
Single glucose molecule transport process revealed by force tracing and molecular dynamics simulations†
Yangang
Pan‡
a,
Yuebin
Zhang‡
c,
Pianchou
Gongpan‡
dg,
Qingrong
Zhang
ae,
Siteng
Huang
c,
Bin
Wang
 f,
Bingqian
Xu
f,
Yuping
Shan
*e,
Wenyong
Xiong
*d,
Guohui
Li
*c and
Hongda
Wang
*abg
f,
Bingqian
Xu
f,
Yuping
Shan
*e,
Wenyong
Xiong
*d,
Guohui
Li
*c and
Hongda
Wang
*abg
aState Key Laboratory of Electroanalytical Chemistry, Research Center of Biomembranomics, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China. E-mail: hdwang@ciac.ac.cn
bLaboratory for Marine Biology and Biotechnology, Qing dao National Laboratory for Marine Science and Technology, Wenhai Road, Aoshanwei, Jimo, Qingdao, Shandong 266237, P. R. China
cState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, Liaoning 116023, P. R. China. E-mail: ghli@dicp.ac.cn
dState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, Yunnan 650201, P. R. China. E-mail: xiong.wenyong@mail.kib.ac.cn
eSchool of Chemistry and Life Science, Advanced Institute of Materials Science, Changchun 130012, China. E-mail: shanyp@ciac.ac.cn
fSingle Molecule Study Laboratory, College of Engineering and Nanoscale Science and Engineering Center, University of Georgia, Athens, GA 30602, USA
gGraduate University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 24th April 2018
Abstract
Transporting individual molecules across cell membranes is a fundamental process in cellular metabolism. Although the crystal diffraction technique has greatly contributed to our understanding of the structures of the involved transporters, a description of the dynamic transport mechanism at the single-molecule level has been extremely elusive. In this study, we applied atomic force microscopy (AFM)-based force tracing to directly monitor the transport of a single molecule, D-glucose, across living cell membranes. Our results show that the force to transport a single molecule of D-glucose across cell membranes is 37 ± 9 pN, and the corresponding transport interval is approximately 20 ms, while the average speed is approximately 0.3 μm s−1. Furthermore, our calculated force profile from molecular dynamics simulations showed quantitatively good agreement with the force tracing observation and revealed detailed information regarding the glucose transport path, indicating that two salt bridges, K38/E299 and K300/E426, play critical roles during glucose transport across glucose transporter 1 (GLUT1). This role was further verified using biological experiments that disrupted these two bridges and measured the uptake of glucose into the cells. Our approaches led to the first unambiguous description of the glucose transport process across cell membranes at the single-molecule level and demonstrated the biological importance of the two salt bridges for transporting glucose across GLUT1.
Conceptual insightsMolecular transports across cell membranes are essential activities of cellular systems. How fast can nutrients, such as single glucose and amino acids, be transported into a living cell? What is the effort the cells need to pay for transporting such molecules? These questions are quite fundamental in cell biology, but have never been revealed at the single molecule level. To address these critical questions, we use the atomic force microscopy (AFM)-based force-tracing technique to monitor the process of single-glucose transport, inspired by a “fishing” concept. We demonstrate that the force-tracing technique enables the single molecule transport process to be followed at a high temporal-spatial resolution. The forces to transport a single molecule of D-glucose across cell membranes and the corresponding transport interval were recorded. Combining with theoretical simulation, the transporting mechanism of glucose is further revealed at an atomic level, which was confirmed by the biological experiments. Our approach leads to the first unambiguous description of the kinetics of the transport process in living cells at the single molecule level, which is significant for understanding how a membrane transporter works. This study provides a new concept of investigating the dynamic function of cell membranes. |
Introduction
Small molecules such as amino acids, sugars, nucleosides, and ions are transported via cellular uptake, which depends on the solute carrier superfamily that includes a major class of cell membrane transporters.1,2 The structure of these membrane transporters has been intensely studied using X-ray diffraction, which facilitates the understanding of molecular interactions between the solute and transporters.3–5 However, molecular transportation is dynamic; therefore, the static structure of the transporter cannot adequately illustrate the dynamic mechanism of molecular transportation. Several ensemble methods have been used to investigate the transport process in cell membranes, such as transverse relaxation-optimized spectroscopy (TROSY) and fluorescence microscopy.6–8 In ensemble experiments, glucose transport is usually presented as an average rate, the number of glucose molecules per cell per minute;9 the main weakness of an ensemble study is the inability to address how a “single” molecule interacts with a heterogeneous cell surface during the transport process. The patch clamp technique is capable of recording single-molecule transportation, but the force information and dynamic details are lacking.10 In general, the transport of a single molecule occurs at the micro to millisecond level,11,12 which challenges the current techniques because the temporal resolution of regular force measurement techniques [e.g., optical tweezers and atomic force microscopy (AFM)-based force spectroscopy] is beyond one millisecond. In this study, we took advantage of a highly sensitive technique (force tracing) to study the complicated process of single-molecule transportation. Utilizing a fine AFM cantilever, the AFM-based force tracing technique allows the detection of piconewton forces13 and rapid processes down to the microsecond scale, which provides a unique opportunity to explore dynamic interactions at the single-molecule or particle level.14In addition to the progress in experimental techniques, molecular dynamics (MD) simulations of membrane proteins have undergone rapid growth in the past decade.15,16 Recent advances in computer hardware and atomistic simulation algorithms have tremendously increased the timescales accessible to MD simulation by several orders of magnitude, from nanosecond to microsecond timescales. The dynamic behaviours of many key biochemical processes, which are rarely observed experimentally, such as protein folding, drug binding, and the permeation or transport of substrates across the cell membrane, can be fully recorded using MD simulations at very fine temporal and spatial resolutions.17–20
We used glucose as an example to illustrate the ability to monitor single-molecule transport. Glucose is the main fuel for most cells, and its importance as an energy source has led to extensive studies on the uptake mechanisms and metabolic processes of glucose.21 The glucose transporter (GLUT) is ubiquitous in eukaryotic cells and plays a pivotal role in glucose uptake in living cells.22 GLUT1, in particular, facilitates the diffusion of glucose into erythrocytes via the glucose gradient and is responsible for supplying glucose to the brain and other organs.23,24 The dysfunction of GLUT1 may lead to a series of diseases, such as De Vivo disease, heart disease, type 2 diabetes and cancer.25–28 Due to these fundamental physiological and pathophysiological significance roles, and the dynamic interactions between glucose and transporter, extensive studies on glucose uptake mechanisms and metabolic processes have been performed.29,30 Unfortunately, the dynamic transport of glucose across cell membranes remains unclear at the single-molecule level. Therefore, we used the force tracing technique to monitor the transportation of a single glucose molecule via GLUT1 in real time. In addition, we also performed MD simulations to investigate the atomic mechanisms of the passage of glucose through GLUT1 and provide a deeper understanding of the experimental observations.
Results and discussion
1. Experimental monitoring via force tracing of the transport of a single glucose molecule
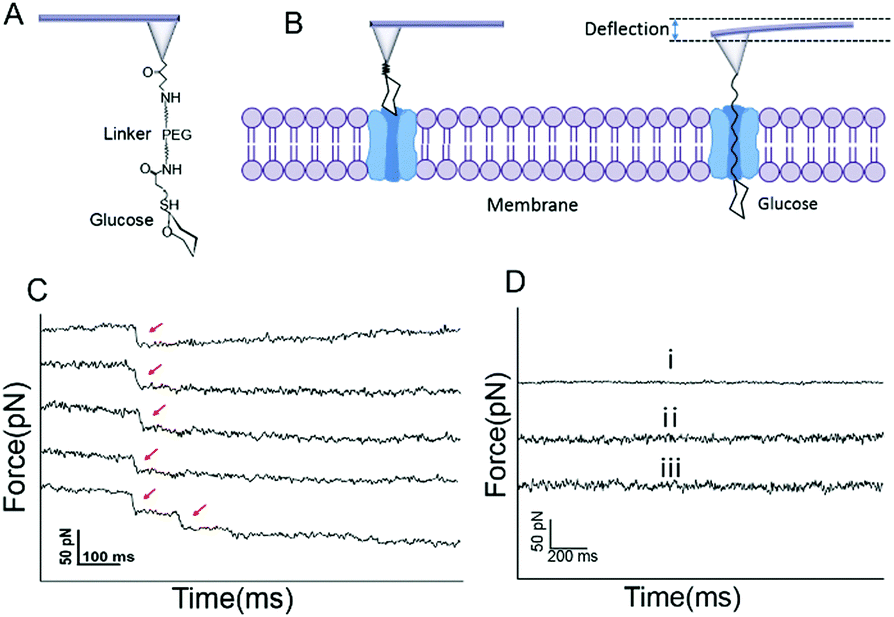 | ||
| Fig. 1 Diagram showing principle and measurement of the force tracing technique. (A) Scheme of the functionalized AFM tip. The 1-thio-D-glucose is covalently coupled to the AFM tip via a heterobifunctional PEG linker. One glucose molecule is attached to one PEG end via an –SH group. (B) Diagram of the force tracing process. The AFM tip is tethered with D-glucose. When the tethered D-glucose (right cantilever) is transported via a GLUT (blue) in a living HeLa cell, a force tracing signal (deflection) is generated. (C) Examples of the force tracing curves showing glucose transport events. The tracing curves start on the left side, and the arrows indicate the transport force signals. (D) The force tracing curves of control experiments were obtained using a GLUT1 antibody tethered AFM tip on cell membranes (i), a glucose molecule modified AFM tip on the plain lipid bilayer (ii), and after adding free excess glucose in the AFM chamber to compete with the D-glucose on the AFM tip (iii). | ||
A schematic diagram of the glucose transport process is shown in Fig. 1B. Initially, the glucose molecules attached onto the AFM tip are statically maintained on the cell surface (the left status in Fig. 1B). When GLUT1 encounters D-glucose on the AFM tip, the channel is opened to capture D-glucose and thereby transport D-glucose into the cell. During this process, the D-glucose goes through GLUT1 and thus stretches the PEG linker, and as a result, the AFM tip bends downwards, as shown in the right status in Fig. 1B. The downwards bend of the AFM tip generates the force signal according to the principle of AFM force spectroscopy, which is recorded in the force tracing curves. The fully dynamic transport process is schematically shown by animation in Movie 1 (ESI†). Typical force tracing curves are shown in Fig. 1C, in which the force curve begins on the left. When glucose on the AFM tip rested on the cell surface, the force curve was flat. Once glucose was captured and transported through GLUT, a sudden force signal resulting from the AFM cantilever bend (indicated by the arrows) was recorded. After the transport event, the cantilever of the AFM tip remained stretched as long as there was a force balance between the cantilever and transporter, as indicated by the right parallel portion of the force tracing curve. In most situations, we observed a single-molecule transport process during one force tracing curve, although rarely a force tracing curve with two force signals was recorded, as shown in Fig. 1C (bottom), which indicated that two D-glucose molecules at different positions on the AFM tip were sequentially transported into the cell via GLUT1.
 | ||
| Fig. 2 Blocking and control experiments. (A) Comparison of transport probabilities before blocking (left bar), after blocking (middle bar) by D-glucose and after the subsequent washout (right bar). The values are indicated as the mean ± standard error (n = 1000). P < 0.05 (one asterisk) for comparisons with the levels in the relevant control group (unblocked, the value from the first bar). (B) Probabilities that the glucose on the AFM tip would be transported under different conditions, including HeLa (on the HeLa cells without glucose in the medium), DMEM (in the glucose-containing medium), MDCK (on the MDCK cells in the glucose-free medium), clean tip (the AFM tip without tip functionalization), and PEG-modified tip (the AFM tip only functionalized with the PEG linker). The values are shown as the mean ± standard error (n = 1000). P < 0.005 (one asterisk) and P < 0.05 (two asterisks) for comparisons with the levels in the relevant HeLa group (the first bar). (C) Distribution of the glucose transport force (n > 150 from 1000 tracing curves). (D) Distribution of the glucose transport time (n > 150). (E) Transport force distribution after the cytochalasin B blocking. The histograms were obtained from approximately 600 randomly chosen tracing curves after the cytochalasin B blocking. | ||
Furthermore, we carefully performed control experiments to exclude the possibility of cellular activities and AFM tip functionalization. Using a clean AFM tip (without any modification) for force tracing on HeLa cells, we observed only a few curves with force signals among thousands of curves in repeated experiments. The probability of obtaining a force signal was only 1.2 ± 0.1% (Fig. 2B), and the force values were less than 20 pN, which is consistent with noise or vibrations from the cell or AFM tip, and these events could be easily distinguished from the transport events by the time scale and shape of the tracing curves.14 The force tracing test with a PEG-modified AFM tip provided results very similar to those of the clean AFM tip (Fig. 2B). Taken together, these results demonstrated that the force signals in our experiments were solely attributed to the D-glucose on the tip undergoing transport via GLUT1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 (6 nm/20 ms). Our results provide the dynamic parameters for the transport of a single glucose molecule, which is helpful for understanding the mechanism of glucose transport.
36 (6 nm/20 ms). Our results provide the dynamic parameters for the transport of a single glucose molecule, which is helpful for understanding the mechanism of glucose transport.
2. Molecular dynamics (MD) simulations of glucose transport through GLUT1
We also performed another set of constant-velocity SMD simulations using a speed as slow as 1 Å ns−1. However, the force experienced by the D-glucose molecule during the constant-velocity SMD simulation was extremely large (Fig. S11, ESI†), indicating that the outward-open conformation prohibited transport, and without considering the conformational changes of GLUT1, investigating the transport process using SMD is not suitable within hundreds of SMD simulations. As mentioned above in the AFM experiments, transporting a single glucose molecule through GLUT occurs on the time scale of milliseconds. Performing such lengthy MD simulations in a single run, on the time scale of milliseconds, is far beyond the computational power available to us. Therefore, our second strategy was to employ the recently developed enhanced sampling methodology, metadynamics simulation,39,40 to simultaneously couple the transport process and the conformational transition event.
Within a 100 ns metadynamics simulation biasing on these two CVs, we observed the transport of the D-glucose molecule through a channel from the extracellular region to the intracellular side of the membrane and the conformational transition of GLUT1 from the outward-open state to the inward-open state. The metadynamics simulation provided a clear view of how GLUT1 facilitates D-glucose transport.
In addition to monitoring the transport process, we also attempted to reproduce experimental observations and thermodynamic properties based on our calculated free energy surface (FES). Unfortunately, only a qualitative estimation of the FES could be obtained from our metadynamics simulation because we observed only a single transport event occurring during the 100 ns metadynamics simulation. In fact, to obtain a quantitatively characterized FES, many re-crossing events should be observed in a single metadynamics run; however, the computational time required to converge the FES can be prohibitively long, especially in large heterogeneous systems. Therefore, we employed a metadynamics-based approach to recover equilibrium properties from a nonequilibrium simulation-sampled conformational space.
In Fig. 3, we show the FES by projecting the conformations onto the spaces of two CVs [CV1 is the conformational transition of GLUT1 from the outward-open state to the inward-open state; CV2 is the z distance between the COM of the D-glucose molecule and GLUT1]. The FES is generated according to the equation F(x,y) = −lnπi(x,y)/kT, where πi is the stationary probability of x, y on the CV1 and CV2 spaces. From the FES in Fig. 3, we can clearly observe four basins on the map that correspond to four metastable states of GLUT1 [basin A for the partially outward-open (POTO) state; basin B for the partially occluded outward-open (POOO) state; basin C for the partially occluded inward-open (POIO) state; and basin D for the partially inward-open (POTO) state].
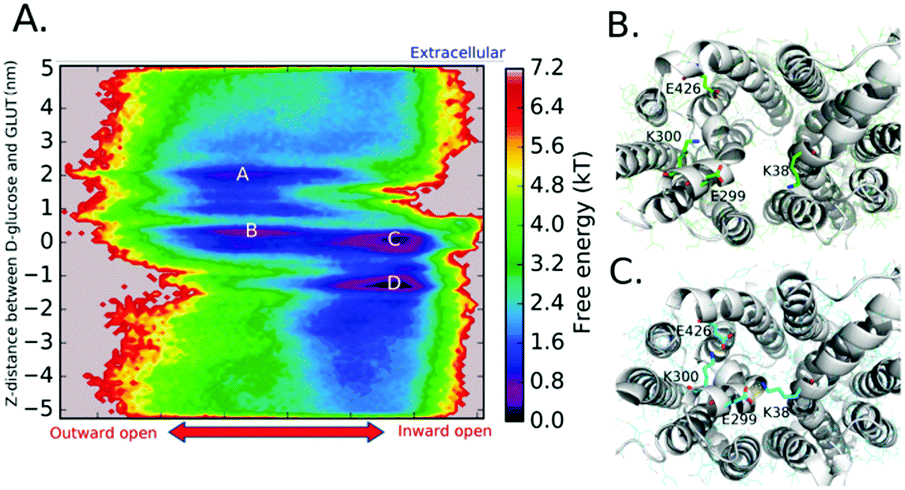 | ||
| Fig. 3 Molecular dynamics simulations of the glucose transport through GLUT1. (A) 2D free energy surface of the glucose transport from the extracellular side to the intracellular side; (B & C) The critical salt bridges (E299/K38 & K300/E426) modulating the conformational transition of GLUT1 at the extracellular glucose entrance. | ||
Once flipping into the entry position, D-glucose is restrained in basin A. Three hydrogen-bond interactions play essential roles in stabilizing the glucose molecules in this basin, as shown in Fig. S15A (ESI†), which are formed between (1) the ring oxygen of glucose and the δ-amide group of Gln37, (2) the C6 OH group and the side chain δ-carboxyl oxygen of Gln37, and (3) the C3 OH group and the carboxyl group of Glu41. In particular, the ring oxygen of glucose is an important hydrogen-bond acceptor that affects the transport activity. Replacing the ring oxygen with sulfur in 5-thio-D-glucose competitively inhibits glucose diffusion.45 In the MD simulation, we found that interactions between the ring oxygen of glucose and the amide groups of the family of conserved asparagine and glutamine residues lining the channel are essential for guiding the diffusion.
After leaving basin A, glucose approaches a metastable binding site in the inner region of GLUT1, corresponding to the POOO state (basin B). In this site, the glucose molecule is tightly surrounded and stabilized by many hydrogen-bond interactions from Asn34, Asn286 and Asn415 (Fig. S15B, ESI†). These asparagine residues are highly conserved in the GLUT family, and Asn34 is an important disease-related residue in GLUT.46 In this analysis, we found that the side chain of Asn34 can act as both the hydrogen-bond donor (the δ-amide group) and hydrogen-bond acceptor (the δ-carboxyl oxygen) and simultaneously contact the C4 and C3 OH groups of glucose. Asn415 plays the same role as the above-mentioned Gln37, which forms a hydrogen bond with the ring oxygen atom of glucose.
The transition from basin B to basin C is a key step for transport. Without the conformational transition of GLUT1 from the POOO state to the POIO state, the transport event would not occur as we have observed in the SMD simulations. After careful analysis of the trajectories obtained from the MD simulations, we found that two pairs of salt bridges formed between K38/E299 and K300/E426 are of particular importance for modulating the conformational transition from POOO to POIO (Fig. 3B and Fig. S16, ESI†). In the outward-open state, these two salt bridges are interrupted. Once the connections are formed between K38/E299 and K300/E426, the conformational transition occurs as the helix segments are tugged closer to each other at the extracellular side of GLUT1.
The final stage corresponds to the escape of glucose from basin C to basin D, which is facilitated by Gln161. The mentioned Gln161, similar to Asn415 and Gln37, plays a key role in guiding diffusion by forming hydrogen bonds between the ring oxygen atom and the C1 OH group of glucose. Moreover, our result also explains the experimental observation that truncating the side chain of Gln161 to Asn or replacing this residue with a non-amide group-containing residue, such as Q161L, dramatically reduces the glucose transport efficiency47 because shortening the side chain of Gln161 may lead to a higher transport free energy barrier from basin C to basin D.
After Gln161 tugs the ligand out of basin C, the orientation of glucose must be readjusted for escape. This process is facilitated by the indole ring of the conserved Trp388, which provides a strong π–π stacking interaction with the glucose plane to force the ligand into an orientation that is perpendicular to the membrane plane (Fig. S15D, ESI†). Consequently, the ligand may easily diffuse to the intracellular side without significant hindrance. Our observation agrees well with the result reported by Kasahara et al.48 that replacing W388 with any of the other 19 native amino acids dramatically impairs the transport activity, indicating the integral role of W338 in modulating the diffusion.
| PMF(z) = −logP(z)/kT, |

As shown in Fig. 4B, the maximum force for D-glucose to overcome is approximately 29.6 ± 1.4 pN at z = 0.5 nm based on our calculation, which is slightly smaller than that observed in the AFM force tracing experiment.
 | ||
| Fig. 4 (A) One dimensional PMF of D-glucose transporting along the z-axis and (B) the corresponding force profiles. 1000 rounds of bootstrap resampling of the unbiased trajectories were used to obtain the profiles. The insert indicates the maximum force experienced by D-glucose, which shows an average value of 30 pN from 1000 rounds of bootstrap resampling. (C) 2D PMF of D-glucose transporting by projecting the center of mass (COM) of the D-glucose and GLUT1 onto the z-axis and xy-plane. | ||
To mimic the transport process of the PEG-conjugated D-glucose, a comparison MD simulation system was also built using 9-nonyl-β-D-glucopyranoside (β-NG) as the ligand, which was co-crystallized in the inward-open conformation of GLUT1 (pdb:4PYP). The calculated PMF and mean force profile of β-NG transport is shown in Fig. S13 (ESI†). Compared with D-glucose, we noted that β-NG experienced a larger force during transport, and the maximum force was approximately 42.5 ± 4.1 pN. In addition, a stronger trapping site was also observed during β-NG transport at z = −2 nm, indicating a slower transport rate. According to the Arrhenius equation, we estimated that the rate constant for D-glucose transport was approximately 4-fold faster than that of β-NG (part 2.6, ESI†).

A volume correction was used to compare the calculated binding free energy to the experimental values20 as follows:
| ΔGbinding = ΔGPMF + ΔGV |
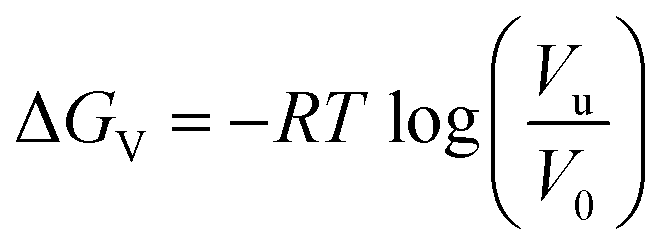 , Vu = 1.142 nm3 is the average system volume of our simulations, and V0 = 1.661 nm3 is the standard volume (1 M concentration).
, Vu = 1.142 nm3 is the average system volume of our simulations, and V0 = 1.661 nm3 is the standard volume (1 M concentration).
To calculate the binding free energy from the experimentally estimated dissociation constant, the following equation was used:
| ΔGbinding = −RTlogKd |
We obtained an overall binding free energy of ΔGbinding = 1.914 kcal mol−1, which is in agreement with the experimental measurements listed in Table 1.50,51
3. Validation of the functional role of the salt bridges using mutation experiments
To further verify our hypothesis and prove the significance of these salt bridges, both the related (K38, K300 and E426) and non-related (G346 and V327) residues of GLUT1 were mutated to alanine (A), and then transfection vectors were constructed.After transiently expressing these mutants and the wild type form of GLUT1 in HEK293 cells, the rates of glucose uptake were estimated. As shown in Fig. 5, the overexpression of K38A, K300A or E426A was able to blunt glucose transport. The rate of inhibition for the K300A, E426A and K38A mutants was 86.8%, 72.9% and 24.4%, respectively. We also performed force tracing measurements on the cells that overexpressed related mutations (K38A and K300A) and non-related mutations (G346A and V327A). The transport probability was 2.0% for cells overexpressing K38A and 0.6% for cells overexpressing K300A (Fig. S18, ESI†), which is clearly lower than that of wild type cells (14.6%). The transport probability was 9% for cells overexpressing G346A and 8% for cells overexpressing V327A (Fig. S18, ESI†), which is close to that of wild type cells (14.6%). In addition, the force value detected on the wild type cells is similar to that detected on the cells overexpressing the non-related mutations (G346A and V327A), as shown in Fig. S19 (ESI†). These results provided sufficient evidence to support our hypothesis that the effects of K38A, K300A and E426A mutations on glucose uptake are specific, which illustrates the critical role of these two salt bridges in modulating glucose transport. In addition, we constructed a double mutant (E299A/K300A) of GLUT1. However, GFP expression of the double mutant was not observed (Fig. S20, ESI†), suggesting that the double mutation might impair the folding of GLUT1.
 | ||
| Fig. 5 Mutations of the K38, K300 or E426 residues of GLUT1 that blunted the uptake of glucose into the cells. (A) Representative images of the HEK293 cell expression of the WT or the K38A, K300A or E426A mutant versions of GLUT1-GFP. (B) Total glucose uptake into the cells. The data were normalized to the WT. (C) Percentage of glucose uptake inhibition observed by expressing the mutants or the WT GLUT1 in the cells. The data were subtracted from the blank and were normalized to the WT. The inhibition values caused by the K300A, E426A or K38A mutants are 86.84%, 72.92% and 24.43%, respectively. The data were subtracted from the blank and were normalized to the WT from B. *P < 0.05, **P < 0.01. | ||
Conclusions
We comprehensively illuminated single-molecule glucose transport using both experimental and theoretical methods. The force tracing experimental results show that the force to transport a single D-glucose molecule across cell membranes is 37 ± 9 pN, and the transport event could be accomplished in approximately 20 ms with an average rate of 0.3 μm s−1. Our calculated force profile from the MD simulations exhibited quantitatively good agreement with the force tracing observation. These results provide the foundation to understand the dynamic mechanisms of glucose transport. Moreover, 4 residues (K38, E299, K300 and E426) on the extracellular side of GLUT were identified to play critical roles in modulating the conformational transition, which is helpful for developing potential therapeutic agents targeting GLUT1.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Stuart Lindsay for providing helpful comments and assistance. This work was supported by the National Key R&D Program of China (No. 2017YFA0505300), the National Natural Science Foundation of China (No. 21727816, 21525314, 21721003 to H. D. W; No. 21573217 to G. H. L.; No. 31330082 and 21673023 to Y. P. S), the Science and Technology Department of Yunnan Province (No. 2017FA044, 2013HA023 to W. X.) and the Jilin Provincial Science Research Foundation of China (No. 20160520133JH to Y. P. S).Notes and references
- H. Krishnamurthy, C. L. Piscitelli and E. Gouaux, Nature, 2009, 459, 347–355 CrossRef CAS PubMed.
- V. Braun, Science, 1998, 282, 2202–2203 CrossRef CAS PubMed.
- P. L. Shaffer, A. Goehring, A. Shankaranarayanan and E. Gouaux, Science, 2009, 325, 1010–1014 CrossRef CAS PubMed.
- Z. L. Johnson, C.-G. Cheong and S.-Y. Lee, Nature, 2012, 483, 489–493 CrossRef CAS PubMed.
- Y. Cao, X. Jin, H. Huang, M. G. Derebe, E. J. Levin, V. Kabaleeswaran, Y. Pan, M. Punta, J. Love, J. Weng, M. Quick, S. Ye, B. Kloss, R. Bruni, E. Martinez-Hackert, W. A. Hendrickson, B. Rost, J. A. Javitch, K. R. Rajashankar, Y. Jiang and M. Zhou, Nature, 2011, 471, 336–340 CrossRef CAS PubMed.
- J. R. Schnell and J. J. Chou, Nature, 2008, 451, 591–595 CrossRef CAS PubMed.
- P. Kolman, A. Pica, N. Carvou, A. Boyde, S. Cockcroft, A. Loesch, A. Pizzey, M. Simeoni, G. Capasso and R. J. Unwin, Am. J. Physiol., 2009, 296, F1227–F1237 CrossRef CAS PubMed.
- P. Hinterdorfer and Y. F. Dufrene, Nat. Methods, 2006, 3, 347–355 CrossRef CAS PubMed.
- S. Rodriguez-Enriquez, A. Marin-Hernandez, J. Carlos Gallardo-Perez and R. Moreno-Sanchez, J. Cell. Physiol., 2009, 221, 552–559 CrossRef CAS PubMed.
- I. A. Belyantseva, H. J. Adler, R. Curi, G. I. Frolenkov and B. Kachar, J. Neurosci., 2000, 20, RC116 CrossRef CAS PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, in Molecular Cell Biology, ed. W. H. Freeman, New York, 4th edn, 2000, Section 15.2 Search PubMed.
- B. Maier, I. Chen, D. Dubnau and M. P. Sheetz, Nat. Struct. Mol. Biol., 2004, 11, 643–649 CAS.
- P. C. Zhang, A. M. Keleshian and F. Sachs, Nature, 2001, 413, 428–432 CrossRef CAS PubMed.
- Y. Pan, S. Wang, Y. Shan, D. Zhang, J. Gao, M. Zhang, S. Liu, M. Cai, H. Xu, G. Li, Q. Qin and H. Wang, Small, 2015, 11, 2782–2788 CrossRef CAS PubMed.
- R. O. Dror, R. M. Dirks, J. P. Grossman, H. F. Xu and D. E. Shaw, Annu. Rev. Biophys., 2012, 41, 429–452 CrossRef CAS PubMed.
- Y. B. Zhang, H. Y. Niu, Y. Li, H. Y. Chu, H. J. Shen, D. L. Zhang and G. H. Li, Sci. Rep., 2015, 5, 8405–8412 CrossRef CAS PubMed.
- N. Plattner and F. Noe, Nat. Commun., 2015, 6, 7653–7662 CrossRef PubMed.
- V. S. Pande, K. Beauchamp and G. R. Bowman, Methods, 2010, 52, 99–105 CrossRef CAS PubMed.
- J. D. Chodera and F. Noe, Curr. Opin. Struct. Biol., 2014, 25, 135–144 CrossRef CAS PubMed.
- I. Buch, T. Giorgino and G. De Fabritiis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10184–10189 CrossRef CAS PubMed.
- B. C. Mulukutla, A. Yongky, T. Le, D. G. Mashek and W.-S. Hue, Trends Biotechnol., 2016, 34, 638–651 CrossRef CAS PubMed.
- D. Deng and N. Yan, Protein Sci., 2015, 25, 546–558 CrossRef PubMed.
- G. W. Gould and G. I. Bell, Trends Biochem. Sci., 1990, 15, 18–23 CrossRef CAS PubMed.
- D. Deng, C. Xu, P. Sun, J. Wu, C. Yan, M. Hu and N. Yan, Nature, 2014, 510, 121–125 CrossRef CAS PubMed.
- D. C. De Vivo, R. R. Trifiletti, R. I. Jacobson, G. M. Ronen, R. A. Behmand and S. I. Harik, N. Engl. J. Med., 1991, 325, 703–709 CrossRef CAS PubMed.
- L. Szablewski, Int. J. Cardiol., 2017, 230, 70–75 CrossRef PubMed.
- I. Stefanidis, K. Kytoudis, A. A. Papathanasiou, D. Zaragotas, L. Melistas, G. D. Kitsios, N. Yiannakouris and E. Zintzaras, Dis. Markers, 2009, 27, 29–35 CrossRef CAS PubMed.
- B. Y. Shim, J. H. Jung, K. M. Lee, H. J. Kim, S. H. Hong, S. H. Kim, D. S. Sun and H. M. Cho, Int. J. Colorectal Dis., 2013, 28, 375–383 CrossRef PubMed.
- M. O. Carayannopoulos, M. M. Y. Chi, Y. Cui, J. M. Pingsterhaus, R. A. McKnight, M. Mueckler, S. U. Devaskar and K. H. Moley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7313–7318 CrossRef CAS.
- I. Neundlinger, T. Puntheeranurak, L. Wildling, C. Rankl, L. X. Wang, H. J. Gruber, R. K. Kinne and P. Hinterdorfer, J. Biol. Chem., 2014, 289, 21673–21683 CrossRef PubMed.
- C. K. Riener, F. Kienberger, C. D. Hahn, G. M. Buchinger, I. O. C. Egwim, T. Haselgrubler, A. Ebner, C. Romanin, C. Klampfl, B. Lackner, H. Prinz, D. Blaas, P. Hinterdorfer and H. J. Gruber, Anal. Chim. Acta, 2003, 497, 101–114 CrossRef CAS.
- C. K. Riener, C. M. Stroh, A. Ebner, C. Klampfl, A. A. Gall, C. Romanin, Y. L. Lyubchenko, P. Hinterdorfer and H. J. Gruber, Anal. Chim. Acta, 2003, 479, 59–75 CrossRef CAS.
- D. Deng, C. Xu, P. Sun, J. Wu, C. Yan, M. Hu and N. Yan, Nature, 2014, 510, 121–125 CrossRef CAS PubMed.
- M. E. Gibbs, D. S. Hutchinson and R. J. Summers, Neuropsychopharmacology, 2008, 33, 2384–2397 CrossRef CAS PubMed.
- T. Sakyo and T. Kitagawa, Biochim. Biophys. Acta, Biomembr., 2002, 1567, 165–175 CrossRef CAS.
- D. Wendell, P. Jing, J. Geng, V. Subramaniam, T. J. Lee, C. Montemagno and P. Guo, Nat. Nanotechnol., 2009, 4, 765–772 CrossRef CAS PubMed.
- J. Schonfelder, R. Perez-Jimenez and V. Munoz, Nat. Commun., 2016, 7, 11777 CrossRef CAS PubMed.
- D. Sharma, O. Perisic, Q. Peng, Y. Cao, C. Lam, H. Lu and H. B. Li, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9278–9283 CrossRef CAS PubMed.
- G. Bussi, A. Laio and M. Parrinello, Phys. Rev. Lett., 2006, 96, 090601 CrossRef PubMed.
- P. Tiwary and M. Parrinello, Phys. Rev. Lett., 2013, 111, 230602 CrossRef PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- X. H. Huang, G. R. Bowman, S. Bacallado and V. S. Pande, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19765–19769 CrossRef CAS PubMed.
- M. K. Scherer, B. Trendelkamp-Schroer, F. Paul, G. Perez-Hernandez, M. Hoffmann, N. Plattner, C. Wehmeyer, J. H. Prinz and F. Noe, J. Chem. Theory Comput., 2015, 11, 5525–5542 CrossRef CAS PubMed.
- G. F. Baker and W. F. Widdas, J. Physiol., 1973, 231, 129–142 CrossRef CAS.
- R. C. Ritter and P. Slusser, Am. J. Physiol., 1980, 238, E141–E144 CAS.
- L. Sun, X. Zeng, C. Yan, X. Sun, X. Gong, Y. Rao and N. Yan, Nature, 2012, 490, 361–366 CrossRef CAS PubMed.
- M. Mueckler, W. F. Weng and M. Kruse, J. Biol. Chem., 1994, 269, 20533–20538 CAS.
- T. Kasahara and M. Kasahara, J. Biol. Chem., 1998, 273, 29113–29117 CrossRef CAS PubMed.
- S. Roblitz and M. Weber, Adv. Data Anal. Classi., 2013, 7, 147–179 CrossRef.
- C. L. Hagglund, I. Gottschalk and P. Lundahl, J. Chromatogr. A, 2004, 1031, 113–116 CrossRef.
- M. Lachaal, R. A. Spangler and C. Y. Jung, Biochim. Biophys. Acta, Biomembr., 2001, 1511, 123–133 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods, Fig. S1–S18 and Movie 1. See DOI: 10.1039/c8nh00056e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |