
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MBene (MnB): a new type of 2D metallic ferromagnet with high Curie temperature†
Zhou
Jiang
,
Peng
Wang
,
Xue
Jiang
 * and
Jijun
Zhao
* and
Jijun
Zhao
Key Laboratory of Materials Modification by Laser, Ion and Electron Beams (Dalian University of Technology), Ministry of Education, Dalian 116024, China. E-mail: jiangx@dlut.edu.cn
First published on 30th January 2018
Abstract
We extend the 2D MXene family into the boride world, namely, MBenes. High-throughput calculations screen twelve MBenes with excellent stability. Among them, 2D MnB MBene exhibits robust metallic ferromagnetism (∼3.2 μB per Mn atom) and high Curie temperature (345 K). After functionalization with the –F and –OH groups, the ferromagnetic ground state of 2D MnB is well preserved. The Curie temperature is increased to 405 and 600 K, respectively, providing a novel and feasible strategy to tailor the TC of 2D magnetic materials.
Conceptual insightsMXenes exfoliated from bulk MAX phases have emerged as promising 2D materials. Inspired by recently fabricated crystalline ternary borides (MAB phases), we extend the MXene family into the boride world, namely MBenes. From ab initio high-throughput search, we identify twelve stable MBene sheets that may exist in reality. Among them, the MnB sheet exhibits robust metallic ferromagnetism and high Curie temperature, making it a promising candidate for spintronics. Remarkably, with the help of the –F and –OH functional groups on the surface, the ferromagnetic ground state can be well preserved and the Curie temperature is even increased by up to 255 K. The thermal and mechanical stabilities of the MnB sheet have been confirmed and its possible mechanical exfoliation from bulk ternary borides has also been assessed by ideal fracture strength. Once the predicted MnB and other MBene sheets are synthesized, they would open a new avenue for 2D materials and devices. |
1. Introduction
Graphene, owing to its flat structure and high surface area, holds great promise for applications in electronics, magnetics, optics, optoelectronics, thermoelectrics, catalysts, and energy storage systems.1,2 The boom of graphene has stimulated the discovery and fabrication of a wealth of other two-dimensional (2D) materials.3 Transition metal carbides, nitrides and carbides/nitrides (namely, MXenes) are attractive additions to the 2D family. MXenes are mainly produced by selectively etching the atomic layers from their layered parents of bulk MAX phases. MAX phases are named according to their compositions, where M is a transition metal, A is an element mostly in the IIIA and IVA columns, and X is carbon or nitrogen.4 So far, more than 70 MXenes have been theoretically predicted and 19 of them have been experimentally synthesized.5 Applications of MXenes in field effect transistors,6 Li ion batteries,7 catalysts,8 and photocatalysts9 have been explored. Even with such a wide variety of MXenes, an intriguing question arises: whether X can be extended to the other elements?Boron is the fifth element in the periodic table and its number of valence electrons is only one/two less than carbon/nitrogen. Its electron deficiency, electronegativity, and atomic size would endow 2D boron materials with distinctly different properties from other 2D materials. For instance, most elementary 2D materials are graphene analogues with flat or buckled honeycomb structures, while borophene has a triangular lattice with different arrangements of hexagonal holes.10,11 The biggest challenge for the synthesis of boron based 2D materials is surplus electron balance, which can be solved by hexagonal hole introduction12,13 and metal substrate passivation.14,15 Another practical method to realize 2D boron based systems is the combination between boron and various transition metals. On one hand, the transition metal would donate electrons to boron to stabilize the boron sheet. On the other hand, the highly coordinated transition metal center may impart novel complexity and diversity to 2D boride materials.
Among transition metal borides, only magnesium diboride16 and titanium diboride17 sheets have been synthesized by exfoliation through ultrasonication and intercalation of alkali metals. However, the conventional chemical etching method to produce MXenes from the corresponding MAX phases has not been reported for 2D transition metal borides. This is mainly limited by the lack of ternary borides having layered crystal structures with easily etchable layers. Recently, MAX analogues, a series of single crystalline ternary borides (MAB phases), have been successfully fabricated, including Cr2AlB2, Cr3AlB4, Cr4AlB6, WAlB, MoAlB, Mn2AlB2, and Fe2AlB2.18 These structures are composed of aluminium layers that are suitable to generate 2D transition metal borides, as demonstrated in previous studies on MXenes.4,19–21 Since these 2D transition metal borides originate from the ternary borides, we name them as MBenes. If any of the MBenes can be verified in experiment, the richness of boron chemistry would open a new avenue with diverse applications. Then it is natural to ask: is it possible to obtain 2D MBenes from MAB phases?
To address this question, here we present ab initio high-throughput calculations to search for potential 2D MBene candidates. From an analysis of the database of binary borometallic molecules, twelve 2D borides (MnB, HfB, ZrB, Au2B, Mo2B, Nb5B2, Nb3B4, Ta3B4, V3B4, OsB2, FeB2, and RuB2) stand out for their satisfactory stability. Among them, 2D metal MnB MBene exhibits robust ferromagnetism (FM). Remarkably, its metallic behavior, ferromagnetic state and high Curie temperature are well retained even after functionalization with the –F or –OH groups. As the first attempt to extend MXenes into boride systems, our findings not only enrich the possible diversity of 2D materials in theory but also bring about new opportunities of spintronic applications.
2. Computational details
Our calculations were carried out using spin-polarized density functional theory (DFT) within the generalized gradient approximation (GGA)22 as implemented in the Vienna Ab initio Simulation Package (VASP),23,24 with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional. The projected augmented wave (PAW) approach was used to describe the ion–electron interaction.25,26 To appropriately account for the strongly correlated electrons, the GGA+U method was used to deal with the partially filled d orbitals of the Mn atoms.27 For the Mn 3d-orbitals, the effective on-site Coulomb interaction parameter (U) and the exchange interaction parameter (J) were set to 3 eV and 1 eV, respectively. The choice of U and J values was carefully tested by optimizing the lattice parameters of Mn2AlB2 and Mn4B4, and the results are given in Table S1 (ESI†). The energy cutoff of the plane-wave basis was 650 eV. During geometry optimization, numerical convergence was achieved with a tolerance of 10−6 eV in energy and 0.001 eV Å−1 in force, respectively. To avoid interaction between a layer and its replica, a large vacuum space of 16 Å was added along the z axis. The Brillouin zone was sampled with a 19 × 19 × 1 Monkhorst–Pack k-point grid for geometry optimization and a set of 23 × 23 × 1 grids for electronic structure calculations. For the equilibrium MBene structures, their phonon dispersions were computed using the direct supercell method as implemented in the Phonopy program.28 Ab initio molecular dynamics (AIMD) simulations were also performed to assess the thermal stability of the 2D MnB structure and its functionalized forms (MnBF and MnBOH) at 600 K, 900 K and 1200 K, respectively.3. Results and discussion
A high-throughput structural search was carried out to identify the stable structures of 2D transition metal borides MxBy, where M stands for transition metals in 3d (Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn), 4d (Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag and Cd) and 5d (Hf, Ta, W, Re, Os, Ir, Pt, Au and Hg), and the x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y ratios are 1:2, 3:4, 1:1, 2:1, and 5:2. Among them, we identified twelve unprecedented 2D MBenes, including MnB, HfB, ZrB, Au2B, Mo2B, Nb5B2, Nb3B4, Ta3B4, V3B4, OsB2, FeB2, and RuB2. Their crystalline structures are displayed in Fig. S1(a) (ESI†). The cohesive energies, bond lengths, and slice thicknesses are listed in Table S2 (ESI†). The cohesive energy is defined as
:y ratios are 1:2, 3:4, 1:1, 2:1, and 5:2. Among them, we identified twelve unprecedented 2D MBenes, including MnB, HfB, ZrB, Au2B, Mo2B, Nb5B2, Nb3B4, Ta3B4, V3B4, OsB2, FeB2, and RuB2. Their crystalline structures are displayed in Fig. S1(a) (ESI†). The cohesive energies, bond lengths, and slice thicknesses are listed in Table S2 (ESI†). The cohesive energy is defined as| Ecoh = (xEM + yEB − EMxBy)/(x + y), |
Fig. 1(a) presents the optimized structure of a 2D MnB sheet. Its unit cell with lattice constants a = 2.881 Å and b = 2.932 Å contains two Mn atoms and two B atoms. Unlike common hexagonal 2D materials such as graphene,29 silicene30 MoS231 and Ti3C2,20 the 2D MnB sheet shows a rectangular lattice with the space group Pmma (No. 51), leading to anisotropy in the x and y directions. Each Mn or B atom forms six bonds with its neighbouring atoms, with a bilayer buckled structure with Mn atoms on the upmost surfaces and the distance between Mn layers being h = 2.125 Å. The intralayer Mn–B bond length is 2.119 Å (d1) and the interlayer one is 2.176 Å (d2). Due to the electron deficiency of boron, the Mn–B bond in MnB is slightly weaker than those in Mn2C32 and MnN233 where the Mn–C and Mn–N bond lengths are 1.956 and 1.949 Å, respectively. Note that in the database of binary boron compounds34 such borides with one dimensional zigzag chains have also been observed in bulk MnB, CrB, and MoB crystals. Our proposed 2D MnB is different from them in terms of the relative position of parallel zigzag chains formed by Mn atoms. This unique configuration suggests that the MnB sheet exhibits distinct physical and chemical properties.
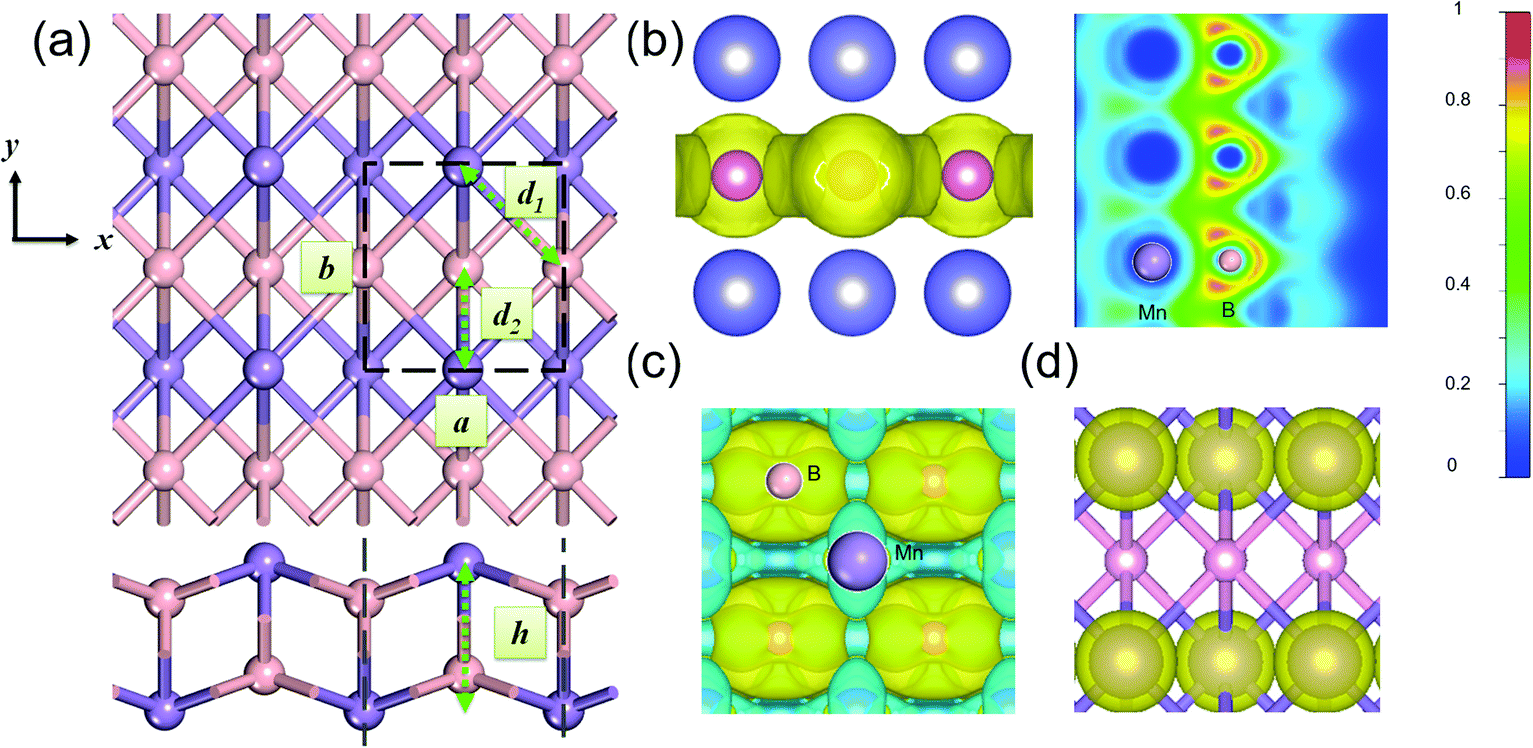 | ||
| Fig. 1 (a) Top and side views of a 2D MnB sheet; a and b are the lattice parameters, and h denotes the thickness of the layer. (b) Maps of the electron localization function (ELF) located on the (0 −1 −6) plane with a scale bar from 0 to 1. (c) The deformation density of the MnB sheet with an isosurface of 0.01 e Å−3. The yellow and cyan regions represent the accumulation and depletion of electrons, respectively. (d) The spin polarization distribution with an isosurface of 0.75 e Å−3. The yellow color indicates a net spin-up polarization, and the spin polarization is mainly on the Mn sites. | ||
The calculated cohesive energy of the MnB sheet is 4.80 eV per atom, which is slightly lower than that of a bulk MnB crystal (5.35 eV) and higher than that of the recently proposed Mn-containing MXene phases such as Mn2C (4.42 eV per atom) and MnN2 (3.45 eV per atom). From our calculations, the 2D elastic constants of C11, C12, C22, and C44 are 197, 50, 158 and 90 N m−1, respectively, satisfying the Born–Huang stability criteria35 and confirming that the MnB layer is mechanically stable. To further check its thermal stability, we used a relatively large 4 × 4 supercell to carry out AIMD simulations within the canonical ensemble (NVT) at temperatures of 300, 600, and 1200 K, respectively. Up to 1200 K, no severe geometrical reconstructions are observed after 20 ps (Fig. S2(a), ESI†) and the average displacement of Mn atoms is about 0.08 Å, which means that the framework of Mn atoms basically remains unchanged.
To explain the origin of high stability and gain deep insight into the bonding character, we calculated the electron localization function (ELF). The homogeneous electron gas is renormalized to values from 0.0 to 1.0, shown in different colours. By definition, the region with a value of 1.0 indicates perfect electron localization, 0.5 represents a fully delocalized electron, and the region with a value close to 0.0 refers to very low charge density.22 From Fig. 1(b), one can clearly see that the ELF for the region around Mn atoms is 0.0, indicating their electron deficiency. At the same time, the B frameworks are fully surrounded by homogeneous electron gas (ELF = 0.5), which is crucial to stabilize the entire 2D sheet via strong B–B bonds. Bader charge analysis shows that each Mn atom donates approximately 0.68 electron to each B atom within the MnB sheet, reflecting the nature of Mn–B ionic bonding. This can be understood by the electronegativities of B (2.04) and Mn (1.55). The electron transfer from Mn to B further contributes to the high stability of the 2D MnB system. As shown in Fig. 1(c), the deformation electron density (DED) also verifies the above bonding characteristics. The transferred electrons are mostly accumulated around the B sites, in agreement with the ELF analysis.
Considering its high stability, it is natural to wonder how the MnB sheet can be synthesized in experiments. There are three feasible pathways to support our hypothetical 2D MnB sheet. Firstly, the Mo2C MXene was prepared with the traditional chemical vapour deposition (CVD) method,36 which is commonly used for growing graphene, MoS2, and many other 2D materials. Thus, monolayer transition borides might also be fabricated using CVD growth. Secondly, the ternary layered phase Mn2AlB2 has already been synthesized by arc-melting.18 The corresponding MnB sheet can be produced by selective chemical etching of the Al metal layer. Such a method has been successfully employed to convert 3D MAX phases Ti2AlC, Nb2AlC, V2AlC, and (Ti0.5Nb0.5)AlC to 2D MXene phases of Ti2C, Nb2C, V2C, and (Ti0.5Nb0.5)C, respectively.19 Thirdly, one can also attempt to mechanically exfoliate MBenes from MAB phases. To further confirm the possibility, we investigated the tensile deformation mechanism of Mn2AlB2. Both the stress–strain curves and ELF counter plots (Fig. 2) show that when the tensile strain increases from 1% to 30% along the z axis the strengths of the B–Mn bonds remain unchanged, while the Mn–Al bonds are significantly weakened as electrons are localized around Al atoms. This implies that the MnB layers are likely to be separated from the Al layers via Mn–Al bond breaking. In addition, the ideal fracture strength for mechanical exfoliation is as low as 32 GPa, which is comparable to the values of W2AlC (36 GPa), Cr2AlC (35 GPa), Mo2AlC (34 GPa), Ta2AlC (32 GPa), and V2AlC (32 GPa).37 This implies that mechanically exfoliating MnB layers from a Mn2AlB2 crystal is feasible.
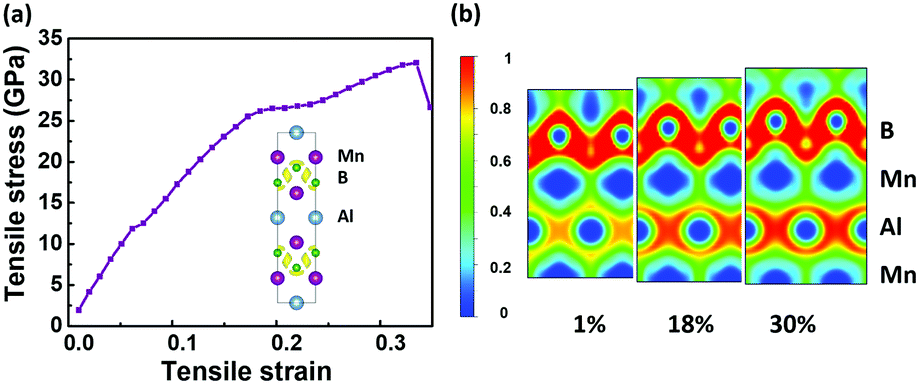 | ||
| Fig. 2 (a) The calculated stress–strain curve of Mn2AlB2 with the ELF contour plots and (b) projected on the plane containing Mn and Al atoms under [0001] tensile strains of 1%, 18%, and 30%. | ||
The spin-polarized band structures in Fig. S3 (ESI†) show that the MnB sheet is metallic with several partially occupied bands crossing the Fermi level in both spin channels. The metallicity is further confirmed by the result of computations using the HSE06 functional.38 Moreover, the atom-projected and orbital-projected bands in Fig. 3 demonstrate that the metallicity is derived from Mn-3d orbitals (Mn-dxy, dx2−y2, dxz) and B-p orbitals near the Fermi energy. As listed in Table 1, the total magnetic moment of 6.04 μB per unit cell is mainly contributed by Mn atoms, which is intuitively shown in the spin-density distribution in Fig. 1(d). Each Mn atom has an on-site moment of 3.2 μB and the spin moments of boron atoms (−0.18 μB) align antiferromagnetically. To further clarify the origin of magnetic moments, Fig. 4 shows the atom-projected and Mn-3d orbital-projected densities of states. It is clear that Mn-3d orbitals (dyz, dxz, dx2−y2) are fully occupied in spin-up states, while the dxy and dz2 orbitals are partially filled in both up and down spin channels. Mn's five d orbitals have non-degenerate energies, which originate from the B induced asymmetric octahedral crystal field around Mn atoms. The strong hybridization between B-p orbitals and Mn-d orbitals is responsible for such a phenomenon. Hence, the electronic structure of MnB can be expected for a formal Mn3+ electronic configuration in a high spin state with three unpaired d electrons (d3↑ spin configuration). The atom- and orbital-projected electronic band structure analyses (Fig. 3) also confirm the p–d hybridization and the difference in the dispersions in the two spin channels associated with the crystal field splitting.
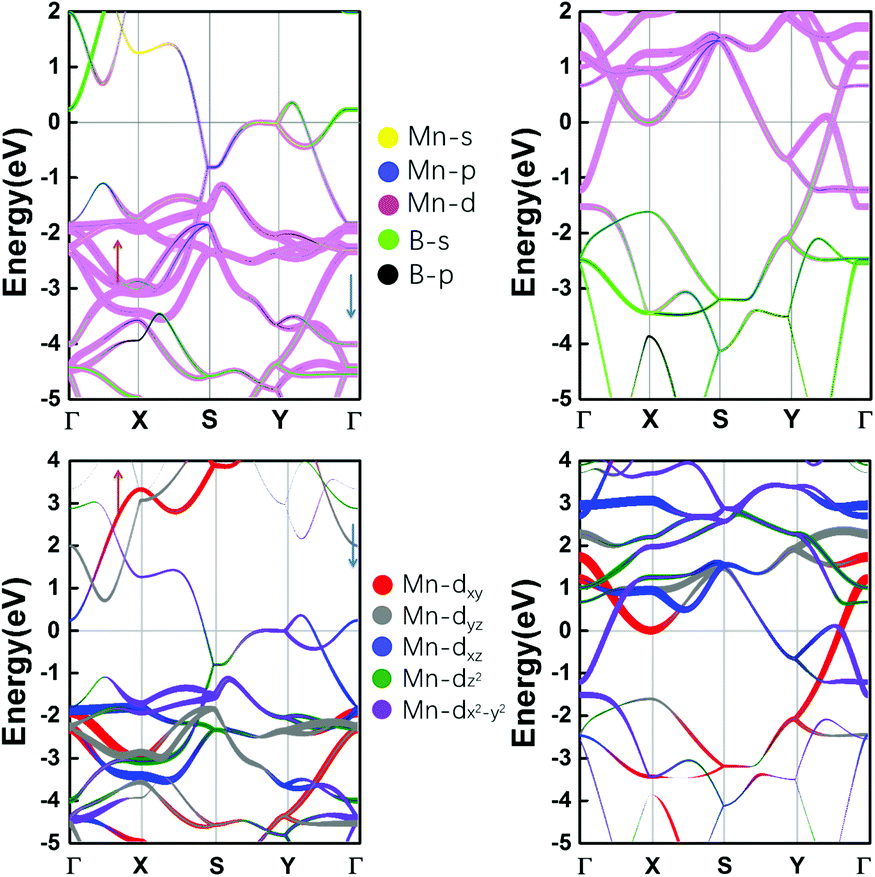 | ||
| Fig. 3 Atom-projected and orbital-projected band structures of MnB sheets in spin-up and spin-down channels, and the Fermi levels are set to zero. | ||
| System | Q Mn/e | Q B/e | Q X/e | M Mn/μB | M Mn-d/μB | M B/μB | M B-p/μB | M X/μB | M T/μB |
|---|---|---|---|---|---|---|---|---|---|
| MnB | −0.68 | 0.68 | — | 3.20 | 3.15 | −0.18 | −0.15 | — | 6.04 |
| MnBF | −1.10 | 0.51 | 0.59 | 3.24 | 3.20 | −0.20 | −0.16 | 0.06 | 6.20 |
| MnBOH | −1.10 | 0.50 | 0.60 | 3.15 | 3.11 | −0.20 | −0.16 | 0.07 | 6.04 |
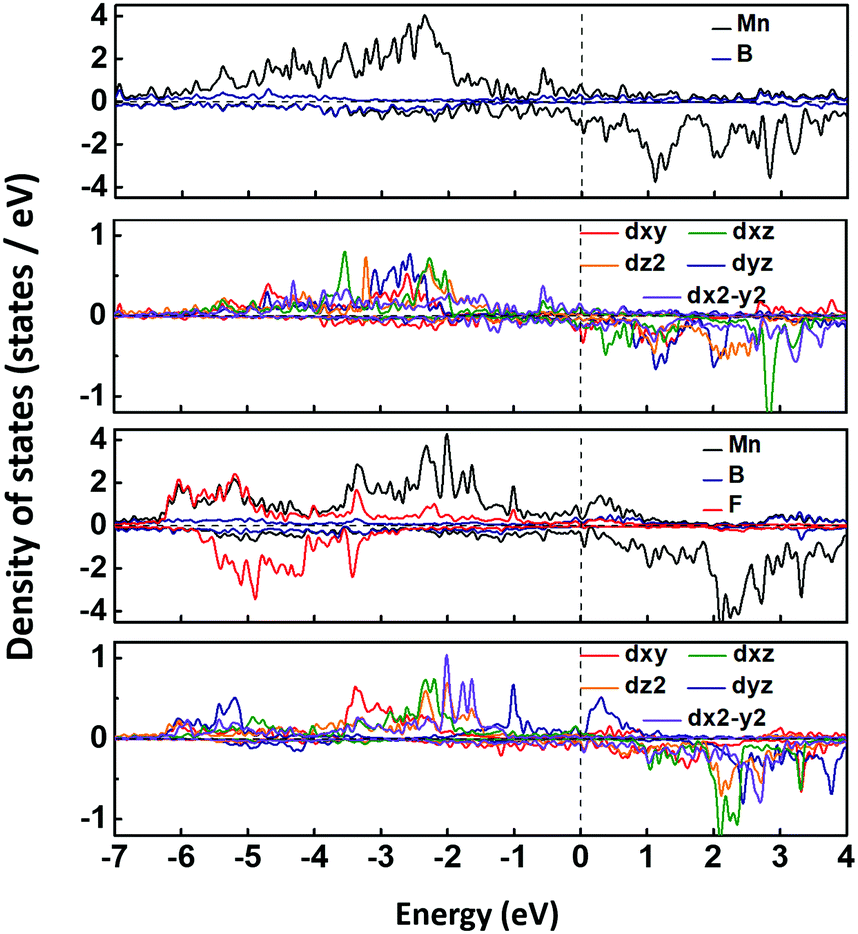 | ||
| Fig. 4 Atom-projected and orbital-projected densities of states for MnB and MnBF sheets, and the Fermi levels are all set to zero. | ||
In order to further explore the preferred magnetic interaction, we optimized a 2 × 2 MnB supercell and considered four possible magnetic configurations (Fig. S4, ESI†): one ferromagnetic (FM) and three antiferromagnetic (AFM). The exchange energy is defined as Eex = (EAFM − EFM) per Mn8B8 formula unit, where EAFM and EFM are the energies of the Mn atoms with AFM and FM coupling in the 2 × 2 MnB sheet. The calculated energy differences between these magnetic configurations are summarized in Table 2. For the MnB sheet, the FM ground state is more stable than the AFM states by at least 180 meV per Mn8B8 formula unit. As discussed above, the indirect p–d exchange interaction plays an important role in the ferromagnetic coupling of MnB sheets.
| E Total/eV | ΔE/meV | M total/μB | |
|---|---|---|---|
| FM | −108.697 | 0 | 25.46 |
| AFM1 | −108.198 | 499 | 0 |
| AFM2 | −108.218 | 479 | 0 |
| AFM3 | −108.517 | 180 | 0 |
One important quantity of ferromagnetism is the Curie temperature (TC) corresponding to the transition from the ferromagnetic to paramagnetic phase. For practical spintronic applications, TC above room temperature is desirable. Here TC is estimated using the 2D Heisenberg model, where the Hamiltonian operator can be written as
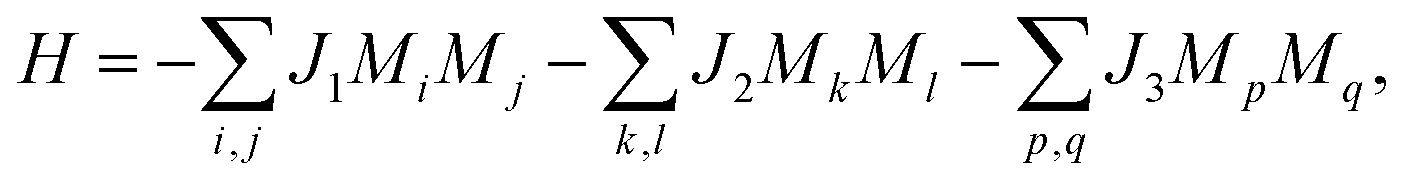
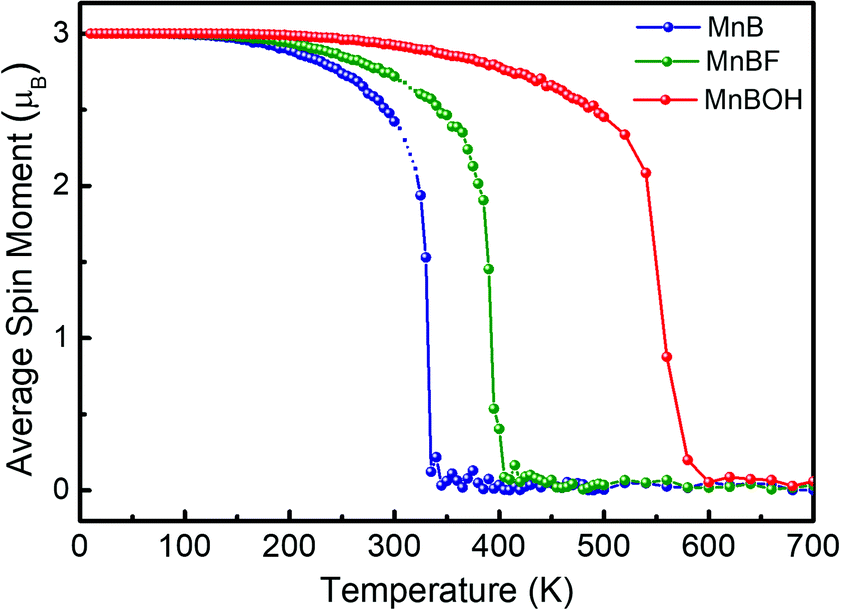 | ||
| Fig. 5 On-site magnetic moment of Mn atoms versus temperature in MnB (blue), MnBF (green), MnBOH (red) sheets. | ||
The magnetic anisotropy energy (MAE) is an important property for high density storage and quantum spin processing. In principle, reducing dimensionality and symmetry would increase MAE relative to the corresponding 3D bulk phase. For the MnB sheet, the easy magnetization axis is the z direction (out-of-plane), along which the magnetization energy is lower than those along the x- and y-directions by 25 and 175 μeV per Mn atom, respectively. These MAE values are comparable to those of reported 2D material Mn2C (25 μeV)32 and bulk Co (65 μeV per Co atom), and are one order of magnitude higher than those of bulk Fe (1.4 μeV per atom) and Ni (2.7 μeV per atom).45 The sizeable MAEs make the MnB sheet potentially useful for magnetoelectronic applications.
Considering that a bare MnB sheet is chemically active and could be exfoliated and terminated with the –F and –OH groups in experiment,19 we further exploited the influence of these functional groups on the structural, electronic and magnetic properties of the MnB sheet. As representatives, we considered two functionalized species, i.e., MnBF and MnB(OH). As shown in Fig. S5 (ESI†), three possible configurations are considered for each MnB sheet, where the functional groups are placed at the top sites of B atoms (H1), hollow sites of Mn atoms (H2), and hollow sites of B atoms (H3). We find that both the –OH and –F functional groups are most energetically favorable at the hollow site B atoms (H3) on both sides. This is understandable since the Mn atoms are the electron donor and the functional groups are more likely to bond with Mn atoms than B atoms. Indeed, significant electron transfer (Table 1) from the Mn-3d orbitals to the –F and –OH functional groups has been observed in MnBF and MnB(OH) by 0.59 e and 0.60 e, respectively, in addition to the electron transfer from Mn to B by 0.5 e on average, whereas no electron transfer was seen between the B atoms and F- and OH- functional groups. As a consequence, such charge transfer slightly elongated the Mn–B bonds; that is, the average Mn–B bond lengths of the OH-terminated and F-terminated MnB sheets are 2.223 Å, and 2.239 Å, respectively. Our AIMD simulations of MnBF and MnBOH indicate that both of them are thermally stable at 600 K after 20 ps (Fig. S2(b), ESI†).
We further investigated the electronic and magnetic properties of those functionalized MnB. Both MnB derivatives still exhibit metallic behavior, as clearly seen from the spin-polarized electronic band structures in Fig. 6. More importantly, the magnetic ground state and magnetic moments on the Mn atoms are well retained after functionalization (Table 1). For MnBF and MnB(OH), the FM state is more stable than the AFM state by 340 meV and 77 meV per 2 × 2 supercell, respectively (Table 3). At the same time, the on-site magnetic moments on Mn atoms are 3.20, 3.24, and 3.15 μB for MnB, MnBF, and MnB(OH), respectively. The total and orbital-projected densities of states of MnB and MnBF sheets are shown in Fig. 4. The almost unchanged magnetic behavior is closely related to the highly split d orbitals of Mn atoms. Initially, the dyz orbitals in the spin-up channel are occupied, while the dx2−y2 orbitals partially occupy the spin-down channel. Upon functionalization, the dyz orbitals in the spin-up channel become partially occupied, while the dx2−y2 orbitals in the spin-up channel become fully occupied. This results in a robust magnetic moment of ∼3 μB per Mn atom for both MnB and MnBF sheets. A similar picture is applicable to the case of the MnBOH system, and the PDOS is shown in Fig. S6 (ESI†).
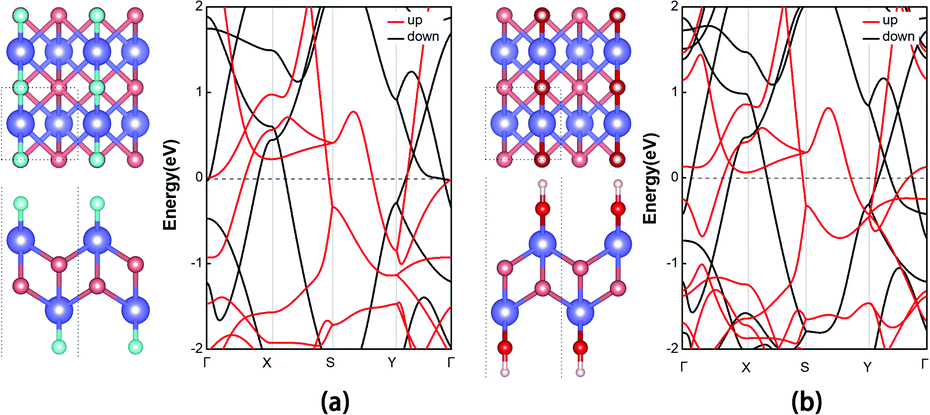 | ||
| Fig. 6 (a and b) Side and top views of MnBX (X = F or OH) structural forms and the corresponding spin-polarized band structures. | ||
| M T/μB | M Mn/μB | ΔE/meV | T C (MFA)/K | T C (MC)/K | |
|---|---|---|---|---|---|
| MnB | 6.04 | 3.20 | 180 | 217–347 | 345 |
| MnBF | 6.20 | 3.24 | 340 | 618–989 | 405 |
| MnBOH | 6.04 | 3.15 | 77 | 333–532 | 600 |
The Curie temperatures of MnBF and MnBOH sheets are then estimated by the MFA method and MC simulation, and the results are listed in Table 1. Fig. 5 shows that the TC is further increased up to 405 K (MC) and 600 K (MC) through F- and OH- functionalization. Note that this value is also comparable to the calculated high TC of a functionalized Mn-based 2D system, i.e., Mn2CF2 (520 K).46 Owing to its insensitive metallic behavior, robust FM state and high Curie temperature, the MnB sheet is superior to most reported 2D MXene materials.47 Taking Ti2C as an example, the magnetic moment of Ti2C is 0.982 μB per atom. However, upon functionalization, the magnetism is quenched, which strongly limits its practical application in spintronics. More excitingly, our results suggest that chemical functionalization is an effective method to tailor TC for 2D magnetic materials. In the present situation, the Curie temperatures of MnBF and MnB(OH) are 60 and 255 K higher than those of the pristine MnB sheet.
4. Conclusions
In summary, motivated by the recent experimentally obtained 3D MAB phases, we explored the geometrical, electronic, and magnetic properties of their 2D MBene analogues. Taking MnB as an example, exfoliation from a 3D crystal to a 2D layer is feasible, as confirmed by its low fracture strength, and high thermal, dynamic, and mechanical stabilities. The 2D MnB MBene sheet and its functionalized products exhibit metallic ferromagnetic behavior with the Curie temperature above room temperature (345–600 K). The ferromagnetism and metallicity mainly arise from the p–d hybridization mechanism and crystal field splitting. Importantly, the characteristic of the high temperature ferromagnetic metal is insensitive to their chemical functionalization. These advantages make 2D MnB MBene a promising material for spintronic devices. Like MXene, transition metal borides can lead to many possible 2D binary compounds; e.g., MnB, HfB, ZrB, Au2B, Mo2B, Nb5B2, Nb3B4, Ta3B4, V3B4, OsB2, FeB2, and RuB2 sheets are predicted as potential metastable phases. Their fruitful applications will be reported in our future study. Our first successful attempt to extend MXenes into boride systems would significantly promote the diversity of 2D materials and encourage scientists to design and synthesize those interesting 2D MBene materials in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (11404050, 11574040) and the Fundamental Research Funds for the Central Universities of China (DUT16RC(4)50, DUT16JJ(G)05, DUT16LAB01, DUT17LAB19). We also acknowledge the Supercomputing Center of the Dalian University of Technology for providing the computing resource.Notes and references
- J. Pan, S. Lany and Y. Qi, ACS Nano, 2017, 11, 7560–7564 CrossRef CAS PubMed.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutierrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- S. Lai, J. Jeon, S. K. Jang, J. Xu, Y. J. Choi, J. H. Park, E. Hwang and S. Lee, Nanoscale, 2016, 8, 1216 RSC.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- G. Fan, X. Li, Y. Ma, Y. Zhang, J. Wu, B. Xu, T. Sun, D. Gao and J. Bi, New J. Chem., 2017, 41, 2793–2799 RSC.
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 CAS.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed.
- H. Tang and S. Ismail-Beigi, Phys. Rev. Lett., 2007, 99, 115501 CrossRef PubMed.
- X. Wu, J. Dai, Y. Zhao, Z. Zhuo, J. Yang and X. C. Zeng, ACS Nano, 2012, 6, 7443–7453 CrossRef CAS PubMed.
- Y. Liu, E. S. Penev and B. I. Yakobson, Angew. Chem., Int. Ed. Engl., 2013, 52, 3156–3159 CrossRef CAS PubMed.
- H. Liu, J. Gao and J. Zhao, Sci. Rep., 2013, 3, 3238 CrossRef PubMed.
- C. Cepek, R. Macovez, M. Sancrotti, L. Petaccia, R. Larciprete, S. Lizzit and A. Goldoni, Appl. Phys. Lett., 2004, 85, 976–978 CrossRef CAS.
- G. Hilz and H. Holleck, Int. J. Refract. Met. Hard Mater., 1996, 14, 97–104 CrossRef CAS.
- P. Chai, S. A. Stoian, X. Tan, P. A. Dube and M. Shatruk, J. Solid State Chem., 2015, 224, 52–61 CrossRef CAS.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Ghidiu, M. Naguib, C. Shi, O. Mashtalir, L. M. Pan, B. Zhang, J. Yang, Y. Gogotsi, S. J. Billinge and M. W. Barsoum, Chem. Commun., 2014, 50, 9517–9520 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467–R5470 CrossRef CAS.
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78, 4063–4066 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- J. Zhao, H. Liu, Z. Yu, R. Quhe, S. Zhou, Y. Wang, C. C. Liu, H. Zhong, N. Han, J. Lu, Y. Yao and K. Wu, Prog. Mater. Sci., 2016, 83, 24–151 CrossRef CAS.
- H. Shu, F. Li, C. Hu, P. Liang, D. Cao and X. Chen, Nanoscale, 2016, 8, 2918–2926 RSC.
- L. Hu, X. Wu and J. Yang, Nanoscale, 2016, 8, 12939–12945 RSC.
- J. Liu, Z. Liu, T. Song and X. Cui, J. Mater. Chem. C, 2017, 5, 727–732 RSC.
- G. Akopov, M. T. Yeung and R. B. Kaner, Adv. Mater., 2017, 29, 1604506 CrossRef PubMed.
- M. Born, K. Huang and M. Lax, Am. J. Phys., 1954, 39, 113–127 Search PubMed.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X. L. Ma, H. M. Cheng and W. Ren, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- Z. Guo, L. Zhu, J. Zhou and Z. Sun, RSC Adv., 2015, 5, 25403–25408 RSC.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- T. Zhao, J. Zhou, Q. Wang, Y. Kawazoe and P. Jena, ACS Appl. Mater. Interfaces, 2016, 8, 26207–26212 CAS.
- M. Kan, J. Zhou, Q. Sun, Y. Kawazoe and P. Jena, J. Phys. Chem. Lett., 2013, 4, 3382–3386 CrossRef CAS PubMed.
- I. Choudhuri, P. Garg and B. Pathak, J. Mater. Chem. C, 2016, 4, 8253–8262 RSC.
- J. Zhou and Q. Sun, J. Am. Chem. Soc., 2011, 133, 15113–15119 CrossRef CAS PubMed.
- J. Jiang, Q. Liang, R. Meng, Q. Yang, C. Tan, X. Sun and X. Chen, Nanoscale, 2017, 9, 2992–3001 RSC.
- F. Matsukura, H. Ohno, A. Shen and Y. Sugawara, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R2037–R2040 CrossRef CAS.
- G. H. O. Daalderop, P. J. Kelly and M. F. H. Schuurmans, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 11919–11937 CrossRef CAS.
- J. He, P. Lyu and P. Nachtigall, J. Mater. Chem. C, 2016, 4, 11143–11149 RSC.
- Y. Xie and P. R. C. Kent, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235441 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Geometries, phonon band structures and other details for twelve MBenes (MnB, HfB, ZrB, Au2B, Mo2B, Nb5B2, Nb3B4, Ta3B4, V3B4, OsB2, FeB2, and RuB2), AIMD simulation results, spin-polarized band structures (PBE and HSE06 methods), four magnetic configurations of a MnB sheet and its functionalized structural forms (MnBF and MnBOH) with their corresponding densities of states. See DOI: 10.1039/c7nh00197e |
| This journal is © The Royal Society of Chemistry 2018 |