
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSystems impact of zinc chelation by the epipolythiodioxopiperazine dithiol gliotoxin in Aspergillus fumigatus: a new direction in natural product functionality†
Aliabbas A.
Saleh
 a,
Gary W.
Jones
ab,
Frances C.
Tinley
a,
Stephen F.
Delaney
a,
Sahar H.
Alabbadi
a,
Keith
Fenlon
a,
Sean
Doyle
*a and
Rebecca A.
Owens
*a
a,
Gary W.
Jones
ab,
Frances C.
Tinley
a,
Stephen F.
Delaney
a,
Sahar H.
Alabbadi
a,
Keith
Fenlon
a,
Sean
Doyle
*a and
Rebecca A.
Owens
*a
aDepartment of Biology, Maynooth University, Co. Kildare, Ireland. E-mail: sean.doyle@mu.ie; rebecca.owens@mu.ie; Tel: +353-1-708-3858 Tel: +353-1-708-3839
bCentre for Biomedical Research, School of Clinical and Applied Sciences, Leeds-Beckett University, Leeds LS1 3HE, UK
First published on 6th June 2018
Abstract
The non-ribosomal peptide gliotoxin, which autoinduces its own biosynthesis, has potent anti-fungal activity, especially in the combined absence of the gliotoxin oxidoreductase GliT and bis-thiomethyltransferase GtmA. Dithiol gliotoxin (DTG) is a substrate for both of these enzymes. Herein we demonstrate that DTG chelates Zn2+ (m/z 424.94), rapidly chelates Zn2+ from Zn(4-(2-pyridylazo)-resorcinol) (Zn(PAR)2) and also inhibits a Zn2+-dependent alkaline phosphatase (AP). Zn2+ addition rescues AP function following DTG-associated inhibition, and pre-incubation of DTG with Zn2+ completely protects AP activity. Zn2+ (1–50 μM) also significantly relieves the potent gliotoxin-mediated inhibition of Aspergillus fumigatus ΔgliT::ΔgtmA (p < 0.05), which infers in vivo dithiol gliotoxin-mediated sequestration of free Zn2+ or chelation from intracellular metalloenzymes as inhibitory mechanisms. Quantitative proteomic analysis revealed that excess Zn2+ alters the effect of gliotoxin on A. fumigatus ΔgliT, with differential abundance of secondary metabolism-associated proteins in the combinatorial condition. GtmA abundance increased 18.8 fold upon co-addition of gliotoxin and Zn2+ compared to gliotoxin alone, possibly to compensate for disruption to GtmA activity, as seen in in vitro assays. Furthermore, DTG effected significant in vitro aggregation of a number of protein classes, including Zn2+-dependent enzymes, while proteins were protected from aggregation by pre-incubating DTG with Zn2+. We conclude that DTG can act in vivo as a Zn2+ chelator, which can significantly impede A. fumigatus growth in the absence of GliT and GtmA.
Significance to metallomicsDithiol gliotoxin is a near-terminal biosynthetic intermediate from the gliotoxin biosynthetic pathway in the human pathogen Aspergillus fumigatus. Chemically reduced gliotoxin, dithiol gliotoxin (DTG), is revealed as a biological zinc chelator, and conversely, zinc can relieve the hitherto cryptic fungal autotoxicity of DTG. There is a systems-wide impact of zinc chelation by DTG on the fungal proteome, and we suggest it is DTG, as opposed to gliotoxin, which chelates zinc from metalloproteins. Since gliotoxin can be sequestered by both fungi and bacteria, our findings infer a new avenue to interfere with, and exploit, cellular zinc homeostasis in microorganisms. |
Introduction
Gliotoxin and holomycin are microbial natural products, which are produced by fungal and bacterial spp., respectively (Fig. 1).1–3 Both are low molecular mass metabolites, and each contains a disulphide bridge formed by the action of oxidoreductases, namely GliT and HlmI, on the respective dithiol precursor (Fig. 1).4–6 Disulphide bridge formation is essential for microbial self-protection against these reactive dithiol intermediates, and is a pre-requisite for the Major Facilitator Superfamily transporter GliA-mediated secretion of gliotoxin by Aspergillus fumigatus.5–8 Both metabolites are also present as bis-thiomethylated forms, and gliotoxin bis-thiomethyltransferase GtmA converts dithiol gliotoxin (DTG) to bis-dethiobis(methylthio)gliotoxin (BmGT) in A. fumigatus, whereas the origin of the cognate activity against dithiol holomycin in Streptomyces clavuligeris is unknown (Fig. 1).9,10 Bernardo et al. have shown that upon uptake by eukaryotic cells, gliotoxin is chemically reduced to the dithiol form by intracellular glutathione (GSH).11 Carberry et al. revealed significantly elevated intracellular GSH in gliotoxin-sensitive A. fumigatus ΔgliT and that Saccharomyces cerevisiae Δgsh1, deficient in intracellular GSH, was resistant to exogenous gliotoxin.12 The epipolythiodioxopiperazine (ETP) gliotoxin autoinduces its own biosynthesis by activating gli biosynthetic gene cluster expression, and BmGT formation has been shown to result in gli cluster attenuation in A. fumigatus, which results in the repression of gliotoxin biosynthesis.9,13,14 Methylation of dithiol holomycin has been proposed as a back-up plan for self-protection, and Dolan et al. revealed that A. fumigatus ΔgliT::ΔgtmA is significantly more sensitive to exogenous gliotoxin than a gliT-deficient mutant.15 This suggests that the combined absence of both self-protection and negative regulation of DTG biosynthesis (or its intracellular presence) results in potent growth retardation – the precise cause of which is unknown.10,15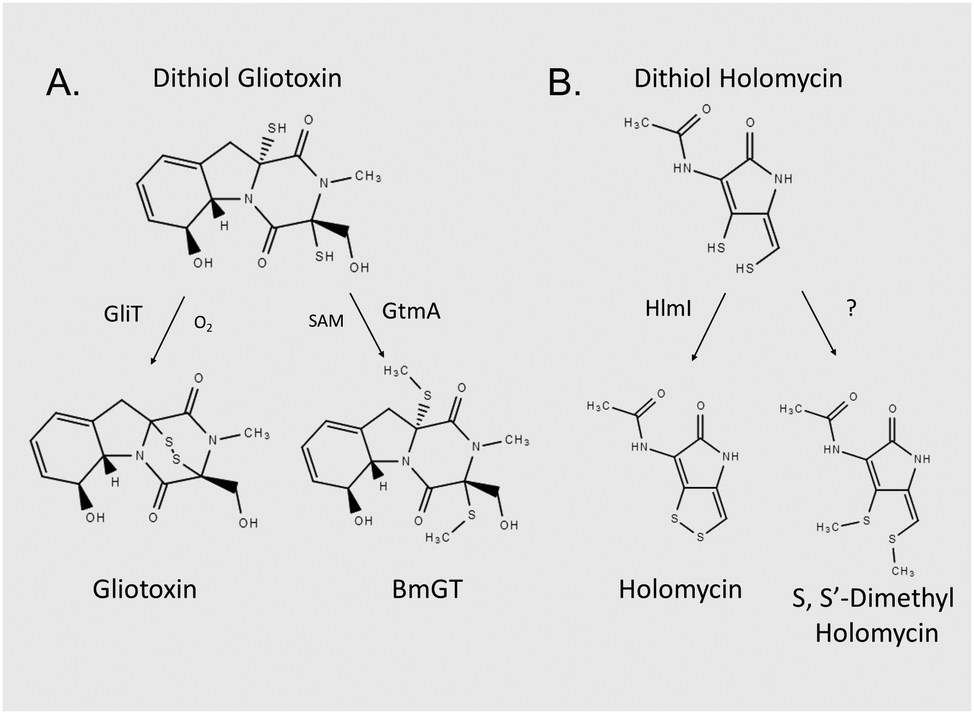 | ||
| Fig. 1 Unrelated microbial dithiol-containing compounds undergo equivalent biotransformations, and appear to be co-substrates for two enzyme functionalities. (A) In Aspergillus fumigatus, DTG is either oxidised to gliotoxin via gliotoxin oxidoreductase GliT or bis-thiomethylated by gliotoxin bis-thiomethyltransferase GtmA. (B) In Streptomyces clavuligeris, dithiol holomycin undergoes oxidoreductase HlmI-mediated conversion to holomycin. The enzyme which catalyses S,S′-dimethyl holomycin formation remains to be identified. | ||
Gliotoxin, other ETPs and dithiolopyrrolones have been shown to inhibit the activity of many enzymes, and functionality of specific proteins.16–21 Several studies have demonstrated that the disulphide moiety of gliotoxin is responsible for most of the associated bioactivity of this molecule, whereas the S-methylated BmGT molecule is relatively inactive.17,22 In general, reactivity of gliotoxin towards protein thiols, damage from redox cycling, and Zn2+ ejection have been proposed as the respective mechanisms whereby protein functionality is altered. Interestingly, addition of the reducing agent, L-dithiothreitol (DTT), significantly augmented the inhibitory activity of gliotoxin towards farnesyltransferase, and either DTT or GSH augmented gliotoxin-mediated inhibition of equine alcohol dehydrogenase.16,17 Notably, neither of the aforementioned studies posited DTG-mediated Zn2+ chelation from either Zn2+-dependent enzyme as the inhibitory mechanism.
In combination, these observations led us to hypothesise that, acting as a potent Zn2+ chelator, DTG (and not gliotoxin), could exhibit an equivalent mechanism of action towards Zn2+-dependent enzyme systems. Moreover, DTG interference with Zn2+ availability or Zn2+-dependent enzyme activity in A. fumigatus ΔgliT::ΔgtmA could be the basis for observed and extreme growth retardation.15 However, it is also essential to consider a mechanistic reciprocity between DTG and Zn2+. Consequently, while DTG may chelate the cation and impede Zn2+-mediated enzyme activities, excess or available metal ion may provide protection against intracellular DTG. Indeed, zinc salts have been successfully used to reverse ovine and bovine facial eczema associated with exposure to fungal ETPs such as sporidesmin A, a related disulphide-containing metabolite secreted by Pithomyces chartarum.23 It is postulated that Zn2+ chelates formed with sporidesmin A may attenuate its unwanted biological effects. Interestingly, Woodcock et al. presented mass spectrometric evidence of Zn2+[sporidesmin] chelates, and noted that Zn2+[gliotoxin] chelates, following NaBH4-mediated reduction, also existed, namely [2gliotoxin + Zn]2−, [2gliotoxin + Zn + Na]− and [gliotoxin + ZnCl]− (m/z 427).24 These authors speculated that that mono-ligands with halide coordination were the preferable complex form, however corresponding mass spectra and fragmentation patterns were not presented.
The ZafA transcriptional regulator, which can induce expression of the Zn2+ transporters zrfA, zrfB and zrfC, regulates zinc homeostasis in A. fumigatus and is essential for virulence.25–28 ZrfC is the key essential transporter which effects Zn2+ acquisition by A. fumigatus, while ZrfA and ZrfB play accessory and non-essential roles.26 Interestingly, transporter ZrfB (AFUA_2G03860) was found to be significantly increased in abundance (log2-fold increase: 1.31) in long-term A. fumigatus ΔgtmA cultures, which suggests interplay between dysregulated gliotoxin biosynthesis and Zn2+ homeostasis.29
Herein, for the first time we reveal DTG as a Zn2+ chelator which can specifically and significantly inhibit Zn2+-dependent metalloenzyme activity. Moreover, we demonstrate that Zn2+ significantly reverses the inhibitory effects of gliotoxin on A. fumigatus ΔgliT::ΔgtmA, which implicates intracellular Zn2+ chelation as a potential growth inhibitory strategy against this pathogen.15 Significant proteomic remodelling in A. fumigatus ΔgliT in response to gliotoxin versus Zn2+/gliotoxin exposure further illuminates a hitherto unanticipated in vivo interaction between DTG and Zn2+.
Methods
High resolution mass spectrometry detection of gliotoxin complexed to Zn2+
Tris(2-carboxyethyl)phosphine hydrochloride (TCEP)-reduced gliotoxin (DTG; 300 μM final) was mixed with ZnSO4·7H2O in the presence of formic acid (0.1% (v/v)) to achieve 0.5, 1 and 3-fold molar equivalents of Zn2+ to DTG. Samples were passed through 0.22 μm spin filters and directly injected onto a Thermo Q-Exactive Mass spectrometer (5 μl min−1). Assessment of DTG-Zn2+ complex formation was evaluated by both positive and negative ESI mode with full MS scan (50–2000.00). The following settings were used in the analysis: spray voltage (negative mode: 3.6 kV, positive mode 4 kV), capillary temperature 320 °C.Assessment of in vitro Zn2+ chelation by DTG
Zinc binding assays were performed as described in Chan et al. with some modifications.30 Titrations were carried out in PBS pH 7.4 (1 ml final volume). 4-(2-Pyridylazo)-resorcinol (PAR) (80 μl; 1.5 mM) was added with 60 μl 1 mM ZnSO4, per ml to yield final ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (PAR:Zn2+). DTG (2.5 mM in methanol) was prepared by 60 min pre-incubation with 12.5 mM TCEP in 100 μl final volume and was then added in increasing concentration to yield 1, 2, 3, 4 and 5 molar equivalents of DTG to Zn(PAR)2 (final concentrations: 60 μM Zn2+, 120 μM PAR, 60–300 μM DTG). 12.5 mM TCEP in methanol and oxidised gliotoxin (2.5 mM) were used as negative controls. After addition of all components, absorbance spectra were recorded between 200–800 nm.
:1 (PAR:Zn2+). DTG (2.5 mM in methanol) was prepared by 60 min pre-incubation with 12.5 mM TCEP in 100 μl final volume and was then added in increasing concentration to yield 1, 2, 3, 4 and 5 molar equivalents of DTG to Zn(PAR)2 (final concentrations: 60 μM Zn2+, 120 μM PAR, 60–300 μM DTG). 12.5 mM TCEP in methanol and oxidised gliotoxin (2.5 mM) were used as negative controls. After addition of all components, absorbance spectra were recorded between 200–800 nm.
Alkaline phosphatase (AP) and GtmA enzyme assays
Zn2+-Dependent AP from bovine intestinal mucosa (AP; 1.44 U ml−1 (Sigma-Aldrich)) in PBS was pre-incubated with the following agents to determine the extent of enzyme inhibition: 5 mM EDTA, 150 μM gliotoxin (GT), 10–150 μM DTG (TCEP-reduced GT), 50 μM TPEN (N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine), 50 μM–5 mM DTT, or 50 μM–5 mM GSH for 15 min in triplicate. Control or treated AP (50 μl per reaction) was added to 1 ml p-nitrophenol phosphate (pNPP) (5 mM; dissolved in PBS with 50 mM glycine pH 9.5) and assayed in triplicate at 37 °C for 30 min. Enzyme reactions were terminated by addition of 20 mM NaOH (5 ml). AP activity was determined by p-nitrophenol (pNP) detection at 405 nm following pNPP hydrolysis. The protective effect of pre-incubating DTG or TPEN with Zn2+ was also evaluated using this assay system. DTG (50 μM) or TPEN (50 μM) were pre-incubated with Zn2+ (25–100 μM) for 15 min prior to addition of AP enzyme and the rest of the assay was performed as before. To test the capacity of Zn2+ to rescue DTP-associated loss of AP activity, AP was pre-incubated with DTG (50 μM) for 15 min, followed by addition of Zn2+ (0.1–1 mM) for a further 15 min, prior to addition of pNPP substrate.GtmA activity was determined as described in Dolan et al. to evaluate the effect of Zn2+ (molar ratio 0.0001–10 Zn2+:1 DTG) on the formation of BmGT.15 Briefly, S-adenosylmethionine (SAM) (1 μM final) and TCEP-reduced gliotoxin (250 μM final) were combined, along with PBS. Zn2+ was added at a range of concentrations (0.025, 0.25, 2.5, 25, 125, 250 μM and 2.5 mM), representing a molar ratio ranging from 0.0001 to 10:1 Zn2+:DTG. A control was also prepared with no Zn2+ added. GtmA (0.5 μM) was added to the mixture followed by incubation at 37 °C for 5 min. Reactions were stopped by protein precipitation using TCA (final 15%) and incubation on ice for 20 min. Clarified mixtures were analysed by RP-HPLC and absorbance monitored at 254 nm to determine the concentration of BmGT and monomethyl gliotoxin (MmGT).
Assessment of the effect of Zn2+ on alkylation of DTG
Iodoacetamide (IAA) was used as an alkylating agent to investigate the effect of Zn2+ on alkylation of DTG. IAA stock (50 mM) was prepared in 50 mM ammonium bicarbonate. 3 mM DTG was prepared by 60 min pre-incubation of gliotoxin with 12.5 mM TCEP. DTG (0.3 mM final) was mixed with ZnSO4 at a range of concentrations (0.1, 0.15, 0.3, 0.6, 0.9 mM) to achieve molar equivalents of Zn2+ to DTG of 0.33, 0.5, 1, 2 and 3, respectively. IAA was subsequently added at a final concentration of 3 mM (ratio IAA:DTG 10:1) and incubated in the dark for 20 min. All reactions were carried out in methanol at 100 μl final volume. Control samples were included to measure levels of DTG and alkylated gliotoxin formed in the absence of Zn2+. Reactions were evaluated by RP-HPLC.15
A. fumigatus phenotypic assays
A. fumigatus ΔgliT and ΔgliT::ΔgtmA strains were grown on MEA agar for 5 days at 37 °C. After incubation, conidia were harvested with PBST and washed three times with PBS and resuspended in PBS. Conidia were counted using haemocytometer and stored at 4 °C for future use. Conidia were serially diluted to 103 μl−1 and 5 μl was spotted on Czapek-Dox agar plates (permissive for endogenous gliotoxin biosynthesis7) containing gliotoxin (0, 15 or 30 μM) and Zn2+, respectively. Plates were then incubated at 37 °C and growth monitored up to 96 h by measuring radial growth (mm) of each colony. Two-way ANOVA analysis was performed to determine the statistical significance between strains at different concentrations of gliotoxin and Zn2+.Extraction and measurement of extracellular and intracellular gliotoxin
A. fumigatus wild-type and ΔgliT::ΔgtmA were grown in Sabouraud-dextrose media for 21 h and gliotoxin was subsequently added for 3 h at 15 μM with and without Zn2+ (n = 3 biological replicates for all specimens). Zn2+ was added at 1 mM final concentration. Supernatant samples were taken after 15, 30, 60, 120 and 180 min and were extracted using chloroform (1:1), as described.31 The organic extracts were subsequently dried down and resuspended in methanol and analysed for gliotoxin content using RP-HPLC with UV detection (Shimadzu), using polar C18 RP-HPLC column (Phenomenex polar C18 Luna Omega column (150 mm × 4.6 mm, 5 μm)) at a flow rate of 1 ml min−1. A mobile phase of acetonitrile and water with 0.1% (v/v) TFA was used under gradient conditions. A. fumigatus wild-type was also grown in Czapek-Dox media for 72 h (gliotoxin-producing conditions) in the presence of either low (0.027 mM) or high (0.5 mM) Zn2+ (n = 3 biological replicates for all specimens). Controls were included, with no Zn2+ added. Aliquots were taken every 24 h and organic extractions and RP-HPLC analyses were performed as outlined above. For intracellular gliotoxin recovery, mycelia collected after 3 h incubation were harvested and snap frozen in liquid N2. Intracellular gliotoxin was extracted as described previously for SAM, using a modified protocol.8 Briefly, mycelia were ground using liquid N2 with a mortar and pestle. 100 mg mycelia were incubated with 0.1 N HCl (250 μl) on ice for 1 h with intermittent vortexing. Protein was removed by addition of 100% TCA to achieve a final concentration of 15% (v/v) TCA. After centrifugation at 16000 × g, supernatants were collected and then analysed by RP-HPLC.
Proteomic analysis of gliotoxin affects, with and without Zn2+, on A. fumigatus ΔgliT
A. fumigatus ΔgliT was cultured (n = 3/condition) for 21 h in Sabouraud-dextrose media (SDM) followed by addition of gliotoxin (15 μM) or methanol (control) with and without ZnSO4 (1 mM) addition for 3 h. Mycelia were then harvested and snap frozen in liquid N2. Mycelia were lysed using buffer (100 mM Tris–HCl, 50 mM NaCl, 20 mM EDTA, 10% v/v glycerol. 1 mM PMSF, 1 μg ml−1 pepstatin A, pH 7.5) with grinding and sonication, and clarified using centrifugation.8 The protein lysates were then subjected to precipitation using TCA/acetone and resuspended in 100 mM Tris–HCl, 6 M urea, 2 M thiourea, pH 8.0. Samples were then reduced by DTT and followed by alkylation using IAA. This was followed by addition of sequencing grade trypsin combined with Protease Max surfactant for overnight digestion. Following sample clean-up using Millipore C18 ZipTips, all peptide mixtures were analysed via a Thermo-Fisher Q-Exactive mass spectrometer coupled to a Dionex RSLCnano.32 LC gradients ran from 4% to 35% ACN over 2 h, and data were collected using a Top15 method for MS/MS scans. Comparative proteome abundance and data analysis was performed using MaxQuant software (v. 1.5.3.30), with PERSEUS used to organize data (v. 1.5.4.0).33,34 Proteins with significant changes in abundance (p < 0.05; fold change ≥2) signified quantitative changes between the sample treatments. Qualitative differences were also noted, where proteins were not detected in one group and were present in at least 2 replicates from the comparator group. Functional analysis was carried out using FungiFun2 (https://elbe.hki-jena.de/fungifun/fungifun.php).35In vitro DTG induced-protein aggregation assays
Protein extracts were prepared from A. fumigatus mycelia from three biological replicates, previously cultured in SDM at 37 °C, for 24 h at 200 rpm. Mycelia were harvested by filtering through Miracloth, snap-frozen and ground in liquid N2 using a pestle and mortar. Lysis buffer (0.1 M Tris–HCl, 50 mM NaCl, 1 mM PMSF pH 7.5; 400 μl) was added to ground mycelia (200 mg) and lysed by sonication. Lysates were clarified by centrifugation at 12000g, 4 °C for 10 min and this was repeated twice to remove insoluble material. Lysates were diluted to 1 mg ml−1 and subjected to the following treatments in triplicate: (a) control: methanol and TCEP were added as a solvent control; (b) DTG:TCEP-reduced gliotoxin was added to the protein extracts to a final concentration of 150 μM and (c) DTG/Zn2+:TCEP-reduced gliotoxin was pre-mixed with ZnSO4·7H2O at a 1:3 molar ratio. The mixture was added to the protein extracts in triplicate to achieve a final concentration of 150 μM DTG and 450 μM Zn2+. All samples were incubated at 50 °C for 30 min prior to addition of solubilisation buffer and analysis by SDS-PAGE. Protein aggregates were observed as a band of Coomassie-stained protein at the interface between the stacking and resolving gels. These bands were excised and subjected to in-gel digestion according to Shevchenko et al.36 Peptide mixtures were de-salted using C18 Zip-tips and analysed using Label-free quantitative proteomics (Thermo Q-Exactive LC-MS). Data analysis was performed using MaxQuant with Perseus for data organisation and statistical analysis.9,32
Results
DTG chelates Zn2+in vitro
High resolution mass spectrometry, spectrophotometry, and chemical and enzyme assays were used to investigate the formation of a complex between DTG and Zn2+. Following TCEP-mediated reduction of gliotoxin and Zn2+ addition, negative mode MS analysis revealed the presence of a Zn[dithiolate gliotoxin] complex with m/z = 424.93851, corresponding to a Cl− adduct of the metal chelate (Fig. 2A and Fig. S1, ESI†). Furthermore, DTG also competitively dissociated Zn2+ from a Zn[(4-(2-pyridylazo)-resorcinol)2] Zn(PAR)2 complex, whereby DTG (60–300 μM) addition resulted in a corresponding decrease in A493nm of Zn(PAR)2, corresponding to Zn2+ chelation (Fig. 2B). Relevantly, gliotoxin had no effect on Zn(PAR)2 interaction which confirmed the specific role of the dithiol moiety of reduced gliotoxin in chelation (Fig. S2, ESI†). To investigate the effect of Zn2+ on the enzymatic S-methylation of DTG, a GtmA activity assay was performed. GtmA bis-thiomethylates DTG to form BmGT using SAM as the methyl source (Fig. 1).9 Gliotoxin S-methylation occurs in a sequential manner, with mono-methylated species (MmGT) appearing initially, followed by formation of BmGT.9,15,37 Zn2+ inhibits GtmA-mediated thiomethylation of gliotoxin in vitro in a concentration-dependent manner, most likely due to an inability to recognise and bind Zn2+[DTG] (Fig. 2C). Support for DTG thiol protection by Zn2+ coordination was provided by an alkylation assay. Although IAA modifies DTG to alkylated gliotoxin, Zn2+ also inhibits alkylation of DTG which further strengthens the concept of thiol occlusion via Zn2+ chelation (Fig. 2D and Fig. S3, ESI†).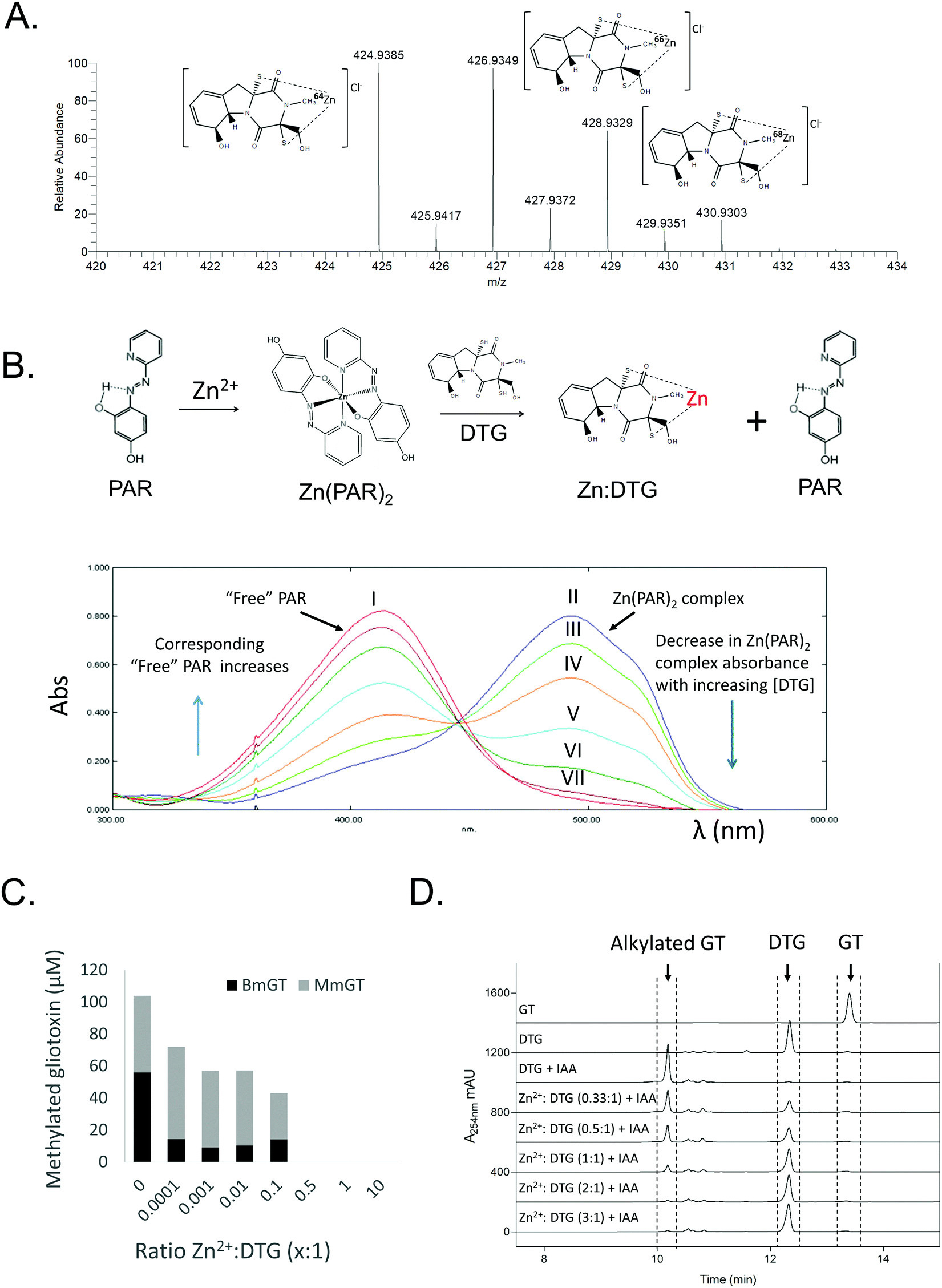 | ||
| Fig. 2 Characterisation of DTG as a Zn2+ chelator. (A) Dithiolate gliotoxin complexed with Zn2+ (Cl− adduct) with accompanying isotopic analysis in negative ion mode MS (direct injection). Structure of DTG:Zn complex is depicted with positioning of the metal ion between the two thiolates, although exact coordination with other groups is unknown. (B) DTG chelates Zn2+ from Zn(PAR)2 (Zn(PAR)2 pKd = 12.15)44 in a concentration-dependent manner. Overlaid UV-vis profiles of PAR alone (I; red), and Zn(PAR)2 (II; blue). DTG added in increased concentration to Zn(PAR)2 1–5 molar equivalents of oxidised gliotoxin to Zn2+; III–VII, respectively. Decrease in A493nm is accompanied by increase in A413nm as free PAR is released. (C) Zn2+ inhibits GtmA-mediated methylation of DTG in a concentration dependent manner. Zn2+ was included in the methylation assay at a molar ratio ranging from 0.0001 to 10:1 (DTG). A control containing no Zn2+ was included. Concentration (μM) of BmGT and MmGT formed after 5 min was determined by RP-HPLC. (D) Zn2+ inhibits IAA-mediated alkylation of DTG in a concentration-dependent manner. RP-HPLC analysis (detection at 254 nm) reveals the inhibitory effect of Zn2+ on DTG alkylation. | ||
DTG inhibits AP by Zn2+ chelation
To investigate the ability of DTG to inhibit the activity of a zinc-dependent metalloenzyme, AP activity assays were carried out. Gliotoxin was reduced to DTG as described above and confirmed by RP-HPLC (data not shown). DTG inhibited AP in a dose-dependent manner (0–150 μM DTG) and time-dependent manner (Fig. 3A and Fig. S4, ESI†). Solvent controls (containing TCEP and methanol) did not show any significant AP inhibition. While 50 μM DTG significantly inhibited AP activity (p < 0.001), pre-incubation of DTG (50 μM) with Zn2+ (25 μM–1 mM) alleviated DTG-associated AP inhibition (Fig. 3B). The Zn2+ chelator TPEN was also tested, but only produced a minor decrease in AP activity (11%) when used at the same concentration as DTG (50 μM) (Fig. 3C). EDTA (5 mM), a metal ion chelator, inhibited AP activity by approx. 30–40% (Fig. S5, ESI†). AP activity was not significantly inhibited by 150 μM GT, 50 μM–5 mM of the cellular reductant GSH or 50 μM–1 mM of the thiol-containing reductant DTT (Fig. S5, ESI†). Lack of inhibition by GT, TCEP, GSH or DTT strongly infers that inhibition is not caused by AP disulphide bridge cleavage or thiol modification. Zinc addition for 30 min reverses DTG-induced inhibition of AP, which further implicates Zn2+ chelation as the mechanism of inhibition (Fig. S6, ESI†).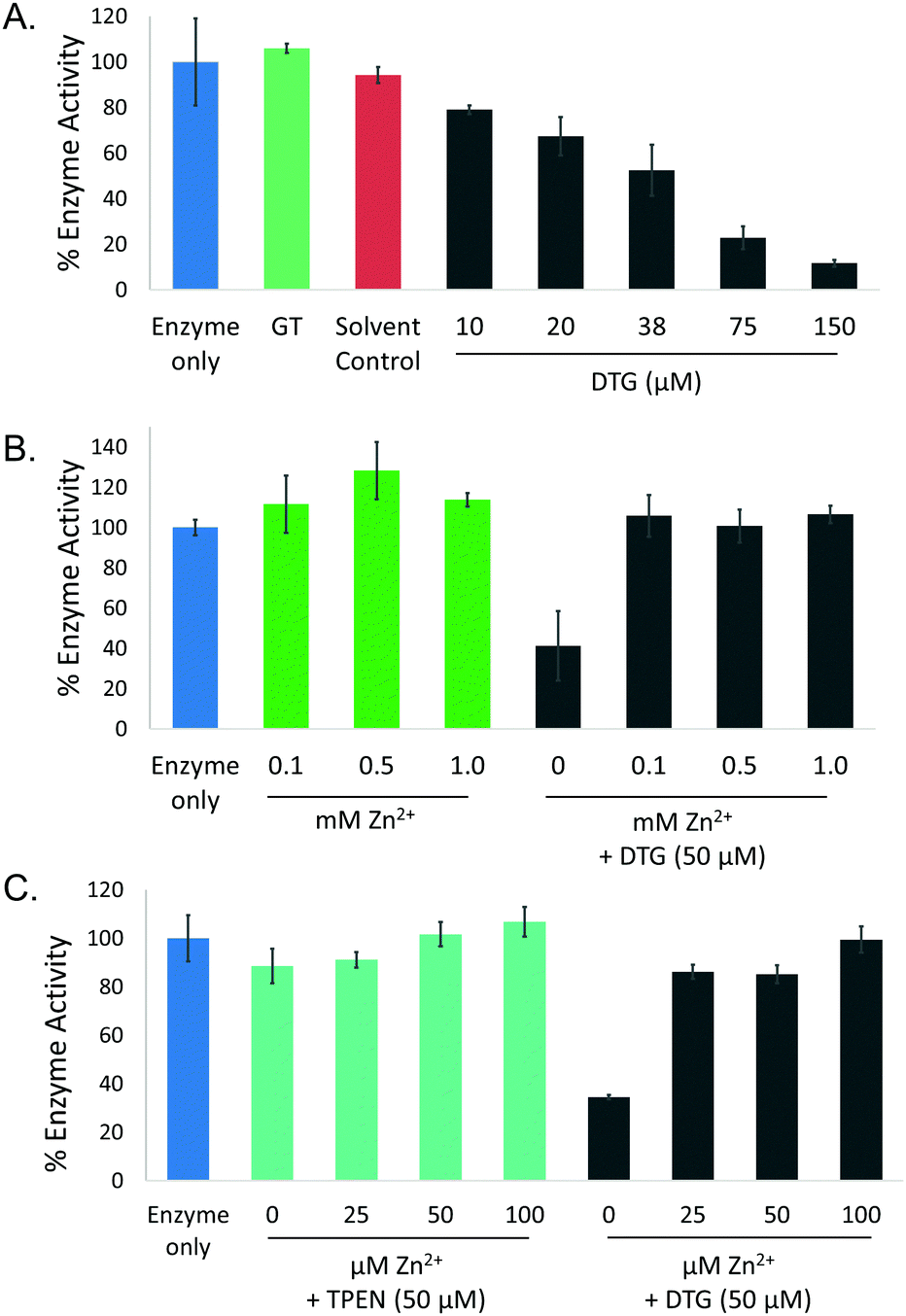 | ||
| Fig. 3 DTG inhibits AP activity by Zn2+ chelation. (A) Dose-dependent (10–150 μM DTG) inhibition of AP activity, with IC50 = 38 μM DTG. Neither GT or the solvent control (12.5 mM TCEP in methanol) inhibit AP activity. (B) Pre-incubation of DTG with Zn2+ (0.1, 0.5 and 1 mM) prevents DTG-mediated enzyme inhibition. (C) TPEN (50 μM) has no significant impact on AP activity (89% activity compared to control). Pre-incubation of DTG with Zn2+ (25–50 μM) significantly recovered AP activity to 86% of the control. | ||
Impact of Zn2+ exposure on A. fumigatus ΔgliT and ΔgliT::ΔgtmA, in the presence of exogenous gliotoxin
In order to determine whether Zn2+ had an impact on gliotoxin-associated growth inhibition, plate assays were carried out using A. fumigatus strains deficient in enzymes that use DTG as their substrate, ΔgliT::ΔgtmA and ΔgliT. Severe radial growth inhibition of A. fumigatus ΔgliT::ΔgtmA in the presence of gliotoxin (15 μM)15 (Fig. 4A) was significantly relieved (p < 0.005) in the presence of 1–10 μM Zn2+, and relief of gliotoxin-mediated growth inhibition was maintained up to 50 μM Zn2+. Radial growth of A. fumigatus ΔgliT::ΔgtmA was unaffected by Zn2+ (1–50 μM). Higher concentrations of Zn2+ significantly inhibited the growth of both A. fumigatus ΔgliT::ΔgtmA (100 μM–1 mM; Fig. 4) and A. fumigatus ΔgliT (1–2 mM; Fig. S7, ESI†). This correlates with a decrease in the rescue effect of Zn2+ on gliotoxin-associated growth inhibition in A. fumigatus ΔgliT::ΔgtmA. The combination of gliotoxin (15–30 μM) and excess Zn2+ (1–2 mM) produced further inhibition of radial growth of A. fumigatus ΔgliT at 96 h (p < 0.005) compared to the growth reduction observed in either the presence of Zn2+ or gliotoxin alone (Fig. S7, ESI†).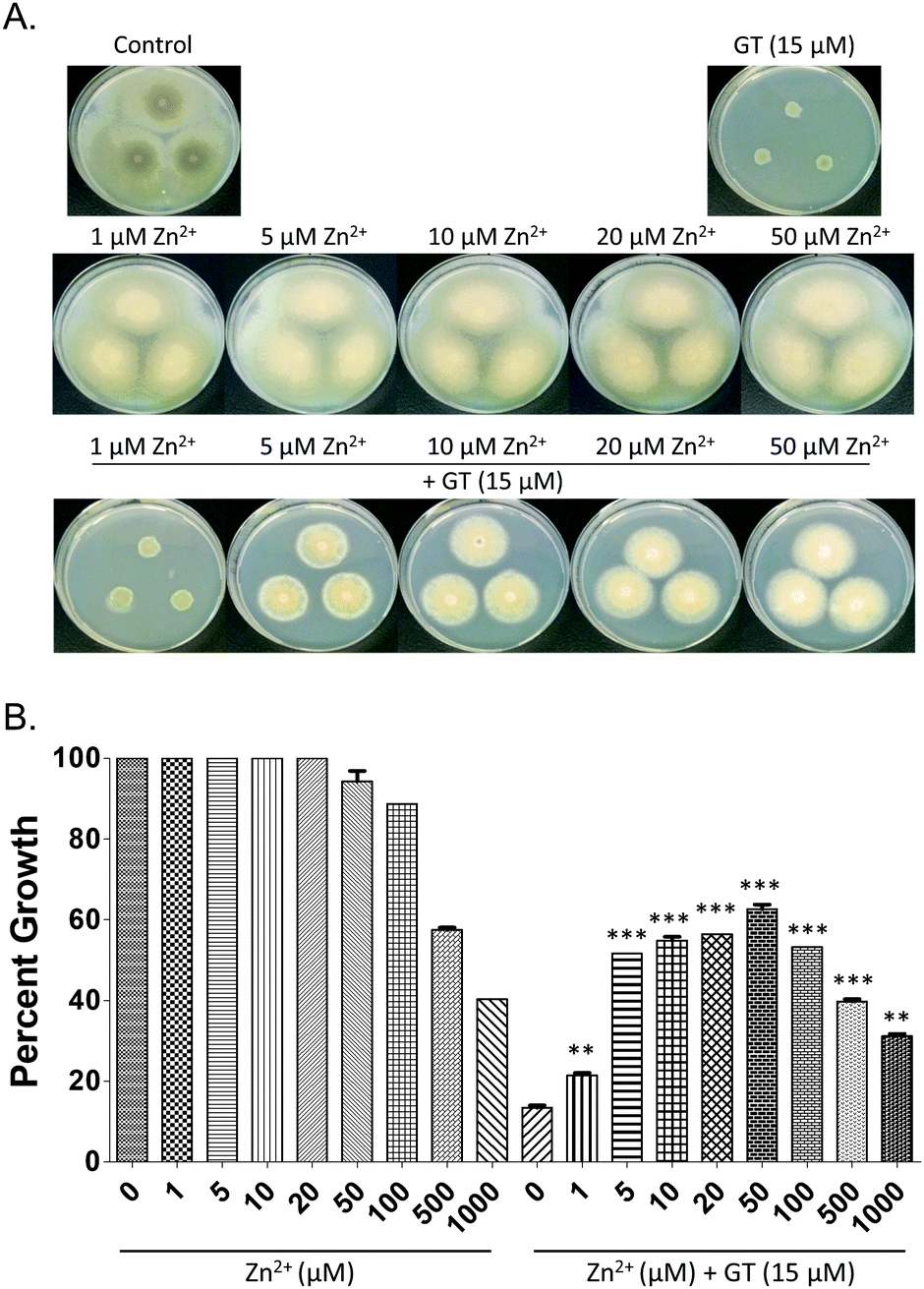 | ||
| Fig. 4 (A) Potent growth inhibitory effect of gliotoxin (15 μM) on A. fumigatus ΔgliT::ΔgtmA is significantly (p < 0.005) reversed by co-addition of Zn2+ (1–50 μM). Zn2+ (0–50 μM) has a negligible effect on mutant growth. (B) Graphical representation of same in terms of relative growth percentages. Note that higher Zn2+ levels (100 μM–1 mM) result in dissipation of the relief, in line with Zn2+-associated toxicity noted in controls. | ||
Zn2+ augments gliotoxin uptake and efflux in A. fumigatus wild-type and ΔgliT::ΔgtmA
Previous studies have revealed that exogenous gliotoxin is sequestered by A. fumigatus.38 To further explore the interaction between Zn2+ and gliotoxin in vivo, both A. fumigatus wild-type and ΔgliT::ΔgtmA were exposed to gliotoxin (15 μM) for 3 h in the presence and absence of Zn2+ (1 mM). In all conditions uptake of gliotoxin by A. fumigatus was evident by decreasing levels detected in culture supernatants across the incubation times, and corresponding increases in intracellular gliotoxin concentration (Fig. S8, ESI†). Fig. S8A–D (ESI†) show that excess Zn2+ augmented gliotoxin uptake in both A. fumigatus wild-type and ΔgliT::ΔgtmA. In the presence of Zn2+ (1 mM), with significantly less gliotoxin detected in A. fumigatus wild-type culture supernatants 30 min after addition (Fig. S8A–C; p < 0.05, ESI†). Zn2+ effected a more marked response in A. fumigatus ΔgliT::ΔgtmA, with a significant decrease in extracellular gliotoxin levels at every time point (15–180 min; Fig. S8B; p <0.05, ESI†). When excess Zn2+ was present in the culture medium, initial rates of gliotoxin uptake increased from 38.32 ± 5 to 55.04 ± 8.78 ng min−1 ml−1 of culture (mean ± SEM) in A. fumigatus wild-type and from 5.06 ± 7.16 to 75.94 ± 7.38 ng min−1 ml−1 of culture (mean ± SEM) in ΔgliT::ΔgtmA. Commensurate increases in intracellular gliotoxin (ng/100 mg mycelia) were observed in both A. fumigatus wild-type and ΔgliT::ΔgtmA, in the presence of Zn2+ (Fig. S8D, ESI†). Intracellular levels of gliotoxin were significantly increased in A. fumigatus wild-type upon Zn2+ co-addition (p < 0.01), likely due to inhibition of GtmA-catalysed BmGT formation and efflux. Elevated intracellular gliotoxin levels in ΔgliT::ΔgtmA correlated with an increased rate of gliotoxin uptake induced by Zn2+ (Fig. S8, ESI†). Intracellular DTG was not detectable due to either spontaneous intracellular or extraction-associated oxidation.LFQ proteomic analysis reveals that Zn2+ alters the effect of gliotoxin on A. fumigatus ΔgliT
To further investigate the nature of the combinatorial inhibition of A. fumigatus ΔgliT during simultaneous exposure to Zn2+ and gliotoxin, compared to gliotoxin alone, comparative LFQ proteomic analysis was carried out. Overall, 47 proteins exhibited unique presence and 23 proteins were significantly increased in abundance in A. fumigatus ΔgliT upon combinatorial exposure (Zn2+/GT) compared to gliotoxin only (Table S1, ESI†). Conversely, 45 proteins were absent and 10 proteins showed significantly reduced abundance in A. fumigatus ΔgliT upon combinatorial exposure compared to gliotoxin only (Table S1, ESI†). Functional analysis revealed that zinc-ion binding proteins were significantly enriched (8/70 proteins; p = 0.005) among proteins with elevated abundance or uniquely detected in the presence of Zn2+/GT compared to gliotoxin alone (Table 1 and Fig. S9, ESI†). Notably, secondary metabolism-associated proteins were altered in abundance, or demonstrated qualitative changes in the combined condition (Zn2+/GT) compared to gliotoxin only (Table 2). These included the ferricrocin synthetase SidC, which was repressed by gliotoxin, but not by the combined condition, as were four members of the fumagillin biosynthetic cluster (Table 2 and Table S1, ESI†). Consequently, when A. fumigatus was grown in gliotoxin-producing conditions, Zn2+ caused a significant increase in fumagillin levels even at lower levels (0.027 mM Zn2+; p < 0.001) (Fig. S10, ESI†).| Protein description | Log2 fold changea (p value) | Uniquely detectedb | Peptides | Gene IDs (AFUA_) |
|---|---|---|---|---|
| a Negative values indicate protein was decreased in abundance in combination treatment (Zn2+/GT) compared to the gliotoxin only treatment, while positive values indicate an increase in abundance. b Uniquely detected: protein uniquely identified in at least 2 biological replicates of either gliotoxin-only treated cultures or the combination condition (Zn2+/GT). | ||||
| Zinc ion binding | ||||
| Zinc knuckle domain protein (Byr3), putative | −1.33 (0.021) | 9 | 1G07630 | |
| Rho GTPase activator activity Rga | N/A | Zn2+/GT | 4 | 1G12680 |
| Putative zinc-binding dehydrogenase family oxidoreductase | 1.55 (0.0018) | 16 | 1G15610 | |
| Putative formaldehyde dehydrogenase | 1.54 (0.0027) | 15 | 2G01040 | |
| Putative carbonic anhydrase CafA | 1.65 (0.0029) | 12 | 4G11250 | |
| C-x8-C-x5-C-x3-H type zinc finger | N/A | Zn2+/GT | 5 | 4G13910 |
| Putative zinc-dependent alcohol dehydrogenase AlcC | 1.62 (0.0004) | 30 | 5G06240 | |
| Cytidine deaminase, zinc ion binding activity | N/A | Zn2+/GT | 2 | 7G01040 |
| Zinc knuckle nucleic acid binding protein, putative | N/A | GT | 2 | 7G02190 |
| Putative carbonate dehydratase, zinc ion binding activity | 1.67 (0.0041) | 7 | 8G06554 | |
| Zinc homeostasis | ||||
| Low affinity plasma membrane zinc transporter ZrfB | N/A | GT | 3 | 2G03860 |
| Allergen Aspf2 Aspf2 | N/A | GT | 7 | 4G09580 |
| Secondary metabolite | Protein description | log2 fold increase (p value) | Uniquely detecteda | Peptides | Gene IDs (AFUA_) |
|---|---|---|---|---|---|
| a Uniquely detected: protein uniquely identified in at least 2 biological replicates of either gliotoxin-only treated cultures or the combination condition (Zn2+/GT). | |||||
| Gliotoxin | Unknown function protein GliH | GT | 3 | 6G09745 | |
| O-Methyltransferase GliM | GT | 4 | 6G09680 | ||
| Gliotoxin thiomethyltransferase GtmA | 4.2361 (0.0052) | 18 | 2G11120 | ||
| NRPS8 (unknown) | NRPS8/Pes3 | GT | 3 | 5G12730 | |
| Ferricrocin | NRPS involved in ferricrocin siderophore biosynthesis SidC | Zn2+/GT | 5 | 1G17200 | |
| Fumagillin | Putative alpha/beta hydrolase FmaC | Zn2+/GT | 11 | 8G00380 | |
| Unknown function protein FmaD | Zn2+/GT | 5 | 8G00400 | ||
| Protein encoded in the fumagillin gene cluster | Zn2+/GT | 10 | 8G00430 | ||
| Putative iron-dependent oxygenase FmaF | Zn2+/GT | 9 | 8G00480 | ||
Low affinity zinc transporter ZrfB, which is ZafA-dependent and normally induced by zinc depletion, was undetectable in the combinatorial exposure condition, which suggests a zinc-limiting environment in gliotoxin-only exposure (Table 1). Likewise, allergen AspF2 which is expressed under zinc-limiting conditions, and shares a divergent promoter with zrfC, was absent upon co-exposure.39 It was also observed that GliM, a component of the gliotoxin biosynthetic capacity was absent, which infers that gliotoxin biosynthesis may be attenuated in the combinatorial exposure scenario (Table 2). Indeed, Zn2+ presence completely abrogated gliotoxin biosynthesis in Czapek-Dox media (Fig. S10, ESI†). These observations pertaining to impeded gliotoxin biosynthesis are further supported by the absence of GpgA (AFUA_1G05210), a G-protein coupled receptor subunit, previously shown to be essential for gliotoxin production in A. fumigatus.40 It is also notable that the nonribosomal peptide synthetase NRPS8/Pes341 (AFUA_5G12730) was absent following co-exposure to Zn2+ and gliotoxin, which suggests that it may contribute to adaptation to zinc-limiting conditions.
Gliotoxin exposure induces elevated GtmA abundance in A. fumigatus ΔgliT,9 and Manzanares-Miralles et al. reported that gliotoxin induces significantly increased abundance of the GtmA ortholog in Aspergillus niger, in which the gli cluster is absent.37 Interestingly, while Zn2+ exposure alone had no significant effect on the abundance of GtmA in A. fumigatus, combinatorial exposure with gliotoxin resulted in significantly elevated GtmA abundance (18.8-fold and p < 0.01) compared to that in the presence of gliotoxin only (Table 2). Allied to GtmA activity data (Fig. 2C), this suggests that in vivo chelation of Zn2+ by DTG may prevent substrate access to GtmA, thereby resulting in a further increased abundance of the enzyme (Fig. 5A).
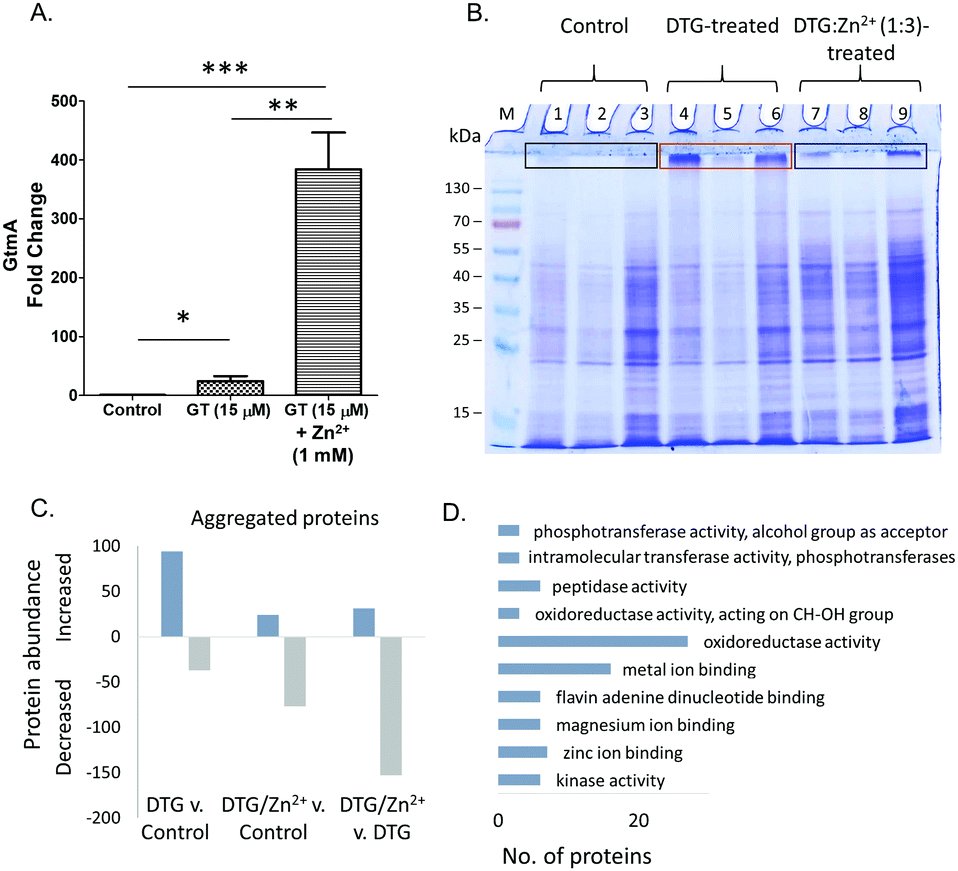 | ||
| Fig. 5 (A) Fold change in GtmA abundance following gliotoxin (GT: 15 μM) and gliotoxin & Zn2+ (15 μM & 1 mM, respectively) exposure compared to controls in A. fumigatus ΔgliT. (B) SDS-PAGE analysis of 3 independently prepared A. fumigatus protein lysates. Lanes 1–3: negative controls: protein lysates treated with TCEP/methanol only. Lanes 4–6: protein lysates treated with DTG. Lanes 7–9: protein lysates treated with DTG:Zn2+ (1:3). M, molecular mass marker. Protein aggregation is evident (boxes) following DTG-exposure. (C) Number of proteins with increased or decreased abundance (qualitative and quantitative) in aggregates recovered from control, DTG or combined DTG/Zn2+ treatments. (D) Functional analysis (FungiFun2) showing GO molecular function categories significantly enriched (p < 0.05) among proteins with increased abundance in DTG-induced aggregates. Arranged in order of ascending p value. | ||
Manifestation of heat-induced in vitro protein aggregation in A. fumigatus mycelial lysates, specifically by DTG
Inhibition of Zn2+-dependent enzyme activities, combined with the observed in vivo interplay between the divalent metal cation and DTG in A. fumigatus, led us to further investigate the interaction between DTG and A. fumigatus proteins. DTG-associated protein destabilisation was investigated by exposing complex lysates to either DTG or a mixture of Zn2+ and DTG, followed by heat treatment. Exposure of A. fumigatus mycelial protein lysates to TCEP/methanol, DTG or a mixture of Zn2+ and DTG followed by SDS-PAGE analysis revealed near-identical electrophoretic patterns in the resolving gel (Fig. 5B). However, uniquely, following protein lysate exposure to DTG, and subsequent heat treatment (50 °C/30 min), either protein aggregation or an inability to disaggregate unfolded proteins was observed (Fig. 5B). Mass spectrometry-based analysis of the proteins retained at the interface between the stacking and resolving gel identified 551 proteins in total from the three conditions tested. Label-free proteomics revealed 94 proteins uniquely detected or significantly elevated in the aggregates following DTG pre-treatment, relative to the control (Fig. 5C and Table S2, ESI†). DTG addition to A. fumigatus lysates elicits an overall increase in aggregated proteins compared to the controls. Functional analysis revealed a significant number of proteins with oxidoreductase activity (n = 27, p = 0.019), metal ion binding (n = 16, p = 0.024) and zinc ion binding (n = 7, p = 0.047) showed increased aggregation in response to DTG treatment (Fig. 5D). Notably, pre-incubation of DTG with Zn2+ prevented this aggregation in the case of 77% of the DTG-responsive proteins (72/94 proteins), whereby the combinatorial condition lead to significantly lower levels of protein aggregation compared to DTG treatment (p < 0.05). Additional stabilisation was observed in the combinatorial condition, as 77 proteins showed lower abundance in the aggregates compared to the control condition (Fig. 5C). Excess Zn2+ used in the pre-mixed DTG/Zn2+ treatment could contribute to this additional stabilising effect.Among the proteins affected by DTG-induced aggregation, was the zinc-dependent alcohol dehydrogenase AlcC, which was only detected in aggregates from lysates pre-incubated with DTG, indicating co-addition of Zn2+ prevented DTG-induced aggregation of this protein. Another protein detected exclusively in aggregates induced by DTG was the putative farnesyltransferase beta subunit Ram1 (AFUA_4G10330) which contains a C-terminal Zn2+ binding pocket, within the active site.42,43 A number metallopeptidases were also increased or uniquely detected in DTG-induced aggregates (AFUA_6G09190, AFUA_4G07910, MepB), as well as the zinc-dependent methionine synthase MetH/D, while co-addition of Zn2+ generated aggregation profiles in line with the control.
Overall, these data suggest that DTG may chelate Zn2+ from selected cellular proteins thereby inducing protein unfolding and consequently resulting in temperature-induced aggregation. It is also possible that DTG inhibits selected components of the proteasome-mediated recognition and/or digestion of protein aggregates; which results in their persistence in DTG-treated lysates. Finally, it is likely that some proteins detected in the aggregates may be present as a result of non-specific physical entrapment, however, these putative, non-specifically aggregated proteins are resistant to the dissolution effects of SDS solubilisation buffer and heat (95 °C/4 min).
Discussion
Herein, we provide evidence that DTG is a Zn2+-chelator. We also reveal Zn2+-metalloenzyme inhibition specifically by DTG, and not by gliotoxin or other reducing agents (DTT or GSH). This suggests Zn2+ chelation, as well as disulphide bridge cleavage or thiol modification, as a key mode of enzyme inhibition by DTG. We propose that DTG, acting as a Zn2+-chelator, can significantly inhibit growth of A. fumigatus completely deficient in essential oxidation or thiomethylation activities. Unbiased LFQ proteomics reveals that Zn2+ can significantly modify the nature and extent of protein abundance alterations caused by A. fumigatus ΔgliT exposure to gliotoxin. This important revelation confirms in vivo interaction between both molecular species. Moreover, additional LFQ proteomic data reveal that in vitro, DTG can cause specific protein aggregation, manifested by heat instability, possibly due to structural alteration to known Zn2+-dependent enzymes. These unexpected observations open a new front in our exploration of the metallo-metabolome in fungi, and possibly other species.High resolution, negative mode MS revealed that DTG complexes Zn2+ and exhibits a monoisotopic peak with m/z 424.93749, which equates to a single Zn2+[DTG] complex detected as a Cl− adduct [(DTG + 64Zn)–2H + Cl]−. Previous work by Woodcock et al. proposed the existence of a similar adduct (m/z 427), following NaBH4-mediated reduction.24 Since no spectra were provided, it is not possible to ascertain the origin of the different observed m/z values, although it seems likely that Woodcock et al. reported the average m/z for the compound or the m/z of the base peak [(DTG + 66Zn)–2H + Cl]−, rather than the monoisotopic peak.24 In addition, we have found that DTG efficiently chelates Zn2+ from Zn(PAR)2 (60 μM), in a dose-dependent manner from 60 to 300 μM DTG. Given that Kocyła et al. have proposed an effective pKd of 12.15 at pH 7.4 for Zn(PAR)2, it is clear that the affinity of DTG for Zn2+ must equate to, or exceed, this value as Zn2+ is removed from the Zn(PAR)2 complex when equimolar amounts of DTG are added to it.44 However, it is not ideal to compare these complexes directly due to differences in stoichiometry. Chan et al. also deployed a PAR-based assay system, which involved chelation of Zn2+ from Zn(PAR)2, and subsequent formation of stable complexes to reveal that Zn2+ is chelated by dithiol holomycin (Fig. 1; termed red-holomycin in Chan et al.) with high affinity.30 Davis et al. previously revealed and characterised alkylation of DTG using 5′-iodoacetamidofluorescein.45 In the present study, we observed that IAA-mediated alkylation of DTG was inhibited by Zn2+, which further underpins our proposal that a Zn[DTG] complex is formed via thiolate coordination and is stable under in vitro conditions used.
DTG acts as a potent inhibitor of the Zn2+-dependent enzyme, AP. Previous studies have reported the inhibitory effects of gliotoxin on mammalian Zn2+-dependent enzymes, without attributing this activity to zinc chelation.16,17 Vigushin et al. studied the inhibition of Zn2+-dependent farnesyltransferase (FTase) and geranyltransferase (GGTase) I by gliotoxin, proposing thiol modification of these enzymes as a possible mechanism of action. Importantly, the authors noted that these assays required the reductant DTT, which aligns with our observations of the inhibition of Zn2+-dependent metalloenzymes (i.e., AP) by DTG rather than gliotoxin. Our data also provides an alternative explanation for previous observations which suggested that reducing agents enhanced the inhibitory activity of gliotoxin against a Zn2+-dependent equine ADH.16 Although these authors posited a redox explanation, it is equally plausible that Zn2+ chelation by DTG effected equine ADH inhibition. This accords with our observations that prior incubation of DTG with Zn2+ prevents inactivation of AP, and also that DTG-associated AP inhibition can be rescued by subsequent addition of Zn2+ (Fig. 3 and Fig. S6, ESI†).
Relevantly, it has been shown that ETPs, in particular gliotoxin, block the interaction between Hypoxia Inducible Factor-α (HIF-α) and the transcriptional coactivator p300 by a Zn2+ ejection mechanism.18 Moreover, these authors noted the antiproliferative effects of ETPs, and provide significant insight into their mechanism of action in animal cells. Cook et al. reference the Zn2+-dependency of many gliotoxin-sensitive enzymes and the Zn2+ requirement of GliZ, the transcription factor essential for gliotoxin biosynthesis.1,18 However, their proposed mechanism of action of Zn2+ ejection from p300 by gliotoxin does not take into account the presence of DTT in assay buffers.18 Thus, consequent to our observations with AP and those of Cook et al., we now speculate that it is DTG, formed due to the presence of equivalent amounts of DTT, and not gliotoxin per se, which causes Zn2+ ejection from p300. Relevantly, it has recently been elegantly demonstrated that dithiol holomycin (Fig. 1) can effect Zn2+ chelation and cause inhibition of a metallo-β-lactamase.30 Our demonstration of DTG-mediated inhibition of AP suggests that the Zn2+ chelation potential of dithiolopyrrolones, like holomycin, extends to ETPs.
Our deployment of A. fumigatus mutants deficient in gliotoxin self-protection (ΔgliT) and self-protection/negative regulation of gliotoxin biosynthesis in combination (ΔgliT::ΔgtmA) reveal a hitherto unknown systems interaction between Zn2+ and gliotoxin biochemistry. We have observed the in vitro inhibition of GtmA-mediated BmGT formation by Zn2+. This mode of inhibition appears to result from DTG chelation of Zn2+, and its subsequent unavailability as a substrate to GtmA, as opposed to direct enzyme inhibition. Interestingly, this observation is in accordance with the effect of Zn2+ (1 and 2 mM) exposure towards A. fumigatus ΔgliT, whereby combinatorial gliotoxin/Zn2+ exposure significantly augments growth inhibition. In effect, we postulate that elevated levels of Zn2+ may augment intracellular Zn(DTG) complex formation, resulting in non-availability to GtmA, which actually significantly increases GtmA abundance (Fig. 5A). Thus, we speculate that Zn2+-mediated disruption of GtmA functionality in A. fumigatus ΔgliT partially creates an inability to dissipate DTG, though not as absolute as pertains in A. fumigatus ΔgliT::ΔgtmA. Indeed, A. fumigatus ΔgliT::ΔgtmA presents an ideal system to explore the functionality of Zn2+-dependent systems in fungi, since the organism lacks both enzymes, GliT and GtmA, which contribute to dissipation of intracellular DTG (Fig. 6), ultimately via either gliotoxin or BmGT efflux.15 The significant, though incomplete, Zn2+-mediated reversal of A. fumigatus ΔgliT::ΔgtmA sensitivity to exogenous gliotoxin exposure implicates either chelation of free Zn2+ or chelation of Zn2+ from cellular metalloenzymes, by DTG as key inhibitory mechanisms (Fig. 6). To our knowledge, this is the first report of an in vivo interaction between gliotoxin biosynthesis, dysregulated DTG presence and growth inhibition due to potential interference with Zn2+-associated growth systems. Moreover, it contributes to explaining the cryptic observation of Dolan et al. that A. fumigatus ΔgliT::ΔgtmA is the most gliotoxin-sensitive mutant observed to date,15 and also why there are two distinct enzymatic activities which can prevent intracellular DTG accumulation. Overall, these data indicate the inhibitory potential of this endogenous, and potent, Zn2+ chelator in A. fumigatus in particular, and possibly microorganisms in general. Indeed, gliotoxin significantly inhibits growth of a range of fungi,12,46 and previously work has also indicated its potent anti-bacterial activity.47
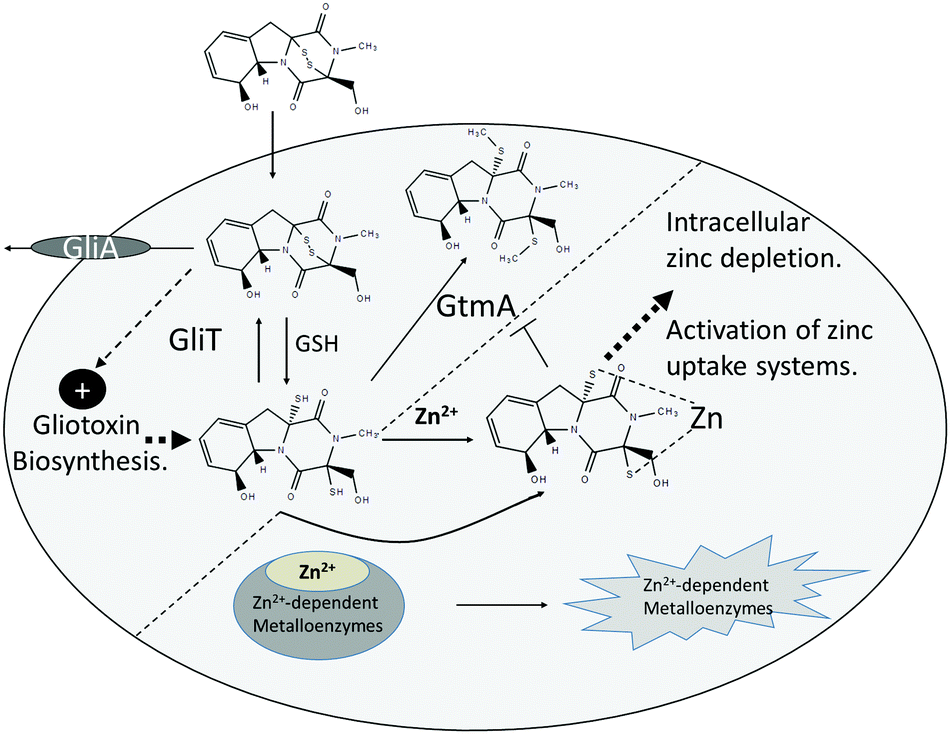 | ||
| Fig. 6 Overview of the proposed mechanism and consequences of Zn2+ chelation by intracellular DTG in A. fumigatus. DTG is produced consequent to gliotoxin uptake, via GSH-mediated chemical reduction, or de novo gliotoxin biosynthesis. Under normal conditions, two enzyme systems, namely GliT-mediated oxidation and GtmA-mediated thiomethylation effect dismutation of DTG to prevent interference with intracellular zinc homeostasis. In the absence of both enzymes, elevated intracellular DTG can either chelate free Zn2+ or chelate it from intracellular Zn2+-dependent metalloenzymes, causing extreme growth inhibition. Additionally, Zn(DTG) cannot be converted to BmGT via GtmA. Either way, disruption of intracellular zinc homeostasis occurs which leads to activation of zinc uptake systems, potentially via ZafA induction of ZrfA-C expression. | ||
It is clear that interference with intracellular DTG levels, via gliotoxin exposure to A. fumigatus ΔgliT impacts on the fungal proteome, which in turn can be modulated by Zn2+ presence. Indeed, it has been estimated that up to 6% (600/10000) of the A. fumigatus proteome comprises Zn2+-binding proteins; interestingly, this includes a prediction of 300 Zn2+ finger transcription factors.48 Relevantly, co-addition of gliotoxin and Zn2+ induced key alterations to abundance of proteins involved in secondary metabolism. Proteins from the gliotoxin biosynthetic cluster, GliM and GliH, were uniquely detected in mycelia exposed to gliotoxin, indicating that combinatorial exposure with Zn2+ prevents activation of gliotoxin cluster expression. While GliM and GliH are induced in response to gliotoxin, these proteins are not detected following BmGT exposure.9 This may implicate Zn2+ chelation in the activation of the gliotoxin transcription factor GliZ and the induction of cluster expression. O’Keeffe et al. noted gliotoxin exposure suppresses fumagillin cluster expression in A. fumigatus ΔgliT.14 This result was reflected in the current study, however co-exposure to gliotoxin and Zn2+ reversed this effect, leading to unique detection of fumagillin biosynthetic proteins. As with gliotoxin, the fumagillin cluster is also regulated by a (Zn2+)2Cys6 transcription factor (FapR/FumR),49,50 possibly implicating Zn2+ chelation in cluster repression, since exogenous Zn2+ blocks gliotoxin-associated repression. These proteomic observations are supported by the switch from gliotoxin to fumagillin production in A. fumigatus upon Zn2+ supplementation (Fig. S10, ESI†).
Incubation of A. fumigatus protein lysates with DTG prior to heat treatment resulted in increased protein aggregation, compared to the solvent control. Oxidoreductases, metal-binding proteins and zinc-binding proteins were significantly enriched amongst these DTG-affected proteins (p < 0.05). DTG-induced aggregation of zinc-binding proteins including two alcohol dehydrogenases, AlcC and AFUA_1G04620, and the farnesyltransferase (AFUA_4G10330) was observed. This is in line with observed inhibition of mammalian homologs of these proteins by gliotoxin in reducing conditions.16,17 AlcC was also observed to increase in abundance in vivo when A. fumigatus ΔgliT was treated with a combination of gliotoxin and Zn2+ compared to gliotoxin alone (Table S1, ESI†), possibly to compensate for loss of activity. AlcC has been identified as the primary hypoxia-responsive ADH in A. fumigatus, with a potential role in pathogenesis,51 while farnesyltransferase has a role in signalling and also contributes to disease.52 Further investigation could elucidate the effect of DTG on the functionality of these proteins in A. fumigatus. Pre-incubation of DTG with excess Zn2+ abrogated this effect, most likely through formation of Zn(DTG) complexes prior to addition to protein lysates, leading to reduced levels of protein aggregation. A number of metallopeptidases (AFUA_6G09190, AFUA_4G07910, AFUA_1G14920 and MepB) were also observed to undergo increased aggregation in response to DTG, while this was prevented in the presence of Zn2+. These peptidases contribute to protein modification and degradation, and so alteration of these processes by DTG has the potential to disrupt protein turnover. Interestingly, gliotoxin has previously been implicated in inhibition of proteolytic activity of human and toxoplasma proteasomes.53,54 The cobalamin-independent methionine synthase MetH/D showed a similar response to DTG treatment, with Zn2+ co-addition blocking aggregation. MetH/D contains a zinc-binding site, required for binding and activation of its substrate homocysteine.55 DTG-mediated aggregation of MetH/D would interrupt an integral part of primary metabolism and potentially affect pathogenesis.56 Interestingly, while metH/D showed no response to gliotoxin in A. fumigatus wild-type, expression was significantly induced in A. fumigatus ΔgliT in response to gliotoxin.14 Persistence of intracellular DTG, in the absence of GliT-mediated oxidation, results in disruption of the methionine cycle, with SAM depletion caused by dysregulation of gliotoxin methylation.8 Added to the extensive SAM consumption, DTG-associated destabilisation of methionine synthase through Zn2+-chelation could place an additional strain on the methionine cycle in A. fumigatus ΔgliT. While further studies are required to confirm if these proteins are directly affected by DTG in vivo, these results present strong targets for future investigations to elucidate the systemic effect of DTG on A. fumigatus. Of course, Zn2+ chelation is likely not the only mechanism by which DTG exerts its effects on the cell, with previous studies illustrating its potential for thiol modification and redox reactions.57,58 This methodology also provides an unbiased discovery-based mechanism allowing for the identification of putative targets of DTG in other complex systems.
Interestingly, Müller et al. have shown that allicin (diallyl thiosulphinate) can cause thiol stress and severe growth inhibition in bacteria.59 Specifically, Muller et al. revealed that allicin induced protein aggregation, likely due to S-allylmercapto protein modification, in crude Escherichia coli cell lysates in a concentration-dependent manner. This is in accordance with our observations of DTG-induced protein aggregation in A. fumigatus protein lysates, although fungal protein destabilisation possibly involves Zn2+ chelation mechanism, as opposed to protein modification. Future work will clarify the relative contribution of either mechanism.
As can be seen in Fig. S8 (ESI†), gliotoxin addition to A. fumigatus results in uptake, followed by conversion to intracellular DTG and induction/augmentation of gliotoxin biosynthesis, as previously reported in Owens et al. and Dolan et al.8,15 Based on our new observations, we now extend this model and provide a mechanistic link between the intracellular presence of DTG and (i) Zn2+ depletion leading to increased ZrfB abundance, as well as (ii) potential Zn2+ chelation and destabilisation of metalloenzymes. Interestingly, the membrane-permeable zinc chelator, TPEN, which has been used in vivo60 and in A. fumigatus studies,61 demonstrated lower AP inhibition than DTG (Fig. 3). The dissociation constant (Kd) of Zn(TPEN) has been reported as 6.4 × 10−16 M (pKd 15.2) at pH 7.4, with the same 1:1 stoichiometry as the Zn(DTG) complex.62,63 Future studies will quantify the affinity of DTG for Zn2+ however, considering DTG caused significantly greater AP inhibition than TPEN, it is reasonable to conclude that DTG is a better Zn2+ chelator than TPEN under the conditions tested. TPEN has been shown to act co-operatively with the antifungal drug caspofungin to significantly improve survival in a mouse model system of Invasive Pulmonary Aspergillosis compared to either caspofungin or TPEN administration alone.64 This exciting development is important because exposure to combinations of antifungal drugs, acting in synergy, may address both the development of pathogen resistance and toxicity of high therapeutic levels of either drug alone to the recipient. Although TPEN administration may not have any immediate harmful effects in animals, its safety profile following co-administration with antifungal drugs is unknown. Our observation of DTG as an intracellular Zn2+ chelator, ideally positions it as a potential endogenous anti-fungal, especially if strategies to interfere with its enzymatic elimination are elucidated in future research.
Conclusions
Overall, new in vivo and in vitro interactions between Zn2+ and DTG, with multiple biological consequences, are revealed. A. fumigatus is a pathogen for which limited therapeutic options exist. Although much recent work has greatly increased the understanding of this and other fungal species, investigation of cryptic interaction between Zn2+ and BGC-encoded metabolite functionality needs urgent study. Any new systems identified could not only represent antifungal drug targets, but also inform on the exploitation of BGC-encoded, dithiol-containing, metabolites to restrict Zn2+ availability in many microbial species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AAS was funded by a Government of Ireland Postdoctoral Fellowship from the Irish Research Council (GOIPD/2015/516). SFD was funded by a SPUR studentship from Maynooth University. Mass spectrometry facilities were funded by Science Foundation Ireland (12/RI/2346 (3)) and the Irish Higher Education Authority.References
- J. W. Bok, D. Chung, S. A. Balajee, K. A. Marr, D. Andes, K. F. Nielsen, J. C. Frisvad, K. A. Kirby and N. P. Keller, Infect. Immun., 2006, 74, 6761–6768 CrossRef PubMed.
- R. A. Owens, G. O’Keeffe, K. A. O’Hanlon, L. Gallagher and S. Doyle, in Human Pathogenic Fungi: Molecular Biology and Pathogenic Mechanisms, ed. D. J. Sullivan and G. P. Moran, Caister Academic Press, 2014, pp. 163–194 Search PubMed.
- B. Li, W. J. Wever, C. T. Walsh and A. A. Bowers, Nat. Prod. Rep., 2014, 31, 905–923 RSC.
- S. K. Dolan, G. O’Keeffe, G. W. Jones and S. Doyle, Trends Microbiol., 2015, 23, 419–428 CrossRef PubMed.
- D. H. Scharf, N. Remme, T. Heinekamp, P. Hortschansky, A. A. Brakhage and C. Hertweck, J. Am. Chem. Soc., 2010, 132, 10136–10141 CrossRef PubMed.
- B. Li and C. T. Walsh, Biochemistry, 2011, 50, 4615–4622 CrossRef PubMed.
- M. Schrettl, S. Carberry, K. Kavanagh, H. Haas, G. W. Jones, J. O’Brien, A. Nolan, J. Stephens, O. Fenelon and S. Doyle, PLoS Pathog., 2010, 6, e1000952 Search PubMed.
- R. A. Owens, G. O’Keeffe, E. B. Smith, S. K. Dolan, S. Hammel, K. J. Sheridan, D. A. Fitzpatrick, T. M. Keane, G. W. Jones and S. Doyle, Eukaryotic Cell, 2015, 14, EC.00055 CrossRef PubMed.
- S. K. Dolan, R. A. Owens, G. O’Keeffe, S. Hammel, D. A. Fitzpatrick, G. W. Jones and S. Doyle, Chem. Biol., 2014, 21, 999–1012 CrossRef PubMed.
- B. Li, R. R. Forseth, A. A. Bowers, F. C. Schroeder and C. T. Walsh, ChemBioChem, 2012, 1–7 Search PubMed.
- P. H. Bernardo, N. Brasch, C. L. L. Chai and P. Waring, J. Biol. Chem., 2003, 278, 46549–46555 CrossRef PubMed.
- S. Carberry, E. Molloy, S. Hammel, G. O’Keeffe, G. W. Jones, K. Kavanagh and S. Doyle, Fungal Genet. Biol., 2012, 49, 302–312 CrossRef PubMed.
- R. A. Cramer, M. P. Gamcsik, R. M. Brooking, L. K. Najvar, W. R. Kirkpatrick, T. F. Patterson, C. J. Balibar, J. R. Graybill, J. R. Perfect, S. N. Abraham and W. J. Steinbach, Eukaryotic Cell, 2006, 5, 972–980 CrossRef PubMed.
- G. O’Keeffe, S. Hammel, R. A. Owens, T. M. Keane, D. A. Fitzpatrick, G. W. Jones and S. Doyle, BMC Genomics, 2014, 15, 894 CrossRef PubMed.
- S. K. Dolan, T. Bock, V. Hering, R. A. Owens, G. W. Jones, W. Blankenfeldt and S. Doyle, Open Biol., 2017, 7, 160292 CrossRef PubMed.
- P. Waring, A. Sjaarda and Q. H. Lin, Biochem. Pharmacol., 1995, 49, 1195–1201 CrossRef PubMed.
- D. M. Vigushin, N. Mirsaidi, G. Brooke, C. Sun, P. Pace, L. Inman, C. J. Moody and R. C. Coombes, Med. Oncol., 2004, 21, 21–30 CrossRef PubMed.
- K. M. Cook, S. T. Hilton, J. Mecinovic, W. B. Motherwell, W. D. Figg and C. J. Schofield, J. Biol. Chem., 2009, 284, 26831–26838 CrossRef PubMed.
- K. M. Reece, E. D. Richardson, K. M. Cook, T. J. Campbell, S. T. Pisle, A. J. Holly, D. J. Venzon, D. J. Liewehr, C. H. Chau, D. K. Price and W. D. Figg, Mol. Cancer, 2014, 13, 91 CrossRef PubMed.
- L. Lauinger, J. Li, A. Shostak, I. A. Cemel, N. Ha, Y. Zhang, P. E. Merkl, S. Obermeyer, N. Stankovic-Valentin, T. Schafmeier, W. J. Wever, A. A. Bowers, K. P. Carter, A. E. Palmer, H. Tschochner, F. Melchior, R. J. Deshaies, M. Brunner and A. Diernfellner, Nat. Chem. Biol., 2017, 13, 709–714 CrossRef PubMed.
- A. Abad, J. V. Fernández-Molina, J. Bikandi, A. Ramírez, J. Margareto, J. Sendino, F. Luis Hernando, J. Pontón, J. Garaizar and A. Rementeria, Rev. Iberoam. Micol., 2010, 27, 155–182 CrossRef PubMed.
- C. Coméra, K. André, J. Laffitte, X. Collet, P. Galtier and I. Maridonneau-Parini, Microbes Infect., 2007, 9, 47–54 CrossRef PubMed.
- J. J. Bennison, R. M. Nottingham, E. L. Key and J. J. Parkins, N. Z. Vet. J., 2010, 58, 201–206 CrossRef PubMed.
- J. C. Woodcock, W. Henderson and C. O. Miles, J. Inorg. Biochem., 2001, 85, 187–199 CrossRef PubMed.
- M. A. Moreno, O. Ibrahim-Granet, R. Vicentefranqueira, J. Amich, P. Ave, F. Leal, J.-P. Latge and J. A. Calera, Mol. Microbiol., 2007, 64, 1182–1197 CrossRef PubMed.
- J. Amich, R. Vicentefranqueira, E. Mellado, A. Ruiz-Carmuega, F. Leal and J. A. Calera, Cell. Microbiol., 2014, 16, 548–564 CrossRef PubMed.
- R. Vicentefranqueira, J. Amich, P. Laskaris, O. Ibrahim-Granet, J. P. Latge, H. Toledo, F. Leal and J. A. Calera, Front. Microbiol., 2015, 6, 160 Search PubMed.
- S. Yasmin, B. Abt, M. Schrettl, T. A. A. Moussa, E. R. Werner and H. Haas, Fungal Genet. Biol., 2009, 46, 707–713 CrossRef PubMed.
- S. Doyle, G. W. Jones and S. K. Dolan, Fungal Biol., 2018, 122, 214–221 CrossRef PubMed.
- A. N. Chan, A. L. Shiver, W. J. Wever, S. Z. A. Razvi, M. F. Traxler and B. Li, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2717–2722 CrossRef PubMed.
- E. B. Smith, S. K. Dolan, D. A. Fitzpatrick, S. Doyle and G. W. Jones, Microb. Cell, 2016, 3, 120–125 CrossRef PubMed.
- N. M. Moloney, R. A. Owens, P. Meleady, M. Henry, S. K. Dolan, E. Mulvihill, M. Clynes and S. Doyle, J. Proteomics, 2016, 136, 99–111 CrossRef PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Mol. Cell. Proteomics, 2014, 13, 2513–2526 Search PubMed.
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein, T. Geiger, M. Mann and J. Cox, Nat. Methods, 2016, 13, 731–740 CrossRef PubMed.
- S. Priebe, C. Kreisel, F. Horn, R. Guthke and J. Linde, Bioinformatics, 2015, 31, 445–446 CrossRef PubMed.
- A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen and M. Mann, Nat. Protoc., 2007, 1, 2856–2860 CrossRef PubMed.
- L. Manzanares-Miralles, Ö. Sarikaya-Bayram, E. B. Smith, S. K. Dolan, Ö. Bayram, G. W. Jones and S. Doyle, J. Proteomics, 2016, 131, 149–162 CrossRef PubMed.
- L. Gallagher, R. A. Owens, G. O’Keeffe, S. K. Dolan, M. Schrettl, K. Kavanagh, G. Jones and S. Doyle, Eukaryotic Cell, 2012, 11, 1226–1238 CrossRef PubMed.
- J. Amich, R. Vicentefranqueira, F. Leal and J. A. Calera, Eukaryotic Cell, 2010, 9, 424–437 CrossRef PubMed.
- K.-S. Shin, N.-J. Kwon and J.-H. Yu, Curr. Genet., 2009, 55, 631–641 CrossRef PubMed.
- K. A. O’Hanlon, T. Cairns, D. Stack, M. Schrettl, E. M. Bignell, K. Kavanagh, S. M. Miggin, G. O’Keeffe, T. O. Larsen and S. Doyle, Infect. Immun., 2011, 79, 3978–3992 CrossRef PubMed.
- M. F. Mabanglo, M. A. Hast, N. B. Lubock, H. W. Hellinga and L. S. Beese, Protein Sci., 2014, 23, 289–301 CrossRef PubMed.
- H.-W. Park, S. R. Boduluri, J. F. Moomaw, P. J. Casey and L. S. Beese, Science, 1997, 275, 1800–1805 CrossRef PubMed.
- A. Kocyła, A. Pomorski and A. Krezel, J. Inorg. Biochem., 2015, 152, 82–92 CrossRef PubMed.
- C. Davis, N. Gordon, S. Murphy, I. Singh, K. Kavanagh, S. Carberry and S. Doyle, Anal. Bioanal. Chem., 2011, 401, 2519–2529 CrossRef PubMed.
- J. J. Coleman, S. Ghosh, I. Okoli and E. Mylonakis, PLoS One, 2011, 6, e25321 Search PubMed.
- W.-L. Liang, X. Le, H.-J. Li, X.-L. Yang, J.-X. Chen, J. Xu, H.-L. Liu, L.-Y. Wang, K.-T. Wang, K.-C. Hu, D.-P. Yang and W.-J. Lan, Mar. Drugs, 2014, 12, 5657–5676 CrossRef PubMed.
- C. C. Staats, L. Kmetzsch, A. Schrank and M. H. Vainstein, Front. Cell. Infect. Microbiol., 2013, 3, 65 Search PubMed.
- P. Wiemann, C. Guo, J. M. Palmer, R. Sekonyela and C. C. C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17065–17070 CrossRef PubMed.
- S. Dhingra, A. L. Lind, H. C. Lin, Y. Tang, A. Rokas and A. M. Calvo, PLoS One, 2013, 8, 1–16 Search PubMed.
- N. Grahl, S. Puttikamonkul, J. M. Macdonald, M. P. Gamcsik, L. Y. Ngo, T. M. Hohl and R. A. Cramer, PLoS Pathog., 2011, 7, e1002145 Search PubMed.
- T. S. Norton, Q. Al Abdallah, A. M. Hill, R. V. Lovingood and J. R. Fortwendel, Virulence, 2017, 8, 1401–1416 CrossRef PubMed.
- M. Kroll, F. Arenzana-Seisdedos, F. Bachelerie, D. Thomas, B. Friguet and M. Conconi, Chem. Biol., 1999, 6, 689–698 CrossRef PubMed.
- A. Paugam, C. Creuzet, J. Dupouy-Camet and P. Roisin, Parasitol. Res., 2002, 88, 785–787 CrossRef PubMed.
- R. W. Wheatley, K. K. S. Ng and M. Kapoor, Arch. Biochem. Biophys., 2016, 590, 125–137 CrossRef PubMed.
- J. Amich, M. Dümig, G. O’Keeffe, J. Binder, S. Doyle, A. Beilhack and S. Krappmann, Infect. Immun., 2016, IAI.01124 Search PubMed.
- R. D. Eichner, P. Waring, A. M. Geue, A. W. Braithwaite and A. Mullbacher, J. Biol. Chem., 1988, 263, 3772–3777 Search PubMed.
- A. M. Hurne, C. L. L. Chai and P. Waring, J. Biol. Chem., 2000, 275, 25202–25206 CrossRef PubMed.
- A. Müller, J. Eller, F. Albrecht, P. Prochnow, K. Kuhlmann, J. E. Bandow, A. J. Slusarenko and L. I. O. Leichert, J. Biol. Chem., 2016, 291, 11477–11490 CrossRef PubMed.
- E. Cho, J.-J. Hwang, S.-H. Han, S. J. Chung, J.-Y. Koh and J.-Y. Lee, Neurotoxic. Res., 2010, 17, 156–166 CrossRef PubMed.
- S. J. Lulloff, B. L. Hahn and P. G. Sohnle, J. Lab. Clin. Med., 2004, 144, 208–214 CrossRef PubMed.
- A. Krężel and W. Maret, Arch. Biochem. Biophys., 2016, 611, 3–19 CrossRef PubMed.
- A. E. Martell and R. M. Smith, Critical Stability Constants, Plenum Press, New York, 1974 Search PubMed.
- P. Laskaris, A. Atrouni, J. A. Calera, C. d’Enfert, H. Munier-Lehmann, J.-M. Cavaillon, J.-P. Latge and O. Ibrahim-Granet, Antimicrob. Agents Chemother., 2016, 60, 5631–5639 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00052b |
| This journal is © The Royal Society of Chemistry 2018 |