
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitative proteomics and systems analysis of cultured H9C2 cardiomyoblasts during differentiation over time supports a ‘function follows form’ model of differentiation†
Cynthia
Kankeu
a,
Kylie
Clarke
a,
Delphi
Van Haver
 bcd,
Kris
Gevaert
bc,
Francis
Impens
bcd,
Anna
Dittrich
e,
H. Llewelyn
Roderick
a,
Egle
Passante‡
f and
Heinrich J.
Huber§
*agh
bcd,
Kris
Gevaert
bc,
Francis
Impens
bcd,
Anna
Dittrich
e,
H. Llewelyn
Roderick
a,
Egle
Passante‡
f and
Heinrich J.
Huber§
*agh
aDepartment of Cardiovascular Sciences, KU Leuven, 3000 Leuven, Belgium
bDepartment of Biochemistry, Ghent University, Ghent, Belgium
cVIB-UGent Center for Medical Biotechnology, Ghent, Belgium
dVIB Proteomics Core, Ghent, Belgium
eDepartment for Systems Biology, Institute of Biology, Otto-von-Guericke University Magdeburg, Germany
fSchool of Pharmacy and Biomedical Sciences, Univ. of Central Lancashire, Preston, UK
gLaboratory for Systems Theory and Automatic Control, Institute for Automation Engineering (IFAT), Otto-von-Guericke University Magdeburg, 39106 Magdeburg, Germany. E-mail: Heinrich.Huber@ovgu.de; Tel: +49 0391 6750219
hTarget Discovery Research, Boehringer Ingelheim Pharmaceutical Research and Development Centre Biberach, Germany
First published on 17th May 2018
Abstract
The rat cardiomyoblast cell line H9C2 has emerged as a valuable tool for studying cardiac development, mechanisms of disease and toxicology. We present here a rigorous proteomic analysis that monitored the changes in protein expression during differentiation of H9C2 cells into cardiomyocyte-like cells over time. Quantitative mass spectrometry followed by gene ontology (GO) enrichment analysis revealed that early changes in H9C2 differentiation are related to protein pathways of cardiac muscle morphogenesis and sphingolipid synthesis. These changes in the proteome were followed later in the differentiation time-course by alterations in the expression of proteins involved in cation transport and beta-oxidation. Studying the temporal profile of the H9C2 proteome during differentiation in further detail revealed eight clusters of co-regulated proteins that can be associated with early, late, continuous and transient up- and downregulation. Subsequent reactome pathway analysis based on these eight clusters further corroborated and detailed the results of the GO analysis. Specifically, this analysis confirmed that proteins related to pathways in muscle contraction are upregulated early and transiently, and proteins relevant to extracellular matrix organization are downregulated early. In contrast, upregulation of proteins related to cardiac metabolism occurs at later time points. Finally, independent validation of the proteomics results by immunoblotting confirmed hereto unknown regulators of cardiac structure and ionic metabolism. Our results are consistent with a ‘function follows form’ model of differentiation, whereby early and transient alterations of structural proteins enable subsequent changes that are relevant to the characteristic physiology of cardiomyocytes.
Introduction
Studying the cellular properties of cardiomyocytes and the process of their differentiation is essential for understanding cardiac molecular physiology and the mechanism by which the heart acquires its function. Additionally, due to the redeployment of the foetal gene programme and physiology in disease, studying cardiomyocyte differentiation is highly valuable to the understanding of disease mechanisms.1 To shed light on these questions, the rat cardiomyoblast cell line H9C2 has become a widely used tool in cardiovascular research2 and remains of broad interest, despite the recent emergence of other models such as inducible pluripotent stem cells3,4 and rodent-derived primary cardiomyocytes. The reason for the ongoing popularity of H9C2 cells is that they have well-characterised properties in terms of morphology and electrochemical physiology,5 can be easily cultured over long time periods and several passages, and enable comparison of results across labs. Moreover, for studying cardiac differentiation, an easy-to-use differentiation protocol to transform H9C2 cells into a cardiomyocyte-like phenotype (henceforth denoted as H9C2 CM) was established that involves their maintenance in minimal serum and exposure to retinoic acid for five days.6 Applying this differentiation protocol subsequently gave rise to several studies related to cardiac development and toxicology.7–10Surprisingly, however, studies that validate the appropriateness of H9C2 as a model for cardiomyocytes by analysing global gene or protein expression changes during their differentiation, and relating these changes to physiological cardiomyocyte function, are rather sparse.11–13 As one example, a recent iTRAQ-based mass spectrometry study by Lenco and colleagues questioned the applicability of H9C2 cells to cardiac research.13 Specifically, they identified a lack of sufficient expression of proteins essential for formation of striated muscle cells and muscular metabolism in H9C2 cardiomyoblasts compared to primary cardiomyocytes. However, their study did not consider (differentiated) H9C2 CM cells. In contrast, Branco and colleagues most recently investigated gene expression profiles in H9C2 CM cells that had been differentiated according to the protocol described above. They confirmed the presence of a cardiomyocyte-like phenotype by identifying mRNAs related to cardiac muscle and mitochondrial metabolism as being upregulated on a transcriptional level relative to those in H9C2.14 However, their study was restricted to investigating changes in the transcriptome, which does not always correlate with the functional proteome;15 therefore, their findings require support from a quantitative and systematic proteomics analysis. This requirement is further reinforced by the pervasive role of micro RNAs in the post-transcriptional regulation of gene expression in cardiomyocytes during differentiation and disease.16 In addition, to validate the appropriateness of H9C2 as a model for cardiomyocyte differentiation and to better understand the experiments performed in these cells, a more rigorous study is required. Such a study would investigate the interplay of differentially regulated proteins by a systems biology approach such as that in reference17 and by exploring the course of differentiation over time. Here, we therefore investigate how and in which temporal sequence H9C2 cells acquire an appropriate cardiomyocyte-like phenotype and physiology.
Results
H9C2 CM population demonstrates increased expression of cardiac structural markers and a cardiomyocyte-like cell shape
The process of H9C2 differentiation was assessed in cells that were differentiated according to the protocol described by Branco, in which cells were cultured in medium containing low serum supplemented with retinoic acid over a period of five days.7 On the 6th day of differentiation, H9C2 CM cells were considered to be differentiated (H9C2 CM D6) compared to cells maintained in standard culture medium without retinoic acid for 6 days (H9C2 D6). Using phase contrast microscopy, we then confirmed that the shapes of the H9C2 CM cells were elongated and partly poly-nucleated, while the H9C2 cells had spindle-like shapes (Fig. 1A-i). | ||
| Fig. 1 Characterisation of the H9C2 differentiation protocol revealing mixed phenotypes and length to diameter ratios. (A) Immunofluorescent detection of cardiac markers for H9C2 D6 cells (upper panels) and for H9C2 CM D6 cells (lower panels) differentiated according to Branco7 are depicted. Phase contrast imaging (left column (i)) reveals spindle-like shapes for H9C2 cells, while H9C2 CM cells are more elongated and partly poly-nucleated (arrows). MYH protein was more highly expressed in the H9C2 CM population (central column (ii)). Moreover, cardiac TNNI was barely expressed in H9C2 cells, while its expression was higher in H9C2 CM cells. There, its expression was predominantly found in the perinuclear region (green area) as demonstrated by confocal microscopy (right column (ii)). (B and C) Immunoblot analysis of MYH and cardiac TNNI confirmed their increased expression in H9C2 CM D6. The expression levels of both proteins were normalized to actin. Data represent the mean ± SD of 3 independent experiments. A one-sided t-test was applied using a significance level of 0.05. (D–F) Morphological analysis revealed that H9C2 CM D6 cells spread to a higher length (D) and a shorter diameter (E), leading to a significantly higher length to diameter ratio as a marker of the differentiated population (F). The individually coloured circles indicate independent differentiation experiments and H9C2 cells, respectively. A two-sided t-test was applied using a significance level of 0.01. (G and H) Numbers of H9C2 cells cultured on day 1 and 3 revealed a doubling time of about 48 h (G). This doubling time was determined by enumerating live cells using a counting chamber under a phase contrast microscope and by excluding dead cells, determined by trypan blue. Proliferation markedly slowed in H9C2 CM cells at days 4 to 7 after the end of the differentiation protocol (H). | ||
We further found that molecular markers associated with cardiomyocyte differentiation were also increased. Specifically, expression of myosin heavy chain (MYH) protein was increased after differentiation (Fig. 1A-ii,18). Further, we examined cardiac troponin I (TNNI), a key protein of the troponin complex that is important for cardiac muscle contraction.19 Because initial experiments using standard fluorescence microscopy suggested the enrichment of TNNI in the perinuclear region relative to the cytosol in H9C2 CM cells (results not shown), we investigated its precise location using confocal microscopy. Using YOPRO (491 nm)/TOPRO (642 nm) staining,20 we confirmed the perinuclear presence of TNNI in H9C2 CM cells (green YOPRO and red TOPRO staining for TNNI and the nucleus, respectively; Fig. 1A-iii). Increased MYH and TNNI protein expression in H9C2 CM cells was also confirmed on a cell population level by immunoblotting using specific antibodies (Fig. 1B and C).
We next characterised the morphological features of H9C2 cells exposed to the differentiation protocol compared to those under buffer control. We found that the H9C2 CM cells tended toward greater lengths and smaller diameters. The length to diameter ratios enabled us to distinguish both cell populations at a significance level of 0.01 (t-test), while length or diameter alone did not meet this criterion (Fig. 1D–F). We finally observed that the duplication time of H9C2 cells was two days, in line with previous results14 (Fig. 1G); meanwhile, after the differentiation protocol, the rate of proliferation decreased (Fig. 1H).
In summary, application of the described differentiation protocol resulted in changes in cell morphology and molecular markers consistent with previously described markers of cardiomyocyte differentiation. We therefore sought to determine whether the entire proteome of H9C2 cells becomes more cardiomyocyte-like following their differentiation.
Shotgun proteomics confirmed differential expression of cardiac markers, sphingolipid metabolism and cell growth between H9C2 and H9C2 CM
The above analysis revealed differential expression of typical cardiac markers and altered morphology between H9C2 CM D6 cells and H9C2 cells. To corroborate these results and to identify functional changes between the two phenotypes, we performed a quantitative proteomic analysis using shotgun LC-MS/MS with data-dependent acquisition21 on three biological replicates. In total, 2105 proteins were identified, of which 41 were found to be expressed at a statistically higher level following differentiation (t-test with FDR = 0.01 and S0 = 1), while 28 proteins were downregulated and hence more highly expressed in the H9C2 population (Fig. 2A and Table S1, ESI†). Proteins that are involved in cardiac muscle contraction, such as proteins of the troponin complex (TNNI1, TNNT2, TNNC1), myosin heavy chain subunits (MYH1, MYH3), myosin light chain subunits and other structural proteins (MYL4, MYL6B, MYLPF, TMP1), were upregulated in H9C2 CM D6 cells, confirming the validity of the differentiation protocol. | ||
Fig. 2 Shotgun proteomics and gene ontology analysis revealed early changes in cardiomyocyte structure. Shotgun proteomics and subsequent gene ontology analysis were used to compare H9C2 D0 cells with H9C2 CM D6 cells. (A) Label-free shotgun proteomics analysis revealed differential expression of proteins associated with cardiac myocyte structure, cell proliferation and metabolism. In total, 2105 proteins were quantified and used for t-testing to generate the volcano plot indicating the log2 (D6/D0) ratio on the X-axis and −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p value on the Y-axis for each protein. 41 proteins were significantly upregulated upon H9C2 differentiation, while 28 proteins were down-regulated (a t-test was performed with FDR = 0.01 and S0 = 1). Components of the MYH, along with troponins and proteins involved in lipid metabolism, were upregulated in H9C2 CM cells (proteins with red marks on the right). In turn, proteins related to cell growth, particularly those related to the helicase complex, were found to be significantly overexpressed in H9C2 (blue marks, left hand side). (B) Gene ontology analysis using GOrilla24 was performed on the gene name list from the shotgun proteomics analysis. The list was ranked according to the strength of the relative expression in H9C2 CM cells. The results confirmed the enrichment of the GO terms that relate to cardiac muscle differentiation and metabolism. p value on the Y-axis for each protein. 41 proteins were significantly upregulated upon H9C2 differentiation, while 28 proteins were down-regulated (a t-test was performed with FDR = 0.01 and S0 = 1). Components of the MYH, along with troponins and proteins involved in lipid metabolism, were upregulated in H9C2 CM cells (proteins with red marks on the right). In turn, proteins related to cell growth, particularly those related to the helicase complex, were found to be significantly overexpressed in H9C2 (blue marks, left hand side). (B) Gene ontology analysis using GOrilla24 was performed on the gene name list from the shotgun proteomics analysis. The list was ranked according to the strength of the relative expression in H9C2 CM cells. The results confirmed the enrichment of the GO terms that relate to cardiac muscle differentiation and metabolism. | ||
Finally, we performed gene ontology (GO) analysis using the GOrilla gene ontology analysis tool.22 We ranked the list of 69 proteins that exhibited statistically significant changes in expression during differentiation from the most upregulated to the most downregulated. In this way, we found differentially regulated functions related to cardiac muscle morphogenesis and sphingolipid metabolism (Fig. 2B). In summary, shotgun proteomics showed that the initial steps of cardiomyocyte differentiation include organization of the contraction machinery and the production of sphingolipids, which are essential for mitochondrial biosynthesis.23
Fractionation-based proteome and gene ontology analysis at D8 confirmed the persistence of muscle morphology development at early differentiation times
Motivated by the results of the shotgun proteomics analysis and to gain insight into the cardiomyocyte differentiation of H9C2 cells, we investigated proteome changes at other time points whilst also aiming to increase the overall coverage of the proteome. We therefore used a strong cation exchange SCX fractionation-based MS approach (3 independent runs24,25) that enabled deeper proteome coverage. We chose to differentiate H9C2 cells for 8 (H9C2 CM D8) and 13 days (H9C2 CM D13) to include processes that occur at both early (H9C2 CM D8) and late (H9C2 CM D13) stages of differentiation. The analyses of H9C2 CM D8 and H9C2 CM D6 days enabled us to gain further insight into the dynamics of early differentiation. By fractionation, we could significantly increase the proteome coverage. We then quantified 5143 proteins; 75 proteins were found to be significantly upregulated and 41 were found to be significantly downregulated at D8 of differentiation. Differential proteome analysis confirmed increased levels of structural cardiac muscle proteins such as troponins and myosins, together with downregulation of glutathione peroxidase 1 (GPX1); these were also found in H9C2 CM D6 and in H9C2 CM D8 (Fig. S1A, ESI†).The GO analysis, as described above, suggested that changes in GO functions relevant to cardiac muscle contraction and morphogenesis were preserved in H9C2 CM D8 compared to H9C2 CM D6 (Fig. S1B, ESI†), suggesting the relevance of these mechanisms especially at earlier time points of differentiation.
Fractionation-based proteomics revealed changes in metabolism and ionic transport at a later time point of differentiation
We next investigated later changes of the proteome of H9C2 cells during differentiation. We therefore investigated protein expression in H9C2 CM D13, which we compared with those in H9C2 CM D8 and H9C2 cells maintained in standard culture medium. Pairwise comparison of H9C2 CM D13 cells with H9C2 cells revealed 119 proteins as being significantly upregulated at this later time point of differentiation (t-test with FDR = 0.01 and S0 = 1), while 82 proteins were downregulated (Table S3 (ESI†), only annotated proteins shown). In contrast, no differential expression of proteins was observed between H9C2 CM D13 and H9C2 CM D8 (Fig. 3A). A subsequent principal component analysis revealed that two major principal components already explain ∼60% of protein variability. These results further demonstrated a clear clustering between biological replicates under the same conditions, validating our differentiation protocol. Moreover, a greater variability between replicates of H9C2 CM D13 versus H9C2 CM D8 and H9C2 cells was observed (Fig. 3B). While this increased heterogeneity prevented the identification of differentially expressed proteins between D8 and D13 at the standard significance level of this study (FDR = 0.01, S0 = 0.1), 11 upregulated and 6 downregulated proteins were revealed when the significance criterion was relaxed (FDR = 0.05, S0 = 0.1, Fig. S2, ESI†). | ||
| Fig. 3 Fractionation-based shotgun proteomics and gene ontology analysis for early versus late differentiated H9C2 CM cells. (A) The volcano plot comparing early differentiated H9C2 CM (H9C2 CM D8) and late differentiated H9C2 CM (H9C2 CM D13) cells indicates that no single protein was found to be differentially regulated with a p value of significance of 0.01. This is likely due to the higher variability of protein expression in the H9C2 CM D13 replicates (as demonstrated in panel B). (B) Principal component analysis was performed on all quantified proteins. Three biological replicates were performed for each condition, comprising H9C2 D0, H9C2 CM D8 and H9C2 CM D13. (C) Explorative gene ontology enrichment analysis employing GOrilla and using the list of proteins from the fractionation-based mass spectrometry analysis of H9C2 CM D13 vs. H9C2 CM D8. Proteins were ranked according to their relative expression at late versus early differentiation times in descending order; a rat gene background was used in GOrilla. GO functions associated with cation homeostasis, cation membrane transport and beta-oxidation were enriched, suggesting that changes in fatty acid metabolism and cardiac ion electrophysiology occur temporally after structural changes have occurred, as shown in Fig. 2B. | ||
Importantly, however, despite the fact that no significant changes between the expression of individual proteins in H9C2 CM D8 and H9C2 CM D13 were observed using a false discovery rate of 0.01 (FDR = 0.01, S0 = 0.1), we identified gene ontology enrichment by GOrilla. We therefore analysed a list ranked according to the expression changes (from highly upregulated to highly downregulated) of all 5143 proteins found using fractionated MS. Specifically, our results revealed highly enriched GO terms related to cation membrane transport, cation homeostasis and beta-oxidation at later time-points of differentiation (Fig. 3C). Our results suggest that these metabolic and cation homeostasis-related changes may be introduced subsequent to changes in muscle apparatus and sphingolipid production, which we found to be already enriched by H9C2 CM D8.
Two-way ANOVA and cluster analysis revealed eight clusters of co-regulated proteins
The previous GO term analysis suggests a different timing of cardiac muscle development compared to metabolic changes and cation homeostasis during differentiation of H9C2 cells. Encouraged by these findings, we considered another approach that allowed a more detailed analysis of the temporal evolution of proteome changes during differentiation on a statistically sounder basis. We specifically sought a method to circumvent the issue of high inter-sample variability observed in H9C2 CM D13, which hampered pairwise comparison of protein expression.We reasoned that protein expression upon differentiation evolves over time, but at different rates in the different cell samples, thereby increasing variability in protein expression over time. We surmised that by accounting for the proposed variance in the rate of differentiation, we can correct for the original lack of statistical significance between differentially expressed proteins. To this end, we performed two-way ANOVA on the intensities of all 5143 identified proteins in the differentiated group (D0 vs. pooled D8 and D13) and the differentiation time group (D0 vs. D8 vs. D13; Fig. 4A). We found 843 proteins to be significantly differentially expressed (p < 0.05). These proteins included some that showed significant expression changes either in the differentiation group (N = 748) or the differentiation time group (N = 650, wherein N = 547 proteins were common). These proteins were further subjected to hierarchical clustering. Through this analysis, we identified eight unique clusters describing co-regulated proteins that were up- or downregulated either early, late, continuously, or transiently upon differentiation (Fig. 4B).
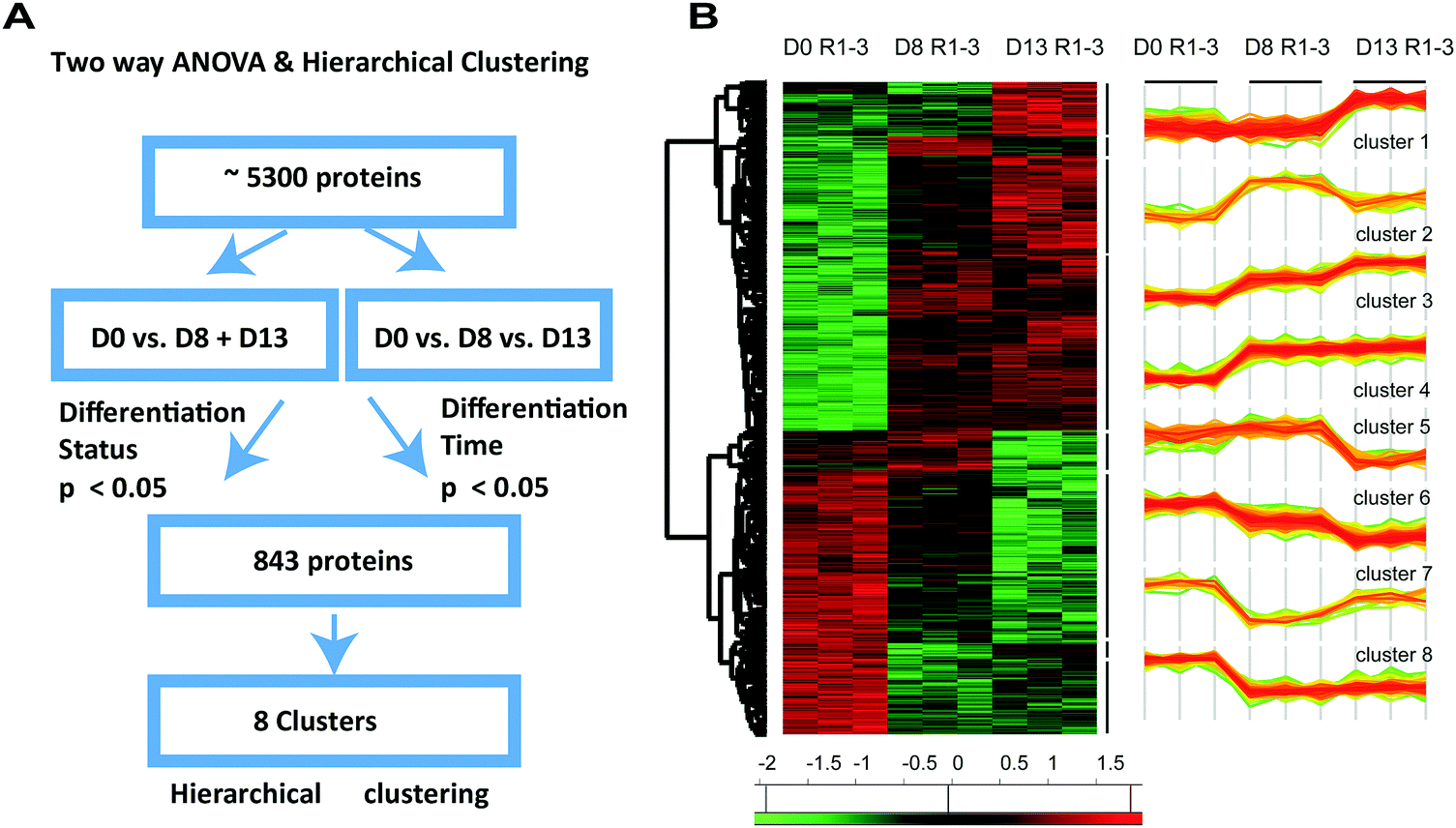 | ||
| Fig. 4 Two-way ANOVA and hierarchical clustering identified sets of statistically significant and co-regulated proteins. (A) To obtain a statistically interpretable pattern of how differentiation time influences differentiation status, two-way ANOVA was performed. We therefore pooled all 5143 identified proteins from H9C2 CM cells (D8 and D13), compared the resulting pool to proteins identified in H9C2 (D0), and analysed their expression vis-à-vis the differentiation times (D13 vs. D8 vs. D0). 843 proteins were identified that showed significant expression changes either in the differentiation group or in the differentiation time group. (B) These proteins were further subjected to unsupervised hierarchical clustering, which revealed eight clusters of co-regulated proteins over time. These eight clusters were defined by proteins that exhibited early, late, continuous and transient upregulation and downregulation of expression following differentiation. | ||
Reactome analysis of proteins from different clusters identified pathways related to different temporal regulation dynamics
Having identified that the proteins that were statistically significantly differentially expressed upon differentiation and according to time of differentiation segregated into 8 clusters based upon their pattern of expression, we analysed the pathways that are associated with these clusters. We subjected the proteins of each cluster to the Reactome pathway analysis tool.26 This allowed us to link the temporal regulation pattern to biological function. The results are shown in Fig. 5 and Fig. S3 (ESI†), wherein clusters from Fig. 4 were rearranged to emphasize the aspects of early vs. long-term and between transient vs. continuous protein regulation. We depicted graphs of the most differentially regulated pathways identified by Reactome. We therefore ranked the identified pathways according to their percentile enrichment, which we defined by the number of proteins from the cluster that were associated with the pathway divided by the total number of proteins related to that pathway by Reactome (both numbers are shown above each bar). | ||
| Fig. 5 Reactome pathway analysis of proteins associated to each cluster. Proteins associated with each cluster of Fig. 4 were linked to signalling pathways using the tool Reactome. Clusters were re-arranged for practical reasons. The output of Reactome was excerpted, and pathways were ranked according to their enrichment. This enrichment, defined by the number of proteins found in the cluster versus the total amount of proteins in the respective pathway, is depicted by the numbers above each bar. Black and grey bars show analyses for clusters with up- or downregulation, respectively, for (A) early, (B) late, (C) continuous and (D) transient regulation. Only pathways containing at least 2% of all proteins of the cluster (* 1% for late-downregulated proteins) were considered. We noted that proteins related to muscle contraction were regulated rather early, while those related to metabolism were regulated rather late. In turn, proteins contributing to extracellular matrix (ECM) organisation were downregulated early, while those relevant to the cell cycle showed transient downregulation. Further details are given in the text. | ||
The cluster comprising early upregulated proteins (C4) was enriched for pathways related to striated muscle and generic muscle contraction (Fig. 5A). In turn, the cluster of early downregulated proteins (C8) was enriched for proteins involved in elastic fiber formation and extracellular matrix (ECM) organization. Together, these results are consistent with the notion that the establishment of cardiomyocyte structure is the hallmark of early differentiation.
In turn, and consistent with our previous GO analysis, we found that proteins that were upregulated at later time points of differentiation (C13) were mainly associated with metabolic pathways (glucose, carbohydrates, fatty acid/beta-oxidation; Fig. 5B, black bars), while proteins that were downregulated at later time points were not significantly enriched for any pathway (Fig. 5B, grey bars). Therefore, the higher pathway enrichment for continuously and late-upregulated proteins compared to those associated with downregulation suggests that long term changes in cell phenotype during differentiation are governed by acquisition of new functions rather than by suppression of existing functions.
As a nuance, we also found proteins involved in metabolic pathways that were enriched within the cluster of continuously upregulated proteins, indicating that the activation of some metabolic functions may have already started at early time points of differentiation (Fig. 5C, black bars). Moreover, mRNA splicing and pre-processing were found to be enriched within the cluster of continuously downregulated proteins (Fig. 5C, black bars). Finally, while enrichment of pathways within the cluster of transiently upregulated proteins was low, we found that pathways related to cell cycle progression and gene expression were associated with the cluster of proteins that were only transiently downregulated. This relatively high proportion of transiently downregulated proteins related to replication in cardiomyocytes may suggest that once proliferation decreases, further changes towards a non-proliferation phenotype are no longer required (Fig. 5D).
Finally, we note that unlike the above GOrilla analysis, Reactome analysis did not reveal information on alterations of ion channels and homeostasis during differentiation. However, manual screening of the cluster results revealed some ion transport-related proteins in the cluster of continuously upregulated proteins (5 transporter proteins of 10 from all clusters; Data S1, ESI†).
Biochemical validation of selected proteins related to cardiomyocyte structure and metabolism
We finally confirmed our proteomics data by immunoblot analysis. We specifically focused on proteins that are involved in cardiac structure and metabolism, which were identified to have high expression values and high significance at least at one time point of differentiation (early or late). Having initially validated TNNI and MYH to be elevated in H9C2 CM (see Fig. 1), we did not further examine the high expression of troponin and myosin subunits that were identified in our proteomics datasets. For four other proteins, selected according to the criteria outlined above, we were able to source antibodies that robustly detected them and exhibited sufficient reproducibility with acceptable inter-experimental variability. For each of those proteins, we compared H9C2 CM D8 and H9C2 CM D13 cells with H9C2 cells that were cultured in parallel (Fig. 6). | ||
| Fig. 6 Immunoblot analysis of differentially expressed proteins. H9C2 CM D8, H9C2 CM D13 (+) and H9C2 cells (−) were maintained in their respective media before performing the analysis. Our proteins of interest were quantified in those cells by western blot. For normalisation of sample loading, total proteins were detected on the blot by a reversible total protein stain (lanes at left to each bar graph). The relative abundance of the proteins of interest between the test and control samples (H9C2 cells cultivated at the same time points D8 or D13, respectively) were then determined by normalising to the total protein. This approach aimed to minimise differentiation state effects. A significant upregulation (*p < 0.05, ***p < 0.01, one-sample t-test of log-transformed data with a hypothetical value of 0) for most proteins and time points was observed, and the results were compared to the proteomics in the Results section. The images are representative of 6 different experimental studies. (A) ACSL3, (B) CAV-3, (C) CA1, (D) SGCD with left and right panels depicting differentiation at D8 and D13, respectively. | ||
First, we quantified ACSL3 (long-chain-fatty-acid–CoA ligase 3), a protein that is involved in lipid biosynthesis and fatty acid degradation.27 From our proteomics analysis (Table 1), ACSL3 was present in cluster 3 of persistently upregulated proteins, with upregulation in H9C2 CM D8 at lower significance (FDR = 0.05) and in H9C2 CM D13 at higher significance (FDR = 0.01). Indeed, immunoblotting confirmed this significant increase at both time points compared to control cells.
| −log(p-value) | log2 diff/undiff | Gene symbol | Protein name | Function | Significance |
|---|---|---|---|---|---|
| D8 vs. D0 | |||||
| 3.15 | 1.14 | ACSL3 | Long-chain-fatty-acid–CoA ligase 3 | Metabolic | FDR: 0.05 |
| 1.64 | 3.09 | CAV-3 | Caveolin-3 | Structure | FDR: 0.01 |
| 1.84 | 1.76 | CA1 | Carbonic anhydrase 1 | Metabolic | FDR: 0.05 |
| 2.54 | 2.75 | SGCD | δ-Sarcoglycan | Structure | FDR: 0.01 |
| D13 vs. D0 | |||||
| 4.05 | 2.17 | ACSL3 | Long-chain-fatty-acid–CoA ligase 3 | Metabolic | FDR: 0.01 |
| 1.60 | 3.31 | CAV-3 | Caveolin-3 | Structure | FDR: 0.01 |
| 3.31 | 4.49 | CA1 | Carbonic anhydrase 1 | Metabolic | FDR: 0.01 |
| 2.11 | 3.19 | SGCD | δ-Sarcoglycan | Structure | FDR: 0.01 |
| D13 vs. D8 | |||||
| 3.54 | 1.03 | ACSL3 | Long-chain-fatty-acid–CoA ligase 3 | Metabolic | FDR: 0.05 |
| 0.16 | 0.22 | CAV-3 | Caveolin-3 | Structure | No sig. |
| 3.90 | 2.73 | CA1 | Carbonic anhydrase 1 | Metabolic | FDR: 0.05 |
| 0.25 | 0.44 | SGCD | δ-Sarcoglycan | Structure | No sig. |
We next focused on Caveolin-3 (CAV-3), a major building block of caveolae, which are specialized microdomains in the sarcolemma membranes of ventricular myocytes involved in regulation of cardiac signalling and function. CAV-3 contributes to the formation of these microdomains through association with several cardiac ion channels and signalling proteins.28 Consistent with our proteomics results, immunoblotting analysis identified significant upregulation of CAV-3 in H9C2 CM D8 and H9C2 CM D13 (Fig. 6B).
We then investigated carbonic anhydrase isoform I (CAI), a metabolic enzyme that is usually found in red blood cells and the gastrointestinal tract29 but, surprisingly, was identified as the fourth most significantly upregulated protein in H9C2 CM D13 vs. H9C2 cells and was also identified as being upregulated in H9C2 CM D8 (Tables S2 and S3, ESI†). Indeed, immunoblotting analysis confirmed significant upregulation at both time points (Fig. 6C). These results indicate that this enzyme may have a yet-to-be-determined role in cardiomyocyte differentiation or function.
We finally investigated the structural protein δ-sarcoglycan (SGCD), a member of the sarcoglycan family, which are single-pass transmembrane proteins localized at the sarcolemmas of muscle fibers.30 Here, immunoblot analysis confirmed a statistically significant upregulation in H9C2 CM D8, as identified by our proteomics data. In contrast, no altered expression in H9C2 CM D13 was identified, which is contrary to the proteomics results but consistent with the association of this protein with the transiently regulated cluster as identified by our cluster analysis.
Discussion
Cellular differentiation involves transformations in the size, morphology, metabolic activity and signal transduction of a cell to acquire the characteristics of its mature phenotype.31 These cellular properties are ultimately determined by changes in the functional proteome. We therefore analyzed the temporal changes of the proteome of H9C2 cells during differentiation to provide a compendium for this frequently used cellular system to study cardiomyocyte biochemistry. Using two independent analytical approaches, our results support a ‘function follows form’ model, whereby initial changes in the proteome provide the basic morphology and contraction apparatus for later development of functions that establish cardiac physiology.First, gene ontology analysis using the GOrilla tool provided us with an overview of the biological functions that are enriched during early and late stages of H9C2 differentiation. This analysis extracts statistically relevant relationships that are included in the interrelation between the identified proteins and their expression, even if some of the proteins are not statistically regulated by themselves. Our results suggested that at earlier times of differentiation, H9C2 CM cells upregulate cardiac structural proteins (such as troponins and myosins) and initiate pathways involved in sphingolipid catalysis and synthesis. Sphingolipids are building blocks of the mitochondrial membranes, where they exert various structural and function-related tasks.23,32 Hence, their production can be seen as a proxy for the generation of mitochondria at early time points. Our results therefore suggest that by producing mitochondria, cardiomyocyte differentiation lays the foundation for enabling beta-oxidation of fatty acids, which indeed was detected at later stages of differentiation by our GO analysis. This reasoning is supported by the observation that mitochondrial mass increases during H9C2 cell differentiation33 and that increased mitochondrial mass is a prerequisite to address the high energy need of H9C2 CM cells.34 Similarly, elevation of cardiac-specific ion transporters was suggested to occur at later time points. Elevation of these transporters as a consequence of H9C2 cell differentiation has been previously reported; however, the concrete timing of their elevation was not investigated in detail.3
As a second approach, we combined two-way ANOVA with a hierarchical cluster analysis. By this approach, we relaxed the stringency for effect detection (while retaining it within accepted limits), which was necessary to account for the detected increase in intra-sample heterogeneity with duration of differentiation. Indeed, this observed heterogeneity is consistent with studies in human embryonic stem cell lines, suggesting that genetic differences progressively amplify during cardiac differentiation.35,36 Our two-way ANOVA approach enabled us to identify proteins that are differentially regulated with respect to differentiation status and timing while identifying eight clusters of co-regulated proteins. Proteins associated with each cluster were then subjected to Reactome pathway analysis, which gave information for each of the clusters on the pathways involved during differentiation at different time points. While the results of both approaches partly overlap, comparison of the two approaches may suffer from the occasionally inconsistent nomenclature of pathway and function terms used by GOrilla and Reactome. Indeed, while the GO analysis revealed regulation of ion transporter processes between H9C2 CM D8 and H9C2 CM D13, Reactome analysis did not prominently highlight these changes. One explanation for this discrepancy is a failure of the Reactome analysis to perform appropriate pathway categorization. More likely, however, the cluster analysis did not detect statistical significance on an individual gene level due to the greater sample variability in H9C2 CM D13 compared to H9C2 CM D8. In contrast, our GO analysis, which shifts away from an individual gene-oriented view to a gene group-based analysis, seems to be more suitable in situations where the individual sample variation is high.37 However, we note that the results of both analyses were clearly consistent with respect to the fact that the development of cardiac muscle morphology preceded changes in metabolism.
The results of our study are consistent with a recent gene expression profiling study of H9C2 cell differentiation by Branco et al.14 in which H9C2 cells were differentiated according to the same protocol used here. Branco and colleagues showed upregulation of mRNAs that encode structural cardiac and metabolic proteins in H9C2 CM cells. Our study adds to this by confirming that these processes are also translated at the protein level and by explaining how the gain of cardiomyocyte function evolves over time. Consistent with the study of Branco and colleagues, we found that lactate dehydrogenase subunit B (LDHB) is significantly upregulated and glutathione peroxidase 1 (GPX1) is significantly downregulated at both early and late differentiation time points. The increased expression of LDHB may enable increased conversion of lactate to pyruvate, hence indicating altered pyruvate/lactate metabolism in response to differentiation.38 In contrast to this, the consequences of the decreased expression of GPX1 in H9C2 CM cells may be more subtle. As GPX1 is often regarded as a harbinger of oxidative stress,39 decreased GPX1 in H9C2 CM cells may be an indicator of decreased anti-oxidant potential; thus, H9C2 CM cells, and potentially also mature primary cardiomyocytes, may be more susceptible to oxidative stress.40 However, we remark that it was not possible to validate the protein regulations of LDHB and GPX1 found in our proteomics analysis by immunoblotting due to the lack of antibody specificity and high variability of the respective antibody signals between replicates (similar to the study by Branco et al.14 in the case of LDHB, results not shown).
Despite the restriction of sourcing appropriate rat antibodies for interesting but nonstandard proteins, we provided several independent validations of structural and potential functional proteins relevant to cardiomyocyte function. Thus, in addition to the validation of the classical cardiac markers TNNI and MYH, we also investigated ACSL3, CAV-3, CA1 and SGCD. As such, the metabolic enzyme ACSL3 was found to be persistently upregulated in the proteomics study and immunoblot validation; it most likely addresses the high energy need in more mature cardiomyocytes. In turn, due to its involvement in lipid synthesis,27 ACSL3 may govern important cardiomyocyte-relevant anabolic processes such as de novo production of mitochondria. To this end, sphingolipid production was already identified as a major function in our gene ontology analysis at earlier time points. Similarly, the proteomics results showing upregulation of the structural proteins SGCD and CAV-3 were previously confirmed at early time points. It has been shown that both proteins are essential for maintaining cardiac structure and that their expression attenuates cardiac dystrophy.41,42 Moreover, CAV-3 has been shown to associate with several cardiomyocyte-membrane ion channels and to be involved in key cardiomyocyte and muscle-specific functions.43 Finally, we also identified CA1, an enzyme that catalyses the rapid interconversion of carbon dioxide and water to bicarbonate and protons (or vice versa), as being highly significantly upregulated, particularly at later time points of differentiation. In addition to CA1, which is key to proton production, other CA isoforms have also been found to be associated with proton exchanger channels and their functions;44 this suggests that CA1 plays a role in cardiac pH regulation. As tight pH regulation is essential for appropriate function of cardiac myofilaments,45 it is therefore tempting to speculate that CA1 has a yet undiscovered role in mediating cardiac contraction or during its malignant deregulation.
We further identified TNNI, one of the three canonical troponin isoforms, as a marker that distinguishes cardiomyocytes from other muscle cells. Interestingly, confocal microscopy suggested that cardiac TNNI was not evenly distributed in the cytoplasm but enriched in the perinuclear area. Consistent with our results, Franklin et al.46 employed mass spectrometry to demonstrate that troponins are present in the nuclei of neonatal rat ventricular cardiomyocytes. Our data further support previous findings of Bergmann et al.47 that suggest a perinuclear location of TNNI.48 While the exact function of perinuclear TNNI is elusive, a potential role of perinuclear cardiac troponins in cell senescence and, hence, in age-related diseases has been proposed.49
Our study has some limitations. First, we did not focus on phosphoproteomics changes. However, while phosphorylation events contribute to signalling, such as the induction of differentiation, these phosphorylation events are generally transient in nature and, with decreasing initial stimulus, would likely be undetected even at day 6. Indeed, an initial shotgun phosphoproteomics approach at day 6 of differentiation was vastly unsuccessful in detecting alterations in the phosphorylation of enzymes related to cardiomyocyte structure or metabolism (results not shown). This view of the lack of influence on phosphorylation at the investigated time point is supported by a recent study by one of our co-authors (LHR). They examined the temporal profile of ERK phosphorylation and immediate gene activation during hypertrophic remodelling in neonatal myocytes and found them be transient, subsiding after 15 to 30 min.50 Similar results have been observed for many other signalling pathways in cardiac hypertrophic remodelling.51 In addition, despite increasing proteome coverage using a fractionation approach, leading to the identification of over 5000 proteins in H9C2 cells, low-abundant proteins such as those related to signal transduction are more difficult to detect compared to those related to morphology or metabolism. As such, these proteins require substantial enrichment for detection.52 This leads to an unavoidable bias toward highly expressed proteins, which is inherent in every current proteomics approach. However, we conclude that proteomic coverage enables the detection and analysis of changes in proteins that govern cardiomycyte structure, function and metabolism. In summary, we confirmed that the applied differentiation protocol drives H9C2 cells towards a cardiac muscle phenotype; moreover, we suggested a temporal differentiation sequence that first establishes the necessary morphology for subsequent metabolic changes.
Methods
Cell culture
H9C2 cells (Sigma, Leuven, Belgium) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, 100 U per ml penicillin and 100 μg per ml streptomycin in humidified air (CO2 5%) at 37 °C. The culture medium was changed every 2 days, and the cells were split upon reaching around 80% confluence to avoid loss of differentiation potential. To differentiate H9C2 cells, they were cultured 5 days in DMEM supplemented with 1% FBS and 1 μM of trans-retinoic acid (ATRA). ATRA was dissolved in DMSO, and the aliquots were stored at −20 °C in the dark to prevent loss of activity.After passaging, H9C2 cells were allowed to adhere overnight in DMEM supplemented with 10% FBS in 75 cm2 flasks at a density of 4666 cells per cm2 for differentiation conditions or 666 cells per cm2 for control conditions. Addition of ATRA to the medium was performed daily for 5 days. After the 5th day, H9C2 CM cells were maintained in culture in 1% FBS-containing medium for 1, 3 or 8 additional days without ATRA. Those cells were respectively denoted as H9C2 CM D6, H9C2 CM D8 and H9C2 CM D13. H9C2 cells were also maintained in culture for 1, 6, 8 or 13 days and were respectively named H9C2 D0, H9C2 D6, H9C2 D8 and H9C2 D13.
Proliferation assay
To determine the doubling times of H9C2 and H9C2 CM, cells were counted after 1, 2, 4 and 7 days in culture. To this end, the supernatant of the cells was collected in a falcon tube. Subsequently, the cells were washed with PBS, trypsinised and combined with the cells from the supernatant. The cells were then centrifuged at 0.2 rcf for 5 min and the supernatant was discarded. The cells were resuspended in PBS and stained with trypan blue to exclude dead cells. Cell proliferation was determined by counting living cells using a counting chamber under a phase contrast microscope (Axiovert 200M inverted microscope, Carl Zeiss NV, Jena, Germany) and by excluding dead cells.Immunoblotting and densitometry
Cellular lysates were prepared from cells harvested from the media and combined with the trypsinised adherent cells, then washed in PBS and re-suspended in lysis buffer (50 mM Tris HCl, pH 7.6; 150 mM NaCl; 0.1% SDS; 0.1% deoxycholate; 1% Triton X100; 1 mM sodium orthovanadate; 1 mM sodium fluoride) for 30 min at 4 °C. Cells were centrifuged at 15700 rcf for 10 min. The protein concentration of samples was determined by the BCA assay (Thermo Fisher Scientific, Ghent, Belgium). Following denaturation at 90 °C for 5 min in Laemmli sample buffer, equivalent amounts of protein (25 μg) were separated by electrophoresis in 8%, 10% or 12% SDS–polyacrylamide gels (SDS–PAGE). The resolved proteins were then transferred to nitrocellulose membranes (0.2 μ pore size, Amersham GE Healthcare, Brussels, Belgium). The membranes were blocked in 5% milk/PBS for 1 h at room temperature and incubated overnight at 4 °C with antibodies directed against MYH (1:1000, Abcam, Cambridge, UK, ab185967) and TNNI (1:1000, Abcam, Cambridge, UK, ab56357). After washing the membranes 3 times with TBST (50 mM Tris–HCl, pH 8; 100 mM NaCl and 0.1% Tween 20), they were further incubated with horseradish peroxidase (HRP) conjugated secondary antibodies, goat anti-mouse IgG (1:10.000, Abcam, Cambridge, UK, ab6789) and goat anti-rabbit (1:10.000, Abcam, Cambridge, UK, ab6721) for 1 h at room temperature. Immunoreactive bands were detected by enhanced chemiluminescence (Amersham GE Healthcare, Brussels Belgium). Expression of proteins was determined by densitometry of the bands using Image Lab software (Bio-Rad, Temse, Belgium).
For validation of ACSL3, CAV-3, CA1 and SGCD, protein expression was determined by fluorescence imaging of immunoreactive bands. To normalize for protein expression between samples, total proteins on the blot were stained using the LICOR REVERT (Leusden, Netherlands) total protein stain kit prior to immunodetection of proteins of interest according to the manufacturer's instructions. After removal of this stain, the membranes were blocked in 5% milk/PBS for 1 h at room temperature and then incubated overnight at 4 °C with antibodies directed against CAV-3 (1:1000, BD Transduction, Erembodegem, Belgium, 610420), CA I (1:5000, Abcam, Cambridge, UK, ab108367), SGCD (1:1000, Cambridge, UK, orb247720), and ACSL3 (1:1000, Abcam, Cambridge, UK, ab151959). After washing the membranes 3 times with TBST (50 mM Tris–HCl, pH 8; 100 mM NaCl and 0.1% Tween 20), they were further incubated with fluorescently-conjugated secondary antibodies: donkey anti-mouse IgG 800 (1:10000, LICOR, Leusden, Netherlands, 925-32210) and Alexa fluor donkey anti-rabbit IgG 680 (1:10.000, Invitrogen, Merelbeke, Belgium, A10043) for 1 h at room temperature. The membranes were washed 3 times with TBST (50 mM Tris–HCl, pH 8; 100 mM NaCl and 0.1% Tween 20) and once with TBS (50 mM Tris–HCl, pH 8; 100 mM). The bands were visualized using a LICOR – Odyssey CLX imaging system, and densitometry was performed using Image Studio software (LICOR, Leusden, Netherlands). The expression levels of proteins were first normalized to the total protein stain and then normalized to H9C2 cells of the same passage and at the same time of culturing (D8 and D13). After logarithmic transformation of the data, a one sample t-test with a hypothetical value of 0 was used to establish statistical significance at a p-value < 0.05, unless indicated otherwise.
Immunocytochemistry
H9C2 CM and H9C2 cells were seeded on coverslips in 6-well plates at numbers of 1066 and 1600 cells per cm2. After overnight incubation, the cells were washed with PBS and fixed in PFA for 20 min. The fixed cells were washed once with PBS and permeabilized with 0.1% Triton X-100 for 15 min. The permeabilized cells were washed 3 times with PBS/0.5% BSA. The cells were blocked for 1 h with PBS/0.5% BSA, then incubated separately with primary antibodies against mouse anti-cardiac MYH (1:1000, Abcam Cambridge, UK, ab185967) or rabbit anti-cardiac TNNI (1:1000, Abcam, Cambridge, UK, ab56357) 1:50 in blocking solution overnight at 4 °C. The cells were washed 3 times for 5 min with TBST. Subsequently, secondary antibodies were added (anti-mouse Alexa Fluor 647, ab150127 and anti-rabbit Alexa Fluor 488, ab150077, 1:100 in blocking solution, Sigma, Leuven, Belgium) for 2 h at room temperature. The coverslips were mounted on glass slides using ProLong anti-fade reagent (Life Technologies, Brussels, Belgium). Cells stained with antibodies against MYH were observed by fluorescence microscopy using a Zeiss microscope with appropriate filter sets (Axiovert 200M inverted microscope, Carl Zeiss NV, Jena, Germany), and cells stained with antibodies against TNNI were observed on the Zeiss LSM 700 confocal microscope using a 60× 1.3 NA oil immersion objective. Images of cells stained with anti-MYH and anti-TNNI antibodies were obtained using Axio vision software and ZEN 2012 software, respectively (Carl Zeiss NV, Jena, Germany).
Proteomics sample preparation
To extract proteins for proteomics analysis, cells were washed twice with PBS and scrapped in lysis buffer (8 M urea; pH 8.0, 20 mM HEPES; 2× phosphor-stop tablets, Sigma, Leuven Belgium). The cells were then sonicated 3 times for 15 s each and centrifuged at 16000 rcf for 15 min. The protein concentrations of the samples were determined by the BCA assay according to the manufacturer's instructions (Thermo Fisher Scientific, Ghent, Belgium). Proteins (2 mg) were frozen at −20 °C prior to digestion.
Proteomics samples were prepared in triplicate for each condition. From each replicate, equal protein amounts were used for further analysis. Proteins were reduced by adding 4.5 mM DTT for 30 min at 55 °C. Proteins were alkylated by addition of 10 mM iodoacetamide at room temperature in the dark for 15 min. The samples were diluted with 20 mM HEPES at pH 8.0 to achieve a urea concentration of 4 M; then, the proteins were digested with 2 μg endoLysC (Wako, Neuss, Germany 1/250, w/w) for 4 h at room temperature. All samples were further diluted with 20 mM HEPES at pH 8.0 to a final urea concentration of 2 M, and the proteins were digested with 5 μg trypsin (Promega, Leiden, Belgium) (1/100, w/w) overnight at 37 °C. The peptides were then purified on a SampliQ SPE C18 cartridge (Agilent, Diegem, Belgium); from each replicate, 100 μg vacuum-dried peptides were used for LC-MS/MS analysis (D0 vs. D6 analysis) or strong cation exchange (SCX) fractionation (D0 vs. D8 vs. D13 analysis).
For SCX fractionation, SCX tips were made by stacking three discs (1.5 mm diameter) of polystyrene divinylbenzene copolymer with sulfonic acid (Empore™, 3M) in a 200 μl pipette tip. First, the SCX tips were rinsed with 100 μl acetonitrile (ACN) by pipetting the solvent in the tip, flicking the tip so the ACN touched the discs, and pushing the solvent through with a syringe. Then, each sample was dissolved in 100 μl loading buffer (1% TFA in water/ACN (95:5, v/v)) and the peptides were loaded on the SCX tips. After washing the tips with 50 μl loading buffer, the peptides were fractionated by subsequently pipetting up and down 20 μl of the following fractionation buffers: 100 mM ammonium acetate, 0.5% FA (fraction 1); 175 mM ammonium acetate, 0.5% formic acid (FA) (fraction 2); 375 mM ammonium acetate, 0.5% FA (fraction 3). The remaining peptides were eluted in 2 × 20 μl of elution buffer (5% NH4OH in water/ACN (20:80, v/v)) (fraction 4). To prevent deamidation in this fraction, 2 μl 10% FA was added. The fractionated peptides were dried by vacuum drying.
LC-MS/MS and data analysis
The peptides were dissolved in 20 μl loading solvent A (0.1% TFA in water/ACN (98:2, v/v)) of which 10 μl was injected for LC-MS/MS analysis on an Ultimate 3000 RSLCnano system (Thermo Fisher Scientific, Ghent, Belgium) in-line connected to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific, Ghent, Belgium) equipped with a Nanospray Flex Ion source (Thermo Fisher Scientific, Ghent, Belgium). Trapping was performed at 10 μl min−1 for 4 min in trapping solvent (0.1% TFA in water/acetonitrile (98:2, v/v)) on a 100 μm internal diameter (I.D.) × 20 mm trapping column (5 μm beads, C18 Reprosil-HD, Dr Maisch, Germany) and the sample was loaded on a reverse-phase column (made in-house, 75 μm I.D. × 400 mm, 1.9 μm beads C18 Reprosil-HD, Dr Maisch, Germany). The peptides were eluted by a non-linear increase from 2% to 56% solvent B (0.1% FA in water/acetonitrile (2:8, v/v)) over 150 min at a constant flow rate of 250 nl min−1, followed by a 10 min wash reaching 99% solvent B and re-equilibration with solvent A (0.1% FA in water/acetonitrile (2:8, v/v)). The column temperature was kept constant at 50 °C (CoControl 3.3.05, Sonation).
The mass spectrometer was operated in data-dependent mode, automatically switching between MS and MS/MS acquisition for the 16 most abundant ion peaks per MS spectrum. Full-scan MS spectra (375 to 1500 m/z) were acquired at a resolution of 60000 in the Orbitrap analyzer after accumulation to a target value of 3000000. The 16 most intense ions above a threshold value of 22000 (non-fractionated samples) or 13000 (fractionated samples) were isolated (window of 1.5 Th) for fragmentation at a normalized collision energy of 32% after filling the trap at a target value of 100000 for maximum 45 ms (non-fractionated samples) or 80 ms (fractionated samples). MS/MS spectra (200 to 2000 m/z) were acquired at a resolution of 15000 in the orbitrap analyser. The S-lens RF level was set at 55, and we excluded precursor ions with single and unassigned charge states from fragmentation selection.
Data analysis was performed with MaxQuant (version 1.5.4.1)53 using the Andromeda search engine with default search settings, including a false discovery rate set at 1% on both the peptide and protein level. The spectra were searched against Rattus norvegicus proteins in the UniProt/Swiss-Prot database (database release version of April 2015 containing 9535 rat protein sequences) as well as the Uniprot/TrEMBL database (database release version of April 2015 containing 27867 rat protein sequences) downloaded from www.uniprot.org. The mass tolerances for precursor and fragment ions were set to 4.5 and 20 ppm, respectively, during the main search. Enzyme specificity was set as carboxy-terminal to arginine and lysine (trypsin), also allowing cleavage at arginine/lysine–proline bonds with a maximum of two missed cleavages. Carbamidomethylation of cysteine residues was set as a fixed modification and variable modifications were set to oxidation of methionine (to sulfoxides) and acetylation of protein amino-termini. Proteins were quantified by the MaxLFQ algorithm integrated in the MaxQuant software.54 Only proteins with at least one unique or razor peptide were retained for identification, while a minimum ratio count of two unique or razor peptides was required for quantification. Further data analysis was performed with Perseus software (version 1.5.5.355) after loading the protein groups file from MaxQuant. Proteins only identified by site, reverse database hits and potential contaminants were removed, and replicate samples were grouped. Proteins with less than three valid values in at least one group were removed, and missing values were imputed from a normal distribution around the detection limit. Then, t-tests were performed (FDR = 0.01 and S0 = 1) for pairwise comparison and to generate the volcano plots depicted in Fig. 2A (D0 vs. D6) and Fig. 3A (D8 vs. D13). A principal component analysis was also performed on the replicate samples from the fractionation analysis to generate the scatter plot shown in Fig. 3B. Furthermore, to reveal proteins whose expression levels were strongly affected during the differentiation process in the fractionation analysis, sample groups were defined based on differentiation (D0 vs. D8 + D13) and differentiation time (D0 vs. D8 vs. D13), and a two-way ANOVA test was performed to compare the intensities of the proteins in the differentiation group with those of the differentiation time group. For each protein, this test calculated a p-value for differentiation and a p-value for differentiation time. 843 proteins with a p-value < 0.05 in at least one of the two groups were considered to be significantly regulated. The intensities of these proteins are shown in a heat map in Fig. 4 after non-supervised hierarchical row clustering. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository56 with the dataset identifier PXD006115.
Gene ontology and reactome analysis
For gene ontology analysis, the tool GOrilla (http://cbl-gorilla.cs.technion.ac.il/) was employed. Therefore, a single list of genes ranked according to the expression levels of their corresponding proteins and starting with the highest expression level in the more differentiated cell system was used and copied into the input panel of the tool. Rattus norvegicus was chosen as the analysis organism, and a process ontology was selected. A p-value threshold of 10−5 was chosen; this is the default choice suggested by the GOrilla tool to manage the trade-off between comprehensiveness and conciseness. Because GOrilla analyzes enrichment by ranking full protein lists from the most upregulated to the most downregulated proteins, ranked input lists were not preselected by fold change or p-value thresholds. Other parameters were set as default.For the Reactome analysis, the respective tool was used (http://www.reactome.org). A list of genes was taken from each of the eight clusters and inserted into the analysis window. The genes were mapped to human pathways and exported to Excel. Enriched pathways were ranked according to the fraction of genes from the list that were associated with a pathway divided by the total number of proteins in the pathway. Only pathways associated with a minimum of 2% of the genes in the cluster were considered.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Bijzonder Onderzoeksfonds of the KU Leuven (PF/10/014) and a personal grant by Life Sciences Research Partners vzw. LSRP_HH_01 (Prof. Collen, Leuven/Belgium) to H. J. H.References
- T. Oka, J. Xu and J. D. Molkentin, Re-employment of developmental transcription factors in adult heart disease, Semin. Cell Dev. Biol., 2007, 18, 117–131 CrossRef PubMed.
- B. W. Kimes and B. L. Brandt, Properties of a clonal muscle cell line from rat heart, Exp. Cell Res., 1976, 98, 367–381 CrossRef PubMed.
- X. Lian, et al., Cozzarelli prize winner: robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1848–E1857 CrossRef PubMed.
- E. Matsa, J. H. Ahrens and J. C. Wu, Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine, Physiol. Rev., 2016, 96, 1093–1126 CrossRef PubMed.
- J. Hescheler, et al., Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart, Circ. Res., 1991, 69, 1476–1486 CrossRef PubMed.
- C. Ménard, et al., Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells, J. Biol. Chem., 1999, 274, 29063–29070 CrossRef.
- A. F. Branco, et al., Differentiation-Dependent Doxorubicin Toxicity on H9c2 Cardiomyoblasts, Cardiovasc. Toxicol., 2012, 12, 326–340 CrossRef PubMed.
- M. Eguchi, Y. Liu, E.-J. Shin and G. Sweeney, Leptin protects H9c2 rat cardiomyocytes from H2O2-induced apoptosis, FEBS J., 2008, 275, 3136–3144 CrossRef PubMed.
- H. M. Korashy, et al., Mitogen-Activated Protein Kinases Pathways Mediate the Sunitinib-Induced Hypertrophy in Rat Cardiomyocyte H9c2 Cells, Cardiovasc. Toxicol., 2015, 15, 41–51 CrossRef PubMed.
- Y. Will, et al., Effect of the Multitargeted Tyrosine Kinase Inhibitors Imatinib, Dasatinib, Sunitinib, and Sorafenib on Mitochondrial Function in Isolated Rat Heart Mitochondria and H9c2 Cells, Toxicol. Sci, 2008, 106, 153–161 CrossRef PubMed.
- G. Y. Bao, Quantitative proteomic study identified cathepsin B associated with doxorubicin-induced damage in H9c2 cardiomyocytes, BioSci. Trends, 2012, 6, 283–287 Search PubMed.
- H.-C. Chou, et al., Proteomics study of oxidative stress and Src kinase inhibition in H9C2 cardiomyocytes: a cell model of heart ischemia-reperfusion injury and treatment, Free Radical Biol. Med., 2010, 49, 96–108 CrossRef PubMed.
- J. Lenčo, et al., Proteomic investigation of embryonic rat heart-derived H9c2 cell line sheds new light on the molecular phenotype of the popular cell model, Exp. Cell Res., 2015, 339, 174–186 CrossRef PubMed.
- A. F. Branco, et al., Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype, PLoS One, 2015, 10, e0129303 Search PubMed.
- B. Zhang, et al., Proteogenomic characterization of human colon and rectal cancer, Nature, 2014, 513, 382–387 CrossRef PubMed.
- E. van Rooij, et al., A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18255–18260 CrossRef PubMed.
- P.-Z. Zheng, et al., Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7653–7658 CrossRef PubMed.
- D. Bader, T. Masaki and D. A. Fischman, Immunochemical analysis of myosin heavy chain during avian myogenesis in vivo and in vitro, J. Cell Biol., 1982, 95, 763–770 CrossRef PubMed.
- J. A. Spudich and S. Watt, The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin, J. Biol. Chem., 1971, 246, 4866–4871 Search PubMed.
- T. Suzuki, K. Fujikura, T. Higashiyama and K. Takata, DNA Staining for Fluorescence and Laser Confocal Microscopy, J. Histochem. Cytochem., 1997, 45, 49–53 CrossRef PubMed.
- R. Aebersold and M. Mann, Mass-spectrometric exploration of proteome structure and function, Nature, 2016, 537, 347–355 CrossRef PubMed.
- E. Eden, R. Navon, I. Steinfeld, D. Lipson and Z. Yakhini, GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists, BMC Bioinf., 2009, 10, 48 CrossRef PubMed.
- M. J. Hernández-Corbacho, M. F. Salama, D. Canals, C. E. Senkal and L. M. Obeid, Sphingolipids in mitochondria, Biochim. Biophys. Acta, 2017, 1862, 56–68 CrossRef PubMed.
- R. Goodacre, et al., Rapid identification of urinary tract infection bacteria using hyperspectral whole-organism fingerprinting and artificial neural networks, Microbiology, 1998, 144, 1157–1170 CrossRef PubMed.
- R. Lavigne, et al., The structural proteome of Pseudomonas aeruginosa bacteriophage KMV, Microbiology, 2006, 152, 529–534 CrossRef PubMed.
- L. Matthews, et al., Reactome knowledgebase of human biological pathways and processes, Nucleic Acids Res., 2009, 37, D619–D622 CrossRef PubMed.
- T. Migita, et al., ACSL3 promotes intratumoral steroidogenesis in prostate cancer cells, Cancer Sci., 2017, 108, 2011–2021 CrossRef PubMed.
- H. H. Patel, F. Murray and P. A. Insel, G-Protein-Coupled Receptor-Signaling Components in Membrane Raft and Caveolae Microdomains, in Protein–Protein Interactions as New Drug Targets, ed. E. Klussmann and J. Scott, Springer Berlin Heidelberg, 2008, pp. 167–184, DOI:10.1007/978-3-540-72843-6_7.
- I. B. Renes, et al., Epithelial proliferation, cell death, and gene expression in experimental colitis: alterations in carbonic anhydrase I, mucin MUC2, and trefoil factor 3 expression, Int. J. Colorectal Dis., 2002, 17, 317–326 CrossRef PubMed.
- K. H. Holt, et al., Functional rescue of the sarcoglycan complex in the BIO 14.6 hamster using delta-sarcoglycan gene transfer, Mol. Cell, 1998, 1, 841–848 CrossRef PubMed.
- G. K. Owens, M. S. Kumar and B. R. Wamhoff, Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease, Physiol. Rev., 2004, 84, 767–801 CrossRef PubMed.
- A. H. Futerman and Y. A. Hannun, The complex life of simple sphingolipids, EMBO Rep., 2004, 5, 777–782 CrossRef PubMed.
- M. Comelli, et al., Cardiac differentiation promotes mitochondria development and ameliorates oxidative capacity in H9c2 cardiomyoblasts, Mitochondrion, 2011, 11, 315–326 CrossRef PubMed.
- R. Ventura-Clapier, A. Garnier, V. Veksler and F. Joubert, Bioenergetics of the failing heart, Biochim. Biophys. Acta, Mol. Cell Res., 2011, 1813, 1360–1372 CrossRef PubMed.
- P. W. Burridge, et al., A Universal System for Highly Efficient Cardiac Differentiation of Human Induced Pluripotent Stem Cells That Eliminates Interline Variability, PLoS One, 2011, 6, e18293 Search PubMed.
- M. Pekkanen-Mattila, et al., Substantial variation in the cardiac differentiation of human embryonic stem cell lines derived and propagated under the same conditions—a comparison of multiple cell lines, Ann. Med., 2009, 41, 360–370 CrossRef PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists, Nucleic Acids Res., 2009, 37, 1–13 CrossRef PubMed.
- D. M. Dawson, T. L. Goodfriend and N. O. Kaplan, Lactic Dehydrogenases: Functions of the Two Types Rates of Synthesis of the Two Major Forms can be Correlated with Metabolic Differentiation, Science, 1964, 143, 929–933 Search PubMed.
- Y. Dotan, D. Lichtenberg and I. Pinchuk, Lipid peroxidation cannot be used as a universal criterion of oxidative stress, Prog. Lipid Res., 2004, 43, 200–227 CrossRef PubMed.
- Y. Octavia, et al., Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies, J. Mol. Cell. Cardiol., 2012, 52, 1213–1225 CrossRef PubMed.
- Y. T. Horikawa, et al., Cardiac-specific overexpression of caveolin-3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling, J. Am. Coll. Cardiol., 2011, 57, 2273–2283 CrossRef PubMed.
- V. Allamand, et al., Early adenovirus-mediated gene transfer effectively prevents muscular dystrophy in alpha-sarcoglycan-deficient mice, Gene Ther., 2000, 7, 1385–1391 CrossRef PubMed.
- G. Whiteley, R. F. Collins and A. Kitmitto, Characterization of the molecular architecture of human caveolin-3 and interaction with the skeletal muscle ryanodine receptor, J. Biol. Chem., 2012, 287, 40302–40316 CrossRef PubMed.
- S. Lindskog, Structure and mechanism of carbonic anhydrase, Pharmacol. Ther., 1997, 74, 1–20 CrossRef PubMed.
- C. H. Orchard and J. C. Kentish, Effects of changes of pH on the contractile function of cardiac muscle, Am. J. Physiol., 1990, 258, C967–C981 CrossRef PubMed.
- S. Franklin, et al., Specialized compartments of cardiac nuclei exhibit distinct proteomic anatomy, Mol. Cell. Proteomics, 2011, 10 DOI:10.1074/mcp.M110.000703.
- O. Bergmann, et al., Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover, Exp. Cell Res., 2011, 317, 188–194 CrossRef PubMed.
- J. Kajstura, M. Rota, T. Hosoda, P. Anversa and A. Leri, Response to Bergmann et al.: Carbon 14 Birth Dating of Human Cardiomyocytes, Circ. Res., 2012, 110, e19–e21 CrossRef PubMed.
- T. Zhang, et al., Troponin T nuclear localization and its role in aging skeletal muscle, Age, 2013, 35, 353–370 CrossRef PubMed.
- C. R. Archer, E. L. Robinson, F. M. Drawnel and H. L. Roderick, Endothelin-1 promotes hypertrophic remodelling of cardiac myocytes by activating sustained signalling and transcription downstream of endothelin type A receptors, Cell. Signalling, 2017, 36, 240–254 CrossRef PubMed.
- A. Clerk, I.-K. S. Aggeli, K. Stathopoulou and P. H. Sugden, Peptide growth factors signal differentially through protein kinase C to extracellular signal-regulated kinases in neonatal cardiomyocytes, Cell. Signalling, 2006, 18, 225–235 CrossRef PubMed.
- S. J. Fuller, et al., Cardiac protein kinases: the cardiomyocyte kinome and differential kinase expression in human failing hearts, Cardiovasc. Res., 2015, 108, 87–98 CrossRef PubMed.
- J. Cox and M. Mann, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification, Nat. Biotechnol., 2008, 26, 1367–1372 CrossRef PubMed.
- J. Cox, et al., Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ, Mol. Cell. Proteomics, 2014, 13, 2513–2526 Search PubMed.
- S. Tyanova, et al., The Perseus computational platform for comprehensive analysis of (prote)omics data, Nat. Methods, 2016, 13, 731–740 CrossRef PubMed.
- J. A. Vizcaíno, et al., 2016 update of the PRIDE database and its related tools, Nucleic Acids Res., 2016, 44, D447–D456 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mo00036k |
| ‡ Egle Passante, PhD is now an employee of Sanofi Genzyme, United Kingdom, Oxford, Oxfordshire, UK |
| § Present address: Target Discovery Research, Boehringer Ingelheim Pharmaceutical Research and Development Centre Biberach, 88400, Biberach an der Riss, Germany, Heinrich_2.Huber@boehringer-ingelheim.com |
| This journal is © The Royal Society of Chemistry 2018 |