
Co/CoP embedded in a hairy nitrogen-doped carbon polyhedron as an advanced tri-functional electrocatalyst†
Yongchao
Hao
abc,
Yuqi
Xu
ac,
Wen
Liu
 *ac and
Xiaoming
Sun
*ad
*ac and
Xiaoming
Sun
*ad
aState Key Laboratory of Chemical Resource Engineering, Beijing, 100029, P. R. China. E-mail: wenliu@mail.buct.edu.cn; sunxm@mail.buct.edu.cn
bSchool of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China
cFaculty of Science, Beijing University of Chemical Technology, China
dBeijing Advanced Innovation Centre for Soft Matter Science and Engineering, College of Energy, Beijing University of Chemical Technology, China
First published on 8th November 2017
Abstract
The design and synthesis of a multi-functional electrocatalyst with low cost and high efficiency is still a great challenge. Herein, we report the rational design and realization of a tri-functional electrocatalyst featuring Co/CoP embedded in a hairy nitrogen-doped carbon polyhedral (Co/CoP–HNC) derived from common metal organic frameworks (MOFs). The components of Co, CoP, and hairy nitrogen-doped carbon in the catalyst respectively render catalytic activity, high electrochemical surface area, and electronic conductivity. The synergy of the tailored Co/CoP–HNC catalyst provides high activity for oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER). The catalytic performance and assembly feasibility are further demonstrated in overall water splitting and Zn–air battery tests, and both revealed high efficiency and long durability of the proposed electrocatalyst.
Conceptual insightsDesign and construction of electrocatalysts for energy-related applications have attracted intensive interest over the past few decades. Despite tremendous efforts on the catalyst design for the specified reaction, catalysts capable of promoting several reactions in the tandem or switchable manner can simplify catalytic electrode design and can be of great importance for practical applications. Although there are some reports on bi-functional catalysts for OER/ORR or OER/HER, to the best of our knowledge, the design and realization of the catalyst with tri-functions is rare. In this study, we present a tri-functional electrocatalyst featuring Co/CoP embedded in hairy nitrogen-doped carbon polyhedral (Co/CoP–HNC), which can provide high activity for ORR, OER, and HER. The great potential for practical applications is further demonstrated by overall water splitting and a Zn–air battery test, and both revealed high efficiency and long durability of the proposed catalyst. Distinct from that of other catalysts derived from MOF structures, the idea of rational design for HER/OER/ORR that combined Co, CoP and hairy nitrogen doped carbon in the catalyst renders high catalytic activity, electrochemical surface area, and electronic conductivity. The design strategy can expand the utilization of MOF-derived materials and is also expected to be applicable for a wide range of catalyst or functional materials. |
Introduction
Ever-growing concerns about global warming and energy shortages demand the expansion of clean and sustainable energy sources and reformation of our schemes toward harvesting and utilizing energy.1–3 Electrochemical reactions are of great importance in energy-related applications such as in PEM electrolyzers, fuel cells, and metal–air batteries.4–7 In all these concepts, the redox reactions of hydrogen gas and oxygen gas inevitably determine the efficiency of the energy conversion process as the HER, HOR, OER, and ORR in the electrochemical reactions.1,8–10 Efficient catalysts are of great importance for these energy-related applications, taking the central role in determining overall energy efficiency for hydrogen gas production and utilization in fuel cells or round-trip efficiency and stability of metal–air batteries.To date, a large number of non-precious catalysts have been investigated and verified as efficient in promoting the redox kinetics.11,12 Traditionally, each catalyst is aimed at a specific reaction owing to the specific interaction between catalytic active sites and intermediate species. However, in quite a number of applications, multiple reactions occur in a tandem or switchable manner; thus, different catalysts are required for different reactions occurring at the same electrode;13–15 for example, for gas electrodes in fuel cells and metal–air batteries, the redox reaction needs both OER and ORR catalysts.16–20 Catalysts capable of promoting several reactions can, therefore, simplify the catalytic electrode design and construction required in these important applications. Moreover, propelling of several catalytic reactions with a certain catalyst can provide a unique way to probe and rationalize the interactions of different reactive intermediate species with catalytic active sites; thus, this would further benefit our understanding of kinetics and catalyst design. In this regard, there has been considerable interest in the development of multifunctional catalysts that exhibit catalytic activities towards several important reactions. However, a multi-functional catalyst design and understanding of the associated mechanisms of activity in several reactions are still at their infancy. For instance, Ni3Fe/N-C sheets, S/N-Fe/N/C-CNT, and Co3O4C-NA exhibited excellent activity in the ORR and OER;7,16,21 Co-P/NC and Cu0.3Co2.7P/NC were derived from MOFs and Janus Co/CoP; and Ni2P nanoparticles demonstrated outstanding HER/OER activities.22–25 To date, the construction of an electrocatalyst based on different components with high efficiency towards multiple reactions is still a great challenge, which requires innovative material design and is of pivotal importance in energy-related applications.
Herein, we report the rational design and construction of an efficient and stable tri-functional catalyst with Co/CoP particles embedded on a hierarchical nitrogen-doped carbon polyhedral (HNC) penetrated with carbon nanotubes (N-CNT). The catalyst architecture is synthesized through a three-step chemical approach, which starts from feasible preparation of cobalt-based MOFs (ZIF-67). The pyrolysis of dicyandiamide and MOFs results in the formation of carbon polyhedrals penetrated with bamboo-like carbon nanotubes, which provide large interface areas for catalytic reactions. The intertwined N-CNT network also boosts the interfacial contact between polyhedrons and offers long range conductivity. The Co nanoparticles embedded inside the N-CNT can provide ORR activity, whereas CoP nanoparticles on the side wall of HNC resulting from phosphorization of uncovered Co nanoparticles offer high catalytic activity towards the OER and HER. Due to these advantageous features, the newly developed Co/CoP–HNC-based catalyst is different from the former MOF-derived catalysts as the Co/CoP–HNC-based catalyst shows outstanding tri-functional catalytic activity towards the ORR, OER, and HER in a KOH solution.17,21,23 In particular, the catalyst exhibits an onset potential of 0.93 V and a half-wave potential of 0.83 V for ORR, which is comparable to the activity of the benchmark Pt/C electrocatalyst, as well as high durability and methanol tolerance. The Co/CoP–HNC also exhibits high activity towards HER in a 1.0 M KOH solution; it only needs an overpotential of 0.18 V and 0.30 V to drive the current densities of 10 mA cm−2 and 100 mA cm−2, respectively; this shows that Co/CoP–HNC is one of the most active non-precious catalysts for the HER. Moreover, the rechargeable Zn–air battery indicates a high round-trip efficiency with a low overpotential and stable voltage plateau after 100 cycles when Co/CoP–HNC is used as an air cathode. The overall water splitting with Co/CoP–HNC requires 1.68 V to drive a 10 mA cm−2 current density with good stability; this is a further evidence of the feasibility and advantages of the tri-functional electrocatalyst.
Experimental
Preparation of the Co/CoP–HNC
Typically, 2.63 g of 2-methylimidazole (2-MIM) was first dissolved in 25 mL methanol; then, the solution was poured into a 25 mL methanol solution containing 1.16 g of Co(NO3)2·6H2O under vigorous stirring. The mixed solution was kept still for 24 h. The violet, powdery precipitates were obtained by centrifugation and washed with methanol several times followed by vacuum drying for 24 h.The as-prepared polyhedrons of ZIF-67 (10 mg) were put in a crucible that was placed in a tube furnace where a porcelain boat containing 3 g of dicyandiamide was placed frontally. The tube furnace was heated to 700 °C under an Ar flow (100 sccm) and maintained for 2 h. After being cooled down naturally, the as-obtained black powders were designated as Co–HNC pre. Subsequently, Co–HNC was phosphatized by 1 g sodium hypophosphite as the P source at 320 °C for 2 h. The obtained product was named as Co/CoP–HNC. The Co–HNC was prepared by acid treatment of Co–HNC pre to remove the exposed unstable metallic Co. The Co/CoP–NC was generated when the ZIF-67 was annealed without dicyandiamide and then phosphatized under the same condition.
Characterization
The morphology of the samples was observed by scanning electron microscopy (SEM) using Zeiss SUPRA 55 at 20 kV. The microstructure of the samples was characterized by high-resolution transmission electron microscopy (HRTEM) using JEOL 2100 at 200 kV. The X-ray powder diffraction patterns were obtained using Rigaku D/max 2500 in the 2 theta range from 10 to 90° at a scan rate of 10° per min. The elemental mapping was carried out by a Bruker Quantax energy dispersive X-ray spectrometer (EDS). The surface chemistry was analyzed by X-ray photoelectron spectroscopy (XPS) using Thermo Electron ESCALAB 250 with an Al X-ray source.Electrochemical measurements
To prepare the catalyst ink, 2.0 mg of catalyst was dispersed by ultrasonication in a mixed solution of 20 μL Nafion (DuPont, 5 wt%) and 0.2 mL DMF for 30 min. Then, 4 μL of the black suspension was dropped on a polished glass carbon (GC) electrode or rotating disk electrode (diameter 5 mm, RDE) and dried slowly under ambient conditions. The catalyst loading was controlled at 0.19 mg cm−2. The ORR tests were conducted using an electrochemical workstation (CHI 660D) connected with a standard three-electrode system. The catalyst-loaded GC electrode or RDE was employed as the working electrode. A Pt plate was employed as the counter electrode, and a saturated calomel electrode (SCE) was used as the reference electrode. The cyclic voltammetry (CV) curves were obtained at a scan rate of 50 mV s−1 in 0.1 M KOH, in which N2 or O2 was bubbled beforehand to saturate the electrolyte. The linear sweep voltammetry (LSV) tests were carried out in O2-saturated 0.1 M KOH with different speed rates (400, 625, 900, 1225, and 1600 rpm) at a scan rate of 5 mV s−1. The electron transfer number n has been calculated at different potentials from the Koutecky–Levich (K–L) plots (j−1vs. ω−1/2), which is based on the following equations:| 1/J = 1/JL + 1/JK = 1/Bω0.5 + 1/JK | (1) |
| B = 0.62nFC0D02/3ν−1/6 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C; C0 and D0 are the O2 concentration and diffusion coefficient in the electrolyte, respectively; and ν is the kinematic viscosity of the electrolyte.
485C; C0 and D0 are the O2 concentration and diffusion coefficient in the electrolyte, respectively; and ν is the kinematic viscosity of the electrolyte.
The ECSA values were evaluated from the double-layer capacitance (Cdl), which was estimated by plotting the Δj (ja − jc) at 1.19V vs. RHE against the scan rates of 5, 10, 15, 20, and 25 mV s−1. The slope is twice the double-layer capacitance Cdl. The ORR durability was tested via the chronoamperometric method at 0.6 V in O2-saturated 0.1 M KOH (ω = 1600 rpm). Then, 3 mL of methanol was added to the solution when the test time reached about 10000 s to evaluate the crossover effect.
The OER and HER measurements were performed in an O2- or H2-saturated 1 M KOH solution, where carbon paper and SCE were employed as the counter and reference electrodes, respectively. The working electrode was prepared as follows: 2.0 mg catalyst was dispersed in a mixed solution of 20 μL Nafion (DuPont, 5 wt%) and 1.98 mL DMF under ultrasonication for 30 min; then, 200 μL of the catalyst ink was coated onto the Ni foam (1 × 1 cm2) electrode and dried. The loading of the catalysts was controlled at 0.2 mg cm−2. To verify the OER and HER stability of the Co/CoP–HNC catalyst, the working electrodes were evaluated with a constant current density of 10 mA cm−2 for 10 h.
The overall water splitting evaluation of Co/CoP–HNC was measured in 1 M KOH using a two-electrode system. The procedure for the preparation of electrodes was the same as that in the OER and HER test, except that the loading amount was 1 mg cm−2. The stability tests were carried out at 1.68 V for continuous water electrolysis.
A Zn–air battery was assembled with a polished zinc plate as an anode and a mixed solution of 6 M KOH and 0.2 M Zn(Ac)2 as an electrolyte. To prepare the air-cathode, the catalyst was mixed with Ketjenblack and a polytetrafluoroethylene (PTFE) binder (60 wt% emulsion, Aladdin) with a weight ratio of 10:3:5 and dispersed in a DMF solution under ultrasonication for 30 min. Then, the catalyst ink was dropped on a Ni foam and dried in air. The cycling performance of the rechargeable Zn–air battery was tested at a constant current of 5 mA cm−2.
Results and discussion
The synthetic strategy for the hairy Co/CoP–HNC catalyst involves three steps, as illustrated in Fig. 1a. The synthesis starts from purple ZIF-67 particles, which have a polyhedron morphology, that are obtained by reacting cobalt ions with 2-methylimidazole (2-MIM) in methanol (Fig. S1a and S2a, ESI†). The zeolitic imidazolate frameworks (ZIF-67) are chosen due to their uniform pore size, well-defined morphology, and abundant C, Co, and N atoms in the structure. | ||
| Fig. 1 Schematic and morphology of the Co/CoP–HNC catalyst. (a) Schematic of the synthesis of the tri-functional Co/CoP–HNC catalyst. (b) SEM image of the Co/CoP–HNC catalyst, (c) regional SEM image to show the hollow structure, (d) TEM image, and (e and f) HRTEM images of the Co/CoP–HNC catalyst. | ||
After subjection to high temperatures with dicyandiamide under an Ar gas flow, the polyhedral shape of the ZIF-67 is maintained, whereas the surface becomes rough with many tiny CNTs (Fig. S1b and S2b, ESI†). Transmission electron microscopy (TEM) was further used to investigate the microstructure of the catalyst, which showed hollow structures composed of CNTs with Co nanoparticles encapsulated on the tips with carbon shells (Fig. S1c, ESI†). The TEM image also reveals Co nanoparticles on the sides of the carbon shells.
The third step features a solid/gas-phase reaction at 320 °C to transform unwrapped Co nanoparticles to CoP. The reactant gas PH3 is generated from thermal decomposition of NaH2PO2·H2O and then reacted with Co–HNC to form the final tri-functional catalyst of Co/CoP–HNC. The morphology and microstructure of the multifunctional catalyst were observed via SEM and TEM, as shown in Fig. 1b–f. The Co/CoP–HNC catalyst is fuzzy particles composed of carbon polyhedrons with carbon nanotubes on the surface (Fig. 1b). From the specifically selected particle with cracks (Fig. 1c), we can see the hollow structure inside the carbon polyhedrons and many short carbon nanotubes on the surface. In contrast, the carbon polyhedrons (Co/CoP–NC) obtained without introducing dicyandiamide at high temperatures are shown in Fig. S3a and c (ESI†): the surface of the catalyst is quite smooth without carbon tubes. The result shows the importance of small organic molecules for the growth of carbon nanotubes. Moreover, the intermediate material (Co–HNC pre) was washed with acid after thermal pyrolysis without phosphorization (Fig. S3b and d, ESI†). The Co–HNC particles show few carbon nanotubes, which may be due to the removal of carbon nanotubes that are connected by Co nanoparticles. In the TEM image of Co/CoP–HNC (Fig. 1d), we can see that the carbon polyhedrons have a hollow structure with CNTs interwoven together forming hairy walls. The carbon nanotubes show a bamboo-like structure, as shown in the upper right corner of the image, which is the typical structure of N-doped CNTs. There are two different lattice fringes observed via the high-resolution TEM (HRTEM). Fig. 1e shows a nanoparticle wrapped with several graphene layers with the d spacing of 2.05 Å, which can be assigned to the Co(111) crystallographic planes. Moreover, as shown in Fig. 1f, there is a lattice spacing of 2.84 Å corresponding to CoP(011) next to carbon; the existence of CoP beside carbon other than that in the coating demonstrates that CoP originates from Co nanoparticles that unwrap with carbon in the second step.
The XRD patterns of the catalyst are shown in Fig. 2a. The diffraction peaks at 44.3° were assigned to Co (PDF# 15-0806) and those at 48.0° were assigned to CoP (PDF# 29-0497). These results, combined with TEM analysis results of the Co/CoP–HNC catalyst, show that the metallic Co exists as nanoparticles inside the carbon nanotubes, wheres CoP exists as nanoparticles on the surface of the catalyst. The surface composition of the catalyst was further examined by SEM-EDS. All the elements Co, P, C, and N were detected in the EDS mapping (Fig. S4, ESI†). To gain further insights into the chemical environment and bonding configuration of the Co/CoP–HNC catalyst, X-ray photoelectron spectroscopy (XPS) was performed on the product. Fig. 2b shows the high-resolution XPS profile of N 1s. The fitted peaks show three types of N species: pyridinic N (398.7 eV), pyrrolic N (400.0 eV), and graphic N (401.1 eV), with a total percentage of N at approximately 7.8% for the catalyst.26,27 The presence of deconvoluted peaks at 129.6 and 130.2 eV in the P 2p XPS profile (Fig. 2c) suggests successful phosphidation of cobalt by forming CoP, whereas a single peak at 133.5 eV represents P–O binding.23,26–28 From Fig. 2d, the Co 2p3/2 can be divided into metallic Co, Co–P, and Co–POx peaks. The appearance of the high oxidation P and Co state may be due to inevitable surface oxidation upon exposure to air, which is often observed for metal phosphides.23,29,30
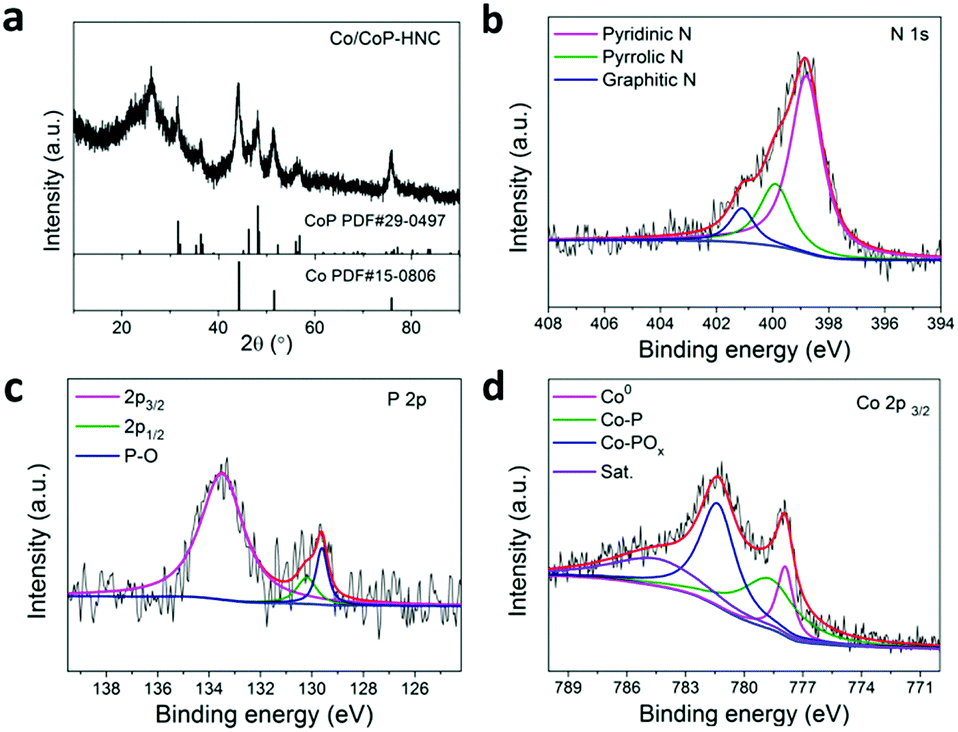 | ||
| Fig. 2 Structural and chemical analysis of the Co/CoP–HNC catalyst. (a) XRD pattern, (b) N 1s XPS spectrum, (c) P 2p XPS spectrum, and (d) Co 2p3/2 XPS spectrum of the tri-functional Co/CoP–HNC catalyst. | ||
The electrocatalytic performance of the catalysts for ORR was measured in a 0.1 M KOH solution with the standard three-electrode configuration. The CV curves in the N2 and O2-saturated electrolyte were obtained and are shown in Fig. 3a. The Co/CoP–HNC catalyst shows an ORR onset potential of 0.94 V and a half wave potential of 0.83 V vs. the reversible hydrogen electrode (RHE). The linear scan voltammogram (LSV) curves with iR correction for different electrocatalysts were obtained at 5 mV s−1 and compared with those of the commercial 20 wt% Pt/C catalyst. As shown in Fig. 3b, although the onset potential of Co/CoP–HNC is 20 mV lower than that of Pt/C (0.96 V), the half wave potential is 23 mV more positive than that of the Pt/C catalyst. Considering the comparative peak current density in LSV, the Co/CoP–HNC actually shows similar or even better ORR activity than the benchmark Pt/C catalyst. In sharp contrast, the Co/CoP–NC shows the lowest ORR activity; this fully demonstrates the importance of a hierarchical structure in the catalyst. The Co–HNC has LSV curves similar to those of Pt/C; this shows that the Co nanoparticles embedded in CNTs are the main contributor to ORR activity; this has also been reported in other studies.31 The Co/CoP–HNC catalysts with different pyrolysis temperatures in the second synthesis step were also compared, and 700 °C was optimized as the best temperature regarding the ORR performance (Fig. S5, ESI†).
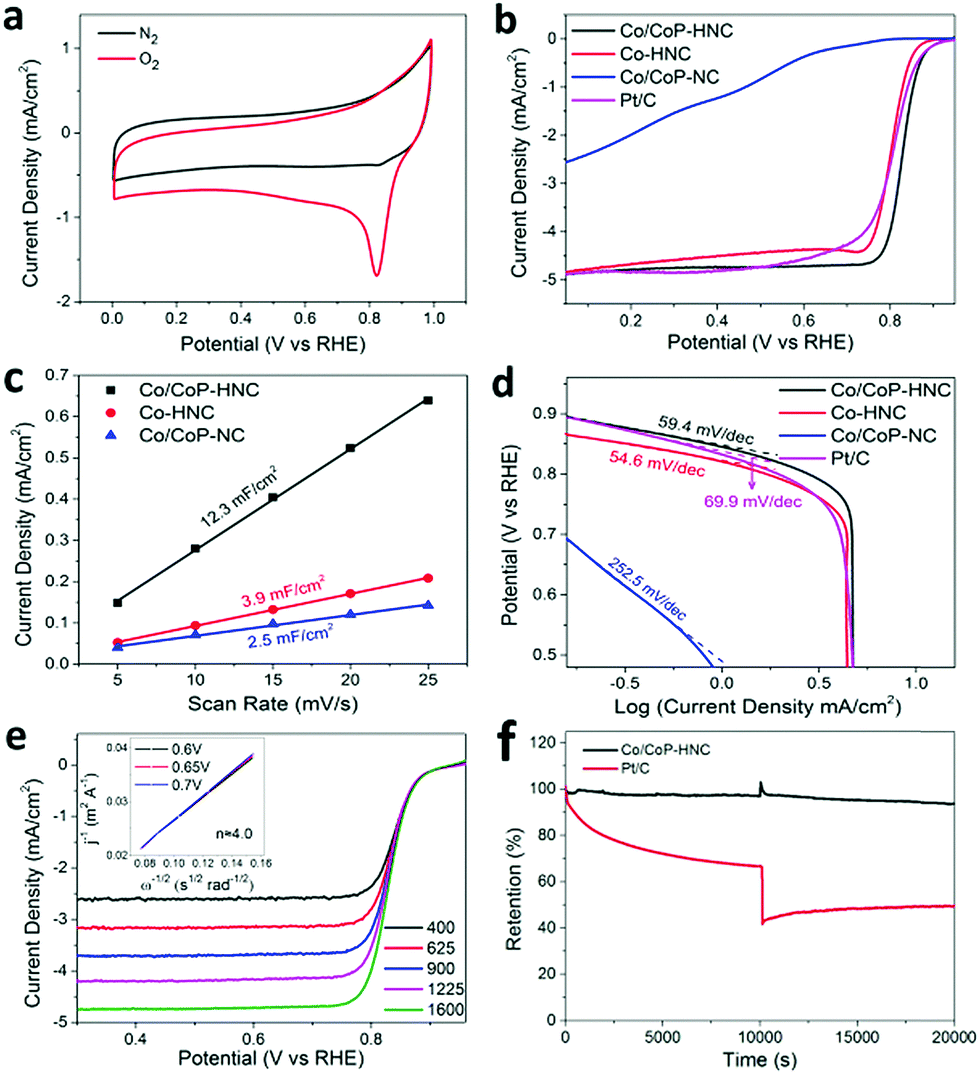 | ||
| Fig. 3 The ORR performance in a 0.1 M KOH solution. (a) CV plots of Co/CoP–HNC in the N2 and O2-saturated electrolytes. (b) LSV plots of electrocatalysts compared with those of Pt/C. (c) Cdl stands for ECSA of the three electrocatalysts. (d) Tafel plots of the three catalysts compared with those of Pt/C. (e) LSV plots at different rotating speeds and K–L plots (insert) of Co/CoP–HNC. (f) The stability and methanol tolerance tests for Co/CoP–HNC at 0.6 V (vs. RHE) compared with those of Pt/C. | ||
Electrochemical surface area (ECSA), an important indicator of catalytic activity reflecting the amount of catalyst in contact with the electrolyte, was obtained by measuring the CV curves with various scan rates (Fig. 3c and Fig. S6, ESI†). The double-layer capacitances of the Co/CoP–NC, Co–HNC, and Co/CoP–HNC catalysts were calculated to be 2.5, 3.9, and 12.3 mF cm−2, respectively.28,32 This can be attributed to different microstructures in the catalysts, as reflected in the SEM images. In Co/CoP–HNC, the formation of interwoven CNTs and a rough surface on the carbon polyhedron increases the electrochemical surface (Fig. 1b–d). After washing with acid, some CNTs drop off the surface of Co–HNC and the catalyst surface becomes much smoother and thus reduces the ECSA (Fig. S3d and d, ESI†). Co/CoP–NC has the smallest surface area possibly due to the smooth surface without the CNT growth and many particles wrapped in the carbon polyhedron (Fig. S3a and c, ESI†). As shown in Fig. 3d, Co/CoP–HNC, Co–HNC, Co/CoP–NC, and Pt/C have the Tafel slopes of 59.4 mV dec−1, 54.6 mV dec−1, 252.5 mV dec−1, and 69.9 mV dec−1, respectively. The better ORR activity of Co/CoP–HNC as compared to that of Pt/C is further confirmed by the smaller Tafel slope for the faster ORR kinetics.33,34
The ORR performances of the Co/CoP–HNC were further evaluated using polarization curves obtained at a rotating disk electrode (RDE). The corresponding Koutecky–Levich (K–L) plots (Fig. 3e) suggest that the electron transfer number is 4.0, with a completely 4e− ORR pathway.35–37 The Co/CoP–HNC retains 93.9% of the initial current density after 20000 s of continuous testing (Fig. 3f); this indicates an excellent ORR catalytic stability. Superior methanol tolerance was also identified while introducing 3 mL of methanol into 75 mL of an O2-saturated 0.1 M KOH solution during the stability test as the Pt/C suffered a dramatic loss in the current, whereas Co/CoP–HNC demonstrated a stable catalytic activity. The results demonstrate that Co/CoP–HNC has great potential as a catalyst in cathode electrodes for fuel cells or metal–air batteries.38,39
The OER properties of Co/CoP–HNC, Co/CoP–NC, and Co–HNC were also determined in a 1.0 KOH solution and compared with those of the commercial Pt/C and IrO2/C. As shown in Fig. 4a, the Co/CoP–HNC shows highest OER activity with only 1.53 V to drive a current density of 10 mA cm−2, which is even comparative to that of the benchmark IrO2/C catalyst. When the current densities increased to 100 mA cm−2, the potentials were 1.58 V, 1.62 V, 1.60 V, 1.69 V, and 1.60 V for Co/CoP–HNC, Co/CoP–NC, Co–HNC, Pt/C, and IrO2/C, respectively. Noteworthy, the Co/CoP–HNC outperformed the IrO2/C catalyst and other carbon polyhedrons in terms of both onset potential and high current densities. To better evaluate the OER activity of Co/CoP–HNC, the catalyst was further investigated using an RDE electrode in an oxygen-saturated 0.1 M KOH electrolyte (Fig. S7, ESI†). The onset potential was 1.54 V for the Co/CoP–HNC, and oxygen bubbles could be seen on the electrode surface after 400 s at 1.56 V (Fig. S7b, ESI†). As shown in Fig. 4c, the Co/CoP–HNC exhibited a much lower Tafel slope (44.2 mV dec−1) as compared to Co/CoP–NC (53.6 mV dec−1), Co–HNC (52.0 mV dec−1), Pt/C (59.6 mV dec−1), and IrO2/C (51.3 mV dec−1). A lower Tafel slope implies faster current density increase with the applied potential, usually suggesting better OER kinetics.40–42 The improved OER performance may originate from the hierarchical structure of CNT-penetrated hollow carbon polyhedron and the formation of metal phosphide particles as CoP is regarded as an efficient OER catalyst in a KOH solution. During the OER reaction, the CoOx formed on the outer surface layer of CoP is responsible for the OER activity.43,44 Owing to the fact that the Co/CoP–NC (1.60 V for 100 mA cm−2) has lower overpotential than Co–HNC (1.62 V for 100 mA cm−2) while having higher Tafel slope (53.6 mV dec−1 for Co/CoP–NC vs. 52.0 mV dec−1 for Co–HNC), we tend to believe that the CoP component increases the activity toward OER, whereas the hierarchical structure further facilitates electrolyte percolation and increases the active interfaces. In the electrochemical impedance spectroscopy (EIS) spectra (Fig. S8a, ESI†), the Co/CoP–HNC catalyst has the lowest impedance indicating the most efficient charge transfer, whereas Co/CoP–NC has larger impedance and Co–HNC has the largest impedance.45,46 The results further confirm that the formed CoP is the dominant active component of the catalyst for the OER. The influence of pyrolysis conditions on OER activity during the second step was also investigated, and 700 °C was optimized as the best temperature for OER performance (Fig. S8c, ESI†). Besides high OER activity, the Co/CoP–HNC also features excellent stability, as revealed by 10 h electrolysis at a controlled constant current of 10 mA cm−2 (Fig. 4e).
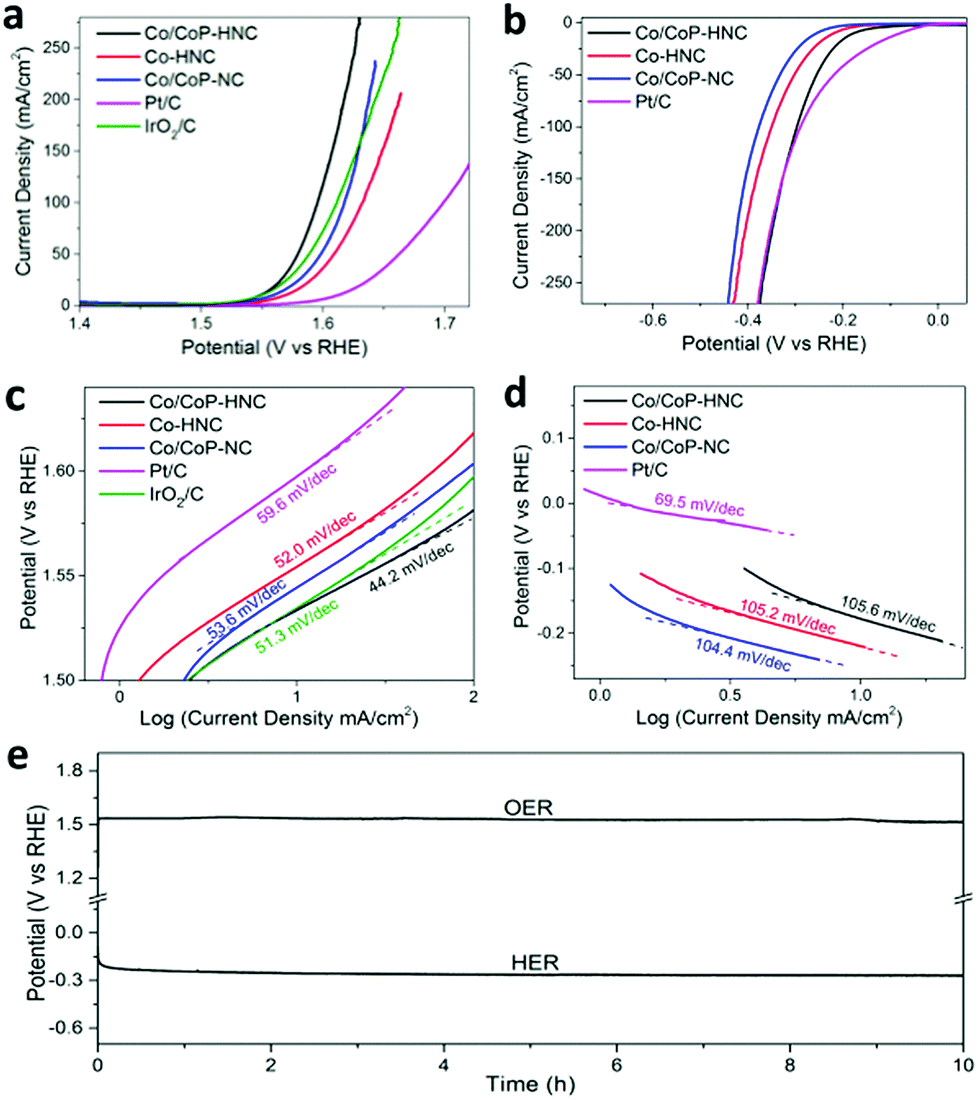 | ||
| Fig. 4 The OER and HER performance in a 1 M KOH solution. (a) The OER performance in an O2-saturated 1 M KOH solution and (c) the corresponding Tafel plots. (b) The HER performance in a H2-saturated 1 M KOH solution, and (d) the corresponding Tafel plots. (e) The OER and HER stability of Co/CoP–HNC under a constant current density of 10 mA cm−2 for 10 h. | ||
The HER activity of the catalysts was also examined in a H2 saturated 1.0 M KOH solution. The LSV curves were obtained alongside with those of the commercial Pt/C catalyst (Fig. 4b). The Co/CoP–HNC catalyst needs a 0.18 V overpotential to drive a current density of 10 mA cm−2, which is better than Co–HNC (0.22 V) and Co/CoP–NC (0.26 V) although inferior to Pt/C (0.04 V). When the current densities increase to 100 mA cm−2, the overpotentials are 0.30 V for Co/CoP–HNC, 0.35 V for Co–HNC, 0.38 V for Co/CoP–NC, and 0.29 V for Pt/C. The Co/CoP–HNC has lower overpotential than Co–HNC; this evidences the great contribution of CoP towards the HER activity; on the other hand, Co–HNC has better HER activity than Co/CoP–NC; this shows the effect of the hierarchical structure. Although HER performance of Co–HNC is explained by modulation of electron density and electronic potential distribution at the carbon layer,47,48 in our study, CoP plays a more important role in the HER activity since only a part of the Co particles have been converted into CoP in the Co/CoP–HNC catalyst, and they have higher HER activity than the original Co–HNC. In Fig. 4d, the corresponding Tafel slopes for carbon polyhedron-based catalysts are 105.6 mV dec−1 for Co/CoP–HNC, 105.2 mV dec−1 for Co–HNC, and 104.4 mV dec−1 for Co/CoP–NC.
The similar Tafel slope values imply similar reaction kinetics, indicating the possible Volmer–Heyrovsky pathway.49–51 In the EIS plots (Fig. S8b, ESI†), Co/CoP–HNC has the lowest impedance as compared to Co–HNC and Co/CoP–NC, which is agreement with the HER catalytic performance and further confirms our speculation that CoP contributes to the intrinsic HER activity, whereas the hairy structure interwoven with CNTs can facilitate charge transfer.52,53 Moreover, 700 °C was optimized as the best pyrolysis temperature for the HER activity (Fig. S8d, ESI†). The HER catalytic stability of Co/CoP–HNC is verified by water electrolysis at a current density of 10 mA cm−2 for 10 h, as shown in Fig. 4e.
Finally, the potential of Co/CoP–HNC to serve as a bifunctional catalyst for overall water splitting or metal–air battery was investigated by assembling a two-electrode electrolyzer and a home-made Zn–air battery (with a catalyst loading of 1 mg cm−2). For the overall water-splitting test, the Co/CoP–HNC was loaded onto nickel foam and employed as an electrocatalyst for both the anode and the cathode electrode. As shown in Fig. 5a, the electrolyzer with Co/CoP–HNC electrodes just needs 1.68 V to drive a current of 10 mA cm−2. When 1.68 V was used on the system as a constant potential (Fig. 5b), the current increased in the first 60 minutes, and afterward, it tended to be stable for the last 240 min; this illustrated extraordinary stability. For the rechargeable Zn–air battery test under ambient conditions (Fig. 5c), a mixed solution of 6 M KOH and 0.2 M Zn(Ac)2 was applied as the electrolyte to form zincate (Zn(OH)42−) to ensure reversible Zn electrochemical reactions at the anode,54 and the Co/CoP–HNC tri-functional catalyst or commercial Pt/C was loaded onto the nickel foam as the air electrode. In the first cycle, the charge–discharge voltage gap of Co/CoP–HNC was just about 0.78 V, and the round-trip efficiency could reach about 61%. After 100 cycles, the Zn–air battery with the Co/CoP–HNC catalyst showed high stability with a voltage gap of 0.96 V, whereas the control Zn–air battery with the Pt/C catalyst suffered a serious decline in the discharge potential after 20 cycles. The results further confirmed the great potential of Co/CoP–HNC in energy-related applications such as in water splitting and metal–air batteries.
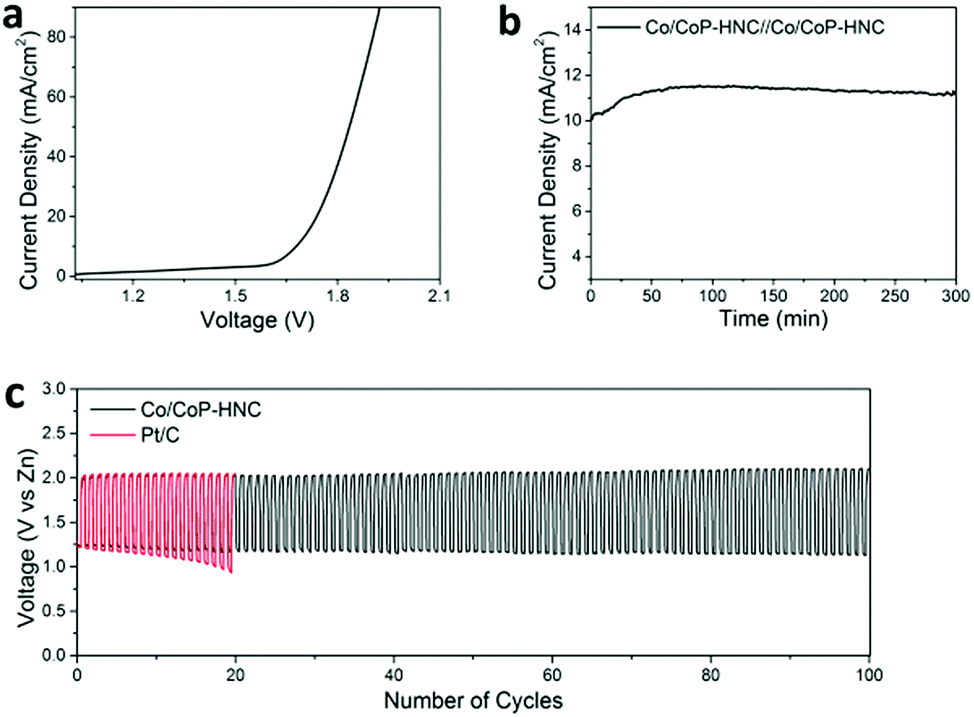 | ||
| Fig. 5 Application of the trifunctional Co/CoP–HNC catalyst. (a) In the overall water splitting, Co/CoP–HNC was used as both the cathode and anode in a 1 M KOH solution without iR correction. (b) Stability test of the Co/CoP–HNC electrodes for water splitting operated at 1.68 V for 300 min. (c) Cycling performance of the rechargeable zinc–air battery with the Co/CoP–HNC and Pt/C catalysts in air electrodes at 5 mA cm−2. The loading for all the catalysts was 1 mg cm−2. | ||
Conclusions
In conclusion, a novel strategy to prepare a tri-functional catalyst assembled with Co/CoP nanoparticles, N-doped carbon nanotubes, and hollow polyhedral carbon was successfully developed. In this catalyst, the metallic Co particles embedded inside N-CNT can provide ORR activity, whereas CoP on the side wall of the HNC offers high catalytic activity towards the OER and HER. Additionally, the interwoven N-CNT network not only enhances the interfacial contact between the Co/CoP–HNC catalyst and the electrolyte but also affords long range conductivity. As compared to that of other MOF-derived materials, the rational design of the hairy structure and the functional components in Co/CoP–HNC improves both the charge transfer efficiency and active sites for electrocatalysis. Due to these advantageous features, Co/CoP–HNC exhibits excellent tri-functional catalytic activity for the ORR, OER, and HER in an alkaline solution; this indicates its great potential in fuel cells, water splitting, and metal–air batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC), the National Key Research and Development Project (2016YFF0204402), the Program for Changjiang Scholars and Innovative Research Team in the University (IRT1205), the Fundamental Research Funds for the Central Universities, and the Long-Term Subsidy Mechanism from the Ministry of Finance and the Ministry of Education of PRC.Notes and references
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 1606459 CrossRef PubMed.
- L. Yu, H. Hu, H. B. Wu and X. W. Lou, Adv. Mater., 2017, 29, 1604563 CrossRef PubMed.
- M. Kuang and G. Zheng, Small, 2016, 12, 5656–5675 CrossRef CAS PubMed.
- G. Fu, Z. Cui, Y. Chen, Y. Li, Y. Tang and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1601172 CrossRef.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29, 1604103 CrossRef PubMed.
- Y. P. Zhu, C. Guo, Y. Zheng and S. Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- J. Yin, Y. Li, F. Lv, Q. Fan, Y. Q. Zhao, Q. Zhang, W. Wang, F. Cheng, P. Xi and S. Guo, ACS Nano, 2017, 11, 2275–2283 CrossRef CAS PubMed.
- J. Masa, W. Xia, I. Sinev, A. Zhao, Z. Sun, S. Grutzke, P. Weide, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2014, 53, 8508–8512 CrossRef CAS PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 610–614 CrossRef CAS PubMed.
- J. Wei, Y. Liang, Y. Hu, B. Kong, J. Zhang, Q. Gu, Y. Tong, X. Wang, S. P. Jiang and H. Wang, Angew. Chem., Int. Ed., 2016, 55, 12470–12474 CrossRef CAS PubMed.
- B. Li, J. Quan, A. Loh, J. Chai, Y. Chen, C. Tan, X. Ge, T. S. Hor, Z. Liu, H. Zhang and Y. Zong, Nano Lett., 2017, 17, 156–163 CrossRef CAS PubMed.
- Z. F. Huang, J. Wang, Y. Peng, C. Y. Jung, A. Fisher and X. Wang, Adv. Energy Mater., 2017, 1500544 Search PubMed.
- Z. Wang, Y. Lu, Y. Yan, T. Y. P. Larissa, X. Zhang, D. Wuu, H. Zhang, Y. Yang and X. Wang, Nano Energy, 2016, 30, 368–378 CrossRef CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- B. You, N. Jiang, M. Sheng, S. Gul, J. Yano and Y. Sun, Chem. Mater., 2015, 27, 7636–7642 CrossRef CAS.
- J. Song, C. Zhu, B. Z. Xu, S. Fu, M. H. Engelhard, R. Ye, D. Du, S. P. Beckman and Y. Lin, Adv. Energy Mater., 2017, 7, 1601555 CrossRef.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Adv. Energy Mater., 2017, 2017(7), 1602355 CrossRef.
- L. A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 CAS.
- W. Xia, R. Zou, L. An, D. Xia and S. Guo, Energy Environ. Sci., 2015, 8, 568–576 CAS.
- Z. Li, M. Shao, L. Zhou, R. Zhang, C. Zhang, M. Wei, D. G. Evans and X. Duan, Adv. Mater., 2016, 28, 2337–2344 CrossRef CAS PubMed.
- H. Tabassum, W. Guo, W. Meng, A. Mahmood, R. Zhao, Q. Wang and R. Zou, Adv. Energy Mater., 2017, 7, 1601671 CrossRef.
- D. Das, A. Das, M. Reghunath and K. K. Nanda, Green Chem., 2017, 19, 1327–1335 RSC.
- C. Tang, R. Zhang, W. Lu, L. He, X. Jiang, A. M. Asiri and X. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef PubMed.
- Y. Hao, Z. Lu, G. Zhang, Z. Chang, L. Luo and X. Sun, Energy Technol., 2017, 5, 1265–1271 CrossRef CAS.
- K. Wan, G.-F. Long, M.-Y. Liu, L. Du, Z.-X. Liang and P. Tsiakaras, Appl. Catal., B, 2015, 165, 566–571 CrossRef CAS.
- G. Zhang, Y. Xu, L. Wang, J. Wang, Y. Kuang and X. Sun, Sci. China Mater., 2015, 58, 534–542 CrossRef CAS.
- J.-S. Lee, G. Nam, J. Sun, S. Higashi, H.-W. Lee, S. Lee, W. Chen, Y. Cui and J. Cho, Adv. Energy Mater., 2016, 6, 1601052 CrossRef.
- H. Wu, J. Wang, G. Wang, F. Cai, Y. Ye, Q. Jiang, S. Sun, S. Miao and X. Bao, Nano Energy, 2016, 30, 801–809 CrossRef CAS.
- Z. Liu, G. Zhang, Z. Lu, X. Jin, Z. Chang and X. Sun, Nano Res., 2013, 6, 293–301 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- Y. Hao, Y. Xu, J. Liu and X. Sun, J. Mater. Chem. A, 2017, 5, 5594–5600 CAS.
- J. Wang, K. Li, H.-X. Zhong, D. Xu, Z.-L. Wang, Z. Jiang, Z.-J. Wu and X.-B. Zhang, Angew. Chem., Int. Ed., 2015, 54, 10530–10534 CrossRef CAS PubMed.
- F. Song and X. L. Hu, Nat. Commun., 2014, 5, 4477 CAS.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- J. Ryu, N. Jung, J. H. Jang, H. J. Kim and S. J. Yoo, ACS Catal., 2015, 5, 4066–4074 CrossRef CAS.
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, ACS Catal., 2015, 5, 6874–6878 CrossRef CAS.
- Y. Pi, Q. Shao, P. Wang, F. Lv, S. Guo, J. Guo and X. Huang, Angew. Chem., Int. Ed., 2017, 56, 4502–4506 CrossRef CAS PubMed.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., 2014, 126, 4461–4465 CrossRef.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- Y. Hou, M. Qiu, G. Nam, M. G. Kim, T. Zhang, K. Liu, X. Zhuang, J. Cho, C. Yuan and X. Feng, Nano Lett., 2017, 17, 4202–4209 CrossRef CAS PubMed.
- Z. Y. Yu, Y. Duan, M. R. Gao, C. C. Lang, Y. R. Zheng and S. H. Yu, Chem. Sci., 2017, 8, 968–973 RSC.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. I. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS PubMed.
- K. Liu, W. Zhang, F. Lei, L. Liang, B. Gu, Y. Sun, B. Ye, W. Ni and Y. Xie, Nano Energy, 2016, 30, 810–817 CrossRef CAS.
- X. Xu, F. Nosheen and X. Wang, Chem. Mater., 2016, 28, 6313–6320 CrossRef CAS.
- Y. Li, M. Guo, Y. Liang, J. Feng, J. E. Kim, H. Wang, G. Hong, B. Zhang. and H. Dai, Nat. Commun., 2013, 4, 1805 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mh00706j |
| This journal is © The Royal Society of Chemistry 2018 |