
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trehalose elevates brain zinc levels following controlled cortical impact in a mouse model of traumatic brain injury
Stuart D.
Portbury
 a,
Dominic J.
Hare
ab,
David P.
Bishop
b,
David I.
Finkelstein
a,
Philip A.
Doble
b and
Paul A.
Adlard
*a
a,
Dominic J.
Hare
ab,
David P.
Bishop
b,
David I.
Finkelstein
a,
Philip A.
Doble
b and
Paul A.
Adlard
*a
aThe Florey Institute of Neuroscience and Mental Health, The University of Melbourne, Parkville, Victoria, Australia. E-mail: paul.adlard@florey.edu.au
bElemental Bio-imaging Facility, University of Technology, Sydney, Broadway, New South Wales, Australia
First published on 2nd June 2018
Abstract
Zinc (Zn) deficiency is a clinical consequence of brain injury that can result in neuropathological outcomes that are exacerbated with age. Here, we present laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) imaging data showing modulation of brain Zn levels by the disaccharide trehalose in aged mice following a controlled cortical impact model of traumatic brain injury. In this proof-of-concept study, trehalose induced an increase in brain zinc levels, providing important preliminary data for larger studies using this simple carbohydrate as a modulator of this essential micronutrient in traumatic brain injury. Our results may have further implications for the treatment of a variety of neurodegenerative diseases and other disorders of the nervous system.
Significance to metallomicsTraumatic brain injury can result in both acute and chronic neurological dysfunction. Zinc is an essential micronutrient involved in a multitude of critical neurological processes, including the biological response to brain injury. In this research manuscript we describe the results of a proof-of-concept study that treated mice with the disaccharide trehalose, delivered orally for 28 days following a controlled cortical impact. Trehalose induced an increase in brain zinc levels, providing important preliminary data for larger studies using this simple carbohydrate as a modulator of this essential micronutrient, which may have implications for the treatment of a variety of neurodegenerative diseases and other disorders of the nervous system. |
Introduction
Traumatic brain injury (TBI) is a major international health concern with growing incidence in high-income countries.1 Whilst TBI is a major cause of death and disability in young people,1a,2 the age at which injury is sustained is a contributing factor to morbidity and mortality.3 Further, the age at which a TBI is sustained has a negative influence on risk of neurodegeneration, including Parkinson's disease,4 dementia (including Alzheimer's disease)5 and amyotrophic lateral sclerosis.6 Consistent with several other age-related neurodegenerative disorders, evidence suggests that disrupted Zn homeostasis is involved in TBI-induced neuropathology both in the acute phase where the liberation of Zn is thought to be excitotoxic,7 and the chronic phase when neuropsychological symptoms arise.8 The disruption of cellular Zn metabolism following injury moderates several biochemical cascades both immediately, and longer term, that results in neuropathology that may be antecedent to the onset of progressive neurological diseases.9 In rodent models, dietary Zn supplementation following TBI improves behavioural outcomes,10 and chelation of bioavailable Zn increases neuronal damage following injury.11 Furthermore, restricted access to Zn reduces neurogenesis within the sub granular zone of the hippocampal dentate gyrus as a response to TBI.12 However, contradictory studies indicate that at 7 days post-TBI Zn chelation is associated with an upregulation of neuroprotective genes in rat brain,13 and a 14-day Zn chelation trial in rat cerebral ischemia was associated with reduced neuronal apoptosis.14 However, these studies were performed in younger animals, and their relevance to aged animals, where there is increased risk of negative outcomes following TBI, remains unknown. There have been two human clinical trials assessing the therapeutic roles of Zn post-TBI,15 both of which have had significant positive patient outcomes. In the first study, in which TBI patients (ranging in age from 18 to 65 years) were prescribed a Zn-adequate or a Zn-supplemented diet, the Zn-supplemented group had improved Glasgow Coma Scale scores two weeks after the start of treatment.15b The second study, a double-blind clinical trial, assessed 100 patients with severe head trauma aged between 18 to 65 years receiving either placebo or 120 mg Zn via nasogastric tube for 15 days. Zinc supplemented patients exhibited significantly improved Glasgow Coma Scale scores, a significantly reduced Sequential Organ Failure Assessment (SOFA), and significantly reduced inflammatory markers. Zinc supplemented patients also had a reduced length of hospitalization and a reduced mortality rate.15a These data strongly support the hypothesis that ensuring a sufficient Zn supply during the chronic stages post-injury may present as an attractive therapeutic target for preventing long-term adverse outcomes of TBI.Trehalose (Fig. 1) is a stable disaccharide found predominantly in lower-order organisms.16 It has been shown to enhance autophagy in neuronal culture models of proteasomal inhibition.17 Dysregulation of autophagy can cause significant changes in cellular Zn homeostasis,18 and impaired autophagy is associated with neuronal cell death after TBI.19 Moreover, autophagy is thought to mediate protective effects of Zn,20 and enhancement of autophagy protects the mouse brain from rampant apoptosis following TBI.21 Furthermore, trehalose has demonstrated therapeutic efficacy in a mouse model of TBI,22 and other neurodegenerative mouse models via a diverse array of mechanisms including autophagy,23 growth factor promotion,22,24 and oxidative stress reduction.25 Given these observations, we examined trehalose as a potential modulator of Zn homeostasis in aged (24 month-old) mice receiving a controlled cortical impact (CCI) TBI. Spatial and temporal concentrations of zinc were assessed in trehalose treated, non-treated and uninjured mouse brains post-TBI utilizing laser ablation-inductively coupled plasma-mass spectrometry (LA-ICPMS) techniques.
 | ||
| Fig. 1 Molecular structure of α-D-glucopyranosyl-(1 → 1)-α-D-glucopyranoside (trehalose). | ||
Experimental
Animal ethics
All procedures were carried out in accordance with protocols approved by the Howard Florey Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as described by the National Health and Medical Research Council of Australia.Brain injury
Controlled cortical impact (CCI) was carried out using a Hatteras PCI 3000 precision cortical impactor (Hatteras Instruments, Cary, NC). 24 month-old male C57Bl6 mice were anesthetised via intraperitoneal injection of 100 mg kg−1 ketamine and 10 mg kg−1 xylazine. Anaesthesia was monitored via respiration and pedal retraction reflexes. Upon adequate anaesthesia, the surgical site was clipped, shaved and cleansed with 70% ethanol. A 10 mm mid-line incision was made over the skull, and the skin and fascia were reflected to make a 4 mm craniotomy on the central aspect of the right parietal bone using a motorised drill (AP-2.25 mm, lateral 2.5). Excised bone was placed in sterile PBS prior to be resituated post-injury. The animal was then positioned in a stereotaxic frame. The 3 mm impact tip of the CCI was lowered to the surface of the exposed dura, signified by an audible contact alarm, and the injury subsequently delivered (3 m s−1 velocity with a 1.5 mm penetration depth). The excised bone was then replaced and glued in position with super glue, the skin similarly glued together and the animal then placed in a 37 °C heated cage until completely ambulatory. Animals did not receive any analgesia post-surgery. An uninjured control group (n = 3) underwent the entire above mentioned procedure excluding the controlled cortical impact.Trehalose dosing
Trehalose dihydrate (Sigma) was prepared as a 2% w/v solution in a standard suspension vehicle (SSV; 0.9% NaCl, 0.5% Na-carboxymethylcellulose, 0.5% benzyl alcohol and 0.4% Tween 80). Animals were gavaged daily with 0.1 ml/10 g bodyweight. Non-trehalose treated mice and non-CCI impacted mice received daily standard suspension vehicle solution excluding trehalose at 0.1 ml/10 g bodyweight.Animal euthanasia
Animals subjected to CCI were euthanised at 24 h (trehalose; n = 5; SSV; n = 4), 72 h (trehalose; n = 4; SSV; n = 3), 7 days (trehalose; n = 3; SSV; n = 3), 14 days (trehalose; n = 5; SSV; n = 4), and 28 days (trehalose; n = 3; SSV; n = 4) post CCI. The uninjured control group (n = 3) were euthanized on day 28. Animals were anesthetised with Nembutal, before a 50 ml pericardial perfusion with ice-cold 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). Brains were rapidly dissected and further incubated in PFA (4% in PBS at 4 °C), followed by immersion in a cryoprotective solution for 7 days (30% sucrose in PBS, 4 °C). Sections were subsequently cryostat sectioned at 30 mm intervals at −20 °C and mounted on standard microscope slides. Sections were stored at −80 °C prior to analysis, when they were then air-dried at room temperature.LA-ICPMS imaging
Analysis was performed using a New Wave Research UP213 laser ablation system with a two-volume large format cell (ablation area 15 × 15 cm). This system was hyphenated to an Agilent Technologies 7500ce ICP-MS fitted with ‘cs’ lenses for enhanced sensitivity. Quantitative data was obtained by representative ablation of matrix-matched tissue standards produced according to the protocol previously reported. Zinc-66 was the selected isotope. The limit of detection for Zn was 0.39 μg g−1 and the limit of quantification was 1.08 μg g−1.Image construction and region of interest selection
Horizontal lines of ablation were drawn across both ipsilateral and contralateral upper quadrants of the selected section. Lines of ablation were spaced apart the same distance as the laser beam diameter. A beam diameter of 30 μm was used, traversing the section at a speed of 120 μm s−1, laser fluence of 0.3 J cm−2 and repetition rate of 20 Hz. Images were produced by reducing multiple ablation lines into ASCII data files via a Python script for importing into ENVI 6.0 (Exelis Visual Information Solutions, Boulder, CO, USA), from which regions of interest (ROIs) were extracted and statistically analysed (Fig. 2). Each ROI represents a rectangular quadrant that radiates outwards from the impact site towards the centre of the brain, or the equivalent region on the contralateral side of the brain (Fig. 3). One brain slice per animal underwent ablation and subsequent analysis. Additionally, the hippocampus was manually extracted as an individual ROI using the Zn map for analysis without adjacent tissue of any ROI region.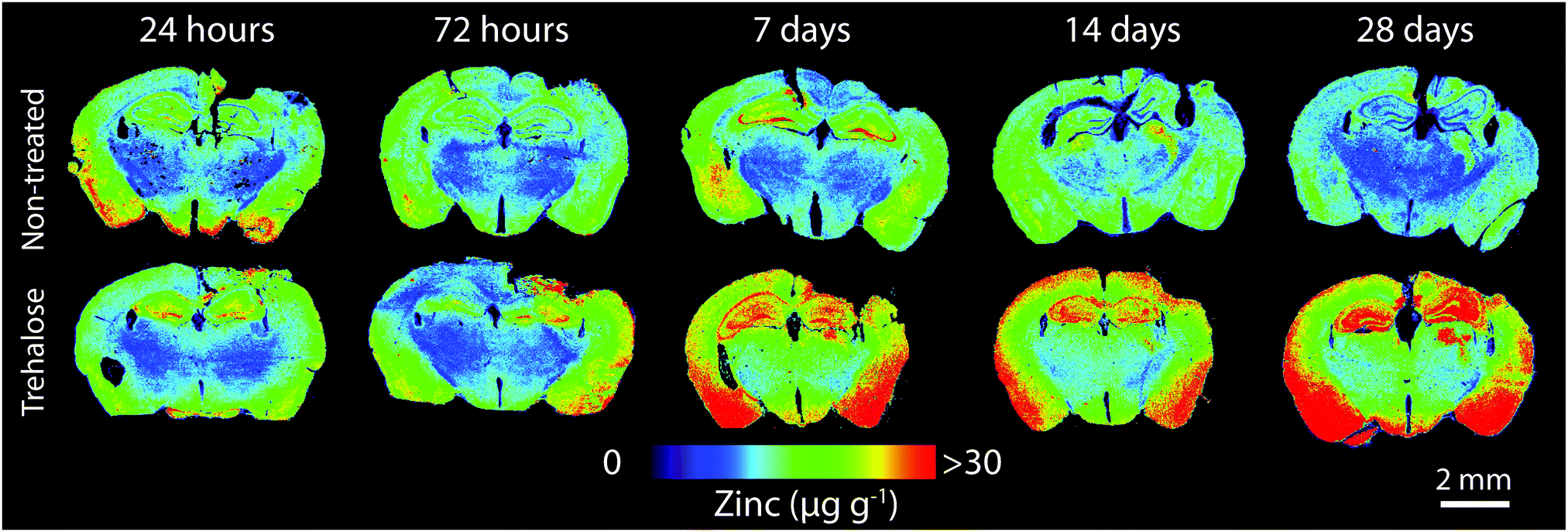 | ||
| Fig. 2 LA-ICP-MS images of quantitative Zn distribution (spatial resolution = 30 μm) in non-treated and trehalose-treated mice from 24 hours to 28 days’ post-injury. | ||
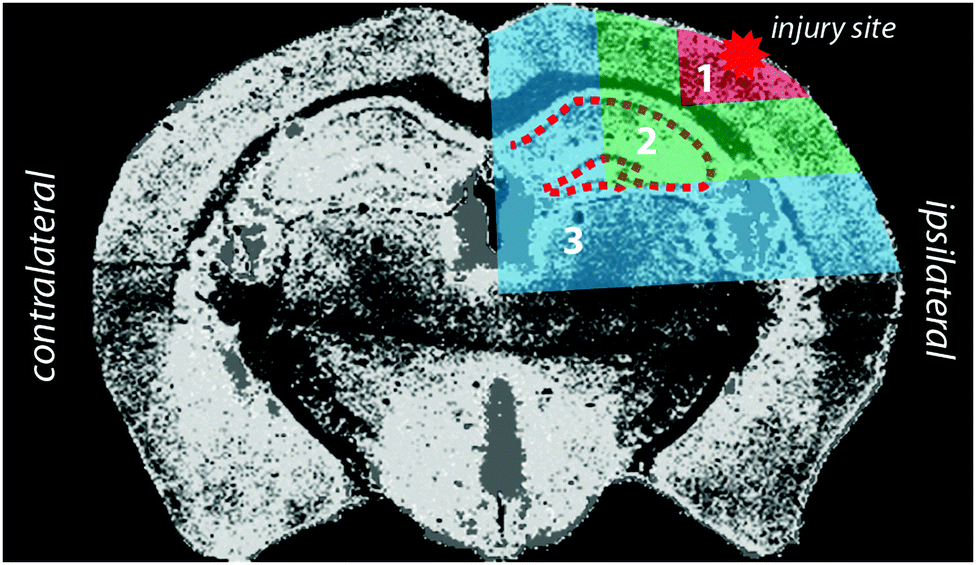 | ||
| Fig. 3 Schematic demonstrating injury site, region of interest (ROI), and hippocampus. Selected ROIs and hippocampus were applied to zinc measurement and extracted for statistical analysis on both the ipsilateral and contralateral hemispheres. | ||
Statistical analysis
Statistical analysis was carried out in Prism 6.0 h (Graph-Pad, La Jolla, CA, USA). Analysis was carried out using either a two-tailed t-test for intraday analysis and a two-way ANOVA to assess differences across the time course, with significance recognised as p < 0.05.Results
In trehalose-treated animals there was an overall significant elevation of Zn concentration in the ipsilateral hemisphere compared to SSV-treated animals across the time course of the experiment within the immediate vicinity of the impact site at region of interest (ROI) 1 (p < 0.0043; Fig. 4a) and in the two additional ROIs extending radially from the lesion site (ROI 2; p < 0.0001; ROI 3: p < 0.0001; Fig. 4b and c), as well as the hippocampus (p < 0.0001; Fig. 4d) leading to a significant overall increase in Zn over the entire ipsilateral hemisphere (p < 0.0001; Fig. 4e). Additionally, time-point specific elevations of Zn were observed in ROI 1 at day 14 (p = 0.0032; Fig. 4a) and day 28 (p = 0.0031; Fig. 4a), in ROI 2 at day 14 (p < 0.0001; Fig. 4b) and day 28 (p < 0.0001; Fig. 4b), in ROI 3 at day 14 (p < 0.0001; Fig. 4b) and day 28 (p < 0.0003; Fig. 4b), in the hippocampus at day 14 (p < 0.0001; Fig. 4d) and day 28 (p = 0.0101; Fig. 4d), and in the entire hemisphere at day 14 (p = 0.0007; Fig. 4e) and day 28 (p = 0.0107; Fig. 4e). when compared to non-trehalose treated mice. We used the Zn concentration of uninjured animals (taken from the 28-day time point) and compared it to the ipsilateral intraday time point Zn concentrations of the post-TBI trehalose treated cohort. Significant time-point specific elevations (dotted significance bars; Fig. 4a–e) of Zn were observed in ROI 1 at day 14 (p = 0.0092; Fig. 4a) and day 28 (p = 0.0160; Fig. 4a), in ROI 2 at day 14 (p = 0.0004; Fig. 4b) and day 28 (p < 0.0001; Fig. 4b) in ROI 3 at day 14 (p = 0.0003; Fig. 4b) and day 28 (p = 0.0013; Fig. 4b), in the hippocampus at day 14 (p = 0.0005; Fig. 4e) and day 28 (p < 0.0001; Fig. 4e), and in the entire hemisphere at day 14 (p = 0.0003; Fig. 4d) and day 28 (p = 0.0012; Fig. 4d). Similarly, the contralateral (uninjured) cortex revealed an elevated Zn concentration profile across the time-course in ROI 1 (p < 0.0001; Fig. 5a), ROI 2 (p < 0.0001; Fig. 5b), ROI 3 (p < 0.0001; Fig. 5c), the hippocampus (p < 0.0001; Fig. 5e), leading to a significant Zn concentration increase in the entire contralateral hemisphere (p < 0.0001; Fig. 5d). Additionally, time-point specific elevations in zinc were observed in the contralateral cortex in ROI 1 at day 14 (p = 0.0030; Fig. 5a) and day 28 (p = 0.0004; Fig. 5a), in ROI 2 at day 14 (p < 0.0001; Fig. 5b) and day 28 (p < 0.0001; Fig. 5b), in ROI 3 at day 14 (p < 0.0001; Fig. 5c) and day 28 (p = 0.0004; Fig. 5c), in the hippocampus at day 14 (p = 0.0001; Fig. 5e) and day 28 (p < 0.0050; Fig. 5e), and in the entire hemisphere at day 14 (p = 0.0005; Fig. 5d) and day 28 (p = 0.0024; Fig. 5d). Comparing the Zn concentrations in the uninjured control brain to the intraday time point Zn concentrations of the post-TBI trehalose treated cohort revealed significant time point specific elevations in Zn concentrations for the trehalose treated mice (dotted significance bars; Fig. 5a–e). Time-point specific elevations of Zn were observed in ROI 1 at day 14 (p = 0.0197; Fig. 5a) and day 28 (p = 0.0025; Fig. 5a), in ROI 2 at day 14 (p = 0.0008; Fig. 5b) and day 28 (p < 0.0021; Fig. 5b) in ROI 3 at day 14 (p = 0.0014; Fig. 5b) and day 28 (p = 0.0037; Fig. 5b), in the hippocampus at day 14 (p = 0.0002; Fig. 5e) and day 28 (p < 0.0001; Fig. 5e), and in the entire hemisphere at day 14 (p = 0.0013; Fig. 5d) and day 28 (p = 0.0017; Fig. 5d).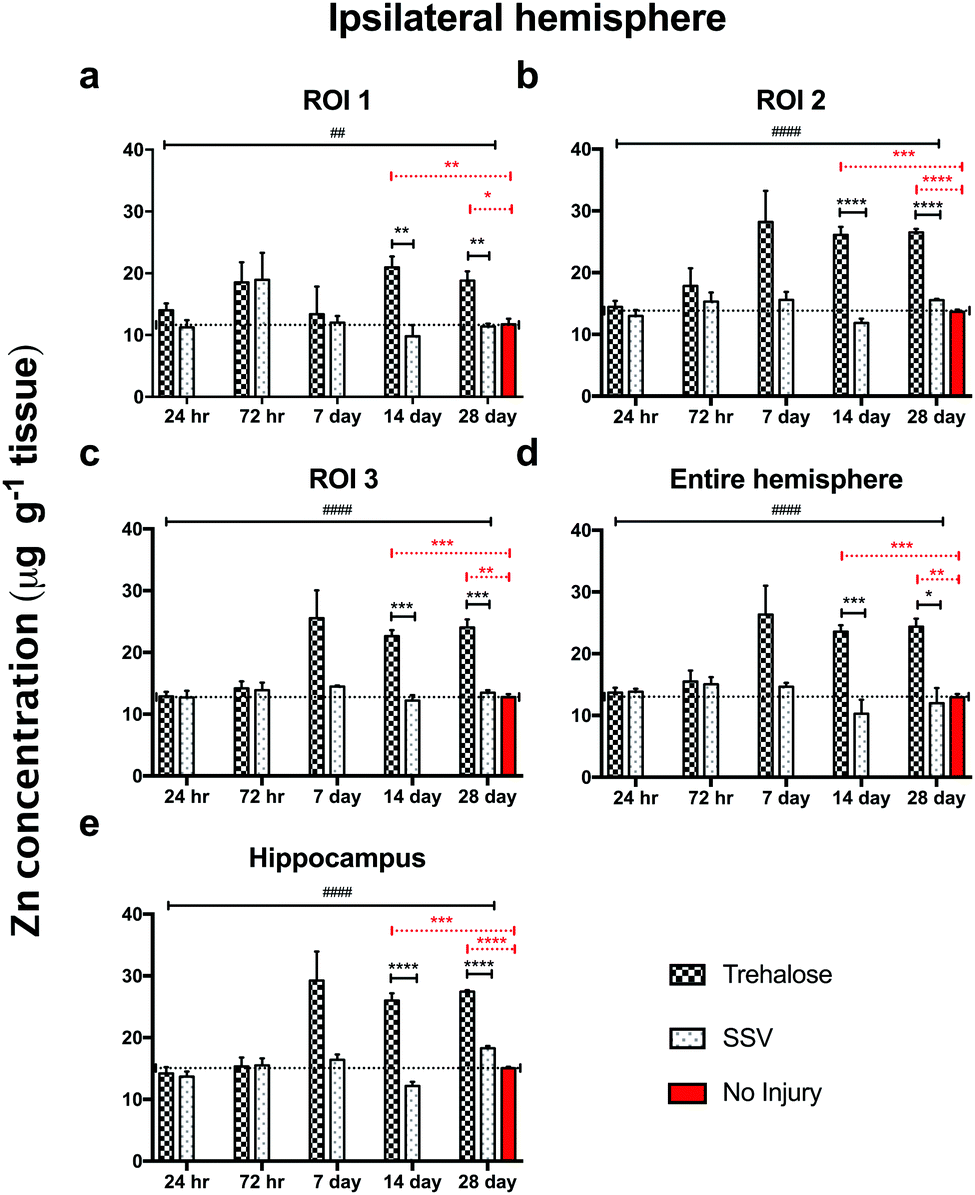 | ||
| Fig. 4 Trehalose significantly increases Zn across the time course post-injury in the CCI (ipsilateral) hemisphere. Post-TBI trehalose treated mice showed a significant increase in Zn across the time course in the ipsilateral ROI 1 (a; two-way ANOVA; ##p < 0.0043), ROI 2 (b; ####p < 0.0001), ROI 3 (c; ####p < 0.0001), the hippocampus (d; ####p < 0.0001) and the entire hemisphere (e; ####p < 0.0001). Intra-day comparisons revealed a significant increase in Zn for trehalose treated mice at day 14 (unpaired t-test; **p = 0.0032) and 28 (**p = 0.0031) in ROI 1; day 14 (****p < 0.0001) and 28 in ROI 2 (****p < 0.0001); day 14 (***p < 0.0001) and 28 (***p = 0.0003) in ROI 3; day 14 (***p = 0.0007) and 28 (*p = 0.0107) in the entire ipsilateral hemisphere, and day 14 (****p < 0.0001) and 28 (****p = 0.0101) in the hippocampus when compared to post-TBI SSV treated mice. Dotted line across the entire time course is a representation of control (uninjured) zinc concentration derived from 28-day time point of uninjured SSV treated mice. Time-point specific significant elevations (dotted significance bars; (a–e)) of Zn above uninjured controls were observed in ROI 1 at day 14 (**p = 0.0092; a) and day 28 (*p = 0.0160: a), in ROI 2 at day 14 (***p = 0.0004; b) and day 28 (****p < 0.0001; b) in ROI 3 at day 14 (***p = 0.0003: b) and day 28 (**p = 0.0013; b), in the hippocampus at day 14 (***p = 0.0005; e) and day 28 (****p < 0.0001; e), and in the entire hemisphere at day 14 (***p = 0.0003; d) and day 28 (**p = 0.0012; d). | ||
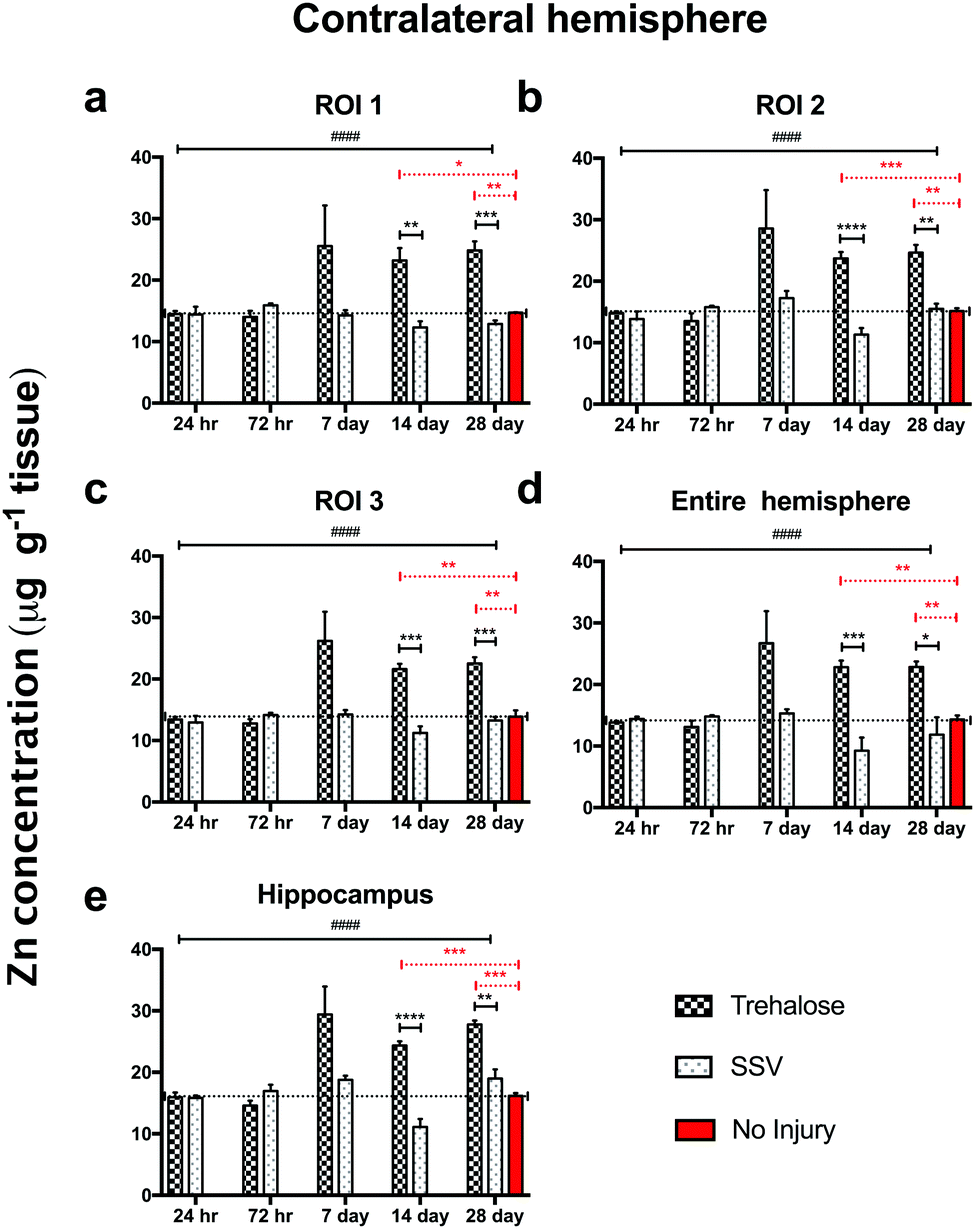 | ||
| Fig. 5 Trehalose significantly increases Zn across the time course post-injury in the uninjured (contralateral) hemisphere. Trehalose treated mice showed a significant increase in Zn across the time course in the contralateral ROI 1 (a; two-way ANOVA; ####p < 0.0001), ROI 2 (b; ####p < 0.0001), ROI 3 (c; ####p < 0.0001), the entire hemisphere (e; ####p < 0.0001), and the hippocampus (d; ####p < 0.0001). Intra-day comparisons revealed a significant increase in Zn for trehalose treated mice at day 14 (unpaired t-test; **p = 0.0030) and 28 (***p = 0.0004) in ROI 1; day 14 (****p < 0.0001) and 28 (**p = 0.0015) in ROI 2; (****p < 0.0001), day 14 (***p < 0.0001) and 28 (***p = 0.0004) in ROI 3; at day 14 (***p = 0.0005) and 28 (*p = 0.00243) in the entire ipsilateral hemisphere, and at day 14 (****p = 0.0001) and 28 (**p < 0.0050) in the hippocampus. Dotted line across the entire time course is a representation of control (uninjured) zinc concentration derived from 28-day time point of uninjured SSV treated mice. Time-point specific significant elevations of Zn (dotted significance bars; Fig. 4a–e) above uninjured controls were observed in ROI 1 at day 14 (*p = 0.0197; a) and day 28 (**p = 0.0025: a), in ROI 2 at day 14 (***p = 0.0008; b) and day 28 (**p < 0.0021; b) in ROI 3 at day 14 (**p = 0.0014: b) and day 28 (**p = 0.0037; b), in the hippocampus at day 14 (***p = 0.0002; e) and day 28 (***p < 0.0001; e), and in the entire hemisphere at day 14 (**p = 0.0013; d) and day 28 (**p = 0.0017; d). | ||
Notably, trehalose treated mice exhibited significantly elevated ipsilateral and contralateral zinc concentrations above the non-injured SSV treated control mice, indicating that trehalose significantly increased whole brain Zn concentrations above normal uninjured control values. The profile of Zn modulation across the time-course in all ROIs was consistent between both the ipsilateral and contralateral cortex (Fig. 6), indicative of whole-brain Zn modulation with trehalose from day 7 post-TBI that was not observed in the post-TBI SSV treated mice nor the uninjured SSV treated mice.
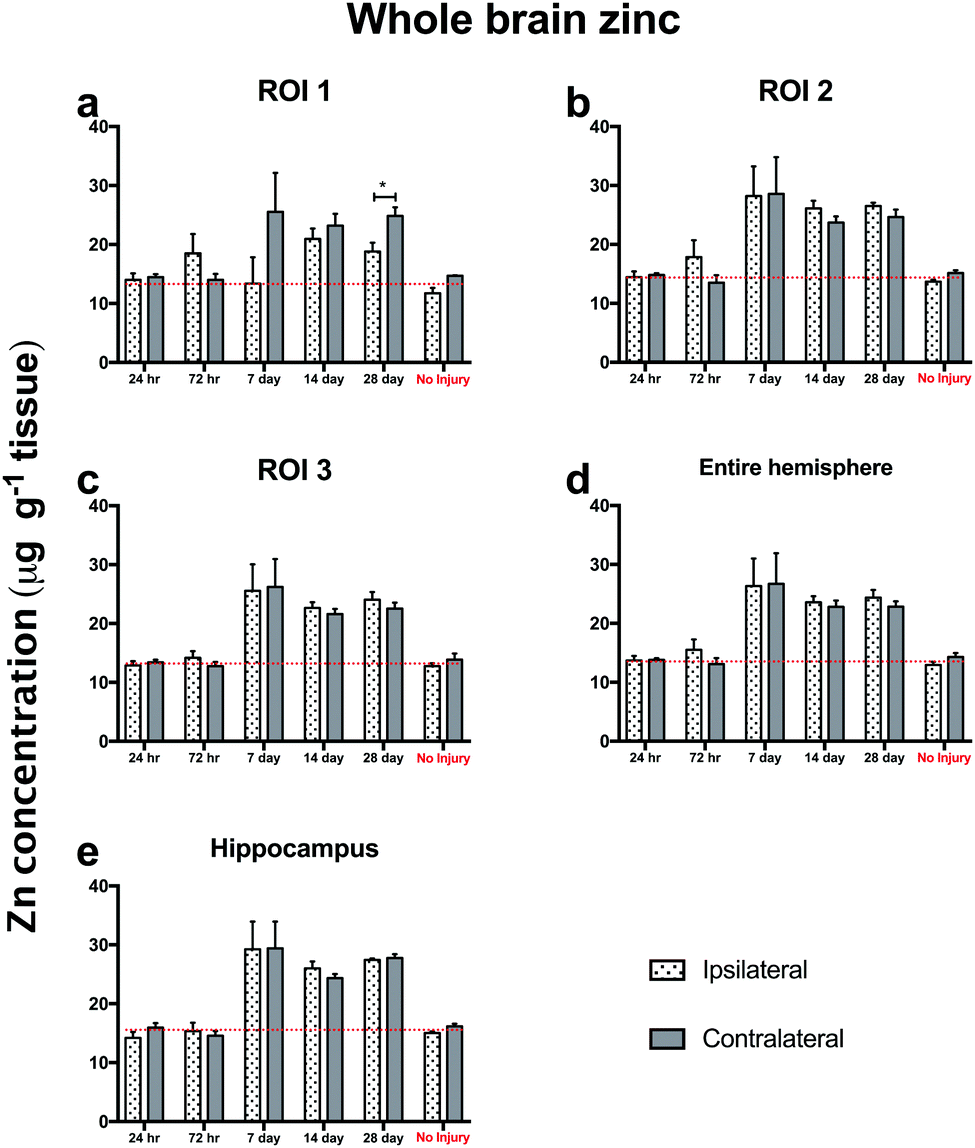 | ||
| Fig. 6 Trehalose treated ipsilateral and contralateral brain regions both have parallel Zn levels post-TBI. Analysis of Zn levels in the ipsilateral and contralateral brain regions post injury are markedly consistent with only one significant contralateral time point specific elevation in ROI 1 at day 28 (unpaired t-test; p < 0.05; *). The brain increase in Zn at day 7 to day 28 is evident in every ROI assessed in both the ipsilateral and contralateral sides, indicating a whole brain modulation of Zn with trehalose after brain injury in aged mice. | ||
Discussion
The data presented herein was intended as a proof of concept study, in which LA-ICPMS was used to assess the ability of trehalose to modulate brain Zn concentrations after a controlled cortical impact TBI. To that end, post-TBI trehalose treatment was shown to significantly elevate brain Zn when compared to post-TBI SSV treated littermates. Additionally, trehalose elevated Zn concentrations to significantly greater levels than SSV treated non-CCI impacted littermates. Whilst the results of this pilot investigation indicate that a simple carbohydrate can modulate an essential micronutrient, it also highlights the need for further work examining the effects of trehalose treatment on behavioural and pathological features of TBI in the aged brain. Nevertheless, the absence of behavioural or biochemical data in this analysis should not detract from the observations that clinical studies and animal models have identified correlations between neuropsychological disorders and reduced brain Zn status including depression and anxiety-like behaviours,8 both common long-term outcomes of TBI, particularly in the elderly.26Supportive of the observation that reduced brain zinc is associated with clinical depression is the evidence of beneficial Zn supplementation for treatment of depression-like behaviours in rats following TBI,10 and as a significant modulator of depression in humans.27 Of further relevance to the data presented herein, a recent study indicated that trehalose exhibited antidepressant-like effects in uninjured mice.28 While improvements in the depression-like behaviour were attributed to enhanced autophagy, our data suggest that Zn modulation by trehalose may also have been an additional or contributing underlying biochemical mechanism of action.
We recently published a study investigating the role of zinc, iron and copper, after a controlled cortical impact TBI in which it was demonstrated that while Zn concentration does not significantly differ between ipsi- and contralateral hemispheres in a unilateral CCI TBI, a reduction in total (that is, entire brain) Zn occurs between seven and 28 days post-injury in aged mice,29 which we hypothesised could potentiate the onset of neuropathology. In this study, whilst trehalose did not significantly alter iron of copper levels post-injury (data not shown), it did completely prevent the reduction in zinc that we have previously shown to occur in a time-dependent manner following injury. Additionally, trehalose elevated the brain zinc concentration above that of uninjured tissue. These results therefore could have clinical implications for both TBI associated neurodegeneration and depression and zinc deficiency in the elderly, which is known to be associated with depressive and psychiatric disorders.30 The observed effect of b rain zinc modulation by trehalose after TBI appears to be an age-related phenomenon, however, as another recent study by our group in which younger (3 month-old) mice treated with trehalose exhibited no zinc modulating effects post TBI.22 It should be noted, nonetheless, that the trehalose treatment improved TBI-induced cognitive decline and was associated with the elevated expression of proteins linked with synaptic activity and neuronal plasticity in the young cohort. Whether trehalose treatment in elderly mice reproduces similar enhancement of synaptic activity and neuronal plasticity observed in conjunction with Zn elevation should be assessed in future studies.
The role of Zn in the onset of pathologies in TBI and dementia has been hypothesised for some time,9 in part due to the effect of Zn on the aggregation, oligomerisation and deposition of pathological proteins.31 It is also noteworthy that in Alzheimer's disease decreased cellular Zn at the cellular level may contribute to neuropathology via Zn sequestration in senile plaques,32 reducing the pool of readily-available synaptic Zn.33 Synaptic Zn is vital for normal memory function and cognition. Mice with an ablation of the slc30a3 gene encoding Zn-transporter-3 (ZnT3) protein that controls synaptic Zn release in the hippocampus show an age-dependent decline in cognitive ability.34 Similarly, slc30a3-null mice subjected to TBI exhibit greater evidence of cellular damage following injury compared to wild-type controls.11 Moreover, studies have shown that the use of Zn ionophores increases brain Zn levels in both ZnT3 knockout35 mice and aged C57Bl/6 mice36 and is associated with improved cognition in both models. These observations clearly indicate that synaptic Zn and cognitive health appear to be intrinsically linked, and that Zn modulation may therefore mitigate neuronal damage and loss of function due to Zn deficiency.
Conclusions
In summary, we have shown that trehalose induces an increase in Zn in the aged mouse brain up to 28 days following CCI. The Zn elevation occurs beyond 7 days post-injury, suggesting a biochemical mechanism separate to the hypothesised acute Zn excitotoxicity stage occurring within 24 hours of injury,37 potentiated by an acute release of glutamate.38 Further experiments are required to elucidate the relevance of this trehalose-mediated increase in Zn following TBI, and studies must be correlated with behavioural and biochemical analyses to determine therapeutic efficacy as it pertains to brain injury and potentially other disorders of the aging nervous system.Conflicts of interest
All authors state that there are no conflicts to declare.Acknowledgements
DJH is supported by an Australian National Health and Medical Research Council Career Development Fellowship (APP1120582) with Agilent Technologies. DJH and PAD are supported by an Australian Research Council Linkage Project (LP120200081) with Agilent Technologies and ESI Ltd. PAA was supported by an Australian Research Council Future Fellowship (FT120100030). In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and the funding from the Operational Infrastructure Support Grant. The Elemental Bio-imaging Facility receives material support from Agilent Technologies. We would like to acknowledge the excellent technical assistance provided by Charlotte J. Sgambelloni during the surgical procedures undertaken for these experiments.Notes and references
- (a) A. I. Maas, N. Stocchetti and R. Bullock, Moderate and severe traumatic brain injury in adults, Lancet Neurol., 2008, 7, 728–741, DOI:10.1016/S1474-4422(08)70164-9; (b) B. Roozenbeek, A. I. Maas and D. K. Menon, Changing patterns in the epidemiology of traumatic brain injury, Nat. Rev. Neurol., 2013, 9, 231–236, DOI:10.1038/nrneurol.2013.22.
- J. Ghajar, Traumatic brain injury, Lancet, 2000, 356, 923–929, DOI:10.1016/S0140-6736(00)02689-1.
- V. E. Johnson and W. Stewart, Traumatic brain injury: age at injury influences dementia risk after TBI, Nat. Rev. Neurol., 2015, 11, 128–130, DOI:10.1038/nrneurol.2014.241.
- R. C. Gardner, J. F. Burke, J. Nettiksimmons, S. Goldman, C. M. Tanner and K. Yaffe, Traumatic brain injury in later life increases risk for Parkinson disease, Ann. Neurol., 2015, 77, 987–995, DOI:10.1002/ana.24396.
- R. C. Gardner, J. F. Burke, J. Nettiksimmons, A. Kaup, D. E. Barnes and K. Yaffe, Dementia risk after traumatic brain injury vs. nonbrain trauma: the role of age and severity, JAMA Neurol., 2014, 71, 1490–1497, DOI:10.1001/jamaneurol.2014.2668.
- H. Chen, M. Richard, D. P. Sandler, D. M. Umbach and F. Kamel, Head injury and amyotrophic lateral sclerosis, Am. J. Epidemiol., 2007, 166, 810–816, DOI:10.1093/aje/kwm153.
- S. W. Suh, J. W. Chen, M. Motamedi, B. Bell, K. Listiak, N. F. Pons, G. Danscher and C. J. Frederickson, Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury, Brain Res., 2000, 852, 268–273 CrossRef PubMed.
- E. C. Cope and C. W. Levenson, Role of zinc in the development and treatment of mood disorders, Curr. Opin. Clin. Nutr. Metab. Care, 2010, 13, 685–689, DOI:10.1097/MCO.0b013e32833df61a.
- (a) C. J. Frederickson, M. P. Cuajungco and C. J. Frederickson, Is zinc the link between compromises of brain perfusion (excitotoxicity) and Alzheimer's disease?, J. Alzheimer's Dis., 2005, 8, 155–160 CrossRef; (b) S. D. Portbury and P. A. Adlard, Traumatic Brain Injury, Chronic Traumatic Encephalopathy, and Alzheimer's Disease: Common Pathologies Potentiated by Altered Zinc Homeostasis, J. Alzheimer's Dis., 2015, 46, 297–311, DOI:10.3233/JAD-143048.
- E. C. Cope, D. R. Morris, A. G. Scrimgeour and C. W. Levenson, Use of zinc as a treatment for traumatic brain injury in the rat: effects on cognitive and behavioral outcomes, Neurorehabil. Neural Repair, 2012, 26, 907–913, DOI:10.1177/1545968311435337.
- P. Doering, M. Stoltenberg, M. Penkowa, J. Rungby, A. Larsen and G. Danscher, Chemical blocking of zinc ions in CNS increases neuronal damage following traumatic brain injury (TBI) in mice, PLoS One, 2010, 5, e10131, DOI:10.1371/journal.pone.0010131.
- B. Y. Choi, J. H. Kim, H. J. Kim, B. E. Lee, I. Y. Kim, M. Sohn and S. W. Suh, Zinc chelation reduces traumatic brain injury-induced neurogenesis in the subgranular zone of the hippocampal dentate gyrus, J. Trace Elem. Med. Biol., 2014, 28, 474–481, DOI:10.1016/j.jtemb.2014.07.007.
- H. L. Hellmich, C. J. Frederickson, D. S. DeWitt, R. Saban, M. O. Parsley, R. Stephenson, M. Velasco, T. Uchida, M. Shimamura and D. S. Prough, Protective effects of zinc chelation in traumatic brain injury correlate with upregulation of neuroprotective genes in rat brain, Neurosci. Lett., 2004, 355, 221–225 CrossRef PubMed.
- Y. Zhao, R. Pan, S. Li, Y. Luo, F. Yan, J. Yin, Z. Qi, Y. Yan, X. Ji and K. J. Liu, Chelating intracellularly accumulated zinc decreased ischemic brain injury through reducing neuronal apoptotic death, Stroke, 2014, 45, 1139–1147, DOI:10.1161/STROKEAHA.113.004296.
- (a) M. Khazdouz, M. Mazidi, M. R. Ehsaei, G. Ferns, A. P. Kengne and A. R. Norouzy, Impact of Zinc Supplementation on the Clinical Outcomes of Patients with Severe Head Trauma: A Double-Blind Randomized Clinical Trial, J. Diet. Suppl., 2018, 15, 1–10, DOI:10.1080/19390211.2017.1304486; (b) B. Young, L. Ott, E. Kasarskis, R. Rapp, K. Moles, R. J. Dempsey, P. A. Tibbs, R. Kryscio and C. McClain, Zinc supplementation is associated with improved neurologic recovery rate and visceral protein levels of patients with severe closed head injury, J. Neurotrauma, 1996, 13, 25–34, DOI:10.1089/neu.1996.13.25.
- A. D. Elbein, Y. T. Pan, I. Pastuszak and D. Carroll, New insights on trehalose: a multifunctional molecule, Glycobiology, 2003, 13, 17R–27R, DOI:10.1093/glycob/cwg047.
- M. J. Casarejos, R. M. Solano, A. Gomez, J. Perucho, J. G. de Yebenes and M. A. Mena, The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells, Neurochem. Int., 2011, 58, 512–520, DOI:10.1016/j.neuint.2011.01.008.
- H. Ni, X. Feng, Z. J. Xiao, L. Y. Tao and M. F. Jin, Dynamic pattern of gene expression of ZnT-4, caspase-3, LC3, and PRG-3 in rat cerebral cortex following flurothyl-induced recurrent neonatal seizures, Biol. Trace Elem. Res., 2011, 143, 1607–1615, DOI:10.1007/s12011-011-8982-4.
- C. Sarkar, Z. Zhao, S. Aungst, B. Sabirzhanov, A. I. Faden and M. M. Lipinski, Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury, Autophagy, 2014, 10, 2208–2222, DOI:10.4161/15548627.2014.981787.
- J. P. Liuzzi, L. Guo, C. Yoo and T. S. Stewart, Zinc and autophagy, BioMetals, 2014, 27, 1087–1096, DOI:10.1007/s10534-014-9773-0.
- K. Ding, J. Xu, H. Wang, L. Zhang, Y. Wu and T. Li, Melatonin protects the brain from apoptosis by enhancement of autophagy after traumatic brain injury in mice, Neurochem. Int., 2015, 91, 46–54, DOI:10.1016/j.neuint.2015.10.008.
- S. D. Portbury, D. J. Hare, D. I. Finkelstein and P. A. Adlard, Trehalose improves traumatic brain injury-induced cognitive impairment, PLoS One, 2017, 12, e0183683, DOI:10.1371/journal.pone.0183683.
- (a) K. Castillo, M. Nassif, V. Valenzuela, F. Rojas, S. Matus, G. Mercado, F. A. Court, B. van Zundert and C. Hetz, Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons, Autophagy, 2013, 9, 1308–1320, DOI:10.4161/auto.25188; (b) J. A. Rodriguez-Navarro, L. Rodriguez, M. J. Casarejos, R. M. Solano, A. Gomez, J. Perucho, A. M. Cuervo, J. Garcia de Yebenes and M. A. Mena, Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation, Neurobiol. Dis., 2010, 39, 423–438, DOI:10.1016/j.nbd.2010.05.014; (c) S. Sarkar, J. E. Davies, Z. Huang, A. Tunnacliffe and D. C. Rubinsztein, Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein, J. Biol. Chem., 2007, 282, 5641–5652, DOI:10.1074/jbc.M609532200.
- (a) C. J. Holler, G. Taylor, Z. T. McEachin, Q. Deng, W. J. Watkins, K. Hudson, C. A. Easley, W. T. Hu, C. M. Hales, W. Rossoll, G. J. Bassell and T. Kukar, Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia, Mol. Neurodegener., 2016, 11, 46, DOI:10.1186/s13024-016-0114-3; (b) S. D. Portbury, D. J. Hare, C. Sgambelloni, K. Perronnes, A. J. Portbury, D. I. Finkelstein and P. A. Adlard, Trehalose Improves Cognition in the Transgenic Tg2576 Mouse Model of Alzheimer's Disease, J. Alzheimer's dis., 2017, 60, 549–560, DOI:10.3233/JAD-170322.
- R. Echigo, N. Shimohata, K. Karatsu, F. Yano, Y. Kayasuga-Kariya, A. Fujisawa, T. Ohto, Y. Kita, M. Nakamura, S. Suzuki, M. Mochizuki, T. Shimizu, U. I. Chung and N. Sasaki, Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage, J. Transl. Med., 2012, 10, 80, DOI:10.1186/1479-5876-10-80.
- J. C. Menzel, Depression in the elderly after traumatic brain injury: a systematic review, Brain Inj., 2008, 22, 375–380, DOI:10.1080/02699050802001492.
- (a) E. Ranjbar, M. S. Kasaei, M. Mohammad-Shirazi, J. Nasrollahzadeh, B. Rashidkhani, J. Shams, S. A. Mostafavi and M. R. Mohammadi, Effects of zinc supplementation in patients with major depression: a randomized clinical trial, Iran. J. Psychiatry, 2013, 8, 73–79 Search PubMed; (b) K. P. Vashum, M. McEvoy, A. H. Milton, P. McElduff, A. Hure, J. Byles and J. Attia, Dietary zinc is associated with a lower incidence of depression: Findings from two Australian cohorts, J. Affective Disord., 2014, 166, 249–257, DOI:10.1016/j.jad.2014.05.016.
- N. Z. Kara, L. Toker, G. Agam, G. W. Anderson, R. H. Belmaker and H. Einat, Trehalose induced antidepressant-like effects and autophagy enhancement in mice, Psychopharmacology, 2013, 229, 367–375, DOI:10.1007/s00213-013-3119-4.
- S. D. Portbury, D. J. Hare, C. J. Sgambelloni, D. P. Bishop, D. I. Finkelstein, P. A. Doble and P. A. Adlard, Age modulates the injury-induced metallomic profile in the brain, Metallomics, 2017, 9, 402, 10.1039/c6mt00260a.
- O. Gronli, J. M. Kvamme, O. Friborg and R. Wynn, Zinc Deficiency Is Common in Several Psychiatric Disorders, PLoS One, 2013, 8, e82793 Search PubMed.
- A. I. Bush, W. H. Pettingell, G. Multhaup, M. D. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Rapid induction of Alzheimer A beta amyloid formation by zinc, Science, 1994, 265, 1464–1467 Search PubMed.
- S. A. James, Q. I. Churches, M. D. de Jonge, I. E. Birchall, V. Streltsov, G. McColl, P. A. Adlard and D. J. Hare, Iron, Copper, and Zinc Concentration in Abeta Plaques in the APP/PS1 Mouse Model of Alzheimer's Disease Correlates with Metal Levels in the Surrounding Neuropil, ACS Chem. Neurosci., 2017, 8, 629–637, DOI:10.1021/acschemneuro.6b00362.
- J. R. Nuttall and P. I. Oteiza, Zinc and the aging brain, Genes Nutr., 2014, 9, 379, DOI:10.1007/s12263-013-0379-x.
- P. A. Adlard, J. M. Parncutt, D. I. Finkelstein and A. I. Bush, Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer's disease?, J. Neurosci., 2010, 30, 1631–1636, DOI:10.1523/JNEUROSCI.5255-09.2010.
- P. A. Adlard, J. Parncutt, V. Lal, S. James, D. Hare, P. Doble, D. I. Finkelstein and A. I. Bush, Metal chaperones prevent zinc-mediated cognitive decline, Neurobiol. Dis., 2015, 81, 196–202, DOI:10.1016/j.nbd.2014.12.012.
- P. A. Adlard, A. Sedjahtera, L. Gunawan, L. Bray, D. Hare, J. Lear, P. Doble, A. I. Bush, D. I. Finkelstein and R. A. Cherny, A novel approach to rapidly prevent age-related cognitive decline, Aging Cell, 2014, 13, 351–359, DOI:10.1111/acel.12178.
- P. Doering, G. Danscher, A. Larsen, M. Bruhn, C. Sondergaard and M. Stoltenberg, Changes in the vesicular zinc pattern following traumatic brain injury, Neuroscience, 2007, 150, 93–103, DOI:10.1016/j.neuroscience.2007.09.066.
- A. I. Faden, P. Demediuk, S. S. Panter and R. Vink, The Role of Excitatory Amino-Acids and Nmda Receptors in Traumatic Brain Injury, Science, 1989, 244, 798–800, DOI:10.1126/Science.2567056.
| This journal is © The Royal Society of Chemistry 2018 |