
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light active nanofibrous membrane for antibacterial wound dressing†
Shuai
Jiang‡
a,
Beatriz Chiyin
Ma‡
a,
Wei
Huang
a,
Anke
Kaltbeitzel
a,
Gönül
Kizisavas
a,
Daniel
Crespy
 ab,
Kai A. I.
Zhang
a and
Katharina
Landfester
*a
ab,
Kai A. I.
Zhang
a and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz 55128, Germany. E-mail: landfester@mpip-mainz.mpg.de
bDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand
First published on 13th April 2018
Abstract
Chronic wound infections, especially due to the emergence of multidrug resistance in bacteria, require the urgent development of alternative antibacterial therapies. Here, we developed a new class of hydrogel nanofibrous membranes that show visible light-induced disinfection. The presented photocatalytic disinfection is based on the generation of reactive singlet oxygen from a conjugated microporous polymer upon visible light irradiation. Therefore, sustained protection of the wound area can be provided in the presence of visible light. Fabrication of the photoactive wound dressing consists of first synthesizing photoactive conjugated microporous polymer nanoparticles by miniemulsion polymerization and subsequently embedding the nanoparticles in polyvinyl alcohol hydrogel nanofibers by colloid-electrospinning. The fibers were then crosslinked in glutaraldehyde/HCl vapor to be water-insoluble. This nanoparticle-in-nanofiber structure allows for a flexible combination of the properties of the nanoparticles and supporting nanofibers. The disinfecting properties of the membranes were evaluated with the inactivation of Escherichia coli K-12 and Bacillus subtilis as model systems of Gram-negative and Gram-positive bacteria, as well as the inhibition of biofilm growth under irradiation of visible light. Cytotoxicity tests on fibroblast cells revealed a high cytocompatibility of the membranes. Furthermore, the good mechanical properties of the membranes allow for their facile removal after use and prevent the leakage of the embedded nanoparticles into the wound, making the photoactive hydrogel membranes a promising candidate for active wound dressing materials.
Conceptual insights(1) A new class of hydrogel nanofibrous membranes that show visible light-induced disinfection has been developed. The nanofibrous membranes could be used as a potential antibacterial wound dressing material. (2) Conventional wound dressing materials rather rely on the release of antiseptic agents. Their effect can be likely inhibited due to the ever-developing multidrug resistance of bacteria. Our study shows a different concept of disinfection mechanism for wound dressing materials, which is based on the generation of reactive singlet oxygen from the photoactive polymers inside the nanofibers upon visible light irradiation. (3) We constructed a “photoactive polymer nanoparticle-in-classical polymer nanofiber” nanofibrous structure via colloid-electrospinning. This unique structure allows a flexible combination of the properties of the nanoparticles and supporting nanofibers. The disinfecting properties of the membranes were evaluated with the inactivation of Escherichia coli K-12 and Bacillus subtilis as model systems of Gram-negative and Gram-positive bacteria, and the inhibition of biofilm growth under irradiation of visible light. The cytotoxicity tests on fibroblast cells showed a high cytocompatibility of the membranes. Additionally, the good mechanical properties of the membranes allow for their facile removal after use and prevent the leakage of the embedded nanoparticles into the wound. |
Introduction
Chronic wound infection by bacteria and other microorganisms is one of the main factors that result in complications during the wound healing process.1 Wound dressings have been traditionally applied to protect open wounds from further injuries and infections, as well as to provide an optimal environment for skin regeneration.2–6 Among the materials being developed for functional wound dressing, special attention has been paid to electrospun polymeric nonwovens that are composed of interconnected nanofibrous networks.3,4 The electrospun membranes exhibit unique advantages as the conformable nonwovens act as an effective physical barrier to protect a wound from further physical damage and contamination while serving as a template for the growth of skin cells during the self-repairing process, which is beneficial for minimize scar formation.7 At the same time, electrospun nanofibrous mats are permeable to moisture and air, therefore, extra exudate can be extracted from the wound area in order to avoid further infections while still keeping a local moist environment.8 Moisture is essential for the acceleration of the wound healing process and reduction of scar formation since in a wet environment skin is regenerated without forming a scab.9A variety of active wound dressings have been reported via loading hydrogel networks with antibacterial agents such as silver nanoparticles, metal oxide nanoparticles, essential oils, antibiotics or their combinations.2,10–16 Such dressing materials allow for a sustained drug release into the wound bed, thus inhibiting infections and accelerating the healing process.3,4 A combination of different antibacterial agents is also often beneficial due to the fact that compounds with different mechanisms of action may act synergistically by targeting multiple sites in the cell,17 hence broadening the antibacterial spectrum, reducing single drug dosage, decreasing side effects, and shortening the duration of treatment, as well as minimizing or delaying the onset of drug resistance.15,18–20 One representative example for this approach is the combination of essential oils with synthetic antimicrobial agents, which showed an enhanced antibacterial effect in vitro.20–22 In our recent work, multiple antibacterial agents with different polarities were able to be loaded into a wound dressing via the design of multicompartment nanofibrous membranes with a nanocapsule-in-nanofiber structure.23 Nevertheless, these materials rely on the release of pre-loaded antibacterial agents from the fibers, and therefore, have a limited service lifecycle. Moreover, despite the fact that such treatments can inhibit the proliferation of pathogens, they can also prevent the proliferation of collagen in the corium and the re-colonization of granulation tissue by keratinocytes, impeding the healing process.24
Recently, photodynamic therapy has emerged as an alternative approach since its effectiveness is based on the generation of reactive oxygen species, e.g. singlet oxygen, upon light irradiation. Diaz-Uribe et al. studied the production of 1O2 from tetra(4-carboxyphenyl)porphyrin adsorbed on SiO2 under visible light irradiation. The photogeneration of 1O2 as the only photoproduct was evidenced by TEMPO EPR spectroscopy and chemical trapping.25 Therefore, it provides a new strategy for treatment against antibiotic-resistant bacteria as well as preventing the selection of resistant strains.26,27 Singlet oxygen has a lifetime of 3.5 μs in aqueous environments28 and its diffusion pathway in polymers and aqueous solutions ranges from tens to hundreds of nanometers.29 Taking this aspect into consideration, the photoinduced disinfecting effect is relatively confined to the skin's surface in comparison to standard antiseptic treatments, and therefore, it holds the advantage of not interfering with surrounding proliferating tissues and deeper tissues.30 Leyland et al. prepared titania coatings that were doped with fluorine and copper.31 The doping was found to provide superior photocatalytic and antimicrobial properties compared to pure titania coatings. These findings were attributed to the higher production of reactive hydroxyl radicals and the toxicity of copper toward pathogenic bacteria. Ag2S/Ag heteronanostructures were found to efficiently inactivate E. coli under UV irradiation.32
Combining photodynamic therapy together with wound dressings represents a promising direction for developing a new generation of active wound dressings. One approach is doping polymer fabrics with hydrophilic photosensitizer (PS), e.g. methylene blue33 and porphyrin.34 The antimicrobial photodynamic activity of the material relies on the generation of reactive oxygen species via irradiating the released PS in the medium. However, the introduction of a PS into the wound issue is not desirable in this case. As an alternative method reported by Mosinger et al., hydrophobic PS tetraphenylporphyrin was encapsulated in polyurethane or polystyrene nanofibers.30,35–37 Therefore, the leakage of the PS could be avoided, but due to the insufficient contact between the hydrophobic fiber surface and the biological targets in an aqueous medium, the photo-oxidation efficiency was significantly decreased. Increasing the surface wettability could improve the photo-oxidation efficiency of the fibers.38 But introducing hydrophilicity to the fibers usually needs additional surface modifications, e.g. chemical or plasma treatments or polymer coating. More efficient photo-oxidation was also achieved by attaching a cationic photosensitizer to the surface of sulfonated nanofibers, where the photosensitizer is more sensitive and again may leak into the surrounding tissues.36,37
Taking the above developments and their limitations into consideration, we present here a new class of visible light-active hydrogel nanofibrous membranes for disinfecting wound dressings (see Scheme 1). This photoactive membrane was fabricated by first synthesizing conjugated microporous polymer nanoparticles in a miniemulsion and subsequently embedding the nanoparticles in polyvinyl alcohol (PVA) hydrogel nanofibers by colloid-electrospinning. The nanofibers were crosslinked in glutaraldehyde/HCl vapor to be water-insoluble. The presented design strategy allows for a flexible combination of the properties of nanoparticles and supporting nanofibers, thereby providing a versatile platform for the fabrication of multifunctional wound dressings. Taking into consideration that the post-crosslinking of the membranes was performed with the vapor of the crosslinker, functional agents, e.g. therapeutic agents and growth factors, could be loaded into the nanoparticles or/and into the fibers without any leakage during the crosslinking process.
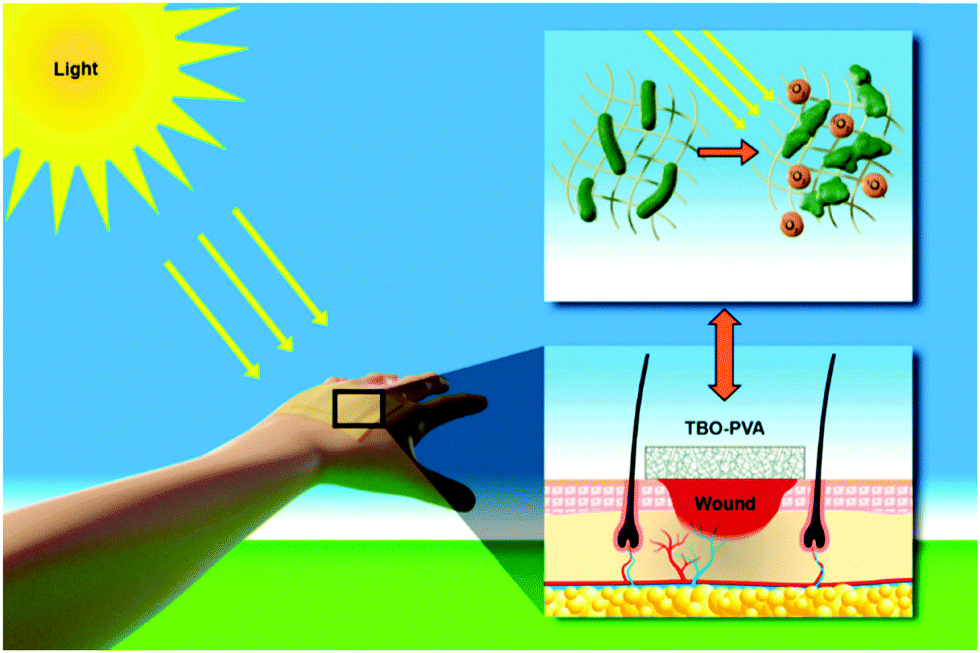 | ||
| Scheme 1 Schematic illustration of visible light active nanofibrous membranes for antibacterial wound dressings. When light is irradiated on the dressing, reactive singlet oxygen is produced that inactivates bacteria present in the wound. The singlet oxygen is produced by photoactive benzo[c]-1,2,5-oxadiazole based conjugated microporous polymer nanoparticles (TBO NPs) embedded in the nanofibers. | ||
Experimental
Materials
Polyvinyl alcohol (Polysciences Inc., 88 mol% hydrolyzed) with a molecular weight Mw of 205![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 was used as received. 1,3,5-Triethynylbenzene (97%), copper(I) iodide (99%), tetrakis(triphenylphosphine) palladium(0) (99%), benzofurazan (97%), triethylamine (99%), iron (99%), 2,2,6,6-tetramethylpiperidine (98%) and crystal violet (90%) were purchased from Sigma Aldrich. Glutaraldehyde (GA, 50 wt% aqueous solution) was purchased from Merck KGaA. Sodium n-dodecylsulfate (99%) was obtained from Alfa Aesar. Magnesium sulfate and sodium bicarbonate were purchased from Fisher Chemical. Escherichia coli K-12 and Bacillus subtilis were purchased from DSMZ Leibniz-Institute (Braunschweig, Germany). Demineralized water was used through all the experiments if not specifically mentioned. All chemicals and solvents were used as received unless otherwise specified.
000 g mol−1 was used as received. 1,3,5-Triethynylbenzene (97%), copper(I) iodide (99%), tetrakis(triphenylphosphine) palladium(0) (99%), benzofurazan (97%), triethylamine (99%), iron (99%), 2,2,6,6-tetramethylpiperidine (98%) and crystal violet (90%) were purchased from Sigma Aldrich. Glutaraldehyde (GA, 50 wt% aqueous solution) was purchased from Merck KGaA. Sodium n-dodecylsulfate (99%) was obtained from Alfa Aesar. Magnesium sulfate and sodium bicarbonate were purchased from Fisher Chemical. Escherichia coli K-12 and Bacillus subtilis were purchased from DSMZ Leibniz-Institute (Braunschweig, Germany). Demineralized water was used through all the experiments if not specifically mentioned. All chemicals and solvents were used as received unless otherwise specified.
Synthesis and purification of benzo[c]-1,2,5-oxadiazole based conjugated microporous polymer nanoparticles (TBO NPs)
The synthesis of TBO NPs was performed using Sonogashira–Hagihara cross coupling reaction in an oil-in-water miniemulsion. First, 1,3,5-triethynylbenzene (50.0 mg, 0.33 mmol), 4,7-dibromobenzo[c]-1,2,5-oxadiazole (139.0 mg, 0.5 mmol), tetrakis(triphenylphosphine) palladium(0) (19.7 mg, 0.017 mmol) and copper(I) iodide (3.2 mg, 0.017 mmol) were dissolved in 2 mL of toluene. Then, triethylamine (1 mL) and an aqueous solution (17 mL) of sodium n-dodecylsulfate (200 mg) were added to the organic phase and vigorously stirred using a Branson W-450D digital sonifier operating at 70% of amplitude for 2 min. The reaction mixture was then stirred and heated at 80 °C overnight. The resulting TBO NP dispersion was transferred to a dialysis tube (MWCO 14000 Da) with continuous water substitution to remove the excess of surfactant and further purified by centrifugation (10000 rpm, 10 min, twice). The resulting final product was collected from the supernatant and characterized. The synthesis of 4,7-dibromobenzo[c]-1,2,5-oxadiazole was carried out according to previous studies and the data are consistent with the literature.39
Fabrication of nanofibrous membranes
2.25 mL of the as-synthesized TBO NP dispersion was added to 2.75 mL of 15 wt% PVA aqueous solution. The mixed solution was magnetically stirred at 500 rpm overnight. In the case of fabrication of the control sample (PVA membrane), the TBO NP dispersion was replaced by demineralized water to mix with PVA solution. The electrospinning process was carried out using a setup fabricated by IME Technologies with an applied voltage of 15 kV, a working distance between the spinneret and the collector of 10 cm, and a feeding rate of 0.2 mL h−1.Crosslinking of PVA-based nanofibers
The electrospun membranes were carefully detached from the aluminum foil collector. The non-wovens were then exposed to GA/HCl vapor in a vacuum desiccator at 6 × 10−2 MPa for 1 h. 1 mL of 50 wt% GA aqueous solution was used as a GA vapor source. 20 μL of 37 wt% HCl aqueous solution was used as the source of HCl vapor which catalyzed the crosslinking reaction between –OH of PVA and –CHO of GA to form acetal bridges.40 Afterwards, the crosslinked membranes were exposed to air flow in a fume hood for 24 h to evaporate unreacted GA and HCl.Photocatalytic antibacterial test
The glassware used was sterilized in an autoclave at 121 °C for 20 min. The bacterial cells were cultured in nutrient broth (Sigma-Aldrich) and agitated at 300 rpm for 16 h at 37 °C and 30 °C for E. coli K-12 and B. subtilis, respectively. The cells were washed with sterilized saline solution (0.9 wt% NaCl), and the final cell density was adjusted to ca. 3.0 × 106 cfu (colony forming units) mL−1. 10 mg of PVA or PVA–TBO nanofibers were first washed 3 times with MilliQ water, and then added to the cell suspension with a final volume of 2 mL and kept in the dark for 60 min. After this, the reaction mixture was bubbled with oxygen and irradiated with visible light (460 nm, 0.1 W cm−2, OSA Opto Lights) for different periods of time. At selected time intervals, aliquots of the sample were collected and serially diluted with sterilized saline solution. An amount of 0.1 mL of the diluted sample was then immediately spread on nutrient agar (Sigma-Aldrich) plates and incubated at 37 °C for 16 h to determine the number of viable cells (in colony forming units, cfu). For comparison, control experiments with only E. coli K-12 and B. subtilis bacterial cells were carried out in the absence of nanofibers and in the dark. Control experiments were also carried out with nanofibers in the absence of light. The survival fraction was determined by dividing the number of cfu of the samples incubated with nanofibers by the number of cfu of the control group in the absence of nanofibers. All treated and control experiments were performed in independent triplicates.Biofilm formation assay
After performing the photocatalytic antibacterial test (2 h light irradiation), treated B. subtilis in a cell density of about 3.0 × 106 cfu mL−1 was added to a final volume of 1 mL in each well of a 24-well polystyrene microtiter plate (Greiner, USA) and incubated at 30 °C for 24 h. After that, the solutions of bacteria were carefully removed. The remaining formed biofilm and wells were rinsed with sterile distilled water. Dark control samples carried out with nanofibers in the absence of light, control samples with bacterial cells in the absence of nanofibers and blank samples without bacteria were also prepared. All samples were then stained with 0.4% (w/v) crystal violet for 30 min, washed with sterile distilled water to remove the excess unbound dye, and eluted with 30% (v/v) acetic acid. Subsequently, the absorbance at 590 nm was measured. The biofilm formation was quantified by measuring the difference between the absorbance of untreated and treated bacteria samples. All treated and control experiments were performed in independent triplicates.Cytocompatibility test of nanofibrous membranes
Cytocompatibility of the membranes was tested in terms of cytotoxicity with NIH 3T3 Fibroblasts. The cells were seeded in a 24-well plate with 100000 cells per well in an RPMI medium supplemented with 10% fetal bovine serum and 1% PenStrep. After incubation for 24 h at 37 °C, the medium was refreshed. The membranes were added in the well and the cells were further incubated for 24 h. After incubation, the medium and the membrane were carefully removed to avoid damage of the attached cells at the bottom. Afterwards, the cells were washed with DPBS. Trypsin–EDTA (0.25%) solution was added to detach the cells. The cell suspensions were centrifuged at 300 g for 5 min. The collected cells were stained with Annexin V and 7-AAD for 30 min at 4 °C and then washed with DPBS. The cell viability was measured by using a Flow Cytometer.
Photooxidation of α-terpinene
To quantify the 1O2 generation efficiency, the photooxidation of α-terpinene was performed using a mixture of α-terpinene (0.1 M), photocatalyst (1 mg mL−1), and mesitylene (0.1 M) as an internal standard in 10 mL acetonitrile. Under an oxygen atmosphere, the mixture was stirred at room temperature and irradiated with visible light for 24 h. Aliquots of the sample were then collected and filtered with a syringe filter and the solution was analyzed by 1H NMR spectroscopy. The 1O2 generation rate was defined as: R(1O2) = (obtained amount of ascaridole [mmol])/(weight of photocatalyst [g])/(reaction time [s]).Analytical methods
The morphologies of the nanoparticles and nanofibers were examined with a Gemini 1530 (Carl Zeiss AG, Oberkochem, Germany) scanning electron microscope (SEM) operating at 0.35 kV and a Jeol 1400 (Jeol Ltd, Tokyo, Japan) transmission electron microscope (TEM) operating at an accelerating voltage of 120 kV. SEM and TEM samples of nanoparticles were prepared by casting the diluted nanoparticle dispersions on silicon wafers and carbon layer-coated copper grids, respectively. The samples of nanofibers for SEM and TEM were prepared by depositing electrospun nanofibers directly on silicon wafers and carbon layer-coated copper grids, respectively. To visualize the distribution of TBO NPs in the fibers, the PVA–TBO samples were stained with RuO4 vapor in a glass-covered dish for 20 min. The diameter of the electrospun nanofibers was estimated by evaluating 100 nanofibers from SEM micrographs. The hydrodynamic diameter of the nanoparticles was evaluated by dynamic light scattering (DLS) using a Nicomp particle sizer (Model 380, PSS, Santa Barbara, CA) at a fixed scattering angle of 90°. The UV/Vis absorption spectra were recorded on a Perkin Elmer Lambda 25 UV-Vis spectrometer. FT-IR spectra were recorded on a Nicolet 730 FT-IR spectrometer. Polymer surface area and pore size distribution were measured by nitrogen adsorption and desorption at 77.3 K using an Autosorb-1 from Quantachrome Instruments. The data were evaluated using QuadraWin software (Quantachrome Instruments). Pore size distributions and pore volumes were derived from the adsorption branches of the isotherms using Quenched Solid Density Functional Theory (QSDFT, N2, assuming carbon as an adsorbent with slit pores). Samples were degassed at 150 °C for 24 h under vacuum before analysis. The BET surface area calculation was based on data points obtained from 0.05 < P/P0 < 0.35 and the nonlinear density functional theory (NLDFT) equilibrium model was used for the BET model fitting. Electron paramagnetic resonance (EPR) was measured on a Magnettech Miniscope MS200 spectrometer with 2,2,6,6-tetramethylpiperidine (TEMP) as the singlet oxygen trapper. Cyclic voltammetry (CV) was recorded on an Autolab Potentiostat PGSTAT204 (Metrohm). A glassy carbon electrode drop-cast with the TBO NPs as the working electrode, Pt wire as the counter electrode, a Hg/HgCl (in saturated KCl solution) electrode as the reference electrode, and Bu4NPF6 (0.1 M in acetonitrile) as the electrolyte were used. The reduction potential was recorded with a scan rate of 100 mV s−1. The mechanical properties of the nanofibrous non-woven were tested by using a ProLine table-top testing machine Z005 from the company Zwick/Roell. The tests were conducted at a crosshead speed of 10 mm min−1 by using dumbbell specimens with the dimensions of 13 mm × 2 mm (gage length × width). The thickness of the membranes was measured by vernier caliper. The water absorption ratio of the membranes was defined as the swelling degree in this study. To evaluate the swelling degree, the membranes were dried at 80 °C under vacuum for 12 h. Subsequently, the dry membranes were soaked in demineralized water in a thermomixer (HLC MKR 23, Ditabis) at 37 °C for 24 h. The weight of the swollen samples was measured after the excessive water on the surface was removed with filter paper. The swelling degree of the membranes is defined as follows: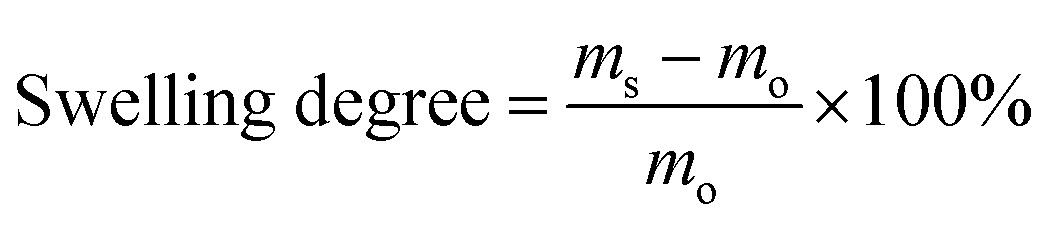 | (1) |
Results and discussion
The preparation of the photoactive wound dressing material consisted of two steps (see Scheme 2). In the first step, benzo[c]-1,2,5-oxadiazole based conjugated microporous polymer nanoparticles (TBO NPs) were synthesized via a palladium-catalyzed Sonogashira–Hagihara cross-coupling polycondensation reaction in an oil-in-water miniemulsion. As a result, a stable dispersion of spherical TBO NPs in an aqueous medium was obtained (see Fig. 1a). The nanoparticles display an average hydrodynamic diameter of 85 ± 37 nm, as measured by dynamic light scattering (see inset in Fig. 1a). The Fourier transform infrared (FT-IR) spectra of the TBO NPs showed a characteristic vibrational signal at ca. 2205 cm−1, corresponding to the alkynes (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) of the polymer (see Fig. S1, ESI†). Vibrational peaks at ca. 1384 cm−1 and 1577 cm−1 are attributed to N–O stretching modes of the BO units, which were incorporated into the conjugated polymer network as strong electron acceptors. The BO units could effectively improve the π-electron delocalization and increase visible light absorption.41 As shown in Fig. 1b, the UV/Vis diffuse reflection (DR) spectrum of the TBO NPs showed a broad absorption range in the visible region up to approximately 520 nm with a maximum at ca. 410 nm. From the Kubelka–Munk-transformed reflectance spectrum, an optical band gap of 2.64 eV could be estimated (see the inset in Fig. 1b).42 To further reveal the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) positions of the TBO NPs, a cyclic voltammetry (CV) measurement was carried out. As shown in Fig. S2 (ESI†), a reduction onset potential of −0.71 V vs. SCE could be determined. Correspondingly, the HOMO level was derived to be +1.93 V (vs. SCE) via the extraction of the LUMO value from the optical band gap. The porosities of the TBO NPs were investigated by nitrogen adsorption/desorption experiments. TBO NPs showed a typical Type I adsorption isotherm (see Fig. S3a, ESI†), which indicated a predominant microporous structure. The Brunauer–Emmett–Teller (BET) method revealed the surface area of the particles to be 412 m2 g−1, with an average pore diameter of ca. 1.4 nm (see Fig. S3b, ESI†).
C–) of the polymer (see Fig. S1, ESI†). Vibrational peaks at ca. 1384 cm−1 and 1577 cm−1 are attributed to N–O stretching modes of the BO units, which were incorporated into the conjugated polymer network as strong electron acceptors. The BO units could effectively improve the π-electron delocalization and increase visible light absorption.41 As shown in Fig. 1b, the UV/Vis diffuse reflection (DR) spectrum of the TBO NPs showed a broad absorption range in the visible region up to approximately 520 nm with a maximum at ca. 410 nm. From the Kubelka–Munk-transformed reflectance spectrum, an optical band gap of 2.64 eV could be estimated (see the inset in Fig. 1b).42 To further reveal the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) positions of the TBO NPs, a cyclic voltammetry (CV) measurement was carried out. As shown in Fig. S2 (ESI†), a reduction onset potential of −0.71 V vs. SCE could be determined. Correspondingly, the HOMO level was derived to be +1.93 V (vs. SCE) via the extraction of the LUMO value from the optical band gap. The porosities of the TBO NPs were investigated by nitrogen adsorption/desorption experiments. TBO NPs showed a typical Type I adsorption isotherm (see Fig. S3a, ESI†), which indicated a predominant microporous structure. The Brunauer–Emmett–Teller (BET) method revealed the surface area of the particles to be 412 m2 g−1, with an average pore diameter of ca. 1.4 nm (see Fig. S3b, ESI†).
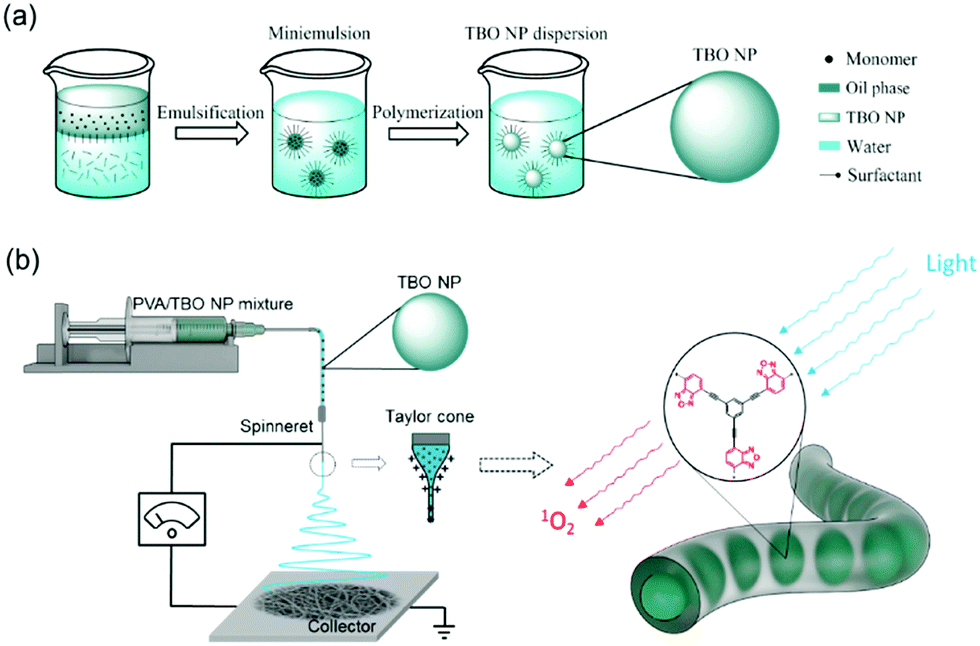 | ||
| Scheme 2 Schematic description of the fabrication process of the visible light active nanofibrous membrane. (a) Synthesis of TBO NPs by miniemulsion polymerization; (b) fabrication of PVA–TBO nanofibers by colloid-electrospinning. | ||
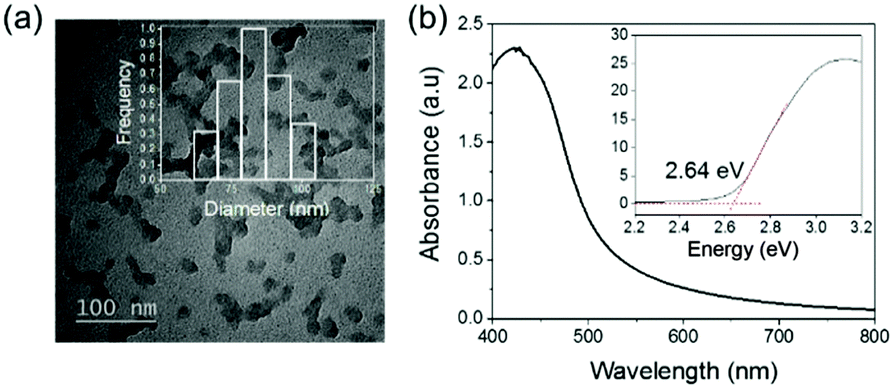 | ||
| Fig. 1 (a) TEM micrograph and the inset is the size distribution of TBO NPs. (b) UV-Vis absorption spectra and the inset displays the Kubelka–Munk-transformed reflectance spectrum. | ||
In the second step, TBO NPs were electrospun with polyvinyl alcohol (PVA) to form PVA–TBO nanofibrous membranes (see Fig. S4, ESI† for the photos of the membranes). Uniform nanofibers with an average fiber diameter of ca. 250 nm were collected as a non-woven (see Fig. S5, ESI†). PVA-based nanofibers were rendered water-insoluble via crosslinking in a glutaraldehyde/HCl vapor.43 In this case, TBO NPs were embedded into the crosslinked fiber matrix, which shall prevent the release of NPs into the wound bed. The crosslinked fiber membranes remained hydrophilic, confirmed by the spreading of water droplets after contacting them with the membranes (see Fig. S6, ESI†). The nanofibrous structure of the membranes was well preserved after immersing in water for 4 days at 37 °C (see Fig. S7, ESI†). The swelling degrees of the crosslinked PVA and PVA–TBO membranes were ∼300%. The high swelling ratio favors the absorption of wound exudate into the dressing material, thus avoiding infections taking place while still keeping a local moist environment. Moisture is beneficial for the wound healing process because it can reduce the formation of scars and allow for a painless removal of the dressing materials.44 The swollen hydrogel membranes are translucent, which allows the penetration of light through the material.30
Good mechanical properties are also essential for membranes in wound dressing applications. We compared the mechanical properties of crosslinked PVA and PVA–TBO membranes with a thickness around 100 μm. As shown in Fig. 2e and Fig. S8 (ESI†), PVA–TBO membranes exhibited an average tensile strength of 11.9 ± 0.7 MPa, which is higher than the tensile strength of PVA membranes (8.8 ± 0.8 MPa). Similarly, there is an increase of ∼18% of the Young's modulus of the PVA-based membrane due to the incorporation of TBO NPs.
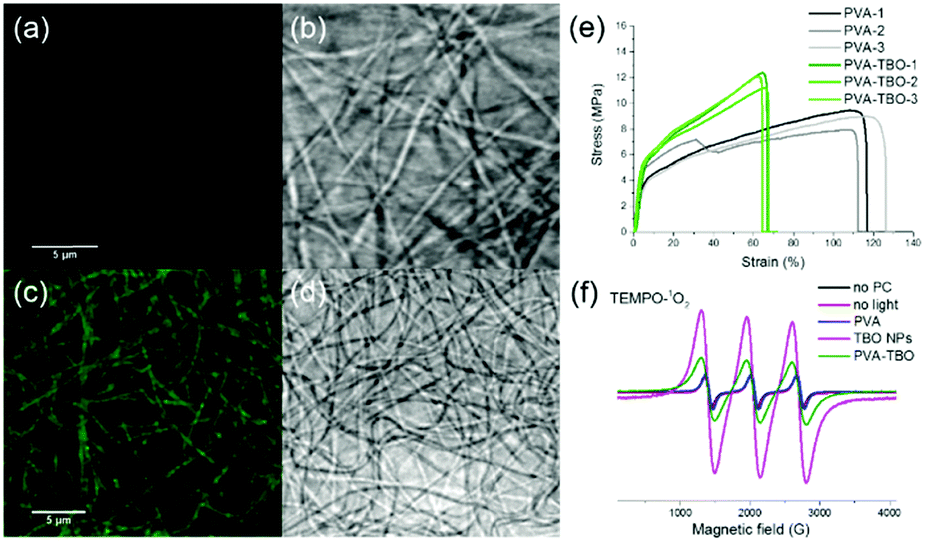 | ||
| Fig. 2 CLSM images of PVA (a and b) and PVA–TBO (c and d) nanofibrous membranes in the fluorescence mode (a and c) and transmission mode (b and d); (e) stress–strain curves of PVA and PVA–TBO membranes from 3 measurements; and (f) EPR spectra of TEMPO–1O2 adducts from PVA, TBO NPs and PVA–TBO without and with light irradiation. | ||
Correspondingly, the elongation at break of the membranes was reduced from 111% (PVA) to 64% (PVA–TBO), indicating the role of incorporated TBO NPs as reinforcing nanofillers to the fibers. This enhancement from the presence of nanoparticles also indicates their uniform distribution in the fiber matrix since defects formed due to particle aggregation are detrimental to the mechanical properties of materials.
The distribution of TBO NPs in the PVA fiber matrix was further investigated. The weight percentage of TBO NPs in PVA–TBO membranes was calculated to be 2.1 wt%. Transmission electron microscopy (TEM) of the nanofibers was used for nanoparticle identification. Composite nanofibers with a nanoparticle-in-nanofiber structure were previously studied by TEM and silica nanoparticles45 and core–shell silica nanocapsules46 could be clearly identified in the PVA nanofibers. However, in the current study, TBO polymer nanoparticles were difficult to distinguish against the PVA matrix due to the insufficient contrast between the matrix and the nanoparticles. This issue is indeed consistent with our previous study, in which polystyrene nanoparticles could not be distinguished from a PVA matrix by TEM.45 After staining the composite nanofibers with RuO4, TBO NPs were clearly visualized in the PVA matrix by TEM (see Fig. S5d, ESI†). An alternative approach is to use confocal laser scanning microscopy. PVA NFs and PVA–TBO NFs were imaged in fluorescence and transmission modes (see Fig. 2a–d). The fiber membranes were placed on a 170 μm coverslip and immersed in water in order to reduce light scattering. Excited by light from an argon laser (wavelengths 458 and 488 nm), the fibers containing TBO NPs showed a weak broad emission. The maximum of the emission spectrum was detected at ∼520 nm (full spectrum in Fig. S9, ESI†). Because this signal was absent in the case of pure PVA membranes, we concluded that the emission signal was generated by the conjugated polymer. The excitation intensity of PVA NFs with PVA–TBO NFs was identical (Fig. 2a and c), the detection window was set to 518–620 nm, and the gain adjusted to 820 V. The resolution limit of confocal fluorescence microscopy using 488 nm excitation was approximately 230 nm. Therefore, the individual NPs were too small to be resolved. However, the emission was visible all along the fibers, i.e. there were no dark regions. The minor variation of the emission along the fibers could be considered as a statistical distribution of TBO NPs inside the fibers.
The disinfecting properties of the PVA–TBO membranes are based on the generation of active oxygen species, especially singlet oxygen (1O2), from the conjugated microporous polymers under light irradiation. The UV/Vis absorption spectrum of the TBO NPs was preserved after their embedding in PVA nanofibers (see Fig. S10, ESI†). To verify the photogeneration of singlet oxygen, electron paramagnetic resonance (EPR) spin trapping experiments were conducted by using 2,2,6,6-tetramethylpiperidine (TEMP) as a singlet oxygen trapping agent. Fig. 2f shows strong 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO–1O2) adduct signals from PVA–TBO membranes under visible light irradiation, whereas much weaker signals were found under dark conditions. To further quantify the 1O2 generation efficiency, an additional experiment was performed with α-terpinene as an 1O2 trapping agent (see Fig. S11–S14 and Table S1, ESI†). With 2.5 mg mL−1 PVA–TBO fiber (PVA fiber containing 4 wt% TBO NPs) in the test medium, an 1O2 generation rate of 6.48 × 10−5 mmol g−1 s−1 was obtained. For comparison, 1 mg mL−1 pure TBO NPs yielded an 1O2 generation rate of 2.89 × 10−4 mmol g−1 s−1.
The disinfecting property of the PVA–TBO membranes was evaluated with E.coli K-12 and B. subtilis as model systems for Gram-negative and Gram-positive bacteria, respectively (see Fig. 3). A suspension of bacteria was incubated with crosslinked PVA–TBO nanofibrous membranes and exposed to visible light irradiation for different time intervals. The viability of bacteria was assessed using the standard plating method. As shown in Fig. 3a, PVA–TBO membranes could achieve 100% cell death against E.coli after 120 min of visible light irradiation. Similarly, PVA–TBO also showed a high disinfection efficiency against B. subtilis (see Fig. 3c), reaching about 100% of cell death after 60 min of visible light irradiation. These results indicated the high efficiency of PVA–TBO membranes in inactivating both Gram-negative and Gram-positive bacteria. The possible mechanism of inactivation is associated with the generation of 1O2 in the photocatalytic process as indicated in Fig. 2f. Remarkably, neither PVA nor PVA–TBO membranes showed any toxicity to bacteria cells without light irradiation. At the same time, pure PVA nanofibers did not present any inactivation of bacteria under light irradiation.
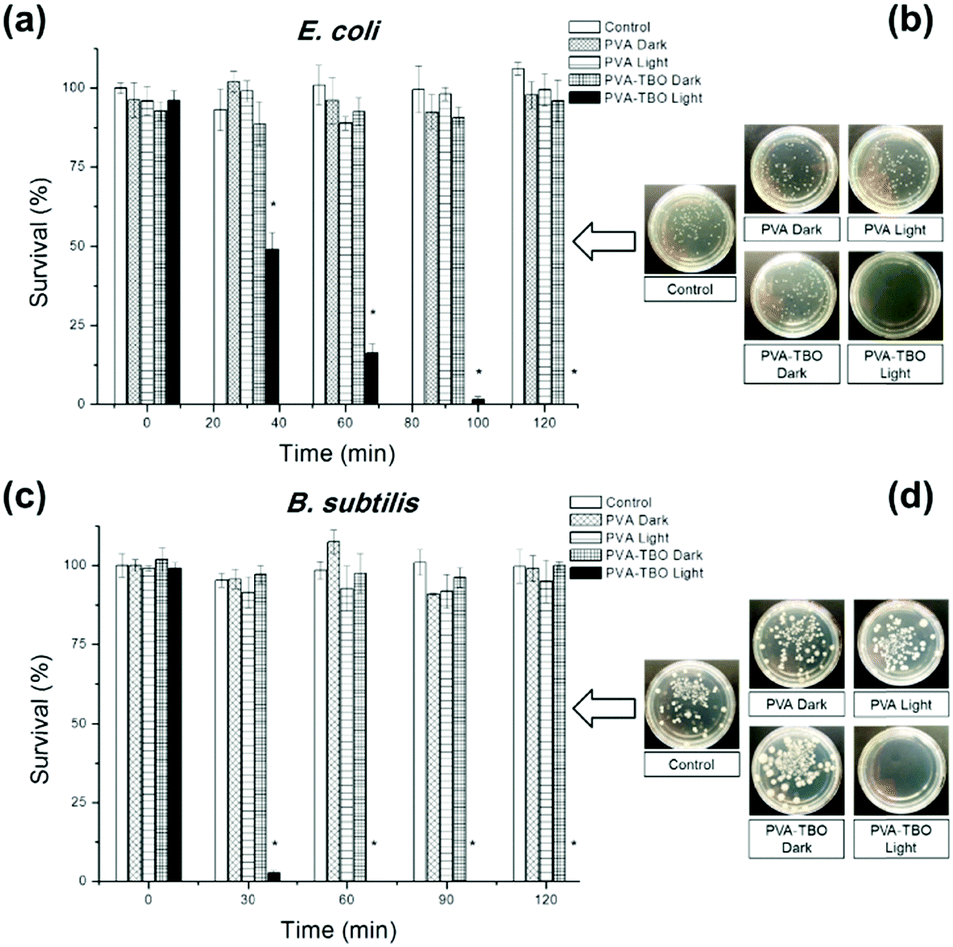 | ||
| Fig. 3 Photocatalytic inactivation of E. coli K-12 (a) and B. subtilis (c) in the presence of PVA and PVA–TBO membranes under dark conditions and visible light irradiation. Photographs of E. coli K-12 (b) and B. subtilis (d) colonies on an agar plate. The control samples contain neither PVA nor PVA–TBO membranes. Data represent the results of three independent experiments (mean ± SE of the mean). *p < 0.01. | ||
Biofilms also play an important role in a wide variety of microbial infections, with an estimate of 80% of association in all infections.47 Bacterial cells living in a biofilm are usually physiologically distinct from planktonic cells of the same species. One of the key aspects in the microenvironment in biofilms is the increased resistance to detergents and antibiotics, as the dense extracellular matrix and the outer layer of cells protect the interior of the community. In some cases antibiotic resistance can be increased 1000-fold.48 A chronic wound infection cannot be effectively managed without controlling the growth of biofilms. Therefore, the antibiofilm properties of the PVA–TBO membranes were further studied. As shown in Fig. 4, PVA–TBO membranes incubated in a solution of bacterial cells and further irradiated under visible light did not present any biofilm formation after 24 h. Comparatively, neither PVA nor PVA–TBO membranes showed any inhibition to the formation of a biofilm in the absence of light irradiation; pure PVA membranes did not present any inhibition to biofilm formation under light irradiation.
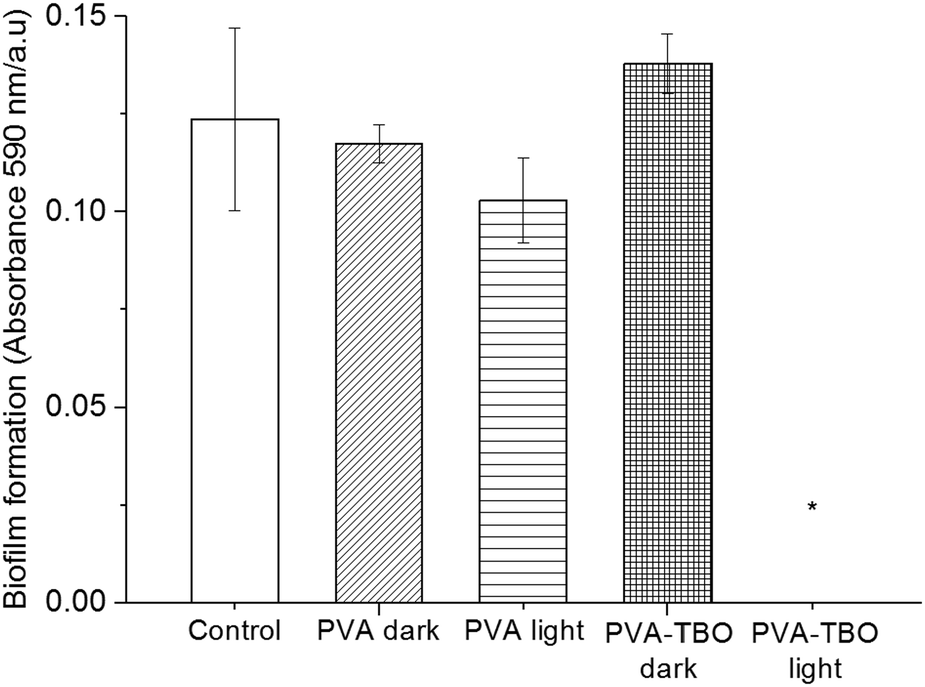 | ||
| Fig. 4 Biofilm formation of Bacillus subtilis expressed as the absorbance at 590 nm of crystal violet-stained cells. Data recorded after 24 h represent the results of three independent experiments (mean ± SE of the mean). *p < 0.01. | ||
Cytocompatibility of the fiber membranes was investigated in terms of cytotoxicity with NIH 3T3 fibroblasts. The cells were incubated with fiber membranes at 37 °C for 2 h under dark or blue light irradiation. The cytotoxicity of the membranes was analyzed by using flow cytometry (see Fig. S15, ESI†). It turns out that >94% of the cells were still alive in both cases of incubation with PVA and PVA–TBO membranes, without and with light irradiation (see Fig. 5). No significant decrease in cell viability was observed in comparison with the negative control sample, indicating the good cytocompatibility of the photoactive membranes.
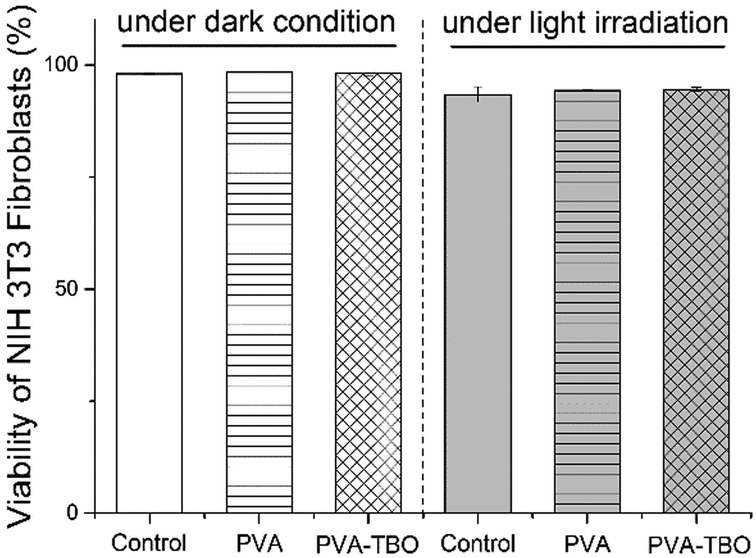 | ||
| Fig. 5 Viability of NIH 3T3 fibroblast cells after incubation with PVA or PVA–TBO membranes under dark and light irradiation. | ||
Conclusions
Nanofibrous membranes with visible-light-responsive disinfecting properties were developed for potential wound dressing materials. The fabrication process of the membranes consisted of first synthesizing photoactive conjugated microporous polymer nanoparticles and subsequently embedding nanoparticles in polyvinyl alcohol nanofibers by colloid-electrospinning. The fiber membranes possess good mechanical properties and cytocompatibility. The antibacterial properties of the photoactive membranes were demonstrated with the efficient inactivation of Gram-negative and Gram-positive bacteria, as well as the inhibition of biofilm formation under visible light irradiation, which shows the potential of the visible light active nanofibrous membrane for future wound dressing applications. Because the material is photoactive, this new generation of wound dressing is efficient for parts of the body that are not covered by clothing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge Stefan Schuhmacher for the scheme design. Open Access funding provided by the Max Planck Society.References
- K. A. Rieger, N. P. Birch and J. D. Schiffman, J. Mater. Chem. B, 2013, 1, 4531–4541 RSC.
- R.-H. Dong, Y.-X. Jia, C.-C. Qin, L. Zhan, X. Yan, L. Cui, Y. Zhou, X. Jiang and Y.-Z. Long, Nanoscale, 2016, 8, 3482–3488 RSC.
- P. Zahedi, I. Rezaeian, S.-O. Ranaei-Siadat, S.-H. Jafari and P. Supaphol, Polym. Adv. Technol., 2010, 21, 77–95 CAS.
- P. J. Rivero, A. Urrutia, J. Goicoechea and F. J. Arregui, Nanoscale Res. Lett., 2015, 10, 1–22 CrossRef CAS PubMed.
- E. Y. Teo, S.-Y. Ong, M. S. K. Chong, Z. Zhang, J. Lu, S. Moochhala, B. Ho and S.-H. Teoh, Biomaterials, 2011, 32, 279–287 CrossRef CAS PubMed.
- Y. Zhou, D. Yang, X. Chen, Q. Xu, F. Lu and J. Nie, Biomacromolecules, 2007, 9, 349–354 CrossRef PubMed.
- M. Abrigo, S. L. McArthur and P. Kingshott, Macromol. Biosci., 2014, 14, 772–792 CrossRef CAS PubMed.
- S. Kim, S.-G. Park, S.-W. Kang and K. J. Lee, Macromol. Mater. Eng., 2016, 301, 818–826 CrossRef CAS.
- G. D. Winter, Nature, 1962, 193, 293–294 CrossRef CAS PubMed.
- R. Jayakumar, M. Prabaharan, P. T. S. Kumar, S. V. Nair and H. Tamura, Biotechnol. Adv., 2011, 29, 322–337 CrossRef CAS PubMed.
- Y. Wang, P. Li, P. Xiang, J. Lu, J. Yuan and J. Shen, J. Mater. Chem. B, 2016, 4, 635–648 RSC.
- L. Chen, L. Bromberg, T. A. Hatton and G. C. Rutledge, Polymer, 2008, 49, 1266–1275 CrossRef CAS.
- A. Jafar, A. B. Kamal, M. M. Mehrdad and A. Asadollah, Protein Pept. Lett., 2015, 22, 940–951 CrossRef.
- F. G. Santos, L. A. Mendonca and H. C. Mantovani, World J. Microbiol. Biotechnol., 2015, 31, 1361–1367 CrossRef CAS PubMed.
- G. Khairunnisa Abdul, M. H. Waleed, G. K. Zeinab, J. C. Robert, S. Mariusz and T. Istvan, Curr. Drug Delivery, 2015, 12, 108–114 CrossRef.
- Y. Liu, W. Ma, W. Liu, C. Li, Y. Liu, X. Jiang and Z. Tang, J. Mater. Chem., 2011, 21, 19214–19218 RSC.
- I. A. Bliziotis, G. Samonis, K. Z. Vardakas, S. Chrysanthopoulou and M. E. Falagas, Clin. Infect. Dis., 2005, 41, 149–158 CrossRef CAS PubMed.
- L. Ejim, M. A. Farha, S. B. Falconer, J. Wildenhain, B. K. Coombes, M. Tyers, E. D. Brown and G. D. Wright, Nat. Chem. Biol., 2011, 7, 348–350 CrossRef CAS PubMed.
- A. Ahmad and A. Viljoen, Phytomedicine, 2015, 22, 657–665 CrossRef CAS PubMed.
- A. Stringaro, E. Vavala, M. Colone, F. Pepi, G. Mignogna, S. Garzoli, S. Cecchetti, R. Ragno and L. Angiolella, J. Evidence-Based Complementary Altern. Med., 2014, 2014, 125904 Search PubMed.
- T. Karpanen, T. Worthington, E. Hendry, B. R. Conway and P. A. Lambert, J. Antimicrob. Chemother., 2008, 62, 1031–1036 CrossRef CAS PubMed.
- S. K. Filoche, K. Soma and C. H. Sissons, Oral Microbiol. Immunol., 2005, 20, 221–225 CrossRef CAS PubMed.
- S. Jiang, B. C. Ma, J. Reinholz, Q. Li, J. Wang, K. A. Zhang, K. Landfester and D. Crespy, ACS Appl. Mater. Interfaces, 2016, 8, 29915–29922 CAS.
- S. L. Groah, A. Libin, M. Spungen, K. L. Nguyen, E. Woods, M. Nabili, J. Ramella-Roman and D. Barritault, Int. Wound J., 2011, 8, 85–95 CrossRef PubMed.
- C. E. Diaz-Uribe, M. C. Daza, E. A. Páez-Mozo, F. Martínez, C. L. B. Guedes and E. Di Mauro, J. Photochem. Photobiol., A, 2013, 259, 47–52 CrossRef CAS.
- S. L. Hopkins, L. Stepanyan, N. Vahidi, A. Jain, B. S. J. Winkel and K. J. Brewer, Inorg. Chim. Acta, 2017, 454, 229–233 CrossRef CAS.
- B. C. Ma, S. Ghasimi, K. Landfester and K. A. Zhang, J. Mater. Chem. B, 2016, 4, 5112–5118 RSC.
- S. Y. Egorov, V. Kamalov, N. Koroteev, A. Krasnovsky, B. Toleutaev and S. Zinukov, Chem. Phys. Lett., 1989, 163, 421–424 CrossRef CAS.
- J. i. Mosinger, K. Lang, J. i. Hostomský, J. i. Franc, J. Sýkora, M. Hof and P. Kubát, J. Phys. Chem. B, 2010, 114, 15773–15779 CrossRef CAS PubMed.
- M. Arenbergerova, P. Arenberger, M. Bednar, P. Kubat and J. Mosinger, Exp. Dermatol., 2012, 21, 619–624 CrossRef CAS PubMed.
- Y. Si, Z. Zhang, W. Wu, Q. Fu, K. Huang, N. Nitin, B. Ding and G. Sun, Sci. Adv., 2018, 4 Search PubMed.
- X. Ma, Y. Zhao, X. Jiang, W. Liu, S. Liu and Z. Tang, ChemPhysChem, 2012, 13, 2531–2535 CrossRef CAS PubMed.
- L. El-Khordagui, N. El-Sayed, S. Galal, H. El-Gowelli, H. Omar and M. Mohamed, Int. J. Pharm., 2017, 520, 139–148 CrossRef CAS PubMed.
- M. A. Castriciano, R. Zagami, M. P. Casaletto, B. Martel, M. Trapani, A. Romeo, V. Villari, M. T. Sciortino, L. Grasso, S. Guglielmino, L. M. Scolaro and A. Mazzaglia, Biomacromolecules, 2017, 18, 1134–1144 CrossRef CAS PubMed.
- L. Plistil, P. Henke, P. Kubat and J. Mosinger, Photochem. Photobiol. Sci., 2014, 13, 1321–1329 CAS.
- P. Kubát, P. Henke, V. Berzediová, M. Štěpánek, K. Lang and J. Mosinger, ACS Appl. Mater. Interfaces, 2017, 9, 36229–36238 Search PubMed.
- P. Henke, K. Lang, P. Kubát, J. Sýkora, M. Šlouf and J. Mosinger, ACS Appl. Mater. Interfaces, 2013, 5, 3776–3783 CAS.
- P. Henke, H. Kozak, A. Artemenko, P. Kubát, J. Forstová and J. Mosinger, ACS Appl. Mater. Interfaces, 2014, 6, 13007–13014 CAS.
- S. Zeng, L. Yin, X. Jiang, Y. Li and K. Li, Dyes Pigm., 2012, 95, 229–235 CrossRef CAS.
- X. F. Wang, X. M. Chen, K. Yoon, D. F. Fang, B. S. Hsiao and B. Chu, Environ. Sci. Technol., 2005, 39, 7684–7691 CrossRef CAS PubMed.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Adv. Mater., 2015, 27, 6265–6270 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- S. Jiang, L.-P. Lv, K. Landfester and D. Crespy, RSC Adv., 2016, 6, 43767–43770 RSC.
- Z. Lu, J. Gao, Q. He, J. Wu, D. Liang, H. Yang and R. Chen, Carbohydr. Polym., 2017, 156, 460–469 CrossRef CAS PubMed.
- S. Jiang, W. He, K. Landfester, D. Crespy and S. E. Mylon, Polymer, 2017, 127, 101–105 CrossRef CAS.
- S. Jiang, L. Lv, Q. Li, J. Wang, K. Landfester and D. Crespy, Nanoscale, 2016, 8, 11511–11517 RSC.
- National Institutes of Health Guide, https://grants.nih.gov/grants/guide/pa-files/PA-03-047.html, accessed December 20, 2002.
- M. A. Biel, Photodynamic Therapy: Methods and Protocols, 2010, pp. 175–194 DOI:10.1007/978-1-60761-697-9_13.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00021b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |