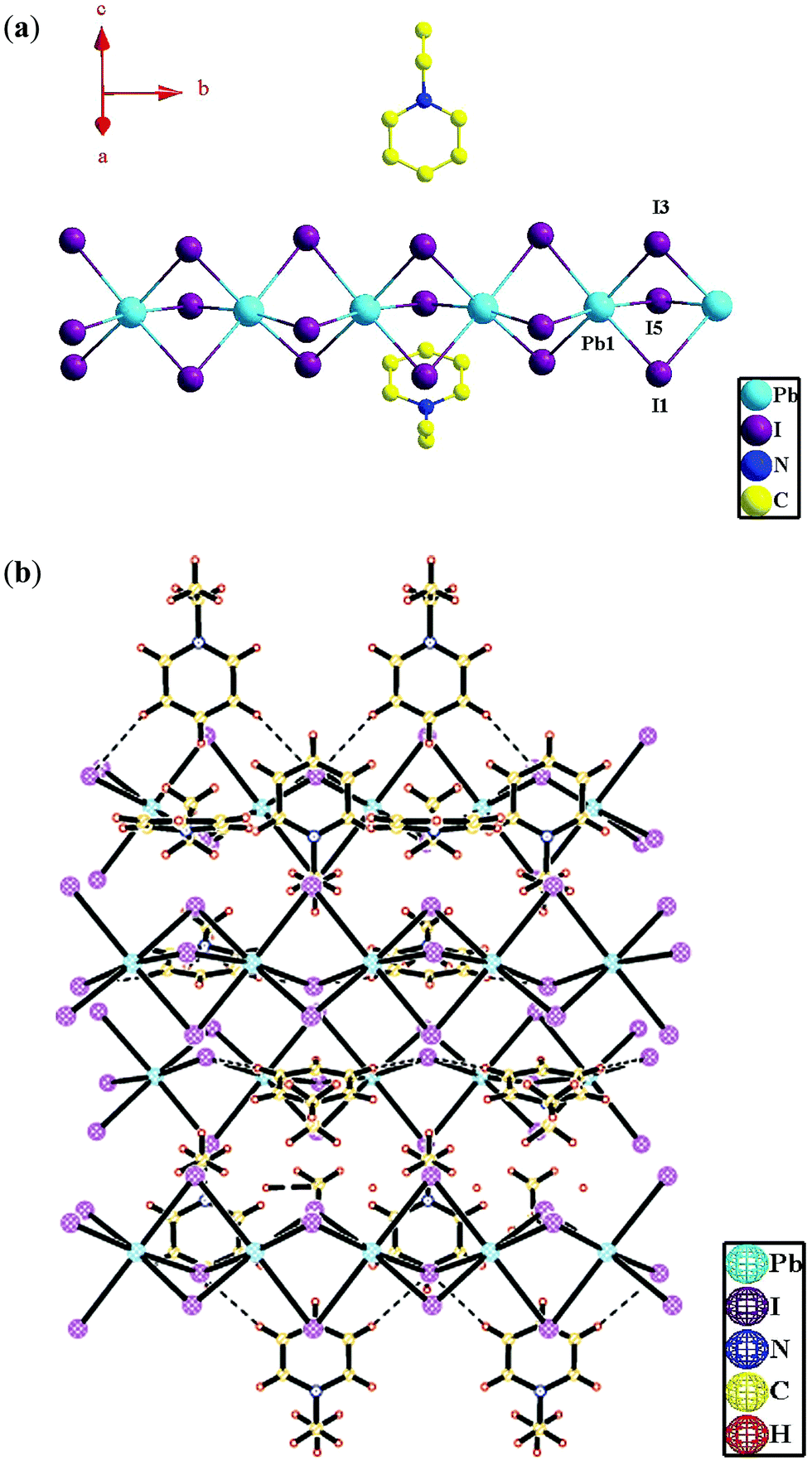 | ||
| Fig. 2 Coordination environment of the Pb(II) ion in compound 2, highlighting the infinite 1D Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains (H atoms omitted for clarity) (a); 3D supramolecular architecture of compound 2 constructed by the H bonds between (N-ethyl-pyridine)+ cations and Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains (H bonds denoted by dotted lines) (b). | ||
 | ||
| Fig. 3 Coordination environment of the Pb(II) ion in compound 3, highlighting the infinite 1D Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains (H atoms omitted for clarity) (a); 3D supramolecular architecture of compound 3 constructed by the H bonds between (Hpyridine)+ cations and Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains (H bonds denoted by dotted lines) (b). | ||
In compounds 2 and 3, three I− ions bridge two Pb(II) ions to form an infinite one-dimensional (1D) Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chain (Fig. 2a and 3a). And different infinite Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains are parallel to each other. The uncoordinated organoamine cations, such as (N-ethyl-pyridine)+ in 2 and (Hpyridine)+ in 3, are linked with the infinite Pb-(μ2-I)3-Pb-(μ2-I)3-Pb chains via H bonds (Table 3) to form a 3D supramolecular architecture (Fig. 2b and 3b).
Band structures and orbital calculations for compounds 1–3 and perovskite CH3NH3PbI3
Based on density functional theory (DFT), the band gaps of compounds 1–3 have been calculated by employing the CASTEP program. As shown in Fig. 4, the calculated band gaps (Egap) of compounds 1–3 are 0.003, 1.842 and 2.778 eV, respectively (Fig. 4a–c). For comparison, the band gap of the perovskite CH3NH3PbI3 is also calculated using the same method. As shown in Fig. 4d, the band gap of CH3NH3PbI3 is 1.493 eV, which is in good accordance with the value (1.5 eV) reported in the previous literature,9 indicating that our calculation method is correct. | ||
| Fig. 4 Band structures for compounds 1 (a), 2 (b), 3 (c) and the perovskite CH3NH3PbI3 (d) near the Fermi energy level (EFermi). The Fermi energy is zero in the energy scale. | ||
In order to further investigate the band structures of compounds 1–3 and the perovskite CH3NH3PbI3, their total and partial density of states (TDOS/PDOS) have also been calculated using the CASTEP program (Fig. 5). As shown in Fig. 5a, it can be found that both the valence band maximum (VBM) and the conduction band minimum (CBM) of compound 1 are dominated by the 2p orbitals of C and N, the 2s orbital of N, the 6p orbital of Bi and the 5p orbital of I (Fig. 5a). And the VBM of compound 2 is mainly constituted by the 5p orbital of I and the 6s orbital of Pb, while its CBM is dominated by the 2p orbitals of C and N atoms (Fig. 5b). Compound 1 possesses a much narrow band gap than compound 2, which is attributed to their different frameworks and different metal centers. As discussed above, compound 1 is constructed by a (Bi2I9)3− cluster and organic cations, (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+, the difference between the DOS of its VBM and CBM is not obvious. Whereas compound 2 is constituted by the Pb–I–Pb chain and the (N-ethyl-pyridine)+ cation, its VBM is mainly dependent on the Pb–I–Pb chain, and its CBM is associated with the organic cation, leading to different band gaps of the two compounds.
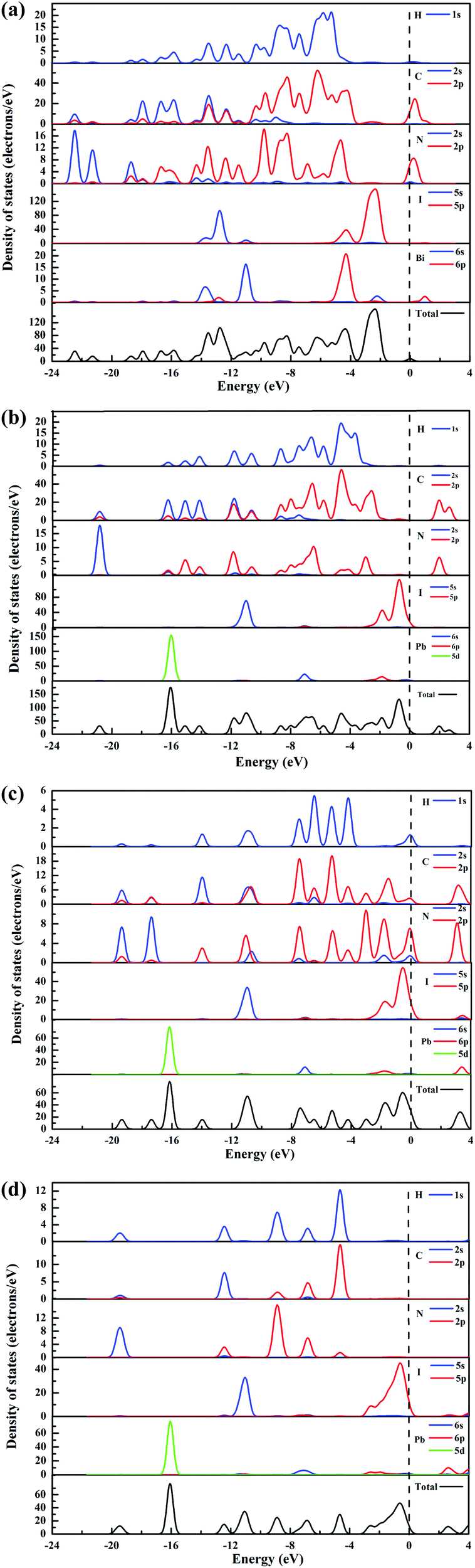 | ||
| Fig. 5 TDOS and PDOS of compounds 1 (a), 2 (b), 3 (c) and the perovskite CH3NH3PbI3 (d). In the PDOS, blue, red, and green lines represent s, p and d orbitals, respectively. | ||
As shown in Fig. 5c, the VBM of compound 3 is mainly ascribed to I 5p, Pb 6s, C 2p, N 2p, N 2s and H 1s states, meanwhile, its CBM is dominated by Pb 6p, I 5p, I 5s, C 2p and N 2p states. As discussed above, compound 3 is also constructed by the Pb–I–Pb chain and the organic cation, indicating that the VBM and the CBM of compound 3 are associated with both the inorganic Pb–I–Pb chain and the organic cation. The organic cation in compound 3 is (Hpyridine)+, which is different from the (N-ethyl-pyridine)+ cation in compound 2. The C, N contents of the two organic cations are different, and the interactions between the organic cations and the Pb–I–Pb chain are different, thus giving rise to the different DOS and band gaps of compounds 2 and 3.
The TDOS and PDOS of the perovskite CH3NH3PbI3 are shown in Fig. 5d. The VBM of CH3NH3PbI3 is dominated by I 5p and Pb 6s states, and its CBM is mainly composed by the Pb 6p state, indicating that the VBM and the CBM of the perovskite are mostly dependent on its 3D inorganic Pb–I–Pb architecture.10 The organic cations, CH3NH3+, occupied the channel in the 3D inorganic framework,11 which does not have obvious contribution to the band gap of the perovskite.
UV-vis absorption (UV) and diffuse reflectance spectra (DRS) for compounds 1–3 and the perovskite CH3NH3PbI3
As shown in Fig. S1 (ESI†), the phase purities of bulk compounds 1–3 and CH3NH3PbI3 in the solid state are characterized by powder X-ray diffraction (PXRD). It was clear that all the peaks in the measured patterns were in good agreement with the corresponding simulated ones, which were derived from single-crystal diffraction data (Fig. S1a–d, ESI†), indicating that each of them is in pure phase.The UV-vis absorption spectra of compounds 1–3 as well as those of the perovskite CH3NH3PbI3 in the solid state were measured at room temperature in order to investigate the probable mechanism of electronic transition. As shown in Fig. S2 (ESI†), in the range of 230–450 nm, compounds 1–3 and CH3NH3PbI3 display absorption peaks at 230/318 (for 1), 230/366 (for 2), 230/390 (for 3) and 230/328//392 nm (for CH3NH3PbI3), respectively, which can be ascribed to the n–π* or π–π* electron transitions. Meanwhile, in the range of 450–850 nm, compounds 1–3 as well as the perovskite CH3NH3PbI3 show absorption peaks at 568 (for 1 and 2), 472/552 (for 3) and 482/568/742 nm (for CH3NH3PbI3), respectively, which are probably attributed to the visible d–d transition.
UV-vis diffuse reflectance spectra (DRS) of compounds 1–3 and CH3NH3PbI3 were also measured. The plot of the reflectance function vs. radiation energy is shown in Fig. S3 (ESI†), and the experimentally determined band gaps (Egap) of compounds 1–3 and CH3NH3PbI3 are 0.2, 2.4, 2.9 and 1.5 eV, respectively (the band-gap extraction method is elaborated in the ESI†).12 As described above, the calculated band gaps (Egap) of compounds 1–3 are 0.003, 1.842 and 2.778 eV, respectively (Fig. 4a–c), which are smaller than the respective experimentally obtained values of ca. 0.2–0.6 eV. The inconsistency is usual for DFT calculations, which generally underestimate the band gap energy of semiconductors.
Photoluminescence properties of compounds 1–3 and the perovskite CH3NH3PbI3
In order to investigate the photo-induced charge transfer and recombination for compounds 1–3 and the perovskite CH3NH3PbI3, the solid-state photoluminescence properties of the samples have been measured at room temperature. As shown in Fig. S4 (ESI†), compounds 1–3 and CH3NH3PbI3 all display a weak emission peak at 519 nm (λex = 320 nm, slit width = 10 nm). Due to the stable 5d106s2 configuration of both the Bi(III) and Pb(II) ions, they are difficult to be oxidized or reduced.13 As a result, the emission of the samples is neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature.14 Consequently, it can be supposed that the n–π* or π–π* electron transition plays an important role in the emission.The electrochemical properties of compounds 1–3
The electrochemical properties of compounds 1–3 were evaluated by cyclic voltammetry (CV) in 0.2 M neutral Na2SO4 aqueous solution, which was carried out in a three-electrode cell with a saturated calomel electrode (SCE) and a Pt foil as the reference and counter electrodes, respectively. The well-polished glassy carbon electrode (GCE) was deposited by the as-synthesized samples to be used as the working electrode. For comparison, the CV of the bare GCE was also measured under the same conditions. However, the CV of the perovskite CH3NH3PbI3 was not performed due to its inherent vulnerability to water.As shown in Fig. 6 and Fig. S5 (ESI†), no redox peak can be observed at the bare GCE in the potential range of −1.8 to 1.8 V vs. SCE at a scan rate of 0.005 V s−1. As shown in Fig. 6 and Fig. S6 (ESI†), a compound 1-modified glassy carbon electrode (1-GCE) exhibits two quasi-reversible redox couples at −0.82/−0.42 and −0.35/−0.02 V vs. SCE, respectively. For a 2-modified GCE (2-GCE), two quasi-reversible redox peaks are found at −0.90/−0.63 and −0.80/−0.44 V vs. SCE, respectively (Fig. 6 and Fig. S7, ESI†). And two oxidation peaks at −0.82 and −0.34 V vs. SCE and two reduction peaks at −0.64 and −0.42 V vs. SCE are observed at a 3-modified GCE (3-GCE) (Fig. 6 and Fig. S8, ESI†). The redox properties of the three compounds are probably associated with the redox of I− rather than the Bi(III) or Pb(II) ions in the structures due to their stable 5d106s2 configuration.15 Notably, the redox peaks of the three compounds are a bit different from each other, which are possibly ascribed to the different organic cations in the three compounds, leading to their different frameworks.
 | ||
| Fig. 6 CVs of the bare GCE (brown), 1-GCE (black), 2-GCE (red) and 3-GCE (blue) as working electrodes in 0.2 M Na2SO4 solution at the scan rate of 0.005 V s−1 (scan directions denoted in black arrows). | ||
As we know, the highest occupied molecular orbital (HOMO) energy level (EHOMO) of a semiconductor can be calculated based on the equation EHOMO = −(4.74 + Eox) eV vs. vacuum (Eox: the onset potential for the first oxidation peak), and the lowest unoccupied molecular orbital (LUMO) level (ELUMO) can be deduced through the formula Egap = ELUMO − EHOMO. As shown in Fig. S6b, S7b and S8b (ESI†), it is found that the Eox values for compounds 1–3 is −0.51, −0.71 and −0.70 V vs. SCE, respectively. Then the HOMO levels (EHOMO) of compounds 1–3 can be calculated as −4.23, −4.03 and −4.04 eV vs. vacuum, respectively (Table 4). And the LUMO levels (ELUMO) of compounds 1–3 can be estimated as −4.03, −1.63 and −1.14 eV vs. vacuum, respectively (Table 4).
| Compounds | 1 | 2 | 3 |
|---|---|---|---|
| The LUMO level vs. vacuum (eV) | −4.03 | −1.63 | −1.14 |
| The HOMO level vs. vacuum (eV) | −4.23 | −4.03 | −4.04 |
| Band gap (eV) | 0.2 | 2.4 | 2.9 |
The photoelectrochemical properties of compounds 1–3
In order to investigate the semiconductor characteristics of compounds 1–3, the transient photocurrent responses of compounds 1–3 were measured in a three-electrode cell at E = 0 V vs. Ag/AgCl using a 300 W Xe lamp (350 nm < λ < 650 nm) applied as the light source, whose light illumination power was 100 mW cm−2. The sample-modified FTO slice was used as the working electrode, and a Pt foil and a Ag/AgCl were used as the counter and reference electrodes, respectively. The morphologies of the sample-modified FTO are characterized by scanning electron microscopy (SEM) (Fig. S9, ESI†). As shown in Fig. S9 (ESI†), the top-view SEM images of the three compounds indicating the samples have been uniformly distributed on the surface of FTO slices. In order to test the “water-resisting” properties of the three compounds, neutral Na2SO4 aqueous solution (0.2 M, 50 mL) was used as the electrolyte and the illumination is set with an interval of 100 seconds in the whole test time of 1800 seconds.As shown in Fig. S10 (ESI†), it is found that the compound 2-modified FTO electrode (2-FTO) and the compound 3-modified FTO electrode (3-FTO) exhibit similar photocurrent behavior when exposed to visible light (350 nm < λ < 650 nm) from the back side, whose largest photocurrent density is 1.5 × 10−4 mA cm−2 approximately. Whereas the compound 1-modified FTO electrode (1-FTO) displays the most energetic photo-induced activity under visible light illumination from the back side with the largest photocurrent density of ca. 4 × 10−3 mA cm−2, which is nearly 25 times more than those yielded on 2-FTO or 3-FTO (Fig. S10, ESI†).
As discussed above, the coexistence of two kinds of organic cations, (N-ethyl-pyridine)+, (N-ethyl-1,3,4-triazole)+ and the (Bi2I9)3− dimer in the structure of compound 1 makes its band gap as low as 0.2 eV, leading to the separation of the photo-induced electron–hole pairs more easier, then the transient photocurrent response of compound 1 is predominant in comparison to 2 and 3.
In order to further analyze the charge transfer and recombination loss at the interfaces between 1-, 2-, or 3-FTO with the electrolyte, the electrochemical impedance spectra (EIS) were measured at E = 0V vs. Ag/AgCl with and without visible light illumination (350 nm < λ < 650 nm). Usually, the high-frequency region in EIS reveals the charge transfer impedance at the interface, and the smaller the arc radius of the high-frequency region in the Nyquist plot, the lower the interfacial resistance.
As shown in Fig. S11 (ESI†), it can be found that the charge transfer impedance at the sample/electrolyte interface under visible light irradiation is lower than that without irradiation regardless of 1-, 2-, and 3-FTO, indicating that visible light irradiation can decrease the charge transfer impedance and inhibit the charge recombination.16 Notably, the charge transfer impedance for 1-FTO is smaller than those for 2-FTO and 3-FTO under illumination (Fig. S11, ESI†), suggesting that compound 1 may yield the strongest photocurrent density with respect to the other samples, which is in good accordance with their photoelectrochemical behaviors (Fig. S10, ESI†).
It is well known that the perovskite CH3NH3PbI3 does not possess “water-shielding” properties due to the gradual loss of CH3NH3+ in the presence of aqueous solution, resulting in the decomposition or inactivation.17 However, compounds 1 and 2 show stable photoresponses in aqueous solution (Fig. S10, ESI†), indicating that they exhibit “water-resisting” properties when immersed in aqueous solution, which is probably due to the hydrophobic aromatic cations such as (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+ in their structures.
Thermogravimetric analyses (TGA) of compounds 1–3
Using thermogravimetric analyses (TGA), the thermal stabilities of the three compounds have been estimated from 25 to 800 °C under the steady flow of nitrogen with a heating rate of 10 °C min−1. The TGA curve of compound 1 displays a weight loss of 11.0% in the range of 25–280 °C, corresponding to the partial loss of the uncoordinated organoamine cations (calcd 16.8%). Then the sample can be stable up to 400 °C (Fig. S12, ESI†).Compound 2 exhibits a gradual weight loss in the range of 25–480 °C, corresponding to the partial removal of organoamine cations (obsd 3.5%; calcd 15.5%). When the temperature is higher than 480 °C, the TG curve of compound 2 shows a sharp weight loss, indicating that the decomposition in the higher temperature range is dramatic (Fig. S12, ESI†).
As for compound 3, a weight loss of 6.5% can be found in the range of 25–70 °C, it is due to the partial release of organic cations (calcd 12.0%) (Fig. S12, ESI†). Then the sample remains stable up to about 300 °C without any weight loss. The decomposition of the sample continues when the temperature is higher than 300 °C (Fig. S12, ESI†).
Conclusion
In conclusion, using 3-(5-(5-(5-(pyridin-3-yl)-2H-1,2,4-triazol-3-yl)thiophen-2-yl)-1H-1,2,4-triazol-3-yl)pyridine (L1), 2,6-di (1H-imidazol-1-yl)pyridin-4-ol (L2), and 2-(5-(5-bromopyridin-3-yl)-2H-1,2,4-triazol-3-yl)pyridine (L3), three inorganic–organic hybrid materials formulated as (N-ethyl-pyridine)2(N-ethyl-1,3,4-triazole)(Bi2I9) (1), [(N-ethyl-pyridine)(PbI3)]n (2) and [(Hpyridine)(PbI3)]n (3) were in situ – synthesized solvothermally and they have been characterized by single-crystal X-ray diffraction.In compound 1, the in situ-formed (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+ cations are linked with the 0 D (Bi2I9)3− cluster via strong H bonds to form a 3D supramolecular architecture. While the in situ-formed (N-ethyl-pyridine)+ cations in compound 2 and (Hpyridine)+ cations in compound 3 are linked with 1D Pb–I–Pb chains via strong H bonds. DFT calculations and DRS reveal that the three compounds have different band structures. And among them, compound 1 possesses the narrowest band gap and it produces the largest photocurrent density under visible light illumination (350 nm < λ < 650 nm). Moreover, compounds 1 and 2 possess good waterproof properties in aqueous solution, which is probably due to the hydrophobic aromatic cations such as (N-ethyl-pyridine)+ and (N-ethyl-1,3,4-triazole)+ in their structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21371184 and 21771028), the large-scale instrument and equipment open foundation in Chongqing University (No. 201606150053), National-Municipal Joint Engineering Laboratory for Chemical Process Intensification and Reaction, and Chongqing Key Laboratory of Chemical Process for Clean Energy and Resource Utilization is gratefully acknowledged.Notes and references
- (a) W. Nie, H. Tsai, R. Asadpour, J. C. Blancon, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam, H. L. Wang and A. D. Mohite, Science, 2015, 347, 522 CrossRef CAS PubMed; (b) B. J. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316 CrossRef PubMed; (c) M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643 CrossRef CAS PubMed; (d) D. Zhong, B. Cai, X. Wang, Z. Yang, Y. Xing, S. Miao, W. H. Zhang and C. Li, Nano Energy, 2015, 11, 409 CrossRef CAS; (e) Y. Deng, E. Peng, Y. Shao, Z. Xiao, Q. Dong and J. Huang, Energy Environ. Sci., 2015, 8, 1544 RSC.
- (a) N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. Seok, Nat. Mater., 2014, 13, 897 CrossRef CAS PubMed; (b) N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Seok, Nature, 2015, 517, 476 CrossRef CAS PubMed; (c) J. H. Im, I. H. Jang, N. Pellet, M. Grätzel and N. P. Park, Nat. Nanotechnol., 2014, 9, 927 CrossRef CAS PubMed; (d) M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395 CrossRef CAS PubMed.
- (a) J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530 CrossRef CAS PubMed; (b) G. Niu, X. Guo and L. Wang, J. Mater. Chem. A, 2015, 3, 8970 RSC.
- Y. Guo, K. Shoyama, W. Sato and E. Nakamura, Adv. Energy Mater., 2016, 6, 1502317 CrossRef.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254 CrossRef CAS.
- (a) W. J. Ke, G. J. Fang, J. Wang, P. L. Qin, H. Tao, H. W. Lei, Q. Liu, X. Dai and X. Z. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 15959 CrossRef CAS PubMed; (b) S. S. Chen, L. Lei, S. W. Yang, Y. Liu and Z. S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 25770 CrossRef CAS PubMed; (c) D. Cui, Z. Yang, D. Yang, X. D. Ren, Y. C. Liu, Q. B. Wei, H. B. Fan, J. H. Zeng and S. Z. Liu, J. Phys. Chem. C, 2016, 120, 42 CrossRef CAS.
- J. Feng and B. Xiao, J. Phys. Chem. Lett., 2014, 5, 1278 CrossRef CAS PubMed.
- N. E. Brese and M. O’keeffe, Acta Crystallogr., 1991, B47, 192 CAS.
- (a) Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan and F. Yu, Adv. Mater., 2015, 27, 5176 CrossRef CAS PubMed; (b) J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764 CrossRef CAS PubMed.
- P. Schulz, E. Edri, S. Kirmayer, G. Hodes, D. Cahen and A. Kahn, Energy Environ. Sci., 2014, 7, 1377 CAS.
- C. Sun, Z. Wu, H. L. Yip, H. Zhang, X. F. Jiang, Q. Xue, Z. Hu, Z. Hu, Y. Shen, M. Wang, F. Huang and Y. Cao, Adv. Energy Mater., 2016, 6, 1501534 CrossRef.
- L. L. Wang, H. M. Noh, B. K. Moon, S. H. Park, K. H. Kim, J. S. Shi and J. H. Jeong, J. Phys. Chem. C, 2015, 119, 15517 CAS.
- (a) S. L. Zheng and X. M. Chen, Aust. J. Chem., 2004, 57, 703 CrossRef CAS; (b) S. R. Zhang, D. Y. Du, J. S. Qin, S. J. Bao, S. L. Li, W. W. He, Y. Q. Lan, P. Shen and Z. M. Su, Chem. – Eur. J., 2014, 20, 3589 CrossRef CAS PubMed.
- (a) L. F. Ma, L. Y. Wang, J. L. Hu, Y. Y. Wang and G. P. Yang, Cryst. Growth Des., 2009, 9, 5334 CrossRef CAS; (b) M. J. Sie, Y. J. Chang, P. W. Cheng, P. T. Kuo, C. W. Yeh, C. F. Cheng, J. D. Chen and J. C. Wang, CrystEngComm, 2012, 14, 5505 RSC.
- (a) M. Zhang, G. Feng, Z. Song, Y. P. Zhou, H. Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241 CrossRef CAS PubMed; (b) X. Zhou, P. Li, Z. Shi, X. Tang, C. Chen and W. Liu, Inorg. Chem., 2012, 51, 9226 CrossRef CAS PubMed.
- (a) J. P. Meng, Y. Gong and J. H. Lin, RSC Adv., 2016, 6, 73869 RSC; (b) P. G. Jiang, P. Zhang, Y. Gong and J. H. Lin, Dalton Trans., 2016, 45, 4603 RSC; (c) P. Zhang, Y. Gong and J. H. Lin, Eur. J. Inorg. Chem., 2016, 322 CrossRef CAS; (d) J. P. Meng, Y. Gong, Q. Lin, M. M. Zhang, P. Zhang, H. F. Shi and J. H. Lin, Dalton Trans., 2015, 44, 5407 RSC; (e) J. P. Meng, Y. Gong and J. H. Lin, Eur. J. Inorg. Chem., 2016, 4928 CrossRef CAS; (f) X. L. Gao, Y. Gong, P. Zhang, Y. X. Yang, J. P. Meng, M. M. Zhang, J. L. Yin and J. H. Lin, CrystEngComm, 2014, 16, 8492 RSC.
- Y. Zhao, J. Wei, H. Li, Y. Yan, W. Zhou, D. Yu and Q. Zhao, Nat. Commun., 2016, 7, 10228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; PXRD patterns; UV-vis absorption and diffuse reflectance spectra; CVs; EIS; SEM and TG curves. CCDC 1524687–1524689. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7nj02815f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |