
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePTMscape: an open source tool to predict generic post-translational modifications and map modification crosstalk in protein domains and biological processes†
Ginny X. H.
Li
 a,
Christine
Vogel
b and
Hyungwon
Choi
*ac
a,
Christine
Vogel
b and
Hyungwon
Choi
*ac
aSaw Swee Hock School of Public Health, National University of Singapore, Singapore. E-mail: hwchoi@nus.edu.sg
bCenter for Genomics and Systems Biology, Department of Biology, New York University, New York, NY 10003, USA
cInstitute of Molecular and Cell Biology, Agency for Science, Technology, and Research, Singapore
First published on 7th June 2018
Abstract
While tandem mass spectrometry can detect post-translational modifications (PTM) at the proteome scale, reported PTM sites are often incomplete and include false positives. Computational approaches can complement these datasets by additional predictions, but most available tools use prediction models pre-trained for single PTM type by the developers and it remains a difficult task to perform large-scale batch prediction for multiple PTMs with flexible user control, including the choice of training data. We developed an R package called PTMscape which predicts PTM sites across the proteome based on a unified and comprehensive set of descriptors of the physico-chemical microenvironment of modified sites, with additional downstream analysis modules to test enrichment of individual or pairs of PTMs in protein domains. PTMscape is flexible in the ability to process any major modifications, such as phosphorylation and ubiquitination, while achieving the sensitivity and specificity comparable to single-PTM methods and outperforming other multi-PTM tools. Applying this framework, we expanded proteome-wide coverage of five major PTMs affecting different residues by prediction, especially for lysine and arginine modifications. Using a combination of experimentally acquired sites (PSP) and newly predicted sites, we discovered that the crosstalk among multiple PTMs occur more frequently than by random chance in key protein domains such as histone, protein kinase, and RNA recognition motifs, spanning various biological processes such as RNA processing, DNA damage response, signal transduction, and regulation of cell cycle. These results provide a proteome-scale analysis of crosstalk among major PTMs and can be easily extended to other types of PTM.
Introduction
Protein post-translational modifications (PTMs) regulate cellular functions in various ways: catalyzing enzymatic activities, conferring substrate specificity to control allosteric interactions, mediating interactions with other molecules such as DNA, co-factors, and lipids, and localizing proteins to organelles.1 With advances in enrichment techniques for PTMs, high-resolution mass spectrometry (MS) has now become the method of choice to experimentally detect and quantify major PTMs at a proteome scale.2 A wealth of PTM data arising from tandem MS/MS experiments has been curated and shared in public databases such as PhosphoSitePlus (PSP),3 PHOSIDA,4 and Uniprot,5 and some major PTMs such as phosphorylation and ubiquitination have been mapped for multiple species. For instance, as of December 2017, the PSP database described ∼240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 phosphorylation and ∼22000 ubiquitination sites for >20000 different human proteins.
000 phosphorylation and ∼22000 ubiquitination sites for >20000 different human proteins.
A comprehensive map of diverse PTMs can help us infer not only the role of individual modifications, but also the complex code of different PTMs localized to the same protein jointly modulating biochemical functions through positive and negative regulatory interactions, also known as the PTM crosstalk.6 A well-known example for such crosstalk is the tumor suppressor gene p53 whose abundant and diverse modifications affect the protein's activity and subsequent cancer formation.7–9 p53 has at least 20 known phosphorylation sites and other types of PTM including acetylation, ubiquitination, methylation, and O-GlcNAc sites. Therefore, a critical first step is to map PTM sites across the entire proteome in an unbiased manner, attaching confidence scores that allow removal of false-positive identifications and incorrect site localizations.
However, even for major PTMs, including acetylation, methylation, and glycosylation, experiments with new enrichment techniques10 often identify novel modification sites, implying that we have not yet reached the coverage to provide the total PTM landscape. While experimental efforts are slowly completing this landscape, there remains the need for statistical frameworks that integrate these diverse modifications at proteome-scale into a unified, comprehensive PTM map with a minimal number of false positives. In addition, such an “atlas” of PTMs should offer annotation of each modification type by predictive descriptors such as physicochemical properties, protein structure information, and sequence motif information such as position specific amino acid propensity (PSAAP) in order to maximize the utility of the resource.
In the literature, there already exists a plethora of computational prediction methods for well-studied PTMs, where the majority of methods rely on complex machine learning algorithms in combination with sequence-level scoring functions that test for single types of PTMs.11 These methods vary by the type of prediction algorithm employed, the use of adjacent residues around candidate sites, the use of three-dimensional structure information, and specificity with respect to kinase families (in the case of phosphorylation). Recently, a number of prediction tools have also been reported for ubiquitination12–14 and arginine methylation,15 in which predictions are made based on amino acid properties rather than sequence characteristics due to the lack of global motifs.
Although these tools provide small-scale predictions in a user-friendly web interface, only a few tools offer batch prediction capability for the whole proteome. This is presumably because of the high computational burden by non-linear prediction algorithms – and are therefore incapable of extracting examples like that of p53 in a comprehensive, genome-wide manner. Moreover, each single PTM prediction tool uses a different set of descriptors (predictive features) and the models are trained using different training PTM data, often leaving the user no freedom to provide input to the construction of the prediction models. More importantly, as these methods rely on non-linear prediction algorithms such as support vector machines (SVM) with radial kernel,16 it remains elusive how each descriptor or a combination of descriptors contributes to the probability of PTM events, challenging the interpretation of the best predictors. Other omnibus tools such as ModPred17 perform batch predictions for multiple types of PTM, but their prediction has limited sensitivity in the whole proteome scale (see below). In addition, those tools’ predictions are made from a pre-trained model, which the user cannot modify or rebuild. Further, whole proteome-scale predictions are extremely time consuming in a standard computing environment, precluding the execution of such analysis for lay users.
To enable researchers to chart a map of various types of PTM across the proteome in addition to the experimental data, here we present PTMscape, a unified, highly sensitive and specific framework for high confidence PTM predictions in a whole proteome scale. PTMscape offers several key advances. First, it is generically applicable to any PTMs and enables the user to train and test predictions using a comprehensive set of descriptors, while operating at the whole proteome scale and in a time-efficient manner. Unlike most existing tools, PTMscape provides a full set of precompiled features and facilitates the construction of new training and test data, allowing the user to control the model building process. With these resources, PTMscape achieves prediction accuracy comparable to the best single-PTM tools currently available. Second, PTMscape performs further downstream statistical enrichment analysis of protein domains in the single type PTMs and their crosstalk in protein domains. Third, PTMscape offers model training and prediction for large-scale data via local installation of the software. PTMscape is packaged for the popular R environment (http://cran.r-project.org) and the user can take full control to create appropriate training and test data for an entire proteome.
To our knowledge, PTMscape is the first open-source, comprehensive statistical framework that helps the user predict novel sites and perform downstream enrichment analysis of protein domains and biological processes in the PTM sites (predicted, experimentally acquired, or a combination). To demonstrate the utility of this tool, we applied PTMscape to ∼17000 human protein sequences and predicted ∼39000 additional PTMs of five different types. To understand the spatial distribution of additionally predicted PTMs in functional units of protein sequences, we tested enrichment of those sites in protein domains and biological processes in which the proteins harboring those PTM site-containing domains are involved. With the addition of predicted sites to the repertoire of experimentally acquired sites in the PSP database, we also illustrate that individual PTMs and some of their combinations preferentially occur in protein domains carrying out specific biological functions.
Results
PTMscape uses linear SVMs and a comprehensive set of predictors to evaluate five major types of PTMs
In PTMscape, we advocate the SVM with linear kernel, i.e. linear classification, for the prediction of each candidate site into a modified site or an unmodified one, although it can be easily replaced by alternative approaches such as SVM with non-linear kernel, random forests,18 and artificial neural networks19 in the implementation. In contrast to PTMscape, most prediction tools use SVM with radial kernel, a non-linear classification method for flexible and highly sensitive classification. The rationale for our choice of linear kernel lies in its fast computation capability and the ease in interpretation of the resulting weight coefficients without sacrifice in prediction performances in large-scale data (see next section for comparison against other machine learning methods). Although the non-linear SVM classifier may indeed be the more sensitive than the linear counterpart, the decision boundaries of the SVMs with non-linear kernel are far too complex for human interpretation. In addition, a non-linear classification method can be easily over-fitted, especially depending on how the user constructs the training data. In contrast, the linear SVM has the advantage that the optimized weight coefficients directly inform on the contribution of each descriptor to the likelihood of the PTM status20 (whether they increase or decrease the likelihood).To evaluate the performance of overall predictions, we first assembled a comprehensive set of physicochemical “descriptors” of the microenvironment of PTM sites, which we derived from a literature survey. In this initial analysis, we aimed to learn how well the positive sites, i.e. PTM sites detected in experiments collected in the PSP database, can be differentiated from the negative sites, i.e. sites not reported in the PSP database, and what information (descriptor) is useful to predict each PTM type.
The descriptors of PTM sites came from three different sources. The first source consisted of 538 amino acid indexes, which have been previously used in prediction methods, e.g. for ubiquitination.13,21 The indexes include physicochemical properties for each amino acid,22 including, for example, hydrophobicity, propensity to be in secondary structures, free energy change, and residue volume. A large number of these properties are highly correlated with one another. Therefore, we grouped the properties into 53 clusters and used their average values as a representative value for each cluster (see Methods). The second source included residue-specific properties such as access to surface area, half sphere exposure, and the probability of being positioned in secondary structures such as coils, sheets, and helix computed by SPIDER3 software, which achieves three-state secondary structure prediction accuracy of 84% with the incorporation of long-range contact information, outperforming other currently available secondary structure prediction tools.23 This information was obtained based on the secondary structure assignment to the protein sequence. The last source was the position specific amino acid propensity (PSAAP) matrix computed for each PTM type based on experimentally acquired sites (positives from the PSP database).11 The final 173 predictors comprised six categories including the average amino acid indexes, four secondary structure features, and the PSAAP scores.
Using PTMscape, we computed these properties for all canonical protein sequences in the human proteome (Uniprot), for window sizes of 11, 15, and 25 amino acids where the center position is a candidate residue. SPIDER3 was able to assign individual residues to secondary structures for approximately 17000 proteins, and we perform all our proteome-scale predictions within this set. Following the convention in other publications,12,21,24–26 we reduced sequence redundancy by removing highly similar sequences for the purpose of prediction accuracy evaluation, resulting in a total of ∼10700 human sequences considered (see Methods). We focused on five different modifications affecting five different residues: phosphorylation (S, T, Y), ubiquitination (K), SUMOylation (K), acetylation (K), and methylation (K, R). We evaluated the performance of PTMscape's linear SVM classifier for each of the different PTM types using 10-fold cross-validation. In the cross-validation, a model was trained on nine folds of the data and tested on the remaining, randomly chosen one-fold, and the same was iteratively applied to all ten folds.
Table 1 shows the overall prediction performance of linear SVMs across the five different PTMs. The area under the curve (AUC) for the receiver-operating characteristic (ROC) was the highest for arginine methylation and lysine SUMOylation (0.79), whereas it was the lowest for lysine ubiquitination (0.64) and acetylation (0.66), for all window sizes (ESI,† Fig. S1). As expected, the modifications known to have clear global sequence motifs were better predicted than those without a sequence motif, and we discuss this point below. For most modifications, the prediction models using the “wide” 25 amino acid window performed the best (ESI,† Table S1), albeit by a small margin. Hence, we used the 25 amino acid windows hereafter.
| PTM type | No. of proteins | No. of PSP sites | Window size 25 | |||
|---|---|---|---|---|---|---|
| AUC | MCC | Sensitivity | Specificity | |||
| Acetylation (K) | 3729 | 10479 |
0.66 | 0.25 | 0.61 | 0.64 |
| Methylation (K) | 1521 | 2566 | 0.74 | 0.39 | 0.61 | 0.76 |
| Ubiquitination (K) | 4874 | 22592 |
0.64 | 0.22 | 0.67 | 0.54 |
| SUMOylation (K) | 1020 | 2996 | 0.77 | 0.42 | 0.63 | 0.79 |
| Methylation (R) | 2301 | 5450 | 0.79 | 0.47 | 0.62 | 0.84 |
| Phosphorylation (S) | 8510 | 76008 |
0.74 | 0.36 | 0.70 | 0.66 |
| Phosphorylation (T) | 6982 | 28359 |
0.72 | 0.33 | 0.66 | 0.66 |
| Phosphorylation (Y) | 6097 | 18645 |
0.70 | 0.30 | 0.72 | 0.58 |
We remark that we treated all sites not reported in the PSP database as negative sites. This assumption may not hold true for all PTM types, since the sites in the PSP database may not cover all true modifiable residues due to relatively strict criteria in the curation of PTM sites. For example, the PTM sites provided in the PSP database are detected based on the mass spectral evidence with at least 95% site localization certainty, and thus their data exclude other sites with site ambiguity. There are other databases such as dbPTM27 and sysPTM28 that often include additional PTM sites, and including their PTM site data may improve the representativeness of positive sites. In this work, however, we considered all unreported sites as negatives as it is the safest option one can take while evaluating predictors. This implies that the sensitivity calculated here, i.e. the prediction of true positives, is likely underestimated and better than what is reported in Table 1.
The importance of specificity cannot be overstated in view of the prediction capability across different methods in major PTMs. To investigate the root cause of lagging AUC values across most prediction methods, we studied the decision power of separating positives and negatives in the feature space in the context of whole proteome-scale prediction. Fig. 1 shows the plots of Partial Least Squares – Discriminant Analysis, a useful tool for projecting high-dimensional data in a supervised way (to separate the positives from the negatives), across all five types of PTMs using all the descriptors we collected. The figure shows that, with the exception of lysine/arginine methylation and SUMOylation, it is difficult to detect a large number of additional true sites at high specificity based on these features. It is likely due to this challenge that many existing methods resorted to complex non-linear prediction methods, attempting to find the sub-space in the feature set that confers increased likelihood of a given PTM event. Nevertheless, the poor overall separation of positives and negatives suggests that it is of paramount importance to maintain a stringent level of specificity in predictions, even at the expense of the sizeable loss in sensitivity. This underscores why we choose a score threshold associated with very high specificity rule (99%) in our predictions below.
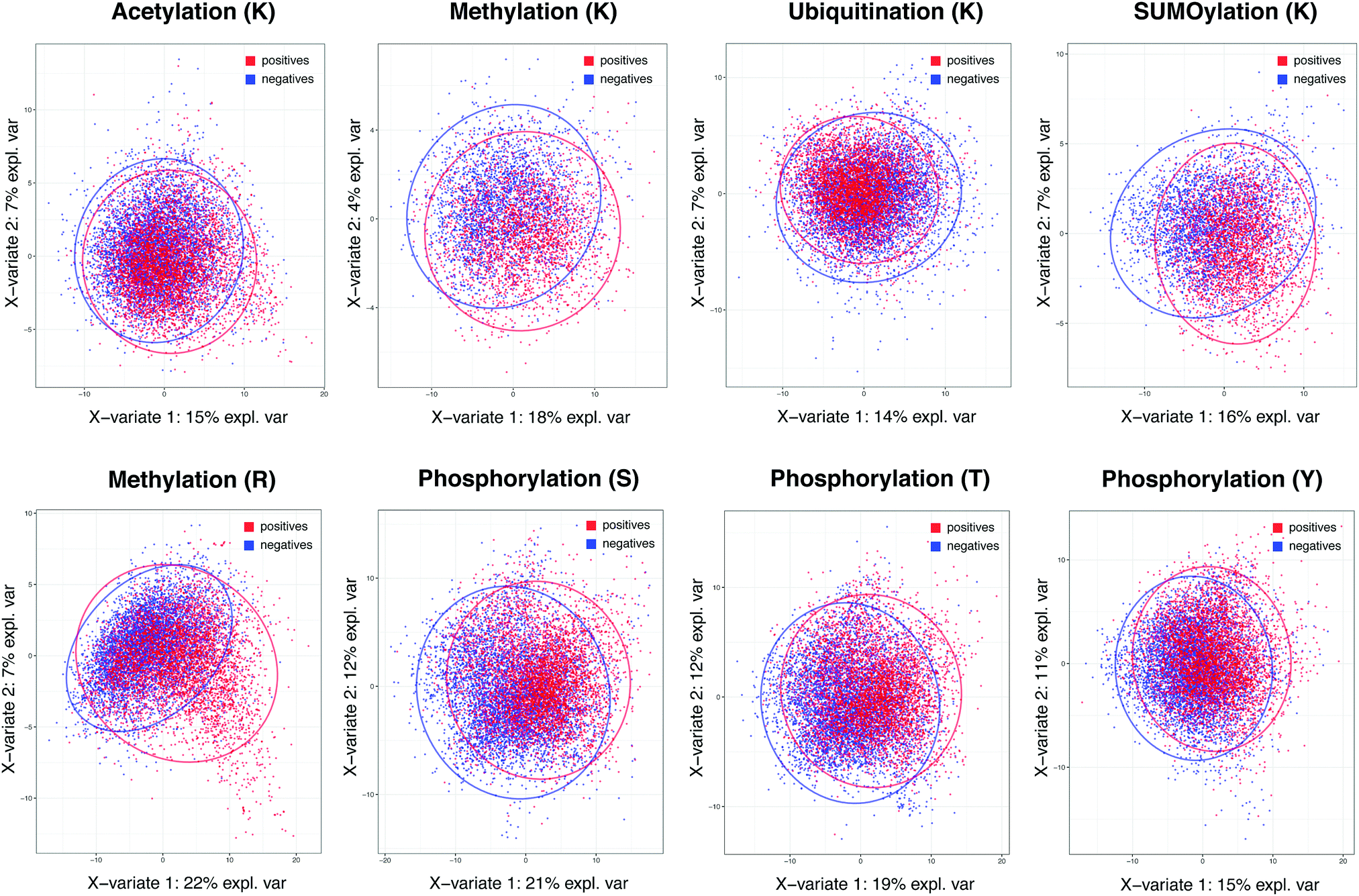 | ||
| Fig. 1 Partial least squares-discriminant analysis (PLS-DA) plot for the five PTM types. Red and blue colors indicate experimentally acquired, positive PTM sites (from the PSP database) and negative sites (the remainder of possible sites). The negatives have been randomly sampled to match the same number of positives in each PTM to prevent a large number of negatives from masking the positives in each plot. | ||
Linear SVM is comparable to the best prediction algorithms of single and multiple PTM prediction methods
Next, we benchmarked the prediction performance of PTMscape's linear SVM against the algorithms leading in prediction of phosphorylation and ubiquitination, the two modifications most widely studied by mass spectrometry-based proteomics. For phosphorylation, PTMscape's linear SVM compared very well with the latest kinase-independent phosphorylation prediction tool called PhosphoSVM, which outperforms the majority of global phosphorylation prediction tools.26 Using PhosphoSVM's test data consisting of 9688 serine, 2919 threonine, and 1269 tyrosine positive sites on 2545, 1499, and 805 protein sequences, we trained PTMscape's linear SVMs using predictive variables computed for 25 amino acid windows with 10-fold cross-validation. This setup was identical to that used by PhosphoSVM. Therefore, the only difference between the two methods was that our classifier (predictor) used more features for prediction and a simpler kernel than that of PhosphoSVM. Notably, PTMscape and PhosphoSVM showed similar performance with the exception of serine phosphorylation, where the AUC was greater by 0.03 in PhosphoSVM (Table 2).| PTM type | Methods | AUC | sd AUC | Choice of threshold | MCC | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|
| Phosphorylation (S) | PhosphoSVM | 0.84 | 0.01 | F-measure | 0.30 | 0.44 | 0.94 |
| PTMscape | 0.81 | 0.01 | MCC | 0.49 | 0.74 | 0.75 | |
| F-measure | 0.47 | 0.86 | 0.60 | ||||
| Matching specificity | 0.37 | 0.36 | 0.94 | ||||
| Phosphorylation (T) | PhosphoSVM | 0.82 | 0.01 | F-measure | 0.25 | 0.37 | 0.95 |
| PTMscape | 0.80 | 0.01 | MCC | 0.48 | 0.71 | 0.75 | |
| F-measure | 0.46 | 0.85 | 0.60 | ||||
| Matching specificity | 0.37 | 0.34 | 0.95 | ||||
| Phosphorylation (Y) | PhosphoSVM | 0.74 | 0.02 | F-measure | 0.21 | 0.42 | 0.87 |
| PTMscape | 0.74 | 0.03 | MCC | 0.41 | 0.66 | 0.73 | |
| F-measure | 0.35 | 0.89 | 0.42 | ||||
| Matching specificity | 0.30 | 0.40 | 0.87 | ||||
| Ubiquitination (K) | ESA-ubiSite | 0.95 | n.a | MCC | 0.48 | 0.66 | 0.94 |
| ESA-SVM-PCPs | 0.83 | n.a | MCC | 0.27 | 0.76 | 0.75 | |
| ESA-5NN | 0.65 | n.a | MCC | 0.17 | 0.69 | 0.66 | |
| Random-SVM | 0.73 | n.a | MCC | 0.17 | 0.67 | 0.67 | |
| PTMscape | 0.82 | n.a | MCC | 0.29 | 0.64 | 0.83 | |
| F-measure | 0.28 | 0.43 | 0.92 | ||||
| Matching specificity | 0.23 | 0.31 | 0.94 | ||||
PTMscape's linear SVM classifier also fared very well with the most state-of-the-art ubiquitination prediction tool called ESA-UbiSite.21 ESA-UbiSite uses an evolutionary screening algorithm coupled with non-linear SVM to address the lack of true negatives by iteratively updating the modification status of negative sites in the model-training phase. Using the training data and the high-confidence test data provided by ESA-UbiSite, PTMscape achieved an AUC of 0.82, comparable to the second best algorithm ESA-SVM-PCPs (AUC 0.83) but worse than the best method ESA-UbiSite (AUC 0.95). It is possible that this difference in AUC most likely arose from the specificity calculation, as the test data is very small with only 645 positive sites on 379 proteins. Therefore, although the evolutionary screening algorithm for identifying better negative sites may make valuable contribution, the calculation of AUC remains to be evaluated on a larger test set.
Next, we show that PTMscape outperforms ModPred, which is one of the few available generic tools for PTM prediction.17 Unfortunately, it was practically infeasible to directly compare PTMscape and ModPred using the same training and test data because we are unable to build the new prediction models for cross-validation within their framework. Therefore, we ran ModPred provided by the developers to predict phosphorylation, ubiquitination, SUMOylation, acetylation, and methylation on ∼12000 non-redundant protein sequences used in our 10-fold cross-validation scheme, and compared their performance with that of PTMscape. This implies that we made predictions on some of the proteins used for training data in ModPred, giving the method a potential advantage over PTMscape. ModPred performed the best with serine phosphorylation at an AUC of 0.74 (ESI,† Table S2) followed by threonine/tyrosine phosphorylation, arginine methylation and lysine acetylation with AUC ranging from 0.65 to 0.7. ModPred performed poorly for other lysine modifications (AUC ≤ 0.6). In contrast, the AUCs of PTMscape were consistently higher, ranging from 0.64 to 0.74 across all modifications (ESI,† Table S2).
Lastly, we compared the performance of linear SVM against non-linear SVM as well as two alternative machine learning approaches, namely artificial neural network (ANN)19 and random forests (RF),18 in terms of the prediction accuracy and computation time in the same 10-fold cross-validation scheme. Given that any proteome-scale prediction must deal with hundreds of thousands of candidate sites, computation time is an important factor in this evaluation. Unfortunately, non-linear SVM with radial kernel did not finish the 10-fold cross-validation within a time period of more than 30 days for any of the PTM types. Table S3 (ESI†) shows the comparison of the remaining three methods. The random forest,18 a prediction method based on ensemble of thousands of classification trees, achieved the highest AUC across the PTM types, yet it was the most time-consuming method among the three, taking hours of computation time. ANN19 consistently performed the worst with the exception of linear SVM (using liblinear implementation). By contrast, linear SVM consistently performed as well as random forest and had the fastest computation time across the PTM types. For this reason, we chose linear SVM as the default prediction method throughout the paper.
Highly predictive features of individual PTM types
In addition to its robust performance, the advantage of PTMscape's linear SVM is the straightforward inference of the most predictive properties for specific PTM types, as extracted from the 173 total predictors. This interpretation is possible because the linear kernel in SVM requires that the decision boundaries be linearly associated with each variable, therefore naturally allowing us to describe the variables as positively or negatively associated with the modification.20Fig. 2(A–E) Shows weight coefficients of the SVM predictors for all PTM types. The amino acid indexes were the strongest predictors for phosphorylation, ubiquitination, acetylation and methylation (Fig. 2A). In general, high hydrophobicity, partition coefficients, low solubility, low probability to be in an alpha helix/helix terminus, and propensity to be in a domain linker tended to be positively predictive of these PTMs. These descriptors are often found in membrane proteins and link to a biased amino acid composition. However, other predictors varied for the different PTM types. For example, hydrophobicity and amino acid composition of intracellular proteins were positively predictive only for phosphorylation, but in no other modifications. By contrast, the propensity of amino acids to be positioned in beta sheets and the properties associated with helical ends (e.g. chain reversal) were positively predictive for several lysine modifications, such as SUMOylation, ubiquitination, and acetylation.
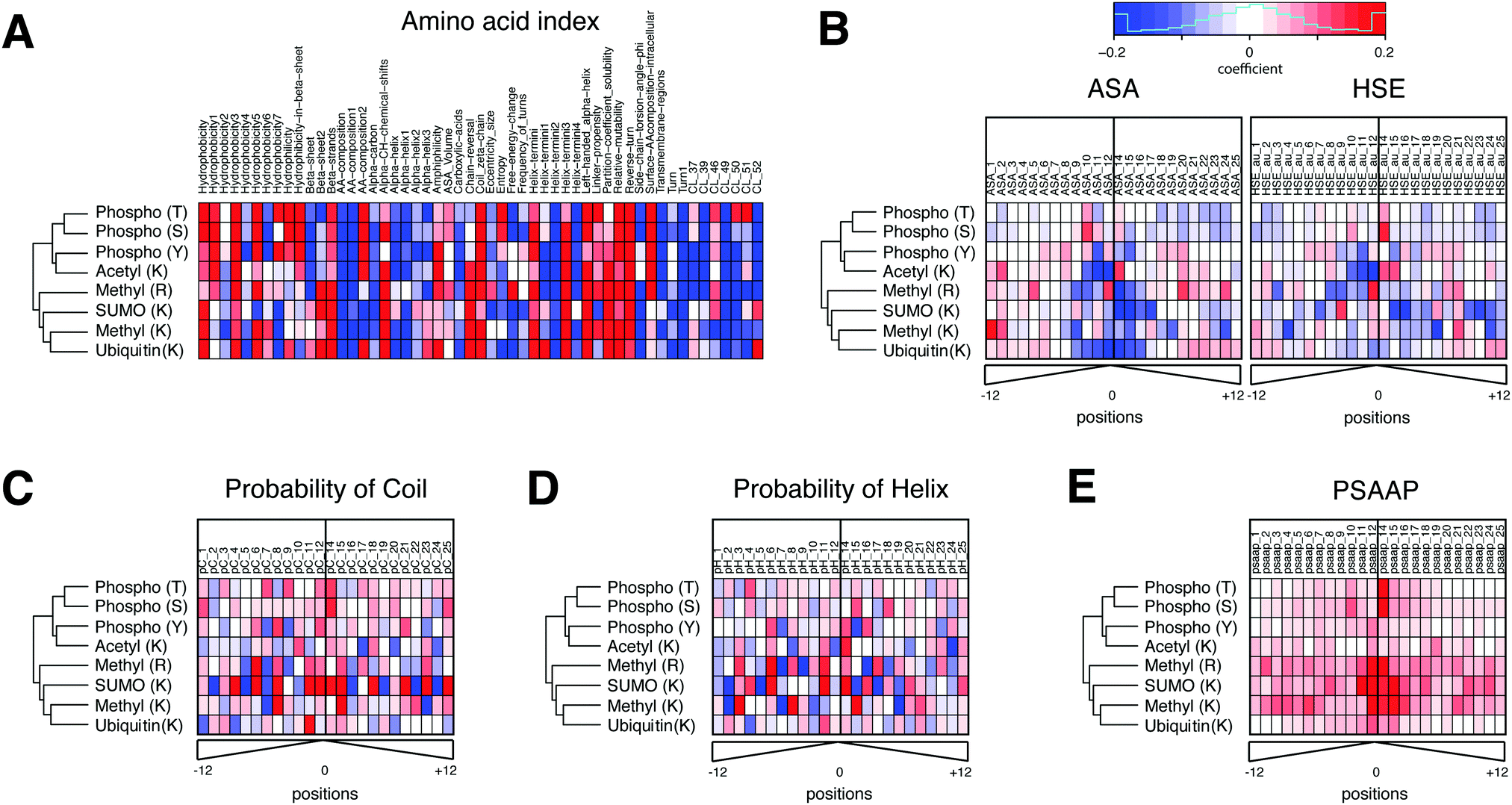 | ||
| Fig. 2 The feature weight coefficients obtained from linear SVM analysis of individual PTMs in heatmaps. The heatmaps were organized into six different sets of features, including (A) amino acid indexes, (B) accessible surface area (ASA) and half-sphere exposure (HSE), (C) probability of coil and (D) probability of helix, and (E) position-specific amino acid propensity (PSAAP) information obtained from known and additionally predicted sites. The names and description of amino acid index clusters can be found in ESI,† Table S6. The hierarchical clustering of five PTM types was performed using all variables. With the exception of amino acid indexes, all features were computed in a position specific manner in a 25aa-long window (distance from a central site). The color scale has been set between −0.2 and 0.2 in coefficient values. Red and blue colors indicate the degree to which they contribute to the probability of modification in each PTM type (red: positive contribution to the probability, blue: negative contribution to the probability). The PSAAP indicate which neighbor residues contribute the most to the likelihood of PTM event. The weight coefficients are computed after scaling all feature variables between −1 and 1, and hence are directly comparable across different features in terms of prediction strength. | ||
Solvent accessibility and secondary structure showed different patterns for different PTM types (Fig. 2B). For example, tyrosine – but not serine or threonine – phosphorylation was more likely in sequences with poor solvent accessibility, i.e. in a pocket shape. This result is consistent with the known higher similarity of serine and threonine phosphorylation compared to tyrosine phosphorylation.29 Lysines modified by ubiquitin, SUMO or acetylation were negatively associated with solvent accessibility. Further, the residues in coiled coils tend to be phosphorylated, arginine methylated, or SUMOylated (Fig. 2C and D). Residues in helices tend to be acetylated, methylated, or SUMOylated.
Lastly, the 25 amino acids-long windows containing the sites predicted by PTMscape had sequence motifs matching those known from literature. The corresponding PSAAP scores were predictive for those PTMs with clear sequence motifs, i.e. lysine and arginine methylation, SUMOylation, and serine/threonine phosphorylation. The sequence logo plots for the experimentally detected and newly predicted sites show the similar relative strength of the motifs in PTMs where predictions accounted for a large proportion (ESI,† Fig. S2). The best examples are the ΨKxE consensus motif for SUMOylation30 and GAR consensus motif for arginine methylation,31 and proline at +1 position of serine/threonine phosphorylation.32 By contrast, there were no clear consensus motifs that could globally predict ubiquitination, lysine acetylation, and tyrosine phosphorylation, as has been demonstrated in previous work.29,33
PTMscape's predictions with ultra-high specificity
Next, we used PTMscape's comprehensive descriptor set and linear SVM classification to map additional sites in a unified statistical framework for the five major types of PTMs, improving coverage on the existing data set (e.g. PSP). We used two-fold cross-prediction (see Methods) so that the training data and the test data are completely exclusive. Predictions were made on the entire human proteome, comprising ∼17000 sequences. The score thresholds were selected so that the prediction gives ultra-high 99% specificity, in contrast to the conventional choice giving the best Matthew's correlation or F-measure, to ensure a low false positive rate account for the varying range of AUCs. A user who is willing to tolerate lower specificity can easily change the score thresholds.
Table 3 shows the experimentally detected sites available in the PSP database (254116 PTM sites) and the sites newly predicted by PTMscape, which amount to a total of 38857 new sites with our choice of score threshold. As expected, the number of newly predicted sites compared to the experimentally detected sites varied across the PTMs. For PTMs with proteome-wide experimental coverage, e.g. phosphorylation and ubiquitination, newly predicted sites accounted for merely <14% of the sites known from PSP. By contrast, for those with incomplete proteome-wide coverage, e.g. SUMOylation and methylation, PTMscape predicted 85–139% new sites even at 99% specificity. The observation above that lysine SUMOylation34–36 and lysine/arginine methylation had the most consistent global sequence motifs between PSP sites and predictions (ESI,† Fig. S2) suggests that those predictions are highly likely true PTM sites, and the number of predicted sites comparable to that of known sites indicates that our current proteome-scale coverage of these PTMs is still incomplete.
| PTM type | Number of unreportedsites | Number of sites in PSP | Number of newly predicted sites | Ratio (predicted/PSP records) |
|---|---|---|---|---|
| Acetylation (K) | 515336 |
17084 |
5148 | 0.30 |
| Methylation (K) | 528563 |
3857 | 5238 | 1.36 |
| Ubiquitination (K) | 497377 |
35043 |
4997 | 0.14 |
| SUMOylation (K) | 526278 |
6142 | 5330 | 0.87 |
| Methylation (R) | 511803 |
7957 | 5072 | 0.64 |
| Phosphorylation (S) | 650342 |
108287 |
6578 | 0.06 |
| Phosphorylation (T) | 436467 |
45302 |
4359 | 0.10 |
| Phosphorylation (Y) | 214390 |
30444 |
2135 | 0.07 |
Further, the protein domains that were already enriched with experimentally acquired sites (PSP) gained more additional PTM sites from the prediction than other domains, including lysine methylation in histone domain and RNA recognition motif domain,37,38 lysine acetylation in protein kinase domain,39,40 and threonine phosphorylation in zinc finger C2H2 domain (ESI,† Fig. S3). In fact, the overall distribution of high confidence predictions in domains agrees with that of PSP sites in most of the PTM types (ESI,† Fig. S4). Therefore, we are confident that PTMscape provides accurate and biologically relevant predictions for protein modification sites to complement the set of experimentally acquired sites.
A global landscape of protein domains and biological processes enriched with PTMs in the human proteome
The combination of experimentally acquired sites in PSP and the sites predicted by PTMscape allows us to explore the protein functions modulated by them. Indeed, for certain modifications such as SUMOylation and methylation (K/R), the number of predicted sites was comparable to that of experimentally validated sites. Using the combined PTM data, we were able to perform an unbiased, in-depth analysis of PTM-enriched proteins and their domains across the human proteome. To do so, we mapped all sites in the combined set to 487 most frequently occurring protein domain families from the Pfam database41 using PTMscape, and tested their enrichment without the bias due to sub-proteomic coverage of detection. We remark that, for the PTMs that already had a proteome-scale coverage such as phosphorylation and ubiquitination, this analysis mostly reflects the enrichment of experimentally acquired sites, rather than predicted sites.More than 159 of these domain families showed statistically significant enrichment of specific types of PTMs (q-value < 0.05). Fig. S5A (ESI†) shows the heatmap of domain enrichment scores of all five PTMs (−logarithm of p-values base 10), an output directly tabulated from PTMscape as a post-prediction module. The domain families with the most statistically significant enrichment include protein kinase domain, zinc finger-C2H2 domain, RNA recognition motif, and histone domain (ESI,† Table S4). Protein kinases are often phosphorylated themselves by other kinases or auto-phosphorylation as part of a signal transduction cascade. In addition, other modifications such as ubiquitination are also well-known to affect the kinase's function.42 Both zinc finger domains and RNA recognition motifs bind nucleic acids, and abundant SUMOylation and phosphorylation events have been described.43,44 Finally, the histone modification ‘code’ is well-known, covering methylation, ubiquitination, and acetylation, for example.45,46
We then tested the enrichment of biological functions in the proteins harboring in-domain PTMs using the GeneMania plug-in in Cytoscape (q-value < 10−20 only).47Fig. 3A shows that mRNA splicing, spliceosome, RNA processing were the most significantly enriched in the domains with modifications of lysine and arginine residues, i.e. SUMOylation, ubiquitination, and methylation – consistent with the enrichment in nucleic acid binding domains. In comparison, functions in signal transduction, DNA damage response, regulation of cell cycle, immune response, and growth factor responses were enriched in domains with a variety of PTMs and affected different residues. Proteins whose domains accumulated threonine and tyrosine phosphorylation sites, and also lysine ubiquitination and acetylation often function in regulation of cell cycle, various receptor signaling pathways, and protein kinase activities – consistent with the abundant role of phosphorylation in signaling cascades. This landscape illustrates the co-existence of different PTMs on the same protein, such as those for p53 mentioned above, and these proteins can have many essential biological functions.
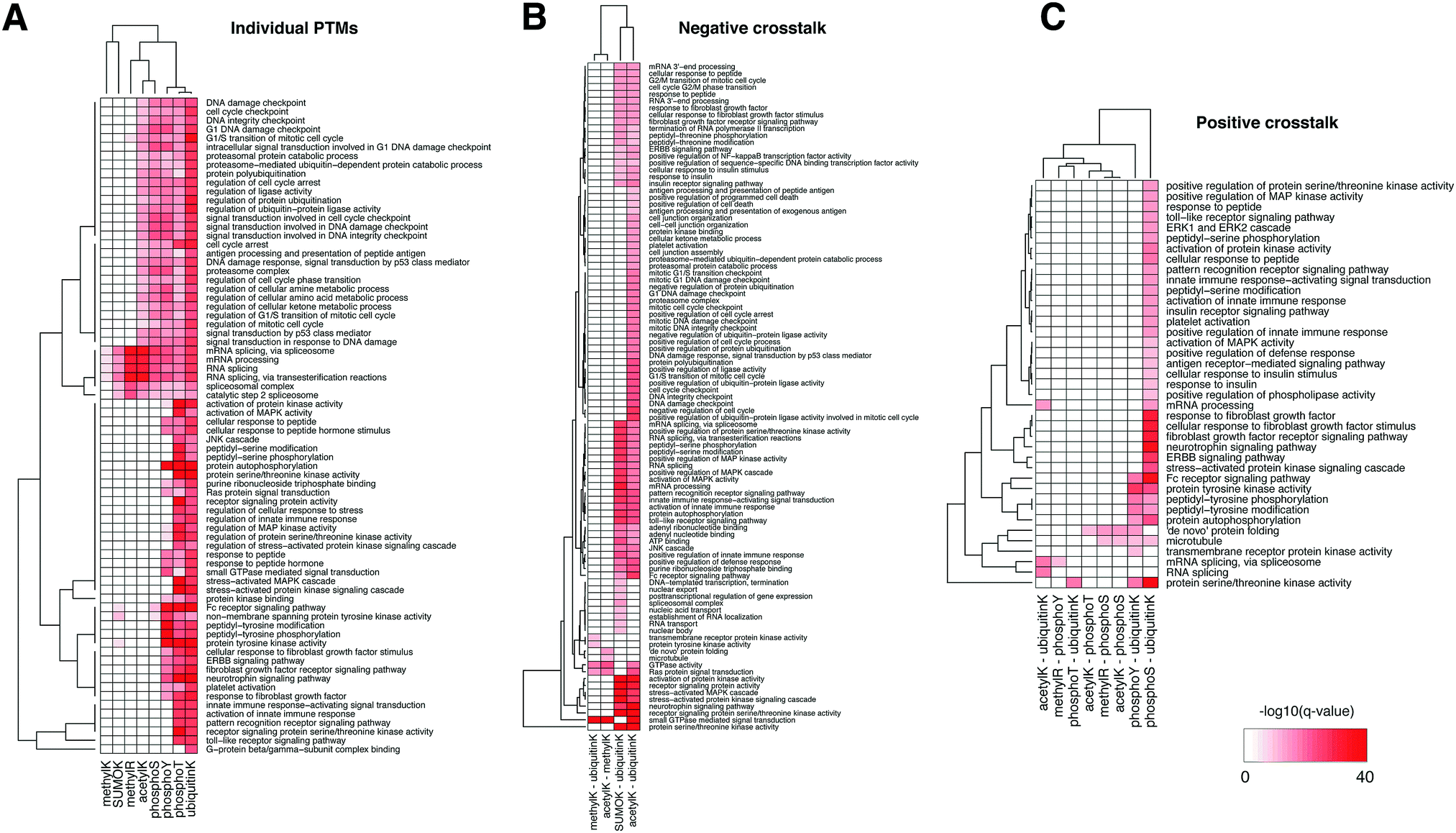 | ||
| Fig. 3 Biological functions enriched in the list of proteins containing (A) individual PTM sites, (B) negative crosstalk sites, and (C) positive crosstalk sites, using GeneMania plug-in in Cytoscape. For individual and crosstalk sites, we included the combined set of experimentally acquired (PSP) and computationally predicted (PTMscape) sites residing in Pfam domains. The color of heatmap was minus logarithm of q-values (base 10) from GeneMania. | ||
A global map of candidates for PTM crosstalk
The current map of all major PTMs, complemented by PTMscape's unified, proteome-scale prediction results, allows for unbiased analysis of interaction between different PTMs. This interaction is known as crosstalk,48 and a recent study reported that the sequence and spatial distances between homo- and heterotypic PTM pairs are non-randomly distributed in resolved protein structures.49 Referring to Hunter,50 we classify crosstalk activities into two types, namely positive crosstalk and negative crosstalk. Positive crosstalks are instances in which several modifications of the same or different type localize to the same sequence region (within five amino acids), but they do not affect the same residue. Many of these modifications might occur simultaneously, e.g. ubiquitination often affects multiple lysine residues within the same structural neighborhood. Positively ‘interacting’ modifications might also have temporal or even causal relationships: one modification might trigger another event on the same protein within the same sequence neighborhood. For example, phosphorylation often triggers subsequent SUMOylation, e.g. as is found for heat shock transcription factor HSF1 for its serine and lysine residues at positions 303 and 298, respectively.51 The p53 tumor suppressor protein can be activated through phosphorylation of serine 46, which promotes acetylation at lysine 382 through positive crosstalk.52,53In comparison, negative crosstalk includes direct competition of two PTMs for the same amino acid or indirect effects when a specific PTM masks the recognition site for second PTM. We only study the former scenario in this work, therefore such negative crosstalk can occur only for lysine residues: for example, lysine can be acetylated, methylated, SUMOylated, or ubiquitinated. However, since these modifications are chemically largely exclusive, we assume that only one can occur at a given time. Therefore, negative crosstalk represents cases in which the PTMs ‘compete’ for the same residue in a temporal or causal manner. Such negative cross occurs, for example, for lysine 382 in p53 mentioned above in which acetylation then competes with methylation.54
Using again the combined set of experimentally acquired sites and the predicted sites, we used the crosstalk analysis module in PTMscape to characterize potential negative crosstalk sites. For example, there are 532420 lysine residues in total across the human proteins considered here, which are candidates for ubiquitination, SUMOylation, acetylation, and methylation. Of these, 9511 lysines (1.8%) are experimentally acquired or predicted to host two or more different modifications. In other words, their sequence context suggested that multiple PTMs compete for the same lysine. As neither PSP nor PTMscape provides temporal resolution for the different PTMs, it remains to future research to resolve their causal relationships.
We then tested enrichment of these negative crosstalk sites in protein domains and biological functions as we did for individual PTMs above (Fig. 5B and Table S5, ESI†). Histone and tubulin domains showed the most pronounced enrichment of lysine residues with multiple PTMs. Further, several zinc finger domain families and the RNA recognition motifs were enriched in negative crosstalk sites for SUMOylation and ubiquitination. Proteins harboring negative crosstalk for acetylation and ubiquitination were involved in processes including DNA damage response, RNA processing, immune response, and signaling cascades (GeneMania q-value <10−10); the proteins with negative crosstalk for SUMOylation and ubiquitination specifically had enrichment of mRNA processing and kinase signaling pathways (Fig. 3B).
Positive crosstalk, i.e. the statistically significant accumulation of simultaneous modifications of different or identical types within the same immediate sequence neighborhood, was much more frequent than negative crosstalk. Since testing all possible combinations of modifications is statistically infeasible, we focused on evolutionarily conserved pairs of modifications.55 Fig. S5C (ESI†) shows the landscape of complex interplay of 80 domain families with statistically significant enrichment of their combined occurrence (Fisher exact test, q-value < 0.05, see Methods). When testing enrichments of combinations of phosphorylation, acetylation, and ubiquitination, we found that positive crosstalk sites had similar biases as the individual PTM analysis, i.e. they were enriched in domain families such as histone and linker histone, ubiquitin, tubulin, protein kinase domains, and the RNA recognition motif, and in functions such as mRNA processing and RNA splicing (Fig. 3C, GeneMania q-value < 10−10). Protein kinase activity, regulation of peptide hormones, receptor signaling pathways were enriched in the crosstalk sites among phosphorylation, acetylation, ubiquitination and SUMOylation events. The full landscape of domain-level and function-level enrichment of crosstalk is provided in ESI,† Table S5.
Features predictive of negative and positive crosstalks
In order to understand the features associated with crosstalk activities between a pair of PTMs, we again used PTMscape to build a prediction model for each crosstalk within the set of sites harboring at least one of the two PTMs, i.e. excluding all candidate sites with neither modification. For example, when negative crosstalk of acetylation and ubiquitination on lysine is investigated, among all the ubiquitinated and acetylated lysines, we investigated the features capturing the difference between the lysines which are experimentally acquired or predicted to be modified in both types and those modified in one of the two only. Similarly, when a positive crosstalk is studied, for example crosstalk of phosphorylation on serine and ubiquitination on adjacent lysine (within ±5 amino acids), we look for features which distinguish the pairs of S–K sites harboring PTMs of both types from the sites modified with one of the two types only.Linear SVMs were built for four negative and eight positive frequently occurring crosstalk types. Fig. S6(A–E) (ESI†) shows the weight coefficients of the models for four types of negative crosstalks. The figure shows that the model for the competition between acetylation and methylation is clearly different from the other crosstalks involving ubiquitination. The classifier for the former type of negative crosstalk shows that various hydrophobicity measures tend to be negatively associated with the chance of crosstalk (ESI,† Fig. S6A). Sites in the both edges of the windows tend to more solvent accessible (ESI,† Fig. S6B), and the probabilities that neighbor residues in specific positions are in a helical structure are also positively or negatively associated (ESI,† Fig. S6C and D).
The predictability of PSAAP matrix reveals strongest sequence motifs for the negative crosstalk between acetylation and methylation and some crosstalks involving ubiquitination (ESI,† Fig. S6E). Fig. S7 (ESI†) shows a [GL]K motif for the crosstalk of acetylation and methylation with depletion of lysines in position −2 and −3 only, KxxE motif for the crosstalk of ubiquitination and SUMOylation (in contrast to KxE motif in SUMOylation), a clear LKxx for the crosstalk of ubiquitination and methylation, and [GLE]KxL for the crosstalk of ubiquitination and acetylation with depletion of lysines. All three crosstalks involving ubiquitination showed clear depletion of lysines in immediately adjacent positions.
Meanwhile, the weight coefficients of models for the eight positive crosstalks are presented in ESI,† Fig. S8(A–E). In general, the partition coefficient solubility and alpha-CH chemical shifts are the mostly positively predictive of these crosstalks, while hydrophilicity and entropy are negatively predictive (ESI,† Fig. S8A). ASA and HSE for sites near the sites were positively predictive of most of these crosstalk types except for the crosstalk between arginine methylation and serine phosphorylation (ESI,† Fig. S8B). The coefficients for the secondary structure information from SPIDER3 did not show any clearly interpretable patterns. Although the coefficients for PSAAP features were the largest in the adjacent positions, the high coefficient values in the neighboring ±5 amino acid positions are an artifact of our current definition of positive crosstalk, i.e. the two target residues are always within such a window. Overall, PTMscape's prediction models for positive crosstalk were the most distinguished by the difference between those for crosstalks involving tyrosine phosphorylation and the others, in which the amino acid properties and solvent accessibility played the biggest role in predicting such sites.
Discussion
In this work, we developed a new computational tool called PTMscape for generic, unified prediction of protein modifications at the whole-proteome scale. To the best of our knowledge, PTMscape is equipped with the most comprehensive set of predictors and uses a fast and robust machine-learning algorithm whose results are easily interpretable for their biological meaning. We demonstrate PTMscape through the example of five types of PTMs analyzed across the entire human proteome, and their potential interactions or co-occurrences.PTMscape moves beyond existing tools for in silico prediction of PTMs in several ways. First and most importantly, it is generic and can be used for any type of the ∼200 currently known PTM types as long as there is training data, such as the data from a single mass spectrometry analysis with enrichment of the given PTM. Second, PTMscape is easy to use at the proteome scale as it is installed locally. It also allows the user to customize the analysis with respect to the types of PTMs that are analyzed, the background sequence file, the types of predictors to be evaluated and, most importantly, the experimental data that is used for training a prediction model. Therefore, PTMscape provides a tool for the expert user who needs a large-scale, comprehensive analysis in the most flexible format. Finally, PTMscape provides substantial additional information that will help interpretation of the results. It evaluates the modification's microenvironment, including the physicochemical properties, site-specific secondary structure properties, and motifs within the sequence neighborhood. PTMscape also includes less commonly used predictors, such as the accessible surface area or secondary structures. Including analysis of such features of the protein three-dimensional structure proved to be highly valuable in particular for modifications such as ubiquitin that lack a defined sequence motif.56,57
These aspects also illustrate future extensions that address some of PTMscape's current limitations. For example, for statistically robust analysis, at least a few thousand experimentally detected sites need to be available. In particular, for PTMs that target a wide range of sequences without the presence of a global motif, such as ubiquitin mentioned above, the training data needs to include a wide range of diverse sites. However, for many types of PTMs, such as O-linked glycosylation, available data is limited at the moment. Therefore, future extensions of PTMscape will address this challenge by exploring additional predictive feature variables extracted from external data, such as information on protein–protein interaction data or features pertaining to each residue's position in the protein structure as obtained from experiments or modeling – moving beyond the current sequence-based prediction. Similarly, future extension might also use windows surrounding possible modification sites based on structural similarity, rather than sequence proximity, or pursue simultaneous prediction of such multiple PTM types in a single model, incorporating the domain and other structural information.6,58
Methods
Overview of the workflow
PTMscape is a R package (http://http:/cran.r-project.org), and it is provided through a GitHub repository, at https://github.com/ginnyintifa/PTMscape under Apache 2.0 license. PTMscape builds sub-sequence windows centered at known or candidate PTM sites. It encompasses a comprehensive set of features describing modification events, which are used as training and test data in the linear kernel SVM library called liblinear.59 The prediction output is further annotated with domain and sub-cellular information. Finally, PTMscape offers statistical tests for the enrichment of protein domains in individual PTM sites and co-occurrence of any two types of PTM events (crosstalk).Source data
From Uniprot Swiss-Prot database, we downloaded 20201 canonical human proteins (as of August 2017).5 Experimentally acquired PTM sites corresponding to five PTM types (Table 3) were gathered from PhosphoSitePlus database.3 A total of 538 numerical indices representing various physicochemical and biomedical properties for 20 amino acids were retrieved from AAindex.22 Position specific secondary structural features, accessible surface area (ASA) and half sphere exposure (HSE) of each residue were predicted with the SPIDER3 tool.23 Information on protein domain families were obtained from the Pfam using the command line tool.41
Removal of sequence redundancy
To obtain a set of non-redundant proteins for classifier performance evaluation, we used the CD-HIT software60 at similarity level 0.3 to reduce the redundancy of the whole proteome (sets of proteins containing at least one modifiable residue for respective PTM type). As a result, prediction of phosphorylation on serine (S) site was conducted for 11490 non-redundant sequences which contained at least one serine. Similarly, we have 12188, 11986, 12222 and 11872 non-redundant sequences for analysis of PTMs on threonine (T), tyrosine (Y), lysine (K), and arginine (R) respectively.
Amino acid index dimension reduction
To reduce feature dimension and minimize correlation within the indices, we hierarchically clustered the features downloaded from amino acid index into 53 clusters using dynamic tree cut algorithm,61 each summarizing a group of similar physicochemical properties for the amino acids such as hydrophobicity, eccentricity size, solubility etc. Detailed information on the clusters is described in ESI,† Table S6. For each window, we calculated the average property of each cluster (excluding the center site). Note that all other features below were position specific around the modified residue (−12, −11,…, −2, −1, +1, +2,…, 12).Position specific structural features
SPIDER3 is a sequence-based prediction tool for local and non-local structural features in proteins using Long Short-Term Memory Neural Networks.23 Prediction output includes probabilities of three secondary structures (helix, strand, and coil), ASA and HSE. For each flanking residue in a window, we extracted four features, namely p_Coil, p_Helix, ASA and HSE. Under the default mode, we constructed 96 dimensional position-specific structural features.Position specific amino acid propensity (PSAAP)
PSAAPs were derived from the frequency of each amino acid in each of the 24 positions within all the windows centered with positive PTM sites (excluding the center residue). Therefore, the matrix is composed of 20 rows and 24 columns. Each column records the probability of 20 amino acids appearing in the corresponding position based on the observed frequency in the positive sites. For instance, for position j, column j can be expressed in the following vector:| (a1j, a2j,…,aij,…,a20j)T |
Note that
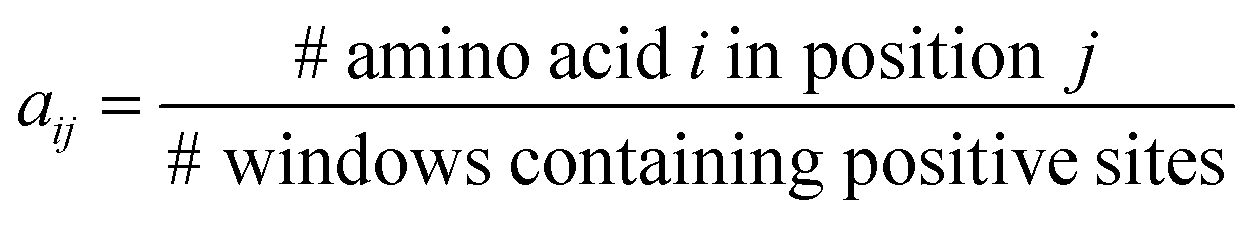
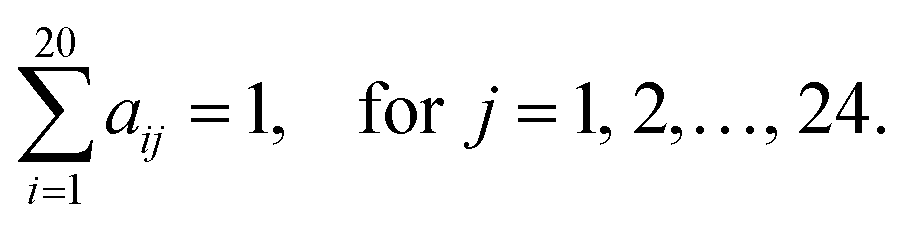
Feature data generation
The data generation module maps protein identifiers and positions of experimentally determined PTM sites (positive sites) to any protein sequences in the user-provided FASTA file, and computes average AA specific features for the k-mer window centered surrounding each candidate site (default k = 25, having 12 amino acids on both sides of each site). The feature variables used in the default mode include 53 clusters of physiochemical properties derived from the AAindex database, 96 (24 × 4) position specific structure properties generated by SPIDER3, and 24 dimensional features formed by position specific amino acid propensity (PSAAP). We refer to the resulting data as the feature data. The program also generates feature data for windows that contain negative sites, i.e. sites without experimental evidence for their modification.Support vector machine with linear kernel
PTMscape uses support vector machine (SVM)16 with linear kernel, to achieve the best classification of PTM sites while avoiding over-training. Moreover, linear SVM provides easy interpretation of how each property contributes to the decision function of the SVM. In a linear kernel setting, the classification is achieved by solving the following problem:where yi equals to 1 or −1 representing the class label of support vector Xi, W represents the weight coefficient for each feature. C is the soft margin tuning parameter. The model fitting is performed using Liblinear library. During training, we used L2-regularized L2-loss support vector classification (-s 2). To obtain the probability score in addition to the classification labels from each prediction, we modified the code of liblinear as suggested in the FAQ page (https://www.csie.ntu.edu.tw/~cjlin/liblinear/FAQ.html).
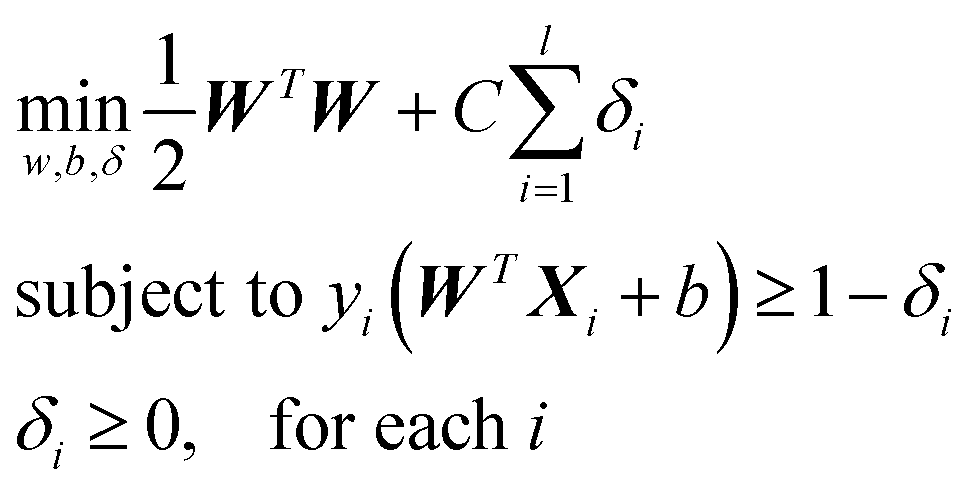
Prediction performance metrics
At a given score threshold, we recorded the true positives (TF), true negatives (TN), false positives (FP) and false negatives (FN). The performance metrics were defined as: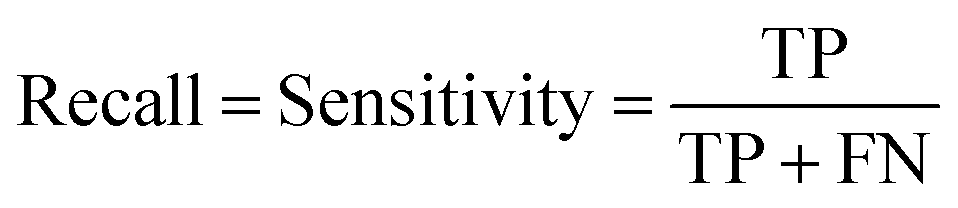
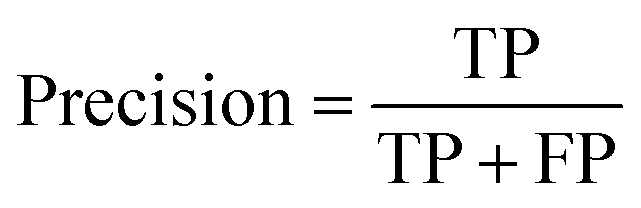
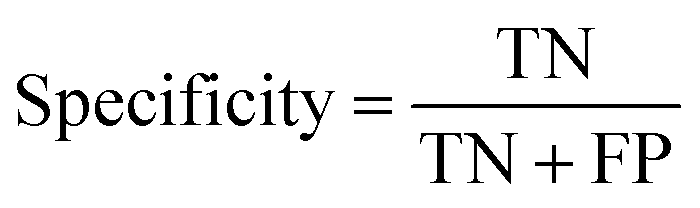
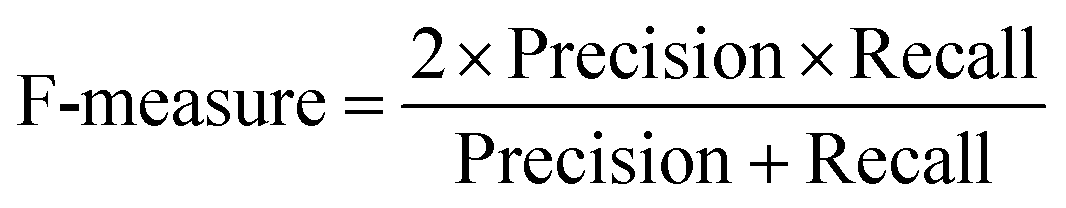
where MCC denotes Matthew's correlation coefficient.
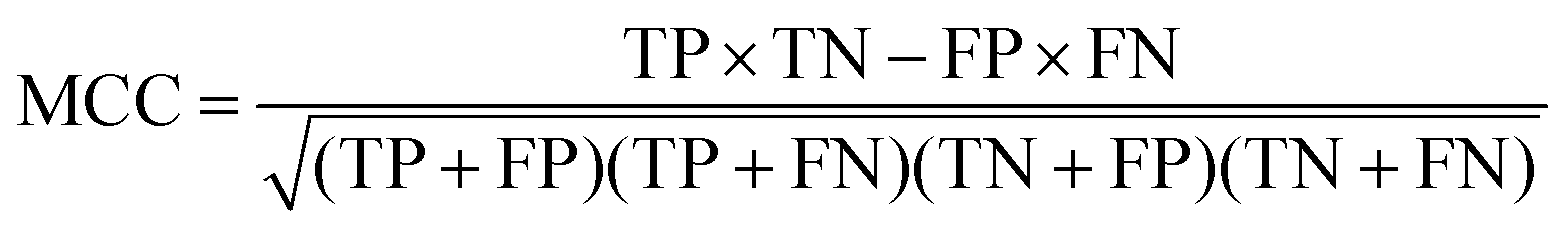
Evaluation of the classifier performance with 10-fold cross validation
To evaluate the performance of the classifier unbiasedly, we implemented a 10-fold cross validation scheme. For each PTM type, we first randomly sampled the same number of negative and the positive windows and randomly divided both sets (positive and negative) into ten folds. Each time we assembled nine out of the ten folds as a training dataset and used the remaining one fold as a validation dataset. Therefore, training and testing was performed on independent datasets. The performance metrics such as Area Under Curve (AUC) and Matthew's Correlation Coefficient (MCC) were calculated by taking the average of the ten iterations.Predictions with PhosphoSVM and ESA-Ubisite
To benchmark our tool against PhosphoSVM, we used the data retrieved from their web portal at http://sysbio.unl.edu/PhosphoSVM/download.php. PTMscape extracted features for 9688, 2919, and 1269 positive PTM sites (on 2545, 1499, and 805 proteins) for serine, threonine and tyrosine phosphorylation, respectively. Then 10-fold cross validation was conducted on the feature data generated. For comparison with ESA-UbiSite, we obtained the data consisting of 645 positive sites and 10336 non-validated sites from 379 protein sequences from their work.21 Specifically, we collected the proteins in the UbiD set for which position specific structural features are available. PTMscape was applied to 355 proteins with 613 positive ubiquitination sites and the same proportion of training and test division was implemented. For model training, we obtained the same number of negative windows as positive windows by randomly sampling the negatives from the whole negative set.
Comparison with ModPred
ModPred predicts as many as 23 different PTM types in different species. The predictor in ModPred is trained with sequence-based properties, physicochemical features and evolutionary conservation information, where ensembles of logistic regression models were built per modification residue type. Using the stand-alone version ModPred_Linux64, we predicted all PTMs on lysine residues for the proteins in the Swiss-Prot database that are longer than 30 residues and hold one or more modifiable lysine (without PSSM features). The AUC of ModPred predictions was calculated by comparing the output prediction scores with known PTM status from the PhosphoSitePlus database.Prediction on the whole human proteome
To predict PTM sites in the whole proteome, we gathered all 17612 sequences for which position-specific structural features can be extracted by SPIDER3. PTMscape was applied in a two-fold cross-prediction scheme, where we used half the data as training data and predicted on the other half, and switched the role of training and test data to make predictions in the former. This ensures that the same data point is not used for training and prediction simultaneously, rendering predictions unbiased. During the training, we randomly sampled from the negative set in a way that the sizes of positive windows and negative windows were the same.
All predicted sites are provided through the website http://137.132.97.109:59739/CSSB_LAB/. The tables list the PTM type, PTMscape score, the score threshold in the respective PTM type associated with 99% specificity, and reports on significant enrichment in protein domain families or protein functions. The table also indicates whether a PTM site is ‘new’, i.e. if it has not been observed in the PSP database that we used for training.
Enrichment analysis of PTM occurrence in protein domains and biological functions
For the enrichment analysis of individual PTMs in each domain, we first counted the number of positive and negative PTM sites in the domain and across the proteome, and performed a chi-square test to test whether the frequency of a PTM type in a domain is significantly higher than the expected frequency across the proteome. We selected domain families with q-value smaller than 0.05.For the analysis of negative crosstalk, we tested association of two competing PTMs as follows. In each domain, we constructed a 2-by-2 contingency table where rows and columns are positive and negative status of the two competing PTM events on all modifiable sites within that domain family. We then tested whether the two PTMs are significantly more frequent in the domain than expected under by Fisher exact test.
For the analysis of positive crosstalk in a domain, we similarly constructed a contingency table, where the sum of the numbers in the four cells is the number of all pairs of modifiable residues within a domain. Here a modifiable pair of residues in a domain refers to two amino acids located within 5 amino acids, where one residue is modifiable in one of the two PTM types and the other residue is modifiable in the other PTM type. Each pair contributes to one of the four cells in the contingency table based on the PTM status of respective types. After the construction of the contingency table, we tested the significance of enrichment of co-occurring two PTMs in a domain by fisher exact test. These significance scores (p-value) were further adjusted for multiple testing (q-value), and the domains with q-value smaller than 0.05 were considered to have a significant positive crosstalk event.
The full table of protein domains enriched in the list of crosstalk can be found in ESI,† Table S5. The tables list the paired modifications, the p-values from the chi-squared test and the q-values (with multiple testing correction) for each protein domain.
Function enrichment analysis of individual PTMs and crosstalk
A list of proteins with individual and crosstalk events in protein domains was analyzed with Genemania47 for function enrichment. In GeneMania, we used 2017-07-13-core version of the human data, with annotation limited to Pathway only, without finding any other top related genes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by a grant from the Singapore Ministry of Education (to HC; MOE2016 T2-1-001) and the United States National Institutes of Health (to CV and HC; 1R01GM113237-01).References
- S. Prabakaran, G. Lippens, H. Steen and J. Gunawardena, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2012, 4, 565–583 CrossRef PubMed.
- C. Choudhary and M. Mann, Nat. Rev. Mol. Cell Biol., 2010, 11, 427–439 CrossRef PubMed.
- P. V. Hornbeck, B. Zhang, B. Murray, J. M. Kornhauser, V. Latham and E. Skrzypek, Nucleic Acids Res., 2015, 43, D512–520 CrossRef PubMed.
- F. Gnad, J. Gunawardena and M. Mann, Nucleic Acids Res., 2011, 39, D253–D260 CrossRef PubMed.
- C. UniProt, Nucleic Acids Res., 2015, 43, D204–D212 CrossRef PubMed.
- P. Minguez, I. Letunic, L. Parca and P. Bork, Nucleic Acids Res., 2013, 41, D306–D311 CrossRef PubMed.
- C. Dai and W. Gu, Trends Mol. Med., 2010, 16, 528–536 CrossRef PubMed.
- J. P. Kruse and W. Gu, Cell, 2008, 133, 930–930.e1 CrossRef PubMed.
- B. Gu and W. G. Zhu, Int. J. Biol. Sci., 2012, 8, 672–684 CrossRef PubMed.
- H. J. Kim, S. Ha, H. Y. Lee and K. J. Lee, Mass Spectrom. Rev., 2015, 34, 184–208 CrossRef PubMed.
- B. Trost and A. Kusalik, Bioinformatics, 2011, 27, 2927–2935 CrossRef PubMed.
- Z. Chen, Y. Zhou, J. Song and Z. Zhang, Biochim. Biophys. Acta, 2013, 1834, 1461–1467 CrossRef PubMed.
- C. W. Tung and S. Y. Ho, BMC Bioinf., 2008, 9, 310 CrossRef PubMed.
- P. Radivojac, V. Vacic, C. Haynes, R. R. Cocklin, A. Mohan, J. W. Heyen, M. G. Goebl and L. M. Iakoucheva, Proteins, 2010, 78, 365–380 CrossRef PubMed.
- J. Shao, D. Xu, S. N. Tsai, Y. Wang and S. M. Ngai, PLoS One, 2009, 4, e4920 Search PubMed.
- C. Cortes and V. Vapnik, Mach. Learn., 1995, 20, 273–297 Search PubMed.
- V. Pejaver, W. L. Hsu, F. Xin, A. K. Dunker, V. N. Uversky and P. Radivojac, Protein Sci., 2014, 23, 1077–1093 CrossRef PubMed.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- A. K. Jain, J. Mao and K. M. Mohiuddin, Computer, 1996, 29, 31–44 CrossRef.
- G. James, D. Witten, T. Hastie and R. Tibshirani, An introduction to statistical learning, Springer, 2013 Search PubMed.
- J. R. Wang, W. L. Huang, M. J. Tsai, K. T. Hsu, H. L. Huang and S. Y. Ho, Bioinformatics, 2017, 33, 661–668 Search PubMed.
- S. Kawashima, P. Pokarowski, M. Pokarowska, A. Kolinski, T. Katayama and M. Kanehisa, Nucleic Acids Res., 2008, 36, D202–D205 CrossRef PubMed.
- R. Heffernan, Y. Yang, K. Paliwal and Y. Zhou, Bioinformatics, 2017, 33, 2842–2849 CrossRef PubMed.
- Y. R. Tang, Y. Z. Chen, C. A. Canchaya and Z. Zhang, Protein Eng., Des. Sel., 2007, 20, 405–412 CrossRef PubMed.
- P. Durek, C. Schudoma, W. Weckwerth, J. Selbig and D. Walther, BMC Bioinf., 2009, 10, 117 CrossRef PubMed.
- Y. Dou, B. Yao and C. Zhang, Amino Acids, 2014, 46, 1459–1469 CrossRef PubMed.
- T. Y. Lee, H. D. Huang, J. H. Hung, H. Y. Huang, Y. S. Yang and T. H. Wang, Nucleic Acids Res., 2006, 34, D622–D627 CrossRef PubMed.
- H. Li, X. Xing, G. Ding, Q. Li, C. Wang, L. Xie, R. Zeng and Y. Li, Mol. Cell. Proteomics, 2009, 8, 1839–1849 Search PubMed.
- R. Amanchy, K. Kandasamy, S. Mathivanan, B. Periaswamy, R. Reddy, W. H. Yoon, J. Joore, M. A. Beer, L. Cope and A. Pandey, J. Proteomics Bioinf., 2011, 4, 22–35 Search PubMed.
- I. Matic, J. Schimmel, I. A. Hendriks, M. A. van Santen, F. van de Rijke, H. van Dam, F. Gnad, M. Mann and A. C. Vertegaal, Mol. Cell, 2010, 39, 641–652 CrossRef PubMed.
- F. M. Boisvert, U. Dery, J. Y. Masson and S. Richard, Genes Dev., 2005, 19, 671–676 CrossRef PubMed.
- K. P. Lu, Y. C. Liou and X. Z. Zhou, Trends Cell Biol., 2002, 12, 164–172 CrossRef PubMed.
- W. Kim, E. J. Bennett, E. L. Huttlin, A. Guo, J. Li, A. Possemato, M. E. Sowa, R. Rad, J. Rush, M. J. Comb, J. W. Harper and S. P. Gygi, Mol. Cell, 2011, 44, 325–340 CrossRef PubMed.
- S. Teng, H. Luo and L. Wang, Amino Acids, 2012, 43, 447–455 CrossRef PubMed.
- G. Beauclair, A. Bridier-Nahmias, J. F. Zagury, A. Saib and A. Zamborlini, Bioinformatics, 2015, 31, 3483–3491 CrossRef PubMed.
- J. Ren, X. Gao, C. Jin, M. Zhu, X. Wang, A. Shaw, L. Wen, X. Yao and Y. Xue, Proteomics, 2009, 9, 3409–3412 CrossRef PubMed.
- M. Lachner and T. Jenuwein, Curr. Opin. Cell Biol., 2002, 14, 286–298 CrossRef PubMed.
- L. Tresaugues, P. M. Dehe, R. Guerois, A. Rodriguez-Gil, I. Varlet, P. Salah, M. Pamblanco, P. Luciano, S. Quevillon-Cheruel, J. Sollier, N. Leulliot, J. Couprie, V. Tordera, S. Zinn-Justin, S. Chavez, H. van Tilbeurgh and V. Geli, J. Mol. Biol., 2006, 359, 1170–1181 CrossRef PubMed.
- X. J. Yang and E. Seto, Mol. Cell, 2008, 31, 449–461 CrossRef PubMed.
- B. L. Parker, N. E. Shepherd, S. Trefely, N. J. Hoffman, M. Y. White, K. Engholm-Keller, B. D. Hambly, M. R. Larsen, D. E. James and S. J. Cordwell, J. Biol. Chem., 2014, 289, 25890–25906 CrossRef PubMed.
- R. D. Finn, P. Coggill, R. Y. Eberhardt, S. R. Eddy, J. Mistry, A. L. Mitchell, S. C. Potter, M. Punta, M. Qureshi, A. Sangrador-Vegas, G. A. Salazar, J. Tate and A. Bateman, Nucleic Acids Res., 2016, 44, D279–D285 CrossRef PubMed.
- B. Mohapatra, G. Ahmad, S. Nadeau, N. Zutshi, W. An, S. Scheffe, L. Dong, D. Feng, B. Goetz, P. Arya, T. A. Bailey, N. Palermo, G. E. Borgstahl, A. Natarajan, S. M. Raja, M. Naramura, V. Band and H. Band, Biochim. Biophys. Acta, 2013, 1833, 122–139 CrossRef PubMed.
- I. A. Hendriks, D. Lyon, C. Young, L. J. Jensen, A. C. Vertegaal and M. L. Nielsen, Nat. Struct. Mol. Biol., 2017, 24, 325–336 Search PubMed.
- A. C. Vertegaal, S. C. Ogg, E. Jaffray, M. S. Rodriguez, R. T. Hay, J. S. Andersen, M. Mann and A. I. Lamond, J. Biol. Chem., 2004, 279, 33791–33798 CrossRef PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef PubMed.
- S. B. Rothbart and B. D. Strahl, Biochim. Biophys. Acta, 2014, 1839, 627–643 CrossRef PubMed.
- D. Warde-Farley, S. L. Donaldson, O. Comes, K. Zuberi, R. Badrawi, P. Chao, M. Franz, C. Grouios, F. Kazi, C. T. Lopes, A. Maitland, S. Mostafavi, J. Montojo, Q. Shao, G. Wright, G. D. Bader and Q. Morris, Nucleic Acids Res., 2010, 38, W214–W220 CrossRef PubMed.
- A. S. Venne, L. Kollipara and R. P. Zahedi, Proteomics, 2014, 14, 513–524 CrossRef PubMed.
- P. Korkuc and D. Walther, Proteins, 2017, 85, 78–92 CrossRef PubMed.
- T. Hunter, Mol. Cell, 2007, 28, 730–738 CrossRef PubMed.
- V. Hietakangas, J. K. Ahlskog, A. M. Jakobsson, M. Hellesuo, N. M. Sahlberg, C. I. Holmberg, A. Mikhailov, J. J. Palvimo, L. Pirkkala and L. Sistonen, Mol. Cell. Biol., 2003, 23, 2953–2968 CrossRef PubMed.
- K. Yoshida, H. Liu and Y. Miki, J. Biol. Chem., 2006, 281, 5734–5740 CrossRef PubMed.
- T. G. Hofmann, A. Moller, H. Sirma, H. Zentgraf, Y. Taya, W. Droge, H. Will and M. L. Schmitz, Nat. Cell Biol., 2002, 4, 1–10 CrossRef PubMed.
- X. Shi, I. Kachirskaia, H. Yamaguchi, L. E. West, H. Wen, E. W. Wang, S. Dutta, E. Appella and O. Gozani, Mol. Cell, 2007, 27, 636–646 CrossRef PubMed.
- P. Minguez, L. Parca, F. Diella, D. R. Mende, R. Kumar, M. Helmer-Citterich, A. C. Gavin, V. Van Noort and P. Bork, Mol. Syst. Biol., 2012, 8, 599 CrossRef PubMed.
- H. M. Dewhurst, S. Choudhury and M. P. Torres, Mol. Cell. Proteomics, 2015, 14, 2285–2297 Search PubMed.
- M. P. Torres, H. Dewhurst and N. Sundararaman, Mol. Cell. Proteomics, 2016, 15, 3513–3528 Search PubMed.
- P. Minguez, I. Letunic, L. Parca, L. Garcia-Alonso, J. Dopazo, J. Huerta-Cepas and P. Bork, Nucleic Acids Res., 2015, 43, D494–D502 CrossRef PubMed.
- R.-E. Fan, K.-W. Chang, C.-J. Hsieh, X.-R. Wang and C.-J. Lin, J. Mach. Learn. Res., 2008, 9, 1871–1874 Search PubMed.
- W. Li and A. Godzik, Bioinformatics, 2006, 22, 1658–1659 CrossRef PubMed.
- P. Langfelder, B. Zhang and S. Horvath, Bioinformatics, 2008, 24, 719–720 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mo00027a |
| This journal is © The Royal Society of Chemistry 2018 |