
Site-specific surface tailoring for metal ion selectivity via under-coordinated structure engineering†
Li
Yu
ab,
Gong
Zhang
c,
Chunlei
Liu
ab,
Huachun
Lan
c,
Huijuan
Liu
 *abc and
Jiuhui
Qu
abc
*abc and
Jiuhui
Qu
abc
aState Key Laboratory of Environmental Aquatic Chemistry, Key Laboratory of Drinking Water Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China. E-mail: hjliu@rcees.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cSchool of Environment, Tsinghua University, Beijing 100084, P. R. China
First published on 25th July 2018
Abstract
Coordinatively unsaturated atoms play an important role in structural and electronic tuning, while their effects on surface site tailoring for selective adsorption have not been well explored. We demonstrate a new concept based on elaborate tuning of the chemical states of lattice atoms to control the accessibility of different surface sites. The under-coordinated manganese structure developed herein not only benefits the formation of hydroxylated lateral edge sites for preferential complexation of lead (Pb) ions, but also favors a decrease in the proportions of metal-ion-nonspecific octahedral vacancy sites. On the basis of this strategy, a common core–shell structure was devised to further assemble highly exposed edge sites, achieving high selectivity coefficients (31.2–172.0) for Pb(II) against various metal cations. Even when these metal cations coexist at higher concentrations, the general specificity is still maintained, with further pH-enabled switchable sorption–desorption for adsorbent recyclability. Different from frequently reported specific ligand-induced selective systems, intrinsic structure modification as described herein will lead to a new paradigm for surface site tailoring that enables a versatile and tunable platform in environmental remediation, resource recovery and analyte sensing.
Conceptual insightsThe adsorption configuration of targeted species and their competitive adsorption with coexisting substances are of primary relevance to many heterogeneous processes. Conventional selective adsorbents are often prepared via surface functionalization with specific ligands, which have difficulty meeting the basic requirements for satisfactory selectivity and recyclability in a facile approach. Different from frequently-reported ligand-effect-induced specific systems, we turn to an alternative strategy of site-specific tailoring via elaborate tuning of the coordinative unsaturation of lattice atoms to control the accessibility of different sites. Although coordinatively unsaturated atoms play a significant role in structural and electronic tuning, knowledge of their effects on surface site tailoring for selective adsorption is limited. We observed a general specificity toward Pb(II) against various metal cations upon changing the stoichiometry of manganese oxide to obtain low-coordinated Mn. This structural change simultaneously allows rational tuning of the relative proportions of lateral edge sites and octahedral vacancy sites, which show different affinities for metal cations. The selective exposure of hydroxylated lateral edge sites can be maximized on a common support for preferential binding and pH-enabled switchable sorption–desorption of Pb(II). This methodology opens a new horizon in understanding and controlling adsorptive specificity correlated with the properties and affinity of structure-intrinsic sites. |
Introduction
Adsorptive specificity can be rationally applied for high-efficiency environmental remediation, resource recovery and analyte sensing unaffected by coexisting substances, even in complex matrices.1,2 The desired adsorbents should typically meet the following basic requirements of satisfactory selectivity, good reusability with facile adsorbate desorption, and ease of preparation at low cost. However, engineering binding sites with strong selectivity (e.g. imprinted polymers) often comes at the expense of high complexity in synthesis3 and unfavorable adsorbate desorption for surface site regeneration,3,4 due to specific surface-memory effects.3 On the other hand, it is otherwise challenging to achieve satisfactory selectivity on a surface with inherently low specificity.In addition to the properties of adsorptive sites, their accessibility and surface exposure also determine the adsorptive performance to a great extent. Surface tailoring for ideal selectivity relies greatly on the identification of functional sites with suitable selectivity and developing a facile approach to prepare a surface dominated by these accessible sites.5 Although surface functional groups are usually recognized as direct adsorption sites, the properties of the underlying atoms to which these groups coordinate greatly affect their interactions with and affinities for adsorbates.6,7 Elaborate tuning of the chemical states of lattice atoms allows an intriguing level in a cooperative structural and electronic regulation, which may bring about new functionalities and behaviors for a material of interest.8 The availability still depends largely on the nature of the tailored sites and their interactions with targeted substances. However, attempts to control the accessibility of different surface sites are lacking, especially for selective sequestration of metal ions. An intrinsic challenge lies in the very similar affinities of metal ions toward the same adsorptive site, causing difficulty in separation from a coexisting mixture.
As an important class of widely used adsorbents, metal oxides usually show high affinities for many adsorbates. Metal oxide-based hybrid aerogels have been recently reported for application in saline water deionization by virtue of the enhanced adsorption ability and facilitated mass (ion) transfer.9 The preferential affinity of targeted species to metal centers can be also tuned for subsequent catalytic conversion and selectivity.10 To realize the desired performance, it is essential to gain a comprehensive understanding of the underlying relationship between structure–surface site-selectivity. Taking naturally prevalent MnO2 as an example, we note that its crystal structure is composed of edge- or corner-sharing [MnO6] octahedra. Structural water and other cations (e.g. H+, K+) can be accommodated in the interstitial lattice, giving rise to excess positive charge in the structure. A fraction of Mn(IV) ions is usually missing for charge compensation.11 The cation vacancies or octahedral vacancy sites (OVSs) meanwhile lower the repulsive forces between adjacent Mn(IV) cations for structural stability.12 At lateral edges along the [MnO6] octahedra, there are also large-surface-area adsorptive sites. Microscopic and spectroscopic investigations indicate that the adsorption of many metal ions (except Pb(II)) mainly takes place above/below OVSs instead of particle edge sites.13,14 In addition to the OVS-occupied configuration, hydroxylated lateral edge sites show a high adsorption affinity toward Pb(II)15,16 with even an equivalent contribution to that of OVSs.14,15 Although this two-site structural model has been proposed, the implications for selective adsorption of Pb(II) have not been well exploited. Previous studies on the potential selectivity of manganese oxides for Pb(II) against mainly Ca(II), Mg(II) or Na(I)17–19 have also failed to relate the selectivity to site affinity. On the other hand, the prevalent OVSs in manganese oxide structures exhibit nonspecific and strong sorption affinities for many metal ions.14 Many other heavy metal ions, such as Cu(II), Zn(II), Cd(II) and Ni(II), tend to be strongly adsorbed on the surface14,20,21 and significantly impact and compete against Pb(II) adsorption. Achieving a general selectivity with high tolerance to competition from various metal ions is still challenging.
An open question is whether site-specific surface tailoring can be realized, to give a maximized exposure of hydroxylated edge sites with the minimum formation of OVSs at the same time. Intrinsic structure modification of manganese oxides may offer many choices to bring about a new functionality or even entirely novel properties.22 A fundamental understanding of the relationship between the intrinsic structure and surface sites is therefore imperative. We herein rationalize the possibility of metal ion selectivity enhancement by defect structure engineering to control the accessibility of different adsorptive sites. We further demonstrate this feasibility via a facile acetic acid (HAc)-assisted KMnO4-engaged redox procedure to elaborately tune the coordinative unsaturation of lattice Mn atoms. The well-tailored under-coordinated Mn structure allows rational tuning of the relative proportions of different sites and selective exposure of hydroxylated lateral edge sites. An unexpectedly general selectivity toward Pb(II) can be achieved, with high selectivity coefficients against various metal cations. This methodology opens a new horizon in the understanding and control of adsorptive specificity for feasible surface modification or other applications.
Results and discussion
The adsorption behaviors of different metal ions on the hydroxylated edge sites of manganese oxides were first investigated using first-principles calculations on the basis of the density functional theory (DFT) method (details in the ESI†). Theoretical calculations indicate that Pb(II) preferentially binds to the hydroxyl-oxygen atoms of manganese oxide with a bond length of ca. 2.0 Å. The most favorable Gibbs free energy of formation among several metal ions (ESI,† Fig. S1) further accounts for the energetically favorable complexation of Pb(II). The electronic analysis gives a distinct overlap between the d-band of Pb and the O 2p band around the Fermi level, suggesting relatively strong hybridization between the Pb 5d-orbitals and O 2p-orbitals. In addition, Pb(II) shows a much lower hydration energy than the other ions, also confirming the underlying preferential adsorption.23,24 In light of these results, we sought to develop a facile approach to first enhance the exposure of hydroxylated edge sites. On the other hand, satisfactory selectivity for Pb(II) relied largely on minimizing the number of OVSs, because of their particularly nonspecific adsorption affinity for many metal ions.We first prepared different manganese oxides (Mn-C, Mn-C-HAc) via a HAc-assisted redox reaction between KMnO4 and carbon, for evaluation of the adsorption performance toward various metal ions. Our synthesis strategy was inspired by both the previous successful coating of MnO2 onto different carbon matrices,25 and the synthetic chemistry of [Mn12O12(O2CCH3)16(H2O)4] clusters with peripheral ligation stabilized by bridging acetate ligands and terminal water molecules in the structure.26 Raman spectra (ESI,† Fig. S2) indicated the presence of the pure manganese oxide structure without coexisting carbon. Because of the presence of adequate KMnO4, only negligible amounts of residual carbon (below 0.5%) were detected by elemental analyses (ESI,† Table S1). On the basis of the determined Mn oxidation states (3.87 for Mn-C and 3.70 for Mn-C-HAc) and elemental contents, the corresponding chemical compositions could be assumed and expressed as H0.35MnO2.11 and H0.44MnO2.07. X-ray photoelectron spectroscopy (XPS) and X-ray absorption near edge structure (XANES) investigations were then conducted to extract the structural information of the local structure of Mn. For Mn-C and Mn-C-HAc, a qualitative spectral comparison with a series of reference compounds first indicated a structural similarity to MnO2. In principle, the energy positions of XPS peaks and XANES absorption edges are valence-dependent and -sensitive for the absorbing atom, as the successive electron loss accounts for an increased binding energy.27 From the slightly negative shift of Mn 2p peaks (0.4 eV) and Mn K-edge energy-rise (Fig. 1a and c), one can easily conclude that the HAc treatment gave rise to an under-coordinated and low-valence state of Mn.
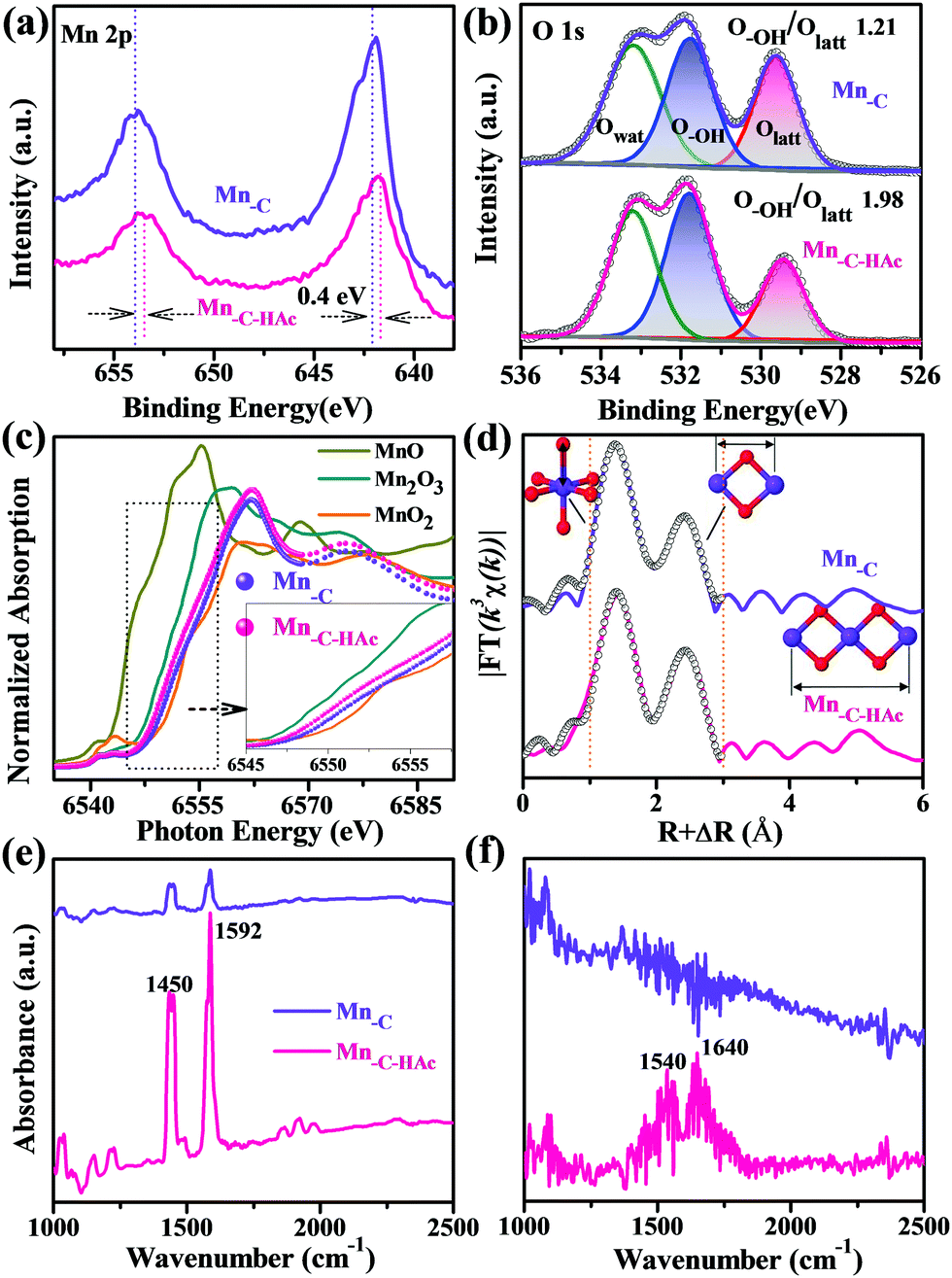 | ||
| Fig. 1 Chemical states of structural Mn and surface sites. (a and b) Mn 2p (a) and high-resolution O 1s (b) XPS spectral interpretation. (c) Mn K-edge XANES spectra. The inset highlights the corresponding energy inflection profiles. (d) Fourier transformed (FT) k3-weighted χ(k)-function of the EXAFS spectra without phase correction (where the black open symbols represent the curve-fitting results). Peak distances are negatively shifted to a lower R by about 0.4 Å. The main FT peaks are assigned to the specific structural configuration (O in red, Mn in purple). (e and f) In situ FTIR spectra of adsorbed pyridine at room temperature (e) and at 150 °C after evacuation (f). | ||
The Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) profiles (Fig. 1d) were further applied for structure elucidation of the local environment around Mn, with mainly a corner- or an edge-sharing Mn–O octahedral configuration. The fitting data, using a starting model consisting of Mn–O and Mn–Mn shells, showed good agreement with the experimental EXAFS patterns in the 1.0–3.0 Å region (not phase corrected). One main FT peak at ∼1.89 Å corresponded to the Mn–O interaction, and its structural motif is schematically depicted in Fig. 1d. The coordination number of the Mn–O first shell in Mn-C-HAc was estimated to be 4.4, compared with the simulated value of 4.6 for Mn-C (Fig. 1d and Table 1), demonstrating the presence of under-coordinated Mn in Mn-C-HAc. The slightly different bond lengths could be attributed to the contribution of elongated Mn(III)–O distances because of Jahn–Teller distortion effects in Mn-C-HAc.28,29 Considering the stabilization of the different chemical states of Mn in the HAc system,30 we believed that acetate ligands facilitated the evolution of such an under-coordinated structure, although they could be easily removed from the surface in aqueous solution.
| Sample | Path | N | R (Å) | σ 2 (10−2 Å2) | R-Factor (%) |
|---|---|---|---|---|---|
| Mn-C, k-range 3.1–9.6 Å−1, R-range 1–3 Å, Nidp = 8, Nvar = 6 | Mn–O.1 | 4.6 | 1.88 | 0.4 | 0.43 |
| Mn–Mn | 3.6 | 2.84 | 1.2 | ||
| Mn-C-HAc, k-range 3.1–9.6 Å−1, R-range 1–3 Å, Nidp = 8, Nvar = 6 | Mn–O.1 | 4.4 | 1.91 | 0.8 | 0.18 |
| Mn–Mn | 4.0 | 2.86 | 1.4 |
In addition to the Mn–O peak, two other peaks were typically assignable to Mn–Mn single- and multiple-scattering (MS) paths (∼2.85 Å and ∼5.60 Å).27 The former was associated with a Mn–Mn interaction between two edge-sharing [MnO6] octahedra, while the latter corresponded to the interaction of three collinearly interconnected Mn–Mn–Mn.29 The EXAFS fitting gave an increased coordination number (4.0) in the Mn–Mn (2.85 Å) shell for Mn-C-HAc, indicating a high cation occupancy ratio and thus a low number of cation vacancies compared with that of Mn-C. However, an attempt at quantitative resolution of the Mn–Mn–Mn MS (5.60 Å) path was quite problematic because of the low peak amplitude.29 We turned to qualitative analysis based on a comparison of the FT peak intensity. In principle, the FT modulus amplitudes of Mn–Mn scattering paths are inversely proportional to the numbers of cation vacancies (OVSs).27 The corresponding peak intensities (IMn–Mn(2.85Å) and IMS(5.60Å)) normalized by the Mn–O peak intensity could provide interspectral assessment on the proportions of OVSs. The intensity ratios were calculated to be 0.68 for IMn–Mn(2.85Å)/IMn–O and 0.19 for IMS(5.60Å)/IMn–O in Mn-C-HAc, while these values were reduced to be 0.58 and 0.14 in Mn-C, respectively. A decrease in the proportions of OVSs was therefore realized simply by HAc treatment and under-coordinated Mn engineering. During the formation of under-coordinated Mn, a fraction of Mn(IV) was replaced by low-valence Mn, which partly compensated for the charge imbalance originating from the excess positive charge due to H+ and K+ incorporation. Compared to Mn(IV), the low-valence Mn also lowered repulsive forces between adjacent Mn cations. The generation of OVSs was not preferred because of the charge neutralization and structural stability induced by low-valence Mn. The results also proved that the number of OVSs was proportional to the average oxidation state of Mn.12,14
In addition, the under-coordinated structure and adjacent oxygen anion pair could dissociatively adsorb water molecules as OH− and H+, giving rise to highly hydroxylated edge surfaces.7,31,32 The increased ratio of hydroxyl oxygen (O–OH) to lattice oxygen (Olatt) (1.21 to 1.98) then confirmed the high density of surface-exposed hydroxyl sites after HAc treatment (Fig. 1b). A relatively high amount of surface hydrogen was also recorded by elemental analyses (ESI,† Table S1). The unsaturated Mn atoms acted as preferential active sites to dissociate water in an energetically favorable manner, contributing to enhanced hydroxylation.33,34 Similar conversion from Lewis acid sites (coordinatively unsaturated metal cations) to Brønsted acid sites (surface –OH) was observed as well in other systems under exposure to water molecules.35,36In situ pyridine (PY)-adsorbed Fourier transform infrared spectra (FTIR) at different temperatures showed a promoted interaction of surface Mn-C-HAc with PY, owing to the coordinatively unsaturated Mn (1450 cm−1) and Brønsted acid sites (Fig. 1e and f). In particular, the characteristic absorption bands could be assigned to hydrogen-bonded PY (1592 cm−1) and the pyridinium ion formed via the proton transfer process (1540 and 1640 cm−1), both from the interaction of PY with surface-activated hydroxyl.35–38 After evacuation at 150 °C, a very weak absorption at 1540 and 1640 cm−1 indicated a moderate Brønsted acidity for the surface of Mn-C-HAc,39 so that switchable sorption–desorption cycles could also be expected in the following adsorption application. The therefore activated Brønsted acid sites facilitated subsequent proton exchange with Pb(II), because of the energetically favorable process in hydrogen transfer.39 As expected, additional metal ion adsorption tests (ESI,† Fig. S3) evidenced an improved selectivity toward Pb(II), due to the exposed acid hydroxyls coordinating to the unsaturated Mn sites and the decreased number of metal-ion-nonspecific OVSs. These activated sites were preferentially occupied by and saturated with Pb(II), showing otherwise poor affinities for competing metal cations.
Even though HAc treatment was beneficial for improved selectivity, the manganese oxides tended to agglomerate into nanosheet-based building blocks (ESI,† Fig. S4) with relatively low adsorption performance. To further engineer highly dispersed ultrathin manganese oxide nanosheets approaching optimal selectivity, magnetic nanospheres were used as cores to ensure well-defined surfaces via a similar HAc-assisted KMnO4-engaged redox procedure. As a starting point, poly(acrylic acid) (PAA)-entangled magnetite (Mag) was prepared via simultaneous thermo-induced polymerization of sodium acrylate during Mag nanosphere formation. Surface covalent binding of PAA on Mag was evidenced from the FTIR spectrum, where the detected characteristic absorption bands near 1555 and 1405 cm−1 could be assigned to the antisymmetric and symmetric stretching modes of carboxylate.40 Two other bands at 1710 and 915 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and O–H out-of-plane deformation vibration from carbonyl groups, while the 1454 cm−1 absorbance comes from C–H bonds (ESI,† Fig. S5). Thermogravimetric analysis (TGA) gave evidence of a dramatic thermal decomposition of ligand layers at ∼200 °C (ESI,† Fig. S6), proving the surface-coating of PAA on Mag. PAA could act as a capping agent to give well-dispersed Fe3O4 (JCPDS 76-1849) (Fig. 2a–c), and then act as a carbon source following the subsequent carbonization step, as revealed by TGA and elemental analyses (ESI,† Table S1). The obtained carbon matrix-encapsulated magnetite (Mag-C) was assembled from loosely packed subunits (Fig. 2d–f), with a high porosity interfacial carbon surface for manganese oxide deposition (Mag-Mn).
O stretching vibration and O–H out-of-plane deformation vibration from carbonyl groups, while the 1454 cm−1 absorbance comes from C–H bonds (ESI,† Fig. S5). Thermogravimetric analysis (TGA) gave evidence of a dramatic thermal decomposition of ligand layers at ∼200 °C (ESI,† Fig. S6), proving the surface-coating of PAA on Mag. PAA could act as a capping agent to give well-dispersed Fe3O4 (JCPDS 76-1849) (Fig. 2a–c), and then act as a carbon source following the subsequent carbonization step, as revealed by TGA and elemental analyses (ESI,† Table S1). The obtained carbon matrix-encapsulated magnetite (Mag-C) was assembled from loosely packed subunits (Fig. 2d–f), with a high porosity interfacial carbon surface for manganese oxide deposition (Mag-Mn).
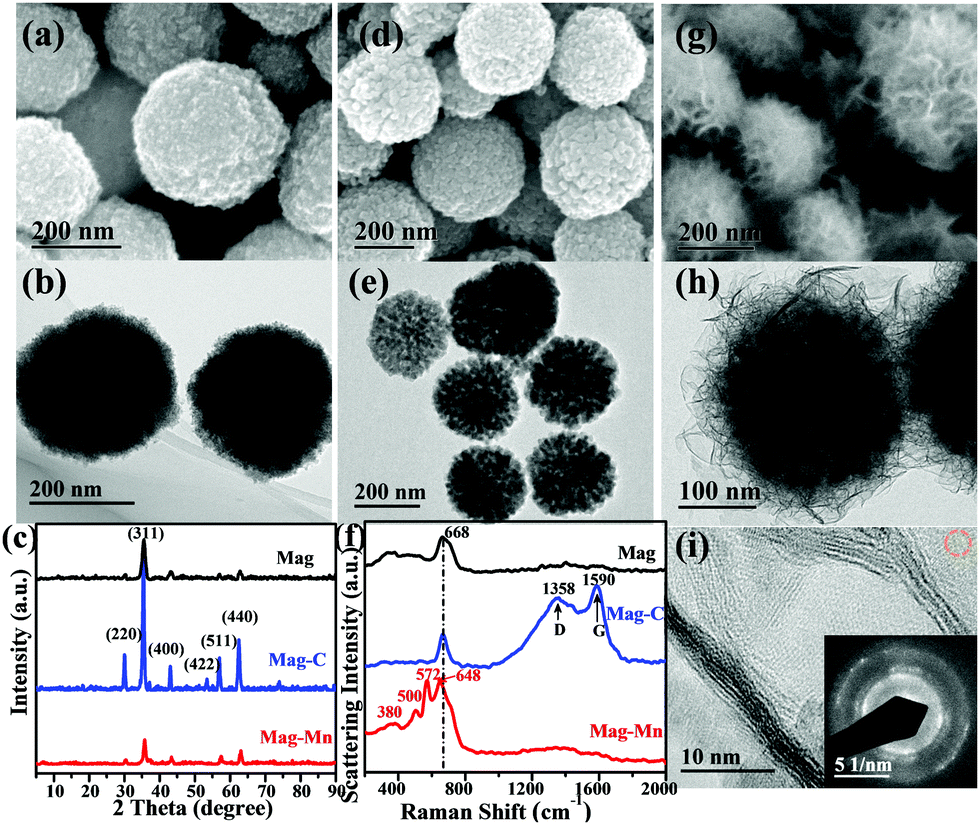 | ||
| Fig. 2 Morphological characterization, phase corroboration and surface properties. (a)–(h) Representative SEM and TEM micrographs for Mag (a and b), Mag-C (d and e) and Mag-Mn (g and h) as well as their corresponding XRD patterns (c) and Raman spectra (f). (i) HRTEM image and SAED pattern (marked area with a dashed circle) of manganese oxide nanosheets. | ||
The deliberately constructed surface carbon was used as a sacrificial template in HAc-assisted KMnO4-involved redox reactions, as shown by the Raman spectra (carbon signal loss at 1358 cm−1 and 1590 cm−1, Fig. 2f) and negligible amounts of residual carbon on Mag-Mn (ESI,† Table S1). The Mag-Mn surface was controllably assembled from large amounts of two-dimensional perpendicularly oriented nanosheets (Fig. 2g and h), with ∼30% deposition of manganese oxide (by weight, ESI,† Table S2) in a homogeneous distribution (ESI,† Fig. S7). HAc herein played a beneficial role in promoting homogeneous manganese oxide deposition, as the reactivity of KMnO4 would be enhanced in a controlled manner under weakly acidic conditions (ESI,† Fig. S8). High-resolution transmission electron microscopy (HRTEM) images revealed that each multilayer stack usually contained 5–10 layers of ultrathin nanosheets (Fig. 2i). The corresponding selected area electron diffraction (SAED) pattern (ESI,†Fig. 2i) indicated short-range crystalline order in (H+, K+)-exchanged MnO2. The newly-emerging Raman bands at 380, 500, 572 and 648 cm−1 corresponded to Mn–O lattice vibrations in [MnO6] octahedra or along the MnO2 framework chains.41 A rather weak signal of Fe 2p meanwhile verified the complete envelopment of stacked MnO2 nanosheets (Fig. 3a).
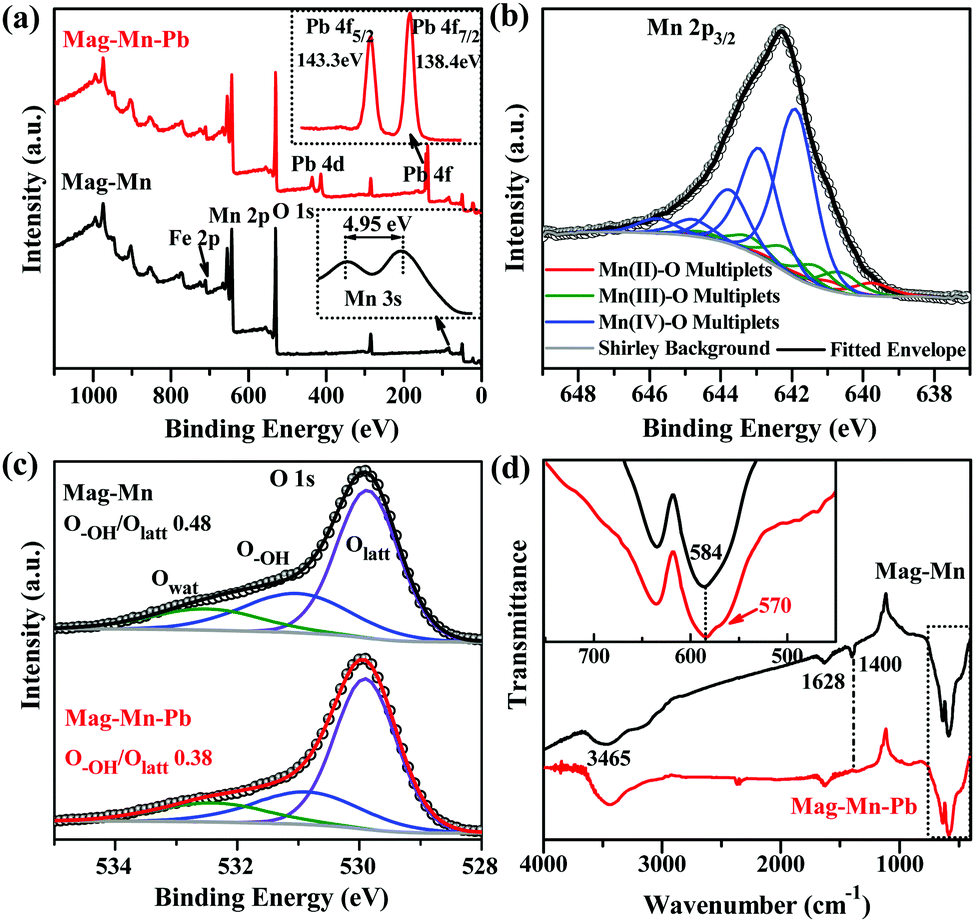 | ||
Fig. 3 Surface structure evolution during adsorption processes. (a) Survey spectra of Mag-Mn before and after adsorption in the multiple metal ion coexisting system with the inset high-resolution Pb 4f spectrum and Mn 3s doublet. (b) XPS Mn 2p3/2 spectral interpretation and multiplet structure resolution for Mag-Mn. For each chemical state, only the lowest-binding-energy peak intensity was allowed to adjust independently, while other peak intensities were set relative to it with fixed ratios. Spectral fitting was processed by constructing the parameter-constrained GL(50) (Gaussian![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Lorentzian 50:50) peaks for the multiplet structure. (c and d) High-resolution core-level O 1s XPS (c) and FTIR (d) analyses for Mag-Mn before and after Pb(II) adsorption. :Lorentzian 50:50) peaks for the multiplet structure. (c and d) High-resolution core-level O 1s XPS (c) and FTIR (d) analyses for Mag-Mn before and after Pb(II) adsorption. | ||
The Mn 2p3/2 XPS multiplet structure resolution consequently gave a near-surface composition of mixed Mn(II), Mn(III) and Mn(IV) oxides and an average Mn oxidation state of 3.62 in Mag-Mn (Fig. 3b). The value was consistent with the experimentally obtained results from oxalic acid–permanganate back-titration.42 The energy splitting (4.95 eV) of the Mn 3s doublet meanwhile confirmed this low-valent state.43,44 Although the under-coordinated Mn limited the evolution of metal-ion-nonspecific OVSs for preferential binding of Pb(II), the selectivity might come at the expense of adsorption quantity, because OVSs are otherwise strong sorption sites for metal ions. However, the higher hydroxylation at under-coordinated Mn generated more accessible sites, i.e. hydroxylated lateral edge sites, for both adsorptive selectivity and quantity. The spatially perpendicular ultrathin layers also provided abundant and discrete adsorptive sites as well as accessible diffusion pathways for the enhanced sequestration of Pb(II). As anticipated, Mag-Mn surfaces featured abundant hydroxyl groups, as shown by the typical FTIR absorption bands near 1400 and 1628 cm−1 from O–H bending vibrations (Fig. 3d). In addition, the broad band around 3465 cm−1 pointed to the existence of hydrogen-bonded –OH or coordinated H2O molecules.24,45 A high content of surface hydrogen of up to ∼1.0% was therefore detected on the hydroxylated surface (ESI,† Table S1). Owing to the preceding carbonization and encapsulation of the uniform two-dimensional nanosheets, Mag-Mn possessed a large specific surface area (101.4 m2 g−1) and a high pore volume (0.183 cm3 g−1, ESI,† Fig. S9 and Table S3). The N2-sorption isotherm and a wide hysteresis loop further proved the presence of pronounced mesoporosity, with the mesopore size centered at 5.4 nm. The rich pore structure ensured highly accessible adsorptive sites on Mag-Mn, making it a promising adsorbent for sequestration of Pb(II).
Prior to adsorption tests, the species distribution modeling and repeatable blank tests first indicated that nearly all Pb(II) existed as soluble ionic Pb2+ at an initial pH of 5.8–6.0 (ESI,† Fig. S10). It should be noted that a quite competitive higher capacity for Pb(II) sequestration (159.8 mg g−1) was observed for Mag-Mn according to the isotherm fitting results using the Langmuir sorption model (Fig. 4a and ESI,† Table S4). The high value of the Freundlich constant (KF) also indicated enhanced adsorption affinity based on the Freundlich model (ESI,† Table S4). Due to the well-tailored surface, the adsorption quantity normalized by manganese oxide content (532.7 mg g−1) was also significantly higher than that of Mn-C-HAc. In particular, the adsorptive performance for Pb(II) was not significantly affected in all the tested binary metal ion systems (with Pb adsorption above 106.0 mg g−1), compared to its independent adsorption (123.0 mg g−1, Fig. 4b). Such an exceptional specificity for Pb(II) could be further reflected by its high distribution coefficients and selectivity coefficients (31.2–172.0, Table 2) against competing metal cations (Mg(II), Ca(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II)). These metal cations, on the other hand, exhibited much poorer affinities toward the Mag-Mn surface (adsorption quantities below 11.0 mg g−1). The adsorbent also showed relatively low equilibrium absorption quantities (below 25.0 mg g−1) for the large-atomic-weight ions (Ag(I), Ba(II), Cs(I)), manifesting its general adsorptive specificity toward Pb(II) (ESI,† Fig. S11).
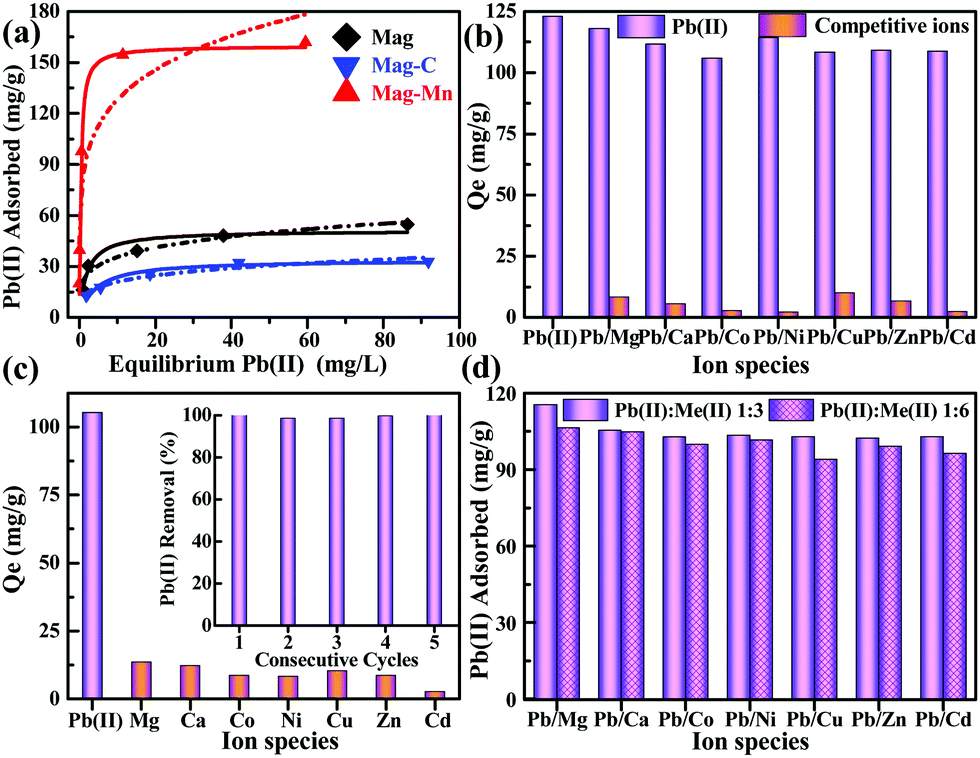 | ||
| Fig. 4 Summary of Pb(II) adsorption behaviors. (a) Comparison of Pb(II) adsorption capacities obtained by the corresponding Langmuir and Freundlich isotherm fittings. (b)–(d) Pb(II) selective adsorption assessment in binary (b) and multiple ion coexisting systems (c) (all individual metal ions 40 mg L−1); and (d) Pb(II) (40 mg L−1) competitive adsorption with the other ions coexisting at high levels (120 or 240 mg L−1). The inset in (c) shows the results of recyclability tests (initial Pb(II) 25 mg L−1). | ||
| Binary metal ions | Distribution coefficient Kda/mL g−1 | Selectivity coefficient kb | |
|---|---|---|---|
| Pb(II) | Competitive ions, Me(II) | ||
| a K d = Qe/Ce, where Qe and Ce represent the equilibrium adsorbed quantity and the aqueous concentration of the tested metal ions, respectively. b k = Kd(Pb)/Kd(Me). | |||
| Pb(II)/Mg(II) | 11238.1 | 221.6 | 50.7 |
| Pb(II)/Ca(II) | 9223.1 | 145.1 | 63.6 |
| Pb(II)/Co(II) | 7851.9 | 71.2 | 110.2 |
| Pb(II)/Ni(II) | 10035.1 | 58.3 | 172.0 |
| Pb(II)/Cu(II) | 8403.1 | 269.5 | 31.2 |
| Pb(II)/Zn(II) | 8598.4 | 177.5 | 48.4 |
| Pb(II)/Cd(II) | 8500.0 | 60.9 | 139.5 |
We then observed concomitant H+ release (pH 4.10–4.40 after equilibrium, ESI,† Table S5) in these systems, potentially suggesting a process that is ion-exchange in nature. Considering that no carboxyl groups were detected at the surface by FTIR, their role in adsorption could be ruled out. The results of XPS interpretation of the O 1s spectra accurately differentiated three oxygen-containing species – the lattice oxygen (Olatt), the mainly hydroxyl oxygen (O–OH) associated with defects and the loosely bound oxygen (mainly adsorbed water OH2O). The decreased surface O–OH/Olatt ratio (0.48 to 0.38, Fig. 3c) confirmed the Pb(II)-exchange with hydroxyl protons. The hydroxyl-engaged complexation could be corroborated as well by the disappearance of the FTIR band at 1400 cm−1 (O–H bending vibration) after adsorption (Fig. 3d).45 In addition, for Pb(II)-adsorbed Mag-Mn (Mag-Mn–Pb), Pb 4f5/2 and Pb 4f7/2 were centered at binding energies of 143.3 eV and 138.4 eV, respectively (Fig. 3a). The remarkably negative shift of 1.2 eV for both peaks relative to the reported binding energies in Pb(NO3)2 implied a strong interaction of Pb(II) on the manganese oxide surface.24 On the other hand, the FTIR absorption peak at 584 cm−1 could be attributed to the deformation vibration mode of Mn–O bonds. It then split into two bands at 584 and 570 cm−1 after Pb(II) adsorption (Fig. 3d). The latter FTIR band originated from the Pb–O bonding, indicating the surface interaction with Pb(II) via inner-sphere complexation.
Owing to the simultaneously enhanced accessibility of the hydroxylated lateral edge sites and the decreased number of metal-ion-nonspecific OVSs, the preferential capture of Pb(II) was still maintained in the fairly complex matrix of a multiple-ion coexisting system (Fig. 4c). This unique selectivity was further tested in binary ion systems with simultaneous presence of a large excess of the competing ions. Even with a concentration (240 mg L−1) about 10–50 fold that of Pb(II) on a molar basis, the coexisting ions still could not adversely affect Pb(II) adsorption markedly (Fig. 4d). In fact, apart from imprinted polymers, this exceptional selectivity toward Pb(II) with extremely high tolerance to competition from other metal ions has rarely been reported for a specific-ligand-free adsorbent. The Pb(II) adsorption herein was a pH-dependent process, with relatively weak adsorption capacities observed under acidic conditions (ESI,† Fig. S12), where the surface oxygen species of adsorbents were mostly protonated and became inactive due to proton competition.15,46 High zeta potentials at low pH indicated a positively charged surface, causing limited and unfavorable Pb(II) adsorption. The adsorbent could be thus regenerated simply in binary 0.1% HNO3 + 0.01 M KNO3 solution and retained superior reusability over five consecutive cycles (inset of Fig. 4c). Because of its stability over a wide pH range, Mag-Mn still maintained the high-efficiency sequestration of Pb(II) without significant capacity decrease.
Magnetic supports provide a versatile and sustainable platform for selectivity enhancement owing to the increased accessibility of well-dispersed surface sites; in addition, a most impressive feature is their magnetic properties, enabling facile magnetic-responsive retrieval.47–50 Magnetic measurements were therefore performed under different applied fields and at different temperatures (ESI,† Fig. S13). Given the surface envelopment by ligand layers (Mag) or manganese oxide layers (Mag-Mn), saturation magnetization reduction was generally observed for Mag (54.4 emu g−1) and Mag-Mn (41.0 emu g−1) with respect to Mag-C (75.3 emu g−1). On the other hand, the departure between zero-field-cooled (ZFC) and field-cooled (FC) magnetization in the investigated temperature range revealed that these nanocomposites remained in the blocked state, with the blocking temperature (TB) beyond the measurement limit (above 350 K).40 The synthesized adsorbents exhibited characteristic ferromagnetic behavior, which could be also reflected in the above hysteresis loops. The plateau region of the FC curves, particularly at a lower temperature, possibly indicated a strong interparticle interaction or the resulting collective magnetic freezing state in assemblies.40,51,52 Interparticle interaction would suppress spin rotation along the applied field and thus stabilize the FC magnetization. The ease of magnetic manipulation and unaffected redispersion properties would then facilitate adsorbent regeneration toward high-feasibility applications in practical adsorption-based water purification.
Conclusions
We have developed a facile HAc-assisted KMnO4-engaged redox strategy to obtain a well-defined under-coordinated manganese structure, which allows site-specific surface tailoring for enhanced accessibility of surface edge sites and a decreased number of nonspecific OVSs. Such a specific ligand-free system shows energetically favorable complexation of Pb(II) with high selectivity coefficients, and pH-enabled switchable sorption–desorption. Elaborate tuning of the accessibility of surface sites with different affinities herein may provide an additional tool for achieving satisfactory selectivity, enabling potential applications in water purification as well as in the fields of analytical chemistry and sensing.Experimental
Material synthesis
Mn-C and Mn-C-HAc were synthesized using graphene oxide (GO) as a sacrificial carbon source following a KMnO4-involved redox synthetic strategy. Typically, GO was first prepared from natural graphite powder via the modified Hummers method.53 The ultrasonically dispersed mixture containing KMnO4 + GO (for Mn-C) or KMnO4+ GO + HAc (for Mn-C-HAc) was maintained at 80 °C for 24 h. The KMnO4, GO and HAc were at concentrations of 5 mM, 0.04 mg mL−1, with a quantity of 2 mL for 80 mL reaction systems, respectively. The final products were collected, washed and subsequently dried at 80 °C in air.PAA-entangled magnetite nanospheres (Mag) were synthesized via a modified solvothermal reaction. Iron(III) chloride hexahydrate (1.08 g), sodium acetate (3.0 g) and sodium acrylate (4.5 g) were dissolved in ethylene glycol (80 mL) under vigorous magnetic stirring. The resulting homogeneous solution was subsequently transferred to a Teflon-lined autoclave and maintained at 200 °C for 12 h. The obtained precipitate was washed repeatedly, dried under vacuum, and then subjected to carbonization at 500 °C for 4 h under high-purity argon to give Mag-C. Mag-C particles were then redispersed ultrasonically into KMnO4 solution (5 mM), followed by the addition of 2 mL HAc. After reaction at 80 °C for 24 h, the products (Mag-Mn) were harvested, washed and subsequently dried at 80 °C in air.
Physicochemical characterization
Morphological and microstructural analyses of the samples were performed on a JEOL JEM-2010F HRTEM and a Hitachi SU-8000 field emission scanning electron microscope (SEM). General characterization techniques including elemental mapping, powder X-ray diffraction (XRD), FTIR, Raman, XPS, zeta-potential, TGA, N2-adsorption and mass and temperature dependent magnetization measurements were further used for exploring the sorbent constituents, crystal structures, surface properties and magnetic behaviors.54 The average manganese oxidation state was determined by a redox-based back-titration method.42 CHNS elemental contents were monitored on a EURO EA 3000 analyzer. The Mn K-edge X-ray absorption fine structure (XAFS) investigation was conducted in transmission mode using radiation from the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF). Photon energy was calibrated with high-purity Mn metal at room temperature. Data collected were processed by first extracting the EXAFS oscillation χ(k) functions. The adopted scattering path phases and amplitudes were ab initio-calculated using FEFF8.4 followed by fitting to the Fourier transformed (FT) k3-weighted χ(k) in the R-space using the Ifeffit 1.2.12 software package. The in situ infrared spectra (IR) of chemisorbed pyridine were recorded using an ultrahigh-vacuum gas sorption system coupled with a Thermo Nicolet 6700 FTIR spectrometer. Samples were first pressed into self-supporting wafers and placed in an in situ cell, and subsequently pretreated at 150 °C for 2 h. The background spectrum was acquired prior to pyridine exposure and was finally subtracted from the subsequently measured IR spectrum. The difference spectra were recorded for surface pyridine species after adsorption and evacuation at different temperatures.Adsorption isotherms and selective sequestration of Pb(II)
Batch adsorption experiments were performed in 40 mL suspensions with a measured amount (0.25 g L−1) of the tested sorbents added. The initial pH of the suspension was adjusted to 5.8–6.0 with small amounts of 0.1 M HNO3 or NaOH solution, with noted exceptions for specific investigation of pH effects. All investigated metal ions (Mg(II), Ca(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), Pb(II), Ag(I), Ba(II) and non-radioactive Cs(I)) were used as their respective nitrate salts and analyzed by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Optima 8300, PerkinElmer). The equilibrium adsorption capabilities were assessed through the corresponding adsorption isotherms as the initial Pb(II) concentration was varied from 5 to 100 mg L−1. The suspension was then oscillated at ambient temperature for 24 h, and the supernatant was subsequently collected under magnetic separation for determination of the remaining Pb(II). Selective sequestration of Pb(II) in a binary metallic ion system was studied by the batch method, where Pb(II) had an initial concentration of 40 mg L−1, while the other competing ions (Mg(II), Ca(II), Co(II), Ni(II), Cu(II), Zn(II), or Cd(II)) coexisted at 40, 120 or 240 mg L−1. The Pb(II) adsorption behavior was also investigated in a mixed solution containing all eight ions with individual initial concentrations of 40 mg L−1. After adsorption equilibrium, the remaining ions in solution were measured and the corresponding adsorption capacities were obtained. Exhausted Mag-Mn was simply regenerated twice with a binary 0.1% HNO3 + 0.01 M KNO3 solution, and its efficiency for Pb(II) removal was then tested in subsequent cycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (Grant No. 51438011, 51708543, and 51722811).Notes and references
- T. P. Rao, R. Kala and S. Daniel, Anal. Chim. Acta, 2006, 578, 105–116 CrossRef PubMed.
- Q. Yuan, P. Li, J. Liu, Y. Lin, Y. Cai, Y. Ye and C. Liang, Chem. Mater., 2017, 29, 10198–10205 CrossRef.
- J. Fu, L. Chen, J. Li and Z. Zhang, J. Mater. Chem. A, 2015, 3, 13598–13627 RSC.
- J. Wang, H. Shen, X. Hu, Y. Li, Z. Li, J. Xu, X. Song, H. Zeng and Q. Yuan, Adv. Sci., 2016, 3, 1500289 CrossRef PubMed.
- P. Christopher and S. Linic, J. Am. Chem. Soc., 2008, 130, 11264–11265 CrossRef PubMed.
- J.-F. Boily and R. D. Lins, J. Phys. Chem. C, 2009, 113, 16568–16570 CrossRef.
- H. Tamura, N. Katayama and R. Furuichi, Environ. Sci. Technol., 1996, 30, 1198–1204 CrossRef.
- X. Zou and B. I. Yakobson, Acc. Chem. Res., 2015, 48, 73–80 CrossRef PubMed.
- H. Yin, S. Zhao, J. Wan, H. Tang, L. Chang, L. He, H. Zhao, Y. Gao and Z. Tang, Adv. Mater., 2013, 25, 6270–6276 CrossRef PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef PubMed.
- P. Ruetschi, J. Electrochem. Soc., 1984, 131, 2737–2744 CrossRef.
- W. Zhao, H. Cui, F. Liu, W. Tan and X. Feng, Clays Clay Miner., 2009, 57, 513–520 CrossRef.
- A. Manceau, M. Lanson and N. Geoffroy, Geochim. Cosmochim. Acta, 2007, 71, 95–128 CrossRef.
- Y. Wang, X. Feng, M. Villalobos, W. Tan and F. Liu, Chem. Geol., 2012, 292-293, 25–34 CrossRef.
- M. Villalobos, J. Bargar and G. Sposito, Environ. Sci. Technol., 2005, 39, 569–576 CrossRef PubMed.
- K. D. Kwon, K. Refson and G. Sposito, Geochim. Cosmochim. Acta, 2010, 74, 6731–6740 CrossRef.
- Q. Su, B. Pan, B. Pan, Q. Zhang, W. Zhang, L. Lv, X. Wang, J. Wu and Q. Zhang, Sci. Total Environ, 2009, 407, 5471–5477 CrossRef PubMed.
- S. Wan, F. He, J. Wu, W. Wan, Y. Gu and B. Gao, J. Hazard. Mater., 2016, 314, 32–40 CrossRef PubMed.
- H. Zhang, L. Gu, L. Zhang, S. Zheng, H. Wan, J. Sun, D. Zhu and Z. Xu, Appl. Surf. Sci., 2017, 406, 330–338 CrossRef.
- E.-J. Kim, C.-S. Lee, Y.-Y. Chang and Y.-S. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 9628–9634 CrossRef PubMed.
- J. Zhao, J. Liu, N. Li, W. Wang, J. Nan, Z. Zhao and F. Cui, Chem. Eng. J., 2016, 304, 737–746 CrossRef.
- T. Zhai, X. Lu, F. Wang, H. Xia and Y. Tong, Nanoscale Horiz., 2016, 1, 109–124 RSC.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113–4116 CrossRef PubMed.
- M. B. Sassin, C. N. Chervin, D. R. Rolison and J. W. Long, Acc. Chem. Res., 2013, 46, 1062–1074 CrossRef PubMed.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- I. Saratovsky, P. G. Wightman, P. A. Pastén, J.-F. Gaillard and K. R. Poeppelmeier, J. Am. Chem. Soc., 2006, 128, 11188–11198 CrossRef PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Chem. Sci., 2012, 3, 2330–2339 RSC.
- J.-E. Jee, O. Pestovsky and A. Bakac, Dalton Trans., 2010, 39, 11636–11642 RSC.
- H. H. Kung, Surface Coordinative Unsaturation, in Studies in Surface Science and Catalysis, ed. H. H. Kung, Elsevier, Netherlands, 1st edn, 1989, vol. 45, pp. 53–71 Search PubMed.
- Z.-Y. Li, J. Zhou, L.-S. Tang, X.-P. Fu, H. Wei, M. Xue, Y.-L. Zhao, C.-J. Jia, X.-M. Li, H.-B. Chu and Y. Li, J. Mater. Chem. A, 2018, 6, 2318–2326 RSC.
- J. Fester, M. García-Melchor, A. S. Walton, M. Bajdich, Z. Li, L. Lammich, A. Vojvodic and J. V. Lauritsen, Nat. Commun., 2017, 8, 14169 CrossRef PubMed.
- F. Dvořák, L. Szabová, V. Johánek, M. Farnesi Camellone, V. Stetsovych, M. Vorokhta, A. Tovt, T. Skála, I. Matolínová, Y. Tateyama, J. Mysliveček, S. Fabris and V. Matolín, ACS Catal., 2018, 8, 4354–4363 CrossRef.
- M. R. Basila, T. R. Kantner and K. H. Rhee, J. Phys. Chem., 1964, 68, 3197–3207 CrossRef.
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef.
- J. Luo, Q. Zhang, J. Garcia-Martinez and S. L. Suib, J. Am. Chem. Soc., 2008, 130, 3198–3207 CrossRef PubMed.
- F. Ouyang, A. Nakayama, K. Tabada and E. Suzuki, J. Phys. Chem. B, 2000, 104, 2012–2018 CrossRef.
- J. An, Y. Wang, J. Lu, J. Zhang, Z. Zhang, S. Xu, X. Liu, T. Zhang, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, P. Fornasiero and F. Wang, J. Am. Chem. Soc., 2018, 140, 4172–4181 CrossRef PubMed.
- L. Yu, H. Liu, C. Liu, H. Lan and J. Qu, Part. Part. Syst. Charact., 2016, 33, 323–331 CrossRef.
- D. Chen, D. Ding, X. Li, G. H. Waller, X. Xiong, M. A. El-Sayed and M. Liu, Chem. Mater., 2015, 27, 6608–6619 CrossRef.
- A. Dyer, M. Pillinger, J. Newton, R. Harjula, T. Möller and S. Amin, Chem. Mater., 2000, 12, 3798–3804 CrossRef.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946–3952 CrossRef.
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef PubMed.
- J. Liu, X. Ge, X. Ye, G. Wang, H. Zhang, H. Zhou, Y. Zhang and H. Zhao, J. Mater. Chem. A, 2016, 4, 1970–1979 RSC.
- M. J. Manos, N. Ding and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3696–3699 CrossRef PubMed.
- M. B. Gawande, A. K. Rathi, J. Tucek, K. Safarova, N. Bundaleski, O. M. N. D. Teodoro, L. Kvitek, R. S. Varma and R. Zboril, Green Chem., 2014, 16, 4137–4143 RSC.
- R. Hudson, Y. Feng, R. S. Varma and A. Moores, Green Chem., 2014, 16, 4493–4505 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- M. Shokouhimehr, Catalysts, 2015, 5, 534–560 CrossRef.
- C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, Chem. Mater., 2006, 18, 3835–3842 CrossRef.
- J. Lu, X. Jiao, D. Chen and W. Li, J. Phys. Chem. C, 2009, 113, 4012–4017 CrossRef.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- L. Yu, G. Zhang, C. Liu, H. Lan, H. Liu and J. Qu, ACS Catal., 2018, 8, 1090–1096 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, theoretical calculations, and supplementary figures, tables and texts. See DOI: 10.1039/c8nh00094h |
| This journal is © The Royal Society of Chemistry 2018 |